
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel magneto-responsive membrane for remote control switchable molecular sieving†
Xi
Lin
ab,
Rong
Huang
ab and
Mathias
Ulbricht
*ab
aLehrstuhl für Technische Chemie II, Universität Duisburg-Essen, 45117 Essen, Germany. E-mail: mathias.ulbricht@uni-essen.de
bCENIDE – Center for Nanointegration Duisburg-Essen, 47057 Duisburg, Germany
First published on 22nd December 2015
Abstract
Stimuli-responsive separation membranes with tunable molecular scale pore size, which are desirable for on-demand sieving of targeted macromolecules, have attracted increasing attention in recent years. In this study, novel magneto-hydrogel pore-filled composite membranes with excellent magneto-responsivity and tunability for molecular sieving have been developed. Such membranes comprising magnetic nanoparticles (MNPs) as localized heater which can be excited by high frequency alternating magnetic field (AMF), poly(N-isopropylacrylamide) (PNIPAAm) hydrogel network as the sieving medium and actuator, and polyethylene terephthalate (PET) track-etched membrane as robust support, have been prepared via in situ reactive pore-filling functionalization. Rheological study has been carried out first to investigate the influence of MNPs and initiation methods on gelation kinetics and microstructure of the MNP–PNIPAAm composite hydrogels, and to identify proper conditions for further pore-filling functionalization. Then AMF distribution of chosen field condition and its heating effectiveness for MNPs and MNP–PNIPAMm composite hydrogel were studied. Pre-functionalization of the PET membranes with linear polymer chains with different composition were compared with respect to their effects for achieving desired MNP loading and fixation of the hydrogel network in the pores. At last, in situ reactive pore-filling functionalization was carried out to immobilize robust magneto-hydrogel in the pores of the membranes. Conditions were investigated and optimized to obtain functionalized membranes with high MNP loading and suited PNIPAM network properties, i.e. good stimuli-responsivity and sieving in the ultrafiltration range. The excellent thermo- and magneto-responsivity of obtained pore-filled membranes was proved by its large and reversible change of water permeability in response to switching on and off the AMF. Finally, it was demonstrated by filtration of dextrans with different molecular weights that the membranes had ultrafiltration properties and that large changes of their molecular sieving performance could be obtained by “remote control” with the external AMF.
1. Introduction
In the last few decades, stimuli-responsive materials including responsive membranes have become a popular topic, and membranes which respond to different signals (e.g. temperature, pH, ions, special molecules or electromagnetic irradiation) have been developed.1–4 Despite the large number of publications, most reported membranes do not have high resolution for the separation of nanoparticular or molecular targets. They either rely on the stimuli-responsiveness of functional polymers blended into a membrane polymer or grafted as brushes on the membrane surface,5–12 on the volume phase transition of blended micro- or nanogel particles,13–16 and on expansion/contraction of responsive substrates with pores from sacrificial or volatile components.17,18 By this means, most previously reported stimuli-responsive membranes achieve micrometer or sub-micrometer range pore size tuning for changing hydraulic permeability5–7,16–18 and molecules and ions diffusion.8,9,13–15 A different strategy is the integration of functional nanoparticles in membranes, as light-responsive local heater to improve the flux through nanofiltration membranes,19 as surface-immobilized magneto-responsive “nano mixer” to reduce concentration polarization20 or as magnetic enzyme carrier in a biocatalytic membrane system.21 As sizes of biomacromolecules are only few nanometers, for applications like drug delivery, biochemical analysis and down-stream processing of bioproducts, it would be very attractive to develop stimuli-responsive separation membranes with tunable pore size in that molecular scale, to achieve on-demand switchable sieving of targeted macromolecules or nanoparticles.22 However, due to the intrinsic rather rough tunability of most membrane functionalizations mentioned above, it is hard to reach precise control over molecular scale transport. Among the very few exceptions were functionalizations of support membranes within sub-micrometer sized pores, where stimuli-responsive separation for macromolecules or small nanoparticles had been achieved.10–12 Another way of preparing responsive membranes with pore sizes in the range below 100 nm as well as narrow pore size distribution and high porosity by using phase separation of self-assembled micelles of stimuli-responsive diblock polymers is also quite attractive.23,24 However, so far only pH-responsive membranes have been obtained with this method and there are concerns about the mechanical properties of such membranes.Because polymeric hydrogels have mesh sizes in the lower nm range and high porosity, they are also used as excellent sieving media for molecules.25,26 Based on the size-exclusion effect, poly(acryl amide) hydrogels have been for long time used for electrophoresis.27 Their adjustable network structure (by cross-linker monomer fraction and total monomer concentration, i.e. via formation of chemical and physical cross-links) and chemical durability would make them more favorable than other materials mentioned above. Moreover, when they are stabilized within a porous (membrane) support, their application could be expanded to pressure-driven molecule filtration.28–30 Among all kinds of hydrogels, stimuli-responsive ones are especially interesting because their mesh sizes could not only be defined by composition and synthesis conditions, but also dynamically tuned by external stimuli.30–33 Previously, novel polymeric hydrogel pore-filled membranes with reversible thermo-responsive molecular sieving effect had been developed by Adrus and Ulbricht.34 Such membranes used the thermo-responsive poly(N-isopropylacrylamide) (PNIPAAm) hydrogel as the sieving medium because the hydrogel had been immobilized inside the pores of a robust polyethylene terephthalate (PET) track-etched microfiltration membrane to enhance its mechanical stability. It had been demonstrated that the membranes have excellent thermo-responsive (macro)molecule size selectivity and stable separation performance under relatively high pressure (up to 4 bar). Upon increasing the temperature above the lower critical solution temperature of PNIPAAm (32 °C), the flux increased very much and the molecular sieving disappeared, which could be ascribed to a micro-syneresis of the pore-immobilized temperature-responsive hydrogel, leading to much larger mesh size compared to room temperature.
However, the conventional temperature actuation has drawbacks like relatively slow response and high energy consumption as it demands the heating of the entire feed. Moreover, there is the risk to denature biomacromolecules which are very often the separation targets. Magneto-heating which had been largely investigated for hyperthermia therapy for tumor treatment35–37 could be adopted to solve those problems. It had been proved that by coupling magnetic nanoparticles (MNP) as localized heaters with thermo-responsive polymers having a transition temperature in the range up to 40 °C, the heat generated by the MNP is effective enough to cause the phase transition of these thermo-responsive polymers. Such mechanism has been used to develop magneto-responsive drug delivery system for pulsed drug release.13–15,37–39 Very recently, it had been shown that permeability through membrane pores containing immobilized MNPs and grafted with linear PNIPAAm chains can be switched reversibly by alternating magnetic field (AMF), but due to large effective pore size (about 500 nm), no size selectivity had been achieved.40
Here, it is proposed to develop a MNP–polymer hydrogel pore-filled composite membrane to achieve magneto-responsive molecule sieving performance. The idea is to use magnetic iron oxide nanoparticles (size 40 nm) as the localized heater, pore-filling PNIPAAm hydrogel as the actuator and sieving medium, and commercial PET track-etched membrane as the robust support, to construct a magneto-responsive hydrogel pore-filled membrane. It is anticipated that by manipulating AMF and thus controlling the heat generation of MNPs, the micro-syneresis of the PNIPAAm network can be locally controlled and magneto-responsive molecule sieving can be achieved (Fig. 1). Because there is no need to heat up the whole feed, it is expected that such membrane would have faster response, be more energy-efficient and maybe can also avoid the denaturing of biomolecules to be released/separated through/by such a membrane.
