
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preparing DNA-mimicking multi-line nanocaterpillars via in situ nanoparticlisation of fully conjugated polymers†
In-Hwan
Lee
a,
Pitchamuthu
Amaladass
a,
Inho
Choi
a,
Victor W.
Bergmann
b,
Stefan A. L.
Weber
b and
Tae-Lim
Choi
*a
aDepartment of Chemistry, Seoul National University, Seoul, 08826, Korea. E-mail: tlc@snu.ac.kr
bMax Planck Institute for Polymer Research, 55128 Mainz, Germany
First published on 19th January 2016
Abstract
A unique hierarchical evolution from single-line nanocaterpillars to multi-line nanocaterpillars and then to multi-line nanocaterpillars bearing a few long-chain branches was demonstrated by in situ nanoparticlisation of fully conjugated poly(2,5-dihexyloxy-1,4-phenylene)-block-poly(3-methylthiophene) (PPP-b-P3MT). PPP-b-P3MTs of various block ratios were successfully synthesised by the Grignard metathesis polymerization method; moreover, these block copolymers underwent spontaneous self-assembly during the polymerization owing to the solvophobicity or strong π–π interactions of the core block, i.e. P3MT. These in situ generated PPP-b-P3MT NPs were quite different from the previously reported NPs generated from poly(2,5-dihexyloxy-1,4-phenylene)-block-polythiophene (PPP-b-PT). AFM and TEM images revealed that PPP-b-P3MTs formed single- to multi-line nanocaterpillars, whereas PPP-b-PTs only formed shorter single-line nanocaterpillars. On the basis of PXRD and UV–vis data, we speculated that this interesting morphology of multi-line nanocaterpillars, resembling duplex DNA, arose from different packing modes and crystallinity as well as improved solubility of the P3MT core compared with the NPs containing the PT core.
Introduction
Self-assembly of conjugated polymers has proved to be a useful approach to construct well-ordered semiconducting nanostructures.1–5 In particular, rod–coil-type block copolymers containing conjugated polymers as the rod segment have attracted considerable attention because they form various nanostructures such as spheres, fibres and vesicles.6–10 However, using these nanostructures as electronic materials may be limited because of the insulating nature of the coil segments. Consequently, recent developments in the synthesis of fully conjugated block copolymers have become interesting because the entire backbone is semiconducting and these copolymers could form various nanostructures such as nanowires,11–13 nanorings,12 helical nanowires,14 vesicles and lamellae in both solution and thin-film states.11–17Conventionally, the self-assembly of block copolymers is induced by post-synthetic treatments such as the slow addition of selective solvents, dialysis, ageing and chemical modification (e.g. charge development16 or adding glue molecules18), which are necessary to impose the driving force for self-assembly upon one segment of the block copolymer. Unfortunately, these additional post-synthetic treatments may take a long time to form the desired nanostructures. Furthermore, the obtained nanostructures are generally unstable against heat, solvent change and mechanical force. Therefore, for improving the process of self-assembly and the stability of the resulting nanostructures, both the rational design of block copolymer architectures and the development of fast and efficient self-assembly processes are highly desirable (e.g. polymerization-induced self-assembly).19–21
Recently, we reported a new strategy called in situ nanoparticlisation of conjugated polymers (INCP), where the direct self-assembly of conjugated polymers spontaneously occurred during their controlled/living block copolymerization.22–28 The main driving force for this step-economic self-assembly process was the high solvophobicity or strong π–π interactions of the second blocks, which comprised insoluble conjugated polyacetylene or polythiophene without any side chain. For example, in situ self-assembly of fully conjugated poly(3-(2-ethylhexyl)thiophene)-block-polythiophene (P3EHT-b-PT)24 and poly(2,5-dihexyloxy-1,4-phenylene)-block-polythiophene (PPP-b-PT)25 afforded highly stable branched nanostructures (nanostars and nanonetworks) and one-dimensional (1D) caterpillar-shaped nanostructures (nanocaterpillars) without any post-synthetic treatments or templates. However, although the previous in situ nanoparticlisation of PPP-b-PT provided 1D nanocaterpillars with narrow length dispersity, the average length of the nanocaterpillars was rather short (ca. 75–120 nm) because the degree