
Atomically thin cesium lead bromide perovskite quantum wires with high luminescence†
Hongwen
Huang‡
a,
Mei
Liu‡
a,
Jing
Li‡
b,
Laihao
Luo
a,
Jiangtao
Zhao
a,
Zhenlin
Luo
a,
Xiaoping
Wang
*a,
Zhizhen
Ye
b,
Haiping
He
 *b and
Jie
Zeng
*a
*b and
Jie
Zeng
*a
aHefei National Laboratory for Physical Sciences at the Microscale, Hefei Science Center, National Synchrotron Radiation Laboratory & Synergetic Innovation Center of Quantum Information and Quantum Physics, Department of Chemical Physics, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China. E-mail: zengj@ustc.edu.cn; xpwang@ustc.edu.cn
bState Key Laboratory of Silicon Materials, School of Materials Science and Engineering, Zhejiang University, Hangzhou, Zhejiang 310027, P. R. China. E-mail: hphe@zju.edu.cn
First published on 29th November 2016
Abstract
We report a room-temperature colloidal synthesis of few-unit-cell-thick CsPbBr3 QWs with lengths over a hundred nanometers. The surfactant-directed oriented attachment growth mechanism was proposed to explain the formation of such CsPbBr3 QWs. Owing to the strong quantum confinement effect, the photoluminescence (PL) emission peak of few-unit-cell-thick CsPbBr3 QWs blue-shifted to 430 nm. The ensemble PL quantum yield (PLQY) of the few-unit-cell-thick CsPbBr3 QWs increased to 21.13% through a simple heat-treatment process. The improvement of PLQY was ascribed to the reduction of the density of surface trap states and defect states induced by the heat-treatment process. Notably, the dependence of the bandgap on the diameter with different numbers of unit cells was presented for the first time in 1-D CsPbBr3 QWs on the basis of the produced few-unit-cell-thick CsPbBr3 QWs.
Introduction
All-inorganic cesium lead halide perovskite (CsPbX3, X = Cl, Br, I) colloidal nanocrystals have recently emerged as promising candidates for a number of optoelectronic applications, including light-emitting diodes, lasers, and quantum emitters, due to their remarkable optical and electronic properties.1–4 Like other colloidal nanocrystals, represented by cadmium chalcogenide nanocrystals, the optical and electronic properties of colloidal CsPbX3 nanocrystals are highly dependent on their sizes and shapes, especially when the size is smaller than their exciton Bohr diameter.5 Recently, the development of colloidal synthesis has enabled exquisite control of the sizes and shapes of CsPbX3 nanocrystals.6–11 Kovalenko and co-workers pioneered the colloidal synthesis of three-dimensionally confined CsPbX3 nanocubes with tunable sizes ranging from 4 to 15 nm at 140–200 °C.6 Both Alivisatos and Manna groups reported the synthesis of two-dimensional (2-D) CsPbX3 nanoplates with finely controlled thickness of a few unit cells.7–9 Yang and co-workers demonstrated the shape evolution of nanocubes to one-dimensional (1-D) nanowires by extending the reaction time at a high temperature of 150–250 °C.10 By further optimization, colloidal CsPbBr3 nanowires with a diameter of 10 ± 2 nm and a purity of 90% were produced.11 These nanowires are believed to serve as promising building blocks for numerous optoelectronic devices owing to their superior transport properties. However, the relatively large diameters and thus the weak quantum confined interactions may impede their uses as ideal platforms to study a myriad of fundamental properties such as the quantum confinement effect. Very recently, the Manna group further reduced the diameter of 1-D nanowires down to 3.4 ± 0.5 nm, which lies in the strong quantum confinement regime.12 In addition, Yang and co-workers demonstrated a complicated stepwise purification method to obtain CsPbBr3 with diameters of 2.2 ± 0.2 nm.13 However, such production is believed to suffer from low efficiency and complicated operation. Moreover, the growth mechanism of such thin nanowires, together with the in-depth understanding of their optical properties, has barely been lucubrated.1-D semiconductor quantum wires (QWs) have received particular attention not only because of their potential applications for a number of quantum devices, but also due to the unique two-dimensionally quantum confined effect and the resulting distinct fundamental properties with regard to the zero/two-dimensional counterparts.14–16 To date, a number of semiconductor QWs, including SnO2 QWs, InP QWs, and cadmium chalcogenide based QWs, have been reported.17–20 Unfortunately, these semiconductor QWs suffered from very low photoluminescence quantum yields (PLQYs), typically lower than 1%, in contrast to the common highly luminescent nanowires. The low PLQYs were generally caused by the high density of surface trap sites in these semiconductor QWs due to their large surface areas.21,22 Although highly luminescent colloidal semiconductor QWs with greatly improved PLQYs reaching the 20%-level have recently been reported, their practical applications are limited because of the complicated surface passivation process which usually relies on the precisely controlled growth of core–shell heterostructures.23,24 Therefore, it is highly desired to develop a simpler approach to the synthesis of highly luminescent colloidal semiconductor QWs.
