
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
s-Block amidoboranes: syntheses, structures, reactivity and applications
Tom E.
Stennett
and
Sjoerd
Harder
*
Friedrich-Alexander-Universität Erlangen-Nürnberg, Inorganic and Organometallic Chemistry, Egerlandstraße 1, 91058 Erlangen, Germany. E-mail: sjoerd.harder@fau.de
First published on 30th November 2015
Abstract
Metal amidoborane compounds of the alkali- and alkaline earth metals have in recent years found applications in diverse disciplines, notably as hydrogen storage materials, as reagents for the reduction of organic functional groups and as catalysts and intermediates in dehydrocoupling reactions. These functions are connected by the organometallic chemistry of the MNR2BH3 group.† This review focusses on central aspects of the s-block amidoborane compounds – their syntheses, structures and reactivity. Well-defined amidoborane complexes of group 2 metals are now available by a variety of solution-phase routes, which has allowed a more detailed analysis of this functional group, which was previously largely confined to solid-state materials chemistry. Structures obtained from X-ray crystallography have begun to provide increased understanding of the fundamental steps of key processes, including amine–borane dehydrocoupling and hydrogen release from primary and secondary amidoboranes. We review structural parameters and reactivity to rationalise the effects of the metal, nitrogen substituents and supporting ligands on catalytic performance and dehydrogenative decomposition routes. Mechanistic features of key processes involving amidoborane compounds as starting materials or intermediates are discussed, alongside emerging applications such as the use of group 1 metal amidoboranes in synthesis. Finally, the future prospects of this vibrant branch of main group chemistry are evaluated.
 Tom E. Stennett | Tom Stennett was born in Preston, England in 1986. He received his master's degree from the University of Durham in 2008 after a project in the lab of Prof. Jörg Sundermeyer at the Phillips-Universität Marburg. He then moved to the University of Bristol to undertake his PhD studies under the supervision of Prof. Duncan Wass on chromium-based catalysts for selective ethylene oligomerisation. After the award of his doctorate in 2013, he had a brief stay with Prof. Paul Pringle in Bristol before moving to the University of Erlangen-Nürnberg, where he is working as a postdoctoral researcher with Prof. Sjoerd Harder on early main-group organometallic chemistry. |
 Sjoerd Harder | Sjoerd Harder, born in The Netherlands, obtained his PhD with Lambert Brandsma (1990, Utrecht) and was awarded the H. J. Backer prize in organic chemistry for his thesis. After post-doctoral stays with Paul Rague von Schleyer (1991, Erlangen), Andrew Streitwieser (1992, Berkeley) and Hans Brintzinger (1993, Konstanz) he received his habilitation in 1998. In 2004 he became a professor of Inorganic Chemistry at the University of Duisburg-Essen and in 2010 he accepted the chair of Molecular Inorganic Chemistry at the University of Groningen. Currently he holds the chair of Organometallic and Inorganic Chemistry at the University of Erlangen-Nürnberg. |
1 Introduction
Although the current interest in hydrogen storage materials has propelled amidoborane compounds to the forefront of research, the story of metal amidoboranes has humble beginnings. In 1938, Schlesinger and Burg were researching the then elusive structure of the “diammoniate of diborane”, B2H6·2NH3.1 As part of their extensive investigations, they found that the reaction of sodium metal with BH3·OMe2 in liquid ammonia produced hydrogen and a compound with an elemental analysis consistent with NaNH2BH3. This was taken as evidence for the structure [NH4+][BH3NH2BH3−] for the diammoniate, on the basis that the presence of sodium inhibits the formation of ammonium ions, and thus the diammoniate does not form under these conditions. This turned out to be an erroneous conclusion: after the discovery of the borohydride anion by the same group,2 Shore and Parry later confirmed the formulation of the diammoniate as [NH3BH2NH3+][BH4−]3 and a crystal structure was reported recently.4 In spite of this, the novel NH2BH3− anion represented an interesting development, and the existence of a BH3NH2BH3− anion was eventually confirmed by the recent report of the thermal rearrangement of NaNH2BH3 to Na(BH3NH2BH3) in refluxing THF.5 Nevertheless, it was over two decades after Schlesinger and Burg's initial discovery before the first amidoborane compounds were truly isolated and characterised. First Aftandilian et al., then Gilje and Ronan, reported syntheses of NaNMe2BH3 by reaction of dimethylamine borane with NaH and Na metal, respectively.6,7 This compound was investigated sporadically over the ensuing years in the broader context of fundamental boron–nitrogen chemistry.8,9The first significant practical application of metal amidoborane compounds came in 1984, when Hutchins et al. reported the use of the sodium amidoborane compounds, NaN(H)tBuBH3 and NaNMe2BH3, as reducing agents for a variety of functional groups, including aldehydes, esters, ketones and certain amides.10 Although the sodium derivatives never found widespread use, the extension of this work to lithium amidoboranes by the group of Singaram11–14 and others15,16 established this class of compound as a useful alternative to classical reducing agents. These so-called ‘LAB reagents’ are more powerful reducing agents than borohydrides, safer and easier to handle than the ubiquitous LiAlH4, and essentially infinitely variable by modification of the nitrogen substituents.17,18 In some cases, unique chemistry can also be accessed by transfer of the amino group of the amidoborane to the substrate, for example the amination of aryl halides.19,20
The resurgent interest in metal amidoboranes of the last decade is largely due to the recognition of the potential of the parent compounds, M(NH2BH3)x, in hydrogen storage. The finite nature of the fossil fuels that are currently the source of the vast majority of the world's energy supplies, and the pollution and associated global warming that result from their combustion, implore us to discover alternatives. A vast amount of research effort is therefore being directed towards the possibility of transition to a hydrogen-based economy.21 Although in terms of production and consumption hydrogen could, at least in principle, be a renewable and pollution-free energy source (the only combustion product being water), there are currently significant obstacles to its storage and transportation.22 The U.S. Department of Energy currently has targets set for 2020 for on-board hydrogen storage/release systems for automobiles that require a gravimetric H2 density of 55 g kg−1 and a volumetric density of 40 g L−1 for the whole system.23 Compressed hydrogen gas requires a pressure of 700 bar to attain such parameters at ambient temperature, before one has even considered the dimensions of the special containers and pressure valves required for its safe handling. Liquid H2 (boiling point: −252.9 °C) has a somewhat higher volumetric density (70 g L−1), but comes with similar problems associated with storage at such low temperature. In addition, both of these solutions come with a large energy penalty for pressurizing or liquefaction, respectively.
Storing hydrogen is especially a volumetric problem and therefore the only realistic approach is chemical storage in a much denser solid or fluid that can release H2 ‘on demand’. Probably the most studied compound in this regard is ammonia-borane (AB, NH3BH3). AB contains 19.6% hydrogen by weight, is a stable, non-flammable solid under standard conditions that does not react with water, and releases hydrogen either thermally or in the presence of a catalyst.22,24,25 While unparalleled in terms of hydrogen content, AB suffers from numerous problems as a storage material. These include slow kinetics, resulting in a higher-than-ideal H2-release temperature (>100 °C), concurrent release of the rather stable borazine as by-product, which acts as a fuel-cell poison, and foaming during hydrogen release.26 Furthermore, the dehydrogenation reaction is highly exothermic, making the prospect of reversibility unlikely.
In 2007, Burrell and co-workers discovered that some of these problems could be overcome, simply by replacing one of the N–H protons with a metal cation.27 They reported that Ca(NH2BH3)2 releases 3.6 equivalents of H2 (7.2 wt%) within the range of 120 °C and 170 °C, with no induction period and no foaming. Nevertheless, traces of ammonia and borazine were still detected. Shortly afterwards, the group of Chen reported further improved properties by using alkali metals. LiNH2BH3 and NaNH2BH3 were both found to release two equivalents of H2, representing 10.9 and 7.5 wt% of the materials, at the considerably lower temperatures of 92 °C and 89 °C, respectively.28 Moreover, there was no evidence of borazine release, and no foaming was observed during the decomposition. Differential scanning calorimetry experiments on the materials also indicated that hydrogen release from these compounds is considerably less exothermic than for AB itself, potentially making fuel regeneration more feasible. This decomposition chemistry was rapidly extended to amidoborane compounds of other s-block metals.29,30 Unfortunately, in all cases, the products of amidoborane thermal decomposition are amorphous solids, and characterisation has generally been limited to elemental analysis and solid-state NMR spectroscopy. Since this breakthrough, efforts within the materials chemistry community have focussed on developing these compounds to improve the available hydrogen content, release temperature and regeneration properties.31–39 From an organometallic chemistry perspective, an interest has also developed in amidoborane moieties as ligands. Solution-phase NMR spectroscopy and single crystal X-ray diffraction techniques have allowed further insight into their unique reactivity and provided mechanistic clues about solid-state processes. The advent of catalytic amine borane dehydrocoupling as a route to new B–N oligo- and polymers has only increased the need for improved understanding of the fundamental steps involved.40–45 The extension of this transition metal dominated chemistry to cheaper, less toxic metals is beginning to be realised.
2 Scope
The aim of this review is to summarise the chemistry of s-block metal amidoborane compounds from the point of view of the main-group organometallic chemist. This enables us to discuss this old but highly relevant class of compound from a perspective in which synthetic methods, detailed structures, reactivity and mechanisms are important. We have chosen to focus on well-defined, soluble species, and predominantly those that have been crystallographically characterised. Due to the wide applicability of metal amidoborane complexes, aspects of their chemistry have previously been covered in review articles from the perspectives of hydrogen storage materials,31,46,47 the catalytic dehydrogenation of amine–boranes48–50 and the reduction of unsaturated organic functional groups.17,18 However, these existing articles either touch only briefly on the s-block amidoboranes in the context of a broader topic, or focus on the applications of the compounds in materials chemistry or organic synthesis. Our emphasis here is on the synthesis, structures and reactivity of s-block metal amidoborane complexes, although applications of the compounds in H2 storage, catalysis and reduction chemistry will be discussed for context where appropriate.3 Bonding in early main-group metal amidoboranes
The bonding situation in amidoborane compounds is interesting, due to the plethora of intra- and intermolecular interactions that can occur. The bonding between the metal cation and the amidoborane anion takes place primarily via interaction of the amide nitrogen with the metal centre. A further contact is typically present between the borohydride moiety and the metal. In the parent amidoborane compounds, Mn+(NH2BH3)n, these two interactions lead to extended polymeric networks in the solid state, while further N–Hδ+⋯Hδ−–B, and even homopolar B–Hδ−⋯Hδ−–B and N–Hδ+⋯Hδ+–N dihydrogen bonding interactions contribute to the stability of the structure, as well as influencing the propensity for H2 release.51–53 In compounds containing larger supporting ligands, intermolecular interactions between amidoborane units are inhibited, and both terminal and bridging amidoborane ligands are possible. Terminal amidoborane ligands display M⋯H–B interactions, which display notable similarities to β-agostic M⋯H–C interactions in metal ethyl complexes, and are believed to play a similar role in β-hydride elimination processes.54,55The bonding of borohydride compounds to transition metals is predominantly a donation from the σ(B–H) orbital to the metal centre, forming a three-centre, two-electron bond.56–58 For the s-block metals, metal–ligand bonding is primarily electrostatic, and any covalent contribution is likely to be minor.59 Mulliken population analysis performed by Wu et al. on LiNH2BH3 and Ca(NH2BH3)2 supported the assignment as largely ionic compounds;60 charges of +0.98 for Li and +1.67 for Ca are close to the formula valences, while in each case the nitrogen atom bears a charge of roughly −1. The slightly negative charges on boron (−0.33 and −0.28, respectively) and the B-bound hydrogens (between −0.09 and −0.17) suggest a weaker, more diffuse interaction between the metal and this group. The labile nature of the M⋯H–B bonding is supported by the observation of a single resonance in the solution phase 1H{11B} NMR spectrum for the BH3 group, for compounds where this data is available. The 11B chemical shifts of an amine–borane and its s-block metal salts are also typically remarkably similar. The 11B–1H J-coupling constant can, however, provide spectroscopic evidence for amidoborane formation, as it typically becomes smaller upon deprotonation at nitrogen (e.g.1JBH(HNMe2BH3) = 96 Hz,611JBH(LiNMe2BH3) = 86 Hz).53 Although the BH3⋯M interaction has yet to be comprehensively investigated for s-block amidoborane compounds, computational studies have shown the related borohydrides, LiBH4 and Ca(BH4)2, to display exclusively ionic interactions between the cations and anions.62,63 The subsequently discussed compounds display a range of BH3⋯M bonding motifs, including the metal being ligated by a single hydride of a BH3 group, by two geminal hydrides (bifurcating), and also the bridging of a hydride between two metal ions. Rather than being determined by orbital interactions, the choice of bonding mode is thus more heavily dependent on steric and conformational factors.
4 Group 1 metals
4.1 Group 1 amidoboranes in hydrogen storage
Despite the prominence of the group 1 metal amidoboranes in materials chemistry and synthesis, there are only a relatively small number of structurally characterised examples. The parent amidoborane complexes, MNH2BH3 (M = Li (1), Na (2), K (3)), are well defined, with structures determined by single crystal and/or powder X-ray diffraction. All of the structures consist of polymeric networks held together by intermolecular interactions. The lithium and sodium compounds are structurally very similar,28,64 with the alkali metal cation in a pseudo-tetrahedral environment, coordinated by one nitrogen atom and three borohydride moieties (Fig. 1). This similarity is presumably partly responsible for the compounds' practically identical decomposition patterns. Conversely, the larger cation in KNH2BH3 is octahedrally coordinated by three nitrogen atoms and three borohydride moieties.30 Despite the obvious size differences between the cations, the structure of the amidoborane fragment remains relatively constant, with all of the B–N distances falling between 1.53 Å and 1.56 Å. As well as conventional B–Hδ−⋯Hδ+–N heteropolar dihydrogen bonds,65 the Li and Na compounds display counterintuitive, homopolar B–Hδ−⋯Hδ−–B and N–Hδ+⋯Hδ+–N dihydrogen bonding interactions.66,67 Although the observation of these latter homopolar interactions may be quite unexpected, there is a notable similarity to the C–H⋯H–C interactions that are becoming increasingly recognised.68–70 Homopolar B–Hδ−⋯Hδ−–B interactions are dominated by van der Waals attraction, but AIM analysis shows that they can be quite significant. For example, calculations on LiNH2BH3 show a bond critical point between hydride atoms. The accumulation of electron density at this point is comparable to that found for Li+⋯Hδ−–B bonding, and nearly twice as high as that found for more conventional N–Hδ+⋯Hδ−–B interactions. Also noteworthy is the observation that LiND2BH3 eliminates some H2 as well as HD upon heating, indicating that such homopolar interactions may play a role in hydrogen release.52 Potassium amidoborane is unique in that it melts before eliminating hydrogen, but the decomposition temperature is nevertheless remarkably similar to that of the Li and Na salts (80 °C).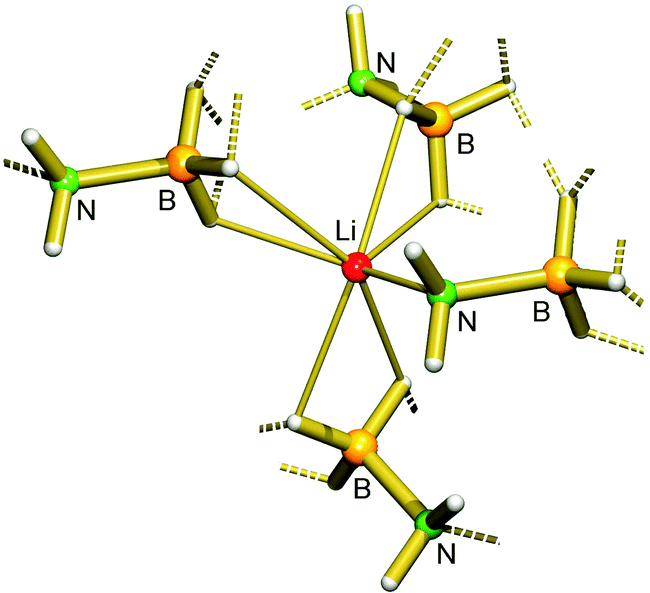 | ||
| Fig. 1 Portion of the crystal structure of LiNH2BH3 (1), displaying the pseudo-tetrahedral coordination environment at Li. | ||
Treatment of compound 1 with ammonia results in formation of the monoammoniate, LiNH2BH3·NH3, which releases up to 11 wt% hydrogen in the remarkably low temperature range of 40–55 °C.33,71 The dehydrogenated material can also be partially regenerated by treatment with hydrazine.72 Unfortunately, facile absorption/desorption of ammonia rules out the use of this compound in fuel cells, and as yet no structure has been reported. The AB adduct of 1, LiNH2BH3·NH3BH3, was found by Chen and co-workers to release 14 wt% of H2, with peak release temperatures of 80 °C and 140 °C – remarkably, lower than both LiNH2BH3 and AB themselves – and with no detectable ammonia or borazine formation.73 The crystal structure of this compound shows alternating LiNH2BH3 and AB layers, with lithium pseudo-tetrahedrally coordinated by one nitrogen atom and three borohydride groups, as in the parent compound.