of polymerization (DP) of the core PT segment was limited.25 Thus, the core was exposed to a lesser extent, which led to limited supramolecular growth. Attempts to further increase the size of the core by increasing the DP of PT resulted in precipitation during block copolymerization because of low solubility. Hence, to increase the length of the nanocaterpillars, we envisioned that poly(3-methylthiophene) (P3MT), which would be more soluble than the previous PT but still insoluble and crystalline enough for the direct in situ self-assembly, might be an excellent candidate for a new core block (Scheme 1). Furthermore, comparing the in situ self-assembly process and the final supramolecular structures between the previous PPP-b-PT and its analogues containing P3MT would be interesting because the PT and P3MT would crystallise with different packing modes in the core (Scheme 1).47–49
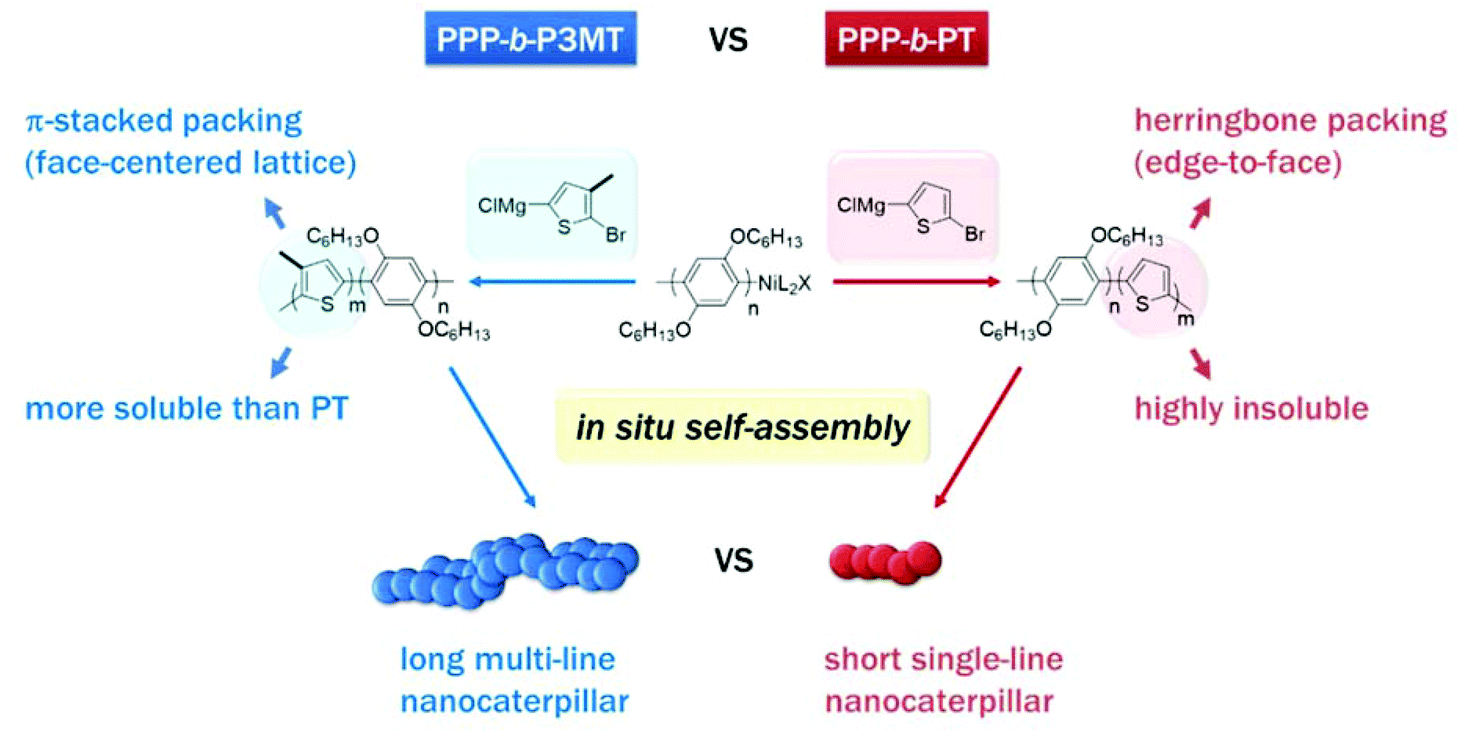 | ||
| Scheme 1 Comparison between in situ nanoparticlisation of PPP-b-P3MT and PPP-b-PT. Note the different solubility and packing modes of P3MT and PT. | ||
Hierarchical supramolecular organization of polymers via a bottom-up approach provides an exciting method to construct a series of well-defined nanoarchitectures from simple polymers.3,9,14,29–37 For example, two single strands of DNA forming a double helix is the basis of life. Similarly, multi-level hierarchical organization of conjugated polymers leads to highly complex functional nanomaterials.3,9,14,30 From our previous reports detailing the construction of various nanostructures such as 0D, 1D, branched and 3D nanostructures via the INCP strategy,22–28 we envisioned that the step-economic and kinetically controlled INCP method could simplify and enrich the hierarchical organization of conjugated polymers. Therefore, we focused our attention on the construction of synthetically challenging but potentially useful nanostructures such as unique supramolecular multi-line nanochains, which are intended to mimic duplex DNA in living systems. Previously, a few reports had demonstrated that small organic molecules such as perylene bisimide38 and inorganic materials such as gold NPs39–41 organised into multi-line nanochains. However, these nanostructures were sensitive to changes in conditions such as solvent, pH and concentration. In addition, the formation of such multi-line nanochains only from polymers had not been realised. Herein, we report the first example of DNA-like multi-line nanochains (or nanocaterpillars) comprising a soft polymeric material. These unique multi-line nanocaterpillars were readily obtained by in situ nanoparticlisation of PPP-b-P3MT, and the longer ones were obtained from the more soluble poly(2,5-di(2-ethylhexyloxy)-1,4-phenylene)-block-poly(3-methylthiophene) (PPP(EH)-b-P3MT). Depending on the DP of P3MT, hierarchical evolution of the nanostructures from nanospheres to single-line nanocaterpillars, multi-line nanocaterpillars and finally to multi-line nanocaterpillars with long chain branches was possible.
Results and discussion
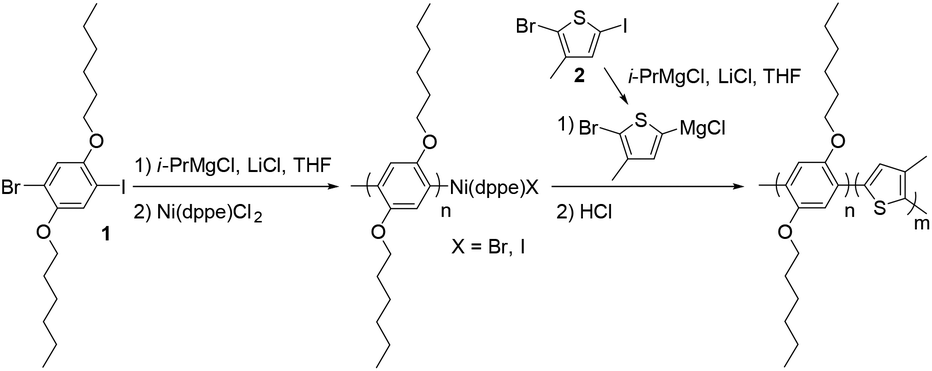
The PPP-b-P3MTs were synthesized by the Grignard metathesis polymerization (GRIM) method.42–44 By fixing the ratio of 1-bromo-4-iodo-2,5-dihexyloxybenzene (1) to (1,2-bis(diphenylphosphino)ethane)dichloronickel(II) (Ni(dppe)Cl2) at 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, the soluble first block PPP with a para-bis(hexyloxy) substituent was produced with a number-average molecular weight (Mn) of 22–26 kg mol−1 and low polydispersity indices (PDI) of 1.04–1.06 (Table 1). Note that compared with our previous GRIM polymerization from 1,4-dibromo-2,5-dihexyloxybenzene to produce PPP,25 using 1 as the monomer resulted in PPP with a higher Mn and lower PDI (Table 1 and Fig. S1a†).43 For the second block, the Grignard reagent from 2-bromo-5-iodo-3-methylthiophene (2) was added to the flask to produce the core of P3MT with high regioselectivity and conversion (Table 1). Moreover, adding one equivalent of lithium chloride (LiCl) during the preparation of the Grignard monomer from 2 was essential to achieve successful block copolymerization of PPP-b-P3MT; otherwise, the block copolymerization was highly inefficient. The DP of the core-forming P3MT block proportionally increased from 36 to 148 upon increasing the feed ratio of monomer 2/Ni(dppe)Cl2 from 50 to 180 (Table 1). The PPP-b-PTs and PPP(EH)-b-P3MTs were prepared by the same GRIM procedure as presented in Table 1 (Fig. S1†).
