
Recent advances in the application of deep eutectic solvents as sustainable media as well as catalysts in organic reactions
Peng Liu
,
Jian-Wu Hao
,
Li-Ping Mo
and
Zhan-Hui Zhang
*
College of Chemistry and Material Science, Hebei Normal University, Shijiazhuang 050024, P. R. China. E-mail: zhanhui@mail.nankai.edu.cn; zhanhui@mail.hebtu.edu.cn
First published on 26th May 2015
Abstract
Deep eutectic solvents (DESs), also known as deep eutectic ionic liquids (DEILs) or low-melting mixtures (LMMs) or low transition temperature mixtures (LTTMs) in the literature, have become more and more attractive in recent years due to their interesting properties and benefits, such as low cost of components, easy to prepare, tunable physicochemical properties, negligible vapor pressure, non-toxicity, biorenewability and biodegradability. These eutectic mixtures have been widely used as green and sustainable media as well as catalysts in many chemical processes. This review focuses on recent advances using DESs in organic reactions including addition reactions, cyclization reactions, replacement reactions, multicomponent reactions, condensation reactions, oxidation reactions, and reducing reactions.
 Peng Liu | Peng Liu was born in 1988 in Hebei, China. He received his Bachelor degree from Cangzhou Normal University, China in 2014. He then moved to Hebei Normal University to continue her graduate study. Under the supervision of Professor Zhan-Hui Zhang, his current work involves the development of new catalytic transformations in deep eutectic solvents. |
 Jian-Wu Hao | Jian-Wu Hao was born in 1987 in Hebei, China. He graduated from Huihua College of Heibei Normal University in 2012 and received her B.Sc degree in Chemistry. Presently, he is working as a postgraduate towards his M.Sc under the supervision of Professor Zhan-Hui Zhang at the Hebei Normal University. Her major research interest focuses on the application of deep eutectic solvents as reaction media in different chemical transformations. |
 Li-Ping Mo | Li-Ping Mo received her Bachelor degree in 1992 from Hebei Normal College. Currently, she is an associate professor of chemistry at Hebei Normal University. |
 Zhan-Hui Zhang | Zhan-Hui Zhang received his PhD degree in organic chemistry under the supervision of Prof. Yong-Mei Wang in 2006 at Nankai University, China. Presently, he is a professor at Hebei Normal University and head of a research group for green chemistry. His current research interests are dedicated towards the development of new green solvents, new reagents and catalysts in organic synthesis. To date he has published around 100 scientific publications with h-index of 37. |
1. Introduction
Developing a cost effective and environmentally benign solvent system is of the utmost importance in chemical industry. One of the proposals is the replacement of conventional hazardous volatile organic solvents by nonvolatile alternatives so that they do not emit toxic or flammable vapors at a wide range of temperatures.1 During the past few years, some green solvents have appeared as innocuous solvents, such as water,2 supercritical fluids,3 perfluorinated solvents,4 glycerol and derived solvents,5 bio-based solvents,6 and ionic liquids (ILs).7 However, the use of these solvents is still limited by many problems. Some compounds, substrates, or reagents have poor solubility or stability in water. Sophisticated equipment is usually required when supercritical fluids are employed. The synthesis process of ILs is complex and expensive and ILs are difficult to purify, which have prevented their widespread usage in the chemical processes.In recent years, deep eutectic solvents (DESs) based on choline chloride (ChCl) first introduced by Abbott and co-workers,8 low-melting mixtures (LMMs) of sugar, urea and inorganic salts first described by König and co-workers,9 natural deep eutectic solvents (NADESs) first recommended by Choi and co-workers,10 low-transition temperature mixtures (LTTMs) proposed by Kroon and co-workers,11 and deep eutectic ionic liquids (DEILs) reported by Hillman' group12 as low-cost eutectic mixtures, with similar physical properties and phase behavior to ILs, are gaining increasing attention in chemistry fields. They are defined as combinations of two or three safe and cheap components which are capable of self-association, through hydrogen bond interactions to form a eutectic mixture, which is a liquid at temperature lower than 100 °C, with a melting point lower than that of the each individual component. The mixing of them creates a disruption on the crystalline structure of the quaternary ammonium salt, triggering a depression in the melting point and thus generating liquids at room temperature. In 2003, Abbott and co-workers first reported that chloline chloride (ChCl) and urea could form a DES by hydrogen-bonding interactions, which appeared as liquid state at room temperature (Scheme 1).8a Compared with ILs, the synthetic processes of these eutectic mixtures are relatively simple, which only mix one or more hydrogen bond donors (HBDs) and one or more hydrogen bond acceptors (HBAs) from natural and readily starting materials in the proper ratio under heating until a homogenous liquid is formed without any purification with 100% atom utilization rate.13 Some typical structures of HBDs and HBAs for DES synthesis are listed in Scheme 2. DESs generally present properties such as low vapor pressure, low toxicity, wide liquid range, water compatibility, biodegradability and non-flammability. The task-specific DESs with different physicochemical properties such as freezing point (Tf), density, viscosity and ionic conductivity can be prepared. Table 1 summarizes main physical properties of some typical DESs.
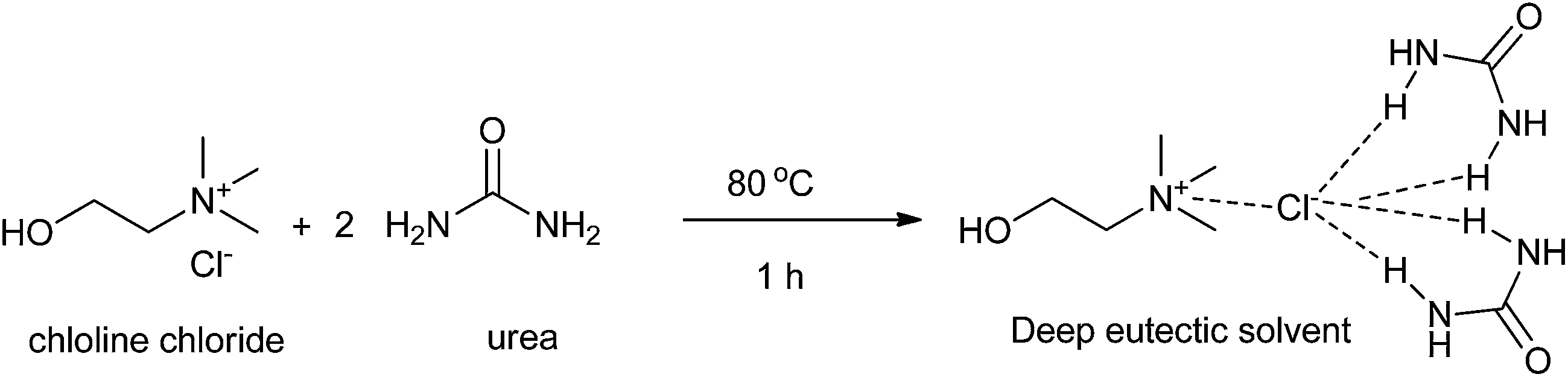 | ||
| Scheme 1 Preparation of DES ChCl/urea from chloline chloride and urea. | ||
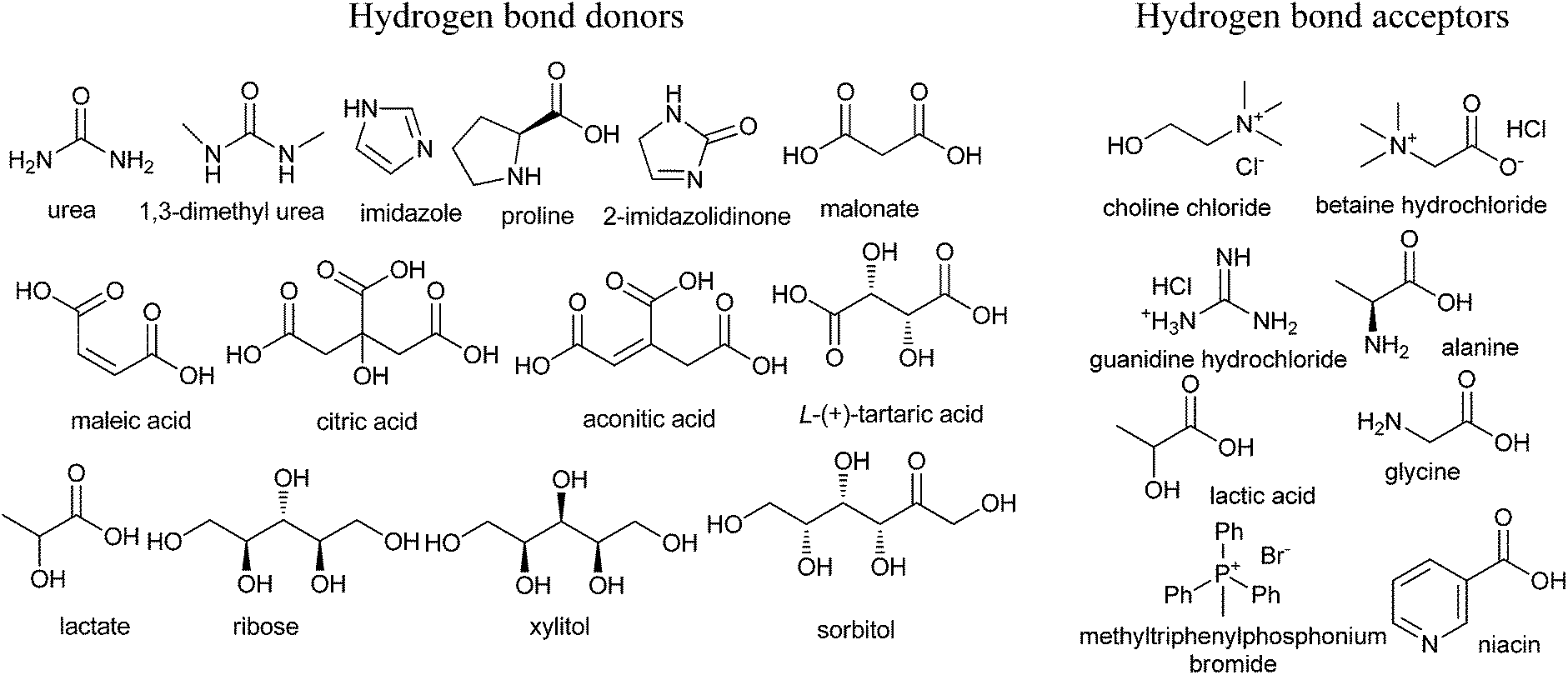 | ||
| Scheme 2 Typical structures of hydrogen bond donors (HBDs) and bond acceptors (HBAs) for DES synthesis. | ||
| Composition (mole ratio) | Tf (°C) | Density (ρ, g cm−3) | Viscosity (cP) | Conductivity (κ, mS cm−1) |
|---|---|---|---|---|
ChCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :urea (1:2) :urea (1:2) |
12 (ref. 13) | 1.25 (ref. 13) | 750 (25 °C)13 | 0.199 (40 °C)13 |
| ChCl:glycerol (1:2) |
−40 (ref. 13) | 1.18 (ref. 13) | 376 (20 °C)13 | 1.05 (20 °C)13 |
| ChCl:imidazole (3:7) |
56 (ref. 13) | 15 (70 °C)13 | 12.0 (60 °C)13 | |
| ChCl:ethylene glycol (1:2) |
−66 (ref. 14) | 1.12 (ref. 14) | 37 (25 °C)14 | 7.61 (20 °C)14 |
| ChCl:malonic acid (1:1) |
10 (ref. 8b) | 3340 (ref. 15) | 0.36 (ref. 15) | |
| ChCl:citric acid (1:1) |
69 (ref. 8b) | |||
| ChCl:oxalic acid (1:1) |
34 (ref. 8b) | |||
| ChCl:succinic acid (1:1) |
71 (ref. 8b) | |||
| ChCl:L-(+)-tartaric acid (1:0.5) |
47 (ref. 16) | |||
| ChCl:itaconic acid (1:1) |
57 (ref. 16) | |||
| ChCl:ethylene glycol (1:2) |
−66 (ref. 14) | 1.12 (ref. 14) | 37 (25 °C)14 | 7.61 (20 °C)14 |
| ChCl:1,4-butanediol (1:3) |
−32 (ref. 14) | 1.06 (ref. 14) | 140 (20 °C)13 | 1.64 (40 °C)13 |
| ChCl:1-methyl urea (1:2) |
29 (ref. 8c) | |||
| ChCl:2,2,2-trifluoroacetamide (1:2) |
1.342 (ref. 14) | 77 (40 °C)14 | 0.286 (40 °C)14 | |
| ChCl:ZnCl2 (1:2) |
24 (ref. 15) | 8500 (25 °C)14 | 0.06 (42 °C)14 | |
| ChCl:FeCl3 (1:2) |
65 (ref. 15) | |||
| ChCl:SnCl2 (1:2) |
37 (ref. 15) | |||
| EtNH3Cl:2,2,2-trifluoroacetamide (1:2) |
1.342 (ref. 13) | 77 (40 °C)13 | 0.286 (40 °C)13 | |
| EtNH3Cl:urea (1:1.5) |
1.14 (ref. 14) | 128 (40 °C)14 | 0.348 (40 °C)14 | |
| ZnCl2:urea (1:3.5) |
1.63 (ref. 14) | 11340 (25 °C)14 |
0.06 (42 °C)13 | |
| Me(Ph)3PBr:glycerol (1:2) |
3–4 (ref. 13) | 1.31 (ref. 13) | ||
| Me(Ph)3PBr:ethylene glycol (1:3) |
−46.25 (ref. 14) | 1.25 (ref. 14) | ||
| Bu4NBr:imidazole (3:7) |
810 (20 °C)13 | 0.24 (20 °C)13 | ||
| Betaine HCl:oxalic·2H2O (1:2) |
33 (ref. 17) | 1.27 (50 °C)17 | ||
| Betaine HCl:citric·H2O (1:1.5) |
48 (ref. 17) |
Up to date, DESs have been applied to diverse fields of research such as polymerizations,18 biomass processing,19 materials preparation,20 biodiesel synthesis,21 enzyme-catalyzed reactions,22 carbon dioxide adsorption,23 electrochemistry,24 extraction,25 nanotechnology,26 and organic synthesis.27 The number of publications on this issue is growing rapidly, especially over the past few years. This review focuses on mainly the applications of this new family of solvents as green media as well as catalysts in organic transformations.