A small number of mixed-metal amidoboranes has been prepared, with the objective of improving the dehydrogenation characteristics of the materials. The first of these, Na[Li(NH2BH3)2], was synthesised by Grochala and co-workers.74 The crystal structure of the compound consists of pseudo-tetrahedral lithium ions coordinated by three N atoms, alongside one Li–borohydride interaction, while the sodium atoms are solely coordinated by borohydride moieties. The compound releases hydrogen, beginning at 75 °C, albeit contaminated with ammonia.37 Shortly afterwards, Wu prepared Na2[Mg(NH2BH3)4], which also produces a significant quantity of H2 (8.4%) starting at 65 °C; in this case, contamination with ammonia and borazine was minimal.38 The structure of this compound contains [Mg(NH2BH3)4]2− dianions held together by sodium ions. A similar structure is also observed for the potassium derivative, K2[Mg(NH2BH3)4].75 The formation of NaMg(NH2BH3)3 has been invoked for the extremely low H2-release temperature (from 45 °C) of ball-milled mixtures of AB and NaMgH3.76 Some of these mixed-metal species display lower hydrogen-release temperatures, and with less contamination from fuel-cell poisons, than their monometallic counterparts. Additionally, it is thought that the presence of a secondary, ‘softer’ cation within the material can improve the thermodynamic stability – a fact that bodes well for the development of a reversible process.77
Nöth and co-workers performed a structural study on the known reducing agent LiNMe2BH3 (4) in combination with various solvents.78 While the compound was found to be poorly soluble in aromatic solvents, addition of coordinating solvents led to a variety of different adducts. 4·TMEDA (TMEDA = N,N,N′,N′-tetramethylethylenediamine) has a dimeric structure containing an 8-membered ring formed by Li+⋯Hδ−–B interactions (Fig. 2). Adducts with 1,4-dioxane, trioxane, and 1,3-dioxolane all form layered polymeric structures, whereas crystallisation from the polyether [12]crown-4 allowed retention of the monomeric structure. More recently, McGrady and coworkers found that both 4 and KNMe2BH3 (5) could be crystallised as solvent-free compounds from THF. Both possess polymeric structures, and the authors found further evidence for B–H⋯H–B interactions in both compounds in the absence of conventional protic hydrogen bond donors.53 The diisopropyl derivative, LiNiPr2BH3, crystallises with [12]crown-4 to produce the salt [Li([12]crown-4)2+][Li(NiPr2BH3)2−].79 This species is unique among lithium amidoboranes for possessing the only structurally characterised example of a ‘side-on’ bound amidoborane ligand, with the nitrogen and borohydride moieties bound to the same metal atom (Fig. 2). Comparison with the structure of 4·[12]crown-4 illustrates the profound impact of the nitrogen substituents on the aggregation of these compounds.
 | ||
| Fig. 2 Crystal structures of TMEDA and [12]crown-4 adducts of LiNMe2BH3 (left) and [Li([12]crown-4)2+][Li(NiPr2BH3)2−] (right). | ||
4.2 Group 1 metals in amine–borane dehydrocoupling
Keller reported in 1975 that the reaction of dimethylamine borane with LiH or KH produces B–N coupled complexes, M(NMe2BH2NMe2BH3), that react further with metal hydride to afford MNMe2BH3 compounds.80 The group of Hill recently went on to report the crystal structure of the potassium complex, K(NMe2BH2NMe2BH3), as its dimeric THF adduct (6, Fig. 3), in this case prepared from KN(SiMe3)2 and Me2NHBH3 (Scheme 1).81 The dimeric structure of 6 is held together by K⋯H–B interactions. They also found that the group 1 amides, MN(SiMe3)2 (M = Li, Na, K), can perform the catalytic dehydrocoupling of dimethylamine borane, producing predominantly the cyclic diborazane (Me2NBH2)2. The rate of reaction was considerably slower than for the equivalent Mg-catalysed process (vide infra), which may be due to the formation of insoluble metal hydride aggregates (MH)∞. Details of the dehydrocoupling process are discussed in more detail in Section 5.2.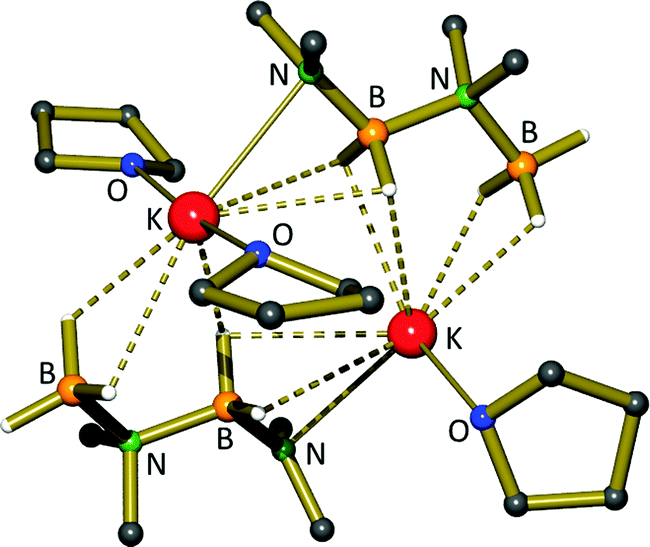 | ||
| Fig. 3 Crystal structure of [K(NMe2BH2NMe2BH3)]2·(THF)3. | ||
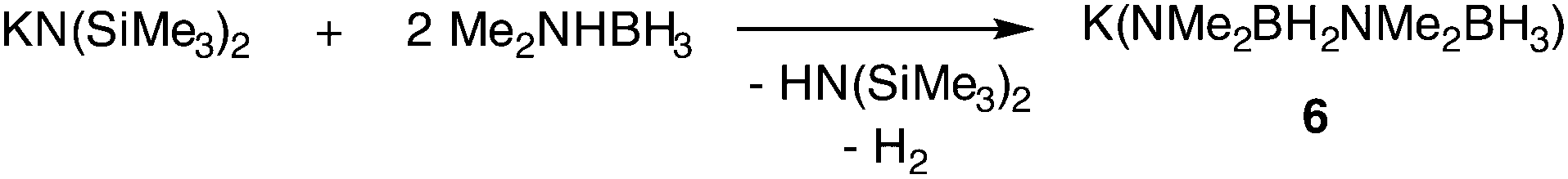 | ||
| Scheme 1 Dehydrogenative on-metal coupling of two amidoborane units. | ||
4.3 Group 1 MNR2BH3 compounds as reagents in salt metathesis reactions
There are a number of examples of group 1 metal amidoboranes being used in salt metathesis reactions with other organometallics (Scheme 2). The first characterised transition metal amidoborane complexes, reported by Roesler and co-workers, were prepared via a salt metathesis route.82 The zirconocene complexes (η5-C5H5)2Zr(H)NH2BH3 and (η5-C5Me5)2Zr(H)NH2BH3 were prepared by the reaction of the corresponding zirconocene dichloride with NH3BH3 and nBuLi (2 equivalents). Use of a single equivalent of AB and base led to the complex (η5-C5H5)2Zr(Cl)NH2BH3. Although lithium amidoborane was not specifically identified, its intermediacy in the reaction is to be assumed. McGrady and co-workers produced crystals of the Ti(III) compound (η5-C5H5)2TiNH2BH3 from the reaction between (η5-C5H5)2TiCl2 and LiNH2BH3 (Scheme 2).54 Similarly, Helten et al. later reported that titanocene dichloride reacts with two equivalents of LiNMe2BH3 to produce the species (η5-C5H5)2TiNMe2BH3 in good yield, with concurrent release of H2.83 The corresponding reaction with (η5-C5H5)2ZrCl2 produces (η5-C5H5)2Zr(H)NMe2BH3.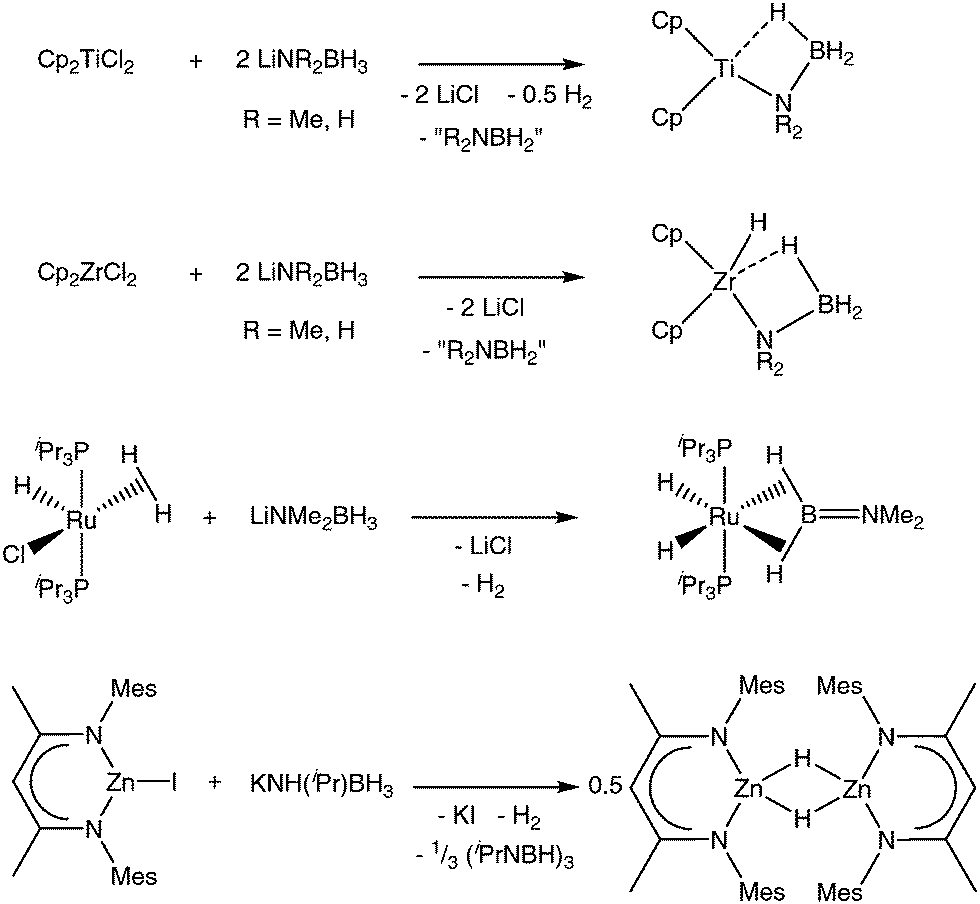 | ||
| Scheme 2 Salt metathesis reactions of group 1 amidoboranes. | ||
In several cases the amidoborane complex is not the final product. The groups of Harder and Schulz exploited this chemistry to gain access to zinc hydride compounds by reaction of mono- and bimetallic diketiminate zinc iodide complexes with KN(H)iPrBH3 (Scheme 2).84,85 Likewise, treatment of the aluminium complex (DIPP-nacnac)AlCl2 (DIPP = 2,6-iPr2C6H3; DIPP-nacnac− = (2,6-iPr2C6H3)NC(Me)C(H)C(Me)N(2,6-iPr2C6H3)−) with KN(H)RBH3 (R = iPr, DIPP) produces (DIPP-nacnac)AlH2 in good yield by presumed double β-hydride elimination.86 NaNMe2BH3 has also been used to convert ReBr2(NO)(η2-H2)(PR3)2 species into tetrahydrides Re(H)4(NO)(PR3)2.87 Quantitative formation of (Me2NBH2)2 suggests an insertion/β-hydride elimination process (vide infra). In a similar reaction, RuHCl(η2-H2)(PiPr3)2 reacts with LiNMe2BH3, in this case forming the ruthenium aminoborane complex RuH2(η2:η2-H2BNMe2)(PiPr3)2.88
Lancaster and co-workers have studied the effect of electron-withdrawing substituents at boron on the reactivity of lithium amidoboranes.89–91 To this end, the compounds LiNH2B(C6F5)n(H)3−n (n = 1–3) were prepared as THF solvates, by reaction of the appropriate ammonia-borane precursor with n-butyllithium or LiN(SiMe3)2.91 Addition of [12]crown-4 to LiNH2B(C6F5)2H·(THF)2 (7·THF2) produced the crown ether adduct ([12]crown-4)LiNH2B(C6F5)2H, which could be structurally characterised by X-ray diffraction. At 1.544(2) Å, the B–N bond is markedly shorter than in the parent amine–borane, NH3B(C6F5)2H (1.6034(19) Å), but unremarkable compared to other amidoborane species. Reactions of these species with group 4 metal complexes produced a variety of interesting products. Combination of 7 with Cp2ZrCl2 led to either Cp2Zr(NH2B(C6F5)2H)2 or Cp2Zr(Cl)(NH2B(C6F5)2H), depending on the stoichiometry, the former of which exists in equilibrium with Cp2Zr(H)(NH2B(C6F5)2H) and aminoborane H2NB(C6F5)2, presumably via a reversible β-hydride elimination/insertion.89 The corresponding reaction with Cp2HfCl2 predominantly yields the N–H activation product Cp2Hf(NHB(C6F5)2H).90 It is proposed that this occurs by the second equivalent of 7 acting as a base to deprotonate the hafnium amidoborane group. Increasing the number of pentafluorophenyl substituents at boron has the expected effect of reducing the nucleophilicity of the amide group. This is demonstrated by the failure of LiNH2B(C6F5)3 (8) to react with Cp2MCl2 (M = Ti, Zr).89 This amidoborane ligand could, however, be transferred to Zr and Hf by reaction of 8 with the zwitterionic compounds Cp2M(Me)(MeB(C6F5)3), with precipitation of the salt Li[MeB(C6F5)3] as by-product. In a similar fashion, the amidoborane group could also be transferred to other Lewis acids, such as E(C6F5)3 (E = B, Al), forming ‘ate’ complexes with a [Li(THF)4]+ counterion.89