:1, the soluble first block PPP with a para-bis(hexyloxy) substituent was produced with a number-average molecular weight (Mn) of 22–26 kg mol−1 and low polydispersity indices (PDI) of 1.04–1.06 (Table 1). Note that compared with our previous GRIM polymerization from 1,4-dibromo-2,5-dihexyloxybenzene to produce PPP,25 using 1 as the monomer resulted in PPP with a higher Mn and lower PDI (Table 1 and Fig. S1a†).43 For the second block, the Grignard reagent from 2-bromo-5-iodo-3-methylthiophene (2) was added to the flask to produce the core of P3MT with high regioselectivity and conversion (Table 1). Moreover, adding one equivalent of lithium chloride (LiCl) during the preparation of the Grignard monomer from 2 was essential to achieve successful block copolymerization of PPP-b-P3MT; otherwise, the block copolymerization was highly inefficient. The DP of the core-forming P3MT block proportionally increased from 36 to 148 upon increasing the feed ratio of monomer 2/Ni(dppe)Cl2 from 50 to 180 (Table 1). The PPP-b-PTs and PPP(EH)-b-P3MTs were prepared by the same GRIM procedure as presented in Table 1 (Fig. S1†).
The initial evidence for the formation of core–shell-type micelles from PPP-b-P3MTs was obtained by 1H NMR measurements. Similar to previous examples of INCP,22–28 the 1H NMR spectra of PPP-b-P3MTs in CDCl3 were identical to that of the PPP homopolymer, without any signals corresponding to P3MT (Fig. S2a†). This was because the core block of P3MT as a result of micellisation was not solvated at all by CDCl3 and became undetectable.
The successful block copolymerization of PPP-b-P3MTs was easily checked by the colour change of the polymerization solution. When the Grignard reagent from monomer 2 was added to a flask containing the living chain of PPP (Table 1), the initial yellow solution turned red and subsequently brick red within 2 min because of the increase in the conjugation length of P3MT (Fig. 1). UV–vis spectra of PPP-b-P3MTs provided more details for the whole polymerization. There were two distinct sets of absorption regions, with the λmax values at 340 nm and 470 nm corresponding to the absorption peaks for PPP and P3MT (Fig. 1). The absorption intensity of P3MT relative to that of PPP increased with the increasing DP of P3MT (Fig. 1). Two vibronic peaks appeared at 545 nm and 596 nm, suggesting the π–π stacking of P3MT in solution;12,45,46 the band gap of PPP-b-P3MTs was 1.9–2.0 eV, as calculated from the onset points of the PPP-b-P3MTs (627–646 nm, Fig. 1). Compared with the UV–vis spectra of the PPP70-b-PTa analogues (a = 50–115), the λmax values of the PPP-b-P3MTs were greatly blue-shifted by 72–81 nm and the intensities of their vibronic peaks were much weaker (Fig. 1 and S3d†). These observations indicated that the chain ordering or packing between PT and P3MT was very different.45,46 In the film state, the UV–vis spectra of the PPP-b-P3MTs were identical to those of the PPP-b-P3MTs in the solution state and did not significantly change after thermal annealing (Fig. S3a–c†). These results implied that the well-ordered packing of the P3MT core in solution was essentially the same as that in the film state.
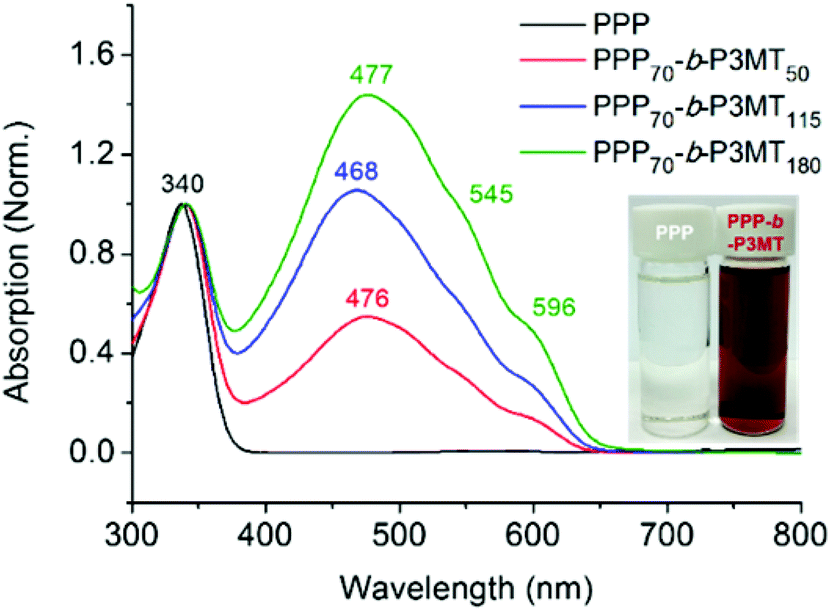 | ||
| Fig. 1 UV–vis spectra of PPP and PPP-b-P3MTs in chloroform at room temperature. | ||
Next, the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energy levels for PPP-b-P3MTs were obtained by cyclic voltammetry and optical band gap measurements. The HOMO and LUMO levels of the PPP-b-P3MTs were −5.4 eV and −3.4 eV, respectively (Fig. S4†). The HOMO level of the PPP-b-P3MTs was similar to that of the previously reported PPP-b-PTs25 but lower than that of poly(3-hexylthiophene) (−4.9 eV) (Fig. S4†), suggesting the high stability of the polymer against air oxidation.
For a detailed examination of the π–π stacking and crystalline domains of PPP-b-P3MTs, the powder X-ray diffraction (PXRD) pattern was analysed. The PXRD pattern of PPP-b-P3MTs showed a combination of peaks from crystalline domains for both PPP ((100), d = 2.1 nm; (001), d = 0.43 nm; (010), d = 0.38 nm, where d is the d-spacing)50 and P3MT ((110), d = 0.52 nm; (020), d = 0.35 nm)48 (Fig. S5a and c†). This demonstrated that the crystalline domains of PPP and P3MT were well separated from each other in the core–shell nanostructure, as shown by the two distinct absorption peaks corresponding to PPP and P3MT in the UV–vis spectrum (Fig. 1). The intensity of the P3MT peak relative to that of the PPP peak increased with increasing DP of P3MT because of the increased crystallinity of P3MT as well as the decreased crystallinity of PPP in the core–shell geometry (Fig. S5a and S6†). In comparison with the PXRD patterns of PPP-b-P3MTs, PPP70-b-PTa (a = 50–115) showed different peak patterns in the region corresponding to the π–π stacking distance ((110), d = 0.45 nm; (200), d = 0.39 nm; (210), d = 0.32 nm)47 (Fig. S5b and c†). This indicates the different stacking modes of PT (herringbone structure) and P3MT (π-stacked structure, face-centred lattice-type packing) in nanostructures.47–49
To obtain information regarding the size and shape of the nanostructures from PPP-b-P3MTs, we analysed them using dynamic light scattering (DLS), atomic force microscopy (AFM) and transmission electron microscopy (TEM). DLS measurements in chloroform at 20 °C confirmed the existence of nanoparticles (NPs) from PPP-b-P3MTs in solution; moreover, their hydrodynamic diameter (Dh) increased from 133 to 251 nm and then to 687 nm (with a minor tailing in DLS profile), according to the DP of P3MT (Fig. 2a). In contrast, the Dh of the nanostructures from PPP70-b-PTa (a = 50–115) under the same DLS measurement conditions only increased from 100 to 204 nm (Fig. S7a†). Note that the Dh of the PPP-b-P3MT NPs increased proportionally with the DP of P3MT (Fig. 2a), whereas that of PPP-b-PT NPs seemed to saturate at ca. 200 nm (Fig. S7a†). This indicated that PPP-b-P3MT could form much larger NPs despite the more soluble P3MT core.
