
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
RuII, IrIII and OsII mesoionic carbene complexes: efficient catalysts for transfer hydrogenation of selected functionalities†‡
Aljoša
Bolje
a,
Stephan
Hohloch
b,
Janez
Košmrlj
*a and
Biprajit
Sarkar
*b
aFaculty of Chemistry and Chemical Technology, University of Ljubljana, Večna pot 113, SI-1000 Ljubljana, Slovenia
bInstitut für Chemie und Biochemie, Anorganische Chemie, Freie Universität Berlin, Fabeckstraße 34-36, D-14195 Berlin, Germany. E-mail: biprajit.sarkar@fu-berlin.de
First published on 7th July 2016
Abstract
Pyridine-appended triazolylidene donors have been recently used as ligands in various homogeneous catalytic processes. We present here a new pyrimidine substituted triazolium salt which was prepared and used in the coordination to RuII and OsII. The new triazolium salt and the obtained complexes were characterized by multinuclear NMR spectroscopy. The molecular composition of the mentioned compounds was confirmed by positive ion electrospray ionization (ESI+) mass spectra. The new pyrimidyl containing complexes, as well as the related pyridyl-triazolylidene containing complexes, were applied in transfer hydrogenation reactions of carbonyls, alkenes, imines and nitroarenes. The pyrimidyl containing complexes reveal an over-all better activity in comparison to their pyridine bearing analogues. The studies of electronic effects of the ligands, as well as mechanistic insights for the reduction of nitrobenzene with three selected precatalysts are presented.
Introduction
The transfer hydrogenation reaction promoted by metal-complexes is currently one of the most commonly investigated homogeneous hydrogenation processes.1 This reaction is highly appealing for several reasons. It does not require hazardous gaseous hydrogen and pressurized reactors, and it employs cheap and sustainable solvents as the source of hydrogen, to name just a few. The wide range of functional groups that can be reduced by transfer hydrogenation is noteworthy. These include carbonyl, olefin, imine, nitrile, and nitro compounds. The need to develop simple and sustainable reaction conditions that require only low catalyst loadings is currently emerging in developing new catalysts and catalytic systems.In the last two decades N-heterocyclic carbenes (NHCs) have been developed as a powerful class of ligands. Despite being used in nearly every field of modern chemical sciences, without a doubt NHCs have most dramatically affected organometallic chemistry and catalysis. Owing to their strong σ-donation ability, air and moisture stability, high structural variability, as well as low toxicity, they have become viable alternatives to widely used phosphines in many catalytic processes. Today, a vast number of transformations are known to proceed only with NHC-based catalysts.2 1,2,3-Triazol-5-ylidenes are a prominent subclass of NHCs possessing a mesoionic carbene (MIC) structure. Due to a lower heteroatom stabilization and mesoionic character, MICs are among one of the best σ-donors among the NHCs.3 The highly efficient and modular nature of click reaction that is generally used to access 1,2,3-triazol-5-ylidenes makes the design and preparation of diverse libraries of these ligands extremely easy.4 Complexes bearing triazolylidenes are increasingly used in homogeneous metal-based catalysis, as well as for photophysical and biochemical purposes.4d Many catalytic systems of this type have been studied, showing comparable or even better efficiency in comparison to the majority of their NHC analogues.5 Metal complexes of MIC ligands have been used as catalysts among others in olefin metathesis,6 oxidations,7 reductions,8 click reactions,9 and C–C/C–N bond formation reactions.10
Recently, three series of complexes consisting of pyridine appended triazolylidenes as ligands with ruthenium(II), iridium(III) and osmium(II) were reported and they were successfully used in transfer hydrogenation of selected carbonyl substrates (Scheme 1).11 All complexes have shown remarkable activity even at very low catalyst loadings with several different substrates investigated. The ruthenium(II) complexes were also used in oxidation of primary and secondary alcohols.7d
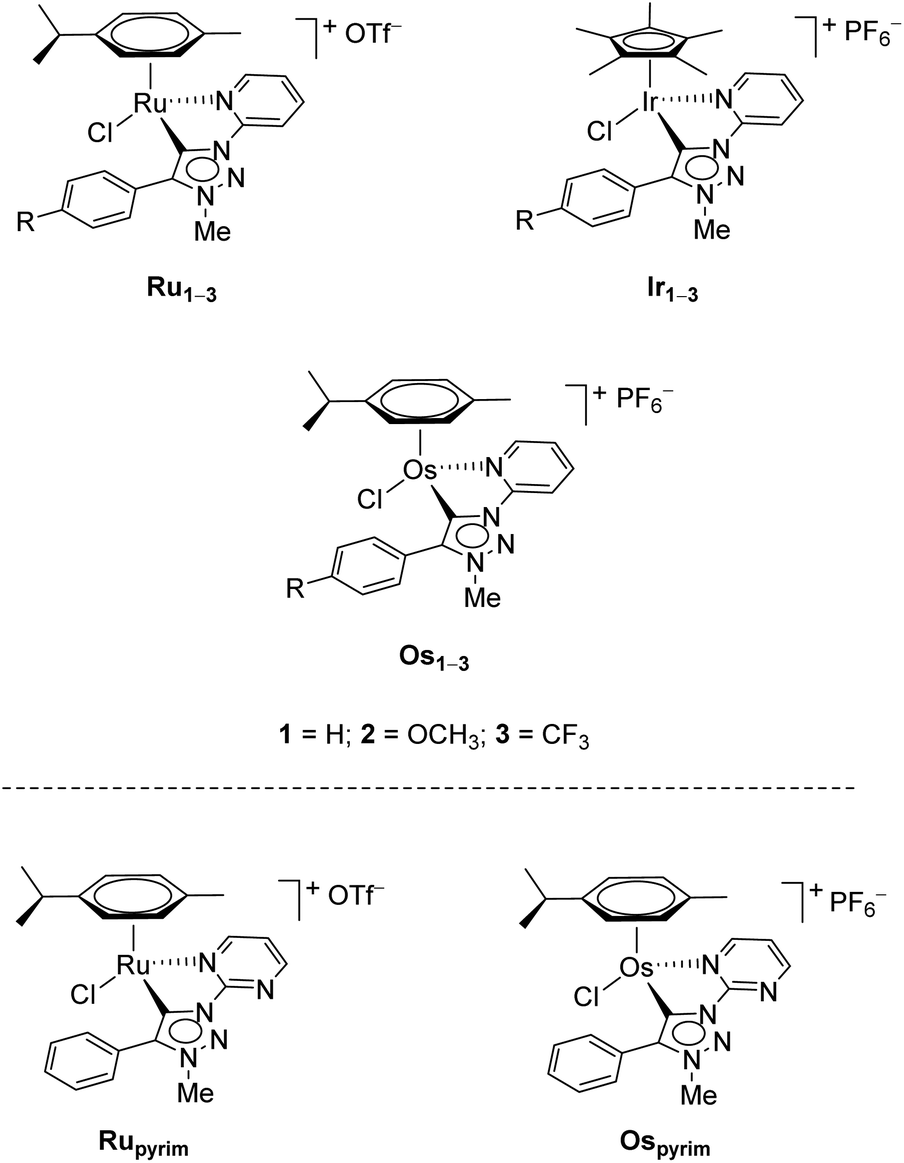 | ||
| Scheme 1 The RuII, OsII and IrIII pyridine-appended MIC complexes (up),11 and RuII and OsII pyrimidine-appended MIC complexes (bottom) used in this work. | ||
Encouraged by these results, we were prompted to test the activity of these complexes also for transfer hydrogenation of substrates bearing other functional groups. In particular, we were interested in different carbonyl, olefin, imine, and nitroarene substrates.
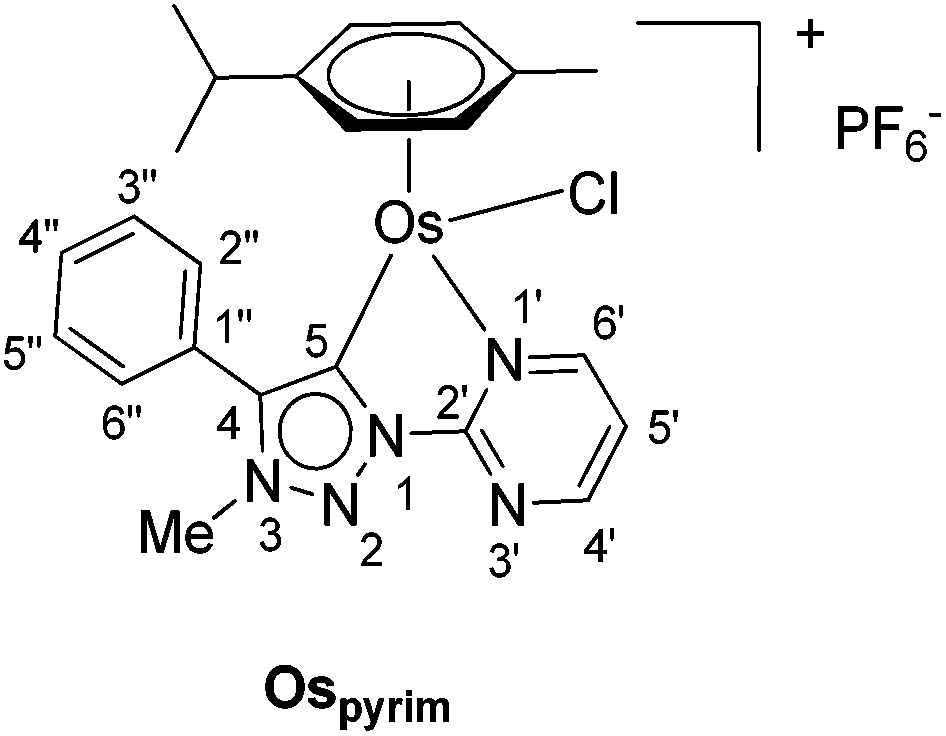
In addition to the pyridine-appended MICs, we decided to design novel MICs that are functionalized with a pyrimidine ring. The starting 1-(2-pyrimidyl)-functionalized triazolium salt was prepared in excellent yields and purity and used to construct the corresponding Ru(II) and Os(II) complexes (Scheme 1). The coordination was achieved by a transmetallation protocol and the corresponding complexes were characterized by 1H, 13C and 15N NMR spectroscopy and high resolution mass spectrometry. Unfortunately, the preparation of the Ir(III) analogue failed. The two pyrimidine-appended MIC complexes of Ru(II) and Os(II) and the previously reported7d,11 pyridine-appended MICs of Ru(II), Ir(III), and Os(II), shown in Scheme 1, were used in transfer hydrogenation of the above mentioned functionalities. This is also the first report on pyrimidine-appended triazolium salts, the corresponding MICs and their transition metal complexes. Some mechanistic insights into the reduction of nitrobenzene with the three selected complexes are presented.