Herein, we report a room-temperature colloidal synthesis of few-unit-cell-thick CsPbBr3 QWs with lengths over a hundred nanometers. The surfactant-directed oriented attachment growth mechanism was proposed to explain the formation of such CsPbBr3 QWs. Importantly, we proposed a simple heat-treatment process to greatly increase the PLQY of such QWs to 21.13%. According to PL measurements, the heat-treatment process induced the reduction of the density of surface trap states and defect states, contributing to the improvement of PLQY. Moreover, we presented the size-dependent bandgap of 1-D CsPbBr3 QWs for the first time.
Results and discussion
In a typical synthesis, a Cs–oleate solution was quickly injected into a mixture solution containing lead bromide (PbBr2), oleylamine (OAm), oleic acid (OA), and octadecene (ODE) in a 20 mL vial at room temperature (25 °C) with vigorous stirring. After the reaction had proceeded for 2 h, the product was collected by centrifugation. Fig. 1A and B show the representative high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images of the product, indicating the formation of uniform QWs. Intriguingly, these QWs tend to be self-assembled into bundles of QWs on TEM grids, which was possibly driven by the high surface energy of ultrathin QWs. The lengths of the QWs were estimated to be more than a hundred nanometers. A histogram of the diameter distribution of the QWs was obtained by counting more than 100 QWs, showing the average diameter of 1.5 ± 0.5 nm. Unfortunately, the detailed structural information of the obtained CsPbBr3 QWs could not be directly visualized by TEM, because the ultrathin QWs were highly vulnerable to the intense electron beam. As shown in Fig. S1,† when the magnification was increased (although we had decreased the accelerating voltage to 80 kV from 200 kV), many bright particles appeared along the QWs. It is believed that the bright particles were Pb particles generated through in situ reduction of CsPbBr3 QWs.8,25 The synchrotron radiation X-ray diffraction (XRD) patterns were recorded to pursue a higher signal-to-noise ratio and resolution. As shown in Fig. 1C, the XRD pattern of the product was consistent with that of a CsPbBr3 cubic phase (ICSD 29073), confirming the formation of cubic CsPbBr3 QWs. According to Bragg's law, the interplanar distance d(200) and the lattice constant were determined to be 0.299 and 0.598 nm, respectively, from the {200} peak at 19.9°. The results demonstrate the formed CsPbBr3 QWs with an average diameter of 2–3 unit cells. It is worth noting that all the other diffraction peaks were broader than those of the {200} family. On the basis of the Scherrer equation, a narrower peak means a larger crystallite size, and vice versa. Because the as-prepared QWs have the largest