2. Organic transformations in deep eutectic solvents
2.1 DES in the addition reactions
Michael addition is one of the most common methods to build a C–C bond, C–N bond, C–S bond and so on.28 In 2014, Azizi et al. disclosed an atom-economic and odorless protocol for carbon–sulfur bond formation via the one-pot reaction of alkylhalides (1), thiourea (2), and electron-deficient olefins (3) using ChCl/urea deep eutectic solvent as both the reaction medium and catalyst (Scheme 3).29 Various additives such as K2CO3, Na2CO3, NaHCO3, triethylamine, NaOH and DBU have also been employed in this thia-Michael addition, but NaOH gave the best results as compared to others. At the same time, the authors investigated the scope of this thia-Michael addition with various combinations of substrates, founding a wide range of α,β-unsaturated compounds underwent the 1,4-addition smoothly with a variety of alkyl halides and thiourea to afford a diverse set of thia-Michael products (4) in high yields.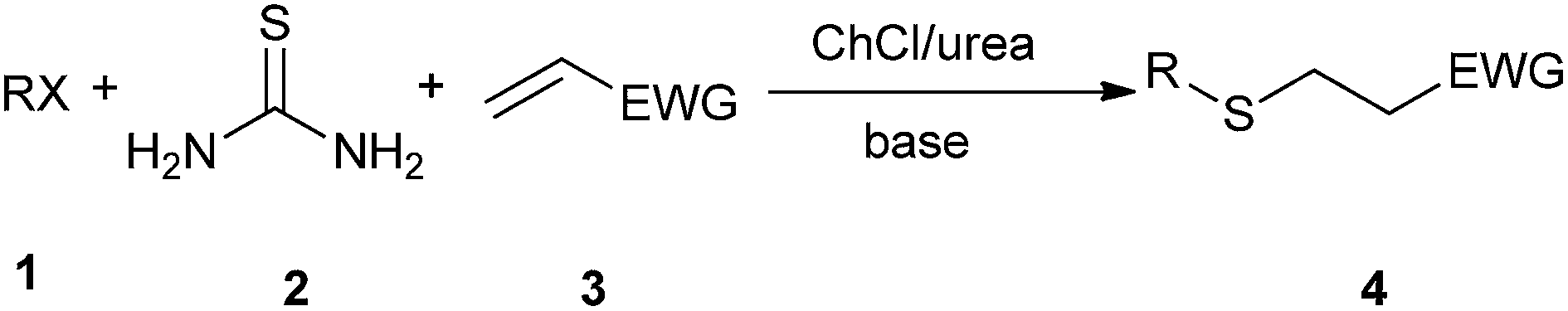 | ||
| Scheme 3 Thia-Michael addition in ChCl/urea. | ||
In 2014, Krishnakumar and co-workers have discovered a novel and green protocol for the Michael addition of N-arylhomophthalimides (5) and chalcones (6) in L-(+)-tartaric acid/DMU low-melting mixture.30 In order to optimize the reaction condition, a series of catalyst–solvent systems and various melt conditions were investigated. The results indicated that L-(+)-tartaric acid/1,3-dimethylurea (DMU) mixture was the most effective reaction medium. Based on the acidity of the melt, the authors suggested a reaction mechanism as shown in Scheme 4. First, the ability of the acidic low-melting mixture to hydrogen-bond plays an important role for activation of the C3 carbonyl group of N-arylhomophthalimide (5) to form the intermediate A. Next, the presence of intermolecular hydrogen bonding provides additional attractive forces between molecules. The low-melting mixture also might assist in improving the reactivity of chalcone. The formed in situ intermediate A attacks the chalcone (6) to form intermediate C. In a final step, the product 7 was obtained and the melt was utilized for further reaction.
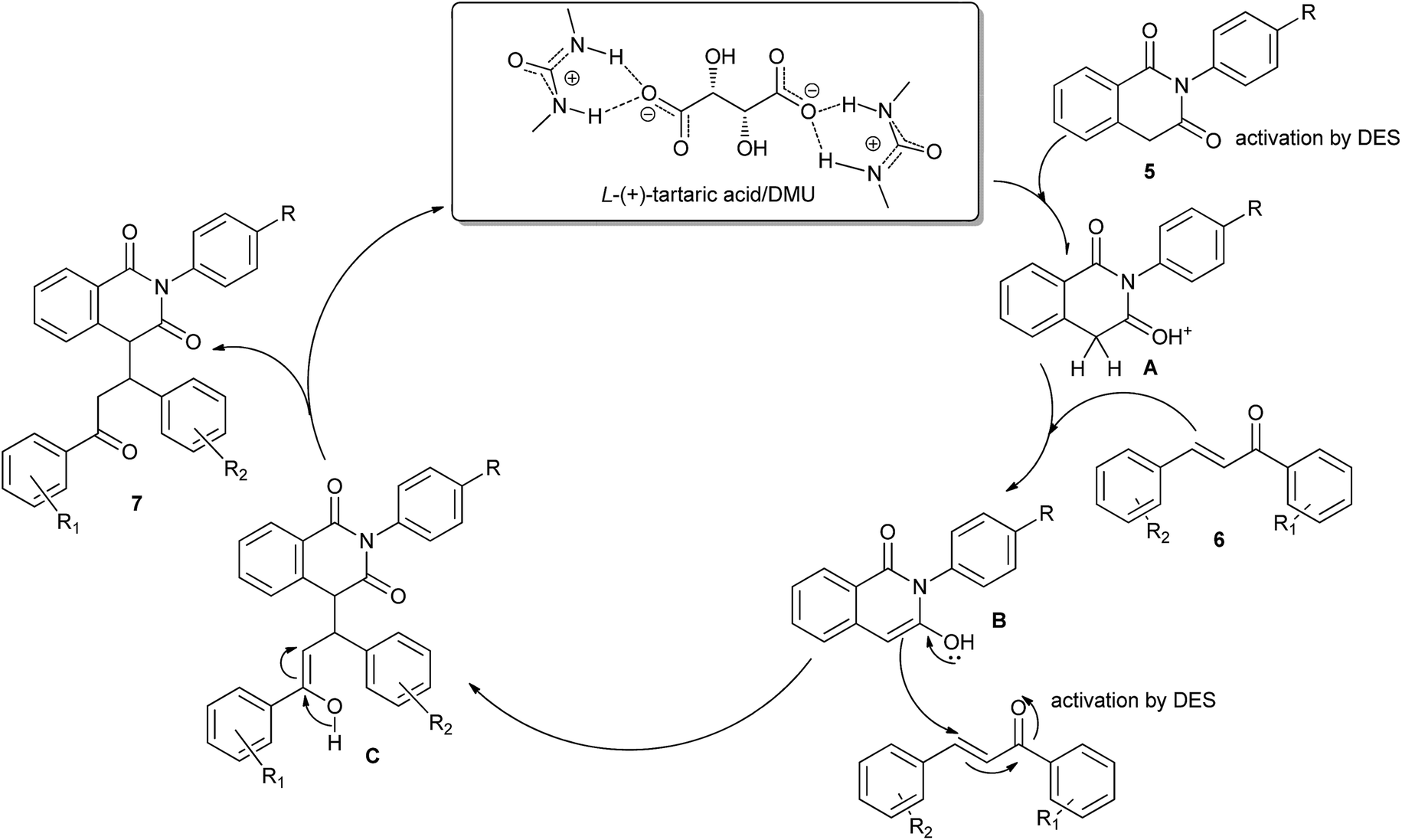 | ||
| Scheme 4 The reaction mechanism for Michael addition of N-arylhomophthalimides with chalcones in L-(+)-tartaric acid/DMU. | ||
In 2014, Yadav and co-workers have developed a green synthesis of β-functionalized ketonic derivatives (10) with the combination of ultrasound and the deep eutectic solvent ChCl/urea.31 In a comparative study, the authors carried out the conjugate addition reaction of nitromethane and 4-fluorobenzylideneacetophenone in different sets of reaction conditions, and it was found that ChCl/urea deep eutectic solvent gave the best product yield. The results demonstrated that the scope of the reaction was broad with regard to various active methylenes such as nitromethane, ethyl cyanoacetate and malononitrile. Higher yields of the products and recoverability of solvent make this method green and environmentally friendly. The proposed mechanism is outlined in Scheme 5. The stronger hydrogen-bonding capabilities of ChCl/urea results in an oxyanion intermediate (D) which on further proton abstraction gives the final addition product 10.
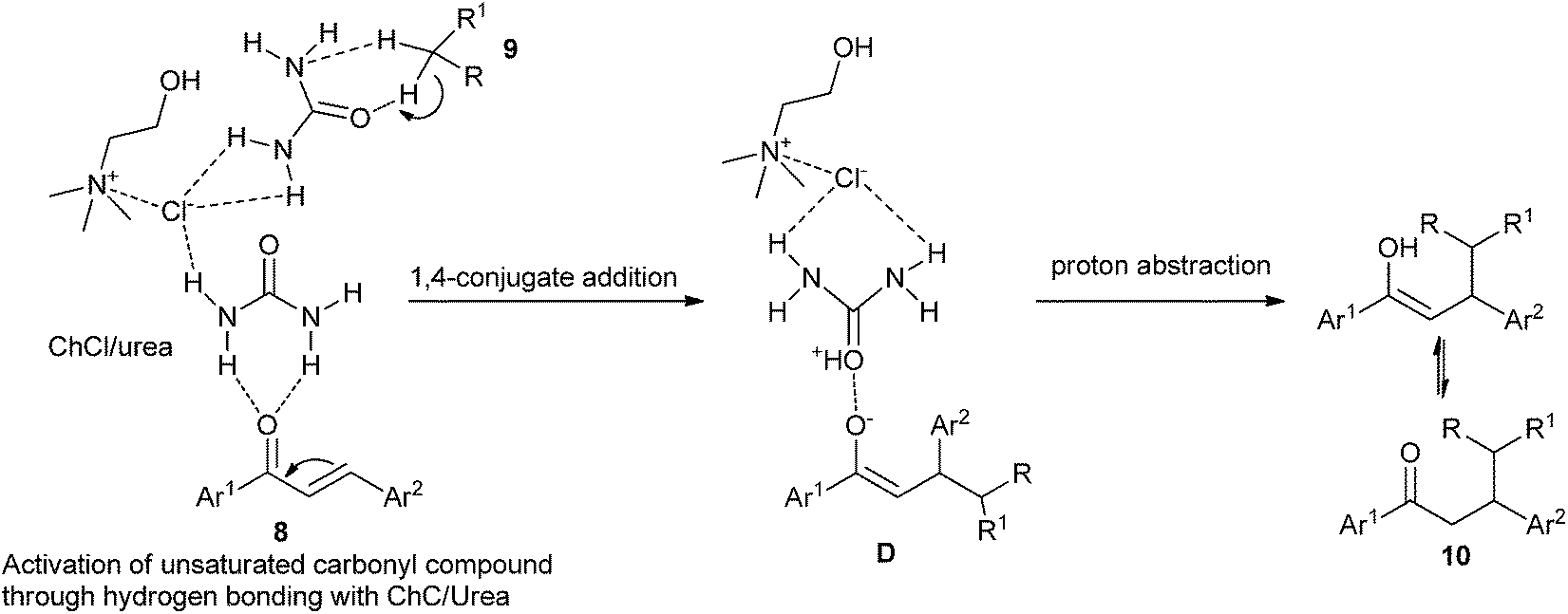 | ||
| Scheme 5 Mechanistic for synthesis of β-functionalized ketonic derivatives catalyzed by ChCl/urea. | ||
Recently, the direct C-3 alkenylation/alkylation of indoles (11) using a deep eutectic solvent of ChCl/oxalic acid was reported by Sanap et al.32 It was found C-3 alkenylation/alkylation of indoles depended on the position of the substituent on the indoles used in this reaction. C-2 substituted indole resulted in the formation of C-3 alkenylated indole derivatives (13), whereas plain indole gave bis(indolyl)carbonyl derivatives (14) instead of the C-3 alkenylation products. A plausible mechanism, as proposed by the authors, is depicted in Scheme 6. In this process, oxalic acid forms hydrogen bonding with ChCl. In the same manner, it also forms hydrogen bond with oxygen atom of electron deficient carbonyl of 1,3-dicarbonyl compounds and increases its electrophilicity, thereby facilitating the attack of indole. Subsequently, the hydroxyl group forms hydrogen bonded with DES. Finally, it facilitates a loss of water molecule to form C-3 alkenylated indole (13). In other case adduct G is prone to be attacked by a plain indole at C-3 carbon atom of alkyl chain to form C-3 alkylated product (14). In these reactions, DES plays dual role of solvent and catalyst. And DES is recovered and reused for several runs without significant loss in catalytic activity.