5 Group 2 metals
5.1 Amidoborane complexes as models for hydrogen storage compounds
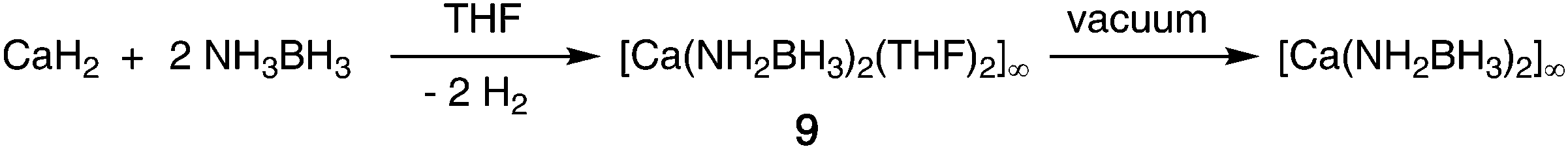 | ||
| Scheme 3 Synthesis of calcium amidoborane coordination polymers. | ||
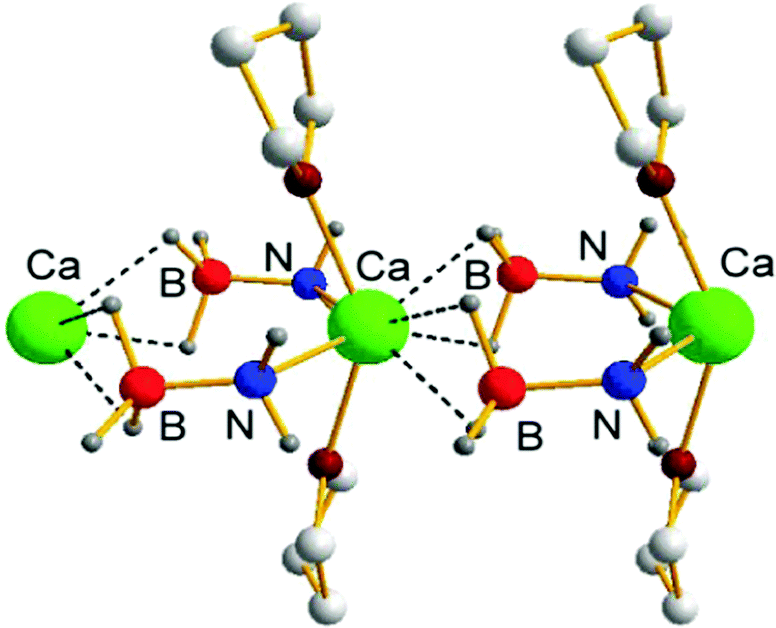 | ||
| Fig. 4 Crystal structure of [Ca(NH2BH3)2(THF)2]∞ (9). | ||
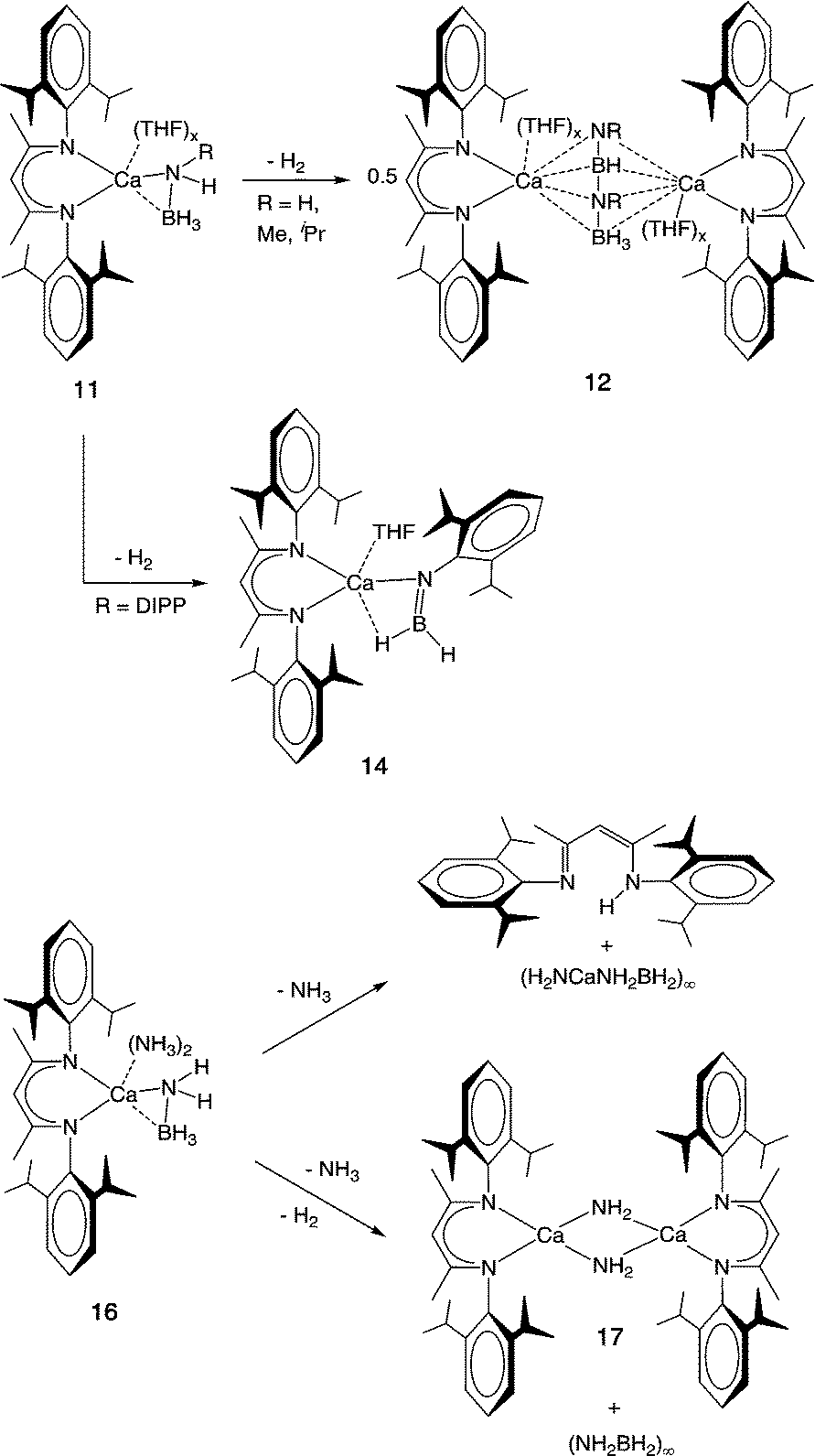
Divalent metals present a unique opportunity for mechanistic study when compared to the alkali metal amidoboranes, in that it is possible to introduce a monoanionic spectator ligand to create a well-defined active site and solubilise the resulting complexes. The strongly coordinating NN-bidentate β-diketiminate ligand, (2,6-iPr2C6H3)NC(Me)C(H)C(Me)N(2,6-iPr2C6H3)− (DIPP-nacnac−) has considerable precedent in alkaline earth metal chemistry. This is due to its ability to stabilise a variety of unusual structures and to disfavour the formation of homoleptic species via Schlenk equilibria.92–96 Harder and co-workers turned to this system to investigate the thermal decomposition of calcium amidoborane complexes under homogeneous conditions. The previously reported, stable, hydrocarbon-soluble calcium hydride complex [(DIPP-nacnac)CaH·(THF)]2 (10)97 was treated with NH3BH3 to produce the monomeric amidoborane (DIPP-nacnac)CaNH2BH3·(THF)2 (11-H) in high yield.98 In sharp contrast to 9, the crystal structure of compound 11-H revealed a ‘side-on’ coordination mode of the amidoborane ligand, in which a B–H hydride atom interacts with the calcium atom of the same molecule rather than containing intermolecular M⋯H interactions or N–H⋯H–B dihydrogen bonds.99
While thermally stable up to 64 °C in the presence of THF, compound 11-H was found to decompose at temperatures as low as 20 °C in benzene solution, with concurrent evolution of H2. The product of the reaction was identified as [(DIPP-nacnac)Ca(THF)]2(HN-BH-NH-BH3) (12-H, Scheme 5). The formation of this unprecedented [HN-BH-NH-BH3]2− dianion implied the doubly-dehydrogenative coupling of two [NH2BH3]− units, and it was interpreted as an intermediate species on the way to fully dehydrogenated [NBH]−. The presence of excess THF presumably inhibits the reaction by preventing the aggregation of two molecules of 11-H.
A selection of other derivatives of 11-H with varied substituents at the nitrogen of the amidoborane was then prepared in order to investigate the influence on the dehydrogenation process. The complexes (DIPP-nacnac)CaN(R)HBH3·(THF) (R = Me (11-Me),98iPr (11-iPr),100 DIPP (11-DIPP)100) were prepared by reaction of the corresponding amine–borane either with [(DIPP-nacnac)CaH·(THF)]2 (10) or the related (DIPP-nacnac)Ca(N(SiMe3)2)·(THF) (13). More recently, Hill et al. also used the latter compound to prepare 11-tBu in the same fashion.101Table 1 shows a selection of structural data and decomposition temperatures for these compounds, from which the following trends can be deduced: (1) increasing steric bulk generally increases both the length of the Ca–N bond and the linearity of the Ca–N–C moiety, (2) the B–N bond distance is essentially independent of the R substituent, and (3) increasing the steric bulk increases the decomposition temperature relating to H2 release.
| Compound | M–NAB/Å | N–Ba/Å | M–NAB–C/° | T dec /°C | Ref. |
|---|---|---|---|---|---|
| a Bond length between the amido nitrogen and adjacent boron atom. b Temperature of initial hydrogen release. | |||||
| NH3BH3 | 0.96(3), 1.07(4); 0.80(6), 0.96(4) | 1.58(2); 1.599(8) | — | 120 | 26, 65 and 102 |
| LiNH2BH3 | 1.973 | 1.560 | — | 92 | 28 |
| LiNH2BH3·NH3BH3 | 2.1(1) | 1.55(9) | — | 80 | 73 |
| Li([12]crown-4)NH2B(C6F5)2H | 2.015(3) | 1.544(2) | — | — | 90 |
| Li([12]crown-4)NH2B(C6F5)3 | 2.052(3), 2.046(4) | 1.552(3), 1.549(2) | — | — | 89 |
| Li([12]crown-4)NMe2BH3 | 1.999(8), 2.005(8) | 1.553(7), 1.561(6) | 108.1(3), 112.3(3), 112.9(3), 116.3(3) | — | 78 |
| [Li([12]crown-4)][Li(NiPr2BH3)2] | 2.052(9), 2.05(1) | 1.546(7), 1.558(8) | 108.5(3), 112.1(4), 120.2(3), 120.7(4) | — | 79 |
| NaNH2BH3 | 2.35 | 1.56 | — | 89 | 28 |
| KNH2BH3 | 2.907(5)–3.364(5) | 1.532(8) | — | 80 | 30 |
| Mg(NH2BH3)2 | — | — | — | 104 | 103 |
| Mg(NH2BH3)2·NH3 | 2.181(6), 2.135(6) | 1.539(8), 1.548(8) | — | 50 | 104 |
| Ca(NH2BH3)2 | — | — | — | 120 | 27 |
| Ca(NH2BH3)2·(THF)2 | 2.069(7), 2.362(7) | 1.52(2) | — | — | 27 |
| Sr(NH2BH3)2 | 2.68 | 1.53 | — | 60 | 29 |
| (DIPP-nacnac)CaNH2BH3·(THF)2 | 2.399(2) | 1.581(4) | — | 20 | 98 |
| (DIPP-nacnac)CaNH(Me)BH3·(THF) | 2.382(4) | 1.581(8) | 125.5(3) | 40 | 98 |
| (DIPP-nacnac)CaNH(iPr)BH3·(THF) | 2.406(4) | 1.582(7) | 133.7(3) | 100 | 100 |
| (DIPP-nacnac)CaNH(DIPP)BH3·(THF) | 2.460(2) | 1.587(4) | 147.6(2) | 120 | 100 |
| (DIPP-nacnac)CaNH(tBu)BH3·(THF) | 2.414(3) | 1.554(6) | 123.8(2) | — | 101 |
| (DIPP-nacnac)CaNH2BH3·(NH3)3 | 2.491(2), 2.528(2) | 1.555(3), 1.566(3) | — | 50 | 105 |
| [(DIPP-nacnac)CaNH(iPr)BH3]2 | 2.394(3) | 1.516(4) | 131.3(2) | — | 105 |
| (DIPP-nacnac)CaNMe2BH3·(THF) | 2.375(3) | 1.497(6) | 119.4(2) | — | 106 |
| (DIPP-nacnac)CaN(CH2)4BH3·(THF) | 2.405(3) | 1.533(6) | 118.2(5), 126.5(5) | — | 107 |
| (DIPP-nacnac)MgNH2BH3·(THF) | 2.056(3) | 1.544(6) | — | 80 | 108 |
| [(DIPP-nacnac)MgNH(iPr)BH3]2 | 2.122(1) | 1.556(2) | 122.57(9) | 110 | 108 |
| (DIPP-nacnac)MgNH(DIPP)BH3 | 2.083(4) | 1.626(9) | 145.2(4) | 120 | 109 |
| PYR-[MgNH(iPr)BH3]2 | 2.123(4) | 1.571(6) | 113.4(2), 113.8(2) | 90 | 108 |
| PYR-[MgNH(DIPP)BH3]2 | 2.121(1), 2.130(1) | 1.591(2), 1.595(2) | 129.64(9), 130.15(9) | 90 | 108 |
| NN-[MgNH(iPr)BH3]2 | 2.093(2) | 1.590(3) | 125.3(2), 131.2(1) | 60 | 108 |
| [K(NMe2BH2NMe2BH3)]2·(THF)3 | 2.8703(17) | 1.503(3) | 98.0(1), 98.5(1), 100.9(1), 102.9(1) | — | 81 |
| Mg(NMe2BH2NMe2BH3)2·(THF) | 2.1667(11) | 1.5647(11) | 111.39(9), 111.57(8), 111.79(8), 112.32(9) | — | 106 |
| (DIPP-nacnac)MgNMe2BH2NMe2BH3 | 2.0929(14) | 1.586(3) | 111.5(1), 112.9(1) | — | 106 |
| (DIPP-nacnac)MgN(CH2)4BH2N(CH2)4BH3 | 2.1146(19) | 1.562(3) | 113.1(2), 113.4(2) | — | 107 |
11-Me and 11-iPr thermally release H2 at 40 and 100 °C, respectively, to afford compounds (12, Scheme 5) containing [RN-BH-NR-BH3]2− dianions. 12-Me displays a similar structure to 12-H, albeit with only one Ca atom bearing a THF ligand (Fig. 5), while in 12-iPr, the steric bulk prevents the terminal nitrogen from bridging both Ca atoms and it only ligates that without a THF ligand. In contrast, the highly bulky 11-DIPP decomposes to form borylamide complex 14. This is likely due to a steric barrier to dimerization, but could also relate to the higher acidity of the remaining N–H proton in comparison to that of alkyl derivatives. Compound 11-tBu was not tested for thermal decomposition, instead having been prepared for catalytic dehydrocoupling investigations (vide infra).
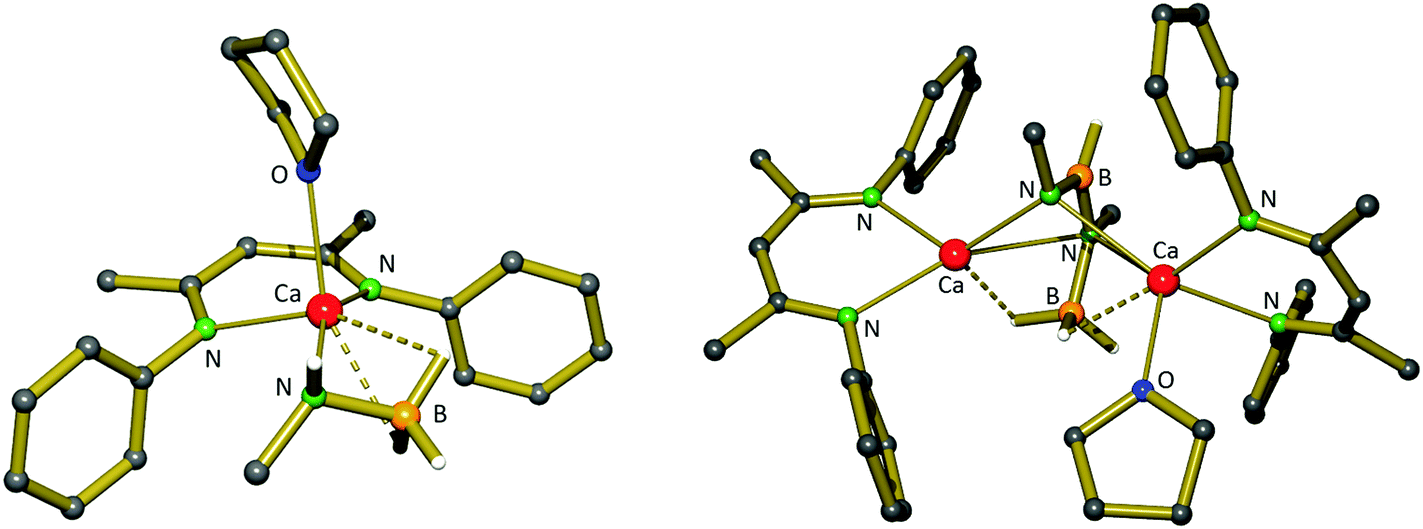 | ||
| Fig. 5 Crystal structure of compound 11-Me (l) and its thermal decomposition product 12-Me (r), displaying the [NMe-BH-NMe-BH3]2− dianion. iPr groups and most H atoms are omitted for clarity. | ||
Complexes of the form (DIPP-nacnac)Ca[NH(R)BH3]·(NH3)x (R = H (16-H), Me (16-Me), iPr (16-iPr), DIPP (16-DIPP); x = 2 or 3) were prepared by the reaction of the precursor [(DIPP-nacnac)Ca(μ-NH2)·(NH3)2]2 with the respective amine boranes. Solid-state structures of the isolated complexes revealed varying connectivity of the ammonia molecules; not only were N–H⋯H–B dihydrogen bonds observed, but in one case there was also an intermolecular interaction between the protic N–H hydrogen atoms and the central carbon of the DIPP-nacnac ligand.
The thermal decomposition of these complexes occurred at the relatively low temperature of 50 °C, but the dehydrogenation was complicated by side-reactions. Loss of NH3 was generally observed upon heating, allowing crystallisation of monoammine and ammine-free products. Nevertheless, hydrogen release was observed in all cases, and evidence for the involvement of NH3 was provided by the formation of [(DIPP-nacnac)Ca(μ-NH2)]2 (17). The protonation of the DIPP-nacnac anion was also found to be a significant side-reaction, and the formation of DIPP-nacnacH was accompanied by the precipitation of an insoluble residue, proposed to be the coordination polymer {H2NCa[NH(R)BH3]}∞. No soluble boron-containing species were formed, supporting the likely formation of polymeric residues after dehydrogenation. Interestingly, and in contrast to the ammine-free derivatives, the decomposition temperature of compounds 16 was independent of the nitrogen substituent, R, of the amidoborane. This was a clear indication that the protic hydrogen originates from ammonia, rather than from the amidoborane unit. This is in agreement with theoretical calculations by Chen and co-workers, which conclude that the lowest-energy hydrogen release process from Ca(NH2BH3)2·(NH3)2 involves combination of ammonia N–H and amidoborane B–H moieties.111 It was proposed that the amidoborane ligand undergoes hydride elimination to a hydride–ammine complex, which then rapidly releases H2 to produce the calcium amide. This, in combination with results from solid-state experiments, suggests that metal amide formation is a feasible decomposition pathway for solid-state amidoborane–ammine materials.33,104
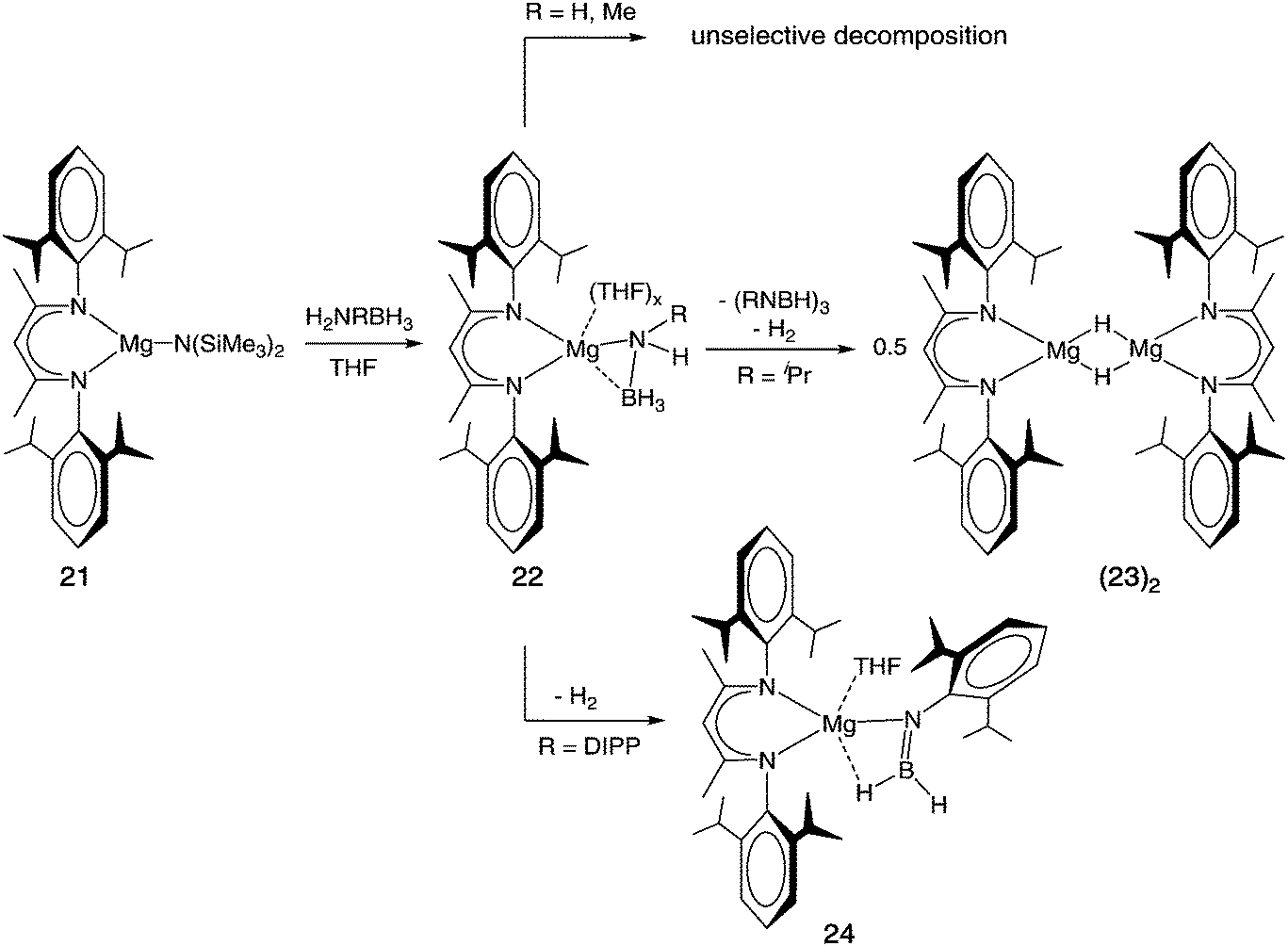
In order to study the influence of the metal on the properties of amidoborane complexes, Harder and co-workers went on to perform experiments with magnesium β-diketiminate complexes. Reaction of (DIPP-nacnac)MgN(SiMe3)2 (21) with primary amine boranes RNH2BH3 produced the corresponding magnesium amidoboranes, (DIPP-nacnac)MgNH(R)BH3 (R = H (22-H), Me (22-Me), iPr (22-iPr),108 DIPP (22-DIPP)109). With the exception of 22-DIPP, the compounds exist as dimers or aggregates, which can be fragmented and solubilised by addition of THF. The increased tendency of the amidoborane to bridge between metal centres is presumably a result of the smaller magnesium dication leading to a more strained four-membered M–N–B–H ring in terminal amidoborane complexes. Structural comparisons are hampered by the differing geometries and coordination numbers of the crystallised products as well as disorder in the amidoborane fragments. Nevertheless, it is evident that the M–N bonds are roughly 0.3 Å shorter with the smaller magnesium dication compared to calcium (see Table 1). An increase in the amidoborane C–N bond length with increasing steric hindrance could also be deduced, as is the case for the calcium analogues. At 1.626(9) Å, 22-DIPP contains the longest B–N bond of any amidoborane compound discussed here. Jones, Stasch and co-workers went on to show that the known amidoborane complexes (22-H)2 and 22-H·THF and the equivalent new compounds based on a bulkier nacnac ligand, (2,6-iPr2C6H3)NC(tBu)C(H)C(tBu)N(2,6-iPr2C6H3)−, could be obtained via a variety of different routes (Scheme 4).114 These included reaction of NH3BH3 with [(DIPP-nacnac)Mg(μ-H)]2 (H2 elimination), [(DIPP-nacnac)Mg(μ-Bu)]2 (butane elimination) and the Mg(I) dimer [{(DIPP-nacnac)Mg}2] (reductive dehydrogenation). Salt metathesis of [(DIPP-nacnac)MgI·(OEt2)] with LiNH2BH3 also afforded the same compounds.
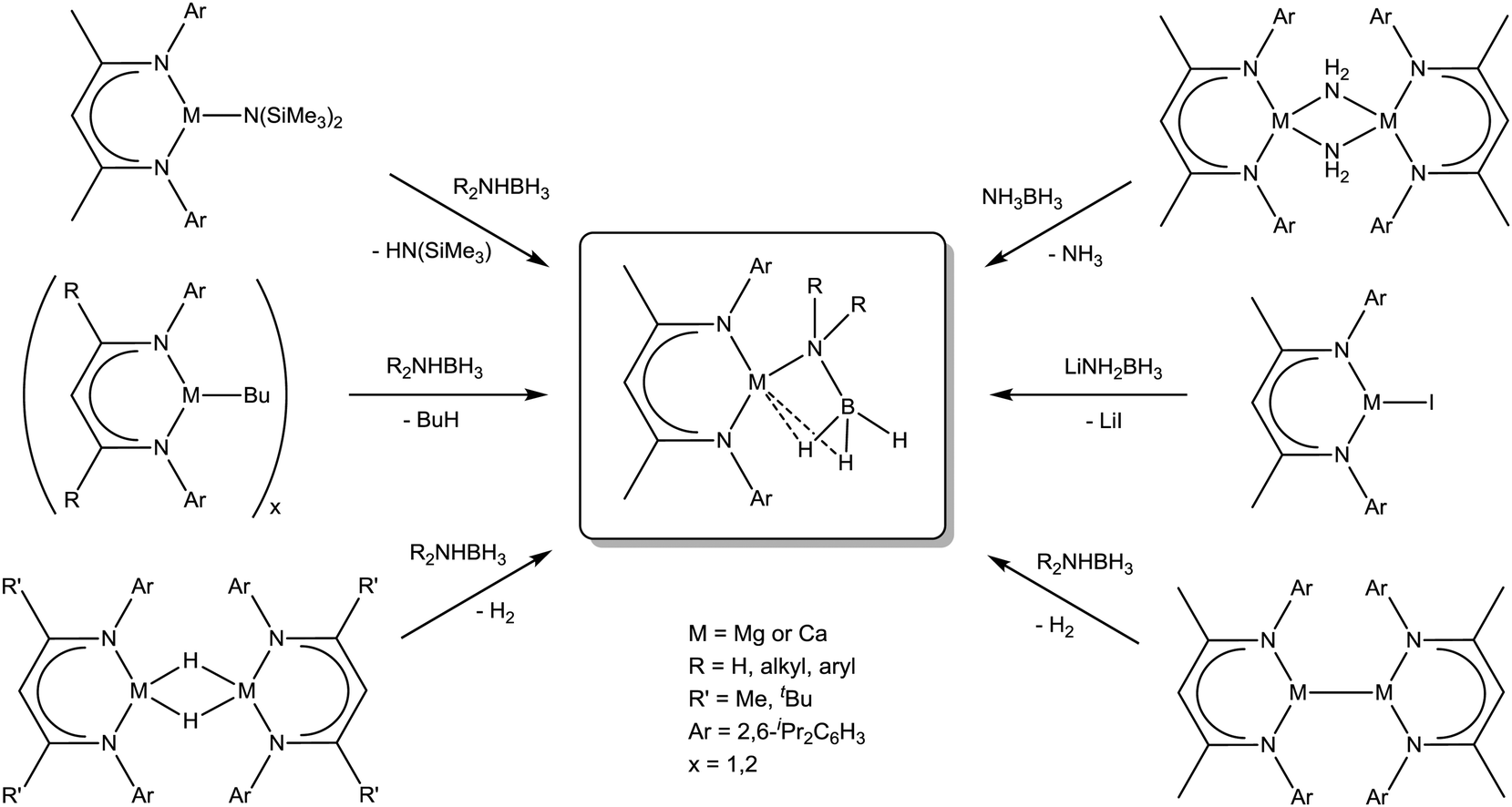 | ||
| Scheme 4 Summary of synthetic routes to β-diketiminate group 2 amidoborane complexes. Coordinated solvent molecules (THF, NH3, Et2O) are present in some cases but are omitted for generality. | ||
 | ||
| Scheme 5 Predominant decomposition pathways of β-diketiminate calcium amidoborane complexes. | ||
Thermal decomposition was again performed by Spielmann et al. for the Mg complexes 22 (Scheme 6).108 The complexes were found to be considerably more stable than their Ca counterparts, requiring temperatures of at least 80 °C to effect decomposition. A clear dependence on the nature of the nitrogen substituent was again observed. The complexes 22-H and 22-Me both decomposed unselectively at 80 °C in benzene. The only defined product identified, albeit in small quantities, was [(DIPP-nacnac)Mg]2(H3B-NMe-BH-NMe)·(THF), in which, unlike its calcium analogue, the BNBN dianion bridges unsymmetrically between the metal centres. 22-iPr decomposed at the higher temperature of 110 °C, but instead of yielding a new amidoborane complex, the known magnesium hydride dimer, [(DIPP-nacnac)Mg(μ-H)]2 ([23]2) was formed in high yield alongside H2 and the borazine (iPrNBH)3. This was proposed to occur by release of iPr(H)NBH2 (via β-H elimination), followed by a magnesium hydride-catalysed oligomerisation to the trimer. The high yield (89%) of the magnesium hydride suggests that amidoborane complexes could be promising precursors to other main-group metal hydride complexes. The bulky 22-DIPP decomposed at 120 °C in the same fashion as its calcium analogue, yielding the borylamide complex (DIPP-nacnac)MgN(DIPP)BH2·(THF) (24).
 | ||
| Scheme 6 Decomposition pathways of monometallic Mg amidoborane complexes. | ||
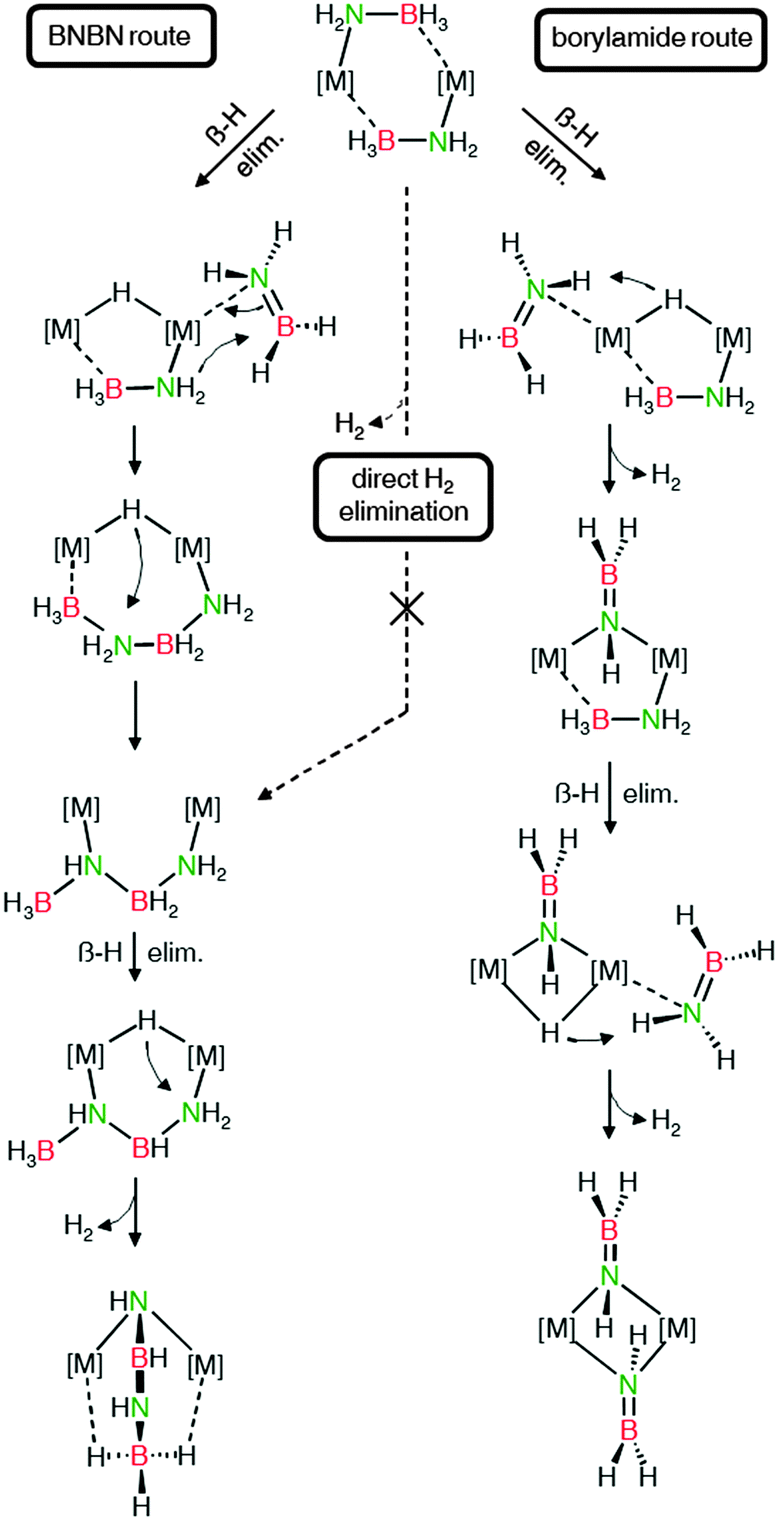
The mechanistic conclusions drawn from these results are summarised in Scheme 7. Mg and Ca amidoborane complexes initially decompose via β-hydride elimination to form metal hydride species and free aminoboranes, R(H)N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) BH2, which may initially coordinate to the metal centre. The subsequent course is dictated by the reactivity of the metal hydride species (Ca > Mg) and the nitrogen substituent. In case of metal hydrides of low reactivity, as is notably the case for zinc hydrides, the aminoborane R(H)NBH2 is simply released and forms oligomers, thus giving the hydride complex as a product.115 In case of more reactive metal hydride complexes (Mg, Ca) the coordinated aminoborane R(H)NBH2 may react. Species with small N-substituents readily dimerise, likely via nucleophilic attack of a bound amide group at the boron of a coordinated aminoborane, leading eventually to the stable [RN-BH-NR-BH3]2− dianion. Larger N-substituents such as DIPP produce aminoboranes that do not dimerise and are then deprotonated by the metal hydride to form borylamide complexes.
BH2, which may initially coordinate to the metal centre. The subsequent course is dictated by the reactivity of the metal hydride species (Ca > Mg) and the nitrogen substituent. In case of metal hydrides of low reactivity, as is notably the case for zinc hydrides, the aminoborane R(H)NBH2 is simply released and forms oligomers, thus giving the hydride complex as a product.115 In case of more reactive metal hydride complexes (Mg, Ca) the coordinated aminoborane R(H)NBH2 may react. Species with small N-substituents readily dimerise, likely via nucleophilic attack of a bound amide group at the boron of a coordinated aminoborane, leading eventually to the stable [RN-BH-NR-BH3]2− dianion. Larger N-substituents such as DIPP produce aminoboranes that do not dimerise and are then deprotonated by the metal hydride to form borylamide complexes.
 | ||
| Scheme 7 Alternative bimetallic decomposition routes of metal amidoborane complexes. | ||
Related computational studies by Kim and co-workers on the decomposition of LiNH2BH3 also advocated a dehydrogenation process involving a coupling of two BN units.32 These authors found that the “BNBN” decomposition route had a considerably lower activation barrier than borylamide formation, which correlates to the decomposition temperatures of group 2 amidoborane model complexes; decomposition via B–N coupling is a more facile process, and thus proceeds at lower temperatures, than formation of borylamide complexes. On the basis of DFT investigations, direct H2 elimination from amidoborane complexes is regarded to be unlikely for both monometallic32 and bimetallic108 decomposition pathways.
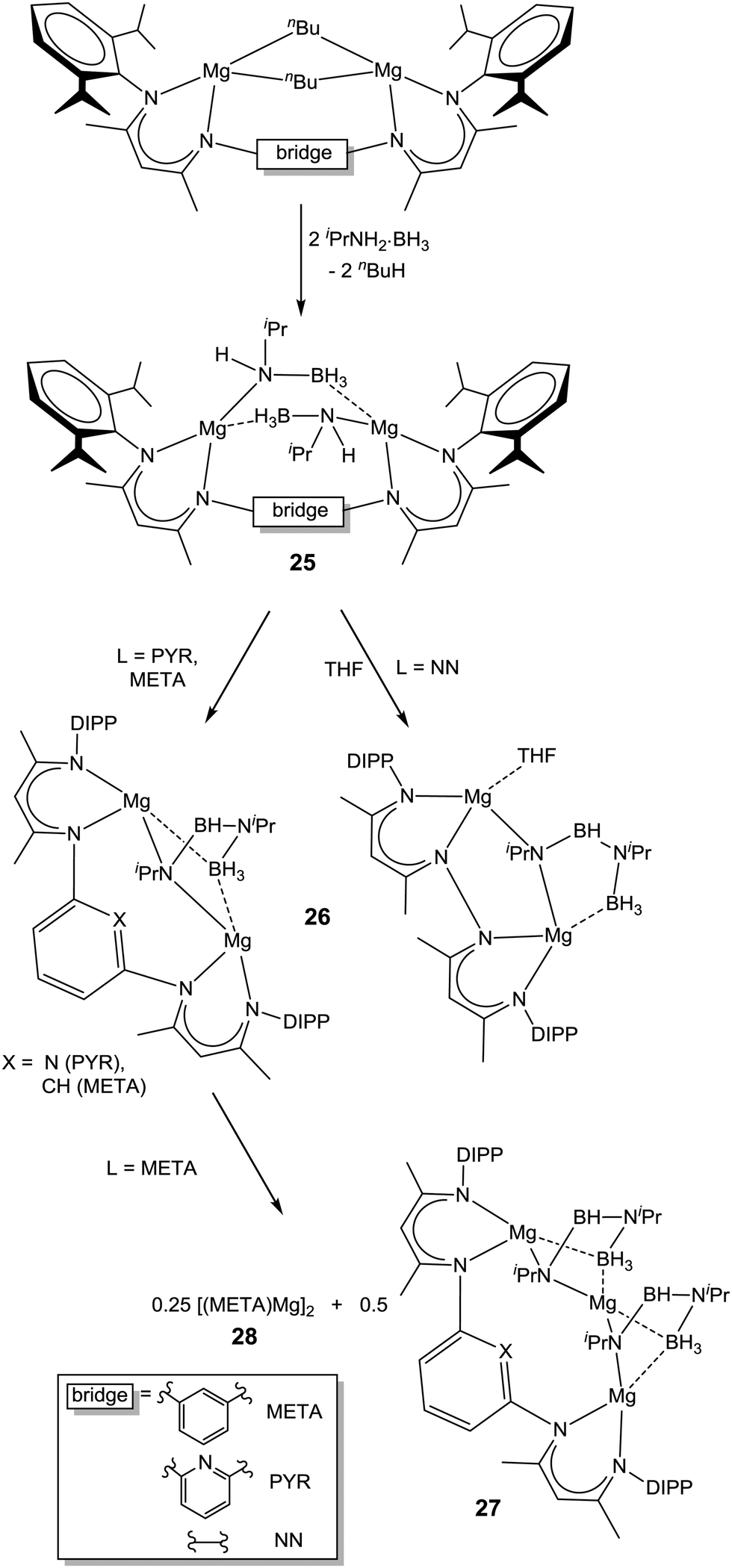 | ||
| Scheme 8 Synthesis and thermal decomposition of bimetallic magnesium amidoborane complexes. | ||
Both 25-NN and 25-PYR were found to decompose (Tdec = 60 °C (25-NN), 90 °C (25-PYR)) with release of H2, producing complexes LMg2[(iPr)NBHN(iPr)BH3] (26) rather than metal hydride species as observed for the monometallic analogue. Clearly, the enforced proximity of two amidoborane ligands favours the formation of the BNBN unit. The complex META-[MgNH(iPr)BH3]2 (25-META) decomposed in a similar fashion at 90 °C, although in this case ligand exchange reactions allowed isolation of the products META-Mg3[(iPr)N-BH-N(iPr)-BH3]2 (27) and (META-Mg)2 (28, Scheme 8). The crystal structure of 27 revealed one Mg atom to be solely bound to two BNBN dianion moieties through both N atoms and one short borohydride contact each. It is distinctly possible that this could be illustrative of the bonding situation in certain decomposition products of solid-state alkaline earth metal amidoboranes. Interestingly, the complex PYR-[Mg(NH(DIPP)BH3)]2 (29) undergoes a single β-hydride elimination upon exposure to THF, yielding the magnesium hydride species PYR-Mg[NH(DIPP)BH3](μ-H)Mg·(THF) (30), in contrast to the borylamide formation from the monometallic analogue 22-DIPP.116
5.2 Group 2 amidoboranes in amine–borane dehydrocoupling
Following extensive research using transition metals, several group 2 metal compounds have recently been applied in the catalytic dehydrocoupling of amine–boranes. This process has attracted considerable attention from the perspectives of both hydrogen storage and materials chemistry, but has traditionally been dominated by metals such as Rh,117–119 Ir,120,121 Ru,122,123 and Ti.83,124–126 While NH3BH3 itself is naturally the most attractive substrate with regard to hydrogen storage, due to its low molecular weight and equal numbers of protic and hydridic hydrogen atoms, study of its dehydrocoupling is somewhat hampered by complicated and/or insoluble product mixtures, caused by polymerisation and multiple dehydrocoupling. Primary and secondary amine–boranes, RNH2BH3 and R2NHBH3, on the other hand, show improved solubility and can typically be selectively dehydrogenated to form monomeric aminoboranes, cyclic dimers or trimers, depending on the nature of the substituent(s).118The first catalytic amine–borane dehydrocoupling reaction by a group 2 metal complex was reported by Harder and co-workers.109 Here, the bulky derivative, DIPPNH2BH3 was found to undergo a catalytic reaction with 5 mol% of (DIPP-nacnac)MgN(SiMe3)2 (21) giving quantitative conversion to the diaminoborane HB[N(H)DIPP]2, since shown to be a competent ligand in f-block chemistry in its doubly deprotonated form,127,128 along with H2 and diborane. The active catalyst was proposed to be (DIPP-nacnac)MgBH4.
The group of Hill has made great strides in the dehydrocoupling of secondary amine–boranes using alkaline earth metals, and they have reported a number of secondary amidoboranes as likely catalytic intermediates. In their initial publication, the simple organometallic magnesium reagent, Mg[CH(SiMe3)2]2·(THF)2 (31), was reported to catalyse the dehydrocoupling of Me2NHBH3. Although slow compared to some transition metal systems (5 mol% cat., 60 °C, 3 days), the catalysis selectively produced the cyclic aminoborane dimer, (Me2NBH2)2. In order to obtain mechanistic insight, stoichiometric reactions were also performed. The starting compound 31 was found to react with four equivalents of Me2NHBH3 to produce a species containing new B–N bonds, Mg(NMe2BH2NMe2BH3)2·(THF) (32, Scheme 9).106 X-ray diffraction revealed the presence of two monoanionic BNBN ligands, each bound to the magnesium centre through a Mg–N bond and two B–H⋯Mg interactions from the δ-BH3 moiety (Fig. 6). Crystallographically determined Mg–N and N–B bond lengths were not significantly different from other magnesium amidoborane species (see Table 1).
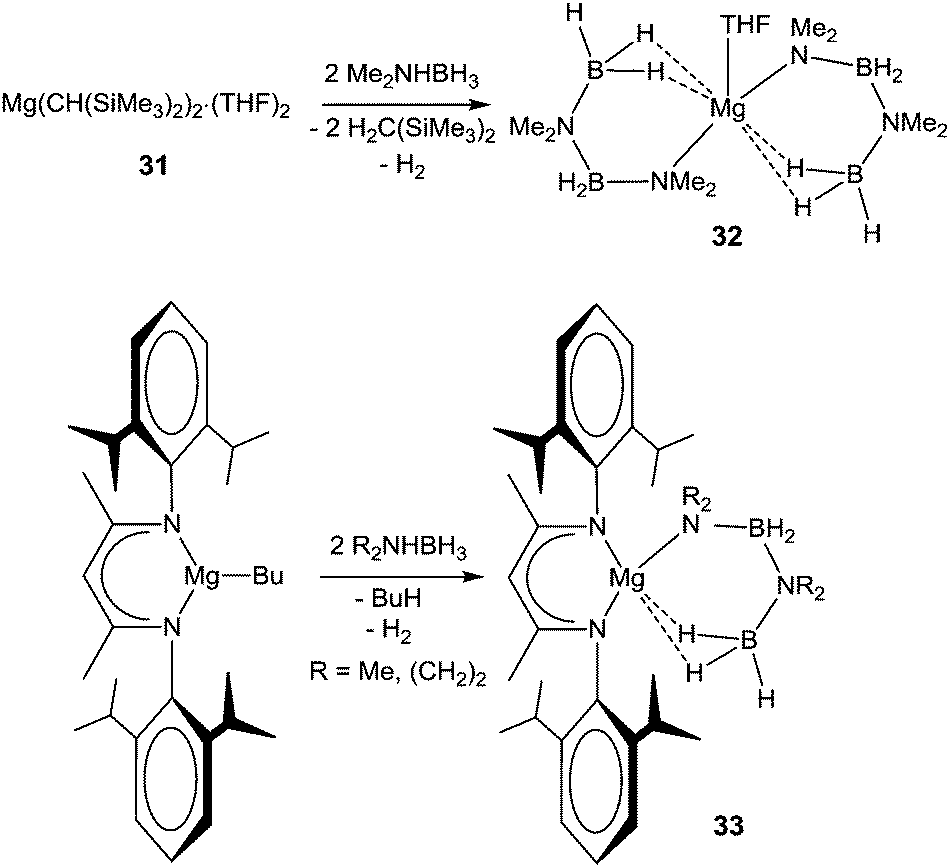 | ||
| Scheme 9 Isolated Intermediates in the Mg-mediated dehydrocoupling of dialkylamine boranes. | ||
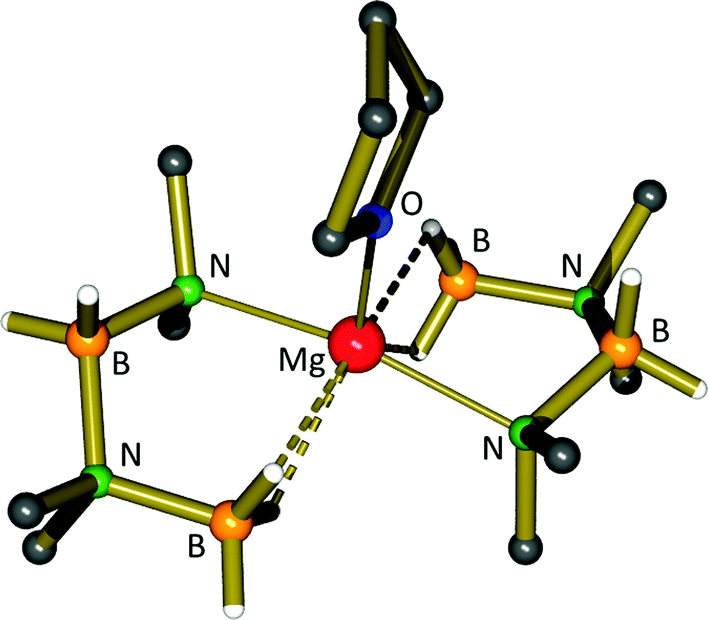 | ||
| Fig. 6 Crystal structure of compound 32, displaying the NMe2-BH2-NMe2-BH3− anion. | ||
Heating compound 32 to 60 °C yielded the cyclic diborazane, (Me2NBH2)2, over the course of several days. This process was proposed to occur via concerted δ-hydride elimination and B–N bond formation, although no direct evidence of magnesium hydride species was observed. In order to test this hypothesis, the complex (DIPP-nacnac)Mg(NMe2-BH2-NMe2-BH3) (33-Me2) was prepared from Me2NHBH3 and (DIPP-nacnac)MgnBu. The NBNB ligand displays similar geometry to that in the homoleptic derivative, and the complex was indeed found to eliminate (Me2NBH2)2, with concomitant production of the previously reported129 hydride species [(DIPP-nacnac)Mg(μ-H)·(THF)]2 (23·THF). A catalytic cycle was therefore proposed (Scheme 10) involving σ-bond metathesis of a magnesium hydride compound with the amine–borane to form an amidoborane complex and H2, followed by β-hydride elimination. The liberated aminoborane, Me2NBH2, was then proposed to insert into an amidoborane M–N bond, before undergoing δ-hydride elimination and releasing the cyclic diborazane.
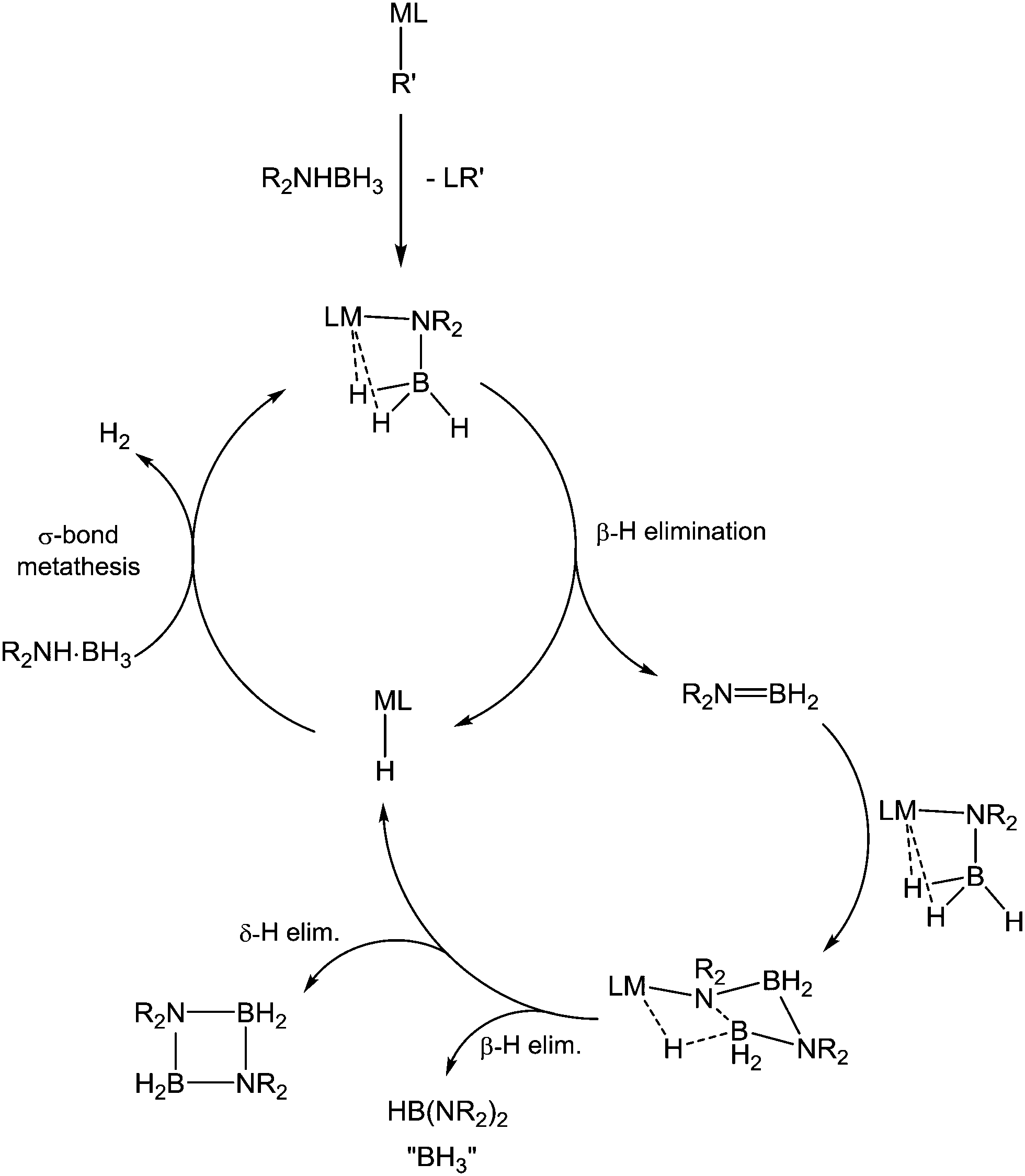 | ||
| Scheme 10 Mechanism of s-block-catalysed amine–borane dehydrocoupling. | ||
The analogous reactions with calcium demonstrated the strong influence of the metal in this chemistry. The reaction of (DIPP-nacnac)Ca[N(SiMe3)2]·(THF) (5) with Me2NHBH3 afforded the amidoborane complex (DIPP-nacnac)Ca(NMe2BH3)·(THF) (34-Me2). In contrast to the magnesium case, no significant B–N coupling could be observed in the presence of excess amine–borane. Heating complex 34-Me2 to 80 °C resulted in slow decomposition via β-hydride elimination, forming a calcium hydride complex, traces of HB(NMe2)2 and the monomeric Me2NBH2. In contrast to the case where the amidoborane has a single alkyl substituent (compounds 3-R), the lack of protic hydrogens allows the highly reactive calcium hydride to be observed.
Very similar reactivity was subsequently reported with pyrrolidine borane, (CH2)4NHBH3, with B–N coupling again observed for Mg but not for Ca.106 The structures of the complexes (DIPP-nacnac)Mg[N(CH2)4BH2N(CH2)4BH3] (33-(CH2)4) and (DIPP-nacnac)Ca[N(CH2)4BH3]·(THF) (34-(CH2)4) displayed similar parameters to their dimethyl-substituted analogues, and the Mg complex decomposed at elevated temperatures to produce the cyclic diborazane [(CH2)4NBH2]2. In the same publication, NMR spectroscopic evidence was also obtained for the formation of a strontium amidoborane complex, (DIPP-nacnac)Sr[N(CH2)4BH3]·(THF) (35); however, this compound could not be crystallised, and was found to be unstable towards Schlenk-type ligand redistribution, complicating its chemistry even further.107
The release of cyclic diborazane from complexes 33-Me2 and 34-Me2 can also be effected by the room-temperature addition of B(C6F5)3 as a hydride abstraction reagent, with the resulting hydrido-borate complexes (36 and 37, Scheme 11) providing a convenient route into CO2 reduction chemistry.130 The mechanism of B–N coupling for Ca amidoboranes is unclear.
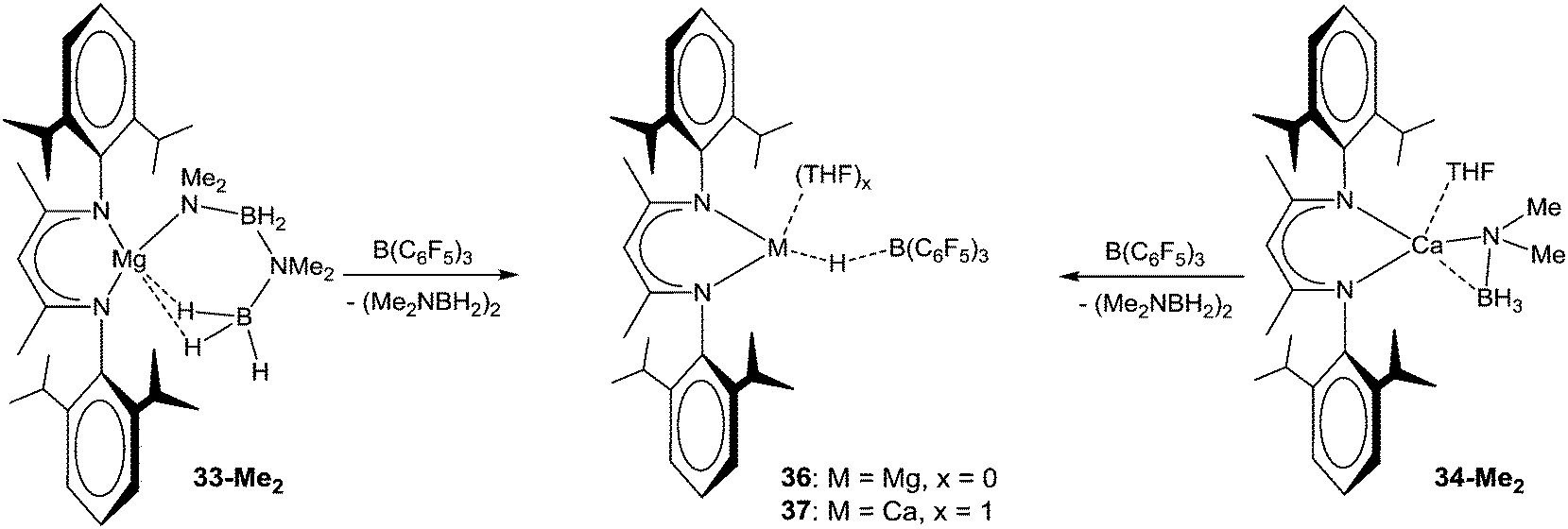 | ||
| Scheme 11 Release of cyclic diborazane from amidoborane complexes with B(C6F5)3. | ||
Subsequent experiments by Hill et al. looked into the reactivity of heteroleptic group 2 amides, M[N(SiMe3)2]2, with respect to secondary amine–boranes.107 Although the absence of a β-diketiminate ligand apparently thwarted attempts to obtain crystalline products for structural analysis, NMR spectroscopy allowed interesting deductions to be made regarding the dimerization process and, in particular, the role of the metal. All of the group 2 metal amides tested (Mg, Ca, Sr, Ba) formed amidoborane complexes upon reaction with Me2NHBH3, as judged by 11B NMR spectroscopy. Rather than mediating dehydrocoupling, the Mg complex underwent β-hydride elimination, eliminating the monomeric aminoborane, Me2NBH2. The analogous reaction with Ca also produced some Me2NBH2, albeit much more slowly. For Sr and Ba, NMR experiments indicated the formation of amidoborane compounds in both cases, but no evidence for Me2NBH2 production was found. Very similar behaviour was observed when using the bulkier substrate, iPr2NHBH3, with the added advantage of rapid catalytic dehydrogenation in the case of Mg[N(SiMe3)2]2. These results demonstrate the stability of amidoborane complexes of the heavier alkaline earth metals, and illustrate the decreasing propensity of amidoborane complexes to undergo β-hydride elimination as the charge density of the metal decreases.
In this context, it is of interest to note that the primary calcium amidoborane complexes (11), containing additional acidic N–H units, react much faster to form BNBN or borylamide products than their magnesium analogues (22). Since these reactions are also proposed to proceed through a metal hydride intermediate (Scheme 7), it may be possible that β-hydride elimination in metal amidoborane complexes to produce metal hydride species is reversible (as proposed for Zr–amidoborane complexes).89 The much more reactive calcium hydride intermediates easily react further, whereas less reactive magnesium hydride intermediates may be present to a larger extent (and can even be isolated) but need much more forcing conditions for further reaction. Only the presence of excess acidic amine-borane (or another proton source), like during catalytic dehydrogenation, may cause fast conversion of magnesium hydride intermediates. Decomposition of the heaviest metal amidoborane complexes (Sr, Ba) by β-hydride elimination would generate the hitherto unknown strontium and barium hydride complexes that would be highly reactive and immediately insert R2NBH2 to reform the metal amidoborane complexes, i.e. the equilibrium lies strongly or completely to the metal amidoborane side. The latter may explain their inactivity in catalytic dehydrogenation.
Hill and co-workers went on to report the dehydrocoupling of the primary amine–borane, tBuNH2BH3, with (DIPP-nacnac)Ca[N(SiMe3)2]·(THF) (13).101 The catalysis was found to proceed through the intermediate amidoborane complex, 11-tBu (Scheme 5). In contrast to catalysis with the aforementioned dialkylamine–boranes, the reaction was rather unselective, affording a complicated mixture of products including the cyclic diborazane [tBu(H)N-BH2]2, the borazine (tBuNBH)3 and the diaminoborane HB[N(H)tBu]2. One likely explanation for this lack of selectivity is the availability of a second protic N–H moiety, which could be deprotonated by the proposed intermediate calcium hydride complex (vide supra). Nevertheless, the operation of different reaction pathways, most notably the proposed dehydrogenation/ring expansion from [tBu(H)N-BH2]2 to (tBuN-BH)3, perhaps reveals a greater depth to this calcium catalysis that warrants further investigation.