crystallite size along their growth direction, the obtained CsPbBr3 QWs are proposed to grow along the [100] direction. The surface chemical composition of the CsPbBr3 QWs was probed by X-ray photoelectron spectroscopy (XPS), as shown in Fig. 1D. In addition to the elemental Cs, Pb, and Br, the presence of elemental N and O on the surface of CsPbBr3 QWs is probably related to the chemisorbed OAm and OA, which served as surfactants for the synthesis of few-unit-cell-thick CsPbBr3 QWs.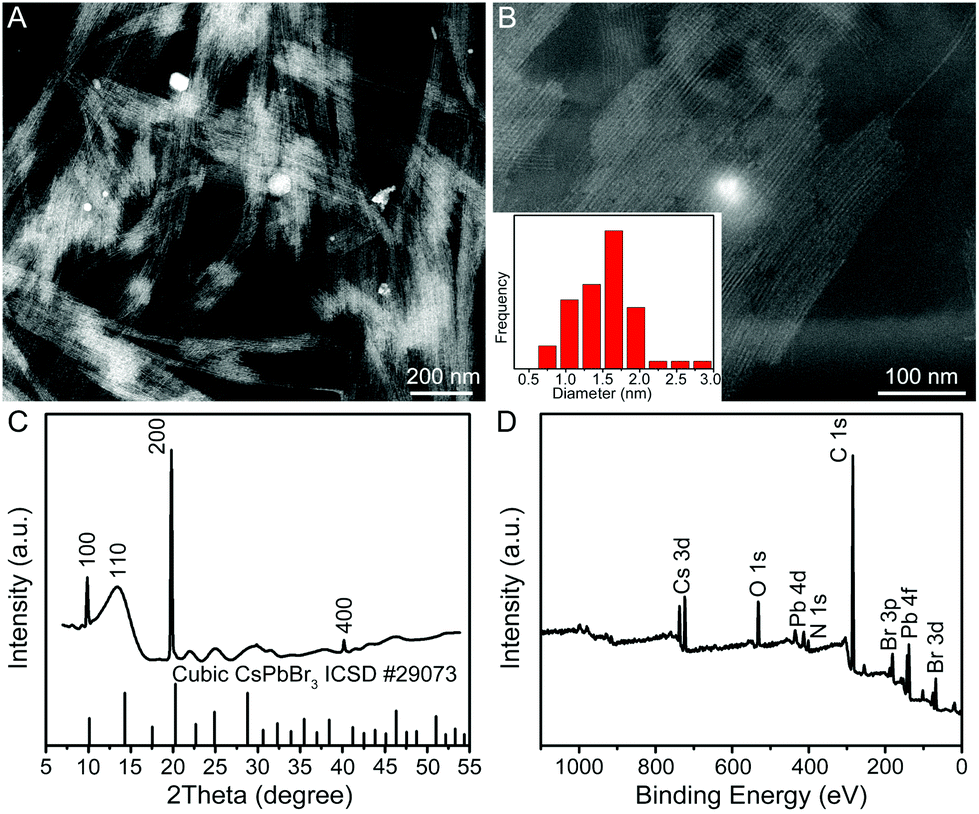 | ||
| Fig. 1 Structural and compositional characterization of the few-unit-cell-thick CsPbBr3 QWs obtained by using the standard procedure. (A, B) Representative HAADF-STEM images with different magnifications, (C) XRD pattern, and (D) XPS survey spectrum. The inset in panel B shows the histogram of diameter distribution for the produced few-unit-cell-thick CsPbBr3 QWs. | ||
In order to figure out the formation process of the CsPbBr3 QWs, we analyzed the products obtained at different reaction stages by PL spectroscopy and TEM. Fig. 2A shows the PL spectra recorded from the time-sequential solutions, which were prepared by diluting the reaction solutions obtained at different reaction times with hexane. From the PL spectra, the intensity of the emission peak located at around 430 nm increased with the reaction time. Given that the intensity of the peak directly reflects the concentration of the produced CsPbBr3, the reaction was almost completed within 2 h. In addition, the positions of emission peaks were barely changed, implying the identical size of the CsPbBr3 nanocrystals produced at different reaction times. The morphology evolution was also probed by TEM, as shown in Fig. 2B–D. At the reaction time of 10 min, a mixture of quantum rods and quantum dots was observed. When the reaction time was prolonged to 60 min, the quantum rods and quantum dots evolved into the QWs. The morphology of the produced QWs was retained after 180 min. In view of these observations, CsPbBr3 QWs were found to be formed through oriented attachment. Specifically, the quantum dots that had been firstly produced at an early reaction stage were subsequently assembled and recrystallized into QWs along a specific orientation.26,27
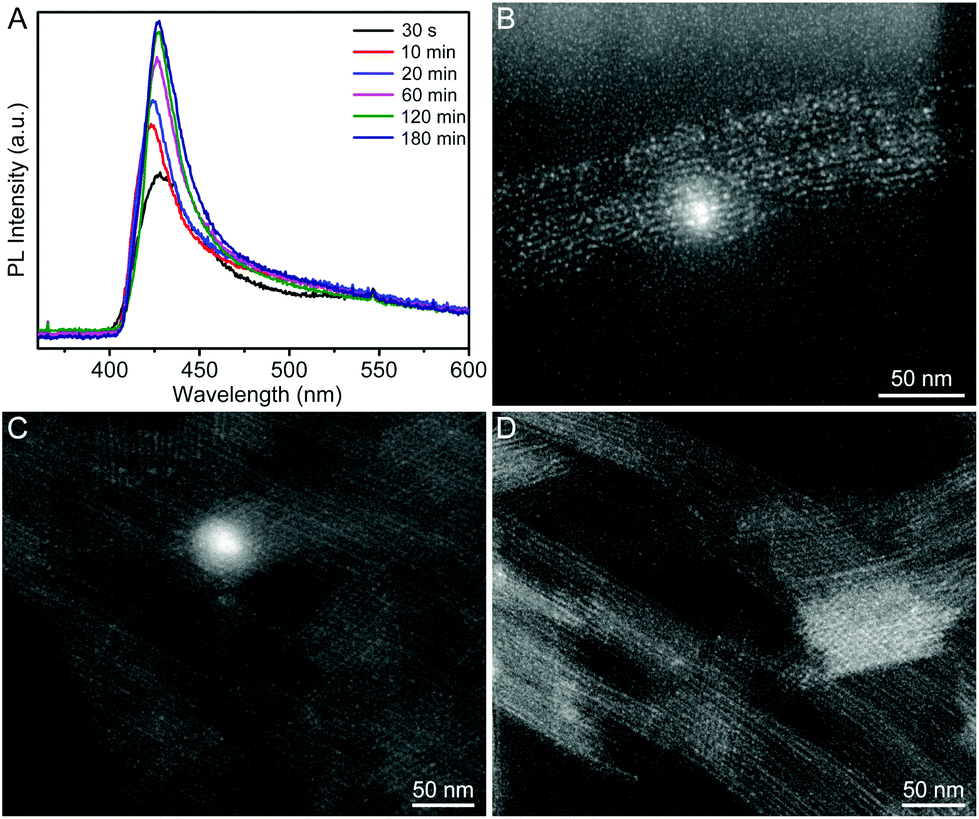 | ||
| Fig. 2 (A) Time-sequential PL spectra recorded from the product solutions, which were prepared by diluting the reaction solutions obtained at different reaction times with hexane. (B–D) The HAADF-STEM images of the products obtained by using the standard procedure except for the different reaction times: (B) 10, (C) 60, and (D) 180 min, respectively. | ||