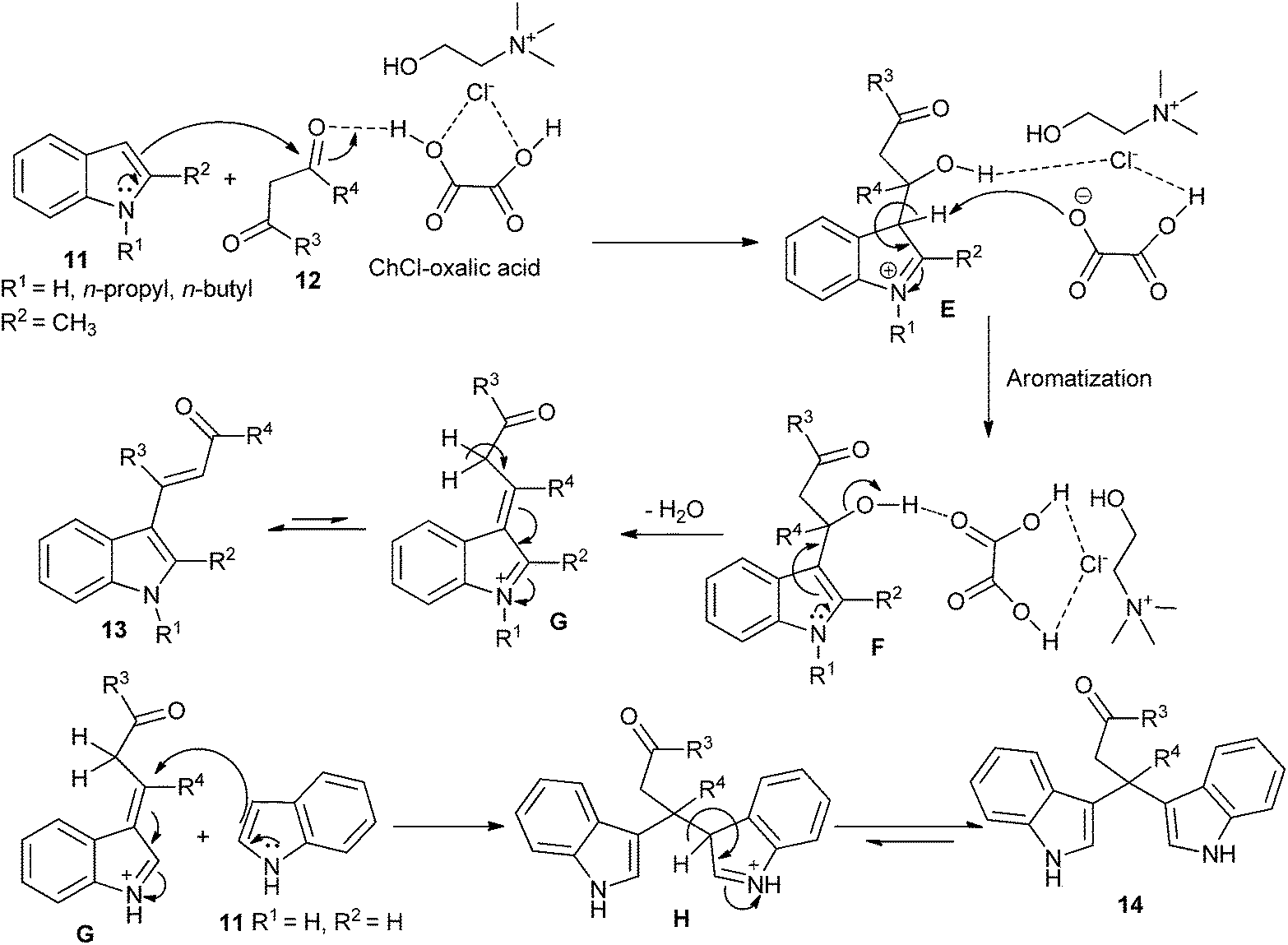 | ||
| Scheme 6 Plausible reaction mechanism for C-3 alkenylated/alkylated indoles in ChCl/oxalic acid. | ||
In 2014, Maugeri and co-workers reported that ChCl/glycerol could be used as reaction medium in lyase-catalyzed reaction. They had successfully explored for the first time the use of benzaldehyde lyase (BAL) and a thiamine-diphosphate dependent lyase (ThDP-lyase) to perform enantioselective C–C bond carboligation reactions in different DES–buffer mixtures (Scheme 7).33 By using ChCl/glycerol DES, BAL remained fully active with excellent enantioselectivity at 60:40 DES–buffer, whereas a significant denaturation was observed at 70:30 mixtures. Remarkably, the use of ChCl/urea DES as reaction media led to full conversions with BAL at such solvent–buffer proportions, suggesting that the design of both the biocatalyst and the neoteric solvent may provide useful novel reactive systems for biocatalysis in non-conventional media.
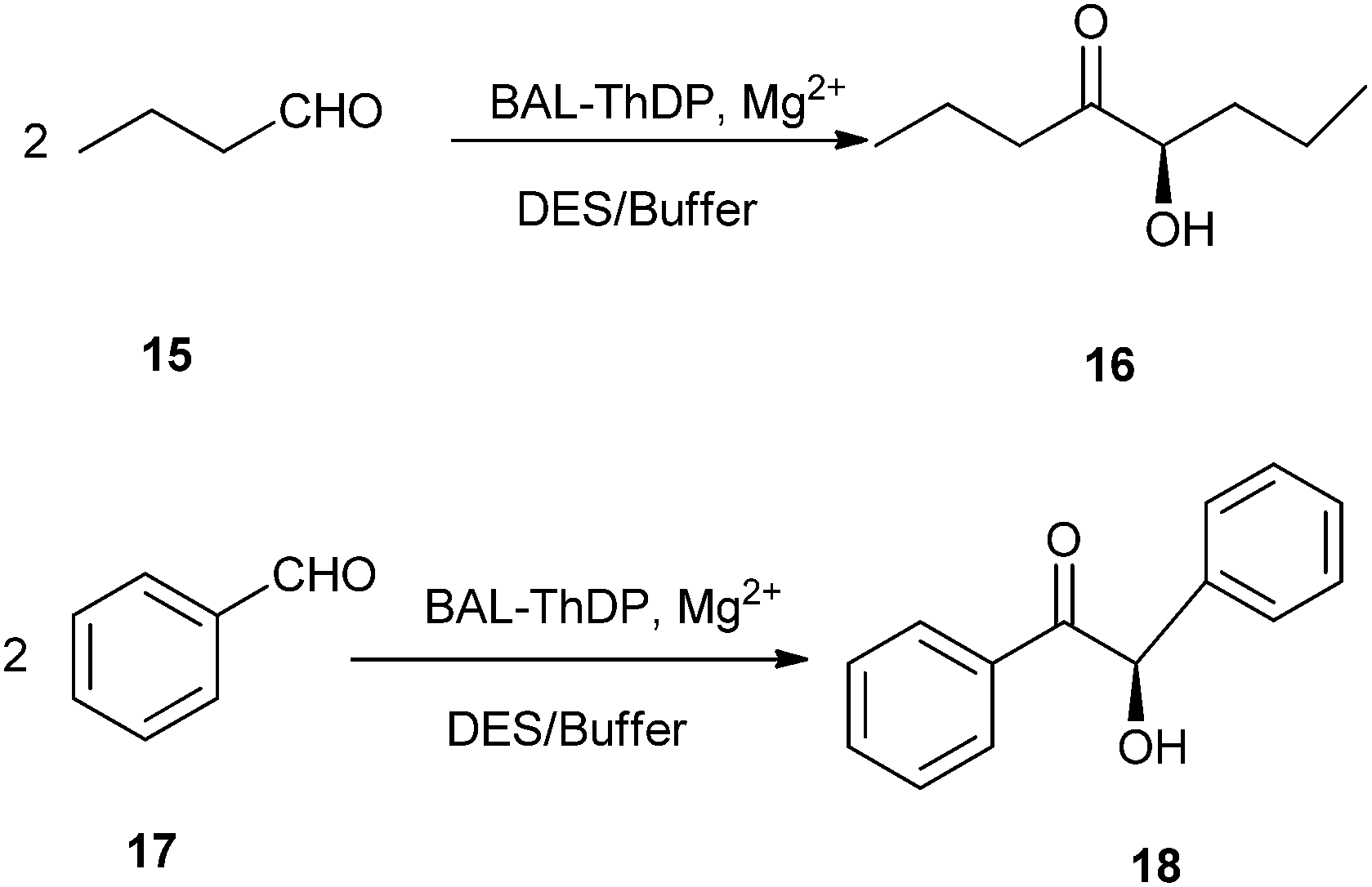 | ||
| Scheme 7 C–C bond carboligation reactions in DES–buffer. | ||
The DES was also used by Sanap et al. in synthesis of aromatic tricyanovinyl compounds (21 and 22) from nucleophilic reagents and tetracyanoethylene (20) (Scheme 8).34 Various DESs and solvents were investigated and yield obtained in ChCl/urea was much higher than those obtained in ChCl/malonic acid, ChCl/oxalic acid, ChCl/urea/ethanol and sole ChCl or urea. The reaction worked well for anilines (19) including substitution at nitrogen and consisted of various alkyl groups such as methyl, ethyl, isobutyl, n-hexyl as well as indoles (11) having substitution at 1 and 2 positions.
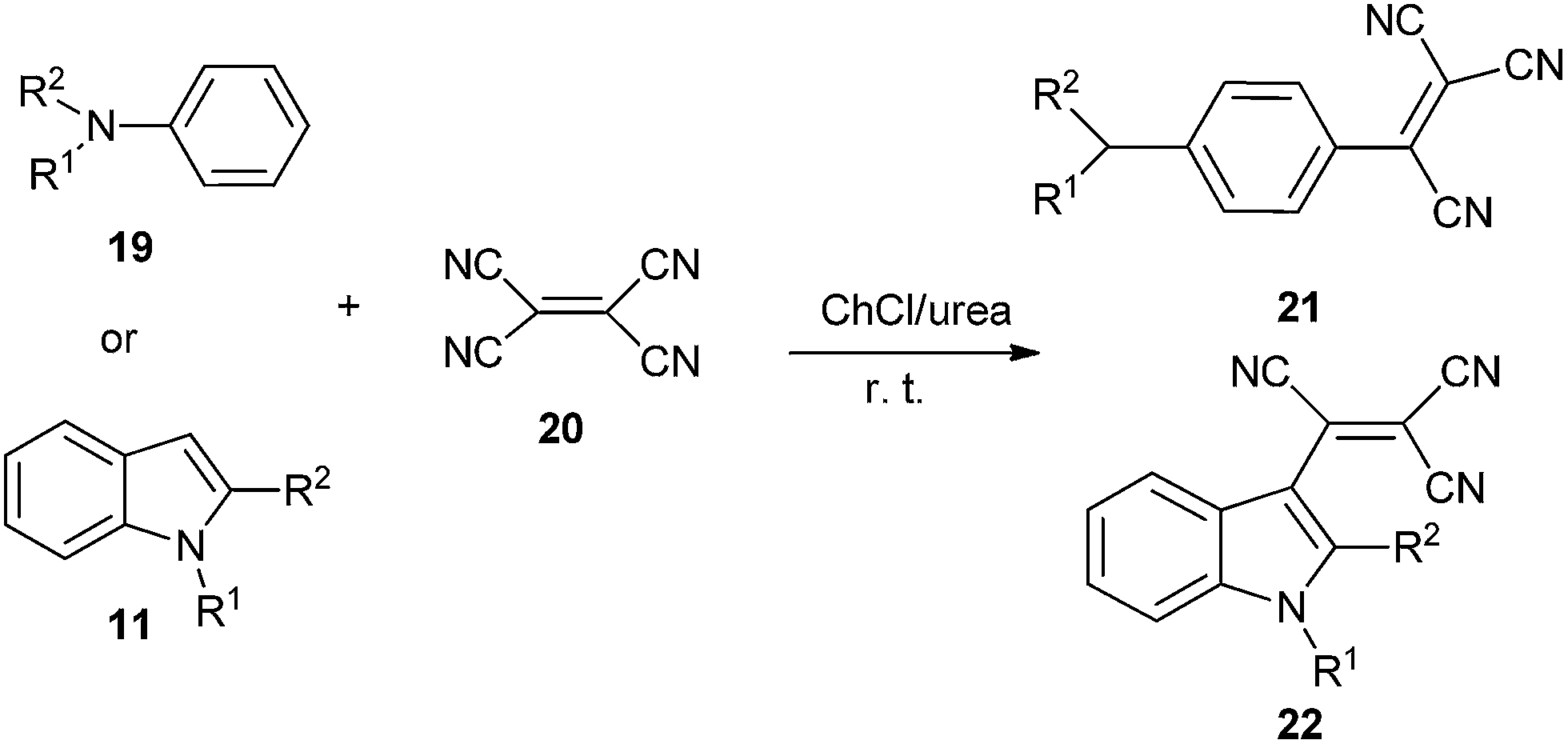 | ||
| Scheme 8 Synthesis of tricyanovinylated aromatics in ChCl/urea. | ||
A simple and efficient method have been described by Singh et al. for the use of ChCl/urea deep eutectic solvent as a catalyst in the synthesis of nitroaldol compounds (23). The effects of various solvents such as hexane, toluene, and methanol have also been studied. It was found that methanol gave the best yields of the products than others. The reaction was also extended towards synthesis of β-hydroxynitriles (24) and β-hydroxy carboxylic acids (25) (Scheme 9).35 It is the first report that DES can effectively catalyze these important C–C bond formation reactions.