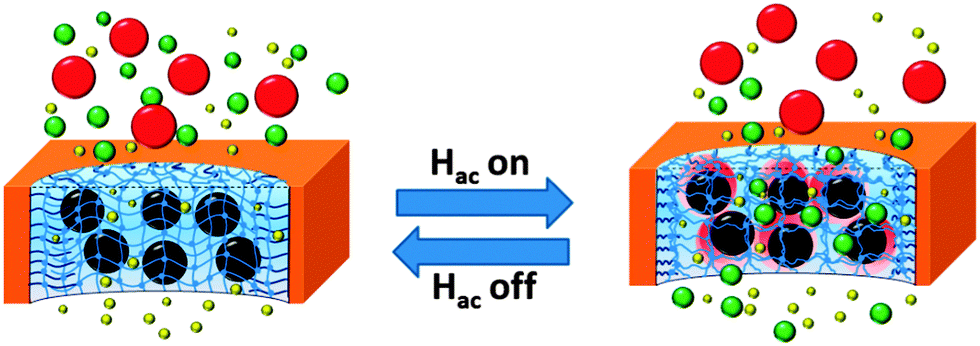 | ||
| Fig. 1 Schematic illustration of magneto-responsive separation membrane: reversible change of molecule sieving through pore-confined polymeric hydrogel network by remote control of immobilized “nano heaters” with alternating magnetic field. | ||
2. Experiments
2.1 Materials
Track-etched PET membranes with a nominal pore diameter of 1000 nm (measured via gas flow/pore dewetting permporometry: 1582 nm34) and a thickness of 23 μm from Oxyphen (Germany) were used. They were washed in ethanol for one hour and dried (40 °C) over night before using for functionalization. Amphiphilic polymer coated magnetic iron oxide nanoparticles (40 nm) with carboxylic acid groups on their surface in aqueous dispersion (5 mg ml−1) from Ocean NanoTech (USA) were used as received. Monomer N-isopropylacrylamide (NIPAAm) was from Acros (Belgium) and recrystallized with hexane before use. Dimethylaminoethyl methacrylamide (DMAEMA) from Polysciences Inc. (USA) was used as comonomer for pre-modification step (“method 2”) as received. N,N′-Methylenebisacrylamide (MBA) from Sigma-Aldrich (Germany) was used as cross-linker for hydrogel syntheses. Irgacure 2959 from Ciba Chemicals (Switzerland) was used as UV initiator for rheological study of gelation. Ammonium persulfate (APS) from Acros (Belgium) was used as redox initiator together with tetramethylethylenediamine (TEMED) from Acros as accelerator, for rheological study of gelation and membrane pore-filling functionalization. The photo-initiator benzophenone from Acros was used for the pre-modification step. Sodium chloride from VWR (Germany) was used to reduce the electrostatic repulsion between negatively charged MNPs during the pore loading process. Dextrans (2000 kg mol−1 from Sigma-Aldrich; 500 kg mol−1 from Pharmacia Fine Chemicals Inc., Sweden, 4 kg mol−1 from Serva, Germany) were used as test solutes for filtration experiment. Ethanol from Fisher Chemicals (Germany) was used for base membrane washing and as solvent for benzophenone coating for pre-modification step. For all experiments, water was purified with a Milli-Q system (Millipore, USA).2.2 Rheological study of bulk MNP–PNIPAM composite hydrogels
To study the influence of MNPs on gelation kinetics and microstructure of the MNP–PNIPAAm composite hydrogels and to choose a suitable polymerization method for pore-filling procedure, rheological studies were carried out for in situ UV- and redox-initiated cross-linking polymerization of NIPAAm with different MNP concentrations, according to a previous work.41Rheometer model MCR-300 (Anton Paar, Austria) equipped with external UV-light source (100 watt mercury lamp, EXFO Omnicure Series 1000, EXFO, Quebec, Canada) and Peltier device for temperature control was used. Cone-plate geometry with an upper plate of cone angle 0.1° and a diameter 25 mm was chosen and different lower plates were used for different polymerization methods. An optically transparent glass-based lower plate was used for UV polymerization while a Peltier plate was used for redox polymerization. First to monitor the gelation process, in situ oscillatory shear measurement with time sweeping mode was carried out. 200 μl reaction mixture was used for each measurement. The concentration of NIPAAm was 1 mol l−1, that of MBA 0.02 mol l−1, that of initiator (Irgacure 2959 for UV and APS for redox polymerization) was 0.02 mol l−1, and the MNP concentrations were varied from 0 to 1 wt% (relative to NIPAAm). To trigger the polymerization, UV light with the intensity 21 mW cm−2 was used for UV polymerization and directly coupled into the rheometer cell via fiber optics. For redox polymerization 0.01 mol l−1 TEMED was added and mixed for 30 seconds before loading the reaction solution onto the plate. The normal force was set at 0 N. An angular frequency of 10 rad s−1 and small strain amplitude of 0.01% were chosen to ensure linear regime of oscillatory deformation. During all measurements, temperature was controlled at 21 °C and the evaporation of solvent was minimized by using a solvent trap. After the hydrogel had been fully polymerized, frequency sweeping was carried out to characterize mechanical properties and to estimate the microstructure of the hydrogels. While the temperature was kept constant at 21 °C, the normal force was reset to 0 N, and the strain amplitude was kept at 1%, an angular frequency in the range of 0.1–100 rad s−1 was applied and the storage modulus of hydrogel was measured. Based on rubber elasticity theory, average mesh size of the hydrogel was estimated from rheological data according to the following equation.41,42
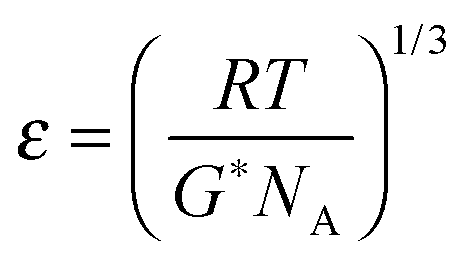 | (1) |
2.3 Investigation of alternating magnetic field (AMF) condition
The field amplitude between the coil sandwich was measured by a self-made pickup probe (2 loops made of fine copper wire with a diameter of 7.3 mm) coupled with an oscilloscope (ESI,† Fig. S1). Due to electromagnetic induction, there will be electrical current induced in the probe, changing simultaneously with alternating magnetic field. By taking coordinate axes along the radius of the coil (r) and height of the coil (h), and placing the probe at different sites between the coils, induced peak voltage at each site was detected by oscilloscope and recorded. To obtain high magnetic field amplitude, here relatively high power input of the generator was chosen: the current was I = 15.6 A, and the frequency f = 745 kHz. The electromagnetic field amplitude (H0) at each site was calculated according to following equations.
