
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFlux-mediated doping of SrTiO3 photocatalysts for efficient overall water splitting†
Yeilin
Ham
a,
Takashi
Hisatomi‡
a,
Yosuke
Goto
a,
Yosuke
Moriya‡
a,
Yoshihisa
Sakata
b,
Akira
Yamakata
c,
Jun
Kubota
d and
Kazunari
Domen‡
*a
aDepartment of Chemical System Engineering, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: domen@chemsys.t.u-tokyo.ac.jp; Fax: +81 03 5481 8838; Tel: +81 03 5481 1652
bGraduate School of Science and Engineering, Yamaguchi University, 2-16-1 Tokiwadai, Ube-shi, Yamaguchi 755-8611, Japan
cGraduate School of Engineering, Toyota Technological Institute, 2-12-1 Hisakata, Tempaku, Nagoya 468-8511, Japan
dDepartment of Chemical Engineering, Fukuoka University, 19-1 Nanakuma 8-Chome, Jonan-ku, Fukuoka, 814-0180 Japan
First published on 25th September 2015
Abstract
SrTiO3 is a photocatalyst that is well known for its activity for the overall water splitting reaction under UV light irradiation. In this study, the effects of SrCl2 flux treatments and Al doping on the photocatalytic properties of SrTiO3 were investigated. The SrTiO3, which showed an apparent quantum efficiency of 30% at 360 nm in the overall water splitting reaction, the highest value reported so far, was prepared by SrCl2 flux treatments in alumina crucibles. Scanning electron microscopy and X-ray diffractometry revealed that the flux-treated SrTiO3 consisted of well-crystalline particles with a cubic shape reflecting the perovskite-type structure. Inductively coupled plasma optical emission spectroscopy revealed that Al ions from the alumina crucibles were incorporated into the SrTiO3 samples. The SrTiO3 that was treated with SrCl2 flux in Al-free conditions showed a marginal improvement in photocatalytic activity despite the high crystallinity and the clear crystal habit. Doping SrTiO3 with Al improved the photocatalytic activity even without SrCl2 treatment. These results suggested that Al doping was a principal factor in the dramatic improvement in the water splitting activity of the flux-treated SrTiO3. The effects of flux treatments and Al doping on the morphology and water splitting activity of SrTiO3 were discussed separately.
1 Introduction
Photocatalytic water splitting is a promising technology for the utilization of solar energy in the production of renewable hydrogen.1–4 In the last four decades, a number of particulate photocatalysts capable of overall water splitting have been developed.1 La-doped NaTaO3 loaded with NiO has reportedly split water at an apparent quantum efficiency of 56% at 270 nm.5 Zn-doped Ga2O3 loaded with Rh2−yCryO3 has also been reported to split water at a high reaction rate.6 Some layered oxides such as K4Nb6O17,7 Rb4Nb6O17,8 and Rb2La2Ti3O10 (ref. 9) are known to show relatively high apparent quantum efficiencies (5%, 10%, and 5%, respectively) at 330 nm when modified with appropriate cocatalysts. However, these materials are active only under UV light, limiting their application to solar energy conversion. The highest apparent quantum efficiency recorded in the visible light region was 5.1% (at 410 nm), attained by Rh2−yCryO3/(Ga1−xZnx)(N1−xOx).3 It is necessary to develop effective methods to improve the apparent quantum efficiency at wavelengths to achieve a solar-to-hydrogen energy conversion efficiency of 5–10%, which has been targeted for cost-competitive production of solar hydrogen via photocatalytic water splitting.3,4,10Given a specific photocatalytic material, controlling charge separation and migration is critical because the band structure and crystallographic character of each semiconducting material are unique. The movement of excited electrons and holes, and thus the photocatalytic activity, can be greatly affected by the crystallinity, particle size, and doping. Higher crystallinity and smaller particle sizes are desirable for charge migration toward surface active sites before recombination. Highly crystalline particles can be obtained by using a flux as a growth medium. The flux method, which allows the growth of crystalline particles via dissolution and recrystallization of solutes driven by supersaturation, has been applied to the synthesis of metal oxides,11 including semiconducting oxides, such as K4Nb6O17,12 KNb3O8,13 Na2Ti6O13,14 K2Ti6O13,15 and SnNb2O6.16 Some of these oxides showed improved photocatalytic activity compared to those prepared by other synthesis methods. Some semiconducting (oxy)nitride photocatalysts such as LaTiO2N,17 C3N4,18 and Ta3N5 (ref. 19) have also been prepared with the aid of a flux. However, the downside of this method is the incorporation of impurities into the target material, a commonly observed phenomenon during flux treatment.20 On the other hand, doping can often change the particle morphology dramatically5 and create mid-gap states essential for visible light activity of some wide-band-gap oxides.21 In fact, the high activity of NaTaO3 and the visible light activity of SrTiO3 photocatalysts rely on such doping effects. Recently, much effort has been made to understand the effect of doping in terms of carrier dynamics.22–24 From the material synthesis standpoint, the challenges in the activation of photocatalysts lie in how to lower defect densities, reduce particle sizes, and incorporate dopants effectively. The flux treatment of photocatalytic materials may offer a solution to such challenges.