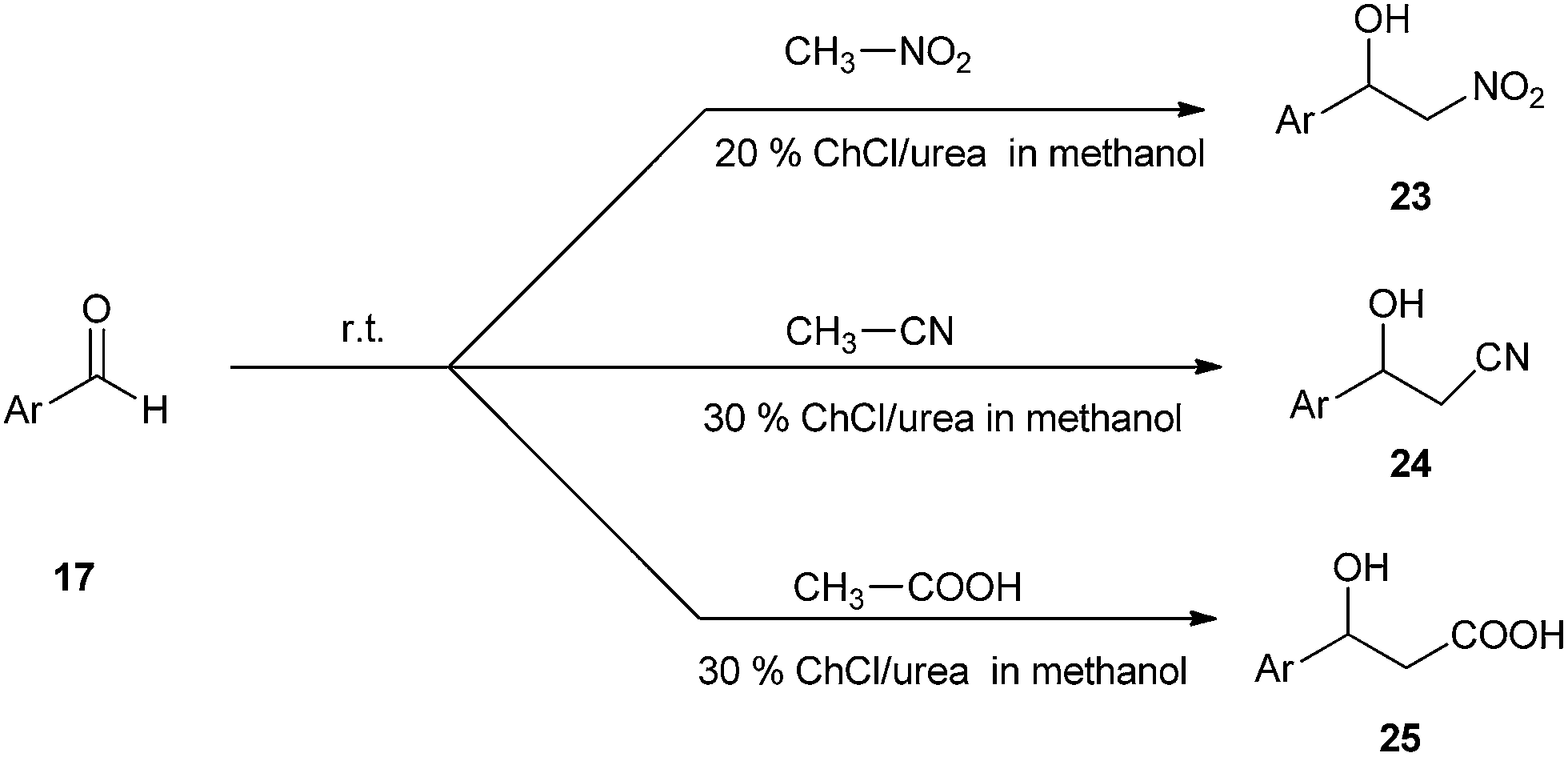 | ||
| Scheme 9 Synthesis of nitroaldol using ChCl/urea. | ||
A choline-based deep eutectic solvent ChCl/SnCl2 was also found to be a new and highly effective catalyst for the ring opening of a variety of epoxides (26) by a range of amines, thiols, alcohols, azide and cyanide, providing 1,2-difunctional ring opening products (28) in good to excellent yields with good chemo, regio, and stereoselectivities with short reaction times. Furthermore, the reactions with unsymmetrical epoxides were regioselective. In the case of aliphatic oxiranes, the reaction likely occurred through an attack by the nucleophile on the less-substituted carbon atom of the epoxide ring. The reaction of different nucleophiles with styrene oxide regioselectively yielded the products derived from the attack on the benzylic position of the epoxide (Scheme 10).36
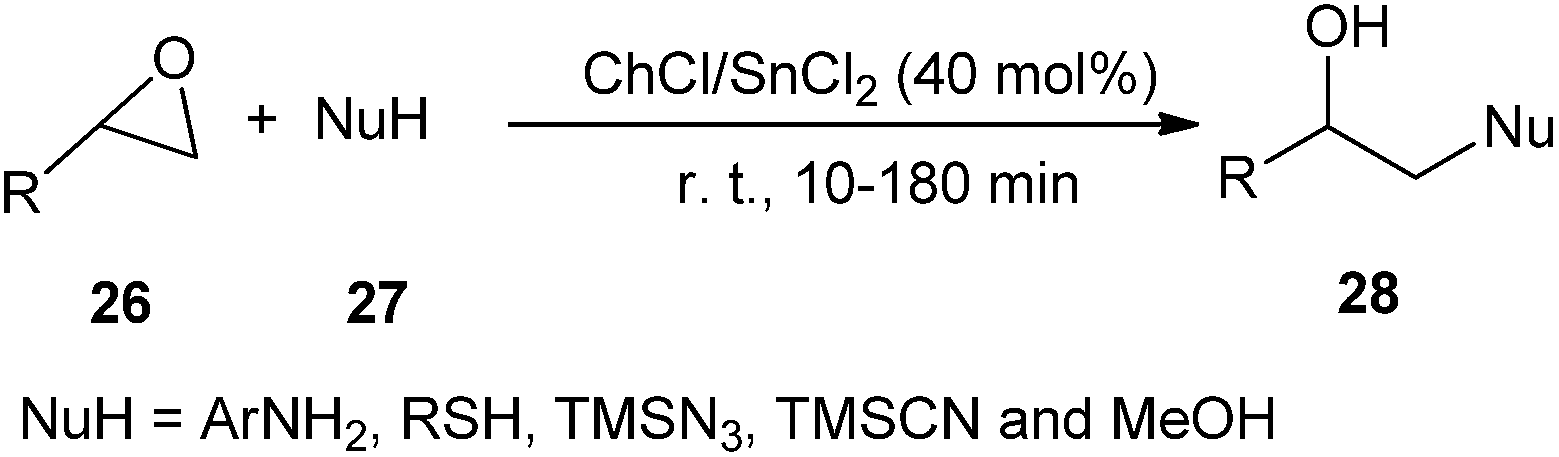 | ||
| Scheme 10 Ring opening of epoxides using /SnCl2. | ||
König's group investigated the Diels–Alder reactions in low melting mixture composed of bulk natural products, such as simple carbohydrates, sugar alcohols or citric acid, with urea and inorganic salts. In a fructose/DMU (7:3) melt, the reaction proceeded well and the Diels–Alder adduct was obtained with a quantitative yield with good endo–exo selectivity ratios (2.9:1) after 8 h of reaction at 71 °C. The addition of Sc(OTf)3 improved the endo–exo selectivity ratios (3.3:1).37 This reaction can be performed in L-carnitine/urea (2:3) melt in the presence of 5 mol% of proline as a catalyst. After 4 hours of reaction at 80 °C, the Diels–Alder adduct was obtained with 93% yield (Scheme 11).38
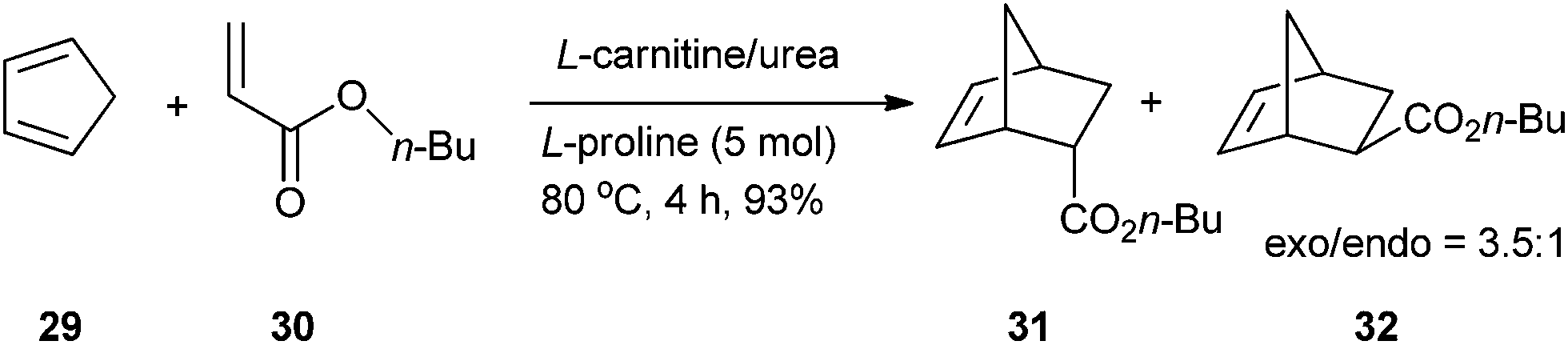 | ||
| Scheme 11 Diels–Alder reaction in L-carnitine/urea. | ||
The addition of Grignard and organolithium reagents to ketones is one of the most versatile and fundamental methodologies to generate new C–C bonds allowing access to tertiary alcohols. However, this reaction is greatly limited by their requirements of low temperatures in order to control their reactivity as well as the need of dry organic solvents and inert atmosphere protocols to avoid their fast degradation of these polar reagents. In 2014, García-Álvarez and co-workers found that the eutectic mixtures ChCl/glycerol and ChCl/H2O may used as superior green and biorenewable reaction media for the chemoselective addition of Grignard or organolithium reagent to ketones in air at room temperature without the need of volatile organic solvents (Scheme 12).39 In this sense, low temperatures are not needed to cool the reaction because DESs have high heat capacities. In all cases, the formation of the desired tertiary alcohols (35) occurred chemoselectively with no side products observed. In this process, ChCl may have a double role, as a component of the DES mixture employed but also as part of organometallic alkylating reagent, being a halide source. The authors suggested that a kinetic activation of the alkylating reagents occurred in DES mixtures, favoring nucleophilic addition over the competing hydrolysis process by comparison of the reactivity profiles of these reagents in DES mixtures with those in pure water. The protocol establishes a bridge between main-group organometallic compounds and DESs.
