
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural insights into the EthR–DNA interaction using native mass spectrometry†
Daniel
Shiu-Hin Chan
a,
Wei-Guang
Seetoh
 a,
Brendan N.
McConnell
a,
Dijana
Matak-Vinković
a,
Sherine E.
Thomas
b,
Vitor
Mendes
b,
Michal
Blaszczyk
b,
Anthony G.
Coyne
a,
Tom L.
Blundell
b and
Chris
Abell
*a
a,
Brendan N.
McConnell
a,
Dijana
Matak-Vinković
a,
Sherine E.
Thomas
b,
Vitor
Mendes
b,
Michal
Blaszczyk
b,
Anthony G.
Coyne
a,
Tom L.
Blundell
b and
Chris
Abell
*a
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB21EW, UK. E-mail: ca26@cam.ac.uk
bDepartment of Biochemistry, University of Cambridge, 80 Tennis Court Road, CB2 1GA Cambridge, UK
First published on 7th March 2017
Abstract
EthR is a transcriptional repressor that increases Mycobacterium tuberculosis resistance to ethionamide. In this study, the EthR–DNA interaction has been investigated by native electrospray-ionization mass spectrometry for the first time. The results show that up to six subunits of EthR are able to bind to its operator.
Tuberculosis (TB) is a contagious disease caused by Mycobacterium tuberculosis (Mtb) that exerts an enormous burden on human health and wellbeing worldwide. The World Health Organization (WHO) has estimated that in 2014, TB killed 1.5 million people, while another 9.6 million people were infected.1 Progress against TB has been challenged by the rise of multidrug resistant (MDR) and extensively drug resistant (XDR) Mtb strains. 3.3% of new cases and 20% of previously treated cases have MDR-TB, and of those, an estimated 9.7% are XDR-TB.1
Ethionamide is a second-line drug used for the treatment of MDR-TB. Mechanistically, ethionamide is a prodrug that is activated in vivo by EthA, a flavin-containing monooxygenase enzyme in Mtb, to form an ethionamide-NAD adduct.2 This adduct inhibits the 2-trans-enoyl reductase enzyme InhA, which in turn leads to the inhibition of the Mtb type II fatty acid synthase system (FAS II).3 However, the potency of ethionamide is reduced by EthR, which is a transcriptional repressor of ethA expression.4 This suggests that inhibitors of EthR activity could function as ethionamide boosters,5 allowing for lower dosages of the drug to be used.
EthR belongs to the TetR/CamR repressor protein family, whose members show high sequence homology between their N-terminal DNA-binding domains, and is expected to bind to the DNA major groove via its helix-turn-helix (HTH) motif.6 Using DNase footprinting assays, Baulard and co-workers showed that EthR recognizes a 55 bp operator sequence within the ethA-R intergenic region.4 Subsequent surface plasmon resonance (SPR) analysis suggested that up to eight units of EthR could bind cooperatively to a 62 bp sequence (DNA62) encompassing the operator site (Fig. S1, ESI†).4 There are numerous X-ray crystal structures of the EthR dimer in complex with various small-molecule ligands,7,8 but the structure of the EthR–DNA complex has not yet been solved.
Native electrospray ionization-mass spectroscopy (ESI-MS) is an ideal technique for the study of the interactions and stoichiometries of macromolecular complexes.9,10 A large body of work has established that the native structure and composition of biomolecular complexes can be adequately maintained as solution species are transferred into the gas phase during the electrospray process. However, compared to the application of ESI-MS for multi-protein complexes, fewer studies on protein–DNA complexes have been reported.11–14 Analysis of protein–DNA complexes containing large DNA sequences using positive-ion native MS is complicated by the heterogeneity of cation adduction and as well as difficulties with achieving a stable electrospray.12,14 In this work, native MS was used to provide structural insights into the EthR–DNA interaction. Our results indicate that up to six subunits of EthR are able to bind to its operator.
Mass spectra of histidine-tagged EthR or EthR–DNA complexes were obtained by nano-electrospray ionization (nESI) from a hybrid quadrupole time-of-flight (qTOF) SYNAPT HDMS (Waters) instrument. The native MS spectrum of EthR alone confirmed the dimeric nature of EthR in solution, with the charge state distribution being centered around the 13+ state (Fig. 1a). The observed mass of dimeric EthR (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 475 ± 97 Da) was consistent with the theoretical mass of the dimer of the construct (50456 Da). A small amount of monomeric EthR was also observed, at about 5% of the total protein content.
