
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergies in lubrication
Andra
Dėdinaitė
 *ab and
Per M.
Claesson
*ab
*ab and
Per M.
Claesson
*ab
aKTH Royal Institute of Technology, School of Chemical Science and Engineering, Department of Chemistry, Division of Surface and Corrosion Science, Drottning Kristinas väg 51, SE-100 44 Stockholm, Sweden. E-mail: andra@kth.se; percl@kth.se
bRISE Research Institute of Sweden, Division of Bioscience and Materials, Stockholm, Sweden
First published on 30th June 2017
Abstract
To slide surfaces against each other with application of a minimum force and minimum wear has been important since ancient times, and it remains equally important today. The use of oil-soluble lubricants is widely spread in technology, whereas living organisms have developed water-soluble lubricants to facilitate sliding motions. In this perspective article we focus on water-based lubrication in the boundary lubrication regime, and particularly lubrication synergies. This focus has, of course, found inspiration from the outstanding lubrication properties of synovial joints. It has ignited significant amount of research, mostly aimed at answering the question: Which molecule is the magic biolubricant? Different research groups have advocated different answers, and the debate has been intensive. In this article we argue that the question in itself is inappropriate. The relevant question is rather the following: How do molecules work in synergy to provide superior lubrication?
 Andra Dėdinaitė | Andra Dėdinaitė is a researcher at the Royal Institute of Technology (KTH) and the Research Institute of Sweden (RISE), Stockholm. She earned her PhD degree in 1999 and was awarded the Akzo Nobel's Nordic Research Prize for the best PhD thesis and scientific activity within Surface and Colloid Chemistry in the Nordic countries 2003. After post-doctoral Marie Curie positions at Unilever Research and Aarhus University she took a research position at KTH, and since then she has focused her research on self-assembly at interfaces, interfacial properties of bio- and biomimetic molecules with particular emphasis on surface forces and lubrication. |
 Per M. Claesson | Per Claesson is a professor in Surface Chemistry at the Royal Institute of Technology, KTH, Stockholm. He is also active at the Research Institute of Sweden (RISE), Stockholm. His scientific interests are focused on surface phenomena, including surface forces and friction, local nanomechanical properties and corrosion. He has led several large collaborative projects involving academic and industrial partners, such as the competence centre “Surfactants Based on Natural Products” and the program on “Microstructure, Corrosion and Friction”. He has been awarded the Akzo Nobel Science Award in 2013 and the Arrhenius medal in 2008. |
1. Introduction
Liquid water has ever since simple life forms appeared on the earth about 3.7 billion years ago been the medium in which the chemistry of life takes place. Thus, even though water from an engineering perspective is a poor lubricant fluid due to low viscosity and low load bearing capacity at a given sliding velocity, it is the medium used in living organisms for providing smooth sliding between moving parts.1 For instance, the friction coefficient found in synovial joints in mammals can be as low as 0.001 as measured with hip function simulator machines, even though values reported in different studies vary significantly.2 A safe statement seems to be that the friction coefficient is well below 0.01, for instance Gale and co-workers quote the range 0.002–0.006.3 Such easy sliding is required under challenging conditions of shear rates (≤106–107 s−1) and pressures (≤25 MPa).4 The fragile joint structure can under optimal conditions function for ≈100 years, even though malfunction of joints is a critical health issue. For instance, it affects 50 million adults in the US.5 Biolubrication is by no means restricted to synovial joints, and friction control is also essential for, e.g., masticatory, speech and lung functions, and effortless blinking is an important issue for contact lens performance. The low friction is achieved by means of biolubricants and sophisticated nanostructured surfaces that work in synergy.Scientists have searched for an outstanding biolubricant that facilitates the excellent lubrication of synovial joints. Phospholipids, bottle-brush glycoproteins, and linear polysaccharides have all been implicated, and the debate between scientists advocating different opinions6,7 has at times been intense. It is nowadays clear that the key is the presence of a fluid water layer between the sliding surfaces that is maintained by the biolubricants,8 and that synergies between different biolubricants need to be considered in order to capture the essence of biolubrication. For instance, it has been reported that addition of hyaluronan and phospholipids together is more effective in alleviating joint disease symptoms than any of these two components alone.9 In model experiments hyaluronan–phospholipid mixtures have also shown synergies,10,11 and the importance of hyaluronan–phospholipid association structures for repaired articular cartilage surfaces12 and for lubrication of hip implants13 has been stressed. Different aspects of biolubrication and biomimetic lubrication have been the topic of several valuable reviews that we recommend to the interested reader.1,8,14
In this perspective article we first discuss some important energy dissipative mechanisms that contribute to the friction forces encountered in liquid media, and identify how such energy dissipation can be minimized. We next consider how this can be achieved by intramolecular synergistic structures and by intermolecular synergistic associations. We focus on measurements carried out on well-defined model surfaces that have the advantage that surface forces and friction can be measured with high precision, and that interpretation of the data is facilitated. However, it should be realized that the complex cartilage surface and the multicomponent synovial fluid add many different aspects to the biolubrication issue that is not captured in these experiments. An important step towards bridging this gap was taken by Israelachvili et al. who measured the friction forces between cartilage surfaces mounted on a Surface Forces Apparatus, SFA.15
2. Energy dissipative mechanisms
In this section we discuss common energy dissipative mechanisms, refer to some scientific reports where these mechanisms have been studied, and comment on how the energy dissipation can be minimized. It should, however, be noted that experimentally it is seldom the case that one energy dissipative mechanism can be varied without affecting others. Thus, the situation is often more complex compared to investigations of surface forces acting in the normal direction where e.g. the double-layer force can be screened by increasing the electrolyte concentration, which often has a minor effect on the van der Waals force and the steric force between uncharged polymer layers.2.1 Chain interpenetration and dragging of chains through the interpenetration zone
When two polymer layers are pressed together under an external load, the chains attached to one surface will partly penetrate the layer attached to the other surface. The region where one finds chains attached to both surfaces is called the interpenetration zone. The inset in Fig. 1, which shows the volume fraction of polymer segments between two identical polymer-coated surfaces as a function of distance from the surface, illustrates how this region is defined. The degree of overlap depends on the energy of interaction and the solvent condition as illustrated in Fig. 1. Clearly, for a given energy of interaction the overlap fraction is higher under poor solvent conditions (blue line) than under good solvent conditions (red line).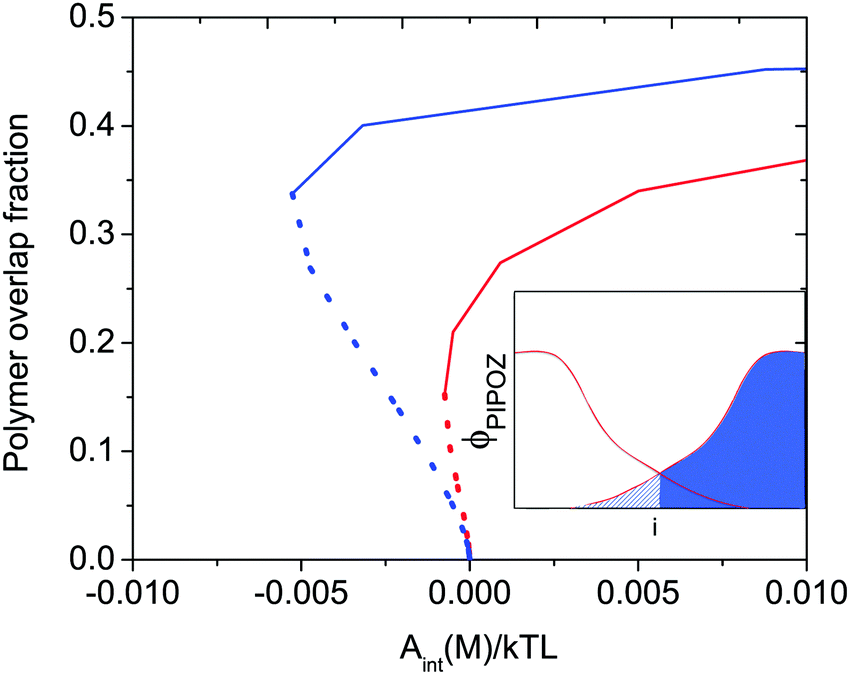 | ||
| Fig. 1 Calculated polymer overlap fraction between two flat surfaces carrying a grafted thermo-responsive polymer layer as a function of the interaction energy Aint(M)/kTL at T = 300 K (red) and T = 325 K (blue) indicated for attractive (dotted curves) and repulsive (solid curves) force branches. The solvent quality decreases with increasing temperature. The polymer overlap fraction is defined as the fraction of polymer segments (hashed area) penetrating into the polymer layer grafted on the opposite surface (see inset). Reproduced with permission from ref. 16. Copyright, 2017 American Chemical Society. | ||
During sliding, the chains in the interpenetration zone need to move past each other, giving rise to viscous energy dissipation, which increases with increasing size of the interpenetration zone and the polymer concentration in this zone. This energy dissipative mechanism has been emphasized by Klein and co-workers in several articles,17,18 and appears to be one of the most important causes for friction between polymer layers.19 Modeling under good solvent conditions shows that the polymer chains become stretched in the sliding direction to an extent that depends on the sliding velocity, and modeling confirms a correlation between the increasing interpenetration zone and increasing friction forces.20 A comprehensive review on theoretical efforts to understand shear forces between polymer-brush coated surfaces has recently been presented,19 and this article is recommended to anyone interested in this subject. It is important to note that solvent present in the brush layer is essential for achieving low friction, as concluded from both theoretical considerations19 and by experiments.21–23 It should also be noted that the osmotic pressure from the counterions present in a polyelectrolyte brush layer counteracts compression and interpenetration, and this makes polyelectrolyte brushes more efficient in reducing friction than comparable brushes of uncharged polymers, as discussed by Zhulina and Rubinstein.24
Thus, to obtain low friction between polymer-bearing surfaces it is important to reduce the interpenetration zone, which can be achieved by having a high graft density and good solvent conditions. It is also predicted that polyelectrolyte brushes should be more efficient in reducing friction forces than brushes of uncharged polymers.