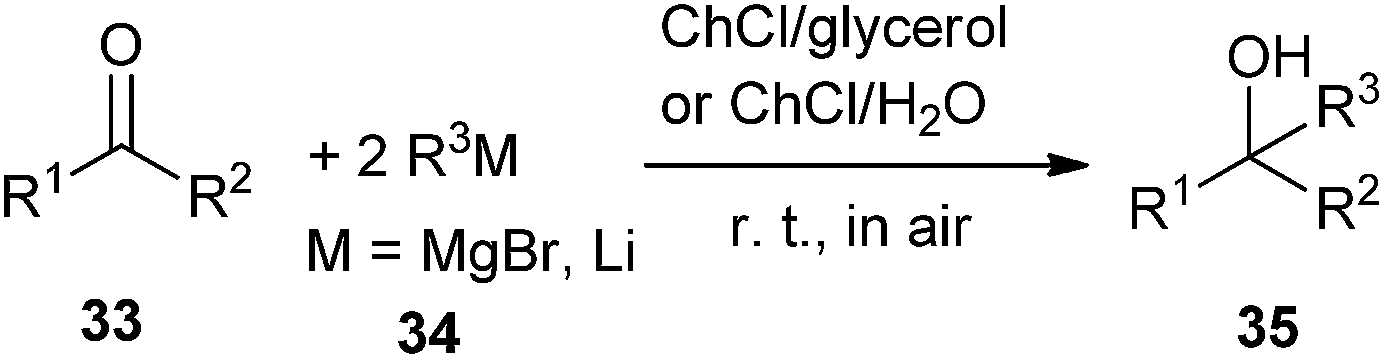 | ||
| Scheme 12 Addition of organolithium or Grignard reagent to ketones in ChCl-based eutectic mixtures. | ||
2.2 DES in the cyclization reactions
Biginelli reaction is a classic ring-forming reaction to synthesize dihydropyrimidinones(DHPMs) derivatives (34). Dihydropyrimidinones and their derivatives have been used as calcium channel blockers and antihypertensive agents. In 2011, Gore established a low melting mixture consisting of L-(+)-tartaric acid and DMU as a new alternative renewable solvent for Biginelli reaction to synthesize dihydropyrimidinones derivatives (Scheme 13).40 It is worth mentioning masked aldehydes have been used as substrates in the Biginelli reaction to give functionalized DHPM derivatives. Furthermore, this method does not require any tedious work up procedures and the DHPMs can be easily obtained in high to excellent yields and high purity.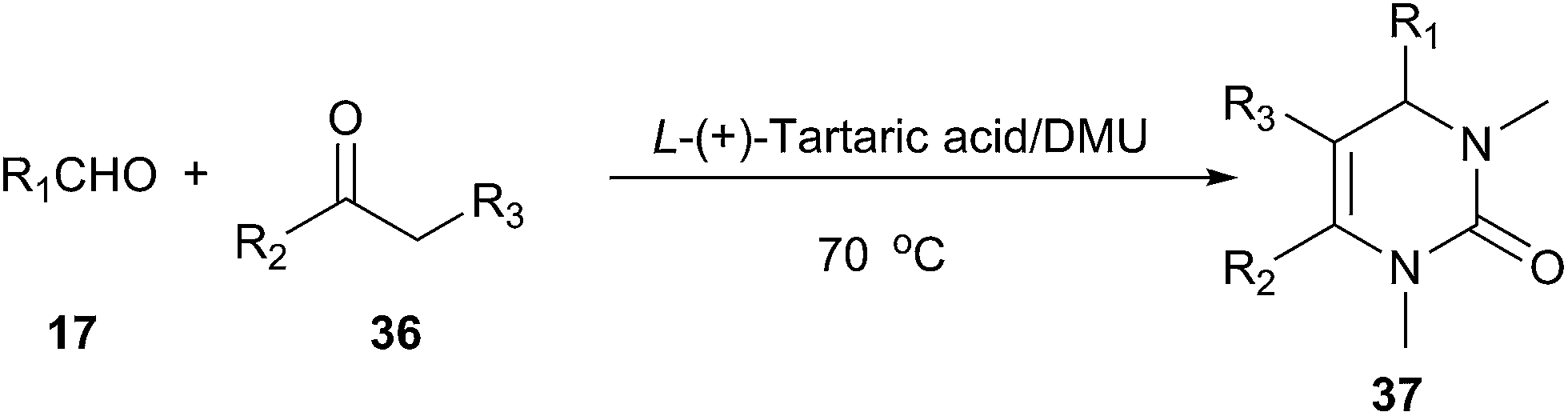 | ||
| Scheme 13 Biginelli reaction in L-(+)-tartaric acid/DMU. | ||
Another similar example described by the same group can be found in synthesis of hydropyrimidopyrimidinediones.41 Exposure of aryl ketones to paraformaldehyde in L-(+)-tartaric acid/DMU resulted in a facile reaction giving pyrimidopyrimidinedione derivatives in good yields (Scheme 14). In these processes, the low melting mixture serves simultaneously as solvent, catalyst and reactant.
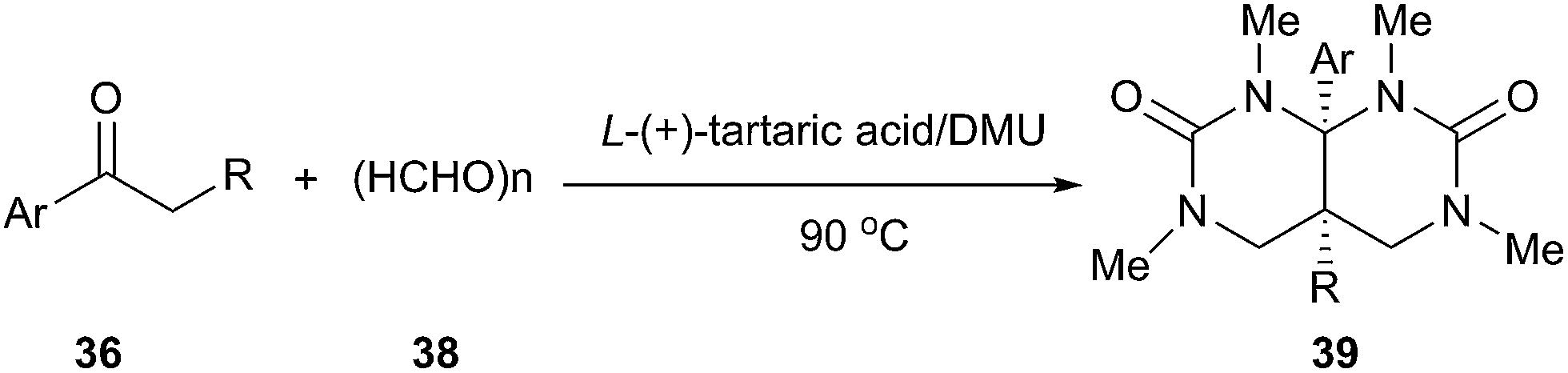 | ||
| Scheme 14 Synthesis of pyrimidopyrimidinediones in L-(+)-tartaric acid/DMU. | ||
The pyrrole ring system is a useful structural element in pharmaceutical and medicinal chemistry. The Paal–Knorr reaction which consists the cyclocondensation of primary amines with 1,4-dicarbonyl compounds to produce substituted pyrroles remains one of the most significant and simple methods.42 In 2013, a highly efficient and facile method for synthesis of substituted pyrroles (42) via Paal–Knorr reaction was presented by Handy and Lavender (Scheme 15).43 This reaction was performed in choline chloride/urea or choline chloride/glycerol. In this process, the weak hydrogen-bonding ability of urea appears to be enhanced due to the high concentration of urea present in the DES, rendering it useful as an organocatalytic solvent.
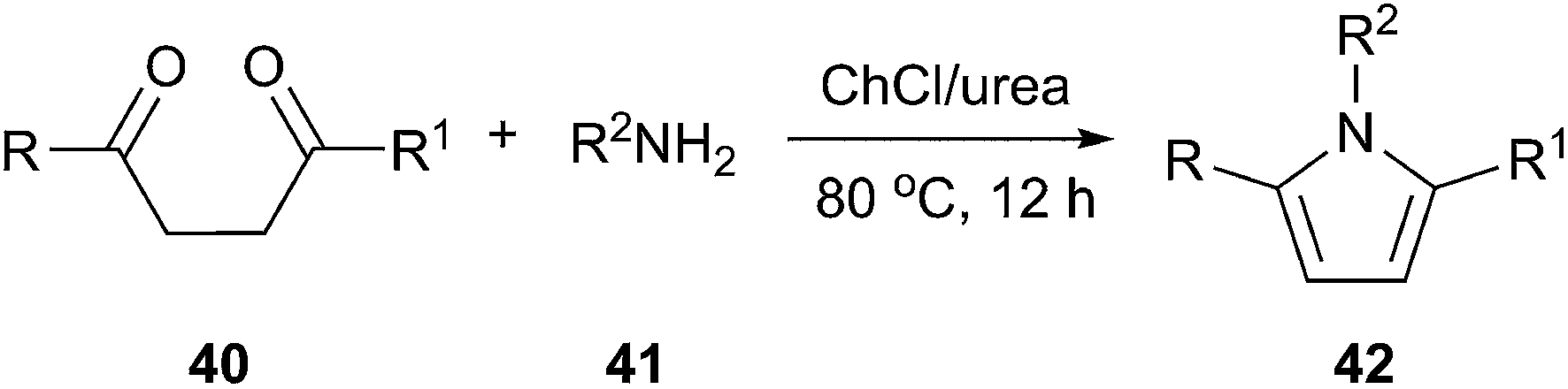 | ||
| Scheme 15 Paal–Knorr reaction in ChCl/urea. | ||
The Pictet–Spengler reaction is a well-known reaction that enables access to tetrahydrocarbolines by β-arylethylamine and carbonyl compound.44 This reaction can be carried out efficiently using ChCl/urea as a solvent reported by Handy and Wright (Scheme 16).45 The corresponding carbolines (44) are obtained in good to excellent yields (64–97%) in short reaction times (2 h). It is interesting to note that the reaction proceeded well with tryptamine (43), however, attempts to perform similar reactions using 3,4-dimethoxyphenethylamine failed to afford the cyclized products, indicating both the limits and the modest activation afforded by this solvent.
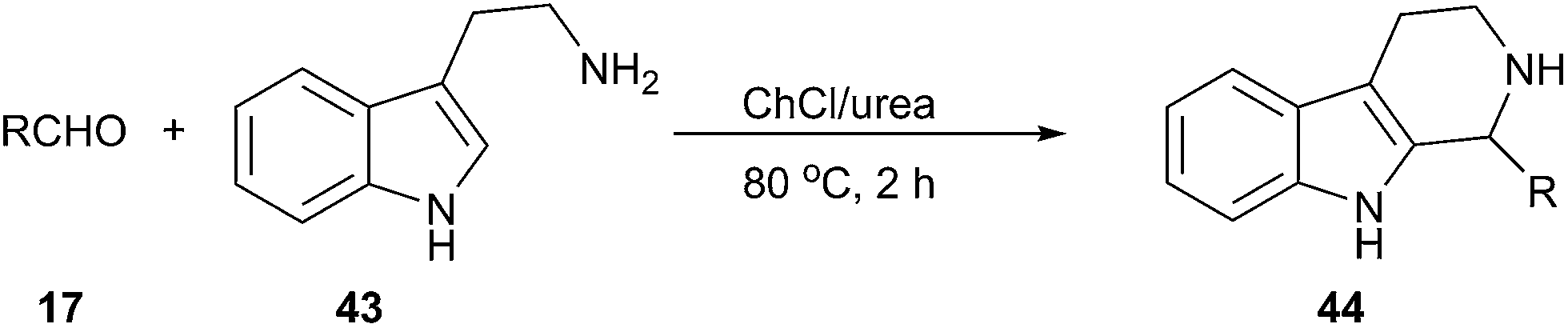 | ||
| Scheme 16 Pictet–Spengler reaction in ChCl/urea. | ||
Indole is a versatile structural framework presents in many natural and unnatural compounds with a broad spectrum of biological activities. Fischer indole synthesis is the most important and versatile approach for the preparation of biologically important indole derivatives.46 In order to avoid the conventional methodologies that using volatile organic solvents, expensive and detrimental metal precursors, and harsh reaction conditions, Morales et al. used DES as a green solvent and catalyst for synthesis of indoles. The Fischer indole synthesis can be performed in ChCl/ZnCl2 and afforded the 2,3-disubstituted indoles by the reaction of alkyl methyl ketones and phenylhydrazine (Scheme 17).47
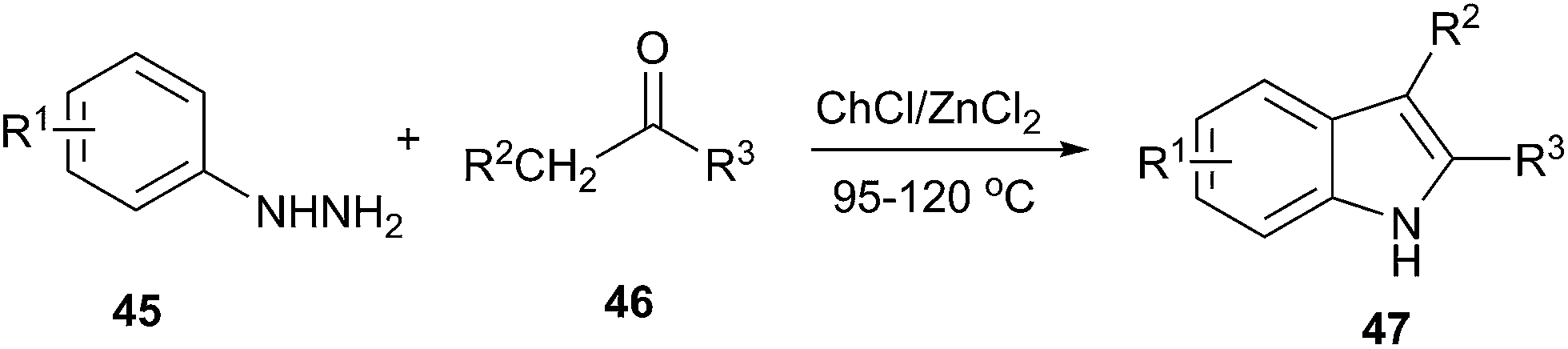 | ||
| Scheme 17 Fischer indole synthesis from aldehyde/ketone in ChCl/ZnCl2. | ||
Very recently, Gore et al. extended this work and have reported that the synthesis of indole derivatives (50, 52) (from phenylhydrazine (48) and α-substituted ketones (51)) can also be conveniently performed via Fischer indole synthesis in low melting L-(+)-tartaric acid (TA)/dimethyl urea (DMU) mixture (Scheme 18).48 The low melting mixture serves as the solvent and as the catalyst. This reaction exhibits a broad substrate scope and sensitive functional groups such as N-Boc, N-Cbz, or azides are stable, and indolenines are obtained regioselectively in excellent yields. At the end of the reaction, water is removed under a vacuum, the low melting mixture is recovered and recycled in the next cycle without any purification.
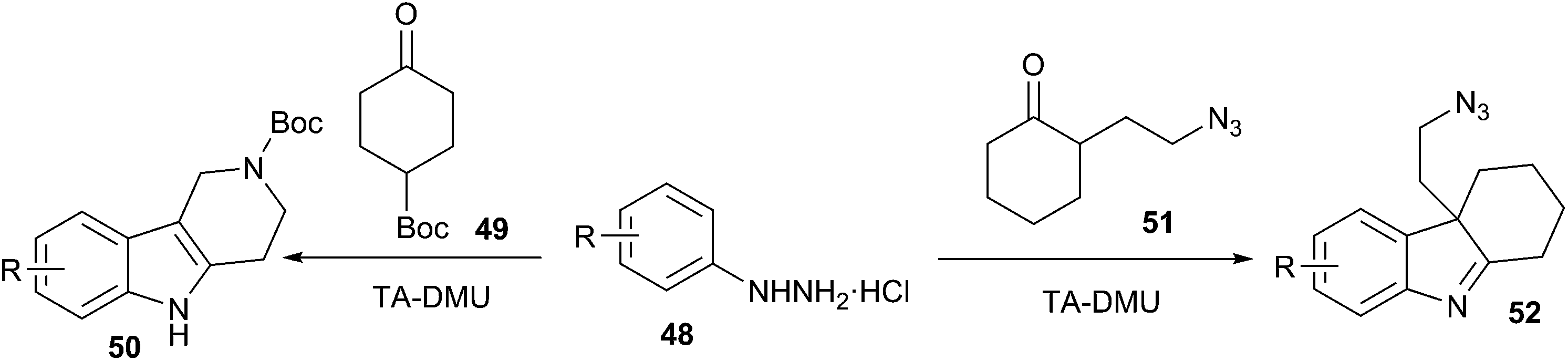 | ||
| Scheme 18 Fischer indole synthesis using α-substituted ketones in TA/DMU. | ||
In 2012, our group investigated the Friedländer heteroannulation reaction of 2-aminoaryl ketones and methylene ketones in various low melting mixtures, such as D-(−)-fructose/DMU, lactose/DMU/NH4Cl, glucose/urea/CaCl2, glucose/urea/NaCl, sucrose/ChCl, and glucose/guanidinium HCl, and L-(+)-tartaric acid/ChCl (Scheme 19).49 In L-(+)-tartaric acid/DMU, the reaction of 2-aminobenzophenone (53) and acetylacetone proceeded well and the product was obtained in 92% yield after 30 min of reaction at 70 °C. Various α-methylene carbonyl compounds such as cycloalkanones, 1,3-diketones, and 1,4-diketones as well as cyclic ketones such as cyclopentanone, cyclohexanone, cycloheptanone, cyclododecanone, 1H-inden-2(3H)-one and dimedone were used to give the respective polycyclic quinolines (54) in high yields. When the unsymmetrical 1,3-diketones such as 1-benzoylacetone was used, excellent regioselectivity was also obtained. Very recently, citric acid/dimethylurea was also reported to be capable of promoting this reaction by Bafti and Khabazzadeh.50 These approaches avoid the use of hazardous acids or bases, toxic organic solvents, and harsh reaction conditions.
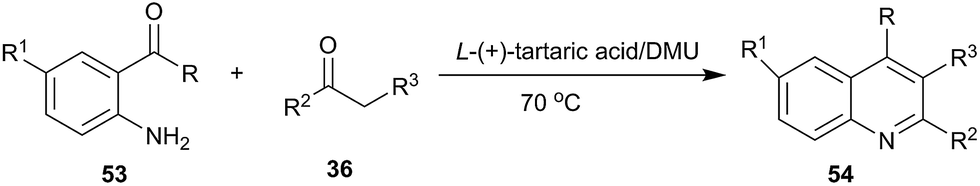 | ||
| Scheme 19 Friedländer reaction in L-(+)-tartaric acid/DMU. | ||
The synthesis of substituted oxazole derivatives has attracted much attention because of their versatile applications, including biological activities such as antibacterial, anti-fungal, anti-tubercular and anti-inflammatory activities as well as their utility as valuable precursors in many useful synthetic transformations. In 2013, Singh designed an efficient procedure for synthesis of novel oxazole compounds (57) by effective combination of ultrasound (US) and deep eutectic solvent (Scheme 20).51 The rate enhancement for the reactions conducted under US irradiation, over conventional heating, was rationalized based on the generation of microscopic internal pressure within the cavitation bubbles. Due to this, extreme microscopic conditions were created within these bubbles such that substrates entering them were converted to highly reactive species. At the same time, urea component in DES might stabilize the oxygen atom of carbonyl group via hydrogen bonding that encouraged the attack of amide on phenacyl bromide derivative as well as promoted cyclization.
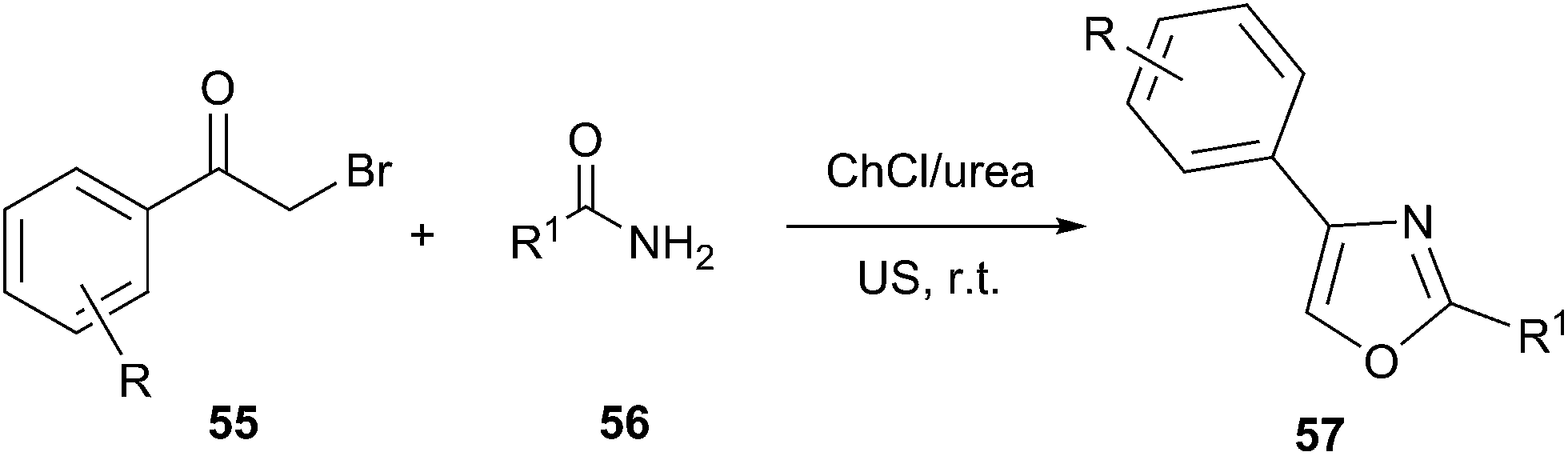 | ||
| Scheme 20 US assisted synthesis of oxazole derivatives in ChCl/urea. | ||
A simple and efficient method has been described by More et al. for the use of ChCl/urea as a solvent for synthesis of oxadiazoles (60) from carboxylic acid (58) and acid hydrazide (59) (Scheme 21).52 Cetyltrimethylammonium peroxodisulphate (CTAPS) in situ generates cetyltrimethylammonium bisulphate ([CTA]HSO4) which efficiently catalyzed cyclodehydration of diacylhydrazide formed from DES mediated condensation of carboxylic acid and acid hydrazide to give 2,5-disubstituted-1,3,4-oxadiazoles with good to excellent yields.
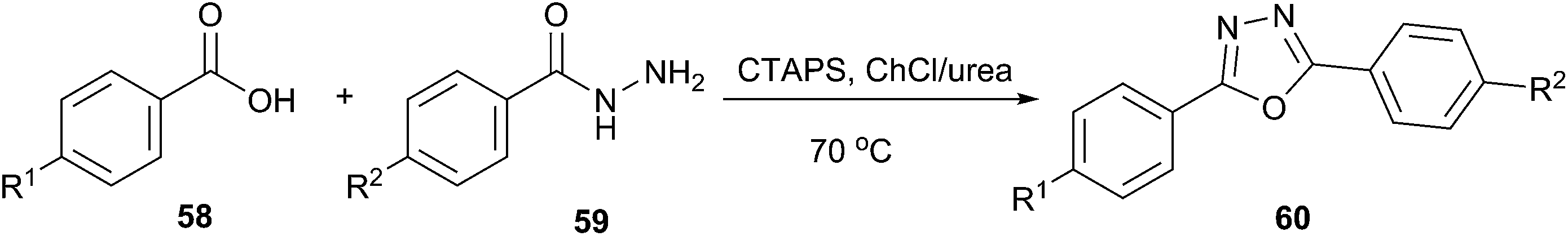 | ||
| Scheme 21 Synthesis of oxadiazole derivatives in ChCl/urea. | ||
The hydantoin moiety is an important structural scaffold in medicinal chemistry. Moreover, substituted hydantoins are valuable intermediates for the synthesis of enantiomerically pure aminoacids. The classic methods for the synthesis of hydantoin include the Bucherer–Bergs synthesis and the reaction of urea with carbonyl compounds. In 2013, Gore et al. reported an efficient domino synthesis of 1,3,5-trisubstituted hydantoins (62) from β,γ-unsaturated ketoacids (61) in TA/DMU (Scheme 22).53 Hence, DMU was used in this example not only as a reactant but also as a component of the low melting mixture. This methodology exhibited a very broad substrate scope and various β,γ-unsaturated ketoacids derived from electron rich as well as electron deficient aldehydes were successfully assembled in TA/DMU to furnish the corresponding substituted hydantoin derivatives in good to excellent yields with good diastereoselectivity.
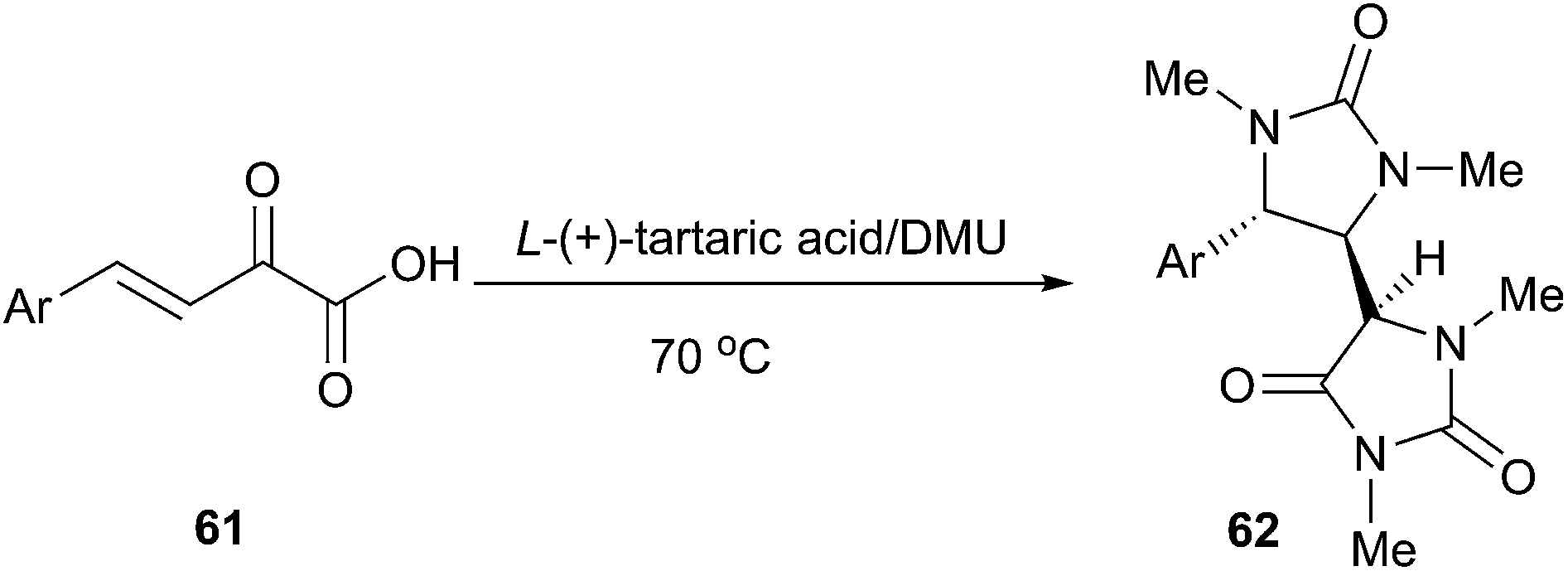 | ||
| Scheme 22 Synthesis of substituted hydantoins in TA/DMU. | ||
In 2014, Rodríguez-Álvarez introduced ChCl/urea deep eutectic solvent as a biorenewable and effective medium for Au(I)-catalysed cycloisomerisation of γ-alkynoic acids (63) into cyclic enol-lactones (65) (Scheme 23).54 In order to optimize the reaction condition, the authors evaluated a series of catalysts, such as [AuCl(PPh3)], Au2O3, Au(I) complex (64) by the cycloisomerisation of 4-pentynoic acid as a model reaction. The authors found Au(I) complex was the most effective catalyst than others. In addition, the effect of various solvents such as ChCl/urea, ChCl/glycerol, ChCl/ethyleneglycol, ChCl/lactic acid, and glycerol had been studied. It found the high yield was observed in the case of DES (ChCl/urea). This catalytic system showed a wide range of applications and tolerance to functional groups in the cycloisomerisation of a variety of terminal alkynes, being compatible with the presence of ester, amino, alkenyl, and alkynyl groups. Furthermore, the catalytic system could be recycled up to four runs. This method is the first report in the literature for the metal-catalyzed cycloisomerisation of γ-alkynoic acids in DESs.
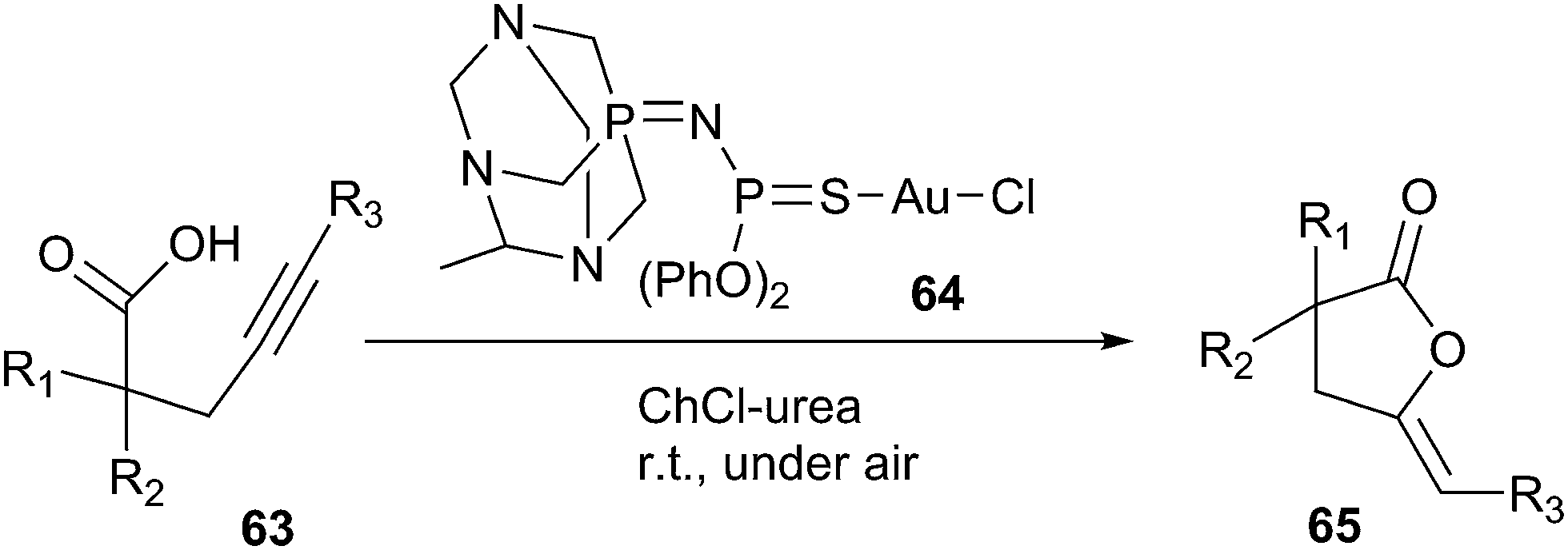 | ||
| Scheme 23 Cycloisomerisation of γ-alkynoic acids in ChCl/urea. | ||
Another example is Clauson-Kaas reaction for the synthesis of N-substituted pyrroles, in which various catalysts have been used to promote this reaction.55 In 2014, we have improved this reaction by using ChCl/L-(+)-tartaric acid deep eutectic solvent as a green medium (Scheme 24).56 In order to optimize the reaction condition, the authors carried out this reaction with a wide range of solvents and found that ChCl/L-(+)-tartaric acid deep eutectic solvent was used as solvent, the yield of the product was highest. Structurally diverse N-substituted pyrroles were obtained by Clauson-Kaas reaction of amines and 2,5-dimethoxytetrahydrofuran (66) in high to excellent yields. The DES can be easily recovered by extracting the reaction crude with ethyl acetate, and then reused in subsequent reactions after drying under vacuum.
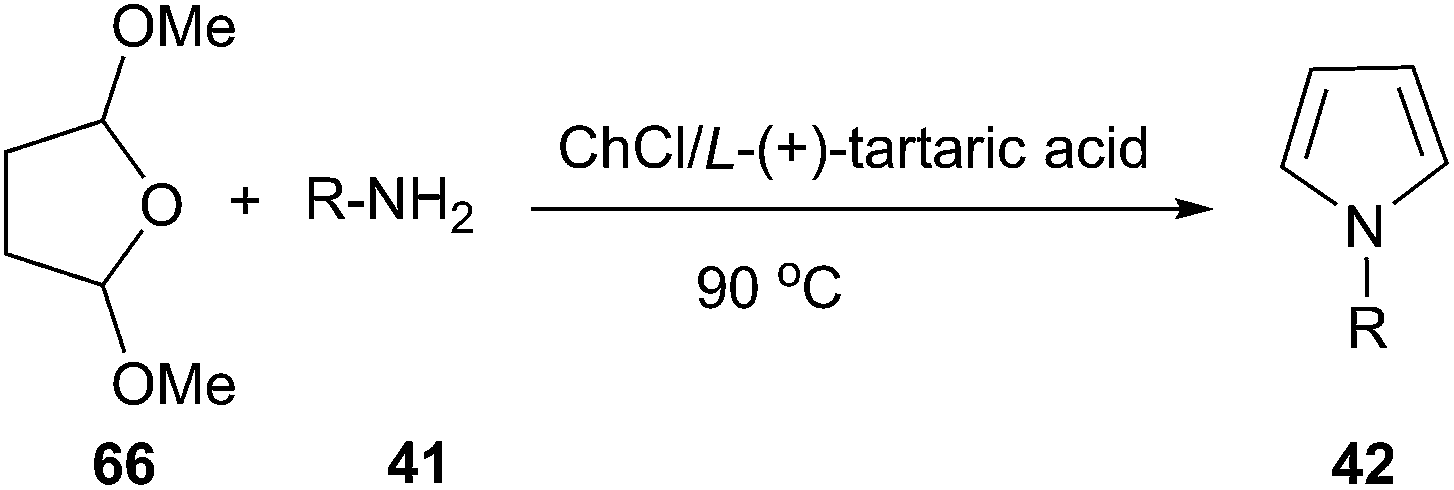 | ||
| Scheme 24 Clauson-Kaas reaction using ChCl/L-(+)-tartaric acid. | ||
2.3 DES in the replacement reactions
The Stille cross-coupling protocol is a powerful and widely used method for the construction of new carbon–carbon bonds. In 2006, Imperato et al. used sugar/urea/salt melts as a green solvent for the Stille alkylation of iodobenzene (67) with tetraalkylstannanes (68) (Scheme 25).57 In order to optimize the reaction condition, the authors carried out the reaction of iodobenzene with tetravinyltin or tetramethyltin and tetrabutyltin in the presence of the tris-(dibenzylideneacetone)dipalladium chloroform complex as palladium source and AsPh3 as ligand in a series of solvents, such as citric acid/DMU, sorbitol/DMU/NH4Cl, maltose/DMU/NH4Cl, lactose/DMU/NH4Cl, mannose/DMU, glucose/urea/NaCl, and fructose/urea/NaCl. Maltose/DMU/NH4Cl was found to be the most promising solvent for the catalytic transfer of butyl and vinyl groups from tin to iodobenzene. From tetramethyltin, the highest yield was obtained in the mannitol/DMU/NH4Cl melt. Catalyst loading may be reduced to 0.001 mol% still achieving a turnover number of 87000. The catalytic system remained active in several reaction cycles and product isolation did not require the use of organic solvents.
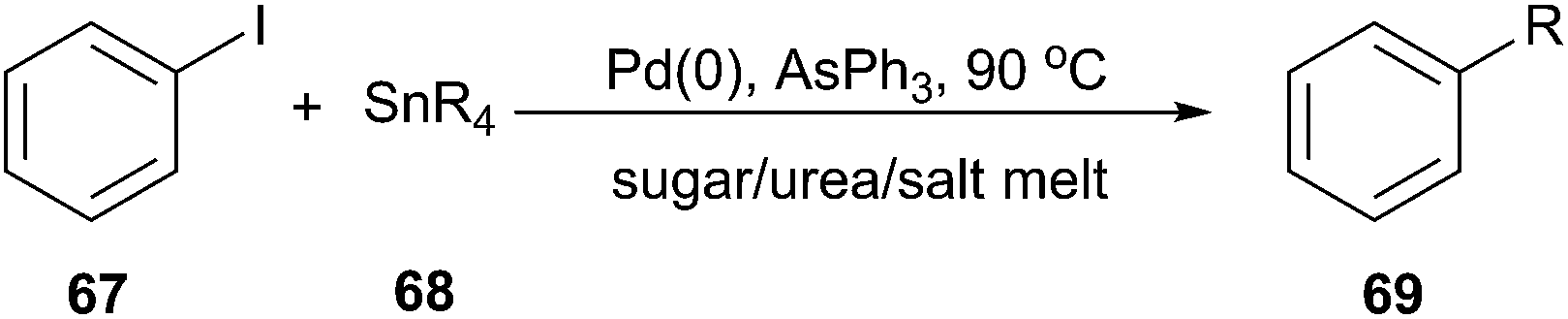 | ||
| Scheme 25 Stille alkylation using sugar/urea/salt melts. | ||
In 2012, Germani's group designed new halogen-free Brønsted acidic deep eutectic solvents by mixing quaternary ammonium methanesulfonate salts such as trimethylcyclohexyl ammonium methanesulfonate (TCyAMsO), trimethylbenzyl ammonium methanesulfonate (TBnAMsO), trimethylbenzyl ammonium methanesulfonate (TBnAMsO), and trimethyloctyl ammonium methanesulfonate (TOAMsO) with p-toluenesulfonic acid (PTSA). These DESs can be used as dual solvent-catalyst for esterification of several carboxylic acids with different alcohols (70) (Scheme 26).58 The use of TCyAMsO–PTSA gave generally good yields (>75%) in 2 h and at 60 °C. Ease of recovery and reusability of DES with high activity make this method efficient and eco-friendly.
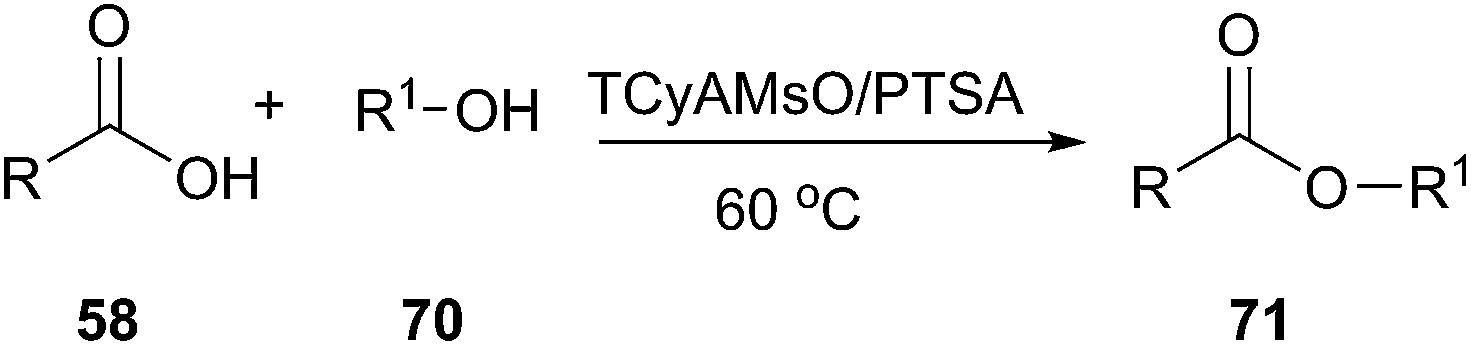 | ||
| Scheme 26 Esterification of carboxylic acids with alcohols in TCyAMsO–PTSA. | ||
Recently, Wang et al. prepared a deep eutectic solvent based on choline chloride and 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) and applied it as the reaction medium for halogenation of boron dipyrromethenes (BODIPY, 72) in the presence of N-halosuccinimide (NBS) (Scheme 27).59 The iodination reaction in the presence of N-iodosuccinimide (NIS) was more active than the bromination in this solvent. The reaction completed within 15–30 min to afford the dihalogenated BODIPYs (73) in good to excellent yields. Other choline-based eutectic mixtures such as ChCl/urea, ChCl/malonic acid, ChCl/glycerol and ChCl/p-toluenesulfonic acid were also evaluated for this reaction. However, BODIPYs were not soluble in these DESs, only trace amount of products were obtained. Ease of recovery and reusability of this DES with high activity make this method efficient and eco-friendly.