475 ± 97 Da) was consistent with the theoretical mass of the dimer of the construct (50456 Da). A small amount of monomeric EthR was also observed, at about 5% of the total protein content.
 | ||
| Fig. 1 Native MS reveals the formation of EthR4–DNA and EthR6–DNA complexes. MS spectra of (a) EthR alone showing that the protein exists predominantly as the dimer, (b) DNA62 alone, and (c) a mixture of EthR and DNA62 in a 8:1 ratio, showing the formation of both EthR4–DNA62 and EthR6–DNA62 complexes. | ||
The native mass spectrum of DNA62 alone showed that while DNA62 existed primarily in its expected duplex state centered around the 10+ charge state, a fraction of the DNA was single-stranded (DNAss62), which could be due to an excess of one of the two complementary oligonucleotides (Fig. 1b). DNA m/z signals were generally broad, which could be a result of the large amount of cations that must be adducted to the polyanionic DNA in order for the nucleic acids to be detected in positive-ion mode.15
Optimization experiments were carried out to investigate the effect of parameters on EthR–DNA formation, including NH4OAc concentration (Fig. S2, ESI†), Mg2+ ion concentration (Fig. S3, ESI†), incubation time (Fig. S4, ESI†), and buffer exchange conditions (Fig. S5 and S6, ESI†). Under the optimized conditions, EthR (20 μM) and DNA62 (2.5 μM) were mixed together in an 8:1 ratio and subjected to nESI-MS. Surprisingly, instead of the expected EthR8–DNA62 complex, only the EthR6–DNA62 complex (192473 ± 44 Da), centered around the 24+ state, and the EthR4–DNA62 complex (140604 ± 23 Da), centered around the 22+ state, were detected (Fig. 1c). The observed masses are about 1% higher than the theoretical masses of 189555 and 139099 Da for the EthR6–DNA62 and EthR4–DNA62 species, respectively, which can be attributed to the adduction of weakly-bound molecules or ions. The EthR6–DNA62 and EthR4–DNA62 complexes are presumably formed by the assembly of three or two EthR dimers, respectively, onto the DNA. In contrast, no protein–DNA complexes were observed by native MS when EthR was mixed with a random 55 bp sequence (DNAR55) in a 8:1 or 12:1 ratio (Fig. S7, ESI†), indicating that the complexes formed between EthR with its operator DNA62 were specific.
The proportion of EthR and DNA62 was varied in order to investigate whether alternative protein–DNA complexes could be formed at different protein to DNA ratios. At a 6:1 or 7:1 ratio of EthR to DNA62, the EthR4–DNA62 and EthR6–DNA62 complexes were detected as the major and minor species, respectively, and some free DNA could also be observed (Fig. S8a and b, ESI†). The relative intensity of free DNA62 in the native mass spectra was much higher than expected for a slight excess of DNA over protein, which could be due to the greater ionization efficiency of DNA compared to the protein–DNA complex, as has been previously reported.11 As the proportion of EthR was increased to 8:1, the EthR6–DNA62 complex became the predominant protein–DNA species, and only free protein could be detected (Fig. S8c, ESI†). Even at a 12:1 ratio of EthR to DNA62, the EthR6–DNA complex was the largest species detected and no evidence of non-specific binding giving rise to higher oligomeric entities could be observed (Fig. S8d, ESI†). A small amount of the EthR5–DNA62 complex was also sometimes observed (Fig. S9, ESI†), which could presumably be formed from the association of a monomeric EthR subunit with a EthR4–DNA62 complex. However, the relatively low abundance of the EthR5–DNA62 complex and the EthR3–DNA37 complex (see below) compared to complexes containing an even number of EthR subunits suggests that their biological relevance may be relatively minor. An alternative interpretation is that these odd-numbered species may represent transient intermediates during protein–DNA complex formation.
The stoichiometric heterogeneity of the protein–DNA species observed by native MS could possibly account for the difficulties in obtaining an X-ray crystal structure of the EthR–DNA complex thus far. A longer 106 bp DNA sequence (DNA106) encompassing the entire ethA-R intergenic region also gave rise to EthR6–DNA106 and EthR4–DNA106 complexes when incubated with EthR (Fig. S10, ESI†), however the quality of the native mass spectra was reduced.