Results and discussion
Ligand synthesis and coordination
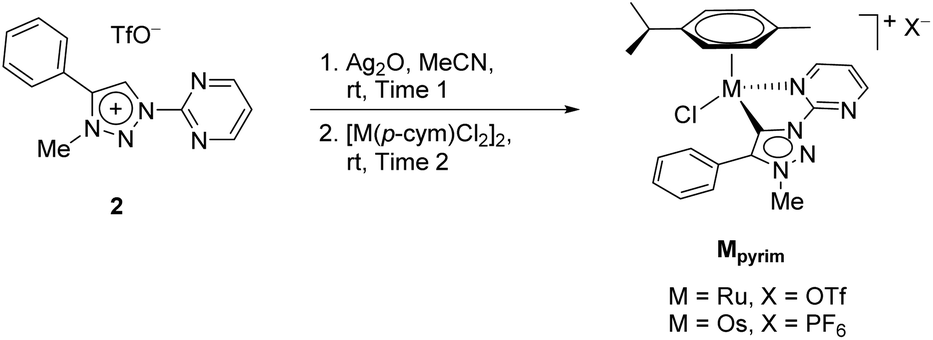
In contrast to earlier reports of unselective methylation of pyridyl-triazoles,12 some of us have recently reported a selective route to generate only NTriazole methylated pyridyl-triazolium salts.13 Some of these salts have already been successfully used to generate ruthenium(II), osmium(II) and iridium(III) complexes.7d,11 Inspired by these reports, we designed and prepared an analogous derivative, that has a pyrimidine ring attached directly to the triazole nitrogen atom N1 (Scheme 2). The starting triazole 1 was prepared by copper-catalyzed cycloaddition of phenylacetylene with 2-azidopyrimidine (tetrazolo[1,5-a]pyrimidine) as reported previously.14 The methylation into 2 was achieved by mixing the triazole with MeOTf in dry dichloromethane at 0 °C for 24 h (Scheme 2). Triazolium salt 2 was isolated as a triflate salt in 91% yield. In analogy to 1-(2-pyridyl)-1,2,3-triazoles,13 1-(2-pyrimidyl)-1,2,3-triazole 1 could be selectively alkylated with MeOTf at triazole nitrogen N3 with no need of pyrimidine ring protection.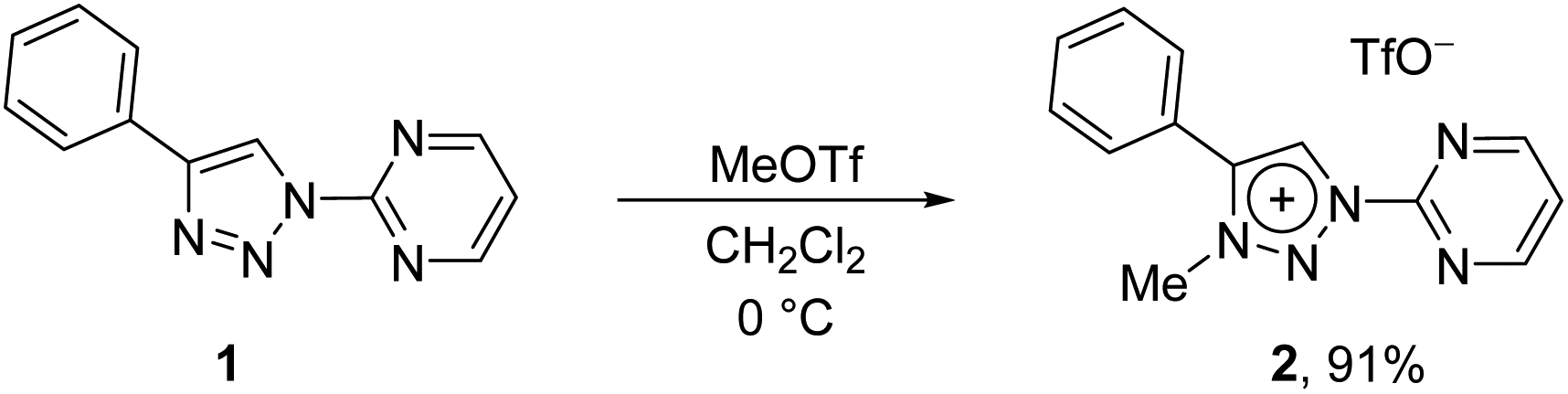 | ||
| Scheme 2 The synthesis of triazolium salt 2. | ||
Next, the coordination ability of triazolium salt 2 towards Ru(II), Ir(III) and Os(II) was examined. To this end, we chose the already established transmetalation protocol via the intermediate carbenic silver(I) complex, shown in Scheme 3.15
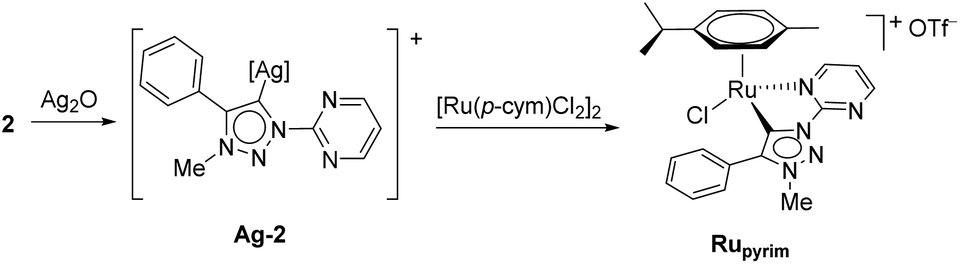 | ||
| Scheme 3 The ruthenation of 2via transmetalation. | ||
The triazolium salt 2 was stirred with Ag2O and in dry acetonitrile at room temperature for 4 days (Scheme 3). The formation of complex Ag-2 was suggested by the disappearance of the triazolium proton H-5 from the 1H NMR spectral analysis in DMSO-d6. Due to the general instability of such silver complexes, we were unable to obtain more experimental data for its structure. Subsequently, Ag-2 was subjected to transmetalation without previous isolation employing [Ru(p-cym)Cl2]2 as a ruthenium source (Scheme 3). The reaction progress was monitored by TLC analysis. The pure complex Rupyrim was isolated as a triflate salt after 2 hours by simple three-step filtration–solvent evaporation–crystallization workup in 85% yield (Table 1). For the preparation of Ospyrim, which involved transmetalation with the OsII precursor [Os(p-cym)Cl2]2 the reaction mixture had to be stirred for a prolonged time of 3 days. The complex Ospyrim was isolated in the pure form as a PF6 salt in a moderate yield of 53% (Table 1). Unfortunately, we were unable to prepare the IrIII analogue. After the addition of [IrCp*Cl2]2 to the reaction mixture of the triazolium salt 2 and Ag2O in acetonitrile, no pure product could be isolated. Instead a mixture of products was isolated. The use of a base such as Cs2CO3 or heating of the reaction mixture did not lead to any improvement. Even though we could identify the product in the mass spectrum of the crude reaction mixture (see the ESI‡), attempts to purify it led to its decomposition. Although it is not clear at this stage as to why the product decomposes at the purification stage, we think that it does not survive either the filtration through Celite, or the salt metathesis with KPF6 that is necessary for its purification.
Transfer hydrogenation catalysis
After having the two complexes Rupyrim and Ospyrim prepared, we examined their activity in transfer hydrogenation catalysis. In this context we also tested some RuII, IrIII and OsII complexes from our previous reports, which were already used for carbonyl group reduction.11 As the model reaction we examined the reduction of benzaldehyde to benzyl alcohol. After testing at room temperature, 60 °C and 80 °C, we observed that the best conversions are obtained at 100 °C. Hence, all transfer hydrogenation reactions (other than nitrobenzene) were carried out at 100 °C. The results for benzaldehyde are presented in Table 2.|
|
|||||
|---|---|---|---|---|---|
| Precatalyst | Conv.b (%) | Precatalyst | Conv.b (%) | Precatalyst | Conv.b (%) |
| a General reaction conditions: benzaldehyde (0.5 mmol), precatalyst, KOH (0.085 mmol), iPrOH (4 mL), 100 °C for 3 h. b Conversion determined by 1H NMR spectroscopy using hexamethylbenzene as an internal standard. c Data collected from ref. 11. | |||||
| Precatalyst loading: 0.5 mol% | |||||
Ru1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c c |
>99 |
Ir1c |
>99 |
Os1c |
>99 |
|
Ru2c |
>99 |
Ir2c |
>99 |
Os2c |
>99 |
|
Ru3c |
>99 |
Ir3c |
>99 |
Os3c |
>99 |
| Rupyrim | >99 | Ospyrim | >99 | ||
| Precatalyst loading: 0.01 mol% | |||||
|
Ru1c |
>99 |
Ir1c |
>99 |
Os1c |
>99 |
|
Ru2c |
>99 |
Ir2c |
>99 |
Os2c |
>99 |
|
Ru3c |
>99 |
Ir3c |
>99 |
Os3c |
>99 |
| Rupyrim | >99 | Ospyrim | >99 | ||
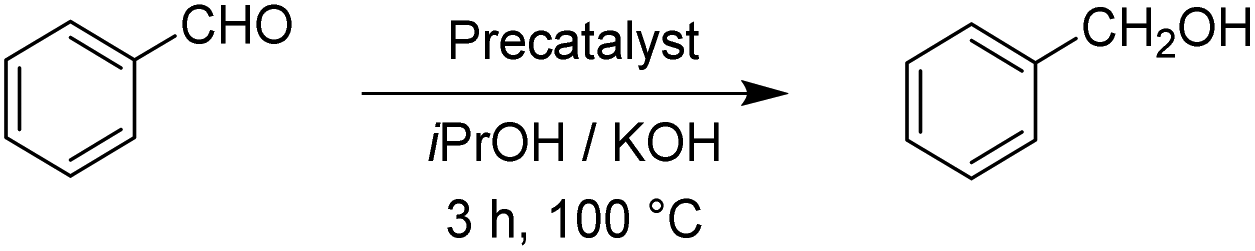
The reduction of benzaldehyde under the applied reaction conditions proceeds smoothly with all tested complexes. Applying 0.5 mol% of the precatalysts, as well as with only 0.01 mol%, the reduction occurred with full conversion of the starting material, reflecting in a TON of about 10000. This is comparable to known ruthenium(II)–NHC complexes16 and in line with the activity of their pyridyl-MIC analogues.11 To investigate further the catalytic performance of the Rupyrim and Ospyrim, acetophenone was used as a substrate. Ketones in general are known to be more challenging substrates than aldehydes for the reduction. The results are outlined in Table 3.