To comprehensively understand the formation mechanism of CsPbBr3 QWs, we further elucidated the effects of surfactants and reaction temperature. By halving the amount of OAm with other conditions being unchanged, a mixture of nanowires and nanoplates was produced, as shown in Fig. S2A.† When the amount of OAm was doubled, CsPbBr3 QWs were generated (Fig. S2B†). Similarly, the amount of OA was also varied to study its influence on the morphology of products. As shown in Fig. S2C and D,† CsPbBr3 QWs with different lengths were obtained by changing the amount of OA. These results testify that OAm and OA indeed play important roles in controlling the morphology and size of the products. Combining the proposed oriented attachment mechanism and the effects of surfactants, we further hypothesize that the OAm and OA in the solution may work by forming a soft template with a 1D structure to drive the 1D oriented attachment growth of CsPbBr3 QWs. By increasing the reaction temperature from room temperature to 60 °C, the morphology of the products shifted from QWs to ultrathin nanoplates (Fig. S3A†). Continually increasing the reaction temperature to 150 °C led to the generation of CsPbBr3 nanocubes (Fig. S3B†). The results suggest that the reaction temperature is crucial to the morphology of the produced CsPbBr3 nanocrystals, which may be due to the different reaction kinetics at different reaction temperatures.
The optical properties of few-unit-cell-thick CsPbBr3 QWs obtained by using the standard procedure were evaluated by using ultraviolet-visible (UV-vis) absorption and PL emission spectra (black lines), as shown in Fig. 3A. The absorption spectrum shows a distinct exciton absorption peak at around 420 nm. The PL spectrum shows a peak at 430 nm, exhibiting a significant blueshift compared with that in most cases of previously reported CsPbBr3 nanocrystals.6–11 However, the emission is rather weak with a low PLQY of less than 1%. Interestingly, it was found that post-growth heat treatment at 120 °C for 2 min could result in an obvious redshift of the emission to 465 nm, and more significantly, improve the PLQY greatly to 21.13% (see Fig. S4† for details). As illustrated by the photograph in Fig. 3A, the 120 °C-treated QW solution emits a bright cyan color under excitation at 325 nm by using a UV lamp. Such a high PLQY exceeds most of the reported values of colloidal semiconductor QWs.21–24 In general, the PLQY of colloidal QWs is less than 1%, because the large surface area increases the probability of locating nonradiative centers for the excitons and charge carriers. Therefore, additional surface passivation or a core–shell heterostructure is necessary to improve the PLQY to the 20%-level, as demonstrated by Liu and Groeneveld et al.23,24 In this regard, it is significant that our high PLQY is achieved simply by a thermal treatment process at a relatively low temperature for a short period of time.