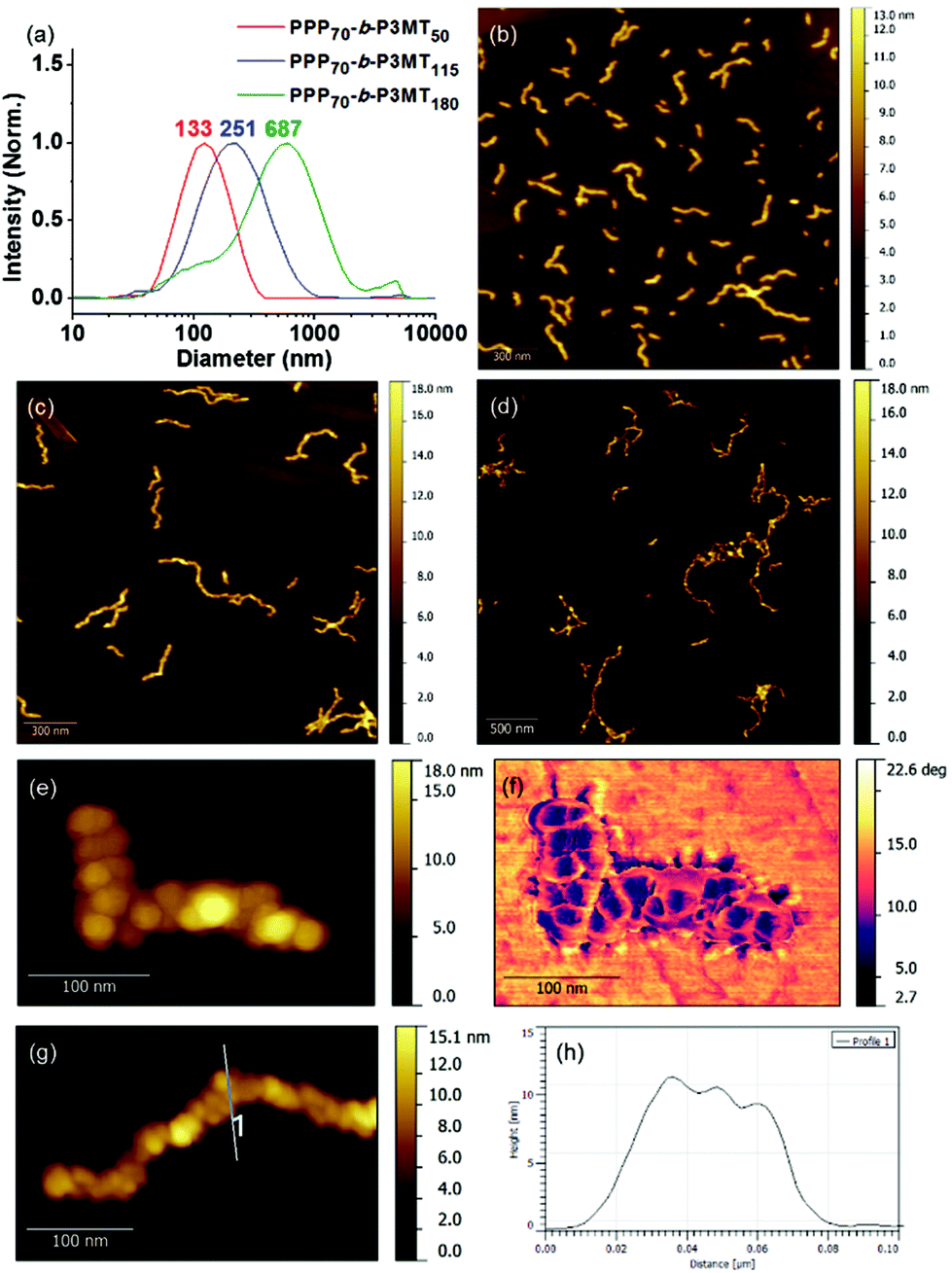 | ||
| Fig. 2 (a) DLS profiles of PPP-b-P3MT nanoparticles in chloroform at 20 °C. AFM images of the nanostructures from PPP-b-P3MTs on HOPG: (b) PPP70-b-P3MT50, (c) PPP70-b-P3MT115 and (d) PPP70-b-P3MT180. High-resolution AFM (e) height and (f) phase images of the nanostructures from PPP70-b-P3MT115. (g) High-resolution AFM image and (h) height profile of the nanostructure from PPP70-b-P3MT115. | ||
The AFM images of the nanostructures from PPP-b-P3MTs on freshly prepared highly ordered pyrolytic graphite (HOPG) revealed an interesting structural evolution as the DP of the P3MT core increased. Initially, PPP70-b-P3MT50 formed mostly short to long 1D undulated nanocaterpillars with a number-average length (Ln) of 130 nm, length dispersity (Lw/Ln) of 1.45 and average height (H) of 6.6 nm (Fig. 2b, S8a, and S9†). There was a small amount of nanospheres as well, indicating that the nanosphere was the basic building block, as found in the previous reports.22–28 In the case of PPP70-b-P3MT115, the nanocaterpillars had grown longer and taller with Ln = 246 nm, Lw/Ln = 1.47 and H = 9.9 nm; this agreed well with the DLS analysis (Fig. 2a, c, S8a, and S9†). Interestingly, high-resolution AFM images of this sample revealed that the nanostructures changed from undulated single-line to undulated multi-line nanocaterpillars (Fig. 2e–g and S8a†). The presence of these intriguing multi-line nanocaterpillars was further supported by the cross-sectional view analysis showing mostly two or maximum three undulations (Fig. 2g and h). Finally, AFM height and phase images of nanostructures from PPP70-b-P3MT180 revealed that the multi-line nanocaterpillars became larger with lengths over 1 μm and H = 11.2 nm (Fig. 2d, S8a, and S9†). Moreover, in some cases, these nanocaterpillars had a small number of branching points (0–3) to give long-chain branching nanocaterpillars (Fig. 2d and S8a†).
The TEM images of these nanostructures provided more definitive evidence for this interesting evolution from single-line to multi-line nanocaterpillars because, even without staining, one could clearly see only the highly crystalline P3MT core of NPs. In TEM images, the core of the nanocaterpillars from PPP70-b-P3MT50 comprised nanospheres arranged in a 1D single line with minimal contact among themselves (Fig. 3a and S8a†), similar to the previous nanocaterpillars from PPP-b-PTs25 (Fig. S8b†). Interestingly, a closer look at the TEM images of NPs from PPP70-b-P3MT115 indeed showed thicker multi-line 1D nanocaterpillars comprising mostly two or three parallel lines of single-line nanocaterpillars resembling duplex DNA (Fig. 3b, d and S8a†). Similarly, the TEM images of NPs from PPP70-b-P3MT180 revealed the arrays of nanospheres arranged in multi-line 1D nanocaterpillars, however, one could see the branching point (Fig. 3c and S8a†). In comparison, the AFM and TEM images of NPs from PPP70-b-PTa (a = 50–115) only showed the short single-line nanocaterpillars even with the largest DP of PT (Fig. S8b†).25 For example, PPP70-b-PT115 formed only single-line nanocaterpillars with Ln = 131 nm, Lw/Ln = 1.16 and H = 7.3 nm (Fig. S8b and S9†); however, the length of multi-line nanocaterpillars from PPP70-b-P3MT115 having similar DPs for both blocks was greater (246 nm vs. 131 nm) and the average height was higher (9.9 nm vs. 7.3 nm) because of the multi-line morphology (Fig. S9†). Note that the length distribution (Lw/Ln) was much broader (1.47 vs. 1.16) for the multi-line nanocaterpillars (Fig. S9†).
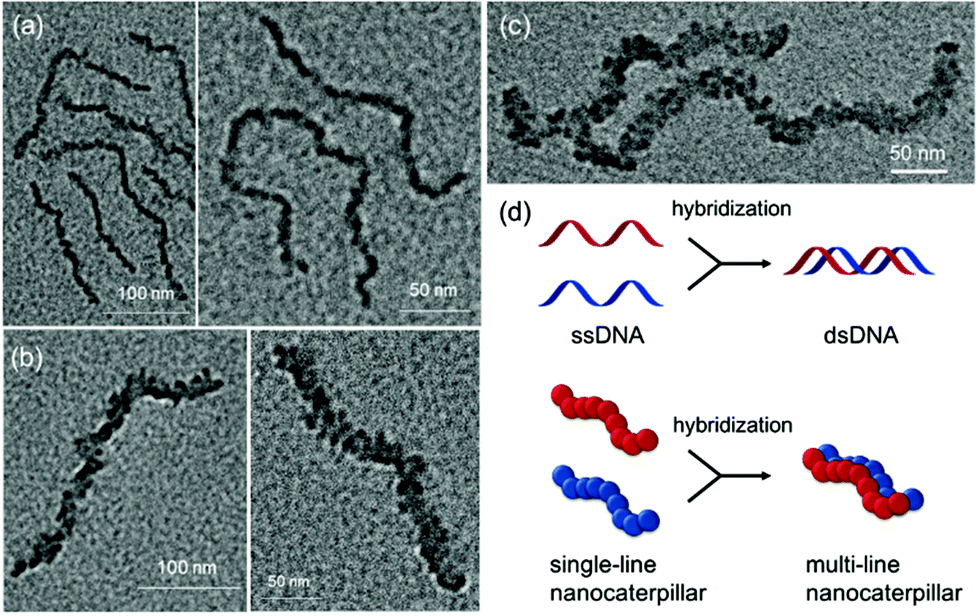 | ||
| Fig. 3 TEM images of the nanostructures from PPP-b-P3MTs on a Cu/C TEM grid: (a) PPP70-b-P3MT50, (b) PPP70-b-P3MT115 and (c) PPP70-b-P3MT180. (d) Schematic comparison between the formation of double-stranded DNA (dsDNA) and multi-line nanocaterpillars. | ||
To unambiguously show the differences in these two nanostructures from PPP-b-PT and PPP-b-P3MT, the PPP70-b-PT90 and PPP70-b-P3MT70 NPs with similar Mn values for PPP and similar DP values of the second blocks (PT and P3MT) were mixed altogether and imaged by AFM and TEM. The AFM images of the NP mixtures on HOPG showed that the PPP70-b-P3MT70 NPs were 2.6 nm taller (Fig. 4a and b) and 188 nm longer than the PPP70-b-PT90 NPs (Fig. S9c and S10†). The TEM image of the mixture also revealed the different core structures and thicknesses of the PPP70-b-PT90 (single-line, thinner) and PPP70-b-P3MT70 (multi-line, thicker) NPs (Fig. 4c and S10†). Again, these results confirmed the different self-assembly behaviour of PPP-b-PTs and PPP-b-P3MTs.