Next, EthR was incubated separately with the DNA duplexes DNA37 and DNA36 (Fig. S1, ESI†), and native mass spectra of the resulting complexes were recorded. In the SPR experiments reported previously,4 each DNA37 duplex bound to an average of 4.5 EthR molecules, whereas with DNA36, protein–DNA binding was greatly decreased. In native MS, a mixture of EthR (15 μM) and DNA37 (2.5 μM) produced mostly the EthR4–DNA37 complex, centered around the 19+ charge state, along with some of the EthR2–DNA37 complex and a small amount of the EthR3–DNA37 complex (Fig. 2a). With DNA36, mainly EthR2–DNA36 complexes were formed, along with a small amount of the EthR4–DNA36 complex (Fig. 2b). Furthermore, significant quantities of both unbound protein and DNA were observed, which is consistent with the greatly diminished binding capacity of EthR to DNA36.
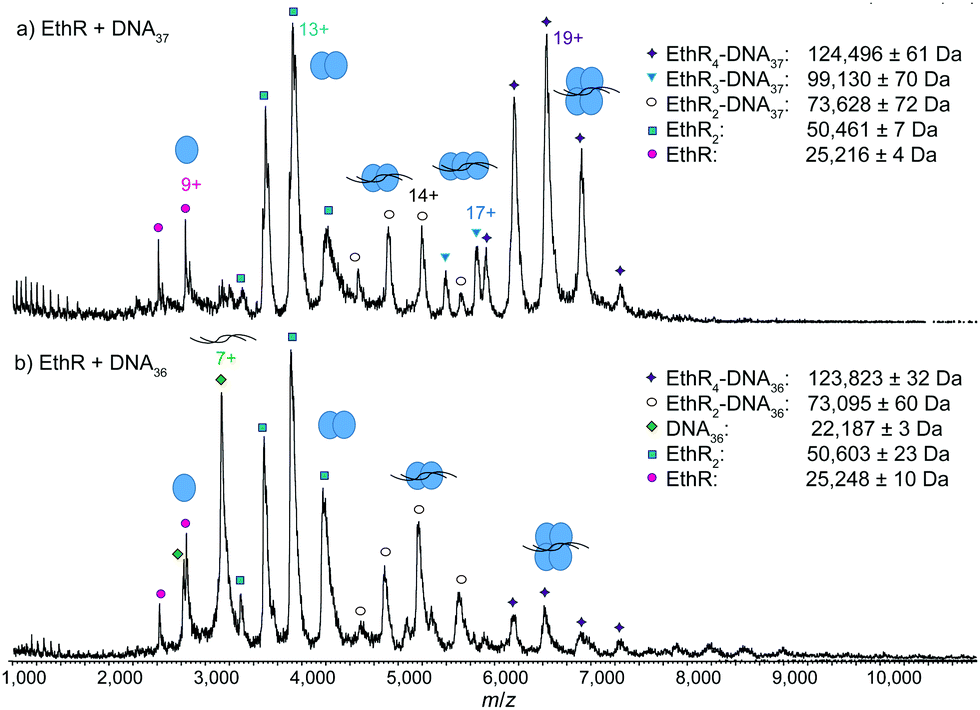 | ||
| Fig. 2 Native MS showing reduced stoichiometry of EthR with the shorter DNA37 and DNA36 sequences. MS spectra of (a) EthR and DNA37 in a 6:1 ratio, showing the formation of mainly EthR4–DNA37 complex, some EthR2–DNA37 complex, and a trace of EthR3–DNA37 complex, and (b) EthR and DNA36 in a 6:1 ratio, showing formation of mainly EthR2–DNA36 complex and some EthR4–DNA36 complex. | ||
To further investigate the EthR–DNA interaction, DNA62 was split into two half-sites, the left-hand site (DNAL31) and the right-hand site (DNAR31) (Fig. S1, ESI†). Each site contained one copy of an imperfect direct repeat that was thought to be responsible for the EthR–DNA binding interaction.4 When EthR (15 μM) was incubated with DNAL31 (2.5 μM), only EthR2–DNAL31 complexes were formed (Fig. 3a). On the other hand, mainly EthR4–DNAR31 complexes were observed with DNAR31, along with a small amount of EthR2–DNAR31 and EthR3–DNAR31 species (Fig. 3b). As before, the significant quantities of both unbound protein and DNA that are observed suggests that the interactions between EthR and the individual half-sites are relatively weak.