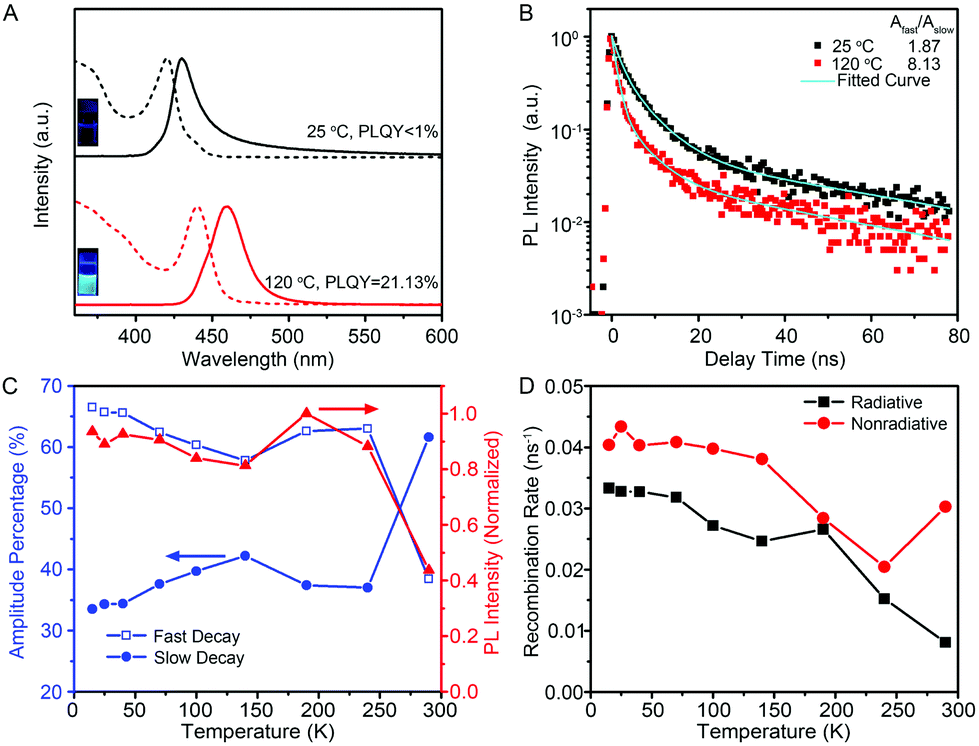 | ||
| Fig. 3 (A) Room temperature optical absorption (dashed lines) and PL spectra (solid lines), and (B) low temperature (15 K) PL decay spectra recorded for the 444 nm emission from the CsPbBr3 QWs synthesized at 25 °C, without (black line) and with (red line) heat treatment at 120 °C. Inset: Photographs of the colloidal QWs solution under UV light illumination. (C) Temperature dependence of the amplitudes of the fast and slow decay (blue lines) for the 472 nm emission from the 120 °C-treated QWs. The temperature-dependent PL intensity (red line) is also plotted. (D) Temperature dependence of the calculated radiative and nonradiative recombination rate for the 120 °C-treated QWs. | ||
The structural variation of the 120 °C-treated QWs was analyzed in detail. As shown in Fig. S5,† the morphology and crystalline phase of the products after heat treatment at 120 °C for 2 min were barely changed. However, the careful examinations for the histogram of diameter indicate that the average diameter of CsPbBr3 QWs increased from 1.5 ± 0.5 to 3.1 ± 0.6 nm after the heat treatment. The increased diameter of CsPbBr3 QWs could account for the higher PLQY from the perspective of smaller specific surface areas and the redshift of the emission peak from the point of the quantum-size effect. Notably, such 120 °C-treated CsPbBr3 QWs could be stored in hexane solution for more than 7 days at room temperature without significant change. However, the CsPbBr3 QWs seem to be more sensitive to the washing process. As shown in Fig. S6,† CsPbBr3 nanowires with larger diameters were obtained after washing with hexane five times possibly due to the rapid ripening process associated with the loss of surfactants on the surface.
An intrinsic study for the improvement of PLQY was conducted by careful PL measurements. Fig. 3B shows the low-temperature PL decay spectra for the 444 nm emission collected from the untreated CsPbBr3 QWs and 120 °C-treated CsPbBr3 QWs. Both of the decay traces can be described by bi-exponential decay with a fast and a slow component. The amplitude ratios of the fast-to-slow component, Afast/Aslow, were estimated to be 1.87 and 8.13 for untreated and 120 °C-treated QWs, respectively, proving the evident decrease of the slow decay component after heat treatment at 120 °C for 2 min. The connection between the fast/slow decay component and PLQY was further revealed by the temperature-dependent PL and PL decay spectra of 120 °C-treated QWs. As shown in Fig. 3C, the temperature-dependent PL intensity and amplitude percentage of Afast and Aslow for the dominant 472 nm emission from 120 °C-treated QWs were extracted from the temperature-dependent PL (Fig. S7†) and decay spectra (Fig. S8†), respectively. Through combining the temperature-dependent PL intensity and temperature-dependent amplitude percentage results, the amplitude percentage of Afast shows a temperature evolution similar to the PL intensity, whereas Aslow shows a contrary trend. The results indicate that the lower portion of the slow decay component corresponds to a higher PLQY. Intrinsically, the slow decay component is considered as the recombination of excitons trapped on the surface states.28,29 It is thus reasonable to ascribe the improvement of PLQY after a heat-treatment process to the lower density of surface trap states, which may be originated from their increased diameter and/or the improved crystal quality after a heat-treatment process.