5.3 Insertion of aminoborane units into M–R bonds
The isoelectronic nature of amidoborane compounds and aminoboranes to alkyl compounds and alkenes, respectively, has led to interest in the extent to which this analogy can be applied to the organometallic chemistry of B–N compounds.56,131–134 The amine–borane dehydrocoupling mechanism (Scheme 10) describes the alkene-like re-insertion of a dehydrogenated aminoborane unit into a metal–nitrogen bond; Hill and co-workers provided support for this mechanism with their report of the insertion of a BN unit of an amidoborane complex into a metal–carbon bond.135 The strontium complexes, [(Me3Si)2HC]Sr(NR2BH3)·(THF)2 (R = Me (38-Me2), (CH2)2 (38-(CH2)4)) were prepared by σ-bond metathesis from homoleptic Sr[CH(SiMe3)2]2·(THF)2. Heating to 80 °C resulted in β-hydride elimination and insertion of the aminoborane unit into the remaining strontium–carbon bond, after which further β-hydride elimination afforded the aminoalkylborane product, [(Me3Si)2HC]B(H)NR2, 39, in both cases (Scheme 12).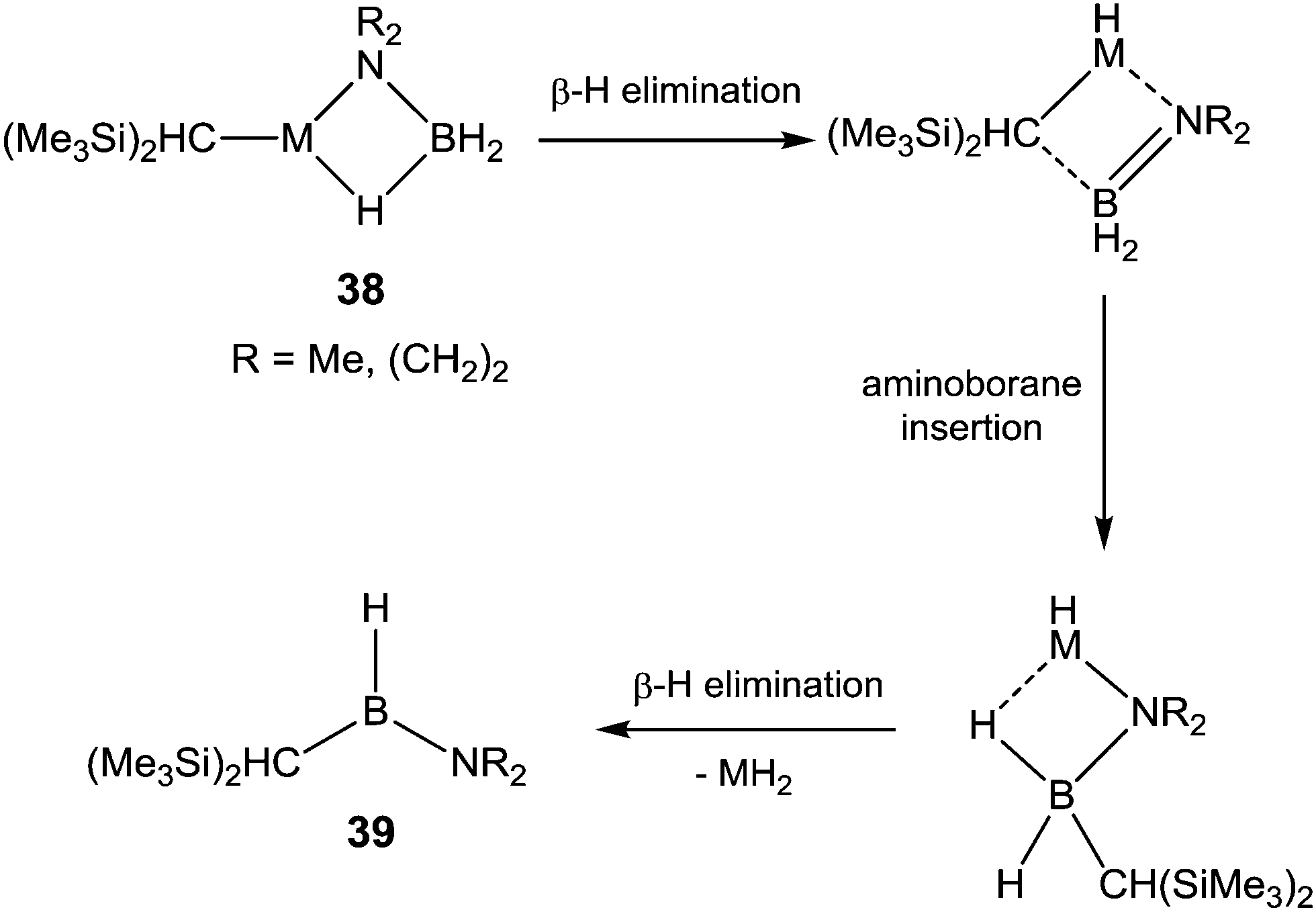 | ||
| Scheme 12 Aminoborane insertion into M–C bonds. | ||
In this case, the reduced tendency of strontium amidoboranes to undergo β-hydride elimination proved advantageous. While some conversion to the aminoalkylborane was observed with Mg[CH(SiMe3)2]2·(THF)2 and Ca[CH(SiMe3)2]2·(THF)2, the reactions were less selective and the low decomposition temperature prevented the isolation of the amidoborane species.136 This publication also contained details of a tetrametallic strontium amidoborane complex, Sr4[CH(SiMe3)2]2[NH(tBu)BH3]6·(THF)4 (40), formed from the equimolar combination of tBuNH2BH3 and Sr[CH(SiMe3)2]2·(THF)2. The mismatch between the number of alkyl and amidoborane ligands in the crystallised product was proposed to be the result of a Schlenk equilibrium. Similar insertion chemistry was also observed with this derivative, affording the aminoalkylborane [(Me3Si)2HC]B(H)NHtBu.
6 Conclusions and outlook
Over the last two decades, the chemistry of metal amidoboranes has progressed from relative obscurity to an active multidisciplinary field. The use of lithium amidoboranes as reducing agents in organic synthesis has reportedly begun to be implemented in undergraduate teaching laboratories,18 and solution-phase amidoborane chemistry has shed some light on the mechanism of potentially lucrative hydrogen storage and release processes.The realisation that early main group MNH2BH3 complexes can release H2 at significantly more convenient temperatures than the hydrogen-rich NH3BH3, with many other merits, such as the absence of foaming and induction periods, and thermoneutrality, clearly puts a spotlight on amidoboranes. The next breakthrough for the practical use of metal amidoboranes in hydrogen storage, however, will only be achieved by finding reversible systems. Efficient fuel regeneration (i.e. rehydrogenation of decomposition products) is an absolute prerequisite for future applications.137
The extension of amine–borane dehydrocoupling to the abundant, cheap and non-toxic metals of the s-block is an interesting development and is likely to be a target for further research, although for group 1 any success likely depends upon the ability to solubilise the metal hydride intermediates. Solution-phase reactivity studies have revealed other possible applications of amidoboranes, in particular for the generation of reactive metal hydride species for both main group and transition metals. There are, nonetheless, significant strides to be made before practical applications of these compounds can be realised. The amidoborane chemistry of the heavier alkaline earth metals also remains in its infancy, while expanding the scope of supporting ligands for solution-phase Mg and Ca amidoborane chemistry beyond the ubiquitous DIPP-nacnac is likely to reveal new reactivity. One thing that can be stated with confidence is that s-block amidoborane compounds will continue to play a role in challenging the notion of transition metal dominance in organometallic chemistry and homogeneous catalysis.
References
- H. I. Schlesinger and A. B. Burg, J. Am. Chem. Soc., 1938, 60, 290–299 CrossRef CAS.
- H. I. Schlesinger and H. C. Brown, J. Am. Chem. Soc., 1940, 62, 3429–3435 CrossRef CAS.
- S. G. Shore and R. W. Parry, J. Am. Chem. Soc., 1958, 80, 8–12 CrossRef CAS.
- M. Bowden, D. J. Heldebrant, A. Karkamkar, T. Proffen, G. K. Schenter and T. Autrey, Chem. Commun., 2010, 46, 8564–8566 RSC.
- S. R. Daly, B. J. Bellott, D. Y. Kim and G. S. Girolami, J. Am. Chem. Soc., 2010, 132, 7254–7255 CrossRef CAS PubMed.
- V. D. Aftandilian, H. C. Miller and E. L. Muetterties, J. Am. Chem. Soc., 1961, 83, 2471–2474 CrossRef CAS.
- J. W. Gilje and R. J. Ronan, Inorg. Chem., 1968, 7, 1248–1249 CrossRef CAS.
- P. C. Keller, J. Am. Chem. Soc., 1969, 91, 1231 CrossRef CAS.
- P. C. Keller, Inorg. Chem., 1971, 10, 1528–1529 CrossRef CAS.
- R. O. Hutchins, K. Learn, F. El-Telbany and Y. P. Stercho, J. Org. Chem., 1984, 49, 2438–2443 CrossRef CAS.
- G. B. Fisher, J. Harrison, J. C. Fuller, C. T. Goraski and B. Singaram, Tetrahedron Lett., 1992, 33, 4533–4536 CrossRef CAS.
- J. C. Fuller, E. L. Stangeland, C. T. Goralski and B. Singaram, Tetrahedron Lett., 1993, 34, 257–260 CrossRef CAS.
- G. B. Fisher, J. C. Fuller, J. Harrison, C. T. Goralski and B. Singaram, Tetrahedron Lett., 1993, 34, 1091–1094 CrossRef CAS.
- G. B. Fisher, J. C. Fuller, J. Harrison, S. G. Alvarez, E. R. Burkhardt, C. T. Goralski and B. Singaram, J. Org. Chem., 1994, 59, 6378–6385 CrossRef CAS.
- A. G. Myers, B. H. Yang and D. J. Kopecky, Tetrahedron Lett., 1996, 37, 3623–3626 CrossRef CAS.
- W. Xu, G. Wu, W. Yao, H. Fan, J. Wu and P. Chen, Chem. – Eur. J., 2012, 18, 13885–13892 CrossRef CAS PubMed.
- L. Pasumansky, C. T. Goralski and B. Singaram, Aldrichimica Acta, 2005, 38, 60–65 Search PubMed.
- L. Pasumansky, C. T. Goralski and B. Singaram, Org. Process Res. Dev., 2006, 10, 959–970 CrossRef CAS.
- S. Thomas, T. Huynh, V. Enriquez-Rios and B. Singaram, Org. Lett., 2001, 3, 3915–3918 CrossRef CAS PubMed.
- S. Thomas, S. Roberts, L. Pasumansky, S. Gamsey and B. Singaram, Org. Lett., 2003, 5, 3867–3870 CrossRef CAS PubMed.
- A. Karkamkar, C. Aardahl and T. Autrey, Mater. Matters, 2007, 2, 6–9 CAS.
- C. W. Hamilton, R. T. Baker, A. Staubitz and I. Manners, Chem. Soc. Rev., 2009, 38, 279–293 RSC.
- Target Explanation Document: Onboard Hydrogen Storage for Light-Duty Fuel Cell Vehicles, United States Department of Energy, 2015.
- T. B. Marder, Angew. Chem., Int. Ed., 2007, 46, 8116–8118 CrossRef CAS PubMed.
- B. Peng and J. Chen, Energy Environ. Sci., 2008, 479–483 CAS.
- M. G. Hu, R. A. Geanangel and W. W. Wendlandt, Thermochim. Acta, 1978, 23, 249–255 CrossRef CAS.
- H. V. K. Diyabalanage, R. P. Shrestha, T. A. Semelsberger, B. L. Scott, M. E. Bowden, B. L. Davis and A. K. Burrell, Angew. Chem., Int. Ed., 2007, 46, 8995–8997 CrossRef PubMed.
- Z. Xiong, C. K. Yong, G. Wu, P. Chen, W. Shaw, A. Karkamkar, T. Autrey, M. O. Jones, S. R. Johnson, P. P. Edwards and W. I. F. David, Nat. Mater., 2008, 7, 138–141 CrossRef CAS PubMed.
- Q. Zhang, C. Tang, C. Fang, F. Fang, D. Sun, L. Ouyang and M. Zhu, J. Phys. Chem. C, 2010, 114, 1709–1714 CAS.
- H. V. K. Diyabalanage, T. Nakagawa, R. P. Shrestha, T. A. Semelsberger, B. L. Davis, B. L. Scott, A. K. Burrell, W. I. F. David, K. R. Ryan, M. O. Jones and P. P. Edwards, J. Am. Chem. Soc., 2010, 96, 4–5 Search PubMed.
- K. Wang, J. G. Zhang, T. T. Man, M. Wu and C. C. Chen, Chem. – Asian J., 2013, 8, 1076–1089 CrossRef CAS PubMed.
- D. Y. Kim, N. Jiten Singh, H. M. Lee and K. S. Kim, Chem. – Eur. J., 2009, 15, 5598–5604 CrossRef CAS PubMed.
- G. Xia, X. Yu, Y. Guo, Z. Wu, C. Yang, U. Liu and S. Dou, Chem. – Eur. J., 2010, 16, 3763–3769 CrossRef CAS PubMed.
- K. Shimoda, Y. Zhang, T. Ichikawa, H. Miyaoka and Y. Kojima, J. Mater. Chem., 2011, 21, 2609–2615 RSC.
- A. T. Luedtke and T. Autrey, Inorg. Chem., 2010, 49, 3905–3910 CrossRef CAS PubMed.
- Y. S. Chua, G. Wu, Z. Xiong, T. He and P. Chen, Chem. Mater., 2009, 21, 4899–4904 CrossRef CAS.
- W. Li, L. Miao, R. H. Scheicher, Z. Xiong, G. Wu, C. M. Araújo, A. Blomqvist, R. Ahuja, Y. Feng and P. Chen, Dalton Trans., 2012, 41, 4754–4764 RSC.
- H. Wu, W. Zhou, F. E. Pinkerton, M. S. Meyer, Q. Yao, S. Gadipelli, T. J. Udovic, T. Yildirim and J. J. Rush, Chem. Commun., 2011, 47, 4102–4104 RSC.
- G. Xia, Y. Tan, X. Chen, Z. Guo, H. Liu and X. Yu, J. Mater. Chem. A, 2013, 1, 1810–1820 CAS.
- A. Staubitz, A. P. Soto and I. Manners, Angew. Chem., Int. Ed., 2008, 47, 6212–6215 CrossRef CAS PubMed.
- J. R. Vance, A. P. M. Robertson, K. Lee and I. Manners, Chem. – Eur. J., 2011, 17, 4099–4103 CrossRef CAS PubMed.
- S. Bhunya, P. M. Zimmerman and A. Paul, ACS Catal., 2015, 5, 3478–3493 CrossRef CAS.
- A. P. M. Robertson, E. M. Leitao, T. Jurca, M. F. Haddow, H. Helten, G. C. Lloyd-Jones and I. Manners, J. Am. Chem. Soc., 2013, 135, 12670–12683 CrossRef CAS PubMed.
- A. N. Marziale, A. Friedrich, I. Klopsch, M. Drees, V. R. Celinski, J. Schmedt auf der Günne and S. Schneider, J. Am. Chem. Soc., 2013, 135, 13342–13355 CrossRef CAS PubMed.
- R. T. Baker, J. C. Gordon, C. W. Hamilton, N. J. Henson, P. H. Lin, S. Maguire, M. Murugesu, B. L. Scott and N. C. Smythe, J. Am. Chem. Soc., 2012, 134, 5598–5609 CrossRef CAS PubMed.
- Y. S. Chua, P. Chen, G. Wu and Z. Xiong, Chem. Commun., 2011, 47, 5116–5129 RSC.
- A. Staubitz, A. P. M. Robertson and I. Manners, Chem. Rev., 2010, 110, 4079–4124 CrossRef CAS PubMed.
- A. Staubitz, A. P. M. Robertson, M. E. Sloan and I. Manners, Chem. Rev., 2010, 110, 4023–4078 CrossRef CAS PubMed.
- R. J. Less, R. L. Melen and D. S. Wright, RSC Adv., 2012, 2, 2191–2199 RSC.
- H. C. Johnson, T. N. Hooper and A. S. Weller, Top. Organomet. Chem., 2015, 49, 153–220 CrossRef.
- X. Chen, J.-C. Zhao and S. G. Shore, Acc. Chem. Res., 2013, 46, 2666–2675 CrossRef CAS PubMed.
- D. J. Wolstenholme, J. T. Titah, F. N. Che, K. T. Traboulsee, J. Flogeras and G. S. McGrady, J. Am. Chem. Soc., 2011, 133, 16598–16604 CrossRef CAS PubMed.
- D. J. Wolstenholme, J. Flogeras, F. N. Che, A. Decken and G. S. McGrady, J. Am. Chem. Soc., 2013, 135, 2439–2442 CrossRef CAS PubMed.
- D. J. Wolstenholme, K. T. Traboulsee, A. Decken and G. S. McGrady, Organometallics, 2010, 29, 5769–5772 CrossRef CAS.
- T. D. Forster, H. M. Tuononen, M. Parvez and R. Roesler, J. Am. Chem. Soc., 2009, 131, 6689–6691 CrossRef CAS PubMed.
- D. Vidovic, D. A. Addy, T. Krämer, J. McGrady and S. Aldridge, J. Am. Chem. Soc., 2011, 133, 8494–8497 CrossRef CAS PubMed.
- M. Shimoi, S. Nagai, M. Ichikawa, Y. Kawano, K. Katoh, M. Uruichi and H. Ogino, J. Am. Chem. Soc., 1999, 121, 11704–11712 CrossRef CAS.
- T. Kakizawa, Y. Kawano and M. Shimoi, Organometallics, 2001, 20, 3211–3213 CrossRef CAS.
- C. Lambert and P. von R. Schleyer, Angew. Chem., Int. Ed. Engl., 1994, 33, 1129–1140 CrossRef.
- H. Wu, W. Zhou and T. Yildirim, J. Am. Chem. Soc., 2008, 14834–14839 CrossRef CAS PubMed.
- T. M. Douglas, A. B. Chaplin, A. S. Weller, X. Yang and M. B. Hall, J. Am. Chem. Soc., 2009, 131, 15440–15456 CrossRef CAS PubMed.
- K. Miwa, N. Ohba, S. Towata, Y. Nakamori and S. Orimo, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 245120 CrossRef.
- K. Miwa, M. Aoki, T. Noritake, N. Ohba, Y. Nakamori, S. Towata, A. Züttel and S. Orimo, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 155122 CrossRef.
- Z. Xiong, G. Wu, Y. S. Chua, J. Hu, T. He, W. Xu and P. Chen, Energy Environ. Sci., 2008, 1, 360–363 CAS.
- W. T. Klooster, T. F. Koetzle, P. E. M. Siegbahn, T. B. Richardson and R. H. Crabtree, J. Am. Chem. Soc., 1999, 121, 6337–6343 CrossRef CAS.
- D. J. Wolstenholme, K. T. Traboulsee, Y. Hua, L. A. Calhoun and G. S. McGrady, Chem. Commun., 2012, 48, 2597–2599 RSC.
- D. J. Wolstenholme, J. L. Dobson and G. S. McGrady, Dalton Trans., 2015, 44, 9718–9731 RSC.
- C. F. Matta, J. Hernández-Trujillo, T.-H. Tang and R. F. W. Bader, Chem. – Eur. J., 2003, 9, 1940–1951 CrossRef CAS PubMed.
- J. Echeverría, G. Aullón, D. Danovich, S. Shaik and S. Alvarez, Nat. Chem., 2011, 3, 323–330 CrossRef PubMed.
- D. J. Wolstenholme and T. S. Cameron, J. Phys. Chem. A, 2006, 110, 8970–8978 CrossRef CAS PubMed.
- S. Bhattacharya, Z. Xiong, G. Wu, P. Chen, Y. P. Feng, C. Majumder and G. P. Das, J. Phys. Chem. C, 2012, 2–7 Search PubMed.
- Z. Tang, Y. Tan, X. Chen and X. Yu, Chem. Commun., 2012, 48, 9296 RSC.
- C. Wu, G. Wu, Z. Xiong, X. Han, H. Chu, T. He and P. Chen, Chem. Mater., 2010, 22, 3–5 CrossRef CAS.
- K. J. Fijalkowski, R. V Genova, Y. Filinchuk, A. Budzianowski, M. Derzsi, T. Jaroń, P. J. Leszczyński and W. Grochala, Dalton Trans., 2011, 21, 4407–4413 RSC.
- Y. S. A. Chua, W. A. B. Li, G. A. Wu, Z. A. Xiong and P. A. Chen, Chem. Mater., 2012, 24, 3574–3581 CrossRef CAS.
- X. Kang, J. Luo, Q. Zhang and P. Wang, Dalton Trans., 2011, 40, 3799–3801 RSC.
- K. Wang, J.-G. Zhang, T. Li, Y. Liu, T. Zhang and Z.-N. Zhou, Int. J. Hydrogen Energy, 2015, 40, 2500–2508 CrossRef CAS.
- H. Nöth, S. Thomas and M. Schmidt, Chem. Ber., 1996, 129, 451–458 CrossRef.
- F. Dornhaus and M. Bolte, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 63, m41–m42 Search PubMed.
- P. C. Keller, Inorg. Chem., 1975, 14, 438–440 CrossRef CAS.
- P. Bellham, M. S. Hill and G. Kociok-Köhn, Dalton Trans., 2015, 3, 3–6 Search PubMed.
- T. D. Forster, H. M. Tuononen, M. Parvez and R. Roesler, J. Am. Chem. Soc., 2009, 131, 6689–6691 CrossRef CAS PubMed.
- H. Helten, B. Dutta, J. R. Vance, M. E. Sloan, M. F. Haddow, S. Sproules, D. Collison, G. R. Whittell, C. L. J. Guy and I. Manners, Angew. Chem., Int. Ed., 2013, 52, 437–440 CrossRef CAS PubMed.
- J. Intemann, P. Sirsch and S. Harder, Chem. – Eur. J., 2014, 20, 11204–11213 CrossRef CAS PubMed.
- S. Schulz, T. Eisenmann, D. Schuchmann, M. Bolte, M. Kirchner, R. Boese, J. Spielmann and S. Harder, Z. Naturforsch., B: J. Chem. Sci., 2009, 64, 1397–1400 CAS.
- S. Harder and J. Spielmann, Chem. Commun., 2011, 47, 11945–11947 RSC.
- Y. Jiang, O. Blacque, T. Fox, C. M. Frech and H. Berke, Organometallics, 2009, 28, 5493–5504 CrossRef CAS.
- U. Helmstedt, L. Vendier, G. Alcaraz, E. Clot and S. Sabo-Etienne, Inorg. Chem., 2011, 11039–11045 Search PubMed.
- A. J. Mountford, W. Clegg, S. J. Coles, R. W. Harrington, P. N. Horton, S. M. Humphrey, M. B. Hursthouse, J. A. Wright and S. J. Lancaster, Chem. – Eur. J., 2007, 13, 4535–4547 CrossRef CAS PubMed.
- E. A. Jacobs, A.-M. Fuller, S. J. Lancaster and J. A. Wright, Chem. Commun., 2011, 47, 5870–5872 RSC.
- E. A. Jacobs, A. Fuller, S. J. Coles, G. A. Jones, G. J. Tizzard, J. A. Wright and S. J. Lancaster, Chem. – Eur. J., 2012, 18, 8647–8658 CrossRef CAS PubMed.
- V. C. Gibson, J. A. Segal, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 2000, 643, 7120–7121 CrossRef.
- S. P. Green, C. Jones and A. Stasch, Science, 2007, 318, 1754–1757 CrossRef CAS PubMed.
- M. R. Crimmin, I. J. Casely and M. S. Hill, J. Am. Chem. Soc., 2005, 127, 2042–2043 CrossRef CAS PubMed.
- J. Spielmann, F. Buch and S. Harder, Angew. Chem., Int. Ed., 2008, 47, 9434–9438 CrossRef CAS PubMed.
- C. Ruspic and S. Harder, Inorg. Chem., 2007, 46, 10426–10433 CrossRef CAS PubMed.
- S. Harder and J. Brettar, Angew. Chem., Int. Ed., 2006, 45, 3474–3478 CrossRef CAS PubMed.
- J. Spielmann, G. Jansen, H. Bandmann and S. Harder, Angew. Chem., Int. Ed., 2008, 47, 6290–6295 CrossRef PubMed.
- T. Richardson, S. de Gala, R. H. Crabtree and P. E. M. Siegbahn, J. Am. Chem. Soc., 1995, 117, 12875–12876 CrossRef CAS.
- J. Spielmann and S. Harder, J. Am. Chem. Soc., 2009, 131, 5064–5065 CrossRef CAS PubMed.
- P. Bellham, M. S. Hill and G. Kociok-Köhn, Organometallics, 2014, 33, 5716–5721 CrossRef CAS.
- M. E. Bowden, G. J. Gainsford and W. T. Robinson, Aust. J. Chem., 2007, 60, 149–153 CrossRef CAS.
- J. Luo, X. Kang and P. Wang, Energy Environ. Sci., 2013, 6, 1018–1025 CAS.
- Y. S. Chua, G. Wu, Z. Xiong, A. Karkamkar, J. Guo, M. Jian, M. W. Wong, T. Autrey and P. Chen, Chem. Commun., 2010, 46, 5752–5754 RSC.
- S. Harder, J. Spielmann and B. Tobey, Chem. – Eur. J., 2012, 18, 1984–1991 CrossRef CAS PubMed.
- D. J. Liptrot, M. S. Hill, M. F. Mahon and D. J. MacDougall, Chem. – Eur. J., 2010, 16, 8508–8515 CrossRef CAS PubMed.
- M. S. Hill, M. Hodgson, D. J. Liptrot and M. F. Mahon, Dalton Trans., 2011, 40, 7783–7790 RSC.
- J. Spielmann, D. E. J. Piesik and S. Harder, Chem. – Eur. J., 2010, 16, 8307–8318 CrossRef CAS PubMed.
- J. Spielmann, M. Bolte and S. Harder, Chem. Commun., 2009, 6934–6936 RSC.
- J. Luo, X. Kang and P. Wang, ChemPhysChem, 2010, 11, 2152–2157 CrossRef CAS PubMed.
- W. Li, G. Wu, Y. Chua, Y. P. Feng and P. Chen, Inorg. Chem., 2012, 51, 76–87 CrossRef CAS PubMed.
- J. H. Luo, X. D. Kang and P. Wang, J. Phys. Chem. C, 2010, 114, 10606–10611 CAS.
- X. Kang, H. Wu, J. Luo, W. Zhou and P. Wang, J. Mater. Chem., 2012, 22, 13174–13179 RSC.
- C. Jones, S. J. Bonyhady, S. Nembenna and A. Stasch, Eur. J. Inorg. Chem., 2012, 2596–2601 CrossRef CAS.
- J. Spielmann, D. Piesik, B. Wittkamp, G. Jansen and S. Harder, Chem. Commun., 2009, 3455–3456 RSC.
- S. Harder, J. Spielmann and J. Intemann, Dalton Trans., 2014, 43, 14284–14290 RSC.
- C. A. Jaska, K. Temple, A. J. Lough and I. Manners, Chem. Commun., 2001, 962–963 RSC.
- C. A. Jaska, K. Temple, A. J. Lough and I. Manners, J. Am. Chem. Soc., 2003, 125, 9424–9434 CrossRef CAS PubMed.
- M. E. Sloan, T. J. Clark and I. Manners, Inorg. Chem., 2009, 48, 2429–2435 CrossRef CAS PubMed.
- M. C. Denney, V. Pons, T. J. Hebden, D. M. Heinekey and K. I. Goldberg, J. Am. Chem. Soc., 2006, 128, 12048–12049 CrossRef CAS PubMed.
- B. L. Dietrich, K. I. Goldberg, D. M. Heinekey, T. Autrey and J. C. Linehan, Inorg. Chem., 2008, 47, 8583–8585 CrossRef CAS PubMed.
- N. Blaquiere, S. Diallo-Garcia, S. I. Gorelsky, D. A. Black and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 14034–14035 CrossRef CAS PubMed.
- M. Käβ, A. Friedrich, M. Drees and S. Schneider, Angew. Chem., Int. Ed., 2009, 48, 905–907 CrossRef PubMed.
- D. Pun, E. Lobkovsky and P. J. Chirik, Chem. Commun., 2007, 3297–3299 RSC.
- T. J. Clark, C. A. Russell and I. Manners, J. Am. Chem. Soc., 2006, 128, 9582–9583 CrossRef CAS PubMed.
- M. E. Sloan, A. Staubitz, T. J. Clark, C. A. Russell, G. C. Lloyd-Jones and I. Manners, J. Am. Chem. Soc., 2010, 132, 3831–3841 CrossRef CAS PubMed.
- S. Harder and D. Naglav, Eur. J. Inorg. Chem., 2010, 2836–2840 CrossRef CAS.
- S. Harder, Dalton Trans., 2010, 39, 6677–6681 RSC.
- S. P. Green, C. Jones and A. Stasch, Angew. Chem., Int. Ed., 2008, 47, 9079–9083 CrossRef CAS PubMed.
- M. D. Anker, M. Arrowsmith, P. Bellham, M. S. Hill, G. Kociok-Köhn, D. J. Liptrot, M. F. Mahon and C. Weetman, Chem. Sci., 2014, 5, 2826–2830 RSC.
- C. Y. Tang, A. L. Thompson and S. Aldridge, Angew. Chem., Int. Ed., 2010, 49, 921–925 CrossRef CAS PubMed.
- C. Y. Tang, N. Phillips, J. I. Bates, A. L. Thompson, M. J. Gutmann and S. Aldridge, Chem. Commun., 2012, 48, 8096–8098 RSC.
- H. C. Johnson, A. P. M. Robertson, A. B. Chaplin, L. J. Sewell, A. L. Thompson, M. F. Haddow, I. Manners and A. S. Weller, J. Am. Chem. Soc., 2011, 133, 11076–11079 CrossRef CAS PubMed.
- A. P. M. Robertson, E. M. Leitao and I. Manners, J. Am. Chem. Soc., 2011, 133, 19322–19325 CrossRef CAS PubMed.
- P. Bellham, M. S. Hill, D. J. Liptrot, D. J. MacDougall and M. F. Mahon, Chem. Commun., 2011, 47, 9060–9062 RSC.
- P. Bellham, M. S. Hill, G. Kociok-Köhn and D. J. Liptrot, Dalton Trans., 2013, 737–745 RSC.
- Y. Tan and X. Yu, RSC Adv., 2013, 3, 23879–23894 RSC.
Footnote |
| † Although the strict definition of the term ‘organometallic‘ refers only to species containing a metal carbon bond, we feel that this term best describes the coordination chemistry and on-metal reactivity reviewed in this article. We refer the reader to the scope of the journal ‘Organometallics’: “…manuscripts dealing with metal-containing compounds that do not contain metal–carbon bonds will be considered if there is a close relationship between the subject matter and the principles and practice of organometallic chemistry…”. |
| This journal is © The Royal Society of Chemistry 2016 |