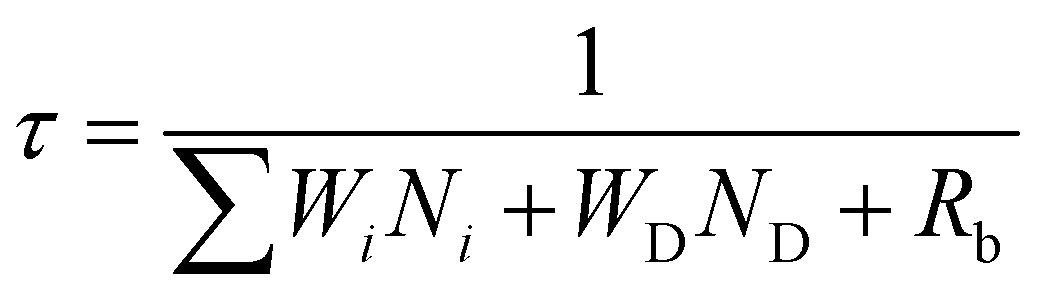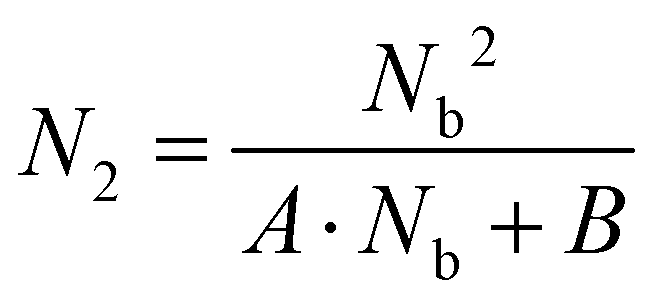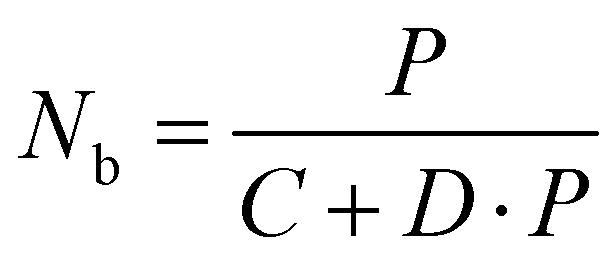DOI:
10.1039/C6NR00812G
(Paper)
Nanoscale, 2016,
8, 6666-6673
Nonlinear spectral and lifetime management in upconversion nanoparticles by controlling energy distribution†
Received
29th January 2016
, Accepted 24th February 2016
First published on 25th February 2016
Abstract
Optical tuning of lanthanide-doped upconversion nanoparticles has attracted considerable attention over the past decade because this development allows the advance of new frontiers in energy conversion, materials science, and biological imaging. Here we present a rational approach to manipulating the spectral profile and lifetime of lanthanide emission in upconversion nanoparticles by tailoring their nonlinear optical properties. We demonstrate that the incorporation of energy distributors, such as surface defects or an extra amount of dopants, into a rare-earth-based host lattice alters the decay behavior of excited sensitizers, thus markedly improving the emitters’ sensitivity to excitation power. This work provides insight into mechanistic understanding of upconversion phenomena in nanoparticles and also enables exciting new opportunities of using these nanomaterials for photonic applications.
Introduction
Lanthanide-doped photon upconversion materials have been studied for several decades owing to their distinct optical properties such as large anti-Stokes’ spectral shift, long luminescence lifetime, and sharp emission band. These attributes make the upconversion materials particularly attractive for use in lighting, lasing, quantum cutting, etc.1,2 In recent years, the rapid development of nanotechnology has enabled immense interest in upconversion nanoparticles (UCNPs) because of their fascinating optical properties and technological potential in many fields of research.3–37 Notably, many strategies, including doping composition/concentration variation, surface ligand coordination and pulse-duration modulation, have been developed to tune the emission profile of the UCNPs.38–45 An important and facile route to control upconversion emission, based on the nonlinear power response of photon upconversion, is to vary the excitation power externally.46–48
In a typical photon upconversion process, the excitation energy is absorbed by sensitizers, such as Yb3+ ions doped in a host matrix, and then transferred to neighboring emitters (or activators), such as Er3+ or Tm3+ featuring ladder-like energy states (Fig. S1 in the ESI†). The sequential photon absorption by the sensitizers permits the emitters to be excited to higher energy levels through energy transfer. It is widely accepted that for multi-step (n) energy transfer, the luminescence intensity I is proportional to n power of the excitation density P.49,50 Thus, the number of energy transfer upconversion (ETU) steps can be derived from the slope (I–P slope) of the luminescence intensity curve plotted against pump power density in a double-logarithmic representation (Fig. S2†). However, the upconversion emission intensity I may not be strictly proportional to n power of the excitation density P. The n value is likely to be affected by a multitude of factors related to energy distribution in UCNPs, such as a crystalline structure, particle size, and dopant concentration. Moreover, the variation of the n value is typically accompanied by a concurrent change in luminescence decay lifetime, meaning that n is highly relevant to the spectral properties and the luminescence dynamics of upconversion materials.
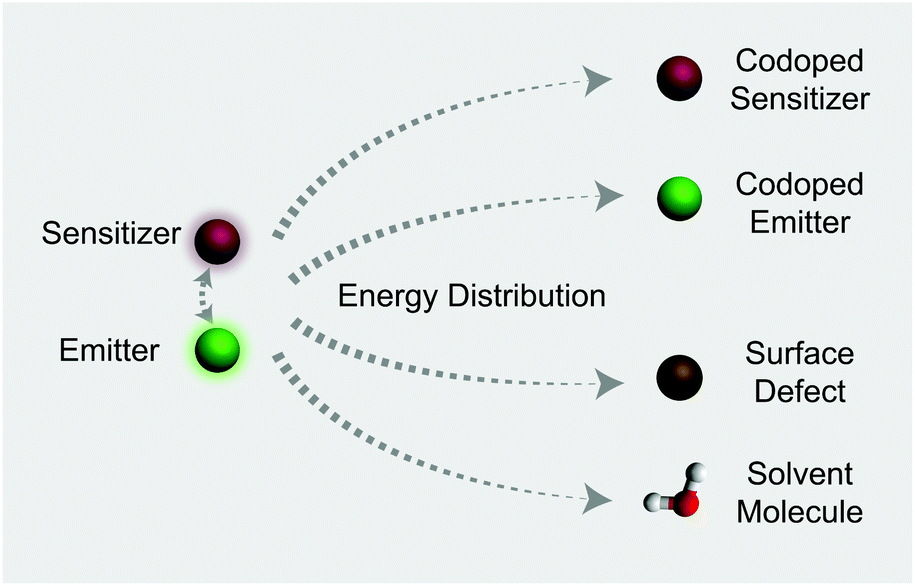
In this work, we investigate the spectroscopic properties and decay behaviors of UCNPs under the influence of different types of energy distributors through surface defect engineering or doping (Fig. 1). Our measured n value correlates well with upconversion emission characteristics in that large numbers of energy distributing pathways increase the sensitivity of the UCNPs to excitation power. Importantly, the decay lifetimes of both sensitizer and emitter ions could be tuned by manipulating the nature of the energy distributors, providing new opportunities for photonic applications in optical switching and security encoding.
 |
| | Fig. 1 Illustration of typical energy distribution processes involving Yb3+-sensitized upconversion. Note that for a particular system at the excited state, possible energy distributors include sensitizer ions, codoped emitters, surface defects, and solvent molecules with high energy vibrational groups. | |
Results and discussion
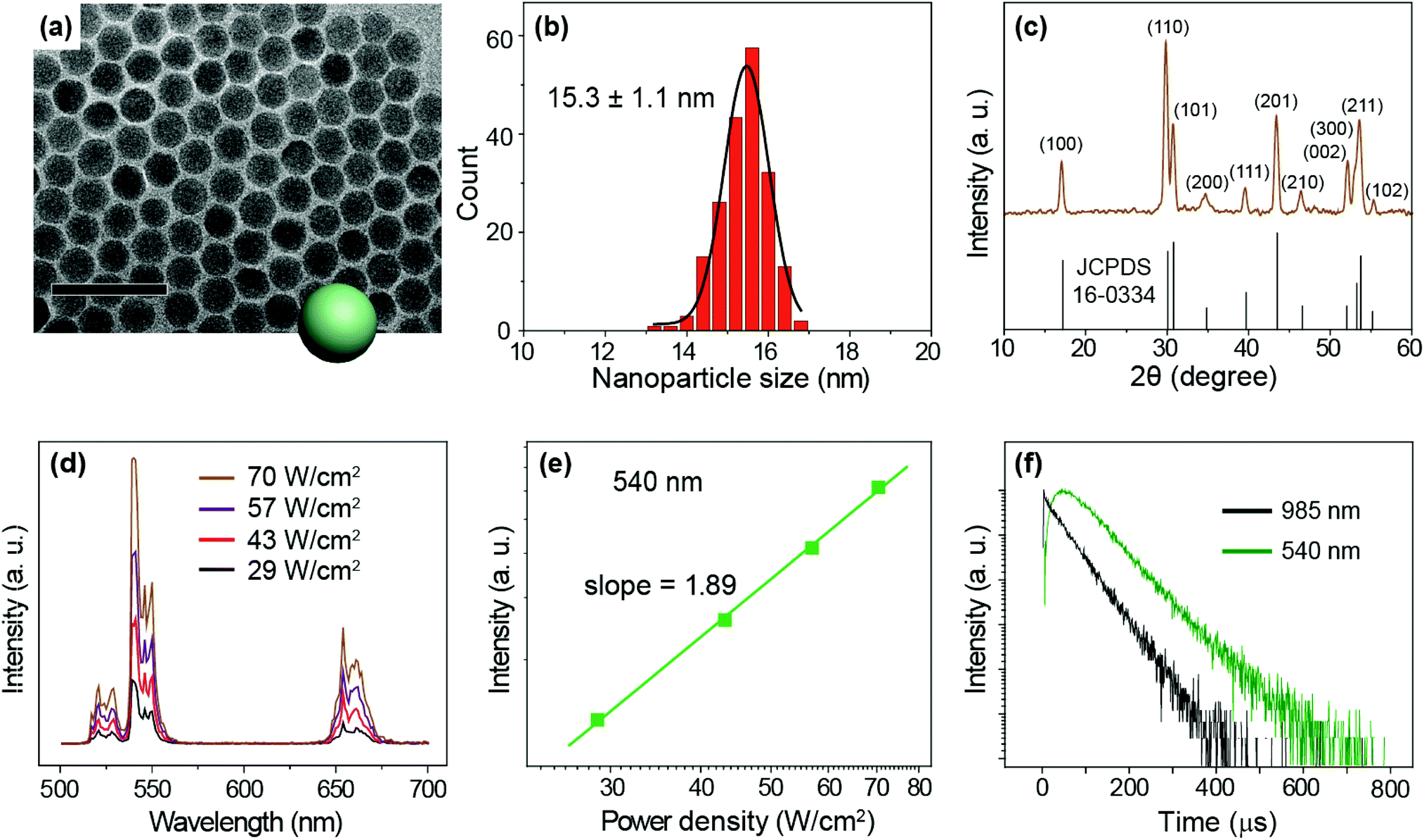
We prepared NaYF4:Yb/Er (20/2%) UCNPs as a model sample by a well-established co-precipitation method.51 As seen under a transmission electron microscope (TEM), the as-synthesized nanoparticles are 15.3 ± 1.1 nm in diameter with a narrow size distribution (Fig. 2a and b). A pure hexagonal phase structure of the nanoparticles was confirmed by X-ray diffraction studies (Fig. 2c). Under the excitation of a 975 nm CW laser, we observed visible emission bands at 540 and 654 nm, typically attributable to the 4S3/2 → 4I15/2 and 4F9/2 → 4I15/2 transitions of Er3+ ions (Fig. 2d). We considered the 540 nm emission as an example to study the excitation power response and decay behaviors of the UCNPs. The power dependent luminescence measurement indicates that in the power density range of 29–70 W cm−2, the 540 nm emission is generated via a typical 2-photon upconversion process (Fig. 2e). On 975 nm pulsed laser excitation, we observed a distinct rise edge in the temporal luminescence curve of the 540 nm emission due to the time consumed by the multi-step energy transfer process.52 We focus on the decay lifetime of the luminescence which is critical to the application of upconversion tuning in the time domain for optical multiplexing.9,23 Notably, the emission of Yb3+ (2F5/2 → 2F7/2 transition) at 985 nm has a decay lifetime of 69 μs, while the decay of Er3+ emission at 540 nm registers a lifetime of 88 μs (Fig. 2f).
 |
| | Fig. 2 (a) A typical TEM image of the as-prepared NaYF4:Yb/Er (20/2%) nanoparticles. Scale bar is 50 nm. (b) Size histogram of the nanoparticles. (c) X-ray diffraction patterns of the nanoparticles, which are in good agreement with the literature reference for hexagonal NaYF4 crystal (Joint Committee on Powder Diffraction Standards file number 16-0334). (d) Emission spectra of the UCNPs recorded with a 975 nm CW laser excitation at different power densities (29–70 W cm−2). (e) The 540 nm (4S3/2 → 4I15/2 transition of Er3+) emission intensity versus pump power density in double-logarithmic representation. (f) Decay curves of 540 nm emissions from Er3+ as well as 985 nm from Yb3+ obtained upon excitation of the nanoparticles at 975 nm. | |
To study the change in the I–P slope and emission lifetime in the presence of varied energy distributors, we adopted different strategies to alter the upconversion luminescence of NaYF4:Yb/Er (20/2%) UCNPs: (i) surface passivation by coating the particles with different thicknesses of a NaYF4 inert shell (Fig. S3†);53–60 (ii) controlling the size of UCNPs (Fig. S4†);61 (iii) coating the particles with a NaYF4:Yb active shell containing different amounts of Yb3+ (Fig. S6†);62,63 and (iv) changing the Er3+ doping concentrations (Fig. S7†). By investigating the luminescence decay lifetime and the pump-power-dependent luminescence intensity as shown in Table 1, it is evident that the I–P slopes of the 540 nm emission enlarge with increasing numbers of energy distributors, indicating the high sensitivity of the luminescence to the excitation power density. Notably, the luminescence decay lifetimes of both Yb3+ and Er3+ are found to diminish as the number of energy distributors increases.
Table 1 Comparative investigations of emission profiles for different types of nanoparticles. Note that decay lifetimes were measured at 985 and 540 nm, and the I–P slope tuning was realized by modulating energy distributors through a variety of strategies, including inert and active shell coating, particle size control, and doping concentration adjustment. All 540 nm I–P slopes were measured under CW laser excitation at 975 nm with a power density range of 20–80 W cm−2
| Structure |
|
Core composition |
Core diameter (nm) |
Shell composition |
Shell thickness (nm) |
985 nm (μs) |
540 nm (μs) |
540 nm I–P slope |
| UCNPs with different inert shells |

|
NaYF4:Yb/Er (20/2%) |
15.3 |
— |
— |
69 |
88 |
1.89 |
| NaYF4:Yb/Er (20/2%) |
15.3 |
NaYF4 |
1.3 |
304 |
187 |
1.77 |
| NaYF4:Yb/Er (20/2%) |
15.3 |
NaYF4 |
1.8 |
403 |
251 |
1.73 |
| NaYF4:Yb/Er (20/2%) |
15.3 |
NaYF4 |
2.8 |
783 |
301 |
1.63 |
| UCNPs with different sizes |

|
NaYF4:Yb/Er (20/2%) |
15.3 |
— |
— |
69 |
88 |
1.89 |
| NaYF4:Yb/Er (20/2%) |
23.8 |
— |
— |
327 |
245 |
1.76 |
| NaYF4:Yb/Er (20/2%) |
27.2 |
— |
— |
506 |
333 |
1.65 |
| UCNPs with different active shells |

|
NaYF4:Yb/Er (20/2%) |
15.3 |
NaYF4 |
1.6 |
379 |
231 |
1.74 |
| NaYF4:Yb/Er (20/2%) |
15.3 |
NaYF4:Yb (10%) |
1.9 |
274 |
180 |
1.78 |
| NaYF4:Yb/Er (20/2%) |
15.3 |
NaYF4:Yb (20%) |
1.7 |
131 |
128 |
1.82 |
| UCNPs with different emitter concentrations |

|
NaYF4:Yb/Er (20/0.5%) |
15.3 |
— |
— |
101 |
123 |
1.83 |
| NaYF4:Yb/Er (20/1%) |
15.5 |
— |
— |
97 |
103 |
1.88 |
| NaYF4:Yb/Er (20/2%) |
15.3 |
— |
— |
69 |
88 |
1.89 |
| NaYF4:Yb/Er (20/4%) |
15.7 |
— |
— |
50 |
65 |
1.92 |
| NaYF4:Yb/Er (20/8%) |
15.2 |
— |
— |
35 |
34 |
1.92 |
| NaYF4:Yb/Er (20/16%) |
15.2 |
— |
— |
19 |
14 |
1.98 |
| NaYF4:Yb/Er (20/32%) |
15.4 |
— |
— |
13 |
7 |
2.09 |
In addition, high levels of vibration energy in solvent molecules are considered to be a dominant factor governing the luminescence profiles of UCNPs. As can be seen in Fig. S8,† ligand-free NaYF4:Yb/Tm (30/0.5%) nanoparticles showed a short decay lifetime (346 μs) in water and a large I–P slope for Tm3+ emission at 800 nm. Indeed, the high vibration nature of water molecules allows the multiphonon relaxation process to be more accessible.64 It is worth noting that with the coating of NaYF4 the core–shell nanoparticles showed essentially the same decay lifetime and I–P slope in cyclohexane and water. Taken together, our results suggest that the vast combinations of different energy distributors (such as surface defects, dopants and solvents) enable precise depletion of the excited states of Yb3+ ions, thereby allowing the emission decay lifetime of the nanoparticles to be controlled within a wide range (Table 1). These strategies could be utilized in generating distinct time-domain codes for optical multiplexing and security applications. Another important note is that without the sensitizer the influence of energy distributors on the luminescence properties of the emitter is virtually negligible (Fig. S9†).
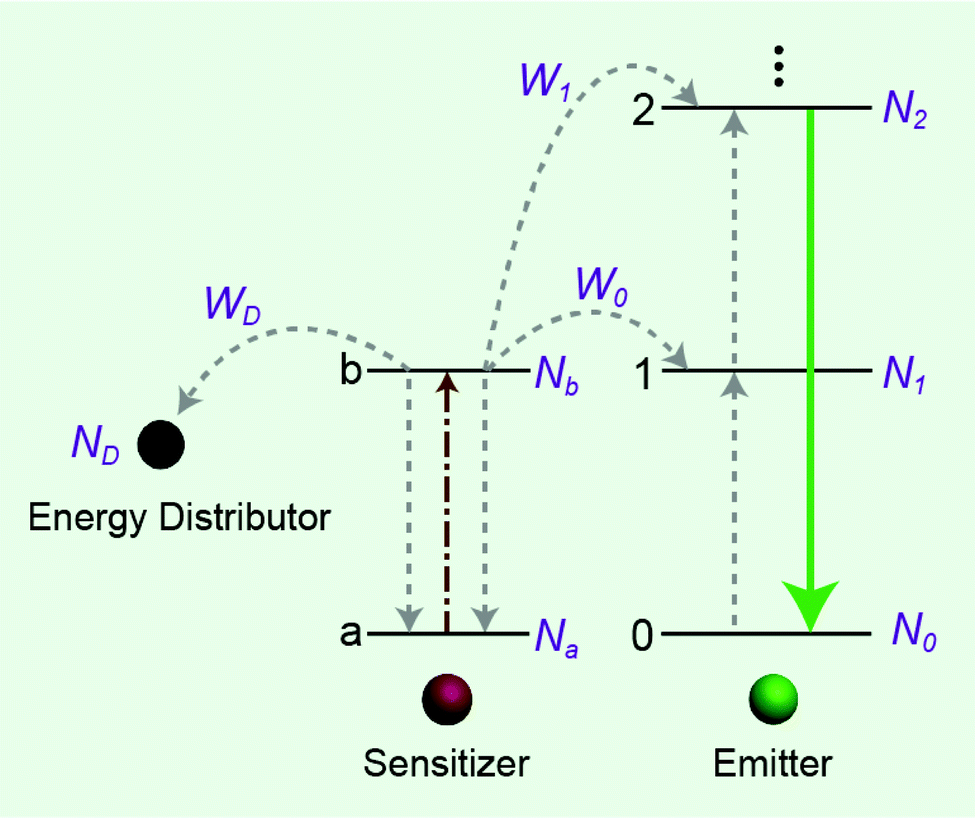
The effect of energy distributors on the measured decay lifetime of Yb3+ is investigated using the rate equations derived from a sensitization upconversion model (Fig. 3):
| | 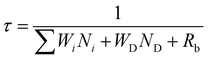 | (1) |
where
Ni (
i = 0, 1, 2…) is the population density of the emitter on level
i and
Wi is the corresponding energy transfer rate from the sensitizer to the emitter.
WD is the energy transfer rate from the sensitizer to the energy distributor.
ND represents the population density of effective energy distributors.
Rb is the radiative decay rate of the sensitizer from the excited state. From
eqn (1), one can conclude that increasing the number of energy distributors accelerates the depletion of excited Yb
3+ ions and thus shortens the measured Yb
3+ decay lifetime.
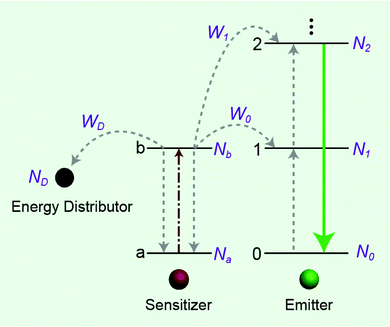 |
| | Fig. 3 Simplified scheme of the energy distributor effect on energy transfer processes involving a sensitization upconversion mechanism. | |
We next carried out theoretical investigations to explore the fundamental principle underlying the spectral and lifetime management of UCNPs through the control of energy distribution within the particles. From the analysis of the rate equations of energy levels in a simplified two-photon upconversion process (see the ESI†), the population density at the emitting level N2, which is proportional to the emission intensity I, can be expressed by eqn (2):
| | 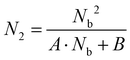 | (2) |
where
A and
B are constants,
Nb denotes the population density of the excited states of the Yb
3+ sensitizer, which determines the population density of the emitting level. From
eqn (2), the
N2–
Nb slope value in double-logarithmic representation is calculated in between 1 and 2.
50 Upon excitation with a power density of
P,
Nb can be further derived from
eqn (3):
| | 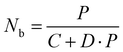 | (3) |
where
C and
D are constants. Notably,
Nb is not simply proportional to the excitation power density
P, as indicated in
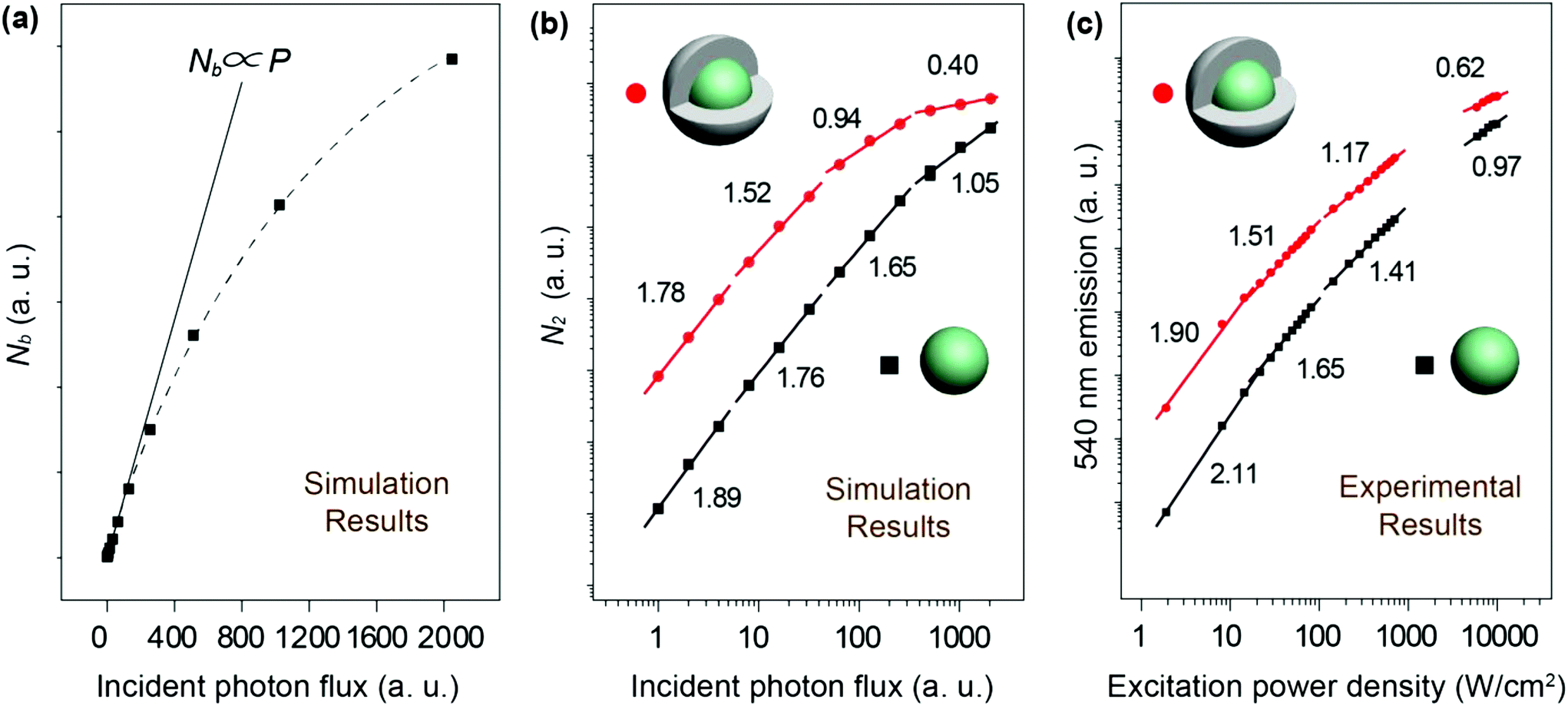
eqn (3). We conducted numerical simulations on NaYF
4:Yb/Er (20/2%) UCNPs to validate the relationship of
Nb with the incident photon flux by introducing the rate equations of a two-photon upconversion process, as shown in
Fig. 4a. We consider that the population density of Yb
3+ at the excited state is nonlinear to the excitation photon flux due to the excitation saturation of Yb
3+. This phenomenon further affects the relationship of
N2–
P, resulting in the deviation of the
I–
P slope away from the normal range. Our numerical simulations on both NaYF
4:Yb/Er (20/2%) and NaYF
4:Yb/Er (20/2%)@NaYF
4 (∼2.8 nm in shell thickness) UCNPs indicated that the slope of
N2–
P diminishes with increasing excitation photons. Compared with the core particles, the shell-coated UCNPs showed a lower value of the
I–
P slope under identical incident photon flux (
Fig. 4b). Remarkably, the simulated value of the
I–
P slope could be reduced to less than 1 at a high power excitation, which agrees well with our experimental results (
Fig. 4c).
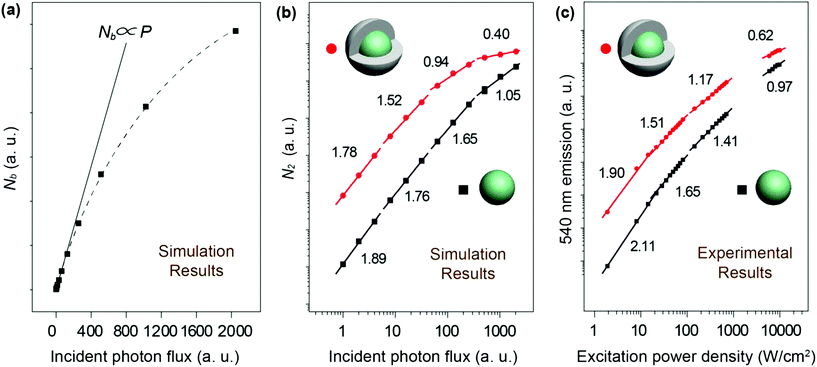 |
| | Fig. 4 (a) Numerical simulations on the population density of the excited state of Yb3+ ions versus the number of excitation photon flux in linear representation using NaYF4:Yb/Er (20/2%) UCNPs. (b) Numerical simulations on the population density of the 540 nm emission level of Er3+ ions versus the number of excitation photon flux in double-logarithmic representation using NaYF4:Yb/Er (20/2%) and NaYF4:Yb/Er (20/2%)@NaYF4 (∼2.8 nm in shell thickness) core–shell UCNPs. (c) Power-density-dependent emission profiles of NaYF4:Yb/Er (20/2%) and NaYF4:Yb/Er (20/2%)@NaYF4 core–shell UCNPs at 540 nm. The excitation power density under study is in the range of 1–104 W cm−2. | |
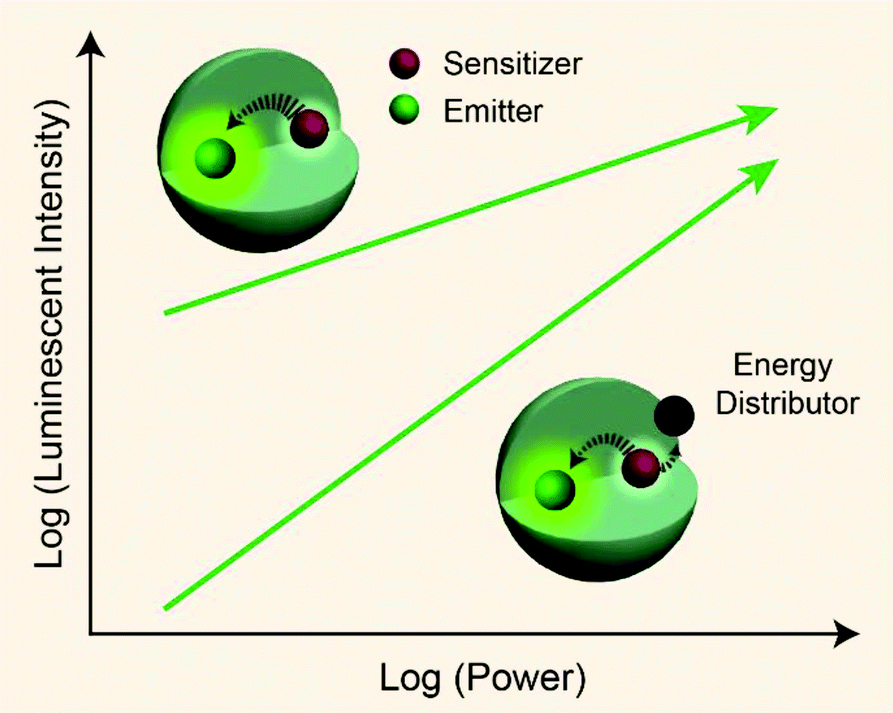
From published work, it can be found that in Yb3+/Er3+/Tm3+ triply-doped upconversion systems, increasing the excitation power density of UCNPs give rise to a larger rate of increase in the blue emission intensity than that of the green emission intensity.46,65–68 This can be attributed to the fact that blue emission from Tm3+ (1G4 → 3H6 and 1D2 → 3F4 transitions) requires more excited photons than green emission from Er3+ (2H11/2/4S3/2 → 4I15/2 transitions) (Fig. S1†). As a result, the blue emission typically features a large I–P slope n, thus its intensity increases rapidly with increasing excitation power density. On the basis of the findings described in Table 1, we reason that placing the emitters in regions close to energy distributors, for example, at the particle surface, might be able to improve the particle's sensitivity to the excitation power density (Fig. 5).
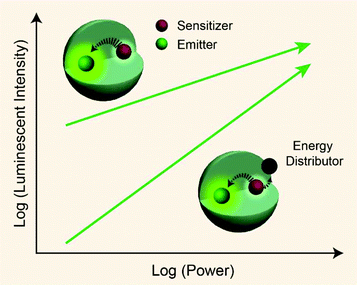 |
| | Fig. 5 Schematic representation showing pump-power-dependent luminescence of a sensitizer/emitter codoped nanoparticle in the presence or absence of an energy distributor. | |
To verify our hypothesis, we prepared two sets of multi-shell structured UCNPs comprising NaYF4:Yb/Tm@NaYF4@NaYF4:Yb/Er@NaYF4:Yb and NaYF4:Yb/Er@NaYF4@NaYF4:Yb/Tm@NaYF4:Yb (Fig. 6a, d and S10†). Note that Tm3+ and Er3+ ions are doped at different layers for direct comparison of their power-dependent emission profiles. An inert layer of NaYF4 was inserted between the two emitting layers to minimize cross-relaxation between Er3+ and Tm3+ (Fig. S12 and S13†).69,70 The outermost NaYF4:Yb shell was utilized to enhance the emission of the outer emitting layer to a level comparable to that obtained from the inner emitting layer (Fig. S14†). As expected, from the obtained emission spectra (Fig. 6b and e), it can be seen that the changes in the blue-to-green emission ratio of these two samples are dramatically different as a function of excitation power density (Fig. 6c, f, and S15a and c†). Consequently, the two colloidal samples exhibited different CIE color profiles with varied excitation power densities (Fig. S15b and d†). As control experiments, we synthesized two similar sets of UCNPs except for replacing the outermost layer of NaYF4:Yb with NaYF4. Interestingly, we observed that both the samples exhibit a similar trend in the blue-to-green emission intensity ratio as measured in response to different excitation powers (Fig. 7). This observation was largely attributed to the elimination of the surface quenching effect through the use of an optically inert NaYF4 layer.
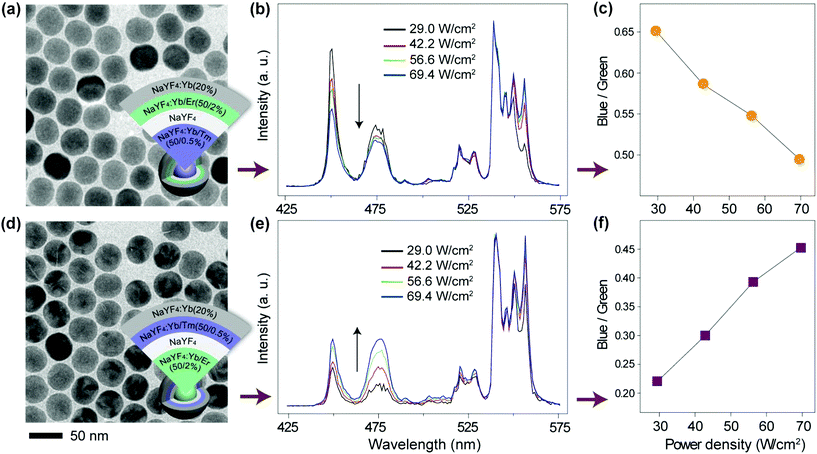 |
| | Fig. 6 (a and d) TEM images of NaYF4:Yb/Tm@NaYF4@NaYF4:Yb/Er@NaYF4:Yb and NaYF4:Yb/Er@NaYF4@NaYF4:Yb/Tm@NaYF4:Yb nanoparticles, respectively. Insets showing the design layout of the core–shell structures. (b and e) Corresponding power-dependent upconversion emission spectra of the two samples under 975 nm excitation. (c and f) Corresponding pump-power-dependence of the blue/green emission ratio obtained from the two samples. | |
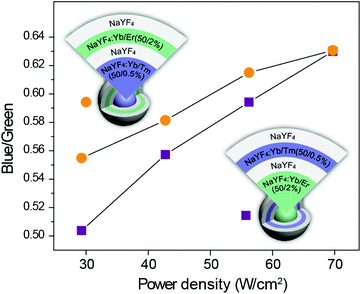 |
| | Fig. 7 Power dependent blue-to-green upconversion emission intensity ratio of NaYF4:Yb/Tm@NaYF4@NaYF4:Yb/Er@NaYF4 and NaYF4:Yb/Er@NaYF4@NaYF4:Yb/Tm@NaYF4 UCNPs upon 975 nm laser irradiation. | |
Conclusions
We have demonstrated a general approach for the design of upconversion nanocrystals with emitters amenable to nonlinear spectral and lifetime management. By controlling the proximity of energy distributors to specific emitters in core–shell structured nanocrystals, power dependent upconversion processes are demonstrated with high temporal tunability. The luminescence lifetime of upconversion nanocrystals can be modified in a wide range, which has potential wide-ranging implications for time-domain optical multiplexing and security applications.
Experimental section
Materials
Yttrium(III) acetate hydrate (99.9%), ytterbium(III) acetate hydrate (99.9%), thulium acetate hydrate (99.9%), erbium(III) acetate hydrate (99.9%), sodium hydroxide (NaOH; >98%), ammonium fluoride (NH4F; >98%), 1-octadecene (90%), oleic acid (90%), and hydrochloric acid were all purchased from Sigma-Aldrich and used as received without further purification. Analytical grades of ethanol and cyclohexane were purchased from Sigma-Aldrich. Water used in the experiment was purified to have a resistivity of 18.2 MΩ.
Synthesis of NaREF4 (RE = Y, Yb, Tm, Er) core nanoparticles
In a typical experiment,51 to a 50 mL flask charged with 3 mL of oleic acid (OA) and 7 mL of 1-octadecene (ODE) was added 2 mL of an aqueous solution containing RE(CH3CO2)3 precursors with a total lanthanide amount of 0.4 mmol. The resulting mixture was heated at 150 °C for 1 h to form a transparent solution and then cooled down to 50 °C. Subsequently, a methanol solution (6 mL) of NH4F (1.6 mmol) and NaOH (1 mmol) was added and stirred for 30 min. The reaction temperature was then increased to 110 °C for another 30 min to remove the methanol from the reaction mixture. After that, the solution was heated to 290 °C and maintained at this temperature under an argon atmosphere for 1.5 h. Then the mixture was cooled down to room temperature. The resulting nanoparticles were precipitated out through addition of ethanol, collected by centrifugation (5000 rpm, 10 min), washed with ethanol twice, and finally dispersed in 4 mL of cyclohexane.
Synthesis of NaREF4@NaREF4 (RE = Y, Yb, Tm, Er) core–shell nanoparticles
The shell precursor was prepared by mixing RE(CH3CO2)3 (0.2 mmol, RE = Y, Yb, Er, Tm), 3 mL OA and 7 mL ODE in a 50 mL flask, followed by heating at 150 °C for 1 h. After cooling down to 80 °C, 0.2 mmol of NaREF4 core nanoparticles dispersed in 2 mL of cyclohexane was added. The mixture was slowly cooled down to 50 °C for 30 min, and a methanol solution (3 mL) of NH4F (0.8 mmol) and NaOH (0.5 mmol) was added. Subsequently, the reaction temperature was increased to 110 °C and kept for 30 min to remove the methanol. After that, the solution was heated to 290 °C and maintained at this temperature under an argon flow for 1.5 h. After cooling to room temperature, the nanoparticles were precipitated out by addition of ethanol, collected by centrifugation, washed with ethanol, and redispersed in 4 mL of cyclohexane. The preparation of multi-shell nanoparticles was carried out by repeating the same procedure except for changing the composition ratio of the lanthanides.
Synthesis of ligand-free nanoparticles
The as-prepared OA-capped nanoparticles were dispersed in the mixture containing 1 mL of ethanol and 1 mL of 0.2 M HCl solution. After removal of the surface ligands by sonication for 1 min, the resulting products were collected by centrifugation (14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 20 min) and were re-dispersed in H2O after washing with a mixed ethanol–H2O solution (1:1, v/v) 3 times.
000 rpm, 20 min) and were re-dispersed in H2O after washing with a mixed ethanol–H2O solution (1:1, v/v) 3 times.
Characterization
Transmission Electron Microscopy (TEM) measurements were carried out on a JEOL-1400 transmission electron microscope (JEOL) operating at an acceleration voltage of 100 kV. Powder X-ray Diffraction (XRD) data were obtained on a Siemens D5005 X-ray diffractometer with Cu Kα radiation (λ = 1.5406 Å). The upconversion luminescence spectra under 975 nm excitation were recorded using an Edinburgh FSP920-C spectrometer equipped with a photomultiplier (PMT), in conjunction with a 975 nm diode laser (1 W). The CW laser beam was applied onto UCNP samples loaded in a quartz cuvette with a path length of 1 cm. The emission from the samples was collected at an angle of 90° to the excitation beam by using a pair of lenses. The decay curves of upconversion emission were measured by using a customized phosphorescence lifetime spectrometer (FSP920-C, Edinburgh) equipped with a nanosecond optical parametric oscillator (OPO) pumped by using a 3.8-ns-pulsed Nd:YAG laser as the excitation source (Ekspla, NT352).
Acknowledgements
The work was supported in part by the Singapore Ministry of Education (grant no. R143000627112), the GSK (Singapore) Research Fund (R143000492592), the National Natural Science Foundation of China (grant no. 21471109 and 21371095), and the Natural Science Foundation of Jiangsu Province (Key Project: BE2015699, BK20131404, and BL2014075).
Notes and references
- F. Auzel, Chem. Rev., 2004, 104, 139–173 CrossRef CAS PubMed.
- S. V. Eliseeva and J.-C. G. Bünzli, New J. Chem., 2011, 35, 1165 RSC.
- M. Haase and H. Schafer, Angew. Chem., Int. Ed., 2011, 50, 5808–5829 CrossRef CAS PubMed.
- H. Schäfer, P. Ptacek, B. Voss, H. Eickmeier, J. Nordmann and M. Haase, Cryst. Growth Des., 2010, 10, 2202–2208 Search PubMed.
- M. Bettinelli, L. Carlos and X. Liu, Phys. Today, 2015, 68, 38–44 CrossRef CAS.
- N. J. Greybush, M. Saboktakin, X. Ye, C. Della Giovampaola, S. J. Oh, N. E. Berry, N. Engheta, C. B. Murray and C. R. Kagan, ACS Nano, 2014, 8, 9482–9491 CrossRef CAS PubMed.
- W. Yang, X. Li, D. Chi, H. Zhang and X. Liu, Nanotechnology, 2014, 25, 482001 CrossRef PubMed.
- S. Guo, X. Xie, L. Huang and W. Huang, ACS Appl. Mater. Interfaces, 2016, 8, 847–853 CAS.
- W. Zheng, P. Huang, D. Tu, E. Ma, H. Zhu and X. Chen, Chem. Soc. Rev., 2015, 44, 1379–1415 RSC.
- D. Liu, X. Xu, Y. Du, X. Qin, Y. Zhang, C. Ma, S. Wen, W. Ren, E. M. Goldys, J. A. Piper, S. Dou, X. Liu and D. Jin, Nat. Commun., 2016, 7, 10254 CrossRef CAS PubMed.
- J. Lai, Y. Zhang, N. Pasquale and K. B. Lee, Angew. Chem., Int. Ed., 2014, 53, 14419–14423 CrossRef CAS PubMed.
- J. Zhou, Q. Liu, W. Feng, Y. Sun and F. Li, Chem. Rev., 2015, 115, 395–465 CrossRef CAS PubMed.
- X. Chen, L. Jin, W. Kong, T. Sun, W. Zhang, X. Liu, J. Fan, S. F. Yu and F. Wang, Nat. Commun., 2016, 7, 10304 CrossRef CAS PubMed.
- B. Zhou, B. Shi, D. Jin and X. Liu, Nat. Nanotechnol., 2015, 10, 924–936 CrossRef CAS PubMed.
- G. Chen, H. Qiu, P. N. Prasad and X. Chen, Chem. Rev., 2014, 114, 5161–5214 CrossRef CAS PubMed.
- Y. I. Park, K. T. Lee, Y. D. Suh and T. Hyeon, Chem. Soc. Rev., 2015, 44, 1302–1317 RSC.
- D. Ni, J. Zhang, W. Bu, H. Xing, F. Han, Q. Xiao, Z. Yao, F. Chen, Q. He, J. Liu, S. Zhang, W. Fan, L. Zhou, W. Peng and J. Shi, ACS Nano, 2014, 8, 1231–1242 CrossRef CAS PubMed.
- A. B. Chinen, C. M. Guan, J. R. Ferrer, S. N. Barnaby, T. J. Merkel and C. A. Mirkin, Chem. Rev., 2015, 115, 10530–10574 CrossRef CAS PubMed.
- L. L. Li and Y. Lu, J. Am. Chem. Soc., 2015, 137, 5272–5275 CrossRef CAS PubMed.
- C. Liu, Z. Gao, J. Zeng, Y. Hou, F. Fang, Y. Li, R. Qiao, L. Shen, H. Lei, W. Yang and M. Gao, ACS Nano, 2013, 7, 7227–7240 CrossRef CAS PubMed.
- X. Ai, C. J. Ho, J. Aw, A. B. Attia, J. Mu, Y. Wang, X. Wang, Y. Wang, X. Liu, H. Chen, M. Gao, X. Chen, E. K. Yeow, G. Liu, M. Olivo and B. Xing, Nat. Commun., 2016, 7, 10432 CrossRef CAS PubMed.
- Y. F. Wang, G. Y. Liu, L. D. Sun, J. W. Xiao, J. C. Zhou and C. H. Yan, ACS Nano, 2013, 7, 7200–7206 CrossRef CAS PubMed.
- Y. Q. Lu, J. B. Zhao, R. Zhang, Y. J. Liu, D. M. Liu, E. M. Goldys, X. S. Yang, P. Xi, A. Sunna, J. Lu, Y. Shi, R. C. Leif, Y. J. Huo, J. Shen, J. A. Piper, J. P. Robinson and D. Y. Jin, Nat. Photonics, 2014, 8, 33–37 Search PubMed.
- Y. Li, J. Tang, L. He, Y. Liu, Y. Liu, C. Chen and Z. Tang, Adv. Mater., 2015, 27, 4075–4080 CrossRef CAS PubMed.
- K. Zheng, G. He, W. Song, X. Bi and W. Qin, J. Mater. Chem. C, 2015, 3, 11589–11594 RSC.
- R. Martín-Rodríguez, S. Fischer, A. Ivaturi, B. Froehlich, K. W. Krämer, J. C. Goldschmidt, B. S. Richards and A. Meijerink, Chem. Mater., 2013, 25, 1912–1921 CrossRef.
- L. Cheng, C. Wang, L. Feng, K. Yang and Z. Liu, Chem. Rev., 2014, 114, 10869–10939 CrossRef CAS PubMed.
- S. Gai, C. Li, P. Yang and J. Lin, Chem. Rev., 2014, 114, 2343–2389 CrossRef CAS PubMed.
- B. Chen, T. Sun, X. Qiao, X. Fan and F. Wang, Adv. Opt. Mater., 2015, 3, 1577–1581 CrossRef CAS.
- J. Lai, B. P. Shah, Y. Zhang, L. Yang and K. B. Lee, ACS Nano, 2015, 9, 5234–5245 CrossRef CAS PubMed.
- W. Xu, X. Min, X. Chen, Y. Zhu, P. Zhou, S. Cui, S. Xu, L. Tao and H. Song, Sci. Rep., 2014, 4 Search PubMed.
- H. H. Gorris and O. S. Wolfbeis, Angew. Chem., Int. Ed., 2013, 52, 3584–3600 CrossRef CAS PubMed.
- J. Wang, R. R. Deng, M. A. MacDonald, B. L. Chen, J. K. Yuan, F. Wang, D. Z. Chi, T. S. A. Hor, P. Zhang, G. K. Liu, Y. Han and X. Liu, Nat. Mater., 2014, 13, 157–162 CrossRef CAS PubMed.
- X. Li, X. Liu, D. M. Chevrier, X. Qin, X. Xie, S. Song, H. Zhang, P. Zhang and X. Liu, Angew. Chem., Int. Ed., 2015, 54, 13312–13317 CrossRef CAS PubMed.
- Y. Wang, W. Cao, S. Li and W. Wen, Appl. Phys. Lett., 2016, 108, 051902 CrossRef.
- Z. Mi, Y. Zhang, S. K. Vanga, C. B. Chen, H. Q. Tan, F. Watt, X. Liu and A. A. Bettiol, Nat. Commun., 2015, 6, 8832 CrossRef CAS PubMed.
- S. Zeng, H. Wang, W. Lu, Z. Yi, L. Rao, H. Liu and J. Hao, Biomaterials, 2014, 35, 2934–2941 CrossRef CAS PubMed.
- E. M. Chan, G. Han, J. D. Goldberg, D. J. Gargas, A. D. Ostrowski, P. J. Schuck, B. E. Cohen and D. J. Milliron, Nano Lett., 2012, 12, 3839–3845 CrossRef CAS PubMed.
- F. Wang and X. Liu, Acc. Chem. Res., 2014, 47, 1378–1385 CrossRef CAS PubMed.
- W. Wei, Y. Zhang, R. Chen, J. Goggi, N. Ren, L. Huang, K. K. Bhakoo, H. D. Sun and T. T. Y. Tan, Chem. Mater., 2014, 26, 5183–5186 CrossRef CAS.
- X. Wu, H. Lee, O. Bilsel, Y. Zhang, Z. Li, T. Chen, Y. Liu, C. Duan, J. Shen, A. Punjabi and G. Han, Nanoscale, 2015, 7, 18424–18428 RSC.
- W. Q. Zou, C. Visser, J. A. Maduro, M. S. Pshenichnikov and J. C. Hummelen, Nat. Photonics, 2012, 6, 560–564 CrossRef CAS.
- F. Wang and X. Liu, J. Am. Chem. Soc., 2008, 130, 5642–5643 CrossRef CAS PubMed.
- R. Deng, F. Qin, R. Chen, W. Huang, M. Hong and X. Liu, Nat. Nanotechnol., 2015, 10, 237–242 CrossRef CAS PubMed.
- A. Sedlmeier and H. H. Gorris, Chem. Soc. Rev., 2015, 44, 1526–1560 RSC.
- J. C. Boyer, C. J. Carling, B. D. Gates and N. R. Branda, J. Am. Chem. Soc., 2010, 132, 15766–15772 CrossRef CAS PubMed.
- C. Zhang, L. Yang, J. Zhao, B. Liu, M. Y. Han and Z. Zhang, Angew. Chem., Int. Ed., 2015, 54, 11531–11535 CrossRef CAS PubMed.
- G. Chen, T. Y. Ohulchanskyy, A. Kachynski, H. Agren and P. N. Prasad, ACS Nano, 2011, 5, 4981–4986 CrossRef CAS PubMed.
- M. Pollnau, D. R. Gamelin, S. R. Lüthi, H. U. Güdel and M. P. Hehlen, Phys. Rev. B: Condens. Matter, 2000, 61, 3337–3346 CrossRef CAS.
- J. F. Suyver, A. Aebischer, S. García-Revilla, P. Gerner and H. U. Güdel, Phys. Rev. B: Condens. Matter, 2005, 71, 125123 CrossRef.
- F. Wang, R. Deng and X. Liu, Nat. Protoc., 2014, 9, 1634–1644 CrossRef CAS PubMed.
- M. T. Berry and P. S. May, J. Phys. Chem. A, 2015, 119, 9805–9811 CrossRef CAS PubMed.
- X. Chen, D. Peng, Q. Ju and F. Wang, Chem. Soc. Rev., 2015, 44, 1318–1330 RSC.
- H. Schäfer, P. Ptacek, O. Zerzouf and M. Haase, Adv. Funct. Mater., 2008, 18, 2913–2918 CrossRef.
- Y. Wang, L. Tu, J. Zhao, Y. Sun, X. Kong and H. Zhang, J. Phys. Chem. C, 2009, 113, 7164–7169 CAS.
- A. Kar and A. Patra, Nanoscale, 2012, 4, 3608–3619 RSC.
- S. Han, R. Deng, X. Xie and X. Liu, Angew. Chem., Int. Ed., 2014, 53, 11702–11715 CrossRef CAS PubMed.
- F. Zhang, R. Che, X. Li, C. Yao, J. Yang, D. Shen, P. Hu, W. Li and D. Zhao, Nano Lett., 2012, 12, 2852–2858 CrossRef CAS PubMed.
- L. Lei, D. Chen, P. Huang, J. Xu, R. Zhang and Y. Wang, Nanoscale, 2013, 5, 11298–11305 RSC.
- Y. Wang, K. Liu, X. M. Liu, K. Dohnalová, T. Gregorkiewicz, X. G. Kong, M. C. G. Aalders, W. J. Buma and H. Zhang, J. Phys. Chem. Lett., 2011, 2, 2083–2088 CrossRef CAS.
- J. Zhao, Z. Lu, Y. Yin, C. McRae, J. A. Piper, J. M. Dawes, D. Jin and E. M. Goldys, Nanoscale, 2013, 5, 944–952 RSC.
- F. Vetrone, R. Naccache, V. Mahalingam, C. G. Morgan and J. A. Capobianco, Adv. Funct. Mater., 2009, 19, 2924–2929 CrossRef CAS.
- F. Wu, X. Liu, X. Kong, Y. Zhang, L. Tu, K. Liu, S. Song and H. Zhang, Appl. Phys. Lett., 2013, 102, 243104 CrossRef.
- J.-C. Boyer, M.-P. Manseau, J. I. Murray and F. C. J. M. van Veggel, Langmuir, 2009, 26, 1157–1164 CrossRef PubMed.
- Y. P. Li, J. H. Zhang, Y. S. Luo, X. Zhang, Z. D. Hao and X. J. Wang, J. Mater. Chem., 2011, 21, 2895–2900 RSC.
- J. M. Meruga, A. Baride, W. Cross, J. J. Kellar and P. S. May, J. Mater. Chem. C, 2014, 2, 2221–2227 RSC.
- G. Y. Chen, Y. Liu, Y. G. Zhang, G. Somesfalean, Z. G. Zhang, Q. Sun and F. P. Wang, Appl. Phys. Lett., 2007, 91, 133103 CrossRef.
- Y. F. Bai, Y. X. Wang, G. Y. Peng, W. Zhang, Y. K. Wang, K. Yang, X. R. Zhang and Y. L. Song, Opt. Commun., 2009, 282, 1922–1924 CrossRef CAS.
- M. Quintanilla, F. Q. Ren, D. L. Ma and F. Vetrone, ACS Photonics, 2014, 1, 662–669 CrossRef CAS.
- H. Wen, H. Zhu, X. Chen, T. F. Hung, B. Wang, G. Zhu, S. F. Yu and F. Wang, Angew. Chem., Int. Ed., 2013, 52, 13419–13423 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nr00812g |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a