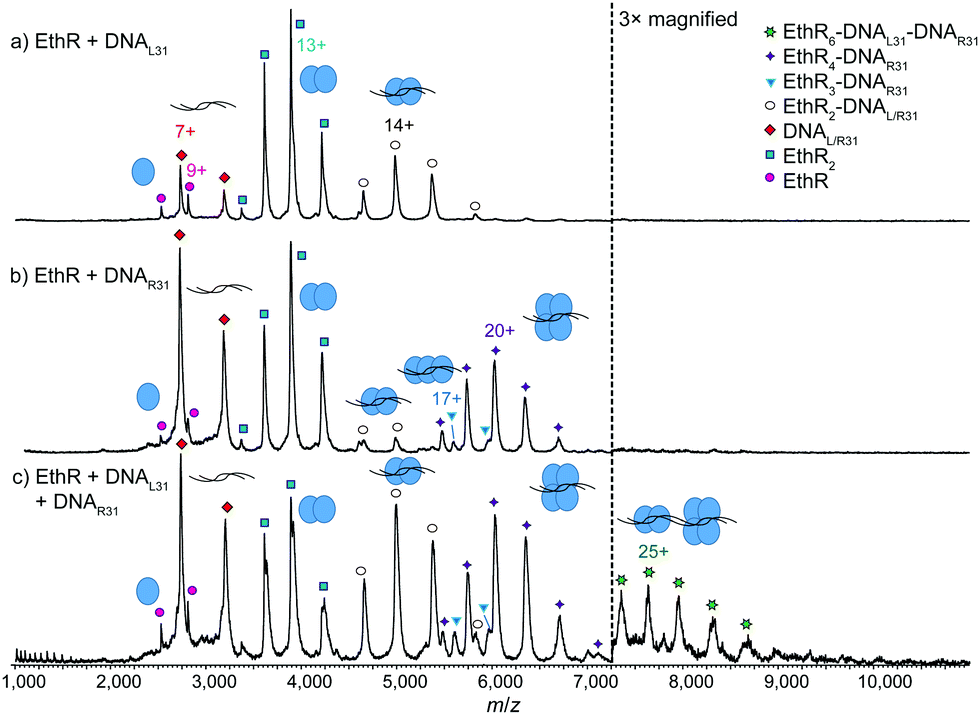 | ||
| Fig. 3 Native MS showing recapitulation of the EthR6–DNA complex from two independent half-sites. MS spectra of (a) EthR and DNAL31 in a 6:1 ratio, showing formation of mainly EthR2–DNA complex, and (b) EthR and DNAR31 in a 6:1 ratio, showing formation of mainly EthR4–DNA complex, along with a small amount of EthR2–DNA and EthR3–DNA complexes. (c) When EthR was mixed with both DNAL31 and DNAR31, a small amount of the putative EthR6–DNAL31–DNAR31 complex was formed. | ||
Intriguingly, when EthR was incubated with both DNAL31 and DNAR31 at the same time, a small amount of higher-order species could be detected that could be putatively assigned as the EthR6–DNAL31–DNAR31 complex (189872 ± 23 Da) (Fig. 3c). This complex could be formed from the association of the EthR2–DNAL31 complex with the EthR4–DNAR31 complex in solution. This interaction appears to be asymmetric, as homodimeric (EthR2–DNAL31)2 or (EthR4–DNAR31)2 species were not detected when EthR was treated with DNAL31 or DNAR31 separately. Additionally, DNAL31 and DNAR31 do not directly associate with each other in the absence of EthR (Fig. S11, ESI†), indicating that some kind of communication must exist between EthR and the DNA in order to form the putative EthR6–DNAL31–DNAR31 complex.