SrTiO3 is a classic photocatalyst that has been reported to be active in overall water splitting under UV light since 1980 (ref. 25 and 26) and is still widely investigated in fundamental studies on the effects of doping,21,23,27,28 particle morphology,29 and cocatalysts.30 In a recent study, we found that doping of lower-valence cations in SrTiO3, such as Na+ into Sr2+ and Ga3+ into Ti4+, dramatically enhanced the photocatalytic activity during the overall water splitting reaction.27 We attributed the positive effect of doping to the lower density of trivalent Ti states. Thus, the effects of the incorporation of even a small amount of impurity into SrTiO3 during the flux treatment should be carefully investigated.
In this paper, we studied the effects of SrCl2 flux treatment and doping on the physical properties and photocatalytic activity of SrTiO3. The crystallinity and water splitting activity of SrTiO3 were dramatically improved by the flux treatment in alumina crucibles. A small amount of Al doped into SrTiO3 from an alumina crucible was found to be responsible for the enhancement in photocatalytic activity. It was found that the external doping of Al in the presence of flux was the most effective for controlled Al doping and produced an apparent quantum efficiency exceeding 30% at 360 nm, the highest value reported so far in this wavelength region and on SrTiO3 powder.
2 Experimental
Sample preparation
Measurement of photocatalytic activity
The activities of the photocatalyst samples were tested in a closed gas circulation system with a top-irradiation-type reactor. The deionized water (100 mL) was evacuated to remove air completely. The reactor was irradiated using a 300 W xenon lamp (λ > 300 nm) through a quartz window or using a 450 W high-pressure mercury lamp through a quartz cooling jacket and, when necessary, a 2 cm-diameter slit, a band pass filter (λ = 360 nm, FWHM = 10 nm), and a series of neutral density filters (OD = 0.3, 0.5, 1.0, and 2.0) to irradiate the sample with monochromatic light with controlled intensity. The evolved gases were analyzed by a gas chromatograph (Shimadzu, GC-8A) equipped with a thermal conductivity detector, using Ar as a carrier gas.Sample characterization
The crystal structures of the products were characterized by X-ray diffractometry (XRD; RINT Ultima III, Rigaku Co.) using Cu Kα radiation at 40 kV and 40 mA. XRD peaks due to Cu Kα1 and Kα2 radiation were deconvoluted and the full width at half maximum (FWHM) of the (110) peak due to the Cu Kα1 radiation was estimated. Specific surface areas were measured with a Belsorp-miniII (BEL Japan Inc.). The morphology of the powder was observed by scanning electron microscopy (SEM; S-4700, Hitachi High-Technologies Co.). Ultraviolet-visible diffuse reflectance spectrometry (DRS; V-670, Jasco Co.) was performed using spectralon (Jasco Co.) as a reference material. Inductively coupled plasma optical emission spectroscopy (ICP-OES; Shimadzu Co., ICPS-8100) was used for elemental analysis. SrTiO3 powder (0.01 g) was melted with 1.0 g of a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of Na2CO3 and B(OH)3 by heating. An aqueous solution of tartaric acid (5%, 10 mL), HCl (1 + 1, 4 mL), and H2O2 (30 wt%, 1 mL) were added to dissolve the melt, and diluted with distilled water to make the total volume 100 mL. The resulting solution was used to measure Al and Y. The solution was further diluted tenfold to measure Sr and Ti.
:1 mixture of Na2CO3 and B(OH)3 by heating. An aqueous solution of tartaric acid (5%, 10 mL), HCl (1 + 1, 4 mL), and H2O2 (30 wt%, 1 mL) were added to dissolve the melt, and diluted with distilled water to make the total volume 100 mL. The resulting solution was used to measure Al and Y. The solution was further diluted tenfold to measure Sr and Ti.