2.2 Breakage and reformation of attractive intralayer physical bonds
In a very interesting work Loveless et al. explored how physical cross-links affect the lateral force needed to drag an AFM tip through a surface-grafted polymer layer.25 They utilized two different types of cross-linkers with similar complexation constants but different on and off rate constants. Both complexing agents resulted in compaction of the layer as judged from a reduced range of the steric forces. Interestingly, the complexing agent with fast kinetics resulted in a reduced lateral force, presumably due to a reduced interpenetration zone. In contrast, the complexing agent with slow kinetics was found to increase the lateral force. Thus, even though the interpenetration zone was also reduced with this complexing agent, a new energy dissipative mechanism associated with stretching of polymer chains between cross-links and disruption and reformation of such cross-links provided the dominating effect. A similar result was obtained by Drummond et al.26 who found that cross-linking of poly(L-lysine)-b-polydimethylsiloxan-b-poly(L-lysine) with sufficiently long dicarboxylic acids increased the cohesion of the adsorbed layer but reduced the lubrication performance. Further, Kampf et al., comparing the friction force between chitosan before and after cross-linking with Graham's salt (predominantly sodium hexamethaphosphate),27 found that addition of this cross-linking agent significantly increased the friction force and the friction coefficient. High friction coefficients (μ ≈ 0.5–1) have also been reported for polyelectrolyte multilayers,28,29 where both breakage of electrostatic interactions and chain stretching within the layers are likely to contribute to the energy dissipation.In the above examples energy dissipation can be caused either by breakage and reformation of the physical cross-links or by viscous stretching of polymer chains between cross-linking points, or both. In a study of friction between a chemically cross-linked chitosan gel and a bare silica surface Liu et al. also found high friction forces (Fig. 2) that were well described by eqn (1) (see below), which is Amontons' first rule modified by an adhesive term.29
| Ff = C + μFn | (1) |
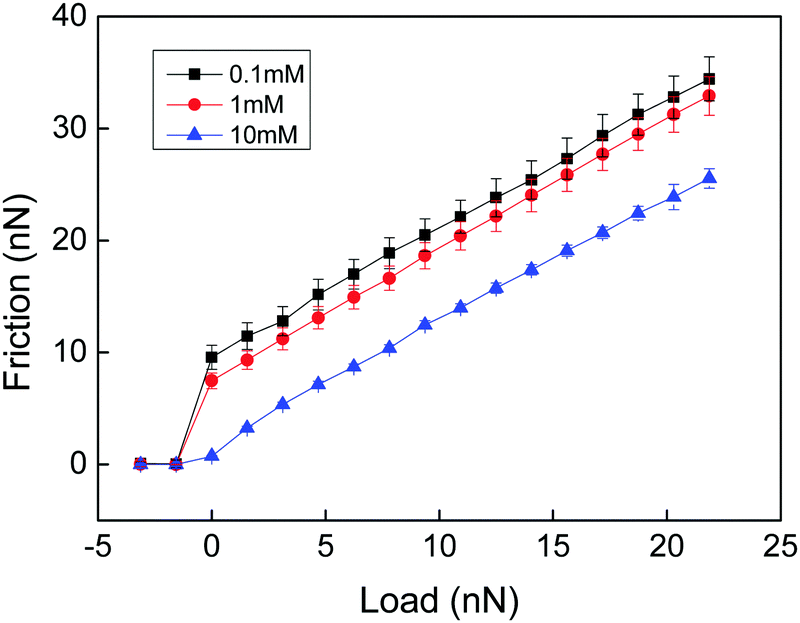 | ||
| Fig. 2 Friction force vs. load measured between a cross-linked chitosan hydrogel and a silica probe across NaCl solutions of different ionic strengths at pH 5.7. There is no hysteresis between the loading and unloading curves. The error bar is calculated from 10 consecutive measurements. The shear velocity was 20 μm s−1. Reproduced with permission from ref. 29. © 2017 Elsevier Inc. | ||
The high friction force observed at low loads (Fig. 2) is due to adhesion between the negatively charged silica surface and the positively charged cross-linked chitosan hydrogel. The friction coefficients (the slope of the lines in Fig. 2) are about 1, independent of the adhesion force. Thus, the energy dissipation that gives rise to increased friction with load was assigned to processes occurring within the layers. Since no breakage of cross-linking points occurs the main energy dissipation mechanism was due to shear-induced stretching and subsequent relaxation of polymer chains between the cross-linking points. Measurements were performed only at two cross-linking densities, and a lower friction force and friction coefficient was found for the higher cross-linking density. To get further insight into how the cross-linking density affects the friction force would require a systematic variation of the distance between chemical cross-links in the hydrogel material.
The effect of cross-linking agents was also investigated by Dunér et al., using grafted poly(acrylic acid), PAA, layers in the absence and presence of calcium ions.30 One PAA-coated surface and a bare silica surface were utilized in their AFM colloidal probe friction studies, and they found very low friction forces in the absence of calcium ions and at low pH in the presence of 10 mM CaCl2. However, increasing the pH to 7.5, which facilitates electrostatic interactions between the divalent cation and carboxylate groups on the polymer, resulted in high friction even though no attractive surface force was present. The increased friction was assigned to formation of bridges between the PAA chains mediated by calcium ions that provided an energy dissipative mechanism as such bridges were broken and reformed during shearing. It was also proposed that as long as this was the dominating energy dissipative mechanism the nature of the bare surface would not affect the friction force. To our knowledge, this prediction has not yet been tested.
In a comprehensive work Wei et al. investigated the effect of different counterions on the state of polyelectrolyte brushes and the friction between such brush layers and a PDMS hemisphere using mainly QCM-D to monitor the state of the brush layer and a pin-on-disc device to determine friction forces.22 They found a correlation between increasing friction and collapse of the brush layer by the counterion and emphasized that counterion exchange is an attractively simple way to tune friction properties. They concluded that high hydration of the polyelectrolyte brush is essential for achieving low friction, but did not specify the energy dissipative mechanism responsible for the increased friction that occurs when the polyelectrolyte brush is collapsed by the counterions. It seems likely to us that the energy dissipative mechanisms discussed above in this section are operative and make a significant contribution.
Attractive interactions between polymer segments within a layer can be induced not only by cross-linking agents, but also by changing the solvent conditions. From the discussion above one would thus expect that making the solvent condition worse would increase the friction force due to increased energy dissipation due to disruption and reformation of physical bonds, and indeed this is what is observed.31,32 However, by changing the solvent condition one also changes interlayer interactions between two polymer-coated surfaces and the degree of interpenetration (see Fig. 1) between the opposing layers.
We conclude that energy dissipation due to intralayer physical cross-links can be minimized if the on and off rate of the complexing agent is fast, and the polymer layer also needs to remain strongly solvated in the presence of the cross-linking agent to facilitate low friction.
2.3 Breakage and reformation of attractive interlayer physical bonds
A change in solvent quality will affect the structure of the adsorbed polymer layer, the surface forces and the friction forces. Generally, one expects the polymer layer to become more compact, the steric force to be less long-ranged and the friction force to be larger as the solvent quality decreases.31 Such a change can, for instance, be induced by varying the solvent composition, or, for thermo-responsive polymers, by changing the solution temperature. In general it is found that for polymers where an increase in temperature leads to worsening of the solvent quality, the friction force also increases with increasing temperature.32,33A recent example is the work by An et al., where the friction forces between negatively charged silica surfaces coated with a cationic–nonionic diblock copolymer were investigated in aqueous solutions.16,34 The non-ionic block consisted of poly(2-isopropyl-2-oxazoline), PIPOZ, which is a thermo-responsive polymer. The surface coverage was relatively low so the adsorbed layer was not in the brush regime. Thus, one would expect a relatively large interpenetration zone and high friction compared to what can be achieved for brush layers, which also was observed. With increasing temperature the solvent condition for the PIPOZ chain decreased and a small attractive force was observed when the surfaces were separated from each other. Under such conditions Amontons' first rule, stating the proportionality between friction force, Ff, and normal load, Fn, needs to be modified according to eqn (1).
Thus, based on eqn (1) a small increase in friction force is expected due to interlayer adhesion, but the slope of the friction force vs. load curve is expected to remain the same (as seen in Fig. 2). Under conditions of constant adsorbed amount the friction force between the surfaces exposing PIPOZ chains was, as expected, found to increase with temperature, but the change was significantly larger than expected from the measured adhesion force. Further, the shape of the force vs. friction curve also changed significantly with temperature and it was never well described by eqn (1), as seen in Fig. 3. Thus, it seems clear that the dominating reason for the change in friction force is not development of an interlayer attraction. Instead, as suggested by modeling,16 for a given load the interpenetration zone increased with increasing temperature (see Fig. 1), and this together with increasing attraction between segments within each layer are more important causes for the increased friction.