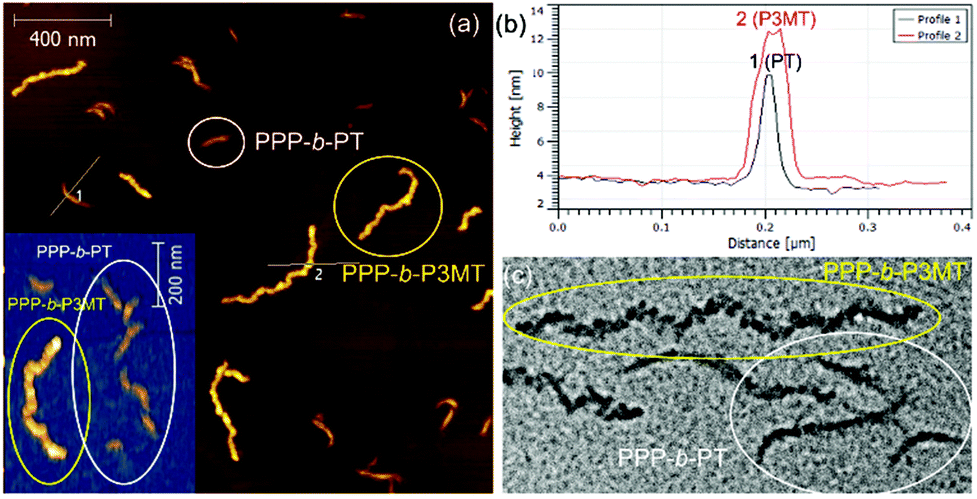 | ||
| Fig. 4 Comparison between (a) AFM images, (b) height profiles, and (c) TEM images obtained from the mixtures of nanostructures from PPP70-b-P3MT70 (Mn = 18.3k and PDI = 1.06 of PPP, actual DP of P3MT = 65, Ln = 315 nm, Lw/Ln = 1.34) and PPP70-b-PT90 (Mn = 20.5k and PDI = 1.05 of PPP, actual DP of PT = 61, Ln = 127 nm, Lw/Ln = 1.17). | ||
During the preparation of PPP-b-P3MTs, it was found that although PPP-b-P3MTs were more soluble than PPP-b-PTs, the diblock copolymers having a high DP of the P3MT core, such as PPP70-b-P3MT180, showed marginal solubility. This led us to change the side chain of PPP from hexyl to branched 2-ethylhexyl (PPP(EH)) to increase the solubility and processability of the NPs (Fig. S1b†). To match the Mn of PPP(EH) with that of the previous PPP, we increased the feed ratio to 100:1 and prepared several analogues, PPP(EH)100-b-P3MT50, PPP(EH)100-b-P3MT90 and PPP(EH)100-b-P3MT130 (Fig. S1b†). All three NPs were analysed by 1H NMR, UV–vis spectroscopy, DLS, AFM, and TEM, and they exhibited identical nanostructures to the PPP-b-P3MTs (Fig. S2b, S3e, S7b, and S8c†). Note that AFM images showed that the lengths of the nanocaterpillars from PPP(EH)-b-P3MTs were significantly longer than those from PPP-b-P3MTs: Ln = 179 nm, Lw/Ln = 1.34 and H = 6.1 nm for PPP(EH)100-b-P3MT50 (Fig. S8c and S9†); Ln = 431 nm, Lw/Ln = 1.76 and H = 7.8 nm for PPP(EH)100-b-P3MT90 (Fig. 5a, S8c, and S9†). PPP(EH)100-b-P3MT130 (H = 14.4 nm) showed much longer nanocaterpillars with a maximum length of over 2 μm and a small degree of branching (Fig. 5b, c, S8c, and S9†). Moreover, the TEM images of the PPP(EH)-b-P3MT NPs revealed that the nanocaterpillar cores changed from single-line (PPP(EH)100-b-P3MT50) to single-line and partially multi-line (PPP(EH)100-b-P3MT90) and finally to fully multi-line (PPP(EH)100-b-P3MT130) upon increasing the DP of P3MT (Fig. 5d and S8c†).
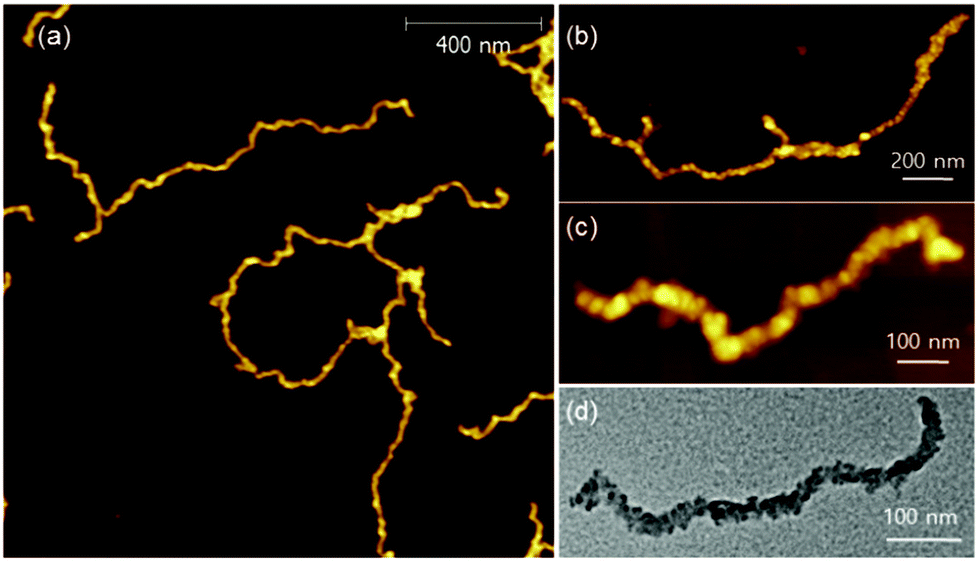 | ||
| Fig. 5 AFM images of the nanostructures from PPP(EH)-b-P3MTs on HOPG: (a) PPP(EH)100-b-P3MT90, (b) PPP(EH)100-b-P3MT130. (c) High-resolution AFM images of the nanostructures from PPP(EH)100-b-P3MT130. (d) TEM images of the nanostructures from PPP(EH)100-b-P3MT130 on a Cu/C TEM grid. | ||
On the basis of all of the experimental data, a plausible mechanism for forming the multi-line nanocaterpillars from PPP-b-P3MTs is proposed. In the early stage, PPP-b-P3MT with a low P3MT DP initially self-assembled into nanospheres by the INCP process, which then underwent supramolecular step-growth polymerization to the single-line 1D nanocaterpillars because of strong π–π interactions or the solvophobic effect of the P3MT core (Fig. 6a). Until this stage, it is exactly the same as the previously suggested mechanism for the formation of nanocaterpillars from PPP-b-PTs.25 As the living polymerization of the Grignard reagent from the monomer 2 continued inside the P3MT core, the core expanded and the nanocaterpillars clung to each other in an end-to-end fashion to generate long single-line nanocaterpillars (Fig. 6a). Further polymerization then led to further expansion of the insoluble core, to the extent that the end-to-end attachment or elongation of the nanocaterpillars was not sufficient for the PPP shell to stabilise the exposed solvophobic P3MT core. Instead, in order to minimize the area of the exposed core more effectively, side-by-side attachment of the nanocaterpillars dominated, resulting in the formation of the multi-line nanostructures, just as single-stranded DNA forms duplex DNA (Fig. 3d and 6b). Finally, a further increase in the DP of P3MT provided a small number of branching points caused by the highly exposed cores on the multi-line nanocaterpillars (Fig. 6c). Although this evolution seemed similar to that for P3EHT-b-PT, which showed nanostar and nanonetwork morphologies,24 the main difference between the nanostructures from P3EHT-b-PT and PPP-b-P3MT is that branching occurred at a much earlier stage for P3EHT-b-PT because of the premature expansion of the PT core,24 whereas the branching occurred at a much later stage for the more soluble PPP-b-P3MT and resulted in long chain branching.