3 Results and discussion
Commercially available SrTiO3 (hereafter STO(pristine)) was mixed with SrCl2 at a molar ratio of SrCl2/SrTiO3 = 10 and heated at 1100 °C for 10 h in yttria crucibles (hereafter STO(flux-Y)) and alumina crucibles (hereafter STO(flux-Al)) to obtain flux-treated SrTiO3. X-ray diffraction (XRD) patterns are presented in Fig. 1. The XRD patterns for STO(flux-Y) and STO(flux-Al) showed only peaks attributable to the SrTiO3 phase and exhibited sharper XRD peaks than STO(pristine), as reported in the literature.24,29 The FWHM of the (110) XRD peak was 0.103 for STO(pristine) and it was reduced to 0.071 for STO(flux-Y) (Table 1). Under the same measurement conditions, the difference in the FWHM of XRD peaks reflects the difference in size and strain of crystallites. The reduction of the FWHM after the flux treatment should reflect the growth of crystallites with weaker strain, which is indicative of higher crystallinity.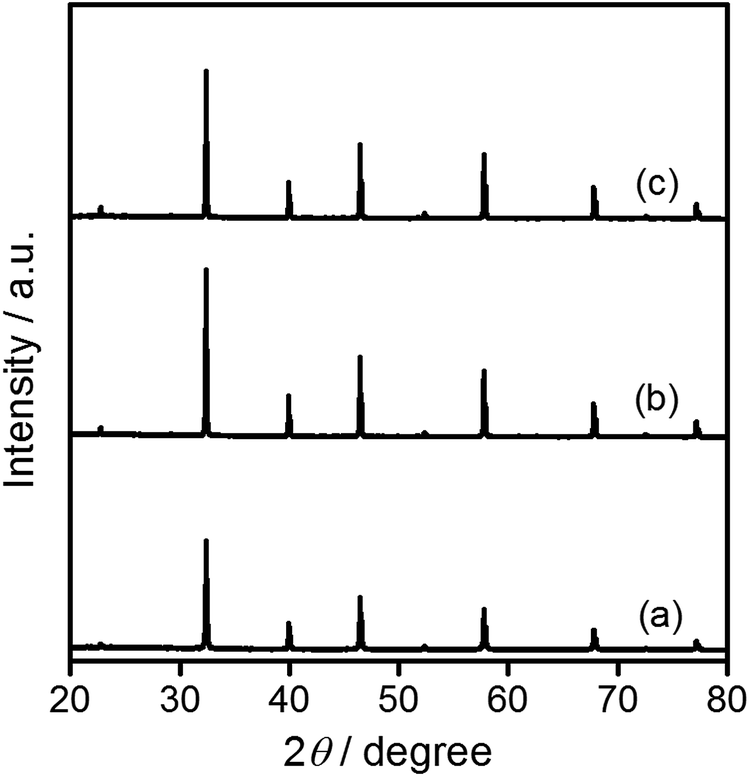 | ||
| Fig. 1 XRD patterns for SrTiO3 particles. (a) STO(pristine), (b) STO(flux-Y), and (c) STO(flux-Al). | ||
| Sample | FWHM of (110) peak/° | BET surface area/m2 g−1 | Molar ratio | |
|---|---|---|---|---|
| 2[Al]/([Sr] + [Ti]) | 2[Y]/([Sr] + [Ti]) | |||
| STO(pristine) | 0.103 | 3.6 | 0.04% | 0.00% |
| STO(flux-Y) | 0.071 | 1.5 | 0.02% | 0.48% |
| STO(flux-Al), 1100 °C | 0.067 | 0.9 | 0.31% | 0.00% |
| STO(flux-Al), 1000 °C | 0.074 | 1.3 | 0.11% | 0.01% |
| STO(flux-Al), 900 °C | 0.084 | 1.9 | 0.12% | 0.00% |
The SEM images of the samples in Fig. 2 show that STO(pristine) consisted of particles with irregular shapes, a few hundred nanometers in size. The flux treatment changed the particle morphology dramatically. The STO(flux-Y) and STO(flux-Al) samples consisted of truncated cubic particles that exhibited a perovskite-type structure. The particles were 0.2–2 μm and 0.2–3 μm in size, respectively. The equilibrium crystal shape of SrTiO3 was reported to be truncated cubic.32 Since the flux treated STO crystals also consisted of truncated cubes, it is considered that these crystals exposed the surfaces with the lowest formation energy. The morphological change agreed well with the change in specific surface area of the samples. The BET surface area decreased upon flux treatment (Table 1). The BET surface area for STO(flux-Al) was slightly smaller than that for STO(flux-Y), agreeing well with the difference in their particle sizes.
 | ||
| Fig. 2 SEM images of SrTiO3 particles. (a) STO(pristine), (b) STO(flux-Y), and (c) STO(flux-Al). | ||
Diffuse reflectance spectra (DRS) of the SrTiO3 samples showed that their band gap was not changed by the flux treatments (Fig. S1 in the ESI†). The absorption edge was located at approximately 390 nm. Their indirect band gaps,33,34 as estimated from the Tauc plots, were 3.2 eV, which well agreed with the reported values.35
Fig. 3 shows the water splitting activities of STO(pristine), STO(flux-Y) and STO(flux-Al). The water splitting activity of STO(pristine) roughly tripled upon SrCl2 flux treatment in an yttria crucible (STO(flux-Y)). This is presumably due to the improvement in crystallinity. When the same SrCl2 flux treatment was carried out on SrTiO3 in an alumina crucible (STO(flux-Al)), the water splitting activity was enhanced much more significantly, reaching 550 H2 μmol h−1 and 280 O2 μmol h−1. Our recent study on carrier dynamics in these SrTiO3 powders conducted by time-resolved absorption (TA) spectroscopy revealed that most photoexcited electrons in STO(pristine) were deeply trapped, while those in STO(flux-Al) were in the conduction band or shallowly trapped.24 This result well explains the enhanced water splitting activity of STO(flux-Al), because deeply trapped electrons would not be readily used for the hydrogen evolution reaction. STO(flux-Al) split water steadily for at least several hours, as shown in Fig. 4. The apparent quantum efficiency of STO(flux-Al) was measured to be 30% at 360 nm. This is much higher than the 4.3% reported for KCl-treated SrTiO3, although the difference in reaction conditions should be taken into account.29 We note that the apparent quantum efficiency was in fact dependent on the intensity of the incident light. As shown in Fig. S2 in the ESI,† the apparent quantum efficiency increased with light intensity under the experimental conditions examined. This result is not consistent with reaction orders to light intensity commonly observed in photocatalytic reactions, which is a first order.36 We suspect that a certain kind of trap state with a limited density may have to be filled with photoexcited carriers to attain sufficient photoconductivity for charge separation. The kinetic interpretation of the light intensity dependence of the photocatalytic water splitting rate will be reported in follow-up studies.