|
|
|||||
|---|---|---|---|---|---|
| Precatalyst | Conv.b (%) | Precatalyst | Conv.b (%) | Precatalyst | Conv.b (%) |
| a General reaction conditions: acetophenone (0.5 mmol), precatalyst, KOH (0.085 mmol), iPrOH (4 mL), 100 °C for 3 h. b Conversion determined by 1H NMR spectroscopy using hexamethylbenzene as an internal standard. c Data collected from ref. 11. | |||||
| Precatalyst loading: 0.5 mol% | |||||
|
Ru1c |
>99 |
Ir1c |
88 |
Os1c |
76 |
|
Ru2c |
>99 |
Ir2c |
71 |
Os2c |
88 |
|
Ru3c |
>99 |
Ir3c |
92 |
Os3c |
65 |
| Rupyrim | >99 | Ospyrim | 70 | ||
| Precatalyst loading: 0.01 mol% | |||||
|
Ru1c |
80 | ||||
|
Ru2c |
89 | ||||
|
Ru3c |
60 | ||||
| Rupyrim | 94 | ||||
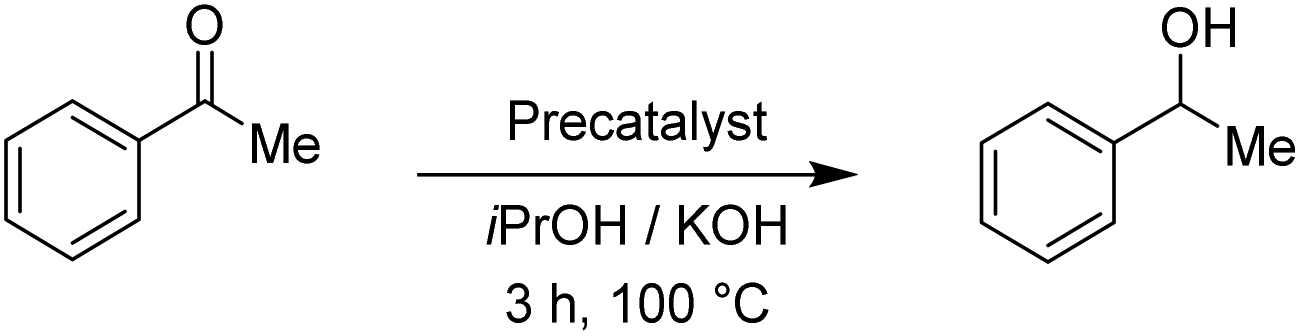
Whereas the reduction of acetophenone with the ruthenium complex Rupyrim at the precatalyst loadings of 0.5 mol% gave full conversion of acetophenone to 1-phenylethanol, the osmium complex Ospyrim gave only 70% of the reduced substrate. By using 0.01 mol%, the pyrimidine analogue Rupyrim has shown the best activity amongst all tested precatalysts. With 0.5 mol% of the Ru2 precatalyst, we were able to isolate the product in 85% yield (conversion >99%). Prompted by very good overall results with acetophenone we decided to continue with a more sterically hindered substrate, benzophenone. The results are collected in Table 4.
|
|
|||||
|---|---|---|---|---|---|
| Precatalyst | Conv.b (%) | Precatalyst | Conv.b (%) | Precatalyst | Conv.b (%) |
| a General reaction conditions: benzophenone (0.5 mmol), precatalyst, KOH (0.085 mmol), iPrOH (4 mL), 100 °C for 3 h. b Conversion determined by 1H NMR spectroscopy using hexamethylbenzene as an internal standard. c Data collected from ref. 11. | |||||
| Precatalyst loading: 0.5 mol% | |||||
|
Ru1c |
>99 |
Ir1c |
65 |
Os1c |
68 |
|
Ru2c |
>99 |
Ir2c |
55 |
Os2c |
88 |
|
Ru3c |
>99 |
Ir3c |
85 |
Os3c |
25 |
| Rupyrim | >99 | Ospyrim | 72 | ||
| Precatalyst loading: 0.1 mol% | |||||
|
Ru1c |
69 | ||||
|
Ru2c |
90 | ||||
|
Ru3c |
59 | ||||
| Rupyrim | 93 | ||||

Benzophenone was fully converted into diphenylmethanol with the Rupyrim precatalyst at 0.5 mol%. The same complex gave an excellent 93% conversion also at 0.1 mol% precatalyst loading, which is the best performance for all tested compounds. With 0.1 mol% of the Ru1 precatalyst, we were able to isolate the product in 60% yield (conversion 69%, Table 4). On the other hand Ospyrim gave only 72% conversion by employing 0.5 mol% of the precatalyst. This result is in line with other Os(II) complexes Os1–3. To finish, we conducted the reduction of cyclohexanone as a representative of an aliphatic substrate. The results are summarized in Table 5.
|
|
|||||
|---|---|---|---|---|---|
| Precatalyst | Conv.c (%) | Precatalyst | Conv.c (%) | Precatalyst | Conv.c (%) |
| a General reaction conditions: cyclohexanone (0.5 mmol), precatalyst, KOH (0.085 mmol), iPrOH (4 mL), 100 °C for 3 h. b Precatalyst (0.05 mol%). c Conversion determined by 1H NMR spectroscopy using hexamethylbenzene as an internal standard. d Data collected from ref. 11. | |||||
| Precatalyst loading: 0.5 mol% | |||||
|
Ru1d |
>99 |
Ir1d |
86 |
Os1d |
>99 |
|
Ru2d |
>99 |
Ir2d |
75 |
Os2d |
>99 |
|
Ru3d |
>99 |
Ir3d |
92 |
Os3d |
91 |
| Rupyrim | >99 | Ospyrim | >99 | ||
| Precatalyst loading: 0.1 mol% | |||||
|
Ru1d |
79 |
Os1d |
62 | ||
|
Ru2d |
94 |
Os2d |
76 | ||
|
Ru3d |
72 |
Os3d |
50 | ||
| Rupyrim | >99, 78b | Ospyrim | 94 | ||
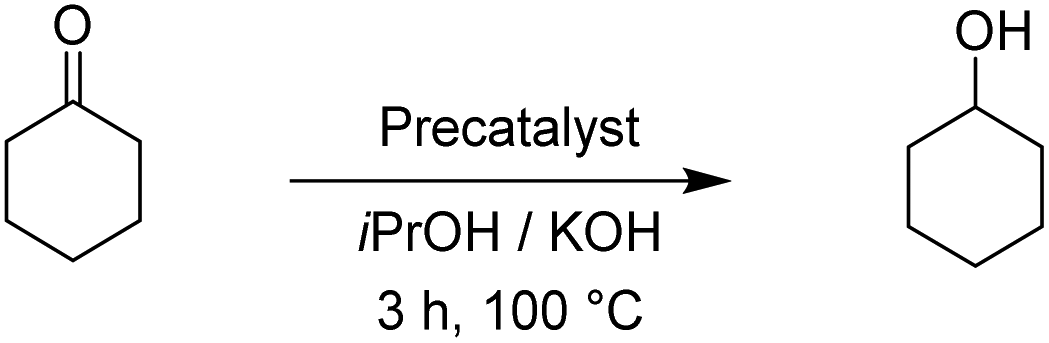
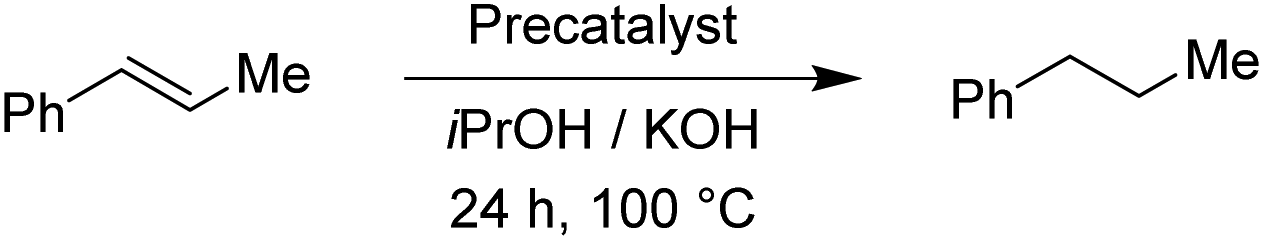
The two pyrimidyl-MIC complexes Rupyrim and Ospyrim have shown the best activity for the transfer hydrogenation of cyclohexanone into cyclohexanol. Employing either 0.5 mol% or 0.1 mol% of the precatalyst resulted in an excellent overall activity. The Rupyrim gave a very good 78% conversion also by using only 0.05 mol% of the precatalyst. Good results of the complexes Rupyrim and Ospyrim in the transfer hydrogenation of the carbonyl group prompted us to test other functional groups. We started with olefin substrates: cyclooctene, methylstyrene and trans-stilbene. The results for the reduction of cyclooctene are presented in Table 6.
|
|
|||||
|---|---|---|---|---|---|
| Precatalyst | Conv.b (%) | Precatalyst | Conv.b (%) | Precatalyst | Conv.b (%) |
| a General reaction conditions: cyclooctene (0.5 mmol), precatalyst, KOH (0.085 mmol), iPrOH (4 mL), 100 °C for 24 h. b Conversion determined by 1H NMR spectroscopy using hexamethylbenzene as an internal standard. | |||||
| Precatalyst loading: 1.0 mol% | |||||
| Ru1 | >99 | Ir1 | 73 | Os1 | 84 |
| Ru2 | >99 | Ir2 | 60 | Os2 | 93 |
| Ru3 | >99 | Ir3 | 80 | Os3 | 59 |
| Rupyrim | >99 | Ospyrim | 89 | ||
| Precatalyst loading: 0.5 mol% | |||||
| Ru1 | 79 | ||||
| Ru2 | 94 | ||||
| Ru3 | 72 | ||||
| Rupyrim | 87 | ||||
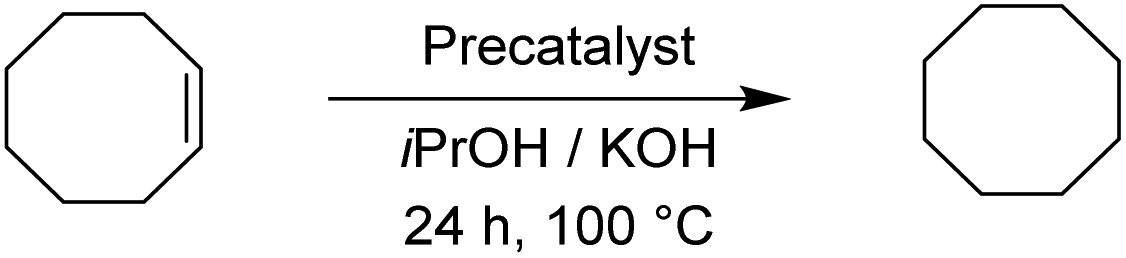
For the reduction of cyclooctene, we used similar reaction conditions as mentioned above. A mixture of cyclooctene, the selected precatalyst and KOH in iPrOH was stirred under an inert atmosphere at 100 °C for 24 h. By using 1.0 mol% of ruthenium-based precatalysts we observed full conversions into cyclooctane with all four tested complexes. Also with the iridium and osmium analogues, the conversions were very good. Scaling down to 0.5 mol% of Ru(II) complexes, we could still reach excellent conversions, better than those described for already known similar systems.17 The best performance was seen with Ru2, bearing the 4-methoxyphenyl donor group at the triazole, which is in line with the results for the reduction of carbonyls. Excellent performance in the reduction of cyclooctene encouraged us to try other such substrates. We took trans-β-methylstyrene and applied the same reaction conditions as for cyclooctene. The results are summarized in Table 7.
|
|
|||||
|---|---|---|---|---|---|
| Precatalyst | Conv.b (%) | Precatalyst | Conv.b (%) | Precatalyst | Conv.b (%) |
| a General reaction conditions: trans-β-methylstyrene (0.5 mmol), precatalyst, KOH (0.085 mmol), iPrOH (4 mL), 100 °C for 24 h. b Conversion determined by 1H NMR spectroscopy using 4-bromobenzaldehyde as an internal standard. | |||||
| Precatalyst loading: 1.0 mol% | |||||
| Ru1 | 65 | Ir1 | 62 | Os1 | 8 |
| Ru2 | 75 | Ir2 | 58 | Os2 | 6 |
| Ru3 | 56 | Ir3 | 67 | Os3 | 4 |
| Rupyrim | 78 | Ospyrim | 0 | ||
| Precatalyst loading: 0.5 mol% | |||||
| Ru1 | 45 | ||||
| Ir1 | 25 | ||||
| Os1 | 4 | ||||
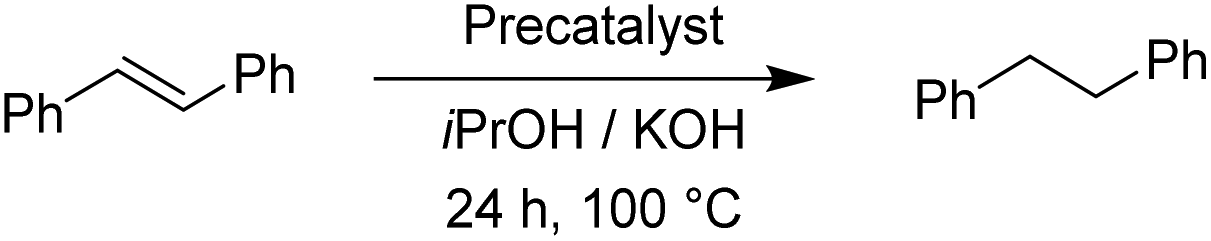
In comparison to cyclooctene, trans-β-methylstyrene proved to be a more challenging substrate. With the exception of the Os(II) representatives, the complexes still performed very well. Nonetheless, our catalysts have shown similar or better performance in this reduction than the previously reported examples.17 The ligand effect trends were similar to the previous observations. For the Ru(II) complexes the electron-donating substituents at the triazole ring worked better, whereas the Ir(III) complexes preferred electron withdrawing groups.11 Furthermore, we investigated the transfer hydrogenation of trans-stilbene, a sterically more hindered alkene. The results from Table 8 confirm the reduction of trans-stilbene into 1,2-diphenylethane as more demanding for our catalytic system, which has been also reported for other such systems.17
|
|
|||||
|---|---|---|---|---|---|
| Precatalyst | Conv.b (%) | Precatalyst | Conv.b (%) | Precatalyst | Conv.b (%) |
| a General reaction conditions: trans-stilbene (0.5 mmol), precatalyst, KOH (0.085 mmol), iPrOH (4 mL), 100 °C for 24 h. b Conversion determined by 1H NMR spectroscopy using hexamethylbenzene as an internal standard. | |||||
| Precatalyst loading: 1.0 mol% | |||||
| Ru1 | 30 | Ir1 | 9 | Os1 | 15 |
| Ru2 | 41 | Ir2 | 11 | Os2 | 16 |
| Ru3 | 28 | Ir3 | 13 | Os3 | 4 |
| Rupyrim | 13 | Ospyrim | 8 | ||
To continue we took an imine substrate and selected N-benzylideneaniline.18 We employed both K2CO3 as well as KOH as bases and stirred the reaction mixture for 18 h at 100 °C. Both bases delivered similar conversions. The results are collected in Table 9.
|
|
|||||
|---|---|---|---|---|---|
| Precatalyst | Conv.b (%) | Precatalyst | Conv.b (%) | Precatalyst | Conv.b (%) |
| a General reaction conditions: N-benzylideneaniline (0.5 mmol), precatalyst, K2CO3 (0.025 mmol), iPrOH (4 mL), 100 °C for 18 h. b Conversion determined by 1H NMR spectroscopy using hexamethylbenzene as an internal standard. | |||||
| Precatalyst loading: 1.0 mol% | |||||
| Ru1 | >99 | Ir1 | 50 | Os1 | 19 |
| Ru2 | >99 | Ir2 | 23 | Os2 | 18 |
| Ru3 | >99 | Ir3 | 17 | Os3 | 17 |
| Rupyrim | >99 | Ospyrim | 30 | ||
| Precatalyst loading: 0.5 mol% | |||||
| Ru1 | 71 | ||||
| Ru2 | 87 | ||||
| Ru3 | 60 | ||||
| Rupyrim | 90 | ||||
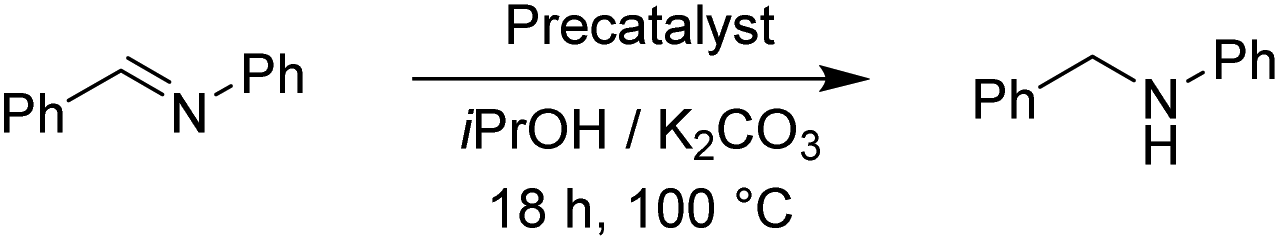
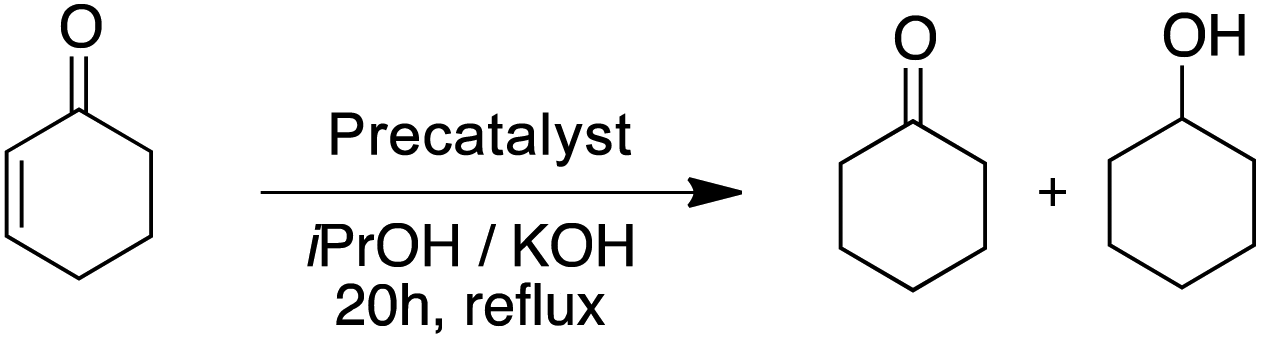
With 1.0 mol% precatalyst loading the ruthenium-based precatalysts converted the N-benzylideneaniline into N-benzylaniline fully. Also by using 0.5 mol% the same complexes gave an excellent overall conversion. The Rupyrim performed best, but also the Ru2 showed excellent activity, confirming the ligand effects for the Ru(II) complex series. On the other hand the Ir(III) and Os(II) precatalysts were less active for the reduction of this type of substrates. The activity of our complexes is comparable to the previously reported ruthenium catalyst in the transfer hydrogenation of N-benzylideneaniline.18 We also tested the reduction of cyclohexenone as an example of an α,β-unsaturated carbonyl. Whereas conversions were high for all tested precatalysts (Table 10), only Os2 displayed relatively high selectivity towards selective reduction of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond over the CO bond.
C bond over the CO bond.
|
|
|||||
|---|---|---|---|---|---|
| Precatalyst | Conv.b (%) | Precatalyst | Conv.b (%) | Precatalyst | Conv.b (%) |
| a Reaction conditions: cyclohexenone (0.5 mmol), catalyst (0.5 mol%), KOH (0.1 mmol), isopropanol (4 mL), 100 °C, 20 h reflux. b Conversion determined by 1H NMR spectroscopy using hexamethylbenzene as an internal standard; >99% conversion of starting meterials: ketone/alcohol product ratio. | |||||
| Ru1 | 0/100 | Ir1 | 21/79 | Os1 | 67/33 |
| Ru2 | 8/92 | Ir2 | 31/69 | Os2 | 83/17 |
| Ru3 | 0/100 | Ir3 | 42/58 | Os3 | 64/36 |
| Rupyrim | 0/100 | Ospyrim | 30/70 | ||
To conclude, we tested the transfer hydrogenation of the nitro group. Recently, an efficient transfer hydrogenation catalytic system for the reduction of nitroarenes into anilines was reported and was based on Ru(II) and Ir(III) MIC precatalysts.8a We carried out an initial screening with nitrobenzene as a substrate, applying 1.5 mol% of the precatalysts and 0.5 equiv. of KOH in iPrOH at 80 °C.8a Azobenzene and azoxybenzene were expected as the side products. The results are presented in Table 11. The screening showed a catalyst dependent conversion, as well as product distribution. In contrast to the previously tested functional groups, the Ir(III) precatalysts were the most active of all the tested complexes, affording aniline as the major product. The Ru(II) complexes gave good conversion into aniline and azoxybenzene with only minute amounts of azobenzene. Once again the pyrimidine analogue Rupyrim showed superior activity. The Os(II) complexes performed similar to the Ru(II) analogues, giving slightly higher conversions. The best performance was seen for Os2, where aniline was the main product.
|
|
||||
|---|---|---|---|---|
| Precatalyst | Anilineb | Starting nitrob | Azob | Azoxyb |
| a General reaction conditions: nitrobenzene (0.5 mmol), precatalyst, KOH (0.21 mmol), iPrOH (4 mL), 80 °C for 24 h. b Conversion in % determined by GC using hexadecane as an internal standard. | ||||
| Precatalyst loading: 1.5 mol% | ||||
| Ru1 | 50 | 10 | 10 | 30 |
| Ru2 | 56 | 26 | 4 | 14 |
| Ru3 | 32 | 40 | 0 | 28 |
| Rupyrim | 71 | 0 | 12 | 17 |
| Ir1 | 93 | 0 | 7 | 0 |
| Ir2 | 85 | 0 | 11 | 4 |
| Ir3 | >99 | 0 | 0 | 0 |
| Os1 | 50 | 31 | 0 | 19 |
| Os2 | 83 | 0 | 3 | 14 |
| Os3 | 50 | 35 | 0 | 15 |
| Ospyrim | 80 | 0 | 3 | 17 |
| Precatalyst loading: 5.0 mol% | ||||
| Ru1 | >99 | 0 | 0 | 0 |
| Ru2 | >99 | 0 | 0 | 0 |
| Ru3 | >99 | 0 | 0 | 0 |
| Rupyrim | >99 | 0 | 0 | 0 |
| Ir1 | 97 | 0 | 3 | 0 |
| Ir2 | 93 | 0 | 7 | 0 |
| Ir3 | >99 | 0 | 0 | 0 |
| Os1 | 64 | 36 | 0 | 0 |
| Os2 | 87 | 10 | 0 | 3 |
| Os3 | 60 | 40 | 0 | 0 |
| Ospyrim | >99 | 0 | 0 | 0 |
As was previously observed,8a when employing 5.0 mol% of the precatalysts, we observed a drastic improvement in catalytic activity. The best results were seen with all four Ru(II) complexes, giving full conversion of nitrobenzene into aniline. The Ir(III) analogues performed very similarly. On the other hand, the Os(II) complexes were less active, except for Ospyrim, which yielded aniline quantitatively.
Mechanistic studies
Recently, a mechanistic study for a similar catalytic system was reported8a where a two-pathway mechanism was postulated based on two previous literature reports.19 The mechanism is outlined in Scheme 4. The metal complex is converted into hydrides “[M]H2” in basic iPrOH. Next, nitrosobenzene is formed, which can be reduced into aniline through two different pathways. Route A proceeds through N-phenylhydroxyamine giving aniline directly. In contrast, path B involves an azoxybenzene intermediate, which gets converted to azobenzene before being reduced to aniline. To determine the reaction pathways for each of the complex families, we ran a time-dependent screening of the product formation. With the chosen precatalysts, we carried out the reduction of nitrobenzene under catalytic conditions with 1.5 mol% loading and analysed aliquots over time. The resulting data are represented in Fig. 1. With ruthenium complex Ru1 54% conversion into aniline was observed after 32 h at 80 °C. While the amount of nitrobenzene decreased over time, the amounts of azoxybenzene and aniline increased. The formation of aniline, as well as azoxybenzene correlated well with the consumption of the starting nitrobenzene. Only minor amounts of azobenzene were traced after 32 h. The results point to pathway B. The iridium complex Ir3 gave 84% conversion to aniline after 24 h with the amount of azoxybenzene less than 18%. The consumption of nitrobenzene correlates very well with the formation of aniline. These results indicate more preference for path A. With Os2 74% conversion into aniline was observed with no azobenzene as a side product. The amount of aniline correlates very well with the consumption of nitrobenzene. The conversion into azoxybenzene was below 8% and quite constant after 24 h. This indicates the preference for pathway A.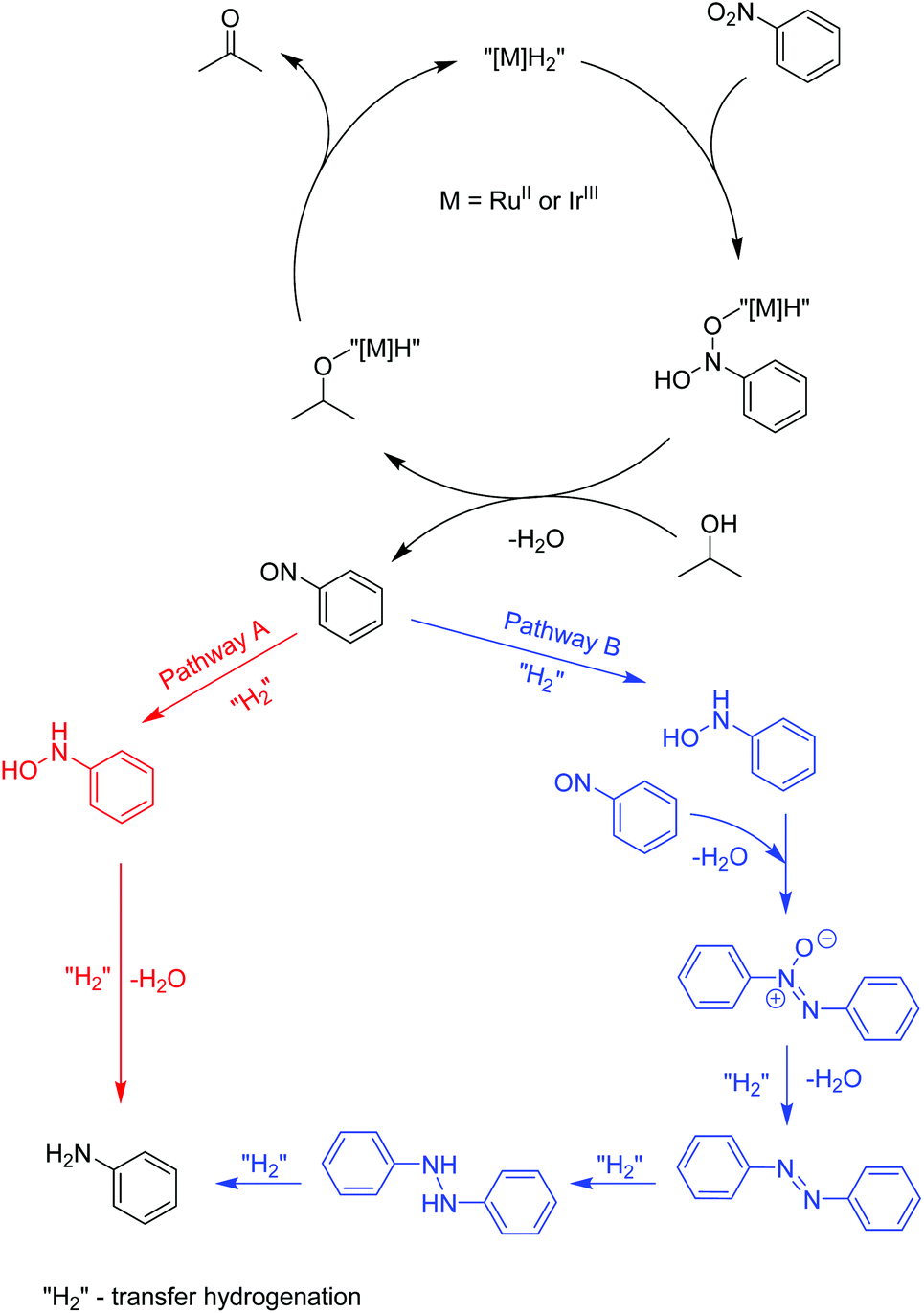 | ||
| Scheme 4 The postulated mechanism for the transfer hydrogenation of nitrobenzene.8a,19 | ||
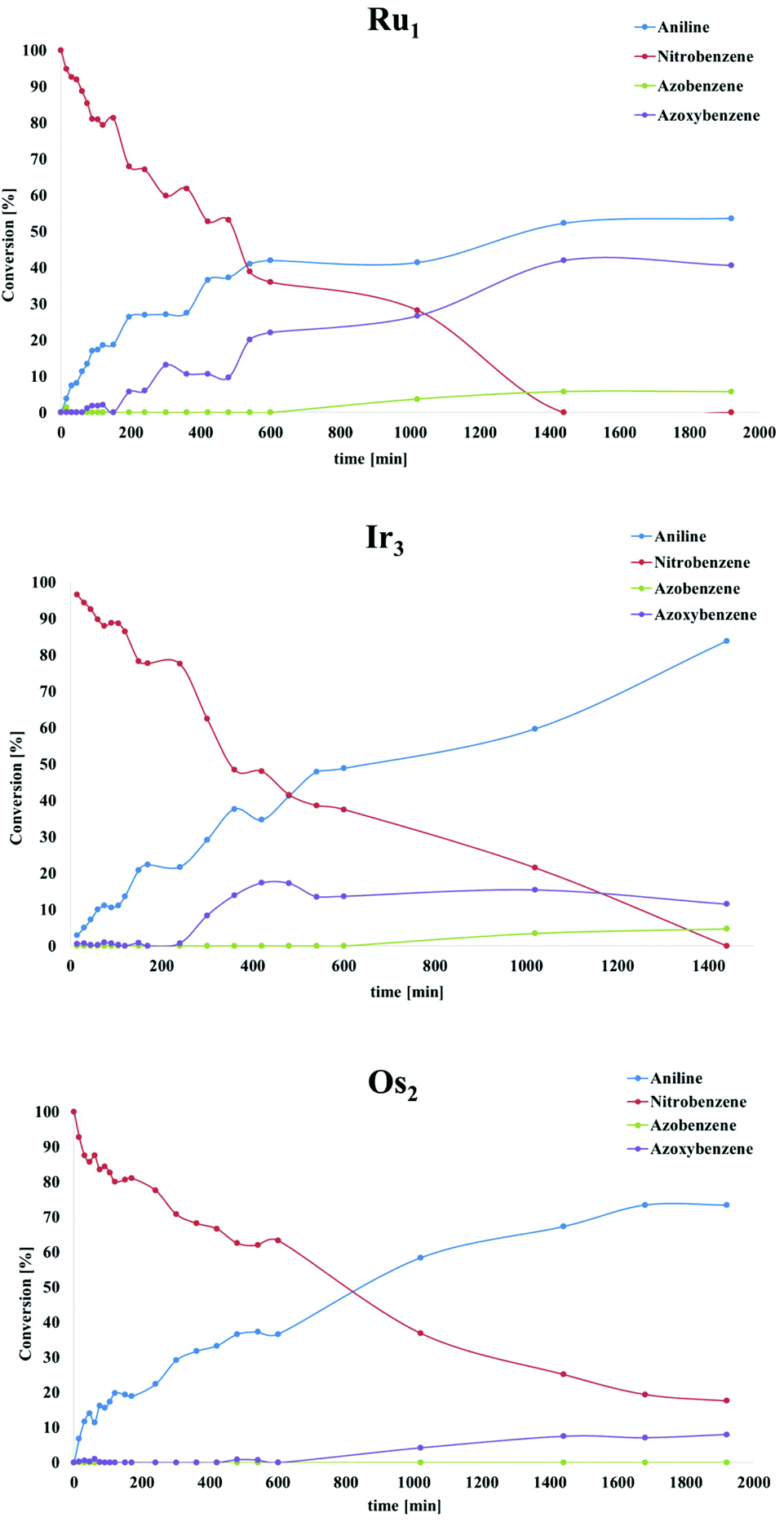 | ||
| Fig. 1 Time-dependence screening for the selected complexes Ru1, Ir3 and Os2. | ||
For the transfer hydrogenation of the nitro functional group we chose four additional substrates: 2-, 3- and 4-nitroanisole, as well as 1-chloro-2-nitrobenzene. We selected Os2 and Ir3 as the precatalysts and conducted the transfer hydrogenation with 1.5 mol% precatalyst loading. Each of the four substrates was stirred with 0.5 equiv. of KOH in iPrOH at 80 °C for 24 h (Table 12). 2-Nitroanisole was converted with both tested complexes into o-anisidine as the only product. The conversions were 65% and 54%, respectively. Both complexes catalysed the reduction of 3-nitroanisole into m-anisidine very well. Ir3 gave full conversion into the amino product. The reduction of 4-nitroanisole into p-anisidine proceeded with both complexes, however, giving lower conversions. By using the complex Ir3, 1-chloro-2-nitrobenzene was fully converted into 2-chloroaniline. In contrast, Os2 gave only 58% of the aniline derivative after 24 h. Overall, the Ir(III) complex Ir3 was more active in the transfer hydrogenation catalysis of the selected nitro substrates. Surprisingly no azo- or azoxy-side products were detected. For the reduction of cyclohexene and for the reduction of nitrobenzene, we performed the Hg-poisoning test by adding Hg during the catalysis. The addition of Hg had no effect on the yield or the rate of formation of the catalytic products. Hence, we believe that catalysis in the present case is likely homogeneous.