The EthR–DNA interaction was also investigated using isothermal titration calorimetry (ITC). EthR was titrated into DNA62, generating a complex, non-sigmoidal binding isotherm (Fig. S12, ESI†) that was analyzed using AFFINImeter software (S4SD). A stoichiometric equilibrium binding model was designed that assumed the stepwise formation of the EthR6–DNA complex from free EthR dimers and DNA62, proceeding through EthR2–DNA and EthR4–DNA intermediates (i.e., EthR2 + DNA62 ⇌ EthR2–DNA62 ⇌ EthR4–DNA62 ⇌ EthR6–DNA62). Fitting of the data to this model revealed a stoichiometry of 2.9 ± 0.1 EthR dimers per duplex of DNA62, which is consistent with the native MS data. The binding affinities of the first and second dimers to DNA62 were identical within experimental error (KD(1) = 3.8 ± 0.8 μM, KD(2) = 3.6 ± 0.6 μM). The third EthR dimer binds to DNA62 with weaker affinity (KD(3) = 10 ± 3 μM). The EthR–DNA62 binding affinities derived from ITC in this work are about an order of magnitude weaker than those previously determined by SPR for the EthR–DNA106 interaction (average KD = 146 nM).4
The calculated thermodynamic parameters indicate that the binding of the first EthR dimer to DNA is entropically favorable (TΔS = +12.1 ± 0.4 kcal mol−1) but enthalpically unfavorable (ΔH = +4.7 ± 0.4 kcal mol−1) (Fig. 4). The favorable increase in entropy may be due to the release of countercations or solvent molecules that were associated with the DNA.16 However, the enthalpic penalty of binding stands in contrast to that observed for most major groove-binding proteins.17 This suggests that significant structural rearrangement takes place in EthR and/or the DNA to accommodate binding of the first EthR dimer. Similar to what has been observed with QacR,18 which binds to DNA as a dimer of dimers, this structural rearrangement could then present the DNA in a conformation that readily accepts the second dimer, thus accounting for the positive cooperativity of the EthR–DNA interaction. Indeed, binding of the second EthR dimer is driven almost entirely by enthalpy (ΔH = −6.2 ± 0.6 kcal mol−1) with only a minor entropic component (TΔS = −1.3 ± 0.6 kcal mol−1). The third dimer binds via a combination of favorable enthalpic (ΔH = −2.1 ± 0.6 kcal mol−1) and entropic (TΔS = −4.7 ± 0.6 kcal mol−1) terms.
 | ||
| Fig. 4 Thermodynamic parameters of the EthR–DNA62 interaction as determined by ITC. ΔG, ΔH and −TΔS values are shown for the binding of the first, second and third EthR dimers with DNA. Moles of EthR are given as the dimer. | ||
The discrepancy in the stoichiometry of the EthR–DNA complex as determined by native MS and ITC versus the previous SPR analysis4 may be a consequence of the correction that was applied to the SPR data. In that experiment, a correction factor of 0.73 was applied to the response of DNA, reflecting the different molar refractive indices of DNA versus proteins.19 However, more recent work has shown that proteins and nucleic acids behave similarly in SPR, and so there may have been no need for this correction factor.20 When this is taken into account, the previous SPR data instead suggest that each DNA62 duplex binds to 6.3 EthR molecules, while each DNA37 duplex binds to 3.3 EthR molecules, both numbers being consistent with the present work.
In conclusion, structural insights into the interaction between EthR and its operator have been obtained by native MS. While EthR was observed to exist as a dimer in solution as expected, the interaction of EthR with the full-length operator produced EthR6–DNA and EthR4–DNA complexes. The stoichiometry of the EthR–DNA complex was confirmed by ITC, which also revealed thermodynamic parameters that were consistent with a cooperative mode of binding. This study also highlights the capability of native MS to provide structural information on macromolecular assemblies, including where heterogeneous mixtures of complexes exist that are intractable to crystallization. Experiments are being conducted to provide structural-level detail of the EthR–DNA complex and to elucidate the precise mechanism of the EthR–DNA interaction.
The authors thank Eva Muñoz (AFFINImeter) for helpful discussion on the ITC analysis. D. S.-H. Chan acknowledges the support of the Croucher Foundation and the Cambridge Commonwealth, European and International Trust for receipt of a Croucher Cambridge International Scholarship. W.-G. Seetoh was supported by the Agency for Science, Technology and Research (A*STAR) Singapore (PhD sponsorship) and the Wellcome Trust Strategic Award (090340/Z/09/Z). B. N. McConnell acknowledges Cambridge Australia Scholarships for the award of a Poynton Scholarship, the Cambridge Philosophical Society and the Access to Learning Fund. S. E. Thomas is supported by the Cystic Fibrosis Trust. V. Mendes and M. Blaszczyk acknowledge the Bill & Melinda Gates Foundation (subcontract by the Foundation for the National Institutes of Health – NIH) (OPP1024021).