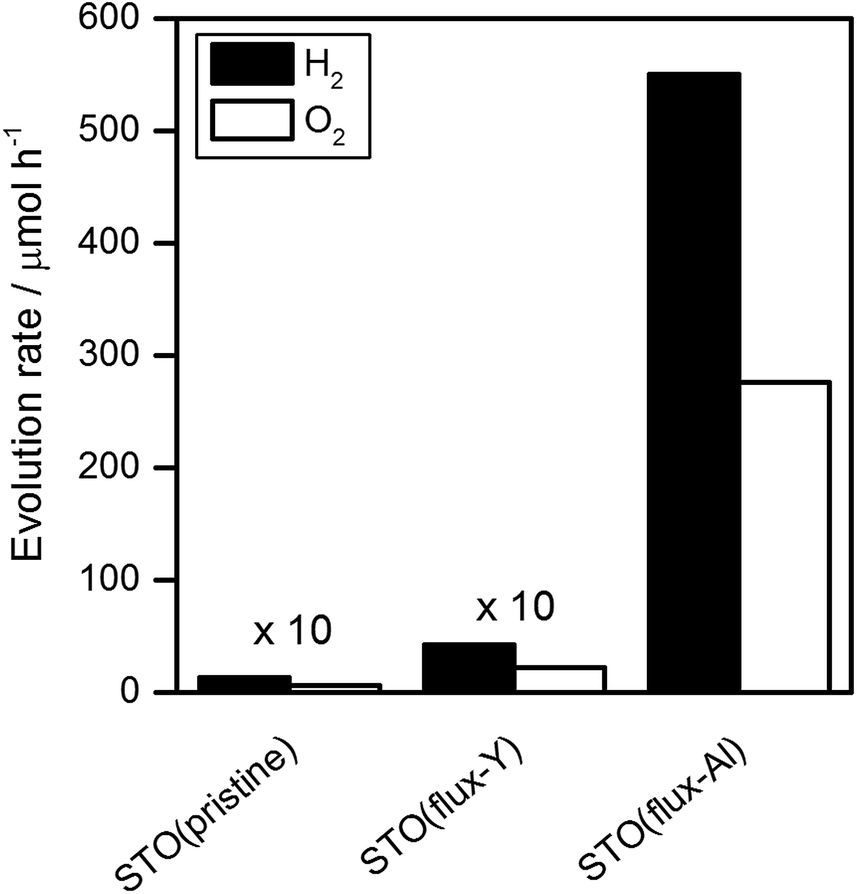 | ||
| Fig. 3 Water splitting activity of SrTiO3 photocatalysts. Reaction conditions: catalyst, 0.1 g; cocatalyst, Rh2−yCryO3 (Rh 0.1 wt%, Cr 0.1 wt%); reaction solution, 100 mL H2O; light source, 300 W Xe lamp (λ > 300 nm). | ||
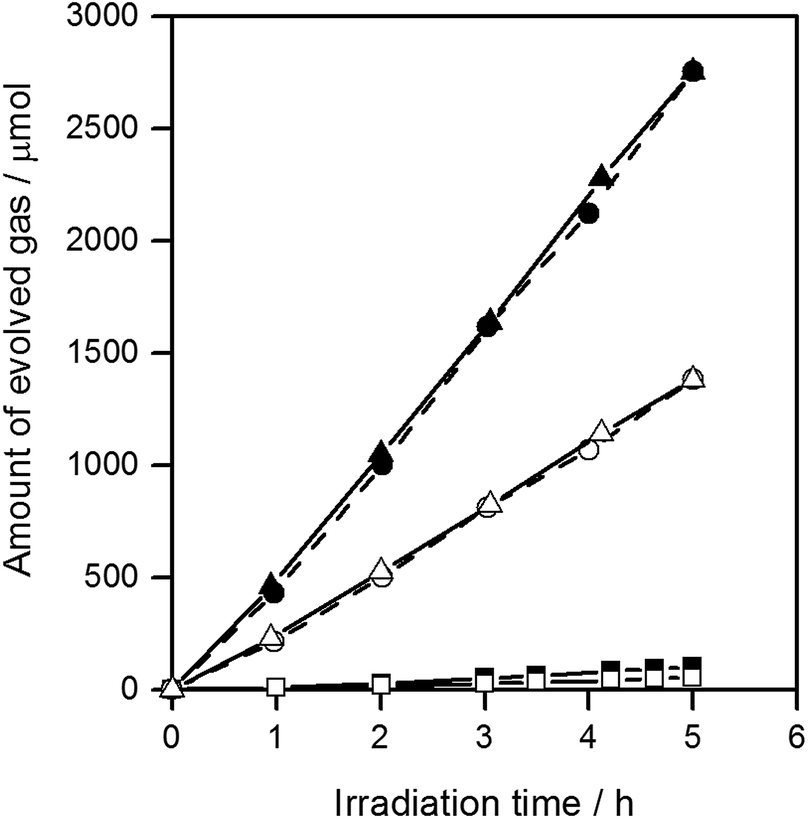 | ||
| Fig. 4 Gas evolution during the water splitting reaction on STO(flux-Al). H2 (■) and O2 (□) on STO(flux-Al) treated at 900 °C, H2 (●) and O2 (○) on STO(flux-Al) treated at 1000 °C, and H2 (▲) and O2 (△) on STO(flux-Al) treated at 1100 °C. Reaction conditions: catalyst, 0.1 g; cocatalyst, Rh2−yCryO3 (Rh 0.1 wt%, Cr 0.1 wt%); reaction solution, 100 mL H2O; light source, 300 W Xe lamp (λ > 300 nm). | ||
The FWHM of the (110) XRD peak for STO(flux-Al) treated at 1100 °C and STO(flux-Y) were 0.067 and 0.071, respectively (Table 1). Therefore, the crystallinity of STO(flux-Al) treated at 1100 °C was higher than that of STO(flux-Y). This might explain the large difference in the enhancement of photocatalytic activity by SrCl2 flux treatment in alumina and yttria crucibles. To determine the influence of crystallinity on the photocatalytic activity of flux-treated SrTiO3, the crystallinity of STO(flux-Al) was controlled by increasing the treatment temperature above the melting point of SrCl2 (874 °C). For treatment temperatures of 900–1100 °C, the XRD patterns for all samples were attributed to single-phase SrTiO3, regardless of the treatment temperature (Fig. S3 in the ESI†). However, the FWHM of the (110) XRD peaks of STO(flux-Al) increased upon lowering the flux treatment temperature and became comparable to that of STO(flux-Y) (Table 1). The water splitting activity of STO(flux-Al) became markedly higher when the treatment temperature increased from 900 to 1000 °C (Fig. 4). This suggested that the improvement in water splitting activity of flux-treated SrTiO3 was associated with improved crystallinity. However, STO(flux-Al) treated at 900 and 1000 °C exhibited a significantly higher water splitting rate than STO(flux-Y), although the crystallinity of the former was inferior. This points to a different, more predominant factor governing the water splitting activity of SrTiO3.
We speculated that Al impurity, derived from the alumina crucibles, became incorporated into SrTiO3 during flux treatment. Indeed, it has been reported that doping SrTiO3 with lower-valence cations can boost its photocatalytic activity.27 Therefore, an elemental analysis of the samples was carried out by inductively coupled plasma optical emission spectroscopy (ICP-OES). Table 1 tabulates the compositions of flux-treated SrTiO3. Al ions, derived from the alumina crucibles, were indeed detected in the STO(flux-Al) samples, where they would replace the Ti4+ sites of SrTiO3. As expected, STO(flux-Y) was free of Al, but instead, it was doped with Y from the yttria crucibles. The higher water splitting activity of STO(flux-Al) compared to STO(flux-Y) is thus most likely attributable to the effect of Al doping. The enhancement of the water