| Precatalyst | Aminob | Nitrob | Azob | Azoxyb |
|---|---|---|---|---|
| a General reaction conditions: nitro substrate (0.5 mmol), precatalyst (1.5 mol%), KOH (0.21 mmol), iPrOH (4 mL), 80 °C for 24 h. b Conversion in % determined by GC using hexadecane as an internal standard. | ||||
| 2-Nitroanisole | ||||
| Ir3 | 65 | 35 | 0 | 0 |
| Os2 | 54 | 46 | 0 | 0 |
| 3-Nitroanisole | ||||
| Ir3 | 100 | 0 | 0 | 0 |
| Os2 | 75 | 25 | 0 | 0 |
| 4-Nitroanisole | ||||
| Ir3 | 54 | 46 | 0 | 0 |
| Os2 | 18 | 82 | 0 | 0 |
| 1-Chloro-2-nitrobenzene | ||||
| Ir3 | 100 | 0 | 0 | 0 |
| Os2 | 58 | 42 | 0 | 0 |
Conclusions
The scope of the pyridine-functionalized triazolylidene complexes of ruthenium, iridium and osmium as precatalysts for transfer hydrogenation was investigated. We have also presented the synthesis of the 1-(2-pyrimidyl)-functionalized triazolium salt and used it as a precursor for the carbenic ligand for coordination to ruthenium(II) and osmium(II). This is the first report of a pyrimidine-appended triazolium salt and also the first examples of ruthenium and osmium complexes with such MIC ligands. All tested complexes are very efficient (pre-)catalysts for the reduction of several functional groups such as carbonyls, alkenes, imines and also nitroarenes. We demonstrated that in general the additional nitrogen atom of the pyrimidine ring is beneficial in terms of catalytic activity. The two pyrimidine-functionalized complexes have shown a better conversion in comparison to pyridine analogues, except for the reduction of CC double bond substrates. The ruthenium and osmium complexes having triazolylidene ligands substituted with an electron-donating group performed better, whereas for the iridium complexes the effects were the opposite: complexes with electron-withdrawing groups are superior catalysts. In the final part we have shown some mechanistic studies for the reduction of nitrobenzene with three selected precatalysts, one for each metal (ruthenium, iridium and osmium). The Cp*–Ir-containing pre-catalysts presented here deliver better conversions for the conversion of CO groups and comparable conversion for the reduction of CC compared to the reported Cp*–Ir complexes with NHC ligands.2l On the other hand, the Cp*–Ir-containing complexes deliver more aniline through the reduction of nitrobenzene compared to related complexes.8a These results are important to understand the behaviour of the complexes during the catalysis. The results also show the potential of the tested complexes with chelating pyridyl- and pyrimidyl-triazolylidenes in the transfer hydrogenation of several functionalities, such as the carbonyl group, CC double bond, imine and also the nitro group. The newly synthesized pyrimidyl-triazolylidenes might be useful ligands for generating both homo- and heterodinuclear complexes.
Experimental section
Materials and physical methods
The reagents and solvents were used as obtained from commercial sources (Sigma-Aldrich, Fluka, Alfa Aesar). Acetonitrile was dried over 3 Å molecular sieves, and dichloromethane over 4 Å molecular sieves. For methylation reactions dry glassware and solvents were used. Triazole 1 was prepared as described in the literature.14 NMR spectra were recorded with a Bruker Avance 300, Bruker Avance III 500 (Ljubljana) and Jeol ECS 400 (Berlin) spectrometers. Proton and carbon spectra were referenced to Si(CH3)4 as the internal standard. Some 13C chemical shifts were determined relative to the 13C signal of the solvent DMSO-d6 (39.5 ppm). 19F NMR and 31P NMR spectra were referenced to CCl3F and 85% phosphoric acid, respectively, as external standards at δ = 0. The nitrogen chemical shifts were extracted from 1H–15N HMBC spectra with 20 Hz digital resolution in the indirect dimension. 15N chemical shifts are rounded to integer numbers because of the digital resolution limits of the experiment. The reported 15N chemical shifts were extracted from 1H–15N HMBC spectra (with 20 Hz digital resolution in the indirect dimension) with respect to external 90% CH3NO2 in CDCl3 and converted to the δ15N (liq. NH3) = 0 ppm scale using the relationship: δ15N (CH3NO2) = δ15N (liq. NH3) + 380.5 ppm. Chemical shifts are given on the δ scale (ppm). Coupling constants (J) are given in hertz. The multiplicities are indicated as follows: s (singlet), d (doublet), t (triplet), q (quartet), sept (septet), m (multiplet) and br (broadened). Assignments of proton, carbon and nitrogen resonances were performed by standard 2D NMR techniques (1H–1H COSY, 1H–13C HSQC, 1H–13C HMBC, and 1H–15N HMBC). The numbering used for the assignment of NMR signals is as follows: 1,2,3-triazole ring, simple figures; pyridine ring, primed figures; phenyl ring, double-primed figures (C-1′′ is attached to the 1,2,3-triazole ring). An Agilent 6224 time-of-flight (TOF) mass spectrometer equipped with a double orthogonal electrospray source at atmospheric pressure ionization (ESI) coupled to an Agilent 1260 HLPC and an Agilent 6210 ESI-TOF spectrometer were used for recording HRMS spectra. Elemental analysis was performed with a Perkin-Elmer 2400 Series II CHNS/O Analyzer. IR spectra were obtained with a Perkin-Elmer Spectrum 100, equipped with a Specac Golden Gate Diamond ATR as a solid sample support. Melting points were determined on a Kofler micro hot stage instrument. The reactions were monitored by TLC on TLC-CARD silica gel, 220–440 mesh. 1H NMR spectra of benzyl alcohol, 1-phenylethanol, diphenylmethanol, cyclohexanol and cyclooctane were compared with those of the authentic samples (Tables 2–6). Propylbenzene20 (Table 7), 1,2-diphenylethane21 (Table 8) and N-benzylaniline22 (Table 9) were identified by comparison of their 1H NMR spectral data with the literature reports. GC-MS analysis was performed on a Varian Saturn 2100C (column: Varian factory four capillary column VF-5 ms; temperature: 50 to 250 °C, heating rate 20 K min−1). The calibration was done as follows: we collected the GC analyses of all the products, using pure samples. We obtained ret. times and also factors for calculating the areas of the GC peaks into conversions, percent yields. Ret. times (min): 6.22 (aniline), 7.36 (nitrobenzene), 11.21 (azobenzene), 12.42 (azoxybenzene); 5.63 (2-anisidine), 6.86 (2-nitroanisole); 5.29 (2-chloroaniline), 6.17 (2-chloronitrobenzene); 6.11 (3-anisidine), 6.73 (3-nitroanisole); 5.96 (4-anisidine), 7.22 (4-nitroanisole). Catalytic reactions were carried out using Schlenk tubes and a pressure-vessel. All catalytic reactions were performed at least in duplicate.Synthesis of the ligand
Pyrimidyl-triazole 1 in dry CH2Cl2 was purged with dry argon and stirred on an ice-bath at 0 °C for a few minutes. MeOTf was added via syringe and the reaction mixture was stirred for 30 min at 0 °C and then 24 h at room temperature. The reaction mixture was concentrated using a rotary evaporator and the residue was column chromatographed on silica gel using a mixture of CH2Cl2 and MeOH (10:1) as the eluent (Rf ≈ 0.3). Re-crystallization from EtOAc–light petroleum afforded analytically pure triazolium salts 2OTf. Quantities used in the above procedure, analytical and spectral data of 2OTf are given below.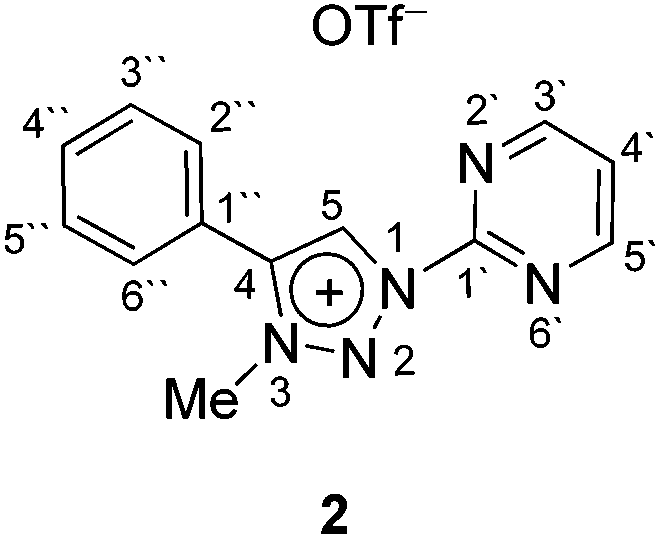 :MeOH = 10:1) = 0.3. Mp 154–156 °C. IR: 3119, 3093, 2972, 1612, 1589, 1424, 1399, 1264, 1228, 1145, 1031, 830, 772, 762, 697, 635 cm−1. 1H NMR (500 MHz, DMSO-d6): δ = 4.48 (s, 3H, CH3-trz), 7.72–7.70 (m, 3H, H-3′′, H-5′′, H-4′′), 7.87 (dd, J = 7.5, 2.0 Hz, 2H, H-2′′, H-6′′), 7.99 (t, J = 4.8 Hz, 1H, H-5′), 9.22 (d, J = 4.9 Hz, 2H, H-6′, H-4′), 10.04 (s, 1H, H-5). 13C NMR (126 MHz, DMSO-d6): δ = 39.8 (CH3-trz), 122.3 (C-1′′), 124.4 (C-5′), 127.2 (C-5), 129.4 (C-3′′, C-5′′), 129.6 (C-2′′, C-6′′), 131.7 (C-4′′), 143.5 (C-4), 152.2 (C-2′), 160.5 (C-6′, C-4′). 15N NMR (51 MHz, DMSO-d6): δ = 245 (N-3), 255 (N-1), 270 (N-1′, N-3′), 340 (N-2). 19F NMR (471 MHz, DMSO-d6): δ = −77.8 (s, 3F, OTf). HRMS (ESI+): calcd for C13H12N5+ [M]+ 238.1087, found 238.1088. Anal. calcd for C14H12F3N5O3S: C 43.41, H 3.12, N 18.08; found: C 43.12, H 3.29, N 18.51.
:MeOH = 10:1) = 0.3. Mp 154–156 °C. IR: 3119, 3093, 2972, 1612, 1589, 1424, 1399, 1264, 1228, 1145, 1031, 830, 772, 762, 697, 635 cm−1. 1H NMR (500 MHz, DMSO-d6): δ = 4.48 (s, 3H, CH3-trz), 7.72–7.70 (m, 3H, H-3′′, H-5′′, H-4′′), 7.87 (dd, J = 7.5, 2.0 Hz, 2H, H-2′′, H-6′′), 7.99 (t, J = 4.8 Hz, 1H, H-5′), 9.22 (d, J = 4.9 Hz, 2H, H-6′, H-4′), 10.04 (s, 1H, H-5). 13C NMR (126 MHz, DMSO-d6): δ = 39.8 (CH3-trz), 122.3 (C-1′′), 124.4 (C-5′), 127.2 (C-5), 129.4 (C-3′′, C-5′′), 129.6 (C-2′′, C-6′′), 131.7 (C-4′′), 143.5 (C-4), 152.2 (C-2′), 160.5 (C-6′, C-4′). 15N NMR (51 MHz, DMSO-d6): δ = 245 (N-3), 255 (N-1), 270 (N-1′, N-3′), 340 (N-2). 19F NMR (471 MHz, DMSO-d6): δ = −77.8 (s, 3F, OTf). HRMS (ESI+): calcd for C13H12N5+ [M]+ 238.1087, found 238.1088. Anal. calcd for C14H12F3N5O3S: C 43.41, H 3.12, N 18.08; found: C 43.12, H 3.29, N 18.51.