As the internal quantum efficiency of PL depends on the contributions of radiative and nonradiative recombination, we therefore extracted radiative (Rr) and nonradiative (Rnr) recombination rates from the temperature-dependent PL intensity and decay to rationalize such a high PLQY for 120 °C-treated QWs.30,31Fig. 3D shows the temperature-dependent Rr and Rnr for the 120 °C-treated QWs, revealing that the nonradiative recombination rate is only slightly larger than the radiative one within the whole temperature range. More significantly, the nonradiative recombination rate shows an increase at T > 240 K, a temperature much higher than those generally observed in other semiconductors for the thermal activation of nonradiative centers.32,33 These intrinsic features contribute to the high PLQY of 120 °C-treated QWs at room temperature.
Because the diameters of the developed few-unit-cell-thick CsPbBr3 QWs are smaller than the Bohr diameter (7 nm for CsPbBr3), it is feasible to uncover the unique 1-D quantum confinement effect for CsPbBr3 colloidal nanocrystals, which has never been reported yet. The low-temperature PL spectra from the few-unit-cell-thick CsPbBr3 QWs were recorded at 15 K to gain detailed optical information, as shown in Fig. 4A. The low-temperature PL spectrum of the few-unit-cell-thick CsPbBr3 QWs obtained by using the standard procedure shows two main sharp peaks located at 425 and 444 nm and a very broad peak with the center around 550 nm, respectively. The appearance of the broad peak could be ascribed to the defect-related emission which also accounts for the lower PLQY of the CsPbBr3 QWs obtained by using the standard procedure.34 By a heat treatment process at 120 °C for 2 min, five discrete peaks at around 425, 444, 459, 472, and 480 nm emerged. Considering the same morphology and crystalline phase of these CsPbBr3 QWs, these discrete emission peaks were reasonably ascribed to the CsPbBr3 QWs with different diameters, which shall vary by an integer number of perovskite unit cells.7 Combining with the histograms of diameters, we thus assigned these successive peaks to the CsPbBr3 QWs with diameters of 2, 3, 4, 5, and 6 unit cells. Fig. 4B shows the comparison of the size-dependent bandgap between 1-D CsPbBr3 QWs and 2-D CsPbBr3 nanoplates recently reported by Bekenstein et al.7 The bandgap of 1-D CsPbBr3 QWs was found to be always larger than that of 2-D CsPbBr3 nanoplates at the same quantum confinement size, presenting the distinct quantum confinement effect.
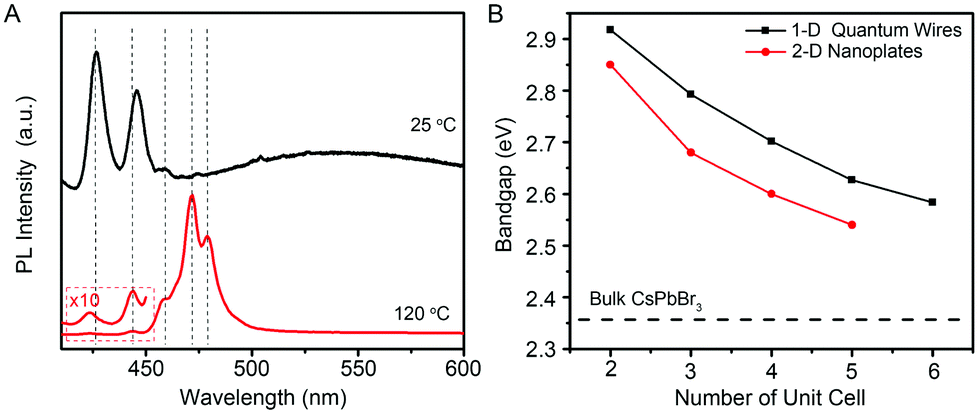 | ||
| Fig. 4 (A) Low-temperature PL spectra of the few-unit-cell-thick CsPbBr3 QWs synthesized at 25 °C, without (black line) and with (red line) heat treatment at 120 °C. The PL spectra were obtained at 15 K. (B) Plot of the band gap energy as a function of the quantum-confined size in unit cell for 1-D CsPbBr3 QWs (black line) and 2-D CsPbBr3 nanoplates (red line). The data for 2-D CsPbBr3 nanoplates are extracted from ref. 7. | ||
Conclusions