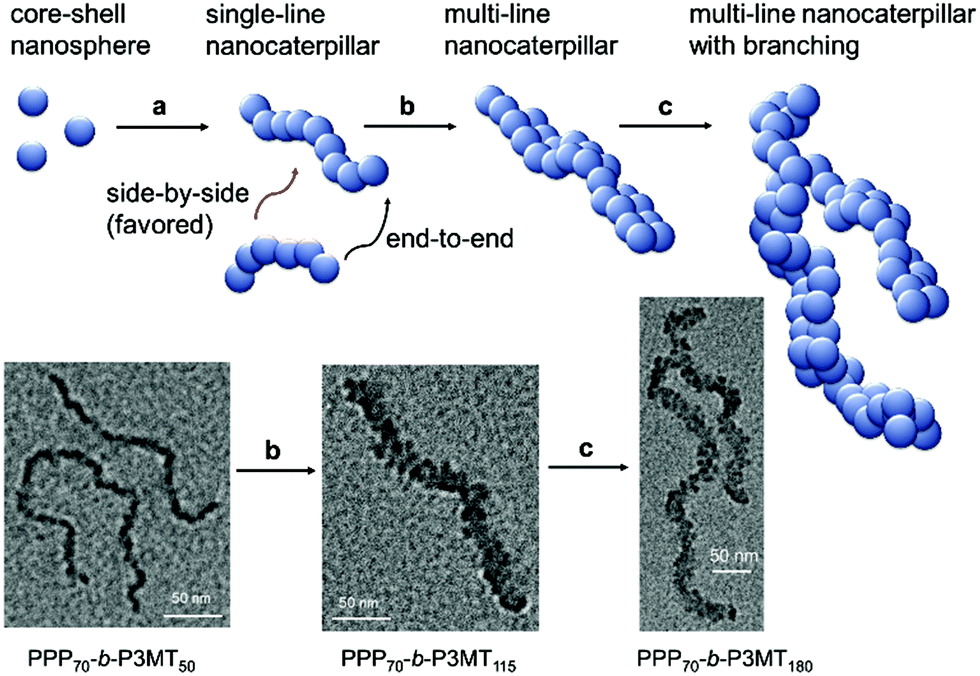 | ||
| Fig. 6 Proposed mechanism for the formation of the multi-line nanocaterpillars from PPP-b-P3MT. | ||
Since the crystallinity and packing mode of P3MT are different from those of PT, the stabilities of the NPs from PPP70-b-P3MT50 and PPP70-b-P3MT115 were examined and compared with that of the NPs from PPP-b-PT. First, their high thermal stability in toluene at high temperatures was confirmed by DLS. There was no significant change in the sizes of NPs from these two PPP-b-P3MTs in toluene at temperatures ranging from 20 °C to 80 °C (Fig. S11a†). In contrast, when their mechanical stability was tested by sonication in chloroform at room temperature, the long multi-line nanocaterpillars fragmented into shorter NPs (Fig. S11b and c†). For example, DLS, AFM and TEM analyses revealed that the size of the PPP70-b-P3MT115 NPs decreased from 238 nm to 99 nm and 74 nm after sonicating the solution for 5 and 45 minutes, respectively (Fig. S11b†). However, the initial core–shell supramolecular structures were still maintained, as shown by 1H NMR (Fig. S11d†). Furthermore, even though the NPs became shorter (about 1/3, 100–200 nm long) after sonication, the TEM images of the fragmented samples from PPP70-b-P3MT115 revealed that their multi-line shape was still maintained even after sonication for one hour (Fig. S11c†). These observations suggested that at least the side-by-side attachment between single-line nanocaterpillars leading to the multi-line ones was quite stable under mechanical stress such as sonication. Despite the high thermal stability of the PPP-b-P3MT NPs, their overall lower mechanical stability implied that the P3MT core was held together by relatively weaker π–π interactions, in contrast to the case of PPP-b-PT, which showed both high thermal and mechanical stability.25 Their weaker vibronic peaks than the PT analogues under UV–vis spectroscopy also supported the weaker π–π interaction. Ironically, this weaker interaction in the P3MT core led to the formation of much longer nanocaterpillars having much more interesting multi-line dimensions.
Conclusion
In summary, in situ self-assembly of fully conjugated PPP-b-P3MTs during GRIM polymerization produced single-line nanocaterpillars, multi-line nanocaterpillars and branched multi-line nanocaterpillars depending on the DP of P3MT. A comparison between PPP-b-P3MT and its analogous PPP-b-PT revealed several different characteristics such as improved solubility, different packing mode (π-stacked packing) and weaker π–π interactions of PPP-b-P3MTs. Furthermore, these differences affected the in situ nanoparticlisation process, resulting in the formation of a unique hierarchical evolution of nanoparticle morphologies. In particular, PPP-b-P3MTs formed longer single to multi-line nanocaterpillars, whereas PPP-b-PTs formed only shorter single-line nanocaterpillars. Changing the side chain from hexyl to branched 2-ethylhexyl increased the length of the nanocaterpillars and led to improved solubility and processability.Acknowledgements
The financial support from the Basic Science Research Program and the Nano-Material Technology Development Program through NRF is acknowledged. I.-H.L. is supported by NRF-2013-Fostering Core Leaders of the Future Basic Science Program. We thank the National Centre for Inter-University Research Facilities at SNU for supporting TEM experiments.Notes and references
- K. J. Ihn, J. Moulton and P. Smith, J. Polym. Sci., Part B: Polym. Phys., 1993, 31, 735 CrossRef CAS.
- H. Sirringhaus, P. J. Brown, R. H. Friend, M. M. Nielsen, K. Bechgaard, B. M. W. Langeveld-Voss, A. J. H. Spiering, R. A. J. Janssen, E. W. Meijer, P. T. Herwig and D. M. de Leeuw, Nature, 1999, 401, 685 CrossRef CAS.
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491 CrossRef CAS.
- F. S. Kim, G. Ren and S. A. Jenekhe, Chem. Mater., 2011, 23, 682 CrossRef CAS.
- J. S. Kim, J. H. Lee, J. H. Park, C. Shim, M. Sim and K. Cho, Adv. Funct. Mater., 2011, 21, 480 CrossRef CAS.
- Z. Li, R. J. Ono, Z.-Q. Wu and C. W. Bielawski, Chem. Commun., 2011, 47, 197 RSC.
- S. K. Patra, R. Ahmed, G. R. Whittell, D. J. Lunn, E. L. Dunphy, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2011, 133, 8842 CrossRef CAS.
- J. B. Gilroy, D. J. Lunn, S. K. Patra, G. R. Whittell, M. A. Winnik and I. Manners, Macromolecules, 2012, 45, 5806 CrossRef CAS.
- A. C. Kamps, M. Fryd and S.-J. Park, ACS Nano, 2012, 6, 2844 CrossRef CAS.
- J. Gwyther, J. B. Gilroy, P. A. Rupar, D. J. Lunn, E. Kynaston, S. K. Patra, G. R. Whittell, M. A. Winnik and I. Manners, Chem. – Eur. J., 2013, 19, 9186 CrossRef CAS.