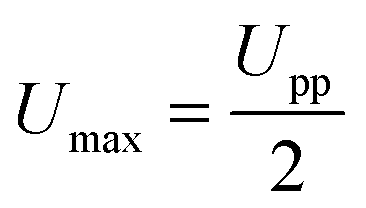
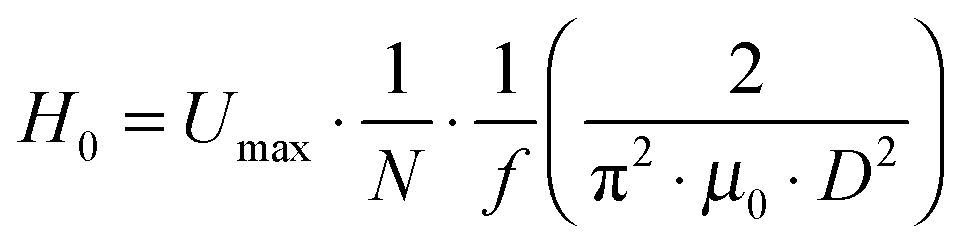 | (2) |
| SLP = c·(ms/mFe)·(ΔTs − ΔTw)/Δt | (3) |
2.4 Membrane functionalization
The membrane functionalization procedure is shown schematically in Fig. 2. PET track-etched membranes were first pre-modified by grafting polymer chains onto pore wall surface as anchors and thereafter pore-filling functionalized by in situ cross-linking polymerization of magneto-hydrogel inside membrane pores. | ||
| Fig. 2 Schematic illustration of methods to fabricate magneto-responsive membranes: as first step surface-grafting of linear polymer chains as anchor for the hydrogel network with iron oxide nanoparticles prepared in second step via in situ cross-linking polymerization. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) for about 60 min before immersing them in degassed aqueous monomer solutions in Petri dish. Then the Petri dish was placed inside a UV system (UVA Cube 2000, Hönle AG, Germany; equipped with a 20 cm long mercury lamp, allowing a homogeneous irradiation of 0.1 m2 area via reflecting walls) with a UVA filter on top (to prevent membrane degradation). UV irradiation was carried out with UV intensity of 40 mW cm−2 for 15 minutes. At last, functionalized membranes were washed with water for 24 hours, dried at 40 °C and cut into 25 mm samples, ready for characterization and further pore-filling functionalization.
:1) for about 60 min before immersing them in degassed aqueous monomer solutions in Petri dish. Then the Petri dish was placed inside a UV system (UVA Cube 2000, Hönle AG, Germany; equipped with a 20 cm long mercury lamp, allowing a homogeneous irradiation of 0.1 m2 area via reflecting walls) with a UVA filter on top (to prevent membrane degradation). UV irradiation was carried out with UV intensity of 40 mW cm−2 for 15 minutes. At last, functionalized membranes were washed with water for 24 hours, dried at 40 °C and cut into 25 mm samples, ready for characterization and further pore-filling functionalization.
In order to obtain a denser hydrogel network for more effective molecular sieving, the concentrations of all chemicals were increased for preparation of the pore-filled membrane used in dextran filtration experiments: 1.1 mol l−1 NIPAAm, 0.022 mol l−1 MBA, 1.5 wt% MNPs (relative to monomer), 0.022 mol l−1 APS, 50 mmol l−1 NaCl, and 0.011 mol l−1 TEMED were used.
2.5 Characterization of functionalized membranes
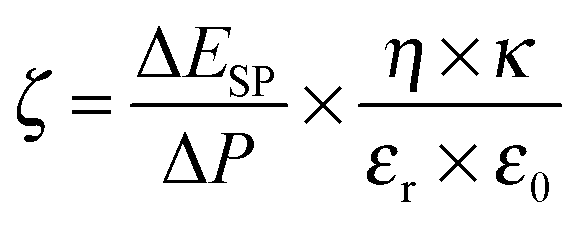 | (4) |
:20) using a K550 sputter coater from Emitech Ltd. (Ashford, UK). Then SEM/EDX was performed with instrument ESEM Quanta 400 FEG from FEI Co. (Hillsboro, USA).

 | (5) |
For the pre-modified membranes, to evaluate the length of the grafted polymer chains, hydrodynamic layer thickness was calculated according to Hagen–Poiseuille equation.44,45

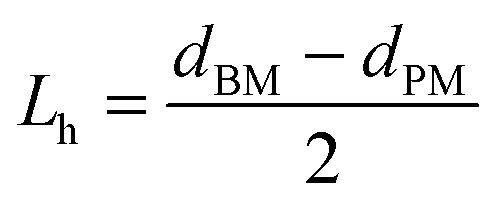 | (6) |
Moreover, gating factor R45/25, which represents the ratio of effective average pore diameter, i.e. permeability, at 45 °C and 25 °C,47 was also introduced and calculated, to quantify the thermo-responsivity of pre-modified membranes.
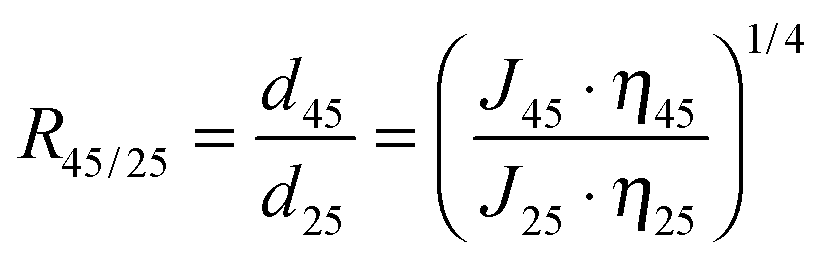 | (7) |
For pore-filled membrane, a thermo-responsivity factor, N45/25, which represents the permeability ratio at different temperatures, without the water viscosity influence, was also introduced:
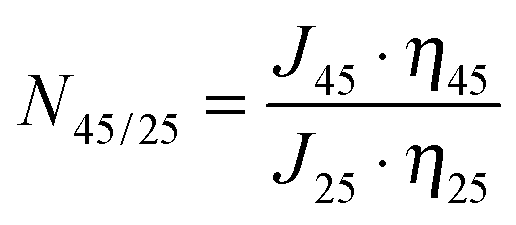 | (8) |
| R = (1 − CP/CF) × 100(%) | (9) |
To eliminate fouling effects, the filtration tests were done in the sequence of 4 kDa, 500 kDa and 2000 kDa. Furthermore, in between the dextran filtrations, the membrane was washed with water for 2 hours with shaking and 3 times of water change. And to monitor possible fouling, water flux tests were carried out to see the effectiveness of washing. In all cases, permeability of membrane was monitored by weighting the permeate.