splitting activity by doping lower valence cations is consistent with the recent studies6,27,37 but contradictory to an earlier work on platinized doped TiO2 systems.38 The difference probably resulted from the cocatalysts used. The Rh2−yCryO3 cocatalyst is known to block the reverse reaction, i.e., formation of water from hydrogen and oxygen molecules, unlike noble metals which readily catalyze the reverse reaction.31,39 Therefore, in this work, the water splitting activity is not obscured by the reverse reactions and closely reflects the change in the behavior of photoexcited charge carriers. Y doping did not have a significant positive effect on the photocatalytic activity. Unlike Al, Y presumably did not substitute well at the Ti sites because of the larger mismatch in ionic radius.
In an attempt to control Al doping in the presence of the SrCl2 flux, SrTiO3 and Al2O3 were mixed at molar ratios of Al/Ti = 0.1%, 1%, and 10%, and heated together with the SrCl2 flux in yttria crucibles. The resulting samples are referred to as x%Al-STO(flux-Y). The amount of Al doping was not directly proportional to the amount of Al2O3 added, but did increase with it (Table 2). In addition, certain amounts of Y were introduced from the yttria crucibles, similar to the case for the STO(flux-Y) sample. However, it is unlikely that Y doping at this level overwhelms the enhancement of photocatalytic activity by Al doping, considering the results for STO(flux-Y) (Fig. 3). The XRD patterns, SEM images, and BET surface areas for the samples and their water splitting activities are presented in Fig. S4 and S5 in the ESI,†Table 2, and Fig. 5, respectively. The 0.1%Al-STO(flux-Y) sample exhibited XRD patterns similar to those for STO(flux-Al). Single-phase SrTiO3 was observed, and the FWHM of the (110) diffraction peak was 0.067. The morphology and BET surface area for 0.1%Al-STO(flux-Y) were also comparable to those for STO(flux-Al). However, the water splitting activity of 0.1%Al-STO(flux-Y) was much lower than that of STO(flux-Al) due to the lower Al content in 0.1%Al-STO(flux-Y) compared to STO(flux-Al). The particle size of 1%Al-STO(flux-Y) and 10%Al-STO(flux-Y) decreased upon Al addition and most of the particles lost their crystal facets, which were distinctive for STO(flux-Al) and STO(flux-Y). This change in morphology is presumably due to suppression of the crystal growth of SrTiO3 by the excess Al2O3, similar to the case for NaTaO3 doped with La.5 A high activity for the overall water splitting reaction, comparable to that for STO(flux-Al), was obtained using yttria crucible when more than 1% of Al was added. This result supports our speculation that Al doping is the controlling factor for the enhancement of photocatalytic activity of SrTiO3. On the other hand, the high activity of 10%Al-STO(flux-Y) despite its comparatively lower crystallinity may have resulted from the small particle sizes, which shorten the time needed for the migration of photoexcited carriers from the interior to the surface of photocatalyst particles.
| Sample | FWHM of (110) peak/° | BET surface area/m2 g−1 | Molar ratio | |
|---|---|---|---|---|
| 2[Al]/([Sr] + [Ti]) | 2[Y]/([Sr] + [Ti]) | |||
| 0.1%Al-STO(flux-Y) | 0.067 | 0.9 | 0.12% | 0.16% |
| 1%Al-STO(flux-Y) | 0.096 | 2.7 | 1.01% | 0.70% |
| 10%Al-STO(flux-Y) | 0.091 | 2.4 | 1.36% | 0.57% |
| 0.1%Al-STO(ssr-Y) | 0.077 | 1.7 | 0.10% | 0.00% |
| 1%Al-STO(ssr-Y) | 0.094 | 3.0 | 1.07% | 0.00% |
| 5%Al-STO(ssr-Y) | 0.095 | 3.7 | 4.24% | 0.01% |
| 10%Al-STO(ssr-Y) | 0.096 | 3.9 | 8.25% | 0.01% |
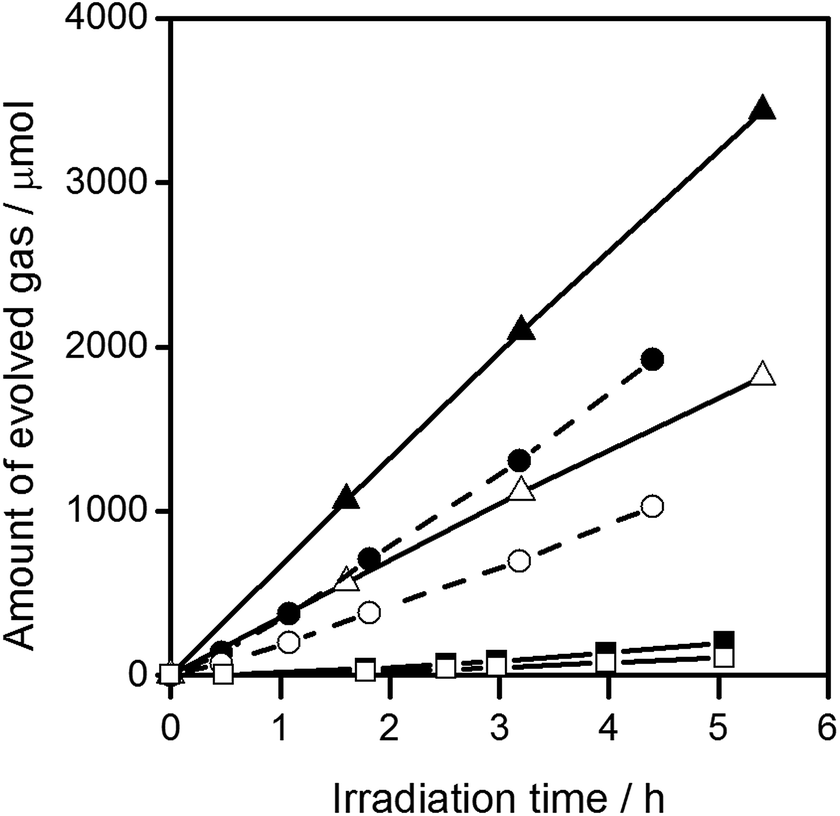 | ||
| Fig. 5 Gas evolution during the water splitting reaction on Al-STO(flux-Y). H2 (■) and O2 (□) on 0.1%Al-STO(flux-Y), H2 (●) and O2 (○) on 1%Al-STO(flux-Y), and H2 (▲) and O2 (△) on 10%Al-STO(flux-Y). Reaction conditions: catalyst, 0.1 g; cocatalyst, Rh2−yCryO3 (Rh 0.1 wt%, Cr 0.1 wt%); reaction solution, 100 mL H2O; light source, 300 W Xe lamp (λ > 300 nm). | ||
To examine the effect of aluminum doping separately, Al2O3 was added as a dopant to SrTiO3 at Al/Ti molar ratios ranging from 0.1% to 10%, and the mixtures were calcined in the absence of the SrCl2 flux for a solid state reaction. The resulting products are referred to as x%Al-STO(ssr-Y), where x% represents the Al/Ti molar ratio in the starting mixture. As tabulated in Table 2, the Al content increased monotonically with increasing amount of Al2O3 addition, although the amounts detected were somehow lower than the amounts added to the starting material for high Al2O3 contents (Al >5%). The amount of Al incorporated into SrTiO3 had a significant influence on the water splitting activity of the resulting samples. The water splitting activity peaked for 0.1%Al-STO(ssr-Y), as shown in Fig. 6. The activity of 0.1%Al-STO(ssr-Y) was two orders of magnitude higher than that of STO(pristine), but lower than that of STO(flux-Al) (Fig. 3). At this doping amount, no impurity phase was detected in the XRD patterns of the sample (Fig. S6†). The BET surface area for 0.1%Al-STO(ssr-Y) decreased to 1.7 m2 g−1 owing to the sintering process. However, it should be noted that no significant difference in morphology was observed between STO(pristine) and x%Al-STO(ssr-Y) (Fig. S7†). These results corroborate the necessity of the SrCl2 flux for morphological change. It is worth mentioning that the impurity concentration that can dramatically improve the water splitting activity of SrTiO3 (∼0.1 mol%) is close to the detection limits of commonly employed methods for elemental analysis, such as energy-dispersive X-ray emission spectroscopy (EDS) and X-ray photoelectron spectroscopy (XPS). Therefore, extra care needs to be taken when the effects of flux treatment on photocatalytic properties are discussed.
 | ||
| Fig. 6 Dependence of the water splitting activity of STO(ssr-Y) on the amount of Al doping. Reaction conditions: catalyst, 0.1 g; cocatalyst, Rh2−yCryO3 (Rh 0.1 wt%, Cr 0.1 wt%); reaction solution, 100 mL H2O; light source, 300 W Xe lamp (λ > 300 nm). | ||
It should be noted that Al doping of SrTiO3 was more effective when SrCl2 was present during the heating. The doping amount of Al in the STO(flux-Al) sample from an alumina crucible may vary depending on the treatment conditions. Nevertheless, this sample showed a higher photocatalytic activity than the Al-STO(ssr-Y) samples containing various and controlled amounts of Al. It is thought that Al was not effectively doped into SrTiO3 during the solid state reaction because Al had to diffuse from the outer surface of the particles. In contrast, a significant portion of the SrTiO3 particles was once dissolved and recrystallized in the presence of SrCl2 flux, together with alumina derived from the crucibles. During this process, some of the Al ions may be doped into the middle part of the SrTiO3 particles and occupy the most stable state thermodynamically. As a result, Al doping can show stronger enhancement of photocatalytic activity when the SrCl2 flux is used. Thus, it is concluded that the dramatic improvement in the photocatalytic activity of STO(flux-Al) was due to Al doping and the enhancement of crystallinity observed upon flux treatment.
4 Conclusions
The photocatalytic activity of SrTiO3 in the overall water splitting reaction was dramatically improved by SrCl2 flux treatment at 1100 °C in an alumina crucible. The improvement in activity was attributed mainly to the doping of Al derived from the crucibles. The morphological change and the enhanced crystallinity also improved the photocatalytic activity, although these factors were found to be less significant than the effect of Al doping on the basis of the results of flux treatment in Al-free conditions. It was confirmed that SrTiO3 doped with Al under a SrCl2 flux showed even higher water splitting activity than SrTiO3 doped with Al by a solid state reaction. It is believed that the flux worked as a medium to dissolve the Al2O3 dopant and the host SrTiO3 particles, facilitating the Al doping of SrTiO3. As a consequence, the apparent quantum efficiency in overall water splitting was increased to 30% at 360 nm. Flux-mediated doping is expected to greatly broaden the possibilities of photocatalytic materials by activating them under visible light irradiation. In addition, the incorporation of impurities into the samples during flux treatment was a common occurrence and could have strong impact on the photocatalytic activity. Therefore, particular attention should be paid to flux treatment of photocatalysts.5 Acknowledgements
This work was financially supported by Grants-in-Aid for Specially Promoted Research (No. 23000009) and for Young Scientists (A) (No. 15H05494), and the A3 Foresight Program of Japan Society for the Promotion of Science (JSPS). The authors thank Dr Taro Yamada in The University of Tokyo for his kind cooperation with ICP-OES.References
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655–2661 CrossRef CAS.
- T. Hisatomi, K. Takanabe and K. Domen, Catal. Lett., 2015, 145, 95–108 CrossRef CAS.
- H. Kato, K. Asakura and A. Kudo, J. Am. Chem. Soc., 2003, 125, 3082–3089 CrossRef CAS PubMed.
- Y. Sakata, Y. Matsuda, T. Nakagawa, R. Yasunaga, H. Imamura and K. Teramura, ChemSusChem, 2011, 4, 181–184 CAS.
- K. Sayama, A. Tanaka, K. Domen, K. Maruya and T. Onishi, Catal. Lett., 1990, 4, 217–222 CrossRef CAS.
- K. Sayama, A. Tanaka, K. Domen, K. Maruya and T. Onishi, J. Catal., 1990, 124, 541–547 CrossRef CAS.
- T. Takata, Y. Furumi, K. Shinohara, A. Tanaka, M. Hara, J. N. Kondo and K. Domen, Chem. Mater., 1997, 9, 1063–1064 CrossRef CAS.
- B. A. Pinaud, J. D. Benck, L. C. Seitz, A. J. Forman, Z. Chen, T. G. Deutsch, B. D. James, K. N. Baum, G. N. Baum, S. Ardo, H. Wang, E. Miller and F. Jaramillo Thomas, Energy Environ. Sci., 2013, 6, 1983–2002 CAS.
- M. Schieber, J. Am. Ceram. Soc., 1964, 47, 537–538 CrossRef.
- K. Teshima, K. Horita, T. Suzuki, N. Ishizawa and S. Oishi, Chem. Mater., 2006, 18, 3693–3697 CrossRef CAS.
- S. Suzuki, K. Teshima, A. Yamaguchi, K. Yubuta, T. Shishido and S. Oishi, CrystEngComm, 2012, 14, 987–992 RSC.
- K. Teshima, K. Yubuta, T. Shimodaira, T. Suzuki, M. Endo, T. Shishido and S. Oishi, Cryst. Growth Des., 2008, 8, 465–469 CAS.
- H. Yoshida, M. Takeuchi, M. Sato, L. Zhang, T. Teshima and M. G. Chaskar, Catal. Today, 2014, 232, 158–164 CrossRef CAS.
- D. Noureldine, D. H. Anjum and K. Takanabe, Phys. Chem. Chem. Phys., 2014, 16, 10762–10769 RSC.
- F. Zhang, A. Yamakata, K. Maeda, Y. Moriya, T. Takata, J. Kubota, K. Teshima, S. Oishi and K. Domen, J. Am. Chem. Soc., 2012, 134, 8348–8351 CrossRef CAS PubMed.
- M. J. Bojdys, J.-O. Müller, M. Antonietti and A. Thomas, Chem.–Eur. J., 2008, 14, 8177–8182 CrossRef CAS PubMed.
- T. Takata, D. Lu and K. Domen, Cryst. Growth Des., 2010, 11, 33–38 Search PubMed.
- G. Muller, A. Ostrogorsky and D. Hurle, Basic Techniques, 1994, vol. 2, pp. 573–574 Search PubMed.
- R. Asai, H. Nemoto, Q. Jia, K. Saito, A. Iwase and A. Kudo, Chem. Commun., 2014, 50, 2543–2546 RSC.
- M. Maruyama, A. Iwase, H. Kato, A. Kudo and H. Onishi, J. Phys. Chem. C, 2009, 113, 13918–13923 CAS.
- K. Furuhashi, Q. Jia, A. Kudo and H. Onishi, J. Phys. Chem. C, 2013, 117, 19101–19106 CAS.
- A. Yamakata, H. Yeilin, M. Kawaguchi, T. Hisatomi, J. Kubota, Y. Sakata and K. Domen, J. Photochem. Photobiol., A, 2015 DOI:10.1016/j.jphotochem.2015.05.016.
- K. Domen, S. Naito, M. Soma, T. Onishi and K. Tamaru, J. Chem. Soc., Chem. Commun., 1980, 543–544 RSC.
- K. Domen, A. Kudo, T. Onishi, N. Kosugi and H. Kuroda, J. Phys. Chem., 1986, 90, 292–295 CrossRef CAS.
- T. Takata and K. Domen, J. Phys. Chem. C, 2009, 113, 19386–19388 CAS.
- Q. Wang, T. Hisatomi, S. S. K. Ma, Y. Li and K. Domen, Chem. Mater., 2014, 26, 4144–4150 CrossRef CAS.
- H. Kato, M. Kobayashi, M. Hara and M. Kakihana, Catal. Sci. Technol., 2013, 3, 1733–1738 CAS.
- T. K. Townsend, N. D. Browning and F. E. Osterloh, Energy Environ. Sci., 2012, 5, 9543–9550 CAS.
- K. Maeda, K. Teramura, H. Masuda, T. Takata, N. Saito, Y. Inoue and K. Domen, J. Phys. Chem. B, 2006, 110, 13107–13112 CrossRef CAS PubMed.
- T. Sano, D. M. Saylor and G. S. Rohrer, J. Am. Ceram. Soc., 2003, 86, 1933–1939 CrossRef CAS.
- A. Kahn and A. Leyendecker, Phys. Rev., 1964, 135, A1321 CrossRef.
- M. Capizzi and A. Frova, Phys. Rev. Lett., 1970, 25, 1298 CrossRef CAS.
- K. van Benthem, C. Elsässer and R. French, J. Appl. Phys., 2001, 90, 6156–6164 CrossRef CAS.
- T. Hisatomi, K. Maeda, K. Takanabe, J. Kubota and K. Domen, J. Phys. Chem. C, 2009, 113, 21458–21466 CAS.
- K. Maeda, D. Lu and K. Domen, Chem.–Eur. J., 2013, 19, 4986–4991 CrossRef CAS PubMed.
- K. E. Karakitsou and X. E. Verykios, J. Phys. Chem., 1993, 97, 1184–1189 CrossRef CAS.
- K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, Angew. Chem., 2006, 118, 7970–7973 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: DR spectra, XRD patterns, SEM images, and photocatalytic activity of the samples. See DOI: 10.1039/c5ta04843e |
| ‡ Japan Technological Research Association of Artificial Photosynthetic Chemical Process (ARPChem), 5-1-5 Kashiwanoha, Kashiwa-shi, 277-8589 Chiba, Japan. |
| This journal is © The Royal Society of Chemistry 2016 |