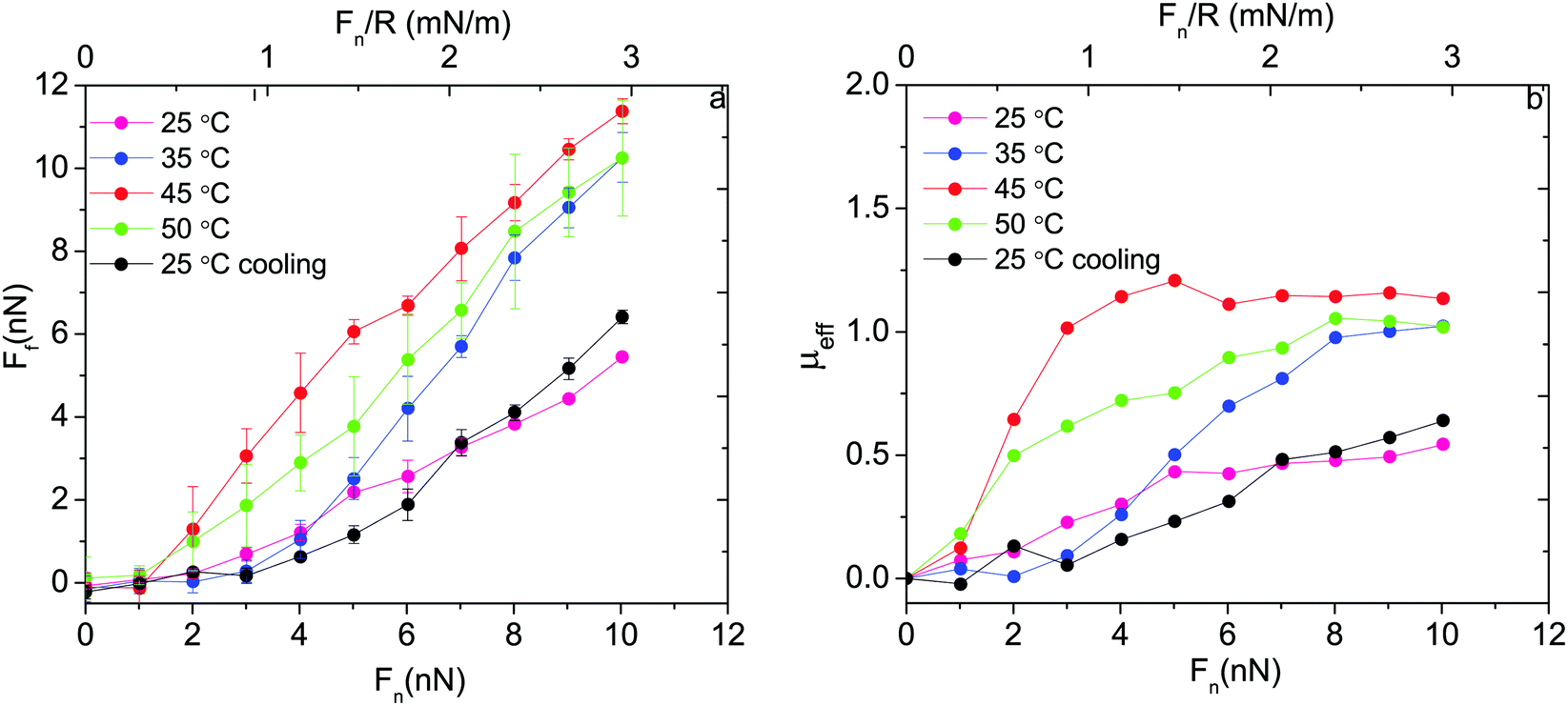 | ||
| Fig. 3 (a) Friction force and (b) effective friction coefficient, μeff = Ff/Fn, as a function of applied normal load between silica surfaces carrying a preadsorbed layer of PIPOZ60-b-PAMPTMA17 measured at different temperatures. Reproduced with permission from ref. 16. Copyright, 2017 American Chemical Society. | ||
The same cationic–nonionic diblock polymer was utilized in another study, but here the adsorption was allowed to vary with temperature.34 Under such circumstances the adsorbed amount increases with increasing temperature due to decreasing repulsion between the PIPOZ chains. In this case, it was found that the friction force decreased with increasing temperature despite the worsening of the solvent quality. This can be rationalized by the higher adsorption density, which reduced the interpenetration zone at higher temperatures. Taken together these two studies suggest that the viscous energy dissipation caused by dragging the polymer chains through the interpenetration zone is more important for the magnitude of the friction force in this system than breakage and reformation of physical bonds between and within the layers. Of course, the extent of and segment density within the interpenetration zone are influenced by the temperature-dependent segment–segment interaction and all effects are coupled.
In another study friction forces between highly stretched surface grafted polystyrene brushes were determined in solutions containing mixtures of toluene (a good solvent) and 2-propanol (a poor solvent).21 It was clearly shown that the friction coefficient and the adhesion force increased as the solvent quality was reduced. The very low friction coefficient observed under good solvent conditions was rationalized by the small interpenetration zone expected for this system.
Based on available data it appears that a small interlayer adhesion is of less importance for the friction coefficient than intralayer attractive interactions and the size of the interpenetration region. Adhesion forces will, however, increase the friction force at low loads.
2.4 Polymer bridging
When the chain density in the adsorbed polymer layer is low, or when a polymer coated surface meets a bare surface, polymers may attach with segments on both surfaces. This gives rise to an attractive force, which normally is referred to as a bridging force. Such polymer bridges have to be broken during shearing and this provides another energy dissipative mechanism. This mechanism explains the high friction force at low loads reported in Fig. 2. A similar but smaller effect on the friction force at zero loads has also been noted between a polyelectrolyte coated surface and a bare surface,35 and between two surfaces carrying mixed adsorbed layers of oppositely charged polyelectrolytes where the surface force curves suggested a weak bridging interaction.28 In another study of friction between polyelectrolyte-coated surfaces, a rapid increase in friction was noted at high loads.36 This was presumably due to erosion of part of the layer that facilitated bridging. Interestingly, the friction force increased markedly, whereas the friction coefficient remained similar. Thus, the effect of bridging was dominantly noticed in the value of the friction at zero load (the value of C in eqn (1)).Clearly, bridging forces should be avoided when low friction is needed, meaning that the adsorbed amount should be high. Bridging forces, as well as other attractive interlayer interactions, appear to increase the friction force most significantly at low loads.
2.5 Lateral motion of molecules along the surface
A boundary lubricant is, of course, only effective as long as it remains at the sliding interface. Thus, it is not enough to achieve a low friction force, but the boundary lubricants also need to have a high load bearing capacity. Experimentally the load bearing capacity is defined as the load at which a significant and sudden increase in friction force is observed, which is suggestive of erosion of the lubricating layer. An example of this behavior is provided in Fig. 4 where the friction force acting between supported dipalmitoylphophatidylcholine, DPPC, bilayers is reported as a function of load. Here the outer sheet of the lubricating phospholipid bilayer is eroded away at an applied load just below 20 nN, which corresponds to a pressure of about 42 MPa.37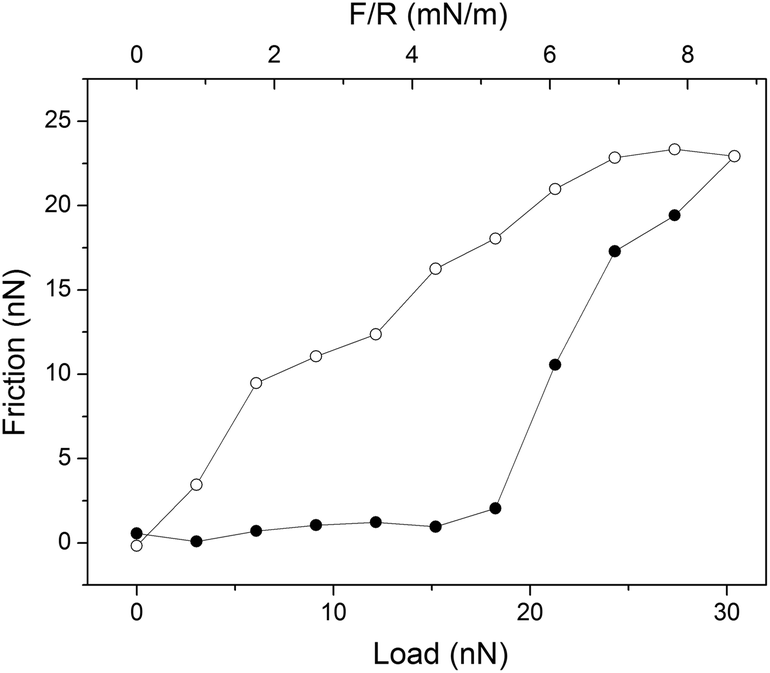 | ||
| Fig. 4 An example of a friction vs. load cycle measured between DPPC bilayers on silica surfaces across 150 mM PBS buffer solution. In this case the bilayer was compromised at a load of just below 20 nN. Friction forces measured on loading and unloading are shown with filled (●) and unfilled (○) symbols, respectively. The temperature was 25 °C. Reprinted with permission from ref. 37. © 2017 Elsevier Inc. | ||
However, before erosion occurs the friction force may increase due to energy dissipation caused by load and shear induced lateral motion of adsorbed molecules along the surface.38 This played a role in the work by Thormann et al., where the friction properties of hydrophobized surfaces coated with methylcellulose, MC, were explored.39 Water becomes a poor solvent for MC at elevated temperatures, and as expected from the discussion above, the friction force at low loads was found to be higher at elevated temperatures. However, the opposite trend was found at high loads. This could be rationalized by the higher attachment strength of the polymer to the hydrophobic substrate at higher temperatures, which resulted in less lateral motion of the polymers along the surface during shearing at the higher temperature.
Electrostatically driven adsorption can provide stronger surface attachment, but also in this case lateral motion along the surface has been noted, particularly at high salt concentrations.40 To further increase the attachment strength catechol anchoring groups on both ends of a poly(ethylene oxide) chain have recently been used to build a layer with predominantly loop structures.41 Very low friction forces and no erosion were observed in these experiments, and this was related to low interpenetration of loops and strong surface anchoring, respectively.
Considering the discussion above one can predict that lubricants that are used for achieving low friction forces in aqueous media should be highly hydrated and strongly attached to the substrate surface, and that the surface should be fully covered by the lubricant. If polymer lubricants are used, their packing density should be high to minimize interpenetration. Some of these requirements are contradictory, for instance a highly hydrated molecule or molecular moiety will have a low driving force for adsorption and thus firm anchoring and high coverage would be difficult to achieve by physical adsorption of such a homopolymer.
3. Intramolecular synergy
Intramolecular synergy is a question of molecular design, where by natural evolution or by synthetic means different functions are included in the molecular structure. The most basic example is amphiphilic molecules combining a polar region and a non-polar region separated in space. This includes phospholipid molecules that self-assemble into membrane structures and synthetic surfactants that are used in everyday products used for, among other things, different cleaning purposes. The hydrophobic moiety drives the surfactants to non-polar interfaces such as the air–water and the oil–water interface. On the other hand, the polar group is strongly hydrated and it is this combination of surface affinity and hydration of different parts of the molecule that makes them good dispersion agents.Similar strategies can be used for polymers, and we have already discussed diblock copolymers that have a designated anchoring group and a highly hydrated buoy block. Blocky polymer structures are also found in nature, where the mucin family consists of block(s) with a high graft density of carbohydrate side-chains and block(s) with low or no side-chains. The mucins cover essentially all internal surfaces in mammals where they provide functions related to hydration, protection and lubrication.42 Further, lubricin, a polymeric mucinous lubricant present in the synovial joint area, consists of a central highly glycosylated region flanked by two globular regions.