Notes and references
- World Health Organization, Global Tuberculosis Report, 2015.
- T. A. Vannelli, A. Dykman and P. R. Ortiz de Montellano, J. Biol. Chem., 2002, 277, 12824 CrossRef CAS PubMed.
- X. Duan, X. Xiang and J. Xie, FEMS Microbiol. Lett., 2014, 360, 87 CrossRef CAS PubMed.
- J. Engohang-Ndong, D. Baillat, M. Aumercier, F. Bellefontaine, G. S. Besra, C. Locht and A. R. Baulard, Mol. Microbiol., 2004, 51, 175 CrossRef CAS PubMed.
- N. Willand, B. Dirie, X. Carette, P. Bifani, A. Singhal, M. Desroses, F. Leroux, E. Willery, V. Mathys, R. Deprez-Poulain, G. Delcroix, F. Frenois, M. Aumercier, C. Locht, V. Villeret, B. Deprez and A. R. Baulard, Nat. Med., 2009, 15, 537 CrossRef CAS PubMed.
- J. L. Ramos, M. Martínez-Bueno, A. J. Molina-Henares, W. Terán, K. Watanabe, X. Zhang, M. T. Gallegos, R. Brennan and R. Tobes, Microbiol. Mol. Biol. Rev., 2005, 69, 326 CrossRef CAS PubMed.
- F. Frénois, J. Engohang-Ndong, C. Locht, A. R. Baulard and V. Villeret, Mol. Cell, 2004, 16, 301 CrossRef PubMed.
- L. G. Dover, P. E. Corsino, I. R. Daniels, S. L. Cocklin, V. Tatituri, G. S. Besra and K. Fütterer, J. Mol. Biol., 2004, 340, 1095 CrossRef CAS PubMed.
- H. Hernandez and C. V. Robinson, Nat. Protoc., 2007, 2, 715 CrossRef CAS PubMed.
- G. R. Hilton and J. L. P. Benesch, J. R. Soc., Interface, 2012, 9, 801 CrossRef CAS PubMed.
- A. Butterer, C. Pernstich, R. M. Smith, F. Sobott, M. D. Szczelkun and J. Tóth, Nucleic Acids Res., 2014, 42, 5139 CrossRef CAS PubMed.
- A. Politis, A. Y. Park, Z. Hall, B. T. Ruotolo and C. V. Robinson, J. Mol. Biol., 2013, 425, 4790 CrossRef CAS PubMed.
- N.-T. Nguyen-Huynh, J. Osz, C. Peluso-Iltis, N. Rochel, N. Potier and E. Leize-Wagner, Biophys. Chem., 2016, 210, 2 CrossRef CAS PubMed.
- V. Gabelica, C. Vreuls, P. Filée, V. Duval, B. Joris and E. D. Pauw, Rapid Commun. Mass Spectrom., 2002, 16, 1723 CrossRef CAS PubMed.
- F. Rosu, S. Pirotte, E. D. Pauw and V. Gabelica, Int. J. Mass Spectrom., 2006, 253, 156 CrossRef CAS.
- D. C. Mikles, V. Bhat, B. J. Schuchardt, C. B. McDonald and A. Farooq, J. Mol. Recognit., 2014, 27, 82 CrossRef CAS PubMed.
- P. L. Privalov, A. I. Dragan, C. Crane-Robinson, K. J. Breslauer, D. P. Remeta and C. A. S. A. Minetti, J. Mol. Biol., 2007, 365, 1 CrossRef CAS PubMed.
- M. A. Schumacher, M. C. Miller, S. Grkovic, M. H. Brown, R. A. Skurray and R. G. Brennan, EMBO J., 2002, 21, 1210 CrossRef CAS PubMed.
- C. Speck, C. Weigel and W. Messer, EMBO J., 1999, 18, 6169 CrossRef CAS PubMed.
- C. Di Primo and I. Lebars, Anal. Biochem., 2007, 368, 148 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Methods and supplementary spectra. See DOI: 10.1039/c7cc00804j |
| This journal is © The Royal Society of Chemistry 2017 |