In summary, we have developed a colloidal synthesis of few-unit-cell-thick CsPbBr3 QWs at room temperature. The CsPbBr3 QWs were found to be formed through a surfactant-directed oriented attachment mechanism. Importantly, the ensemble PLQY of the few-unit-cell-thick CsPbBr3 QWs could be increased to 21.13% after a simple heat-treatment process. The improvement of PLQY was ascribed to the reduction of the density of surface trap states and defect states because of the heat treatment. On the basis of the developed few-unit-cell-thick CsPbBr3 QWs, we further demonstrated the distinct size-dependent bandgap of 1-D QWs relative to 2-D nanoplates because of the different quantum confinement effect. We believe that the reported few-unit-cell-thick CsPbBr3 QWs not only represent a promising candidate for optoelectronic devices, but also provide an ideal platform for a myriad of fundamental studies.Acknowledgements
This work was supported by the Collaborative Innovation Center of Suzhou Nano Science and Technology, Hefei Science Center CAS (2015HSC-UP016), the China Postdoctoral Science Foundation (2015M580536, 2016T90569), the National Natural Science Foundation of China (21603208, 51372223, 91333203, 11374010, 11434009), and the Fundamental Research Funds for the Central Universities. The authors also thank the staff at beamline BL19U of SSRF for their support.Notes and references
- J. Song, J. Li, X. Li, L. Xu, Y. Dong and H. Zeng, Adv. Mater., 2015, 27, 7162–7167 CrossRef CAS PubMed.
- S. Yakunin, L. Protesescu, F. Krieg, M. I. Bodnarchuk, G. Nedelcu, M. Humer, G. D. Luca, M. Fiebig, W. Heiss and M. V. Kovalenko, Nat. Commun., 2015, 6, 8056 CrossRef CAS PubMed.
- Y. Wang, X. Li, J. Song, L. Xiao, H. Zeng and H. Sun, Adv. Mater., 2015, 27, 7101–7108 CrossRef CAS PubMed.
- Y. Park, S. Guo, N. S. Makarov and V. I. Klimov, ACS Nano, 2015, 9, 10386–10393 CrossRef CAS PubMed.
- X. Peng, L. Manna, W. Yang, J. Wickham, E. Scher, A. Kadavanich and A. P. Alivisatos, Nature, 2000, 404, 59–61 CrossRef CAS PubMed.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- Y. Bekenstein, B. A. Koscher, S. W. Eaton, P. Yang and A. P. Alivisatos, J. Am. Chem. Soc., 2015, 137, 16008–16011 CrossRef CAS PubMed.
- Q. A. Akkerman, S. G. Motti, A. R. S. Kandada, E. Mosconi, V. D'Innoscenzo, G. Bertoni, S. Marras, B. A. Kamino, L. Miranda, F. D. Angelis, A. Petrozza, M. Prato and L. Manna, J. Am. Chem. Soc., 2016, 138, 1010–1016 CrossRef CAS PubMed.
- J. Shamsi, Z. Dang, P. Bianchini, C. Canale, F. D. Stasio, R. Brescia, M. Prato and L. Manna, J. Am. Chem. Soc., 2016, 138, 7240–7243 CrossRef CAS PubMed.
- D. Zhang, S. W. Eaton, Y. Yu, L. Dou and P. Yang, J. Am. Chem. Soc., 2015, 137, 9230–9233 CrossRef CAS PubMed.
- D. Zhang, Y. Yang, Y. Bekenstein, Y. Yu, N. A. Gibson, A. B. Wong, S. W. Eaton, N. Kornienko, Q. Kong, M. Lai, A. P. Alivisatos, S. R. Leone and P. Yang, J. Am. Chem. Soc., 2016, 138, 7236–7239 CrossRef CAS PubMed.
- M. Imran, F. D. Stasio, Z. Dang, C. Canale, A. H. Khan, J. Shamsi, R. Brescia, M. Prato and L. Manna, Chem. Mater., 2016, 28, 6450–6454 CrossRef CAS.
- D. Zhang, Y. Yu, Y. Bekenstein, A. B. Wong, A. P. Alivisatos and P. Yang, J. Am. Chem. Soc., 2016, 138, 13155–13158 CrossRef CAS PubMed.
- A. I. Hochbaum and P. Yang, Chem. Rev., 2010, 110, 527–546 CrossRef CAS PubMed.