3.1 Amphiphilic molecules
In a series of reports Drummond and co-workers have explored the friction forces between surfactant coated mica surfaces using the SFA. The friction response is in general quite complex,43,44 but here we focus only on the case where the highly hydrated polar moiety of the surfactant is directed towards solution and thus provides low friction force due to hydration lubrication. The hydration lubrication effect arises from the strong affinity between polar groups and water, giving rise to a short-range and strong repulsive surface force – the hydration repulsion – that keeps the surfaces apart even under high forces. This, combined with the fact that the confined water molecules retain their fluidity, facilitates sliding with minimal energy dissipation. This lubrication mechanism was emphasized and discussed in two seminal publications by Raviv, Klein and Laurat,45,46 and more recently in a perspective article by Jahn and Klein.47The double-chained cationic surfactant didodecyldimethylammonium bromide adsorbs strongly onto the highly negatively charged mica surface to form a bilayer structure. In SFA experiments it has been demonstrated that this surfactant bilayer has sufficient cohesion to withstand shear under high loads, and the friction force was found to be so small that it could not be measured with the very accurate and sensitive SFA tribometer.48
Phospholipids are naturally occurring bilayer-forming amphiphiles. They are readily deposited on solid surfaces, and their headgroups are strongly hydrated. Considering the findings by Drummond et al., it is conceivable that deposited phospholipid bilayers also may be able to facilitate effortless sliding via the hydration lubrication mechanism. Indeed, several reports have described very low friction forces between phospholipid bilayers up to high pressures.11,37,49,50 Perhaps counter-intuitively, it was found that DPPC bilayers in the fluid state have a higher load bearing capacity than such layers in the frozen state.37 This is illustrated in Fig. 5, which shows the fraction of experiments where the layer has been compromised at or below a load of 20 nN.
 | ||
| Fig. 5 Left panel: The percentage of friction experiments performed at a given temperature where the load bearing capacity was found to be less than 20 nN. The number shown in each column is the total number of experiments performed at a given temperature. Right panel: AFM topographical image of a DPPC bilayer with defects in PBS buffer solution. Height lines over the regions marked with the corresponding colors on the topographical image are shown below the image. The temperature was 32 °C. Reproduced with permission from ref. 37 © 2017 Elsevier Inc. | ||
This finding was rationalized by the lateral mobility of the molecules in the fluid state that allows self-healing of small disturbances in the bilayer structure induced by the combined action of load and shear. It has also been found that phospholipid vesicles adsorbed to solid surfaces can provide low friction, but in this case the frozen state of the phospholipid performed better than the fluid state.51 These are important findings considering that phospholipids are present in the synovial fluid at concentrations6 of 0.1–0.2 mg mL−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) and multilayers of phospholipids have been reported to exist on the cartilage surface.52 Thus, naturally, phospholipids have been implied as important biolubricants52 even though other studies stress the importance of biomacromolecules for the superior lubrication of synovial joints with phospholipids providing less benefits.6
and multilayers of phospholipids have been reported to exist on the cartilage surface.52 Thus, naturally, phospholipids have been implied as important biolubricants52 even though other studies stress the importance of biomacromolecules for the superior lubrication of synovial joints with phospholipids providing less benefits.6
Based on the data above it is clear that the hydration lubrication mechanism can provide superior lubrication up to high loads on flat surfaces. It remains to be explored if such thin layers as formed by surfactant and phospholipid bilayers also can provide favorable lubrication on rough surfaces.
3.2 Copolymers
Block copolymers that are used for achieving favorable lubrication properties should have one or more anchor blocks that provide firm anchoring to surfaces, and one or more blocks that extend from the surface and experience good solvent conditions. The lengths of the different blocks need to be balanced in order to allow formation of a compact layer that counteracts interpenetration. In the previous sections we have already provided examples of friction properties attained with different diblock and triblock copolymers. Here we will instead focus on bio- and biomimetic bottle-brush polymers consisting of a main chain decorated with a high density of side-chains. Biopolymers of this type include the mucin family and the mucinous glycoprotein lubricin, where the main chain is a polypeptide and the side chains consist of oligosaccharides. Biomimetic polymers of this type often have a cationic main chain that provides electrostatic anchoring to negatively charged surfaces, and hydrophilic side chains often made of poly(ethylene oxide), also known as poly(ethylene glycol), or oligosaccharides.Many mucins have an overall train-of-brushes structure, containing several heavily glycosylated regions linked by short non-glycosylated stretches (see Fig. 6).
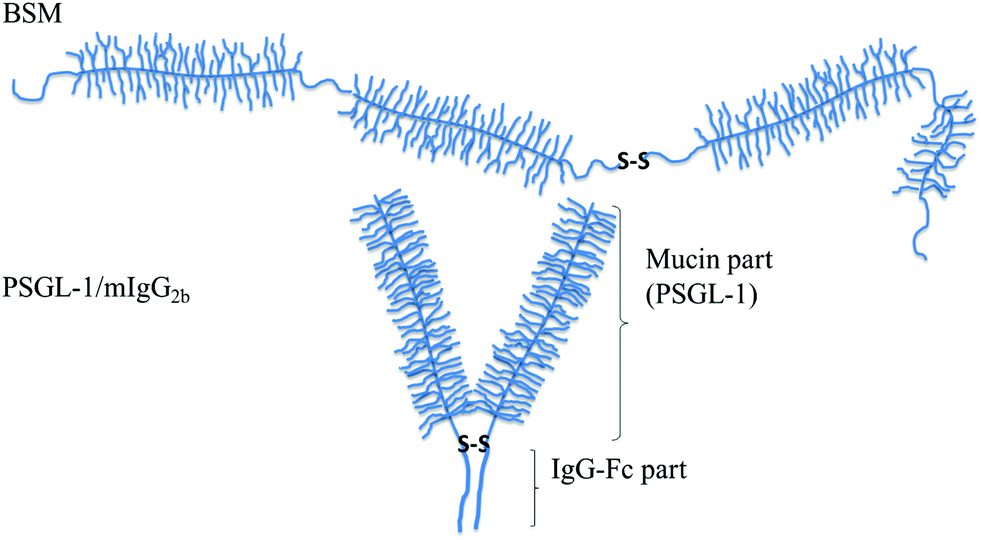 | ||
| Fig. 6 Schematic illustration of mucin structures (not drawn to scale). Top: Secreted bovine submaxillary mucin (BSM) consists of alternated non-glycosylated protein regions (thin lines) and heavily glycosylated regions (with side chains). The overall structure can be characterized as a train-of-brushes. Bottom: PSGL-1/mIgG2b produced as a dimer containing two glycosylated brush regions attached to the Fc part of mIgG (the anchor block). The overall structure can be characterized as a brush-with-anchor. Reproduced with permission from ref. 53 Copyright, 2017 American Chemical Society. | ||
The lubricating ability of different types of mucins has been investigated on a range of different surfaces,28,53–56 and the lubrication performance reported is found to vary significantly depending on the mucin used and the nature of the substrate surface. In cases where anchoring is strong and the adsorbed amount is high, low friction forces are observed due to the presence of a high density of strongly hydrated oligosaccharide chains that counteract interpenetration and facilitate hydration lubrication. An and co-workers compared the lubrication performance of a typical train-of-brushes mucin with that of a recombinant mucin consisting of an anchor region from which two glycosylated brush structures are extended (brush-with-anchor structure, Fig. 6). The non-glycosylated regions provide anchoring to hydrophobic surfaces, and both mucin types were found to adsorb to poly(methyl methacrylate), PMMA. However, the brush-with-anchor mucin adsorbed with higher affinity and at higher amount to the PMMA surface. As a result the friction force was significantly lower between the PMMA surface coated with the brush-with-anchor mucin as illustrated in Fig. 7.53
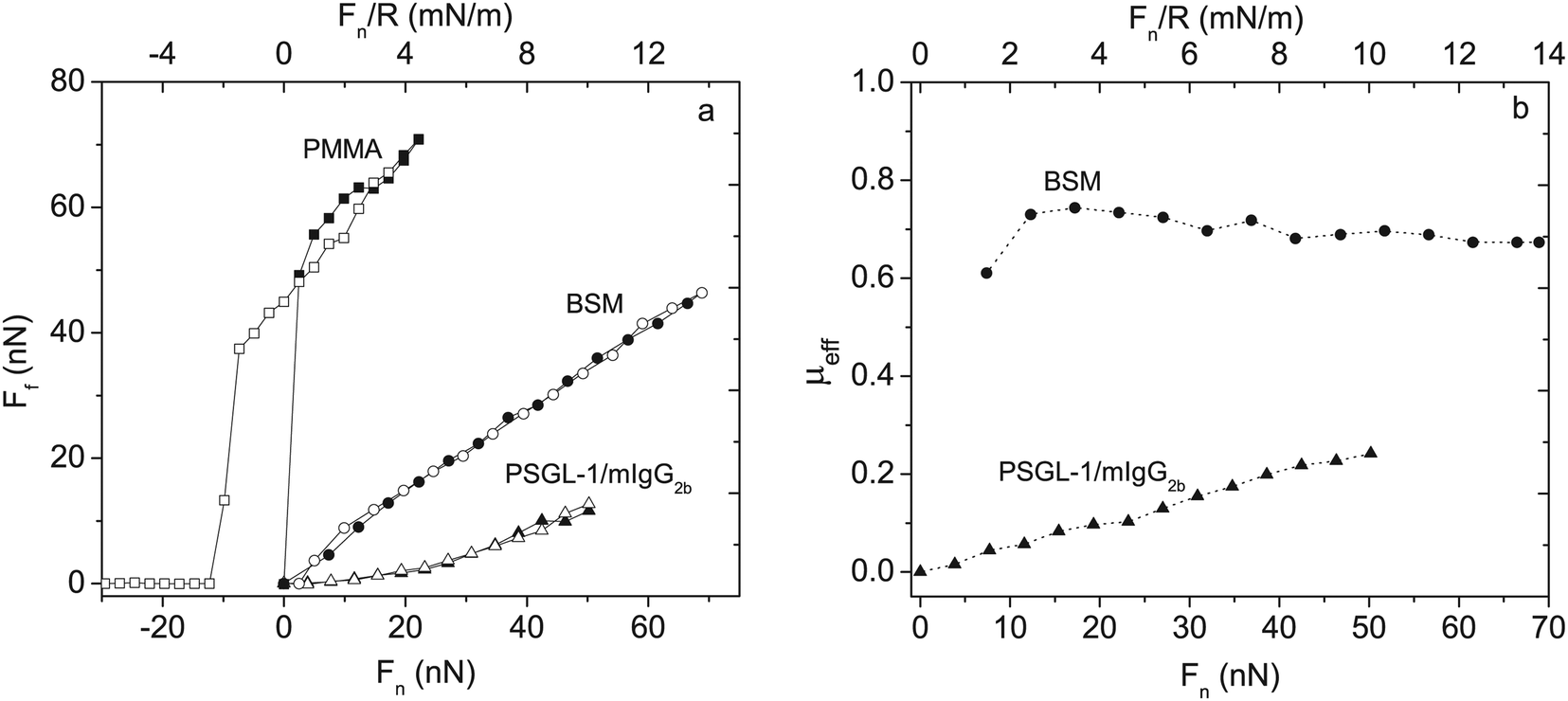 | ||
| Fig. 7 Left panel: Friction force Ffvs. load, Fn and Fn/R, between two bare PMMA surfaces (squares) across 155 mM NaCl solution, BSM-coated PMMA (circles) and PSGL-1/mIgG2b-coated PMMA layers (triangles) across 100 ppm mucin solutions in 155 mM NaCl. Filled and unfilled symbols represent data obtained upon loading and unloading, respectively. BSM has a train-of-brushes structure and PSGL-1/mIgG2b a brush-with-anchor structure. Right panel: The effective friction coefficient μeff = Ff/Fnvs. load for BSM (circles) and PSGL-1/mIgG2b (triangles). Reproduced with permission from ref. 53 Copyright, 2017 American Chemical Society. | ||
It is generally acknowledged that it is the highly hydrated oligosaccharide side chains that facilitate lubrication between mucin layers, but only recently was it attempted to elucidate how the nature of the side-chains affects the lubrication performance between recombinant brush-with-anchor mucins.56 It was found that the recombinant mucin with longer oligosaccharide side chains performed better at low loads, and this was interpreted as being due to higher hydration. However, at higher loads the reverse was observed and the mucin with shorter oligosaccharide side chains provided more efficient lubrication. It was suggested that this was due to the smaller interpenetration zone leading to less entanglement during shearing.
Considering the importance of biomacromolecules with brush-structures for biolubrication, it has been natural to explore biomimetic polymers of similar architecture for achieving low friction forces in aqueous media. With such aims a range of structures, including random copolymers containing positive charges and side chains of poly(ethylene oxide),57–59 diblock copolymers with a cationic anchoring block and a block carrying high density of grafted poly(ethylene oxide),40 brush-on-brush structures with cationic anchoring sites,60 diblock polymers with a hydrophobic anchoring block and a lubricating polyelectrolyte block,61 have, among others, been utilized. The lubrication performances reported in several studies are summarized in the work of Liu et al.40 Here, we only note that in several studies a friction coefficient below 0.05 and a load bearing capacity in excess of 20 MPa are reported. Thus, it seems clear that such biomimetic structures can elicit favorable lubrication in aqueous media provided the anchoring is strong and the density of the exposed and highly hydrated side chains is large enough to efficiently counteract interpenetration. However, it is essential to optimize the polymer architecture to perform in an optimal manner on a given substrate. In this context one should mention the work by Perry et al. who studied a range of poly(L-lysine)-graft-poly(ethylene glycol) structures and found that the friction force was reduced with increasing graft density of the PEG side chains and increasing length of the PEG chains.59 Likewise, Pettersson et al. explored a range of cationic polyelectrolytes with grafted side-chains and found an optimal lubrication performance when 90% of the segments carried a grafted PEO side chain and the remaining 10% a cationic charge.57
4. Intermolecular synergy
In this section we discuss how the combination of two different molecules can provide lubrication synergy. Lubrication synergy can occur for several different reasons. It may affect the adsorption strength or the orientation of the adsorbed molecules, it may promote accumulation of the lubricants in the interfacial region and it may provide lower friction forces and higher load bearing capacity. In the following we discuss some reports that have provided evidence for such synergy mechanisms.4.1 Anchoring and structuring
Lubricin is one of the biomacromolecules that has been advocated as being essential for synovial joint lubrication. As such, its lubrication ability has been investigated on model surfaces, but the results have indicated poor performance in the absence of other biomacromolecules. One reason for this is that lubricin does not adsorb strongly enough onto the model surfaces used, and adjuvant biomacromolecules are needed to provide anchoring and structuring of the lubricin layers.The mode of attachment of molecules to surfaces is of decisive importance for the lubricity of molecules and molecular aggregates. In studies of lubricins' performance on hydrophilic charged surfaces Zappone et al. noticed that lubricin adsorbed as extended loops and tails, but the lubrication performance of lubricin was limited to low pressures, below 0.6 MPa, and on hydrophobic surfaces low friction forces were never observed.62 This led to the conclusion that simple physisorption of lubricin alone is not sufficient for achieving low friction and high resistance to wear. Thus, they concluded that contributions of “complementary cross-linking interactions between lubricin and some other components in the synovial fluid and/or on the cartilage surface” are required.62
Such anchoring of lubricin to surfaces seems to be partly successfully mediated by fibronectin. The attachment of lubricin through its C-terminus (PEX-like domain) to fibronectin-coated mica was reported to yield remarkable wear protection with no damage to the surfaces up to pressures of 14 MPa.63 However, the friction forces observed for the fibronectin–lubricin synergistic pair were rather high, exhibiting a friction coefficient of 0.18 in the best case, which is far above what is observed in healthy synovial joints. Thus, lubricin’s attachment to fibronectin via the C-terminus leaves the N-terminus free for self-aggregation of lubricin into dimers.64 This provides a synergistic effect in terms of wear protection, but it is not sufficient in eliciting low friction.
Macromolecules such as collagen and hyaluronan that are inherent parts of the cartilage and synovial fluid are able to anchor and keep lubricin on surfaces, in a mode that favors low friction on soft surfaces, as shown by Majd et al.65 Though it is not clear how lubricin precisely attaches to the collagen type II and hyaluronan surfaces, it does so spontaneously from solution and the resulting friction forces on the combined hyaluronan and collagen II surface in the presence of lubricin are reported to be low (μ = 0.01 ± 0.0004). However, the pressure applied to these soft surfaces was at the most 0.013 MPa, which are orders of magnitude lower than the 25 MPa pressures that can be encountered in the articular joint area. On the other hand, Chang et al.66 using chemically grafted collagen type II on gold surfaces also showed that this pair of molecules is efficient in reducing friction, and friction coefficients of 0.1 or less were observed.66 Interestingly, normal forces between such lubricin-coated surfaces exhibited relatively long-range attractive forces. This is in contrast to what was observed for lubricin adsorbed on fibronectin63 or cartilage oligomeric matrix protein (COMP)67 coated surfaces.
Das et al.68 demonstrated lubrication and wear protection synergy between hyaluronan chemically grafted on a mica surface and infused with lubricin, yielding low friction, μ = 0.09, up to pressures of ≈4 MPa. It was clearly shown that robust chemical attachment of hyaluronan is necessary in order to keep the synergistic biomacromolecular pair between the surfaces, as a physically adsorbed hyaluronan–lubricin mixture laden with a negative charge is easily expelled from between the mica surfaces.
Cartilage oligomeric matrix protein (COMP) is one of the biomacromolecules that promotes anchoring of lubricin in a fashion that facilitates lubrication. Flowers et al. identified COMP–lubricin complexes in arthritic synovial fluid and showed that the C-terminal of COMP bound non-covalently to the N-terminal of lubricin.69 This is important as it seems that COMP is the protein in the cartilage that is able to direct the adsorption of lubricin such that the lubrication ability of lubricin is expressed optimally. Some data obtained for the COMP–lubricin pair are provided in Fig. 8.67 Lubricin adsorbed directly on PMMA reduces friction but the friction force remains high, and the lubricin layer is easily eroded during shearing as illustrated by the hysteresis in the friction vs. load curve. The adsorption of COMP to PMMA is more robust and no hysteresis is observed in the friction vs. load curve, but the friction forces remain relatively high. However, if lubricin is allowed to adsorb on the COMP-coated PMMA surface a durable layer is obtained, which also reduces friction significantly. In this case a friction coefficient of 0.06 was obtained up to the highest load applied, corresponding to a pressure of 7 MPa, with no sign of erosion.67