Synthesis of complexes
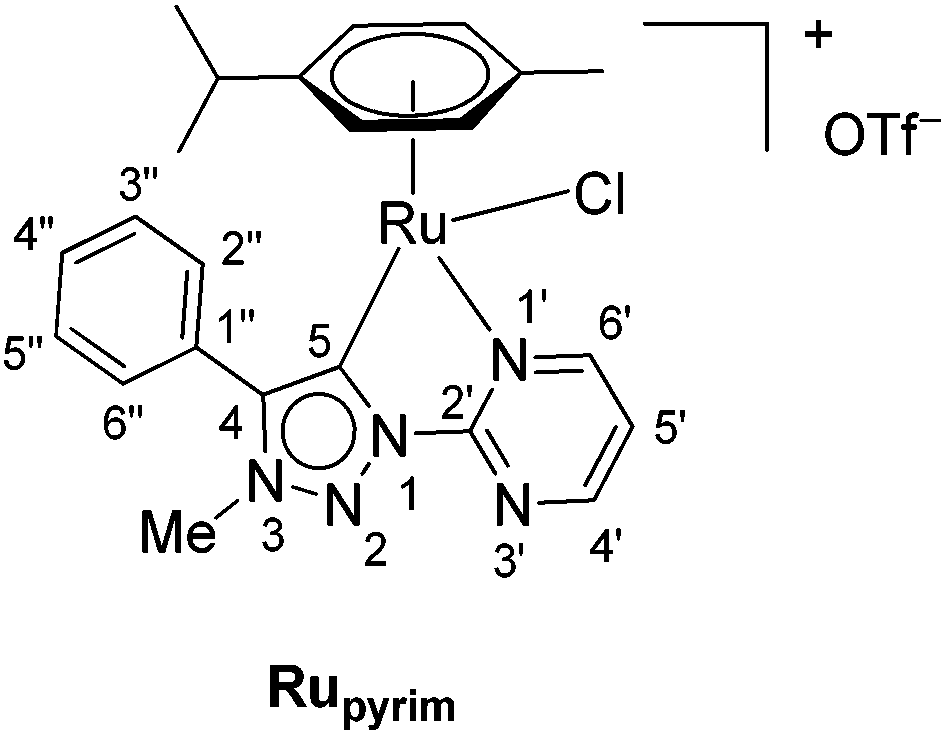 :MeOH = 10:1) = 0.8. Mp 71–73 °C. IR: 2965, 1611, 1555, 1472, 1258, 1222, 1151, 1028, 803, 775, 710, 636 cm−1. 1H NMR (500 MHz, CDCl3): δ = 0.98 (d, J = 7.0 Hz, 3H, (CH3)2CH), 1.08 (d, J = 6.9 Hz, 3H, (CH3)2CH), 2.04 (s, 3H, CH3Cym), 2.54 (septet, J = 6.9 Hz, 1H, (CH3)2CH), 4.34 (s, 3H, CH3–N-3), 5.21 (dd, J = 6.2, 0.7 Hz, 1H, ArHCym), 5.58 (dd, J = 6.2, 0.8 Hz, 1H, ArHCym), 5.67 (t, J = 6.4 Hz, 2H, ArHCym), 7.72–7.71 (m, 3H, H-3′′, H-4′′, H-5′′), 7.83–7.81 (m, 3H, H-5′, H-2′′, H-6′′), 8.96 (dd, J = 4.8, 2.0 Hz, 1H, H-4′ or H-6′), 9.84 (dd, J = 5.7, 2.0 Hz, 1H, H-6′ or H-4′). 13C NMR (126 MHz, CDCl3): δ = 18.9 (CH3Cym), 21.7 ((CH3)2CH), 22.7 ((CH3)2CH), 31.2 ((CH3)2CH), 38.7 (CH3–N-3), 83.5 (ArCym), 85.7 (ArCym), 88.3 (ArCym), 89.9 (ArCym), 104.2 (ArCym), 110.4 (ArCym), 123.3 (C-5′), 125.9 (C-1′′), 129.8 (C-3′′, C-5′′), 130.5 (C-2′′, C-6′′), 131.6 (C-4′′), 147.7 (C-4), 155.0 (C-2′), 160.0 (C-4′ or C-6′), 166.5 (C-6′ or C-4′), 170.7 (C-5). 15N NMR (51 MHz, DMSO-d6): δ = 218 (N-1′ or N-3′), 242 (N-3), 268 (N-3′ or N-1′), 339 (N-2). 19F NMR (471 MHz, DMSO-d6): δ = −78.3 (s, 3F, OTf). HRMS (ESI+): calcd for C23H25ClN5Ru+ [M]+ = 508.0842, found 508.0834.
:MeOH = 10:1) = 0.8. Mp 71–73 °C. IR: 2965, 1611, 1555, 1472, 1258, 1222, 1151, 1028, 803, 775, 710, 636 cm−1. 1H NMR (500 MHz, CDCl3): δ = 0.98 (d, J = 7.0 Hz, 3H, (CH3)2CH), 1.08 (d, J = 6.9 Hz, 3H, (CH3)2CH), 2.04 (s, 3H, CH3Cym), 2.54 (septet, J = 6.9 Hz, 1H, (CH3)2CH), 4.34 (s, 3H, CH3–N-3), 5.21 (dd, J = 6.2, 0.7 Hz, 1H, ArHCym), 5.58 (dd, J = 6.2, 0.8 Hz, 1H, ArHCym), 5.67 (t, J = 6.4 Hz, 2H, ArHCym), 7.72–7.71 (m, 3H, H-3′′, H-4′′, H-5′′), 7.83–7.81 (m, 3H, H-5′, H-2′′, H-6′′), 8.96 (dd, J = 4.8, 2.0 Hz, 1H, H-4′ or H-6′), 9.84 (dd, J = 5.7, 2.0 Hz, 1H, H-6′ or H-4′). 13C NMR (126 MHz, CDCl3): δ = 18.9 (CH3Cym), 21.7 ((CH3)2CH), 22.7 ((CH3)2CH), 31.2 ((CH3)2CH), 38.7 (CH3–N-3), 83.5 (ArCym), 85.7 (ArCym), 88.3 (ArCym), 89.9 (ArCym), 104.2 (ArCym), 110.4 (ArCym), 123.3 (C-5′), 125.9 (C-1′′), 129.8 (C-3′′, C-5′′), 130.5 (C-2′′, C-6′′), 131.6 (C-4′′), 147.7 (C-4), 155.0 (C-2′), 160.0 (C-4′ or C-6′), 166.5 (C-6′ or C-4′), 170.7 (C-5). 15N NMR (51 MHz, DMSO-d6): δ = 218 (N-1′ or N-3′), 242 (N-3), 268 (N-3′ or N-1′), 339 (N-2). 19F NMR (471 MHz, DMSO-d6): δ = −78.3 (s, 3F, OTf). HRMS (ESI+): calcd for C23H25ClN5Ru+ [M]+ = 508.0842, found 508.0834.
General procedure for the catalysed transfer hydrogenation
Acknowledgements
The financial support from the Ministry of Education, Science and Sport, Republic of Slovenia, the Slovenian Research Agency (Grant P1-0230; Young Researcher Grant to A. B.), and the Slovene Human Resources Development and Scholarship Found, Public Call for Scholarships or Grants for the Research Cooperation of Doctoral Students Abroad in 2012, No.: 11012-16/2013 is acknowledged. We are grateful to the Fonds der Chemischen Industrie (FCI) and the Freie Universität Berlin is also kindly acknowledged for financial support.Notes and references
- For selected examples see: (a) R. Noyori, M. Yamakawa and S. Hashiguchi, J. Org. Chem., 2001, 66, 7931 CrossRef CAS PubMed; (b) G. Zassinovich, G. Mestroni and S. Gladiali, Chem. Rev., 1992, 92, 1051 CrossRef CAS; (c) J. S. M. Samec, J. E. Bäckvall, P. G. Andersson and P. Brandt, Chem. Soc. Rev., 2006, 35, 237 RSC; (d) T. Ikariya, K. Murata and R. Noyori, Org. Biomol. Chem., 2006, 4, 393 RSC; (e) P. Braunstein and F. Naud, Angew. Chem., Int. Ed., 2001, 40, 680 CrossRef CAS; (f) J. E. Bäckvall, J. Organomet. Chem., 2002, 652, 105 CrossRef; (g) W. Baratta, F. Benedetti, A. Del Zotto, L. Fanfoni, F. Feluga, S. Magnolia, E. Putihano and P. Rigo, Organometallics, 2010, 29, 3563 CrossRef CAS; (h) W. Baratta, G. Bossi, E. Putignano and P. Rigo, Chem. – Eur. J., 2011, 17, 3474 CrossRef CAS PubMed; (i) Y. Jiang, Q. Jiang and X. Zhang, J. Am. Chem. Soc., 1998, 120, 3817 CrossRef CAS; (j) S. E. Clapham, A. Hadzovic and R. H. Morris, Coord. Chem. Rev., 2004, 248, 2201 CrossRef CAS; (k) L. T. Ghoochany, S. Farsadpour, Y. Sun and W. R. Thiel, Eur. J. Inorg. Chem., 2011, 3431 CrossRef; (l) C. P. Casey, S. W. Singer, D. R. Powell, R. K. Hayashi and M. Kavana, J. Am. Chem. Soc., 2001, 123, 1090 CrossRef CAS PubMed; (m) D. Gnanamgari, E. L. O. Sauer, N. D. Schley, C. Butler, C. D. Incarvito and R. H. Crabtree, Organometallics, 2009, 28, 321 CrossRef CAS; (n) N. Gürbüz, E. O. Özcan, I. Özdemir, B. Cetinkaya, O. Sahin and O. Büyükgüngör, Dalton Trans., 2012, 41, 2330 RSC; (o) H. Türkmen, T. Pape, F. E. Hahn and B. Cetinkaya, Organometallics, 2008, 27, 571 CrossRef; (p) F. E. Hahn, C. Holtgrewe, T. Pape, M. Martin, E. Sola and L. A. Oro, Organometallics, 2005, 24, 2203 CrossRef CAS; (q) S. Burling, M. K. Whittlesey and J. M. J. Williams, Adv. Synth. Catal., 2005, 347, 591 CrossRef CAS; (r) P. L. Chiu and H. M. Lee, Organometallics, 2005, 24, 1692 CrossRef CAS.