- R. Ambigapathy, I. Bar-Joseph, D. Y. Oberli, S. Haacke, M. J. Brasil, F. Reinhardt, E. Kapon, B. Deveaud, A. P. Higginbotham, F. Kuemmeth, T. W. Larsen, M. Fitzpatrick, J. Yao, H. Yan, C. M. Liber and C. M. Marcus, Phys. Rev. Lett., 2014, 112, 216806 CrossRef.
- T. Schumacher, H. Giessen and M. Lippitz, Nano Lett., 2013, 13, 1706–1710 CrossRef CAS PubMed.
- X. Xu, J. Zhuang and X. Wang, J. Am. Chem. Soc., 2008, 130, 12527–12535 CrossRef CAS PubMed.
- H. Yu, J. Li, R. A. Loomis, L. Wang and W. E. Buhro, Nat. Mater., 2003, 2, 517–520 CrossRef CAS PubMed.
- H. Yu, J. Li, R. A. Loomis, P. C. Gibbons, L. Wang and W. E. Buhro, J. Am. Chem. Soc., 2003, 125, 16168–16169 CrossRef CAS PubMed.
- V. L. Wayman, P. J. Morrison, F. Wang, R. Tang, W. E. Buhro and R. A. Loomis, J. Phys. Chem. Lett., 2012, 3, 2627–2632 CrossRef CAS PubMed.
- F. Vietmeyer, P. A. Frantsuzov, B. Janko and M. Kuno, Phys. Rev. B: Condens. Matter, 2011, 83, 115319 CrossRef.
- V. Protasenko, S. Gordeyev and M. Kuno, J. Am. Chem. Soc., 2007, 129, 13160–13171 CrossRef CAS PubMed.
- Y. Liu, F. Wang, J. Hoy, V. L. Wayman, L. K. Steinberg, R. A. Loomis and W. E. Buhro, J. Am. Chem. Soc., 2012, 134, 18797–18803 CrossRef CAS PubMed.
- E. Groeneveld, S. van Berkum, M. M. van Schooneveld, A. Gloter, J. D. Meeldijk, D. J. van den Heuvel, H. C. Gerritsen and C. de Mello Donega, Nano Lett., 2012, 12, 749–757 CrossRef CAS PubMed.
- L. Dou, A. B. Wong, Y. Yu, M. Lai, N. Kornienko, S. W. Eaton, A. Fu, C. G. Bischak, J. Ma, T. Ding, N. S. Ginsberg, L. Wang, A. P. Alivisatos and P. Yang, Science, 2015, 349, 1518 CrossRef CAS PubMed.
- H. Liao, L. Cui, S. Whitelam and H. Zheng, Science, 2012, 336, 1011–1014 CrossRef CAS PubMed.
- D. Li, M. H. Nielsen, J. R. I. Lee, C. Frandsen, J. F. Banfield and J. J. De Yoreo, Science, 2012, 336, 1014–1018 CrossRef CAS PubMed.
- X. Wang, L. Qu, J. Zhang, X. Peng and M. Xiao, Nano Lett., 2003, 3, 1103–1106 CrossRef CAS.
- F. Zhang, H. Zhong, C. Chen, X. Wu, X. Hu, H. Huang, J. Han, B. Zou and Y. Dong, ACS Nano, 2015, 9, 4533–4542 CrossRef CAS PubMed.
- K. Okamoto, I. Niki, A. Shvartser, Y. Narukawa, T. Mukai and A. Scherer, Nat. Mater., 2004, 3, 601–605 CrossRef CAS PubMed.
- M. Gurioli, A. Vinattieri, M. Colocci, C. Deparis, J. Massies, G. Neu, A. Bosacchi and S. Franchi, Phys. Rev. B: Condens. Matter, 1991, 44, 3115–3124 CrossRef CAS.
- D. Rosales, T. Bretagnon, B. Gill, A. Kahouli, J. Brault, B. Damilano, J. Massies, M. V. Durnev and A. V. Kavokin, Phys. Rev. B: Condens. Matter, 2013, 88, 125437 CrossRef.
- I. C. Robin, B. Gauron, P. Ferret, C. Tavares, G. Feuillet, Le Si Dang, B. Gayral and J. M. Gérard, Appl. Phys. Lett., 2007, 91, 143120 CrossRef.
- D. Priante, I. Dursun, M. S. Alias, D. Shi, V. A. Melnikov, T. K. Ng, O. F. Mohammed, O. M. Bakr and B. S. Ooi, Appl. Phys. Lett., 2015, 106, 081902 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details. See DOI: 10.1039/c6nr08250e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |