
A high-spin nickel(II) borohydride complex in dehalogenation†
Hyeonwoo
Tak
,
Hyunjoo
Lee
,
Joongoo
Kang
and
Jaeheung
Cho
*
Department of Emerging Materials Science, DGIST, Daegu 42988, Korea. E-mail: jaeheung@dgist.ac.kr
First published on 23rd November 2015
Abstract
A nickel(II)–borohydride complex bearing a macrocyclic tridentate N-donor ligand, [Ni(Me3-TACN)(BH4)(CH3CN)]+ (Me3-TACN = 1,4,7-trimethyl-1,4,7-triazacyclononane), was prepared, isolated, and characterized by various physicochemical methods, including UV-vis, ESI-MS, IR and X-ray analyses. The structural and spectroscopic characterization clearly shows that the borohydride ligand is bound to the high-spin nickel(II) center in an η2-manner. Density functional theory calculations provided geometric information of 2, showing that the η2-binding of borohydride to the nickel center is more favorable than the η3-binding mode in CH3CN. The complex is paramagnetic with an effective magnetic moment of 2.9μB consistent with a d8 high-spin system. The reactivity of the high-spin nickel(II)–borohydride complex was examined in dehalogenation with numerous halocarbons. A kinetic isotope effect value of 1.7 was observed in the dehalogenation of CHCl3 by the nickel(II)–borohydride complex. Kinetic studies and isotopic labeling experiments implicate that hydride ion or hydrogen atom transfer from the borohydride group is the rate determining step. The positive Hammett ρ value of 1.2, obtained in the reactions of [Ni(Me3-TACN)(BH4)(CH3CN)]+ and para-substituted benzoyl chloride, indicates that the dehalogenation by the nickel(II)–borohydride species occurs via a nucleophilic reaction.
 Jaeheung Cho | Dr Jaeheung Cho received his B.S. (in 2000) and M.S. (in 2002) degrees in Chemistry from Pukyong National University in Korea and his PhD degree in Inorganic Chemistry from Kanazawa University in Japan under the guidance of Professor Masatatsu Suzuki in 2005, in which he examined the dinuclear metal-active oxygen species in aliphatic C–H bond functionalization. After graduation, he moved to the group of Professor Charles G. Riordan at the University of Delaware in USA as a Postdoctoral Fellow. Then, he joined Ewha Womans University in Korea as a Full-Time Lecturer and a Professor by special appointment where he investigated models of mononuclear metal-active oxygen intermediates, in collaboration with Professor Wonwoo Nam. In 2010, he got the Distinguished Lectureship Award from the Chemical Society of Japan. In 2012, he began his independent career at DGIST in Korea as an Assistant Professor. His research interests focus on the synthesis, characterization and reactivity of biomimetic complexes, especially those mimicking active sites of metalloenzymes. |
Introduction
The nickel–hydrogen interactions are of significant importance in industrial and biological catalytic systems. Nickel–hydrogen complexes are involved in catalytic hydrogenation for organic syntheses,1–4 nickel hydrogen batteries for rechargeable cells,5 and hydrogen storage for transportation applications.6 In nickel-containing enzymes such as hydrogenases, which catalyse the reversible two electron reduction of protons to hydrogen gas, nickel–hydrogen adducts are key intermediates in the proposed mechanisms of the enzymatic reactions.7–9 One of the eight known nickel enzymes, methyl coenzyme M reductase (MCR), catalyzes the reductive cleavage of the thioether cofactor methyl coenzyme M to produce methane.10–13Among the nickel–hydrogen adducts, much attention has recently been focused on nickel–borohydride species, since such adducts are potential materials for nickel-catalyzed hydrogenation.14 In synthetic chemistry, numerous nickel–borohydride complexes have been prepared and characterized by various spectroscopic and X-ray crystallographic methods, which provided considerable information. The well characterized mononuclear nickel–borohydride species show a variety of hapticities for the borohydride ligand (Scheme 1).1,15 Cramer and co-workers reported the first example of a single crystal structure of the nickel–borohydride complex bearing a tridentate monoanionic N-donor macrocyclic ligand, [Ni(Tp*)(BH4)], in an η3-binding mode, where the importance of ancillary ligands in stabilizing nickel–borohydride species was emphasized.16 Its reactivity was also investigated for dehalogenation reactions.
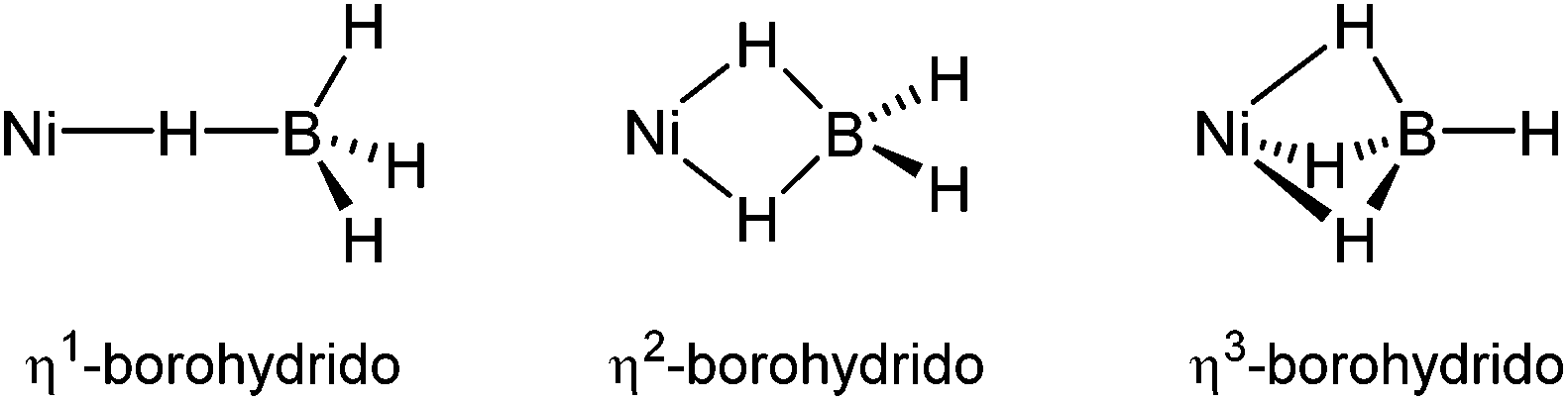 | ||
| Scheme 1 Mononuclear nickel–BH4 adducts with three different coordination modes. | ||
With PCP pincer ligands, Guan et al. described (η2-BH4) nickel(II) complexes by the reaction of the corresponding nickel(II)–hydride complexes and BH3.17 Recently, the reactivity of a similar complex reported by Kirchner and co-workers has been examined in CO2 reduction.18 McGrady's group on the other hand reported the (η2-BH4) nickel(I) complex with a tridentate P-donor ligand.19 Churchard and co-workers were able to isolate two isomers of nickel(II)–borohydride complexes bearing a tetradentate N-donor macrocycle where such complexes were suggested as hydrogen storage materials.20 Herein we report the preparation and characterization of a mononuclear high-spin (η2-BH4) nickel(II) complex bearing a tridentate N-donor ligand, [Ni(Me3-TACN)(BH4)(CH3CN)]+ (Me3-TACN = 1,4,7-trimethyl-1,4,7-triazacyclononane). To the best of our knowledge, the present study reports not only the first example of a high-spin nickel borohydride complex that has an η2 binding mode but also the first well investigated kinetics in dehalogenation reactions of nickel–borohydride species.
Results and discussion
Nickel(II) precursor complex
The starting complex, [Ni(Me3-TACN)(CH3CN)2](ClO4)2 (1-(ClO4)2), was synthesized by reacting Ni(ClO4)2·6H2O and Me3-TACN in CH3CN. The UV-vis spectrum of 1 in CH3CN exhibits several d–d bands at 340 (ε = 25 M−1 cm−1), 540 (ε = 16 M−1 cm−1), 800 (ε = 23 M−1 cm−1), and 880 nm (ε = 28 M−1 cm−1) (Fig. 1a). The electrospray ionization mass spectrum (ESI-MS) of 1 shows intense isotope envelopes at a mass-to-charge (m/z) of 155.6 for {Ni(Me3-TACN)(CH3CN)2}2+, 176.2 for {Ni(Me3-TACN)(CH3CN)3}2+ and 369.2 for {Ni(Me3-TACN)(ClO4)}+ (see Fig. S1, ESI†). The spin state of 1 in CH3CN solution was determined using the 1H NMR spectroscopy method of Evans, and the room temperature magnetic moment of 3.2μB indicates a high-spin state (S = 1) of the nickel(II) species.21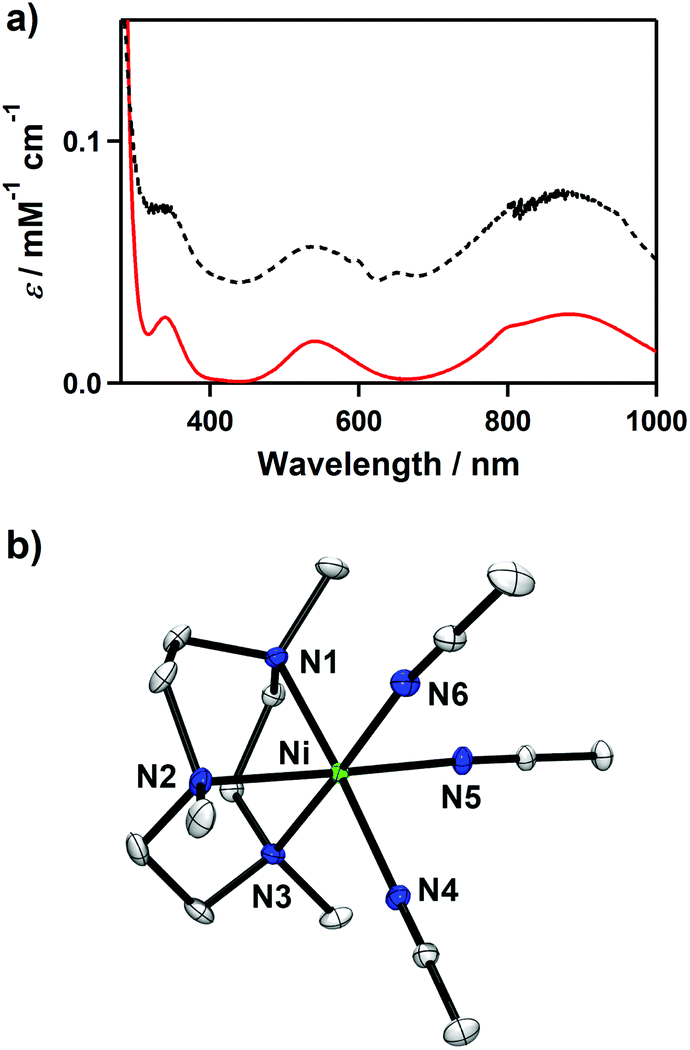 | ||
| Fig. 1 (a) UV-vis spectrum of 1 in acetonitrile (red) and the reflectance spectrum of a powdered sample of 1 (black, intensity scale is arbitrary). (b) X-ray structure of the [Ni(Me3-TACN)(CH3CN)3]2+ cation in 1-(BPh4)2 showing 30% probability thermal ellipsoids. Hydrogen atoms are omitted for clarity. | ||
Violet crystals were obtained upon addition of NaBPh4 to a solution of 1-(ClO4)2 in CH3CN. The crystal structure of the cationic part of 1-(BPh4)2 is shown in Fig. 1b and Table 1, and selected bond distances and angles are listed in Table S1 (ESI†). Complex 1 has a distorted octahedral geometry based on facially coordinated Me3-TACN and three CH3CN solvent molecules.