3. Results and discussion
3.1 AMF condition
When placing the coordinate axis along the radius (r) and height of the coils (h) as shown in Fig. 3a, the electromagnetic field distribution between the inductor coils, calculated from the induced peak voltage (eqn (2)), was as shown in Fig. 3b. The field amplitude distribution was not even; it was quite high near to surface (h = ±40 mm) and in the centre (r = 0 mm) of the coils and relatively low along the outer circle of the coils (r = ±10 mm). Moreover, results of a computer simulation which had been done to describe the amplitude distribution based on the inductor coil geometry (Fig. 3c) match well with the experimental results and further prove the uneven distribution of field amplitude. Therefore, in order to ensure constant field conditions throughout all experiments, the same current and frequency inputs (I = 15.6 A, f = 745 kHz) were always used and the samples (hydrogels and membranes) were always placed in the centre of the middle plane (h = 0 mm), where the AMF amplitude was 20 ± 10 kA m−1.
 | ||
| Fig. 3 Electro-magnetic field amplitude distribution between the two inductor coils; (a) geometry of the inductor coils and placing of coordinates; (b) measured field amplitude distribution with current input of 15.6 A and frequency of 745 kHz; (c) computer simulation of field amplitude distribution (highest intensities red, lowest blue; no scale used because simulation had been done with other current and frequency input parameters than in experiments). | ||
The temperature changes for water and MNP dispersion after 5 minutes of AMF exposure are shown in Table 1. The SLP calculated using eqn (3) is 672 W g−1 and it correlates well with the reported SLP of iron oxide nanoparticles with similar size and under similar field conditions (magnetosome: diameter 30–40 nm, f = 410 kHz, H = 10 kA m−1, SLP = 960 W g−1),36 indicating the relatively good heating efficiency of the particles and AMF conditions used in this study.
| Sample | Time [minutes] | Temperature change [°C] |
|---|---|---|
| Water | 5 | 2 ± 0.4 |
| MNP dispersion | 5 | 4.4 ± 0.7 |
3.2 Investigations of bulk magneto hydrogels
To verify the good dispersion of MNP in the monomer solution, its zeta potential and stability of particle size in the monomer solution have been investigated. It was shown that the commercial MNP which had been surface functionalized with carboxylic acid groups are strongly negatively charged (ESI,† Fig. S3b) and are stable in the monomer solution (ESI,† Fig. S4).
UV-initiated gelation was studied first, because a previous investigation had shown that UV polymerization is a better option for in situ preparation of tailored functional hydrogels as well as fabrication of hydrogel pore-filled membranes.34,41 However, the results for composite hydrogels were not as ideal as expected. As shown in Fig. 4a, similar to the results of previous investigations, gelation during UV polymerization of monomer mixture without particles started soon (within 50 seconds) and was complete early (within 400 seconds). However, with the increase of MNP concentration, the gelation was systematically delayed and the storage modulus of the resulting hydrogel systematically decreased (by 20% when comparing 0% and 1% MNP fraction). This may be because the MNPs can block the penetration of UV light, which is responsible for the generation of radicals; this results in reduced extent of polymerization. Therefore, UV initiation was not considered a suited method to obtain a well-defined magneto-hydrogel and was not further studied here.
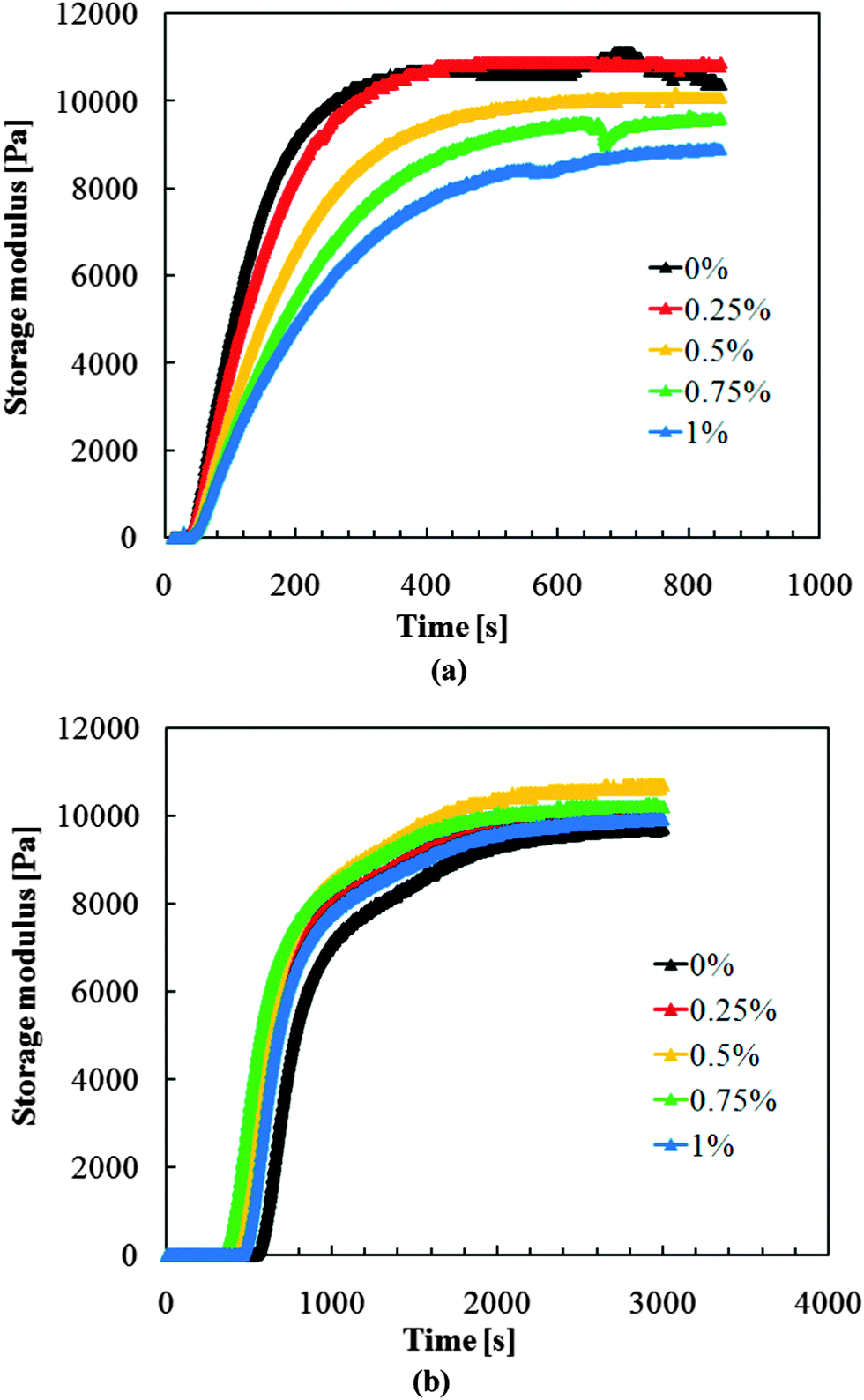 | ||
| Fig. 4 Gelation kinetics study by in situ rheology. (a) UV-initiated polymerization; (b) redox polymerization; both with different MNP concentrations in the reaction mixture comprising NIPAAm and MBA in water. | ||
Redox polymerization was evaluated as another option. As shown in Fig. 4b, redox polymerization took longer than UV-initiated polymerization to lead to gelation (300–500 seconds) and to reach full conversion (more than 2000 seconds). However, there was no apparent influence of the MNP concentration on the course of polymerization and the mechanical properties of the resulting hydrogels. Variations of storage modulus after full conversion were less than 3% around the average value for all data. The MNP–PNIPAAm composite hydrogels had similar storage modulus as the pure PNIPAAm hydrogel, indicating that redox polymerization is applicable to synthesize magneto-hydrogels at full conversion for the entire range of MNP loading. In addition, longer gelation time would make pore-filling preparation procedure easier to perform, as there will be more time to wait for all the chemicals, including the accelerator TEMED, to reach equilibrium in the membrane pores. Therefore, further investigations of the microstructure were carried out with hydrogels obtained after redox-initiation.