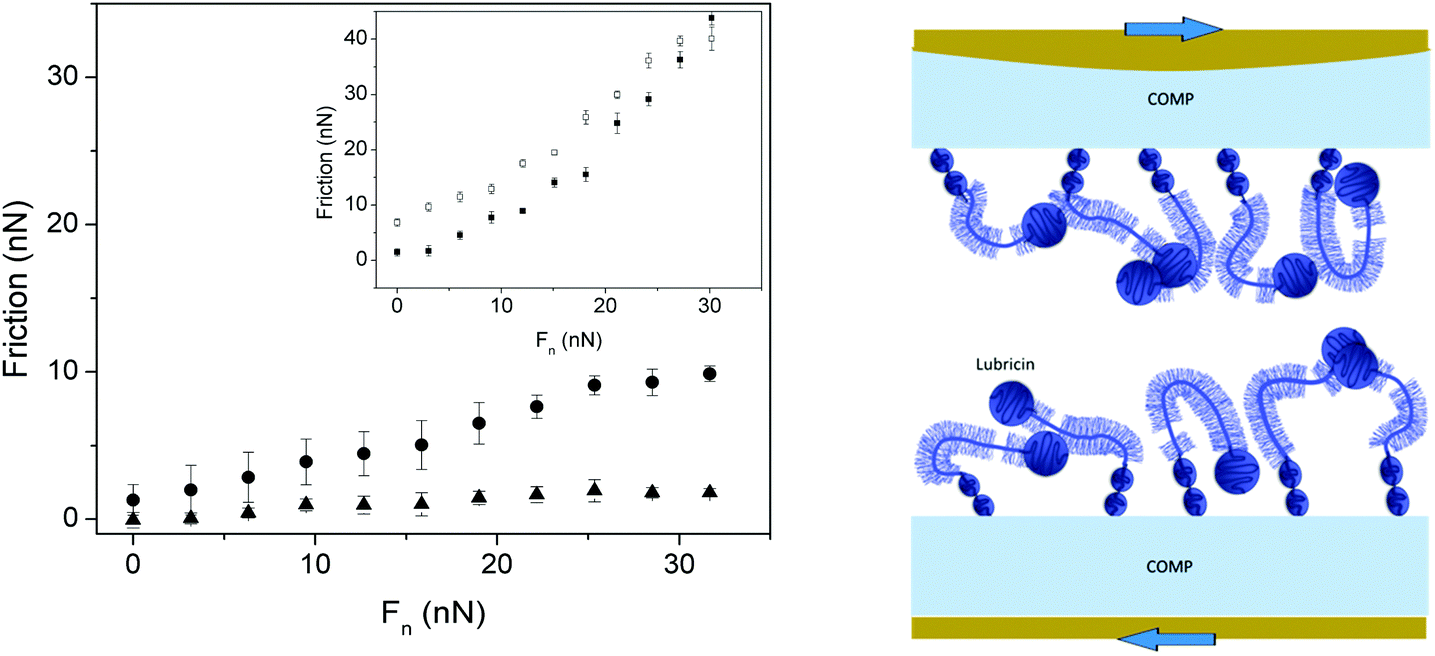 | ||
| Fig. 8 Left Panel: Friction forces as a function of load. Main figure: Measurements performed between two PMMA surfaces coated with COMP (filled circles), and COMP–lubricin (filled triangles). Inset: Measurements performed between two PMMA surfaces coated with lubricin (filled and open symbols – loading and unloading, respectively). Right panel: A sketch of the COMP–lubricin layer. Reproduced with permission from ref. 67 © 2017 Elsevier Inc. | ||
From the data discussed above it appears that lubricin is able to interact synergistically with other biomacromolecules to facilitate strong anchoring and low friction. However, the friction coefficients reported for such synergy pairs remain well above the values reported for cartilage in the synovial fluid (down to 0.001) at high pressures. The discussion and analysis of the data obtained in studies of intermolecular synergy between biopolymers are burdened by the fact that experiments with various synergistic combinations of biopolymers are performed on a variety of surfaces exhibiting a broad range of stiffness values, leading to widely different applied loads and contact areas for a given pressure, which makes the comparison of the synergistic efficiency reported in different studies difficult. In passing, we note that the value of the elastic modulus of cartilage depends on species and age, and it also depends on the set-up used for the measurements. For instance, it has been reported to be a few MPa when probed by mm or μm sized objects, but when the surface is probed locally by sharp AFM tips the modulus is about two orders of magnitude smaller.70 It is also known that the modulus of human cartilage increases significantly with age with mean values ranging from 1 MPa to 15 MPa.71
4.2 Accumulation
Phospholipids and hyaluronan are two important components in the synovial fluid and both have been suggested to participate in lubrication in synovial joints. Early work of Hills demonstrated the presence of oligolamellar layers of phospholipids on cartilage72 and on other surfaces in the body where lubrication is needed.73 This strongly implies that it is advantageous to accumulate many layers of a lubricant in order to ensure supply and self-healing ability in case of local damage and wear of the lubricant layer. One way of achieving this in model systems is by simply adsorbing tightly packed rigid phospholipid vesicles on surfaces,51 which assures accumulation of several phospholipid bilayers between surfaces upon compression.Experiments with mixtures of hyaluronan and phospholipids showed that the two components readily self-assemble both in bulk and at interfaces and the lubrication ability of such mixed layers is excellent.10,11,49,74 The association between hyaluronan and DPPC allows accumulation of a high amount of the DPPC lubricant at the interface, either through layer-by-layer deposition49 or through adsorption of preformed aggregates.11 The data shown in Fig. 9 illustrate the adsorption from a mixed solution of DPPC vesicles and hyaluronan.11 The first rapid adsorption step corresponds to bilayer formation, and if DPPC vesicles in the absence of hyaluronan is used, no further adsorption will occur.10 However, from the mixed solution, adsorption of hyaluronan-decorated vesicles proceeds until rinsing. When the mixed solution is reinjected the adsorption process starts again. The layers formed by adsorption from the mixed solution are heterogeneous, but they nevertheless provide low friction forces (μ < 0.01).
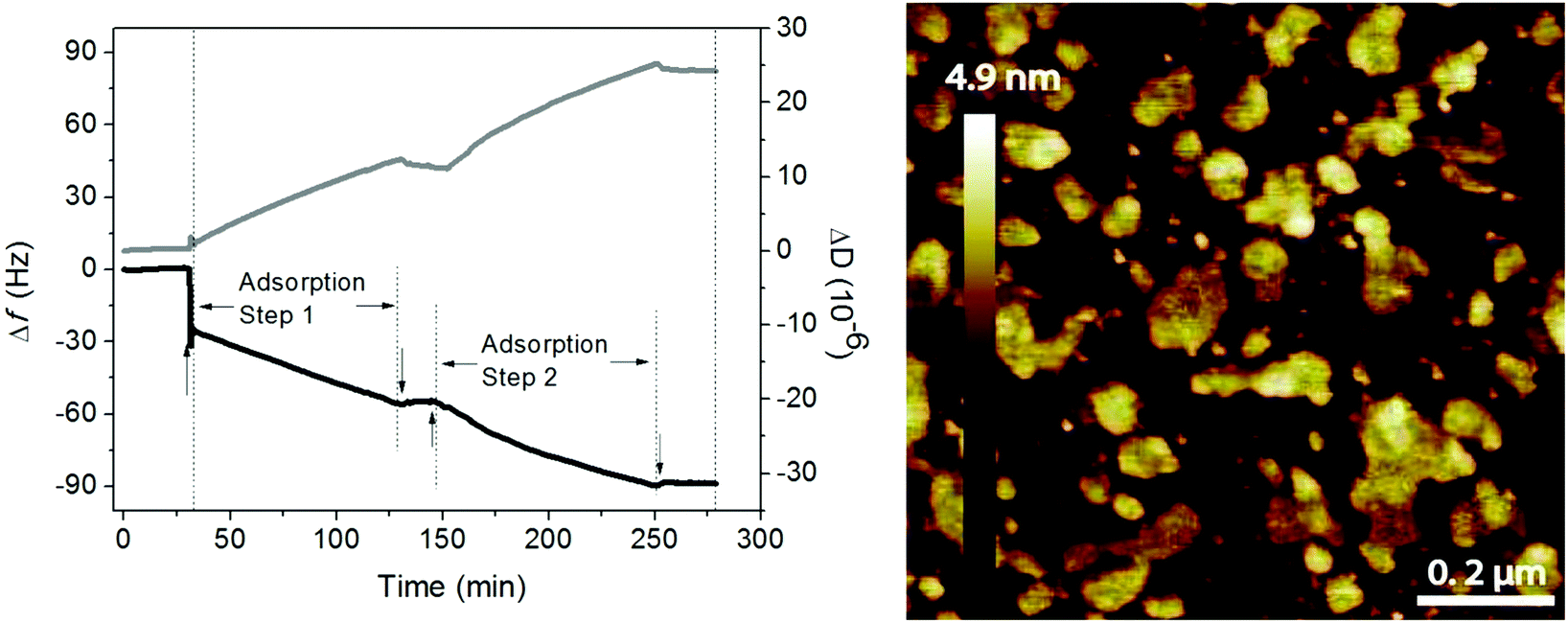 | ||
| Fig. 9 Left Panel: Adsorption from a solution containing 0.5 mg mL−1 DPPC and 0.5 mg mL−1 hyaluronan (start marked by↑) on a silica surface in 155 mM NaCl solution at 55 °C monitored by QCM-D (frequency (black line) and dissipation (grey line) change). Rinsing (start marked by↓) was done with 155 mM NaCl solution at 55 °C. Right panel: AFM topography image of the adsorbed layer formed after 40 minutes of adsorption from solutions containing 0.5 mg mL−1 DPPC and 0.5 mg mL−1 hyaluronan in 155 mM NaCl on a silica surface. After 40 minutes the solution was exchanged with pure 155 mM NaCl solution, and the resulting layer was imaged in this solution at a temperature of 47 °C. Image size 1 × 1 μm2. Note that the AFM tip will deform and possibly disrupt the adsorbed aggregates, so the height of these aggregates when undisturbed by the tip is underestimated in the image. Adopted with permission from ref. 11 © 2017 Elsevier Inc. | ||
Association is observed between many polymer–surfactant pairs, and the association is particularly strong between highly charged polyelectrolytes and oppositely charged surfactants.75 In many such cases the association leads to formation of insoluble aggregates with well-defined internal organization,76 and it has also been observed that association between a preadsorbed highly charged cationic polyelectrolyte on mica and the anionic surfactant sodium dodecylsulphate, SDS, leads to formation of an adsorbed layer with similar internal organization.77 SDS does not adsorb onto bare mica and silica surfaces, but the presence of the preadsorbed cationic layer allows accumulation of the surfactant at the interface. The friction forces between such mixed polyelectrolyte–surfactant layers have been reported and shown to be very low except for some pronounced friction peaks (Fig. 10).35
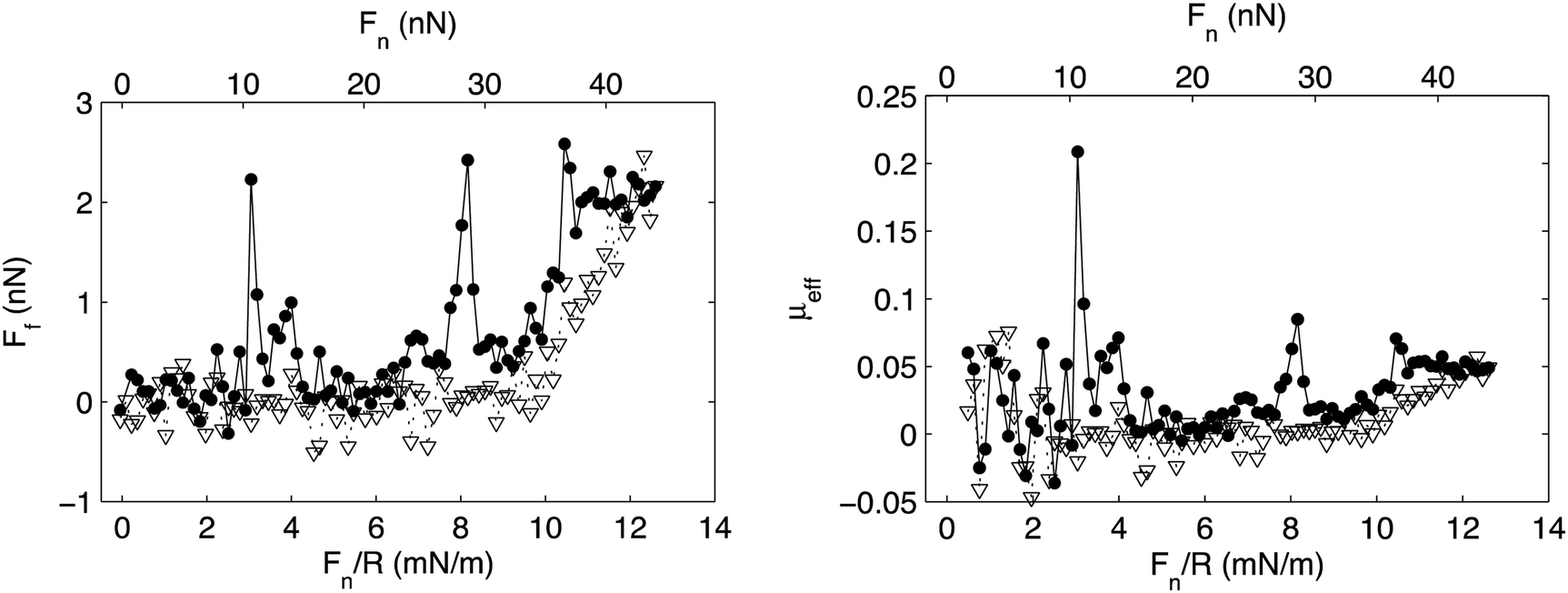 | ||
| Fig. 10 Left panel: Friction force, Ff, vs. load, Fn/R, and Fn, between a mica and a silica surface coated with the cationic polyelectrolyte PMAPTAC across a 1 cmc SDS solution. Filled and unfilled symbols represent data obtained upon loading and unloading, respectively. Right panel: Effective friction coefficient, μeff, vs. load. The symbols correspond to the same experimental conditions as described for the left panel. Reproduced from ref. 35 with permission from the Royal Society of Chemistry. | ||
The low friction force was suggested to be due to the presence of an easily sheared water layer, whereas the friction peaks were assigned to structural rearrangements within the layer due to the combined action of load and shear. Interestingly, after rearrangement the layers return to a low friction state until they finally are compromised at a pressure of about 20 MPa (load Fn just above 30 nN), which thus is the load bearing capacity. Similar transient friction peaks have also been observed between lubricating layers formed by adsorption from mixed hyaluronan–DPPC solutions, and again related to structural rearrangements in the layer.11
It seems clear that mixtures of polymers and surface active agents in many cases allow accumulation of the surfactants at the interface in amounts that exceed what can be achieved with the surfactant alone. For instance, multilayers of phospholipids and synthetic cationic polyelectrolytes have been shown to form and the effect of shear rate on the structures has been elucidated by means of neutron reflectivity and ATR-FTIR.78 However, there are very few reports that have attempted to elucidate under which circumstances mixed polymer–surfactant layers are able to provide low friction and high load bearing capacity.