| 1-(BPh4)2 | 2-(BPh4) | |
|---|---|---|
| Empirical formula | C63H70B2N6Ni | C35H48B2N4Ni |
| Formula weight | 991.58 | 605.10 |
| Temperature (K) | 100(2) | 100(2) |
| Crystal system | Monoclinic | Orthorhombic |
| Space group | P21/c | Pbca |
| Unit cell dimensions | ||
| a (Å) | 18.0900(2) | 20.1159(6) |
| b (Å) | 11.65560(10) | 15.8862(5) |
| c (Å) | 25.8637(3) | 20.4047(6) |
| β (°) | 90.1750(10) | 90 |
| Volume (Å3) | 5453.33(10) | 6520.6(3) |
| Z | 4 | 8 |
| d calc (g cm−3) | 1.208 | 1.233 |
| μ (mm−1) | 0.401 | 0.625 |
| Reflections collected | 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 457 457 |
110259 |
| Independent reflections | 13591 |
8116 |
| Refinement method | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 |
| F(000) | 2112 | 2592 |
| Goodness-of-fit on F2 | 1.030 | 1.070 |
| Final R indices [I > 2σ(I)] | R 1 = 0.0303 | R 1 = 0.0524 |
| wR2 = 0.0775 | wR2 = 0.1251 | |
| R indices (all data) | R 1 = 0.0361 | R 1 = 0.0573 |
| wR2 = 0.0775 | wR2 = 0.1291 | |
Nickel(II)–borohydride complex and characterization
The nickel(II)–borohydride complex, [Ni(Me3-TACN)(BH4)(CH3CN)]+ (2), was prepared by adding 1.5 equiv. of NaBH4 to a solution containing 1 in CH3CN at −20 °C under an N2 atmosphere, where the color changed from violet to purple (Scheme 2). Complex 2 is thermally stable in CH3CN under N2, which allowed us to isolate crystals by adding NaBH4 that were used in structural and spectroscopic analyses and reactivity studies. The crystalline solid is stable to air and moisture for several days. The UV-vis spectrum of 2 in CH3CN shows three absorption bands at 355 (ε = 23 M−1 cm−1), 545 (ε = 18 M−1 cm−1) and 881 nm (ε = 17 M−1 cm−1) (Fig. 2a), which are characteristic bands expected for a high-spin d8 nickel(II) in a distorted octahedral geometry and can be attributed to three possible d–d transitions, 3A2g → 3T1g (P), 3A2g → 3T1g (F) and 3A2g → 3T2g, respectively.22,23 The reflectance spectrum of a powder sample prepared from crystals of 2 shows that the spectral feature of 2 in the solid state is the same as in the solution state (Fig. 2a). The room temperature magnetic moment of 2.9μB, determined using the 1H NMR Evans method, is consistent with a high-spin state (S = 1) of the nickel(II) species.21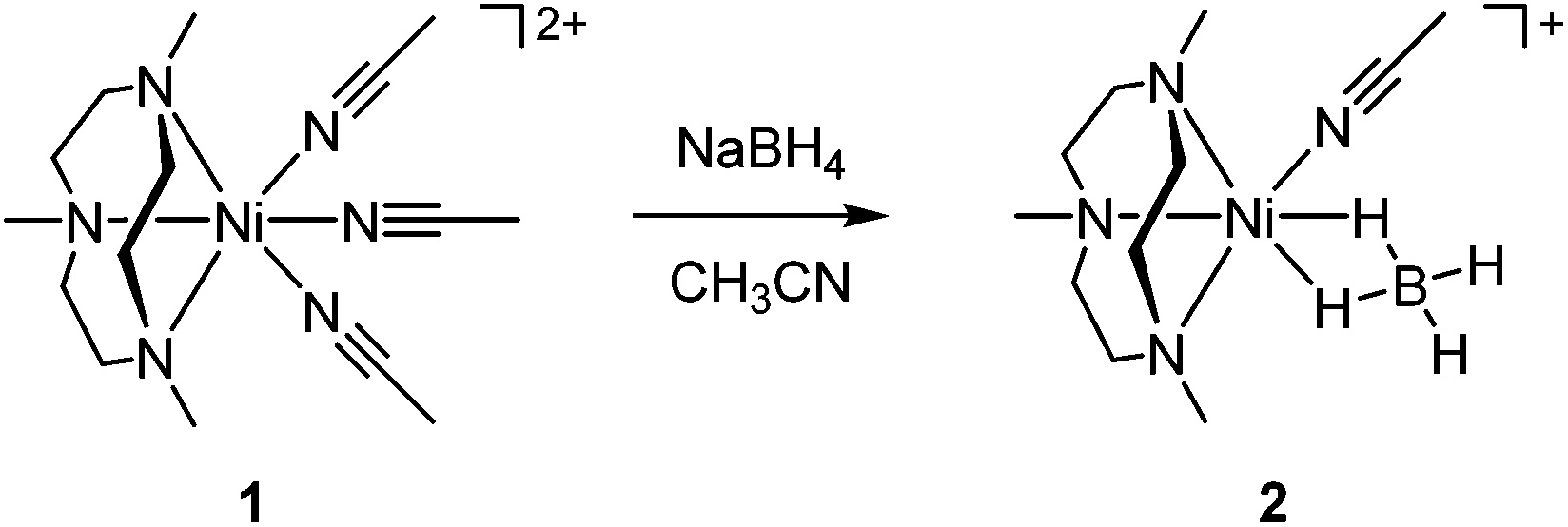 | ||
| Scheme 2 A synthetic route for a mononuclear nickel(II)–borohydride complex. | ||
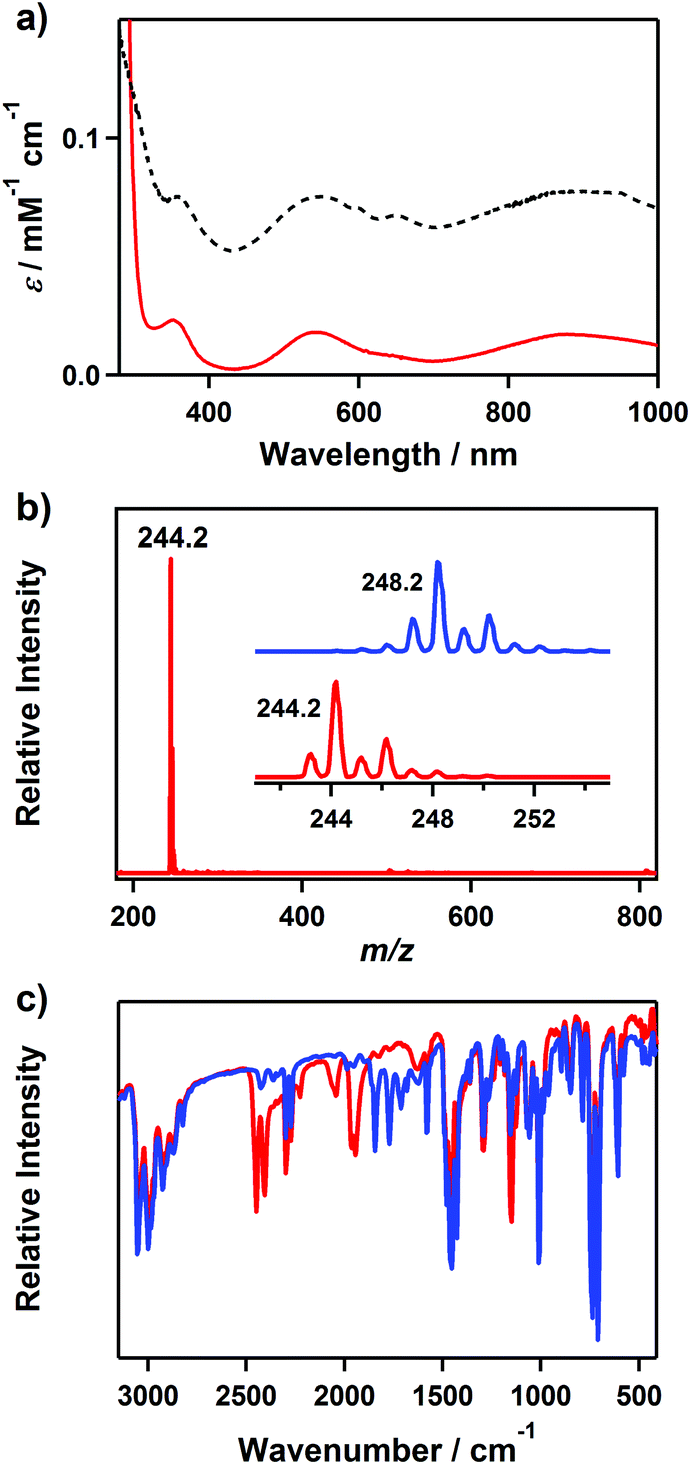 | ||
| Fig. 2 (a) UV-vis spectrum of 2 in acetonitrile (red) and the reflectance spectrum of a powdered sample of 2 (black, intensity scale is arbitrary). (b) ESI-MS of 2 in CH3CN under N2. Inset shows observed isotope distribution patterns for {Ni(Me3-TACN)(BH4)}+ at m/z 244.2 (lower) and {Ni(Me3-TACN)(BD4)}+ at m/z 248.2 (upper). (c) FT-IR spectra of 2 prepared with BH4− (red line) and BD4− (blue line). | ||