| MNPs concentration [%] | Storage modulus [Pa] | Mesh size [nm] |
|---|---|---|
| 0 | 10000 ± 500 |
7.3 |
| 0.25 | 10200 ± 500 |
7.2 |
| 1.0 | 10500 ± 500 |
7.2 |
 | ||
| Fig. 5 Thermal images of magneto-hydrogel and reference material without MNP after 1 minute of AMF exposure (I = 15.6 A, f = 745 kHz, H ∼ 20 kA m−1); inset shows photographs of the samples. | ||
3.3 Pre-modification of PET membranes
Successful pre-modifications of base membrane with the two different grafted polymers were verified by water flux measurements including thermo-responsivity and zeta-potential measurements.As shown in Fig. 6a, the much lower water permeability of modified membranes proved the successful grafting of polymer chains onto the track-etched pore walls. Further, the much higher water permeability at 45 °C than at 25 °C and the corresponding high gating factors of pre-modified membranes indicated excellent thermo-responsive permeability which was attributed to the grafted PNIPAAm. Very similar permeabilities have been obtained for “method 1” and “method 2”, indicating similar degree of grafting for the homo- and the copolymer. However, a very large decrease of water permeability had been observed with the increase of DMAEMA fraction in the reaction mixture beyond the concentration of 0.01 mol l−1, relative to the NIPMAAm concentration of 0.2 mol l−1 (see ESI,† Fig. S6, and related discussion). According to Hagen–Poiseuille law (eqn (6)), effective hydrodynamic layer thickness of grafted polymers was calculated from the water flux data and found to be within the range of 400 to 600 nm. This is smaller than the pore radius of ∼790 nm. Because photo-initiated free radical polymerization had been used, a gradient in polymer segment density from the pore wall to the interior can be expected, but the relatively large thickness should provide robust anchoring of the cross-linked hydrogel from the same polymer, in situ formed during the pore-filling step.
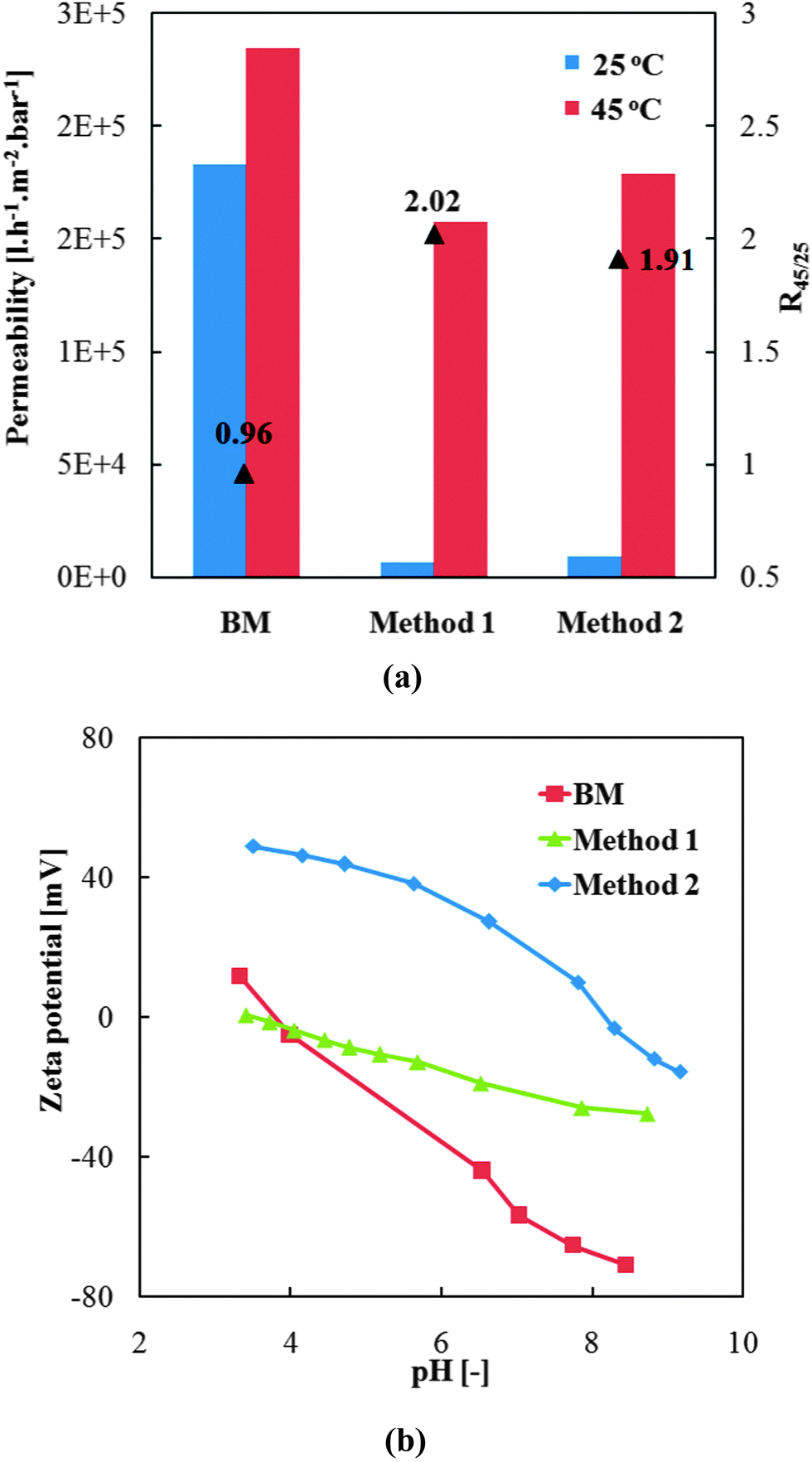 | ||
| Fig. 6 Thermo-responsivity of water flux (a) and zeta potential (b) of pre-modified PET track-etched membranes obtained by methods 1 and 2 (cf.Fig. 2). | ||
Furthermore, the pre-modification of base membrane should also facilitate the loading of MNPs by influencing the electrostatic interactions between the membrane surface and the MNPs. The zeta potential of the pre-modified membranes was characterized and the result is shown in Fig. 6b. First, the PET base membrane was negatively charged as expected because of the carboxylic acid groups generated by the alkaline hydrolysis step during the track-etching process in the PET. Surface of pre-modified membrane from “method 1” was less negative compared with the base membrane, which can be attributed to screening of PET surface charge by the neutral PNIPAAm hydrogel layer. The membrane functionalized by “method 2” showed much more positive zeta potential in the analyzed pH range, indicating the successful incorporation of PDMAEMA segments into the grafted PNIPAAm chains. Hence, considering also the results of the DMAEMA concentration variation (cf. ESI,† Section S6), only 0.01 mol l−1 of DMAEMA was added on the basis of 0.2 mol l−1 of NIPAM for “method 2”, as such ratio offers moderate positive charge density and comparable layer thickness as the “method 1”.
3.4 MNP loading of hydrogel pore-filled membrane
Several factors would influence the MNP loading: the pore size of the functionalized membranes, the electrostatic interactions between membrane surface and the MNPs, and the mutual electrostatic repulsion between the negatively charged MNPs. To enhance the loading of the MNP, pre-modifications with moderate chain lengths and in one case with positive charges (“method 2”) were adopted (cf. Section 3.3), a step-wise loading procedure for MNPs with long loading times was used, and small amounts of NaCl were also added to reduce the mutual repulsion between the MNPs. The results of the thus optimized procedure according to the two methods were characterized by SEM and EDX (Fig. 7). The images reveal that the outer and inner surface of the membrane is tightly covered with particles and that the hydrogel is rather hard to distinguish from the other features. It must be noted that the samples had been analyzed in dry state where the hydrogel is collapsed; consequently, the visualization of PNIPAAm in SEM had also been complicated in previous work.34 After optimization of the loading procedure, both routes yield high MNP amounts on and in the membrane, which had been a great improvement compared with first experiments where the loading had been done in one step and without addition of NaCl (cf. ESI,† Fig. S7). For” method 1”, the MNP loading on the surface estimated by EDX is 6 times higher than inside the pores. For “method 2”, loading of MNPs inside the pore is significantly higher than for “method 1” which can be attributed to the positively charged groups provided by grafted poly(NIPAAm-co-DMAEMA). That the loading with MNPs inside the pores was smaller than on the outer membrane surface could be due to sterical hindrance between particles and pores which are partially filled with grafted PNIPAAm (cf.Fig. 6a). However, it is also possible that a μm-thin hydrogel layer formed on the two outer surfaces of the membrane is contributing to that effect. This had been observed before,34 and it can be explained by the preparation procedure where the membrane which is filled with reaction mixture is then tightly sandwiched between two glass plates; the laminar film between membrane surface and glass will also be cross-linked to form a (composite) hydrogel. | ||
| Fig. 7 Visualization by SEM (left: top surface; right: cross-section; insets show details at higher magnification) and quantification by EDX of MNP loading (shown in red letters) for hydrogel pore-filled membranes obtained by the two different methods (cf. Fig. 2). | ||
3.5 Responsive water permeability of nanoparticle hydrogel pore-filled composite membranes
Thermo-responsive water permeability of pore-filled membranes was evaluated by water filtration at different temperatures (25 °C and 45 °C). In the previous work by Adrus and Ulbricht,34 for PET track-etched membrane with pore size of about 1600 nm and using 1.3 mol l−1 of NIPAM and 0.05 mol l−1 of MBA in reaction solution, water permeability of pore-filled membranes was around 100 l h−1 m−2 bar−1. Here relatively lower concentration of monomer NIPAAm (1 mol l−1) and cross-linker MBA (0.02 mol l−1) were used, and the water permeabilities of various individual membranes were ranging from 50 to 1000 l h−1 m−2 bar−1. The broad range of water permeability and the partially higher absolute values indicated relatively poor homogeneity of the hydrogel network (or the pore-filling) and/or a rather loose network structure. This had possibly been caused by the relatively low concentration of monomer and cross-linker, but an effect of the relatively large MNP (diameter 40 nm) on the network structure cannot be excluded. Despite the relatively poor reproducibility of water permeability, all membranes showed clearly significant temperature responsivity (thermo-responsivity factor N45/25 in the range 3 to 25; for unmodified membrane R or N factors are ∼1; cf.Fig. 6a). It had been noticed that the thermo-responsivity factor also has a broad range, but values correlated with water permeability: membranes with high water permeability had low thermo-responsivity factor and membranes with low permeability had high thermo-responsivity. This indicates clearly that pore-filled membranes with high permeability have looser hydrogel network or uneven pore-filling leading to poor barrier properties even at 25 °C, so that the change upon increase of temperature is only small. Overall, no difference with respect to achievable membrane quality could be seen when comparing preparations according to “method 1” and “method 2”.Therefore, for the verification of the magneto-responsivity, two specific membranes from both preparation methods with relatively high thermo-responsivity factor (indicating good function of PNIPAAm network) were chosen (see Fig. 8a). The membrane from “method 2” had a lower water permeability and higher thermo-responsivity factor than the one from “method 1” only because a representative membrane with excellent thermo-responsivity had been chosen here while the membrane from “method 1” represents a membrane with good thermo-responsivity. The magneto-responsive water permeability is shown in Fig. 8b. When the AMF was off in the first 15 minutes, both membranes showed relatively low permeability. After the AMF was switched on, the permeability of both membranes increased significantly within 20 minutes and reached a plateau value. This plateau permeability indicated the stationary state between heat generation by the MNPs and cooling by convective flow of the feed (kept at 25 °C) through the pores. The absolute increase in water flux due to AMF was significantly larger for the membrane from “method 2”, which can be explained by its lower permeability allowing only smaller flux and, hence, leading to less convective cooling compared to membrane from “method 1”. Thus, the membranes behaved analogous to the magneto-responsive valve membranes reported earlier.40 For both membranes, the plateau water permeability was lower than their permeability at 45 °C (cf.Fig. 8a), therefore the temperature in the membrane pores under AMF condition was in the range of the lower critical solution temperature of PNIPAAm, but lower than 45 °C. It must be noted that the estimation of the actual temperature in the pores from the measured permeability is not straight forward because of the largely sigmoidal shape of the viscosity-normalized flux vs. temperature curve.40 On the one hand, this indicates that the heat generation under the used conditions is limited; on the other hand, this would provide favorable conditions for the separation of biomacromolecules as the temperature is still within a mild range. Finally, when AMF was switched off again, the permeability returned to the value at the beginning of the experiment. This proved the heat generated by the MNPs can largely change the conformation of PNIPAAm network under the used AMF condition and that the permeability of the membranes can be fully reversibly manipulated by external AMF. The relatively slow response can be attributed to a large extent to the larger external systems volume relative to the very small membrane volume; cf. discussion in ref. 40.
 | ||
| Fig. 8 Thermo-responsive (a) and magneto-responsive (b) water permeability of two representative pore-filled membranes. | ||
Furthermore, systems with conventional thermo-responsive membranes (comprising feed container, tubing/pipes, and membrane module) need time to heat up the whole feed and then also to equilibrate the membrane to the new conditions. In laboratory work with an analogous system, e.g. a conventional PNIPAAm hydrogel pore-filled membrane, the total time for changing the feed water temperature from 25 to 45 °C and obtaining constant fluxes under the new conditions was at least 30 minutes,55i.e., longer than in this study. And the entire feed volume would constantly dissipate heat to the environment. For magneto-responsive membranes with localized heaters, only the very small fraction of feed volume passing through the membrane pores needs to be heated up to so that much less energy will be dissipated. These arguments illustrate the potential of the novel membranes with pore-integrated “nano heaters” for faster response and higher energy efficiency compared to conventional temperature-responsive membranes. However, it should be noted that the efficiency of the conversion of electrical to AMF energy is not considered here. Analogous conclusions with respect to potential for efficiency improvement had been reached in the work of Vanherck et al.19 when comparing laser-induced heating of gold nanoparticles embedded in a nanofiltration membrane with conventional heating.
3.6 Magneto-responsive molecule sieving by pore-filled membrane
Finally, magneto-responsive molecule sieving performance of the magneto-hydrogel pore-filled membranes was investigated. Because the membranes functionalized with 1 mol l−1 NIPAAm had relatively high and poorly reproducible water permeability, also dextran rejection had typically been low, and magneto-responsive switching of rejection could only be demonstrated semi-quantitatively with dextran 2000 kDa (data not shown). Therefore, the NIPAAm concentration during preparation according to “method 1” had been increased to 1.1 mol l−1 (and concentrations of all other components were also increased accordingly), aiming to obtain a denser network for more effective molecular sieving. It turned out that the water permeability of obtained membranes decreased significantly, and the values at 25 °C narrowed down to the range 2 to 10 l h−1 m−2 bar−1 and thermo-responsive factor N40/25 was in the range of 16 to 20. Compared with the previously reported thermo-responsive PNIPAM hydrogel pore-filled membrane,34 such hydrogel pore-filled composite membranes have clearly lower water permeability. This is probably because the embedded MNPs reduced the free space in the hydrogel network. As shown in ESI,† Fig. S8, two pore-filled membranes prepared under those conditions demonstrated also very good magneto-responsivity of water flux.Thermo- and magneto-responsive water permeability and dextran sieving behaviour are shown in Fig. 9. As can be seen in Fig. 9a, the chosen membrane had a rather dense and even pore-filling hydrogel network, therefore its permeability at 25 °C was quite low and responsivity was quite high (thermo-responsive factor, N40/25 = 16). During dextran filtration tests, it demonstrated largely different rejection for different sizes of dextran molecules at 25 °C. For small dextran with average molecular weight of 4 kDa and average Stokes diameters of 3 nm,34 only 26% was rejected; for medium dextran with molecular weight of 500 kDa and Stokes diameter of 28 nm, a higher percentage, 58%, was rejected; for dextran with molecular weight of 2000 kDa and Stokes diameter of 54 nm, 94% was rejected. Hence the molecular weight cut-off, defined at 90% rejection, was clearly below 2000 kDa/50 nm. Average mesh size estimated for bulk gels, prepared at slightly lower monomer concentration and directly after synthesis was much smaller, i.e. ∼7 nm (cf.Table 2), and cut-off values for only hydrogel pore-filled membranes had also been lower (≤100 kDa).34 This implies that the relatively large MNP may introduce heterogeneities into the network structure which reduce its size-selective fractionation performance. However, when the AMF was switched on, the membrane permeability increased more than 10 times, and molecular sieving properties changed accordingly. The rejection for dextrans 4 kDa and 500 kDa decreased to 0%, and for dextran 2000 kDa, the rejection was also largely reduced, from 94% to 30%. This indicates that the average mesh size had been very much enlarged and that the molecular weight cut-off had been largely shifted to values beyond 50 nm, because of the magneto-actuation. Therefore, such MNP–PNIPAm hydrogel pore-filled composite membrane has demonstrated magneto-switchable molecular-sieving behaviour. Also, as shown in Fig. 9(c), such stimuli-responsive change of molecular sieving effect is fully reversible, because one membrane could be used for three cycles of ultrafiltration experiments, always including switching AMF on and off, and the initial membrane permeability was fully recovered.
 | ||
| Fig. 9 Responsive water permeability (a) and molecular sieving behaviour (b) of a specific magneto-hydrogel pore-filling composite membrane as well as water and dextran filtrate permeabilities during a step-wise experiment with different dextran solutions and interim water washing (c). | ||
4. Conclusions
For the first time, a smart membrane with magneto-responsive molecular sieving effect has been developed. Its molecular sieving behaviour can be remotely controlled by manipulating external alternating magnetic field, so that heating the feed solution for stimulating the membrane can be avoided. The structure of the network is more complex than that of pure cross-linked PNIPAm, because of the incorporation of the nanoparticles which have a larger diameter than the average mesh size of the bulk hydrogel. Attempts to characterize and optimize this composite network have been made because the polymeric network structure determines the sieving effect while nanoparticle loading or nanoparticle size affect the magnitude and rate of magneto-responsivity. The obtained membranes have excellent switchable molecular sieving behaviour. However, absolute permeabilities are low compared to established (anisotropic) ultrafiltration membranes with similar molecular weight cut-off; this is mainly because of the much larger barrier thickness and lower barrier porosity of the used macroporous substrates which had been pore-filled completely. In future work, the structure of the composite network will be further characterized in detail as it is of particular importance. And original size of hydrogel mesh and its stimuli-responsive effect could be tuned by varying monomer concentration and cross-linker proportion during the preparation. To increase the permeability of such membranes, adapting the method to porous membranes with smaller barrier thickness and higher porosity is another target of further development. Also, the responsivity of the system during the filtration process could be increased by manipulating the magnetic field amplitude. However, this should be combined with attempts to achieve a more homogenous field distribution; work to redesign the inductor coils is in progress. Overall, because of their interesting responsive molecule sieving behaviour, such novel smart membranes are expected to have potential applications in biomedical and microfluidics fields.Acknowledgements
The authors gratefully acknowledge the financial support of China Scholarship Council (CSC), the technical support by Inge Danielzik and Tobias Kallweit, Technical Chemistry II, University Duisburg-Essen (UDE), Jürgen Leistikow, Physical Chemistry, UDE, the computer simulation kindly provided by Prof. Daniel Erni's group at Department of Electrical Engineering, UDE, and SEM and EDX-SEM analyses conducted by Smail Boukercha, Center for Scanning Electron Microscopy at UDE.References
- L. Zhai, Chem. Soc. Rev., 2013, 42, 7148–7160 RSC.
- D. Wandera, S. R. Wickramasinghe and S. M. Husson, J. Membr. Sci., 2010, 357, 6–35 CrossRef CAS.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Mueller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- M. Ulbricht, Polymer, 2006, 47, 2217–2262 CrossRef CAS.
- Y. Sugawara, T. Tamaki and T. Yamaguchi, Polymer, 2015, 62, 86–93 CrossRef CAS.
- H. Ohashi, S. Ebina and T. Yamaguchi, Polymer, 2014, 55, 1412–1419 CrossRef CAS.
- H. H. Himstedt, Q. Yang, X. Qian, S. R. Wickramasinghe and M. Ulbricht, J. Membr. Sci., 2012, 423–432, 257–266 CrossRef.
- L. Y. Chu, Y. Li, J. H. Zhu, H. D. Wang and Y. J. Liang, J. Controlled Release, 2004, 97, 43–53 CrossRef CAS PubMed.
- S. J. Lue, C. H. Chen, C. M. Shih, M. C. Tsai, C. Y. Kuo and J. Y. Lai, J. Membr. Sci., 2011, 379, 330–340 CrossRef CAS.
- T. Ito, T. Hioki, T. Yamaguchi, T. Shinbo, S. Nakao and S. Kimura, J. Am. Chem. Soc., 2002, 124, 7840–7846 CrossRef CAS PubMed.