4.3 Load bearing capacity
We have seen that mixtures of polymers and surfactants under some circumstances are able to provide low friction and high load bearing capacity. Drummond and co-workers have explored this in some detail, using surfactants that adsorb onto the substrate surface mixed with a diblock copolymer to modify the lubrication performance.44,48,79,80 In their initial studies, using a double-chained surfactant which in itself provided low friction and high load bearing capacity, it was found that coadsorption of a hydrophobically end-modified poly(ethylene oxide) compromised the lubrication performance.48 In later studies oligomeric cationic surfactants were utilized, and the layers formed by these surfactants alone had relatively low load bearing capacity. However, co-adsorption of a poly(acrylic acid)-poly(acrylamide) diblock copolymer significantly enhanced the cohesion of the adsorbed layer and thus increased the load bearing capacity.79 It was also noted that there was an optimal copolymer concentration range that should not be exceeded in order to retain the excellent lubricity.79,80 The structure of the surfactant played an important role in the enhancement of the load bearing capacity achieved by addition of the diblock copolymer. It was significantly enhanced by the poly(acrylic acid)-poly(acrylamide) diblock copolymer for the cationic trimeric and dimeric surfactants with C12 hydrocarbon chains and C3 spacers, whereas layers composed of the monomeric surfactant cetylammonium chloride and the co-polymer did not display a high load bearing capacity.44We also note that the load bearing capacity of lubricin was significantly higher on COMP-coated PMMA compared to when lubricin was adsorbed directly on PMMA (see Fig. 8), whereas layer-by-layer deposition of DPPC and hyaluronan reduced the load bearing capacity compared to DPPC alone.10
The studies reported above demonstrate the great possibility of using mixtures of surfactants and polymers to achieve low friction and high load bearing capacity in aqueous media. However, more studies are needed before a clear picture emerges on the structure–lubrication synergy relation of polymer–surfactant and polymer–polymer systems, including biopolymers and phospholipids.
5. Conclusions and outlook
It is clearly possible to achieve low friction forces in aqueous media using a range of different molecular structures combining different functions within the molecule. It spans from bilayer forming surfactants and phospholipids that on smooth surfaces provide friction forces that are so low that they are on the limit of what can be measured. The lubrication mechanism – hydration lubrication – is nowadays well established. It is, however, not established that such thin layers also can perform well on rough surfaces where one could expect that the load bearing capacity would be compromised. Different blocky polymer structures and grafted polymer brushes also provide low friction in the boundary lubrication regime provided the chain density is high enough to counteract interpenetration and the extended chains are highly hydrated. Molecular design to fully utilize intramolecular synergies needs to be optimized for different surfaces, and despite a few scientific reports in this area it is judged that much more research is needed, including modeling where molecular structures and interactions can be varied at will. A few recent reports also emphasize lubrication synergy between different types of molecules, and we have discussed several aspects of such synergistic actions. Nature is an expert in using synergistic effects to achieve desired properties, and we mean that it is highly unlikely that biolubrication would be an exception. Thus, we believe and hope that future studies in the biolubrication area will pay increasing attention to intermolecular synergies. For the boundary lubricants to fulfill their lubricating function they obviously need to remain on the sliding surfaces. Thus, their load bearing capacity is essential and self-healing of lubricating layers is a necessity that is favored by excessive accumulation at the interfaces. We mean that there is a lack of systematic studies focusing on erosion of lubricating layers, and on how the erosion resistance can be improved by molecular design and intermolecular synergistic effects. Thus, future studies focusing on load bearing capacities to an equal extent as on the friction forces prior to erosion would be of high value.Acknowledgements
PC acknowledges financial support from the Swedish Research Council, VR People Programme (Marie Curie Actions) of the European Union's Seventh Framework Programme FP7/2007–2013/under REA grant agreement no. 290251 is acknowledged for financial support. The authors also would like to thank all talented PhD-students in our group that over the years have provided some of the data mentioned in this article. Special thanks go to Ali Naderi, Joseph Iruthayaraj, Torbjörn Pettersson, Xiaoyan Liu, Min Wang, Junxue An and Akanksha Raj. It was great working with you!References
- W. H. Briscoe, Curr. Opin. Colloid Interface Sci., 2017, 27, 1–8 CrossRef CAS.
- H. Forster and J. Fisher, Proc. Inst. Mech. Eng., Part H, 1996, 210, 109–119 CrossRef CAS PubMed.
- L. R. Gale, R. Coller, D. J. Hargreaves, B. A. Hills and R. Crawford, Tribol. Int., 2007, 40, 601–606 CrossRef CAS.
- G. Spahn and R. Wittig, Zentralbl. Chir., 2003, 128, 78–82 CrossRef CAS PubMed.
- L. Murphy and C. G. Helmick, Am. J. Nurs., 2012, 112, S13–S19 CrossRef PubMed.
- T. A. Schmidt, N. S. Gastelum, Q. T. Nguyen, B. L. Schumacher and R. L. Sah, Arthritis Rheum., 2007, 56, 882–891 CrossRef CAS PubMed.
- B. A. Hills, Proc. Inst. Mech. Eng., Part H, 2000, 214, 83–94 CrossRef CAS PubMed.
- J. Klein, Friction, 2013, 1, 1–23 CrossRef CAS.
- T. Kawano, H. Miura, T. Mawatari, T. Moro-Oka, Y. Nakanishi, H. Higaki and Y. Iwamoto, Arthritis Rheum., 2003, 48, 1923–1929 CrossRef CAS PubMed.
- M. Wang, C. Liu, E. Thormann and A. Dedinaite, Biomacromolecules, 2013, 14, 4198–4206 CrossRef CAS PubMed.
- A. Raj, M. Wang, T. Zander, D. C. F. Wieland, X. Liu, J. An, V. Garamus, R. Willumeit-Römer, M. Fielden, P. M. Claesson and A. Dedinaite, J. Colloid Interface Sci., 2017, 488, 225–233 CrossRef CAS PubMed.
- S. Jung, A. Petelska, P. Beldowski, W. K. Augé II, T. Casey, D. Walczak, K. Lemke and A. Gadomski, Colloid Polym. Sci., 2017, 295, 403–412 CAS.
- J.-B. Park, C.-T. Duong, H.-G. Chang, A. R. Sharma, M. S. Thompson, S. Park, B.-C. Kwak, T.-Y. Kim, S.-S. Lee and S. Park, Biointerphases, 2014, 9, 031007 CrossRef PubMed.
- A. Dedinaite, Soft Matter, 2012, 8, 273–284 RSC.
- D. W. Lee, X. Banquy and J. N. Israelachvili, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E567–E574 CrossRef CAS PubMed.
- J. An, X. Liu, P. Linse, A. Dedinaite, F. M. Winnik and P. M. Claesson, Langmuir, 2015, 31, 3039–3048 CrossRef CAS PubMed.
- J. Klein, Annu. Rev. Mater. Sci., 1996, 26, 581–612 CrossRef CAS.
- J. Klein, E. Kumacheva, D. Perahia and L. J. Fetters, Acta Polym., 1998, 49, 617–625 CrossRef CAS.
- T. Kreer, Soft Matter, 2016, 12, 3479–3501 RSC.
- T. Kreer, K. Binder and M. H. Müser, Langmuir, 2003, 19, 7551–7559 CrossRef CAS.
- A. Nomura, K. Okayasu, K. Ohno, T. Fukuda and Y. Tsujii, Macromolecules, 2011, 44, 5013–5019 CrossRef CAS.
- Q. Wei, M. Cai, F. Zhou and W. Liu, Macromolecules, 2013, 46, 9368–9379 CrossRef CAS.
- J. Klein, E. Kumacheva, D. Mahalu, D. Perahia and L. J. Fetters, Nature, 1994, 370, 634–636 CrossRef CAS.
- E. B. Zhulina and M. Rubinstein, Macromolecules, 2014, 47, 5825–5838 CrossRef CAS PubMed.
- D. M. Loveless, N. I. Abu-Lail, M. Kaholek, S. Zauscher and S. L. Craig, Angew. Chem., Int. Ed., 2006, 45, 7812–7814 CrossRef CAS PubMed.
- C. Drummond, P. Richetti, J. Rodriguez-Hernandez and S. Lecommandoux, J. Adhes., 2007, 88, 431–448 CrossRef.
- N. Kampf, U. Raviv and J. Klein, Macromolecules, 2004, 37, 1134–1142 CrossRef CAS.
- T. Pettersson and A. Dedinaite, J. Colloid Interface Sci., 2008, 324, 246–256 CrossRef CAS PubMed.
- C. Liu, E. Thormann, E. Tyrode and P. M. Claesson, J. Colloid Interface Sci., 2015, 443, 162–169 CrossRef CAS PubMed.
- G. Dunér, E. Thormann, O. Ramström and A. Dedinaite, J. Dispersion Sci. Technol., 2010, 31, 1285–1287 CrossRef.
- J. Klein, E. Kumacheva, D. Perahia, D. Mahalu and S. Warburg, Faraday Discuss., 1994, 98, 173–188 RSC.
- A. Dedinaite, E. Thormann, G. Olanya, P. M. Claesson, B. Nyström, A.-L. Kjøniksen and K. Zhu, Soft Matter, 2010, 6, 2489–2498 RSC.
- N. Nordgren and M. W. Rutland, Nano Lett., 2009, 9, 2984–2990 CrossRef CAS PubMed.
- J. An, X. Liu, A. Dedinaite, A. Korchargina, F. M. Winnik and P. M. Claesson, J. Colloid Interface Sci., 2017, 487, 88–96 CrossRef CAS PubMed.
- A. Dedinaite, T. Pettersson, B. Mohanty and P. M. Claesson, Soft Matter, 2010, 6, 1520–1526 RSC.
- M. A. Plunkett, A. Feiler and M. W. Rutland, Langmuir, 2003, 19, 4180–4187 CrossRef CAS.
- M. Wang, T. Zander, X. Liu, C. Liu, A. Raj, D. C. F. Wieland, V. M. Garamus, R. Willumeit-Römer, P. M. Claesson and A. Dedinaite, J. Colloid Interface Sci., 2015, 445, 84–92 CrossRef CAS PubMed.
- U. Raviv, R. Tadmor and J. Klein, J. Phys. Chem. B, 2001, 105, 8125–8134 CrossRef CAS.
- E. Thormann, R. Bodvik, L. Karlson and P. M. Claesson, Colloids Surf., A, 2014, 441, 701–708 CrossRef CAS.
- X. Liu, E. Thormann, A. Dedinaite, M. Rutland, C. Visnevskij, R. Makuska and P. M. Claesson, Soft Matter, 2013, 9, 5361–5371 RSC.
- T. Kang, X. Banquy, J. Heo, C. Lim, N. A. Lynd, P. Lundberg, D. X. Oh, H.-K. Lee, Y.-K. Hong, D. S. Hwang, J. H. Waite, J. N. Israelachvili and C. J. Hawker, ACS Nano, 2016, 10, 930–937 CrossRef CAS PubMed.
- A. Dedinaite, Encyclopedia of Surface and Colloid Science, Taylor and Francis, 2nd edn, 2010, pp. 1–17 Search PubMed.
- C. Drummond, J. Israelachvili and P. Richetti, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2003, 67, 0066110 CrossRef CAS PubMed.
- J.-M. Lagleize, P. Richetti and C. Drummond, Tribol. Lett., 2010, 39, 31–38 CrossRef CAS.
- U. Raviv, P. Laurat and J. Klein, Nature, 2001, 413, 51–54 CrossRef CAS PubMed.
- U. Raviv and J. Klein, Science, 2002, 297, 1540–1543 CrossRef CAS PubMed.
- S. Jahn and J. Klein, Macromolecules, 2015, 48, 5059–5075 CrossRef CAS.
- A. Blom, C. Drummond, E. J. Wanless, P. Richetti and G. G. Warr, Langmuir, 2005, 21, 2779–2788 CrossRef CAS PubMed.
- C. Liu, M. Wang, J. An, E. Thormann and A. Dedinaite, Soft Matter, 2012, 8, 10241–10244 RSC.
- A.-M. Trunfio-Sfarghiu, Y. Berthier, M. H. Meurisse and J.-P. Rieu, Langmuir, 2008, 24, 8765–8771 CrossRef CAS PubMed.
- R. Goldberg, A. Schroeder, Y. Barenholz and J. Klein, Biophys. J., 2011, 100, 2403–2411 CrossRef CAS PubMed.
- B. A. Hills, Intern. Med. J., 2002, 32, 242–251 CrossRef CAS PubMed.
- J. An, A. Dedinaite, A. Nilsson, J. Holgersson and P. M. Claesson, Biomacromolecules, 2014, 15, 1515–1525 CrossRef CAS PubMed.
- I. C. Hahn-Berg, L. Lindh and T. Arnebrant, Biofouling, 2004, 20, 65–70 CrossRef CAS PubMed.
- N. M. Harvey, G. E. Yakubov, J. R. Stokes and J. Klein, Biomacromolecules, 2011, 12, 1041–1050 CrossRef CAS PubMed.
- J. An, C. Jin, A. Dedinaite, J. Holgersson, N. G. Karlsson and P. M. Claesson, Langmuir, 2017, 33, 4386–4395 CrossRef CAS PubMed.
- T. Pettersson, A. Naderi, R. Makuska and P. M. Claesson, Langmuir, 2008, 24, 3336–3347 CrossRef CAS PubMed.
- M. T. Müller, X. Yan, S. Lee, S. S. Perry and N. D. Spencer, Macromolecules, 2005, 38, 5706 CrossRef.
- S. S. Perry, X. Yan, F. T. Limpoco, S. Lee, M. Müller and N. D. Spencer, ACS Appl. Mater. Interfaces, 2009, 1, 1224–1230 CAS.
- T. Krivorotova, R. Makuska, A. Naderi, P. M. Claesson and A. Dedinaite, Eur. Polym. J., 2010, 46, 171–180 CrossRef CAS.
- U. Raviv, S. Giasson, N. Kampf, J.-F. Gohy, R. Jerome and J. Klein, Nature, 2003, 425, 163–165 CrossRef CAS PubMed.
- B. Zappone, M. Ruths, G. W. Green, G. D. Jay and J. Israelachvili, Biophys. J., 2007, 92, 1693–1708 CrossRef CAS PubMed.
- R. C. A. Eguiluz, S. G. Cook, C. N. Brown, F. Wu, N. J. Pacifici, L. J. Bonassar and D. Gourdon, Biomacromolecules, 2015, 16, 2884–2894 CrossRef PubMed.
- A. R. C. Jones, J. P. Gleghorn, C. E. Hughes, L. J. Fitz, R. Zollner, S. D. Wainwright, B. Caterson, E. A. Morris, L. J. Bonassar and C. R. Flannery, J. Orthop. Res., 2007, 25, 283–292 CrossRef CAS PubMed.
- S. E. Majd, R. Kuijer, A. Kowitsch, T. Groth, T. A. Schmidt and P. K. Sharma, Langmuir, 2014, 30, 14566–14572 CrossRef CAS PubMed.
- D. P. Chang, F. Guilak, G. D. Jay and S. Zauscher, J. Biomech., 2014, 47, 659–666 CrossRef PubMed.
- A. Raj, M. Wang, C. Liu, L. Ali, N. G. Karlsson, P. M. Claesson and A. Dedinaite, J. Colloid Interface Sci., 2017, 495, 200–206 CrossRef CAS PubMed.
- S. Das, X. Banquy, B. Zappone, G. W. Greene, G. D. Jay and J. N. Israelachvili, Biomacromolecules, 2013, 14, 1669–1677 CrossRef CAS PubMed.
- S. A. Flowers, S. Kalamajski, L. Ali, L. I. Bjorkman, J. R. Raj, A. Aspberg, N. G. Karlsson and C. Jin, Osteoarthritis Cartilage, 2017 DOI:10.1016/j.joca.2017.03.016.
- M. Stolz, R. Raiteri, A. U. Daniels, M. R. VanLandingham, W. Baschong and U. Aebi, Biophys. J., 2004, 86, 3269–3283 CrossRef CAS PubMed.
- J. K. Rains, J. L. Bert, C. R. Roberts and P. D. Pare, J. Appl. Phys., 1992, 72, 219–225 CAS.
- B. A. Hills, J. Rheumatol., 1989, 16, 82–91 CAS.
- B. A. Hills, Proc. Inst. Mech. Eng., Part H, 2000, 214, 83–94 CrossRef CAS PubMed.
- J. Seror, L. Zhu, R. Goldberg, A. J. Day and J. Klein, Nat. Commun., 2015, 6, 6497 CrossRef CAS PubMed.
- B. Lindman and K. Thalberg, in Interactions of Surfactants with Polymers and Proteins, ed. E. D. Goddard and K. P. Ananthapadmanabhan, CRC Press, Boca Raton, 1993 Search PubMed.
- L.-M. Bergström, U. R. M. Kjellin, P. M. Claesson and I. Grillo, J. Phys. Chem. B, 2004, 108, 1874–1881 CrossRef.
- A. Dedinaite, P. M. Claesson and M. Bergström, Langmuir, 2000, 16, 5257–5266 CrossRef CAS.
- F. Schwörer, M. Trapp, M. Ballauff, R. Dahint and R. Steitz, Langmuir, 2015, 31, 11539–11548 CrossRef PubMed.
- C. Drummond, G. Marinov and P. Richetti, Langmuir, 2008, 24, 1560–1565 CrossRef CAS PubMed.
- J.-M. Lagleize, P. Richetti and C. Drummond, Langmuir, 2009, 25, 11472–11479 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2017 |