The ESI-MS of 2 exhibits a prominent ion peak at a m/z of 244.2, whose mass and isotope distribution patterns correspond to {Ni(Me3-TACN)(BH4)}+ (calculated m/z of 244.2) (Fig. 2b). When the reaction was carried out with isotopically labeled NaBD4, its mass corresponding to {Ni(Me3-TACN)(BD4)}+ appeared at a m/z of 248.2 (calculated m/z of 248.2) (Fig. 2b, inset). The four mass unit shift on substitution of BH4− with BD4− proves that 2 contains a borohydride unit. The Fourier transform infrared (FT-IR) spectrum of 2 shows several isotopically sensitive features (Fig. 2c). [Ni(Me3-TACN)(BH4)(CH3CN)]+ (2H) exhibits a strong band at 2428 cm−1 (doublet, 41 cm−1 splitting), which shifts to 1809 cm−1 (doublet, 74 cm−1 splitting) in [Ni(Me3-TACN)(BD4)(CH3CN)]+ (2D). This band has been tentatively assigned to be v(B–H)terminal and the observed doublet is characteristic of an η2 manner of BH4− binding to the metal center as previously reported.1,18,20 The observed v(B–H)/v(B–D) ratio of 0.75 for v(B–H)terminal is in good agreement with the calculated ratio of 0.74 based on Hooke's law. According to the literature,1,20 the bidentate binding of BH4− is of a broad band at 1650–2150 for v(B–H)bridging. In 2H, three isotopically sensitive bands at 2043, 1964 and 1943 cm−1 are observed in the region of v(B–H)bridging, which shift to a spectral region obscured by the ligand fingerprint bands in 2D. It was also difficult to observe the lower frequencies for 2 due to the vibrational modes of the supporting ligand. The solution IR spectrum of 2 recorded in CH3CN exhibits that the spectral behavior of 2 in solution is the same as that in the solid state (Fig. S2, ESI†), consistent with the result from UV-vis spectroscopy (vide supra).
The X-ray crystal structure of [Ni(Me3-TACN)(BH4)(CH3CN)](BPh4) (2-(BPh4)) reveals that the borohydride ligand coordinates in a symmetrical η2-fashion (Fig. 3). The presence of a counter anion, BPh4−, supports the nickel(II) oxidation state assignment. The distorted octahedral nickel center of 2 is bound by three nitrogen atoms of Me3-TACN, two bridging hydrogen atoms of BH4−, and one nitrogen atom of CH3CN. To the best of our knowledge, this is the first crystal structure of a high-spin nickel(II)–borohydride complex in an η2-manner. The Ni⋯B distance of 2 (2.225(3) Å) is longer than that of the tridentate borohydride nickel(II) complex, [Ni(Tp*)(η2-BH4)] (2.048 Å);16 and slightly shorter than that of the bidentate borohydride nickel(I) complex, [Ni(triphos)(η2-BH4)] (2.24 Å).19 However, the Ni⋯B distance of 2 is comparable to that of the bidentate nickel(II) complex, [Ni(PCPMe–iPr)(η2-BH4)] (2.218 Å).18 The average Ni–H bond length of 2 (1.745 Å) is comparable to that of [Ni(PCPMe–iPr)(η2-BH4)] (1.78 Å) and lies well within the nickel–borohydride category (1.59–1.94 Å). Based on the structural and spectroscopic characterization, complex 2 is assigned as a high-spin nickel(II)–borohydride complex in an η2-fashion.
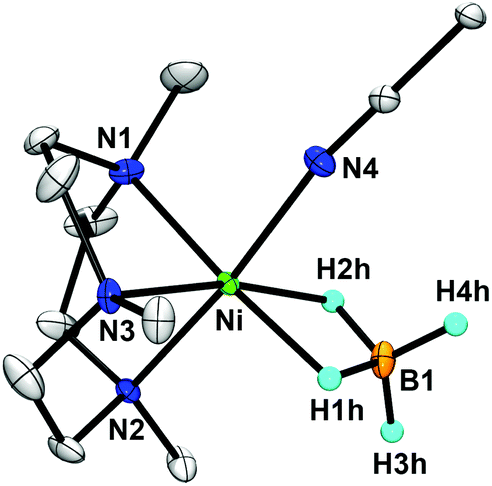 | ||
| Fig. 3 X-ray structure of the [Ni(Me3-TACN)(BH4)(CH3CN)]+ cation in 2-(BPh4) showing 30% probability thermal ellipsoids. Hydrogen atoms except those attached to boron are omitted for clarity. Selected bond lengths (Å) and angles (deg): Ni–N1 2.083(2), Ni–N2 2.1430(18), Ni–N3 2.0701(18), Ni–N4 2.0945(19), Ni–H1h 1.72(3), Ni–H2h 1.77(3), Ni⋯B1 2.225(3), N1–Ni–N2 83.84(7), N2–Ni–N3 84.40(7), N3–Ni–N1 84.92(8). | ||
Reactivity
It is known that mononuclear metal–borohydride complexes are capable of performing reductive dehalogenation of halocarbons.16,19,24,25 In order to examine the dehalogenation reactivity of 2, we carried out the reaction of CHCl3 with 2. Upon the addition of CHCl3 to 2 in CH3CN at 25 °C, the UV-vis absorption bands of 2 disappeared with a pseudo-first-order decay (Fig. 4a, inset). Product analysis of the final reaction mixture revealed the formation of CH2Cl2 and very little H2, where the volatile products were confirmed by 1H NMR.16,26 In addition, [Ni(Me3-TACN)(Cl)]+ was found in the reaction solutions as a product of 2 (Fig. S3 for ESI-MS analysis, ESI†). Although several attempts to isolate the product as crystals have not been successful, an intense band at 342 nm of the product solution is similar to that of previously reported tetrahedral Ni(II)–Cl complexes (Fig. 4a).27 These bands have been assigned to the Cl pπ → Ni dπ* LMCT band on the basis of theoretical calculations (Fig. S4, ESI†).28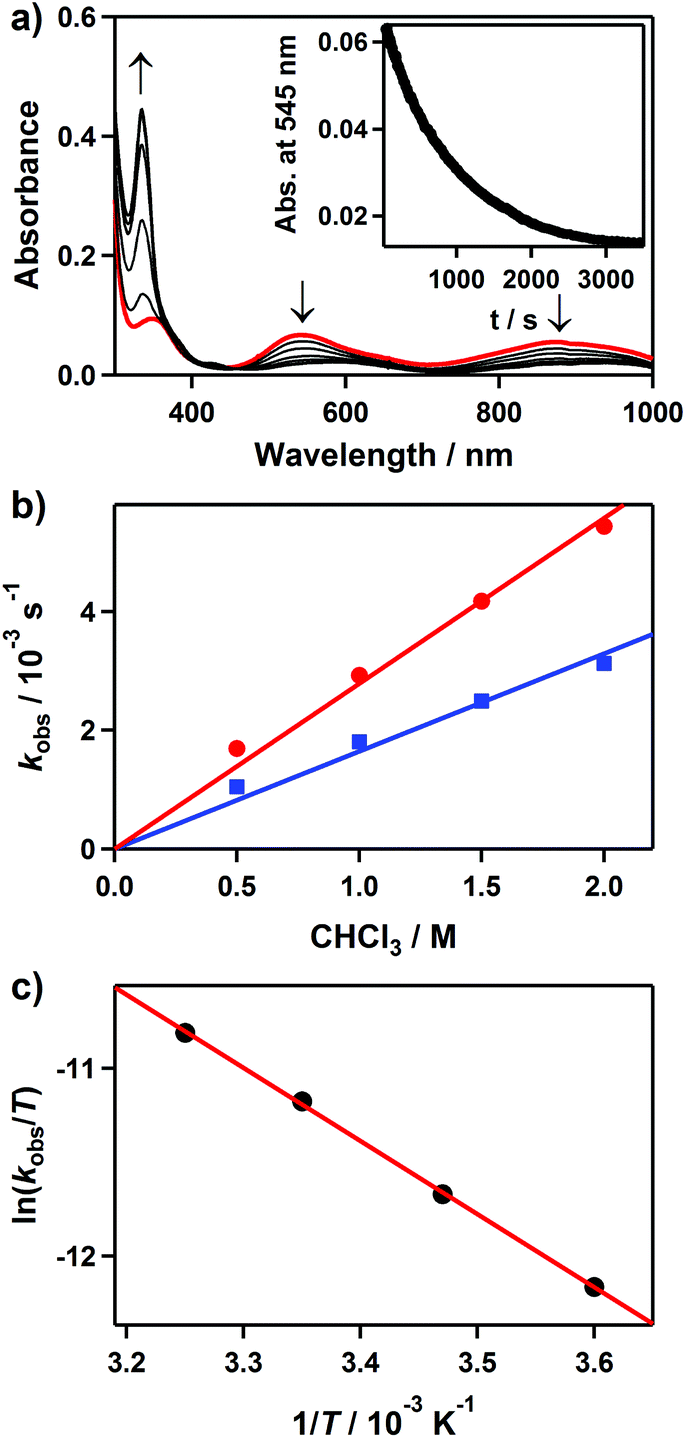 | ||
Fig. 4 Reactions of 2 with chloroform (CHCl3) in CH3CN. (a) UV-vis spectral changes of 2 (4 mM) upon addition of 125 equiv. of CHCl3 at 25 °C. Inset shows the time course of the absorbance at 545 nm. (b) Plots of kobs against the concentration of CHCl3 to determine second-order rate constants for the reactions of 2H (red line with ●) and 2D (blue line with  ) at 25 °C. (c) Plot of second-order rate constants against 1/T to determine activation parameters for the reaction of 2 and CHCl3. ) at 25 °C. (c) Plot of second-order rate constants against 1/T to determine activation parameters for the reaction of 2 and CHCl3. | ||
The pseudo-first-order rate constants, monitored at 545 nm, increased proportionally with the CHCl3 concentration, giving a second-order rate constant (k2) of 2.7(1) × 10−3 M−1 s−1 (Fig. 4b). The rates were dependent on the reaction temperature, where a linear Eyring plot was observed in the range of 278–308 K with activation parameters of ΔH‡ = 33 kJ mol−1 and ΔS‡ = −182 J mol−1 K−1 (Fig. 4c). The observed negative entropy value and second-order kinetics suggest that a bimolecular mechanism is performing in the reduction of CHCl3 by 2 (Scheme 3). The reactivity of 2 in dehalogenation is much higher than that of the low-spin (η3-BH4) nickel(II) complex, [Ni(Tp*)(BH4)], where the latter reacted with CHCl3 only at elevated temperatures (>50 °C).16,18
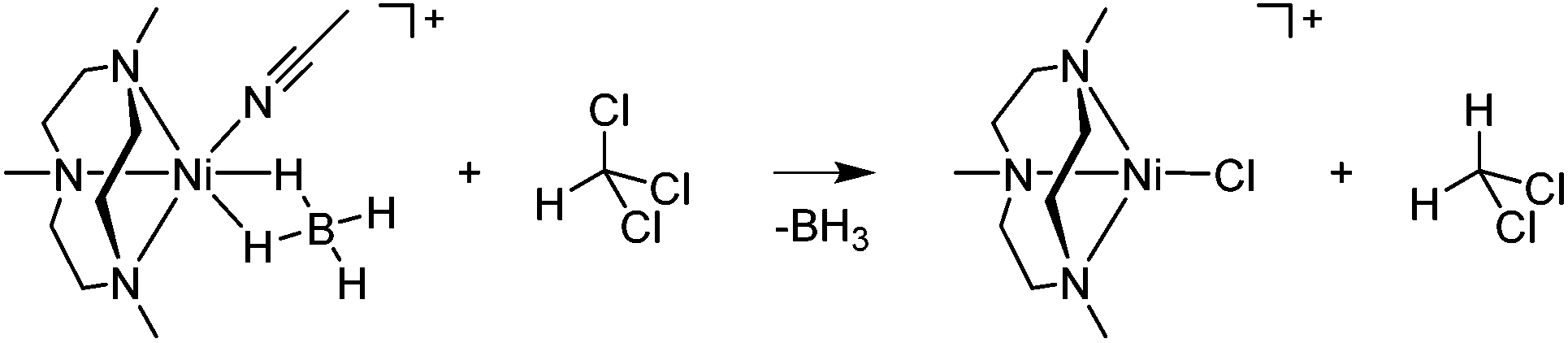 | ||
| Scheme 3 Proposed mechanism of the dehalogenation of CHCl3 by 2. | ||
We also observed a kinetic isotope effect (KIE) value of 1.7 in the reduction of CHCl3 by 2 when 2H was replaced with 2D (Fig. 4b). This KIE value is comparable to that of [Ni(Tp*)(BH4)] (kH/kD = 3),16 in which hydride ion or hydrogen atom transfer was proposed to be rate limiting.
The reactivity of 2 was further investigated using para-substituted benzoyl chloride, which has a series of electron-donating and -withdrawing substituents at the para-position of the phenyl group (para-X-Ph-COCl; X = MeO, tBu, H, Cl, NO2). 2 disappeared immediately upon addition of benzoyl chloride (10 equiv.) in CH3CN at −40 °C. Thus, the reactivity of 2 with para-substituted benzoyl chloride was examined using a stopped-flow spectrometer. The Hammett plot of the second-order rate constants vs. σp+ gave a ρ value of 1.2 (Fig. 5). The positive ρ value is consistent with the process involving a nucleophilic character. Product analysis of the final reaction mixture revealed the formation of para-substituted benzaldehydes.
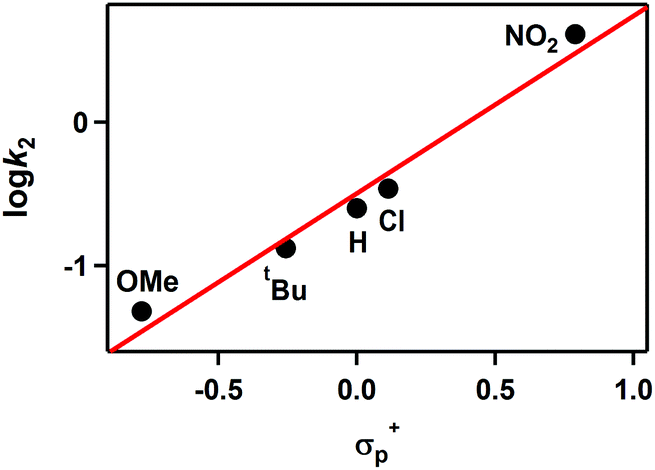 | ||
| Fig. 5 Hammett plot of logk2 against σp+ of benzoyl chloride derivatives in the reactions of 2 with para-substituted benzoyl chloride, para-X-Ph-COCl (X = OMe, tBu, H, Cl, NO2) in CH3CN at −40 °C. | ||
Density functional theory results
To compare the stability of the Ni(II)–borohydride complexes in different coordination modes at a quantitative level, we performed spin-polarized density functional theory (DFT) calculations of the complexes in the gas phase using a non-local hybrid exchange–correlation functional (B3LYP).29 Two different coordination modes, η2 and η3, were considered (Fig. 6). Both complexes are in a high-spin state (S = 1). For the [Ni(Me3-TACN)(BH4)(CH3CN)]+ cation in an η2-fashion, the Ni⋯B distance is calculated to be 2.19 Å and the average Ni–H bond length is 1.75 Å, in good agreement with the experimental values. For the [Ni(Me3-TACN)(BH4)]+ cation in an η3-fashion, the Ni⋯B distance is shortened to 1.97 Å, while the Ni–H bonds are elongated with the average value of 1.85 Å. We found that the η2-complex is 35 kJ mol−1 more stable than the η3-complex, indicating that the Ni(II)–borohydride will exist in an η2-fashion in a solution at room temperature.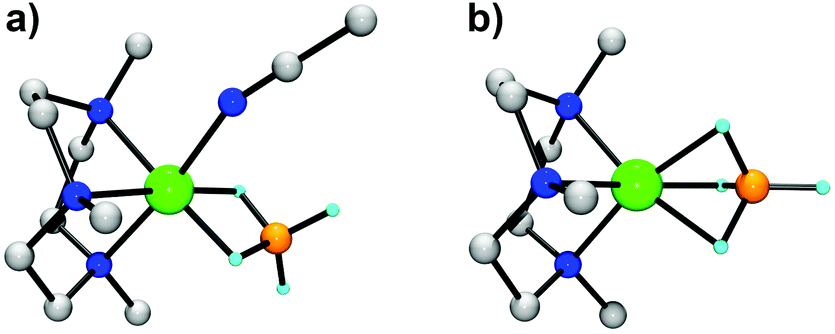 | ||
| Fig. 6 (a) DFT structure of 2 in an η2-fashion (gray, C; blue, N, cyan, H; orange, B; green, Ni). Hydrogen atoms except those attached to boron are omitted for clarity. Selected bond lengths (Å) and angles (deg) are listed below for a direct comparison with the result in Fig. 3: Ni–N1 2.14, Ni–N2 2.20, Ni–N3 2.15, Ni–N4 2.17, Ni–H1h 1.76, Ni–H2h 1.75, Ni⋯B1 2.19, N1–Ni–N2 83.26, N2–Ni–N3 82.95, N3–Ni–N1 83.85. (b) DFT structure of the metastable [Ni(Me3-TACN)(BH4)]+ cation in an η3-fashion. | ||
Conclusions
A mononuclear high-spin (η2-BH4) nickel(II) complex, [Ni(Me3-TACN)(BH4)(CH3CN)]+ (2), has been successfully prepared and characterized by various physicochemical methods. In particular, FT-IR spectroscopy together with X-ray crystallography clearly shows that a borohydride is bound to the nickel center in an η2-fashion. 2 is capable of performing dehalogenation reactions. Kinetic studies and isotope labelling experiments suggest that bimolecular collision between 2 and halocarbons is the rate-limiting step in dehalogenation reactions.Experimental section
Materials
All chemicals obtained from Aldrich Chemical Co. were of the best available purity and used without further purification unless otherwise indicated. Solvents were dried according to published procedures and distilled under Ar prior to use.30Physical methods
UV-vis spectra were recorded on a Hewlett Packard 8453 diode array spectrophotometer equipped with UNISOKU Scientific Instruments for low-temperature experiments or with a circulating water bath. Fast reactions were monitored using a Hi-Tech Scientific SF-61 DX2 cryogenic stopped-flow spectrometer equipped with a Xe arc lamp and a KinetaScan diode array rapid scanning unit. Electrospray ionization mass spectra (ESI-MS) were collected on a Waters (Milford, MA, USA) Acquity SQD quadrupole mass instrument, by infusing samples directly into the source using a manual method. The spray voltage was set at 2.5 kV and the capillary temperature at 80 °C. Infrared spectra were recorded on a Thermo Scientific Nicolet iS10 FT-IR spectrometer equipped with a diamond attenuated total reflectance (ATR) accessory. The effective magnetic moments were determined using the modified 1H NMR method of Evans at room temperature.21,31,32 A WILMAD® coaxial insert (sealed capillary) tube containing the blank acetonitrile-d3 solvent (with 1.0% TMS) only was inserted into the normal NMR tubes containing the complexes dissolved in acetonitrile-d3 (with 0.03% TMS). The chemical shift of the TMS peak (and/or the solvent peak) in the presence of the paramagnetic metal complexes was compared with that of the TMS peak (and/or the solvent peak) in the inner coaxial insert tube. The effective magnetic moment was calculated using the equation, μ = 0.0618(ΔνT/2fM)1/2, where f is the oscillator frequency (MHz) of the superconducting spectrometer, T is the absolute temperature, M is the molar concentration of the metal ion, and Δv is the difference in frequency (Hz) between the two reference signals. Product analysis was performed on a Thermo Fisher Trace 1310 gas chromatograph system equipped with a flame ionization detector (GC). 1H NMR spectra were recorded with a Bruker AVANCE III-400 spectrometer at CCRF in DGIST.Syntheses
X-ray crystallography
Single crystals of [Ni(Me3-TACN)(CH3CN)3](BPh4)2 (1-(BPh4)2) and [Ni(Me3-TACN)(BH4)(CH3CN)](BPh4) (2-(BPh4)) were picked from solutions by using a nylon loop (Hampton Research Co.) on a handmade copper plate mounted inside a liquid N2 Dewar vessel at ca. −40 °C and mounted on a goniometer head in a N2 cryostream. Data collections were carried out on a Bruker SMART APEX II CCD diffractometer equipped with a monochromator in the Mo Kα (λ = 0.71073 Å) incident beam. The CCD data were integrated and scaled using the Bruker-SAINT software package, and the structure was solved and refined using SHELXTL V 6.12.33 Hydrogen atoms were located in the calculated positions except those of borohydride, which were found from the Fourier difference map. All non-hydrogen atoms were refined with anisotropic thermal parameters. Crystal data for 1-(BPh4)2: C63H70B2N6Ni, monoclinic, P21/c, Z = 4, a = 18.0900(2), b = 11.65560(10), c = 25.8637(3) Å, β = 90.1750(10)°, V = 5453.33(10) Å3, μ = 0.401 mm−1, ρcalcd = 1.208 g cm−3, R1 = 0.0303, wR2 = 0.0775 for 13591 unique reflections, 655 variables. Crystal data for 2-(BPh4): C35H48B2N4Ni, orthorhombic, Pbca, Z = 8, a = 20.1159(6), b = 15.8862(5), c = 20.4047(6) Å, V = 6520.6(3) Å3, μ = 0.625 mm−1, ρcalcd = 1.233 g cm−3, R1 = 0.0524, wR2 = 0.1251 for 8116 unique reflections, 399 variables. The crystallographic data for 1-(BPh4)2 and 2-(BPh4) are listed in Table 1, and Table S1† lists the selected bond distances and angles. CCDC 1423623 for 1-(BPh4)2 and 1423622 for 2-(BPh4) contain the supplementary crystallographic data for this paper.