- T. Luo, S. Lin, R. Xie, X. J. Ju, Z. Liu, W. Wang, C. L. Mou, C. Zhao, Q. Chen and L. Y. Chu, J. Membr. Sci., 2013, 450, 162–173 CrossRef.
- S. Frost and M. Ulbricht, J. Membr. Sci., 2013, 448, 1–11 CrossRef CAS.
- T. Hoare, B. P. Timko, J. Santamaria, G. F. Goya, S. Irusta, S. Lau, C. F. Stefanescu, D. Lin, R. Langer and D. S. Kohane, Nano Lett., 2011, 11, 1395–1400 CrossRef CAS PubMed.
- T. Hoare, B. P. Timko, J. Santamaria, G. F. Goya, S. Irusta, D. Lin, S. Lau, R. Padera, R. Langer and D. S. Kohane, Nano Lett., 2009, 10, 3651–3657 CrossRef PubMed.
- I. Csetneki, G. Filipcsei and M. Zrinyi, Macromolecules, 2006, 39, 1939–1942 CrossRef CAS.
- G. Wang, R. Xie, X. J. Ju and L. Y. Chu, Chem. Eng. Technol., 2012, 35, 2015–2022 CrossRef CAS.
- H. Kuroki, C. Islam, I. Tokarev, H. Hu, G. Liu and S. Minko, ACS Appl. Mater. Interfaces, 2015, 7, 10401–10406 CAS.
- I. Tokarev, M. Orlov and S. Minko, Adv. Mater., 2006, 18, 2458–2460 CrossRef CAS.
- K. Vanherck, S. Hermans, T. Verbiest and I. Vankelecom, J. Mater. Chem., 2011, 21, 6079–6087 RSC.
- H. H. Himstedt, Q. Yang, L. P. Dasi, X. Qian, S. R. Wickramasinghe and M. Ulbricht, Langmuir, 2011, 27, 5574–5581 CrossRef CAS PubMed.
- A. Y. Gebreyohannes, M. R. Bilad, T. Verbiest, C. M. Courtin, E. Dornez, L. Giorno, E. Curcio and I. F. J. Vankelecom, J. Membr. Sci., 2015, 487, 209–220 CrossRef CAS.
- G. Jeon, S. Y. Yang and J. K. Kim, J. Mater. Chem., 2012, 22, 14814–14834 RSC.
- S. P. Nunes, A. R. Behzad, B. Hooghan, R. Sougrat, M. Karunakaran, N. Pradeep, U. Vainio and K. V. Peinemann, ACS Nano, 2011, 5, 3516–3522 CrossRef CAS PubMed.
- X. Qiu, H. Yu, M. Karunakaran, N. Pradeep, S. P. Nunes and K. V. Peinemann, ACS Nano, 2013, 7, 768–776 CrossRef CAS PubMed.
- L. C. Dong, A. S. Hoffman and Q. Yan, J. Biomater. Sci., Polym. Ed., 1994, 5, 473–484 CrossRef CAS PubMed.
- M. L. White, J. Phys. Chem., 1960, 64, 1563–1565 CrossRef CAS.
- P. G. Righetti and C. Gelfi, J. Chromatogr. B: Biomed. Sci. Appl., 1997, 699, 63–75 CrossRef CAS.
- V. Kapur, J. C. Charkoudian and J. L. Anderson, J. Membr. Sci., 1997, 131, 143–153 CrossRef CAS.
- V. Kapur, J. C. Charkoudian, S. B. Kessler and J. L. Anderson, Ind. Eng. Chem. Res., 1996, 35, 3179–3185 CrossRef CAS.
- Q. Yang, N. Adrus, F. Tomicki and M. Ulbricht, J. Mater. Chem., 2011, 21, 2783–2811 RSC.
- I. Tokarev and S. Minko, Adv. Mater., 2009, 21, 241–247 CrossRef CAS.
- I. Tokarev and S. Minko, Soft Matter, 2009, 5, 511–524 RSC.
- J. J. Kim and K. Park, Bioseparation, 1999, 7, 177–184 CrossRef CAS.
- N. Adrus and M. Ulbricht, J. Mater. Chem., 2012, 22, 3088–3098 RSC.
- J. Thevenot, H. Oliveira, O. Sandre and S. Lecommandoux, Chem. Soc. Rev., 2013, 42, 7099–7116 RSC.
- R. Hergt, S. Dutz, R. Mueller and M. Zeisberger, J. Phys.: Condens. Matter, 2006, 18, S2919–S2934 CrossRef CAS.
- C. S. Brazel, Pharm. Res., 2009, 26, 644–656 CrossRef CAS PubMed.
- N. S. Satarkar and J. Z. Hilt, J. Controlled Release, 2008, 130, 246–251 CrossRef CAS PubMed.
- N. S. Satarkar and J. Z. Hilt, Acta Biomater., 2008, 4, 11–16 CrossRef CAS PubMed.
- A. Gajda and M. Ulbricht, J. Mater. Chem. B, 2014, 2, 1317–1326 RSC.
- N. Adrus and M. Ulbricht, React. Funct. Polym., 2013, 73, 141–148 CrossRef CAS.
- J. Wang and V. M. Ugaz, Electrophoresis, 2006, 27, 3349–3358 CrossRef CAS PubMed.
- M. G. Weimuller, M. Zeisberger and K. M. Krishnan, J. Magn. Magn. Mater., 2009, 321, 1947–1950 CrossRef PubMed.
- C. Geismann and M. Ulbricht, Macromol. Chem. Phys., 2005, 206, 268–281 CrossRef CAS.
- C. Geismann, A. Yaroshchuk and M. Ulbricht, Langmuir, 2007, 23, 76–83 CrossRef CAS PubMed.
- C. Geismann, F. Tomicki and M. Ulbricht, Sep. Sci. Technol., 2009, 44, 3312–3329 CrossRef CAS.
- R. Xie, L. Y. Chu, W. M. Chen, W. Xiao, H. D. Wang and J. B. Qu, J. Membr. Sci., 2005, 258, 157–166 CrossRef CAS.
- D. L. Huber, Small, 2005, 1, 482–501 CrossRef CAS PubMed.
- S. B. Campbell, M. Patenaude and T. Hoare, Biomacromolecules, 2013, 14, 644–653 CrossRef CAS PubMed.
- S. Ghosh and T. Cai, J. Phys. D: Appl. Phys., 2010, 43, 415504 CrossRef.
- J. A. Galicia, F. Cousin, E. Dubois, O. Sandre, V. Cabuil and R. Perzynski, Soft Matter, 2009, 5, 2614–2624 CAS.
- J. Fan, Z. Shi, M. Lian, H. Li and J. Yin, J. Mater. Chem. A, 2013, 1, 7433–7443 CAS.
- D. Ma and L. M. Zhang, J. Phys. Chem. B, 2008, 112, 6315–6321 CrossRef CAS PubMed.
- J. D. Ferry, Viscoelastic Properties of Polymers, John Wiley & Sons, Canada, 1980 Search PubMed.
- N. Adrus, Stimuli-responsive hydrogels and hydrogel pore-filled composite membranes, PhD thesis, Universität Duisburg-Essen, 2012 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5tb02368h |
| This journal is © The Royal Society of Chemistry 2016 |