- P.-T. Wu, G. Ren, C. Li, R. Mezzenga and S. A. Jenekhe, Macromolecules, 2009, 42, 2317 CrossRef CAS.
- M. He, L. Zhao, J. Wang, W. Han, Y. Yang, F. Qiu and Z. Lin, ACS Nano, 2010, 4, 3241 CrossRef CAS.
- B. A. G. Hammer, F. A. Bokel, R. C. Hayward and T. Emrick, Chem. Mater., 2011, 23, 4250 CrossRef CAS.
- E. Lee, B. Hammer, J.-K. Kim, Z. Page, T. Emrick and R. C. Hayward, J. Am. Chem. Soc., 2011, 133, 10390 CrossRef CAS.
- U. Scherf, A. Gutacker and N. Koenen, Acc. Chem. Res., 2008, 41, 1086 CrossRef CAS.
- U. Scherf, S. Adamczyk, A. Gutacker and N. Koenen, Macromol. Rapid Commun., 2009, 30, 1059 CrossRef CAS.
- M. J. Robb, S.-Y. Ku and C. J. Hawker, Adv. Mater., 2013, 25, 5686 CrossRef CAS.
- H. Cui, Z. Chen, S. Zhong, K. L. Wooley and D. J. Pochan, Science, 2007, 317, 647 CrossRef CAS.
- S. Sugihara, A. Blanazs, S. P. Armes, A. J. Ryan and A. L. Lewis, J. Am. Chem. Soc., 2011, 133, 15707 CrossRef CAS.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581 CrossRef CAS.
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753 CrossRef CAS.
- K.-Y. Yoon, I.-H. Lee, K. O. Kim, J. Jang, E. Lee and T.-L. Choi, J. Am. Chem. Soc., 2012, 134, 14291 CrossRef CAS.
- J. Kim, E.-H. Kang and T.-L. Choi, ACS Macro Lett., 2012, 1, 1090 CrossRef CAS.
- I.-H. Lee, P. Amaladass, K.-Y. Yoon, S. Shin, Y.-J. Kim, I. Kim, E. Lee and T.-L. Choi, J. Am. Chem. Soc., 2013, 135, 17695 CrossRef CAS.
- I.-H. Lee, P. Amaladass and T.-L. Choi, Chem. Commun., 2014, 50, 7945 RSC.
- K.-Y. Yoon, I.-H. Lee and T.-L. Choi, RSC Adv., 2014, 4, 49180 RSC.
- K.-Y. Yoon, S. Shin, Y.-J. Kim, I. Kim, E. Lee and T.-L. Choi, Macromol. Rapid Commun., 2015, 36, 1069 CrossRef CAS.
- S. Shin, K.-Y. Yoon and T.-L. Choi, Macromolecules, 2015, 48, 1390 CrossRef CAS.
- O. Ikkala and G. ten Brinke, Science, 2002, 295, 2407 CrossRef CAS.
- H. Frauenrath and E. Jahnke, Chem. – Eur. J., 2008, 14, 2942 CrossRef CAS.
- J. Hu, G. Liu and G. Nijkang, J. Am. Chem. Soc., 2008, 130, 3236 CrossRef CAS.
- T. Gädt, N. S. Ieong, G. Cambridge, M. A. Winnik and I. Manners, Nat. Mater., 2009, 8, 144 CrossRef.
- A. Walther, M. Drechsler, S. Rosenfeldt, L. Harnau, M. Ballauff, V. Abetz and A. H. E. Müller, J. Am. Chem. Soc., 2009, 131, 4720 CrossRef CAS.
- L. E. R. O'Leary, J. A. Fallas, E. L. Bakota, M. K. Kang and J. D. Hartgerink, Nat. Chem., 2011, 3, 821 CrossRef.
- A. H. Gröschel, F. H. Schacher, H. Schmalz, O. V. Borisov, E. B. Zhulina, A. Walther and A. H. E. Müller, Nat. Commun., 2012, 3, 710 CrossRef.
- A. H. Gröschel, A. Walther, T. I. Löbling, F. H. Schacher, H. Schmalz and A. H. E. Müller, Nature, 2013, 503, 247 Search PubMed.
- Z. M. Hudson, C. E. Boott, M. E. Robinson, P. A. Rupar, M. A. Winnik and I. Manners, Nat. Chem., 2014, 6, 893 CrossRef CAS.
- X. Zhang, D. Görl, V. Stepanenko and F. Würthner, Angew. Chem., Int. Ed., 2014, 53, 1270 CrossRef CAS.
- M. G. Warner and J. E. Hutchison, Nat. Mater., 2003, 2, 272 CrossRef CAS.
- H. Wang, L. Chen, X. Shen, L. Zhu, J. He and H. Chen, Angew. Chem., Int. Ed., 2012, 51, 8021 CrossRef CAS.
- W. Li, I. Kanyo, C.-H. Kuo, S. Thanneeru and J. He, Nanoscale, 2015, 7, 956 RSC.
- R. Miyakoshi, K. Shimono, A. Yokoyama and T. Yokozawa, J. Am. Chem. Soc., 2006, 128, 16012 CrossRef CAS.
- R. Miyakoshi, A. Yokoyama and T. Yokozawa, Chem. Lett., 2008, 37, 1022 CrossRef CAS.
- S. Wu, L. Bua, L. Huang, Y. Xinhong, Y. Han, Y. Geng and F. Wang, Polymer, 2009, 50, 6245 CrossRef CAS.
- P. J. Brown, D. S. Thomas, A. Köhler, J. S. Wilson, J.-S. Kim, C. M. Ramsdale, H. Sirringhaus and R. H. Friend, Phys. Rev. B: Condens. Matter, 2003, 67, 064203 CrossRef.
- S. Samitsu, T. Shimomura, S. Heike, T. Hashizume and K. Ito, Macromolecules, 2008, 41, 8000 CrossRef CAS.
- Z. Mo, K.-B. Lee, Y. B. Moon, M. Kobayashi, A. J. Heeger and F. Wudl, Macromolecules, 1985, 18, 1972 CrossRef CAS.
- T. Yamamoto, D. Komarudin, M. Arai, B. L. Lee, H. Suganuma, N. Asakawa, Y. Inoue, K. Kubota, S. Sasaki, T. Fukuda and H. Matsuda, J. Am. Chem. Soc., 1998, 120, 2047 CrossRef CAS.
- S. Tsuzuki, K. Honda and R. Azumi, J. Am. Chem. Soc., 2002, 124, 12200 CrossRef CAS.
- H. Yang, L. Wang, J. D. Zhang, X. H. Yu, Y. H. Geng and Y. C. Han, Macromol. Chem. Phys., 2014, 215, 405 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and further characterization. See DOI: 10.1039/c5py01967b |
| This journal is © The Royal Society of Chemistry 2016 |