- For selected reviews and examples see: (a) W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290 CrossRef CAS; (b) M. Melaimi, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 8810 CrossRef CAS PubMed; (c) M. C. Jahnke and F. E. Hahn, Top. Organomet. Chem., 2010, 30, 95 CrossRef CAS; (d) P. de Frémont, N. Marion and S. P. Nolan, Coord. Chem. Rev., 2009, 253, 862 CrossRef; (e) C. C. Loh and D. Enders, Chem. – Eur. J., 2012, 18, 10212 CrossRef CAS PubMed; (f) N. Marion, S. Díez-González and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2988 CrossRef CAS PubMed; (g) V. Cesar, S. B. Laponnaz and L. H. Gade, Chem. Soc. Rev., 2004, 33, 619 RSC; (h) F. A. Glorius, Top. Organomet. Chem., 2007, 21, 1 CrossRef CAS; (i) E. A. B. Kantchev, C. J. O'Brien and M. G. Organ, Angew. Chem., Int. Ed., 2007, 46, 2768 CrossRef CAS PubMed; (j) A. Kumar and P. Ghosh, Eur. J. Inorg. Chem., 2012, 3955 CrossRef CAS; (k) J. D. Egbert, C. S. J. Cazin and S. P. Nolan, Catal. Sci. Technol., 2013, 3, 912 RSC; (l) U. Hintermair, J. Campos, T. P. Brewster, L. M. Pratt, N. D. Schley and R. H. Crabtree, ACS Catal., 2014, 4, 99 CrossRef CAS.
- (a) K. F. Donnelly, A. Petronilho and M. Albrecht, Chem. Commun., 2013, 49, 1145 RSC; (b) R. H. Crabtree, Coord. Chem. Rev., 2013, 257, 755 CrossRef CAS; (c) J. M. Aizpurua, R. M. Fratila, Z. Monasterio, N. Perez-Esnaola, E. Andreieff, A. Irastorza and M. Sagartzazu-Aizpurua, New J. Chem., 2014, 38, 474 RSC; (d) J. D. Crowley, A. Lee and K. J. Kilpin, Aust. J. Chem., 2011, 64, 1118 CrossRef CAS; (e) G. Guisado-Barrios, J. Bouffard, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 4759 CrossRef CAS PubMed; (f) D. Schweinfurth, N. Deibel, F. Weisser and B. Sarkar, Nachrichten aus der Chemie, 2011, 59, 937 CrossRef CAS; (g) P. Mathew, A. Neels and M. Albrecht, J. Am. Chem. Soc., 2008, 130, 13534 CrossRef CAS PubMed.
- (a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS; (b) C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef CAS PubMed; (c) J. D. Crowley and D. McMorran, in Topics in Heterocyclic Chemistry, ed. J. Košmrlj, Springer, Berlin/Heidelberg, 2012, vol. 22, p. 31 Search PubMed; (d) B. Schulze and U. S. Schubert, Chem. Soc. Rev., 2014, 43, 2522 RSC; (e) Y. Hua and A. H. Flood, Chem. Soc. Rev., 2010, 39, 1262 RSC; (f) Y. H. Lau, P. J. Rutledge, M. Watkinson and M. H. Todd, Chem. Soc. Rev., 2011, 40, 2848 RSC.
- For a recent example from our laboratory, see: M. Gazvoda, M. Virant, A. Pevec, D. Urankar, A. Bolje, M. Kočevar and J. Košmrlj, Chem. Commun., 2016, 52, 1571 RSC.
- B. K. Keitz, J. Bouffard, G. Bertrand and R. H. Grubbs, J. Am. Chem. Soc., 2011, 133, 8498 CrossRef CAS PubMed.
- For selected examples see: (a) R. Lalrempuia, N. D. McDaniel, H. Müller-Bunz, S. Bernhard and M. Albrecht, Angew. Chem., Int. Ed., 2010, 49, 9765 CrossRef CAS PubMed; (b) D. Canesco-Gonzalez and M. Albrecht, Dalton Trans., 2013, 42, 7424 RSC; (c) A. Prades, E. Peris and M. Albrecht, Organometallics, 2011, 30, 1162 CrossRef CAS; (d) A. Bolje, S. Hohloch, D. Urankar, A. Pevec, M. Gazvoda, B. Sarkar and J. Košmrlj, Organometallics, 2014, 33, 2588 CrossRef CAS; (e) S. Hohloch, L. Hettmanczyk and B. Sarkar, Eur. J. Inorg. Chem., 2014, 3164 CrossRef CAS; (f) S. Hohloch, S. Kaiser, F. L. Dücker, A. Bolje, R. Maity, J. Košmrlj and B. Sarkar, Dalton Trans., 2015, 44, 686 RSC; (g) R. Maity, T. Tichter, M. van der Meer and B. Sarkar, Dalton Trans., 2015, 44, 18311 RSC.
- For selected examples see: (a) S. Hohloch, L. Suntrup and B. Sarkar, Organometallics, 2013, 32, 7376 CrossRef CAS; (b) M. Delgado-Robollo, D. Canseco-Gonzalez, M. Hollering, H. Müller-Bunz and M. Albrecht, Dalton Trans., 2014, 43, 4462 RSC; (c) R. Maity, S. Hohloch, C.-Y. Su, M. van der Meer and B. Sarkar, Chem. – Eur. J., 2014, 20, 9952 CrossRef CAS PubMed; (d) R. Maity, M. Van der Meer, S. Hohloch and B. Sarkar, Organometallics, 2015, 34, 3090 CrossRef CAS; (e) R. Maity, A. Mekic, M. van der Meer, A. Verma and B. Sarkar, Chem. Commun., 2015, 51, 15106 RSC; (f) M. van der Meer, E. Glais, I. Siewert and B. Sarkar, Angew. Chem., Int. Ed., 2015, 54, 13792 CrossRef CAS PubMed; (g) S. Hohloch, F. L. Duecker, M. van der Meer and B. Sarkar, Molecules, 2015, 20, 7379 CrossRef CAS PubMed.
- (a) T. Nakamura, T. Terashima, K. Ogata and S. Fukuzawa, Org. Lett., 2011, 13, 620 CrossRef CAS PubMed; (b) S. Hohloch, C.-Y. Su and B. Sarkar, Eur. J. Inorg. Chem., 2011, 3067 CrossRef CAS; (c) S. Hohloch, B. Sarkar, L. Nauton, F. Cisnetti and A. Gautier, Tetrahedron Lett., 2013, 54, 1808 CrossRef CAS; (d) S. Hohloch, D. Scheiffele and B. Sarkar, Eur. J. Inorg. Chem., 2013, 3956 CrossRef CAS; (e) S. Hohloch, L. Suntrup and B. Sarkar, Inorg. Chem. Front., 2016, 3, 67 RSC.
- For selected examples see: (a) R. Saravanakumar, V. Ramkumar and S. Sankararaman, Organometallics, 2011, 30, 1689 CrossRef CAS; (b) D. Canseco-Gonzalez, A. Gniewek, M. Szulmanowicz, H. Müller-Bunz, A. M. Trzeciak and M. Albrecht, Chem. – Eur. J., 2012, 18, 6055 CrossRef CAS PubMed; (c) T. Terashima, S. Inomata, K. Ogata and S. Fukuzawa, Eur. J. Inorg. Chem., 2012, 1387 CrossRef CAS; (d) S. Hohloch, W. Frey, C.-Y. Su and B. Sarkar, Dalton Trans., 2013, 42, 11355 RSC; (e) E. C. Keske, O. V. Zenkina, R. Wang and C. M. Crudden, Organometallics, 2012, 31, 456 CrossRef CAS; (f) J. Huang, J.-T. Hong and S. H. Hong, Eur. J. Org. Chem., 2012, 6630 CAS; (g) R. Maity, A. Verma, M. van der Meer, S. Hohloch and B. Sarkar, Eur. J. Inorg. Chem., 2016, 111 CrossRef CAS; (h) D. Mendoza-Espinosa, R. Gonzalez-Olvera, C. Osornio, G. E. Negron-Silva, A. Alvarez-Hernandez, C. I. Bautista-Hernandez and O. R. Suarez-Castillo, J. Organomet. Chem., 2016, 803, 142 CrossRef CAS; (i) J. R. Wright, P. C. Young, N. T. Lukas, A.-L. Lee and J. D. Crowley, Organometallics, 2013, 32, 7065 CrossRef CAS PubMed.
- A. Bolje, S. Hohloch, M. van der Meer, J. Košmrlj and B. Sarkar, Chem. – Eur. J., 2015, 21, 6756 CrossRef CAS PubMed.
- R. Lalrempuia, N. D. McDaniel, H. Mìller-Bunz, S. Bernhard and M. Albrecht, Angew. Chem., Int. Ed., 2010, 49, 9765 ( Angew. Chem. , 2010 , 122 , 9959 ) CrossRef CAS PubMed.
- A. Bolje and J. Košmrlj, Org. Lett., 2013, 15, 5084 CrossRef CAS PubMed.
- A. Bolje, D. Urankar and J. Košmrlj, Eur. J. Org. Chem., 2014, 8167 CrossRef CAS.
- J. C. Garrison and W. J. Youngs, Chem. Rev., 2005, 105, 3978 CrossRef CAS PubMed.
- For selected examples see: (a) N. Gürbüz, E. O. Özcan, I. Özdemir, B. Cetinkaya, O. Sahin and O. Büyükgüngör, Dalton Trans., 2012, 41, 2330 RSC; (b) H. Türkmen, T. Pape, F. E. Hahn and B. Cetinkaya, Organometallics, 2008, 27, 571 CrossRef; (c) F. E. Hahn, C. Holtgrewe, T. Pape, M. Martin, E. Sola and L. A. Oro, Organometallics, 2005, 24, 2203 CrossRef CAS; (d) S. Burling, M. K. Whittlesey and J. M. J. Williams, Adv. Synth. Catal., 2005, 347, 591 CrossRef CAS; (e) P. L. Chiu and H. M. Lee, Organometallics, 2005, 24, 1692 CrossRef CAS.
- S. Horn and M. Albrecht, Chem. Commun., 2011, 47, 8802 RSC.
- For selected examples see: (a) G.-Z. Wang and J.-E. Bäckvall, J. Chem. Soc., Chem. Commun., 1992, 980 RSC; (b) J. S. Samec and J.-E. Bäckvall, Chem. – Eur. J., 2002, 8, 2955 CrossRef CAS PubMed.
- (a) R. V. Jagadeesh, G. Wienhöfer, F. A. Westerhaus, A.-E. Surkus, H. Junge, K. Junge and M. Beller, Chem. – Eur. J., 2011, 17, 14375 CrossRef CAS PubMed; (b) S. P. Annen and H. Grützmacher, Dalton Trans., 2012, 41, 14137 RSC.
- J. J. Eisch and S. Dutta, Organometallics, 2005, 24, 3355 CrossRef CAS.
- P. J. Black, M. G. Edwards and J. M. J. Williams, Eur. J. Org. Chem., 2006, 4367 CrossRef CAS.
- D. B. Bagal, R. A. Watile, M. V. Khedkar, K. P. Dhake and B. M. Bhanage, Catal. Sci. Technol., 2012, 2, 354 CAS.
Footnotes |
| † Dedicated to Prof. Bernhard Lippert on the occasion of his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c6dt01324d |
| This journal is © The Royal Society of Chemistry 2016 |