Reactivity
All reactions were run in a 1 cm UV cuvette by monitoring UV-vis spectral changes of reaction solutions, and rate constants were determined by fitting the changes in absorbance at 545 nm for [Ni(Me3-TACN)(BH4)(CH3CN)]+ (2). For the stopped-flow experiments, all reaction traces were collected at −40 °C. The raw kinetic data were treated with KinetAsyst 3 (Hi-Tech Scientific) and Specfit/32 Global Analysis System software from Spectrum Software Associates. Reactions were run at least in triplicate, and the data reported represent the average of these reactions. Crystalline samples of 2 were used in kinetic studies, such as the reduction of chloroform in CH3CN at 25 °C. After the completion of reactions, pseudo-first-order fitting of the kinetic data allowed us to determine kobsvalues. Products formed in the reduction of chloroform by 2 in CD3CN at 25 °C were analyzed by 1H NMR. Products were identified by comparing with authentic samples.Computational details
Spin-polarized DFT calculations were performed using a non-local hybrid exchange–correlation functional (B3LYP).29 We used the projector-augmented wave (PAW) method34 and a plane wave cutoff of 400 eV, as implemented in the VASP code.35,36 Atomic structures of the Ni(II)–borohydride complexes were relaxed within 0.03 eV Å−1. Geometry optimization and time-dependent DFT (TD-DFT) calculations for [Ni(Me3-TACN)(Cl)]+ were performed with the Gaussian 09 package.Acknowledgements
The research was supported by NRF (2014R1A1A2056051), the Ministry of Science, ICT and Future Planning (DGIST R&D Program 15-BD-0403 & 15-HRLA-02, KCRC 2014M1A8A1049320, and 2015M3D3A1064890) and the Ministry of Oceans and Fisheries (Marine Biotechnology Program 20150220) of Korea (J.C.). We are grateful to Prof. Kiyoung Park (KAIST) and Mr Donghyun Jeong (DGIST) for valuable discussions on TD-DFT. This research used capabilities of the DGIST Supercomputing and Bigdata Center.Notes and references
- H. D. Kaesz and R. B. Saillant, Chem. Rev., 1972, 72, 231–281 CrossRef CAS.
- S. Chakraborty, J. Zhang, J. A. Krause and H. Guan, J. Am. Chem. Soc., 2010, 132, 8872–8873 CrossRef CAS PubMed.
- H. W. Lee, J. B. Ahn, J. H. Lee, S. K. Kang, S. K. Ahn and S. J. Lee, Heterocycles, 2005, 65, 1843–1856 CrossRef CAS.
- S. Yakabe, M. Hirano and T. Morimoto, Tetrahedron Lett., 2000, 41, 6795–6798 CrossRef CAS.
- T. B. Reddy, Linden Handbook of Batteries, McGraw-Hill, New York, 2002 Search PubMed.
- U. Häussermann, H. Blomqvist and D. Noréus, Inorg. Chem., 2002, 41, 3684–3692 CrossRef.
- S. W. Ragsdale, J. Biol. Chem., 2009, 284, 18571–18575 CrossRef CAS PubMed.
- W. Lubitz, M. V. Gastel and W. Gärtner, in Nickel and Its Surprising Impact in Nature, ed. A. Sigel, H. Sigel and R. K. O. Sigel, John Wiley & Sons, Ltd, United Kingdom, 2007, ch. 7, vol. 2, pp. 279–322 Search PubMed.
- P. M. Vignais and B. Billoud, Chem. Rev., 2007, 107, 4206–4272 CrossRef CAS PubMed.
- T. A. Bobik, K. D. Olson, K. M. Noll and R. S. Wolfe, Biochem. Biophys. Res. Commun., 1987, 149, 455–460 CrossRef CAS PubMed.
- J. Ellermann, R. Hedderich, R. Böcher and R. K. Thauer, Eur. J. Biochem., 1998, 172, 669–677 CrossRef.
- C. Holliger, G. Schraa, E. Stupperich, A. J. M. Stams and A. J. B. Zehnder, Bacteriol., 1992, 174, 4427–4434 CAS.
- C. Holliger, S. W. M. Kengen, G. Schraa, A. J. M. Stams and A. J. B. Zehnder, Bacteriol., 1992, 174, 4435–4443 CAS.
- B. Ganem and J. O. Osby, Chem. Rev., 1988, 86, 763–780 CrossRef.
- T. J. Marks and J. R. Kolb, Chem. Rev., 1977, 77, 263–293 CrossRef CAS.
- P. J. Desrochers, S. Lelievre, R. J. Johnson, B. T. Lamb, A. L. Phelps, A. W. Cordes, W. Gu and S. P. Cramer, Inorg. Chem., 2003, 42, 7945–7950 CrossRef CAS PubMed.
- S. Chakraborty, J. Zhang, Y. J. Patel, J. A. Krause and H. Guan, Inorg. Chem., 2013, 52, 37–47 CrossRef CAS PubMed.
- S. Murugesan, B. Stöger, M. Weil, L. F. Veiros and K. Kirchner, Organometallics, 2015, 34, 1364–1372 CrossRef CAS.
- M. Kandiah, G. S. McGrady, A. Decken and P. Sirsch, Inorg. Chem., 2005, 44, 8650–8652 CrossRef CAS PubMed.
- A. J. Churchard, M. K. Cyranski, Ł. Dobrzycki, A. Budzianowski and W. Grochala, Energy Environ. Sci., 2010, 3, 1973–1978 CAS.
- D. F. Evans and D. A. Jakubovic, J. Chem. Soc., Dalton Trans., 1988, 2927–2933 RSC.
- J. Cho, Y. M. Lee, S. Y. Kim and W. Nam, Polyhedron, 2010, 29, 446–450 CrossRef CAS.
- F. A. Cotton, G. Wilkinson, C. A. Murillo and M. Bochmann, Advanced Inorganic Chemistry, Wiley, New York, 1999 Search PubMed.
- M. Cha, C. L. Gatlin, S. C. Critchlow and J. A. Kovacs, Inorg. Chem., 1993, 32, 5868–5877 CrossRef CAS.
- J. D. Crane, Annu. Rep. Prog. Chem., Sect. A: Inorg. Chem., 1997, 93, 593–610 RSC.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- P. J. Desrochers, J. Telser, S. A. Zvyagin, A. Ozarowski, J. Krzystek and D. A. Vicic, Inorg. Chem., 2006, 45, 8930–8941 CrossRef CAS PubMed.
- T. Deb, C. M. Anderson, H. Ma, J. L. Petersen, V. G. Young Jr. and M. P. Jensen, Eur. J. Inorg. Chem., 2015, 458–467 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- W. L. F. Armarego and D. D. Perrin, Purification of Laboratory Chemicals, Elsevier, Oxford, 1997 Search PubMed.
- D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC.
- J. Löliger and R. Scheffold, J. Chem. Educ., 1972, 49, 646–647 CrossRef.
- G. M. Sheldrick, SHELXTL, Version 6.12, Bruker AXS, Inc., Madison, WI, 2001 Search PubMed.
- P. B. Blöchl, Phys. Rev. B: Condens. Matter, 1994, 50, 17953–17979 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: [Ni(Me3-TACN)(CH3CN)3](BPh4)2 and [Ni(Me3-TACN)(BH4)(CH3CN)](BPh4). CCDC 1423622 and 1423623. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qi00206k |
| This journal is © the Partner Organisations 2016 |