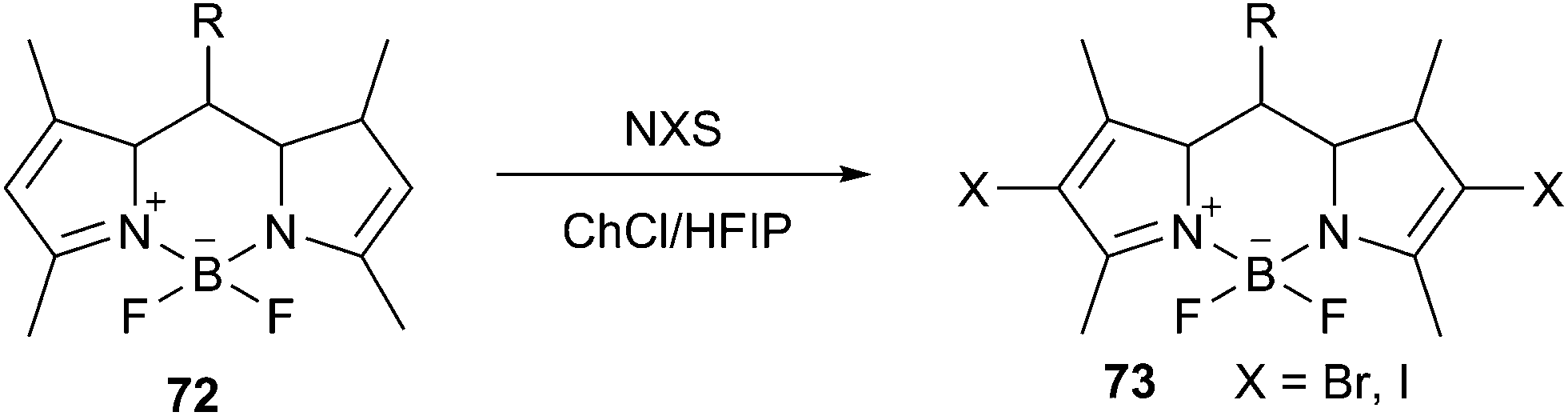 | ||
| Scheme 27 Dihalogenation of BODIPYs in ChCl/HFIP. | ||
DES prepared from choline chloride and p-TsOH has been successfully used for preparation α-fluoroacetophenones (Scheme 28). This method took advantage of chlorination in DES and nucleophilic fluorination to prepare α-fluoroacetophenones (75) directly from acetophenones (74) in one pot and the yields were higher than some examples of electrophilic fluorination using NF reagents. In this process, 1,3-dichloro-5,5-dimethylhydantoin (DCDMH) was selected as the chlorinating reagent, and tetrabutylammonium fluoride (TBAF) was used as the fluorinating reagent.60
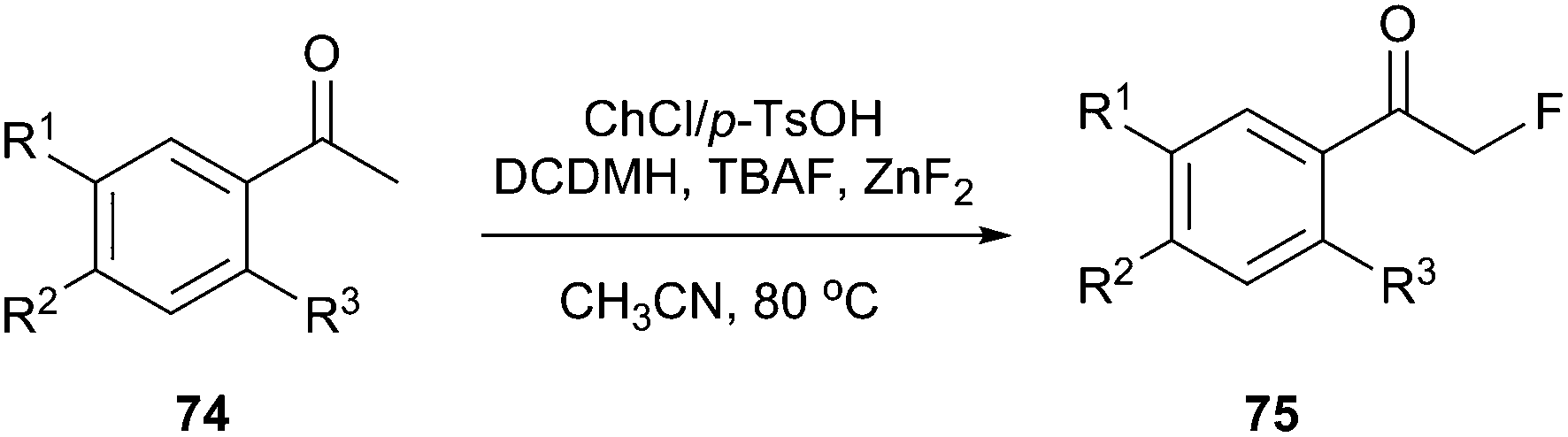 | ||
| Scheme 28 One-pot fluorination of acetophenones in ChCl/p-TsOH. | ||
In 2009, Zou and co-workers have reported the acid-catalyzed synthesis of the α,α-dichlorination of acetophenone derivatives (33) in an acidic DES composed of ChCl and p-toluene sulfonic acid mixed in a ratio of 1:1. When the ratio DES/acetophenone was 2:1, the α,α-dichlorinated adduct (70) was obtained with 86% yield at room temperature in the presence of 1,3-dichloro-5,5-dimethylhydantoin (DCDMH, 69) as a chlorinating agent. Various α,α-dichlorinated acetophenones and β-ketoesters were obtained in high yields (86–95%) with high selectivity (Scheme 29). In this process, NCS was not more efficient than DCDMH.61
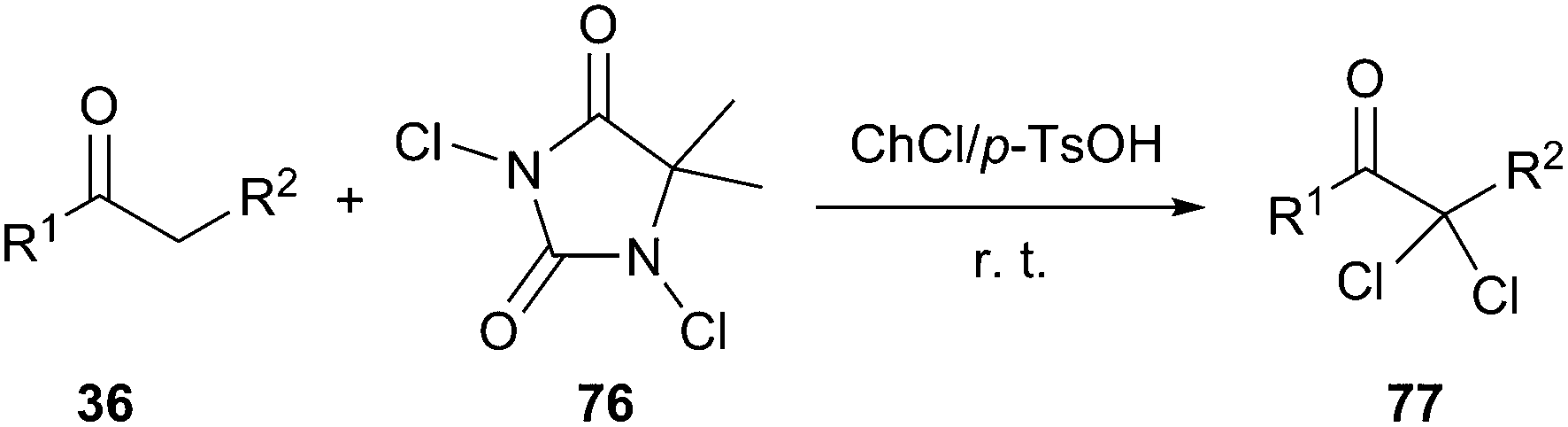 | ||
| Scheme 29 Selectively prepare α,α-dichloro ketones and β-ketoesters in DES/acetophenone. | ||
In 2011, mono N-alkylation of various aromatic primary amines using ChCl/urea as catalyst and solvent with broad scope was developed by Singh and co-workers.62 N-Alkylation reactions of aromatic amines with electron donating groups gave good yields and their reaction times were shorter in comparison to aromatic amines with electron withdrawing groups. The reactions gave selectively mono products (79) as a major component and avoided complexity of multiple alkylations. The proposed mechanism of this reaction is displayed in Scheme 30. The DES was formed by intramolecular hydrogen bonds between choline chloride and urea, while the DES on the urea also forms hydrogen bond with aromatic amino group and increases the nucleophilicity of aromatic amines thus leading to their faster attack on alkyl bromides.
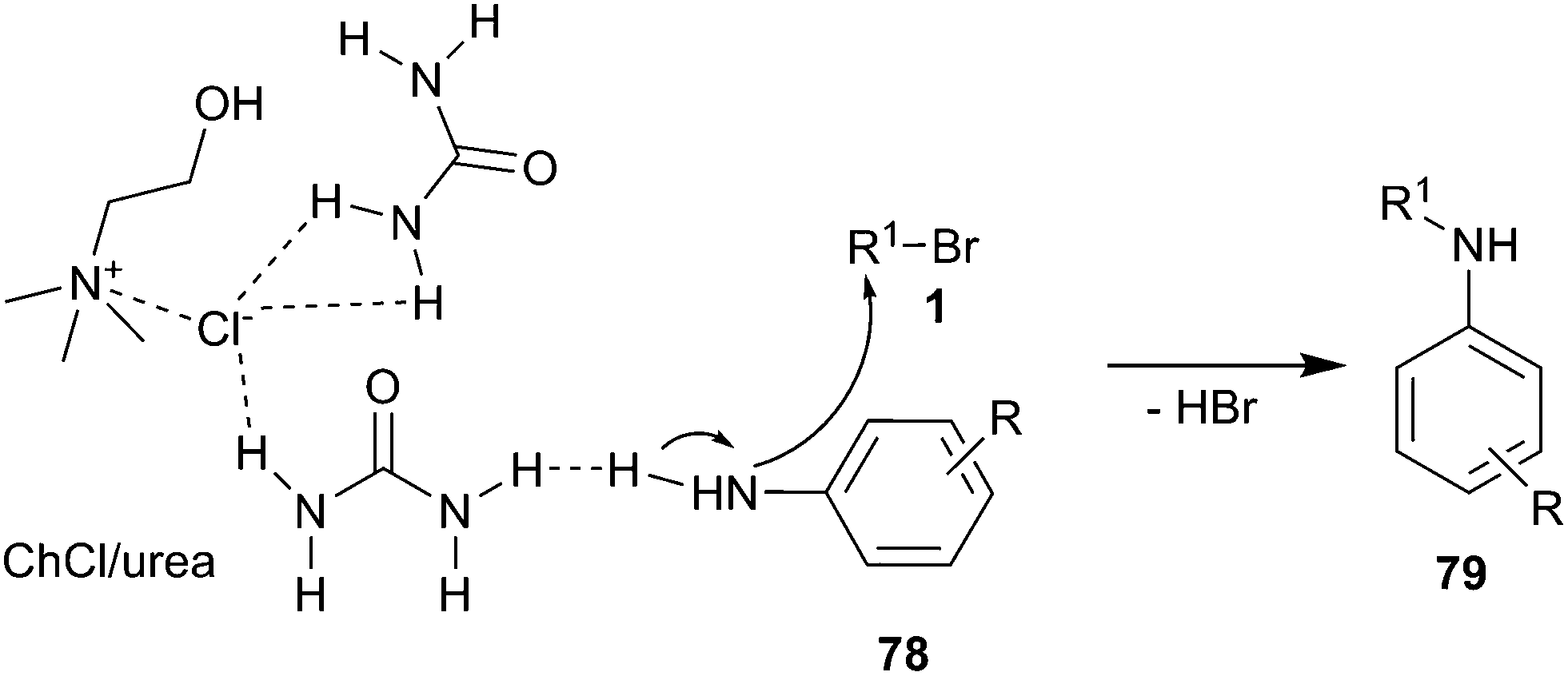 | ||
| Scheme 30 Proposed mechanism for N-alkylation of aromatic amines in ChCl/urea. | ||
Indole and its derivatives are important because their structural subunits exist in many natural products with various physiological properties and pharmacological activities. Consequently, they have attracted considerable attention in the realm of synthetic organic chemistry.63 Bis(indolyl)methanes (81) have been often obtained by electrophilic substitution reaction of indole with aldehydes or ketones in the presence of a Brönsted or a Lewis acid catalyst.64 However, these methodologies have suffered from disadvantages including leaching of metals and harsh reaction conditions. Yadav et al. demonstrated that the condensation of indole with various aromatic and heterocyclic aldehydes could be proceed in the presence of 30 mol% of ChCl/oxalic acid at room temperature (Scheme 31).65 The reaction was clean and the rates of the reaction were faster (10–25 min) with high yields (88–95%) when performed in EDS as compared to already existing protocols in which organic solvents were utilized with catalysts.
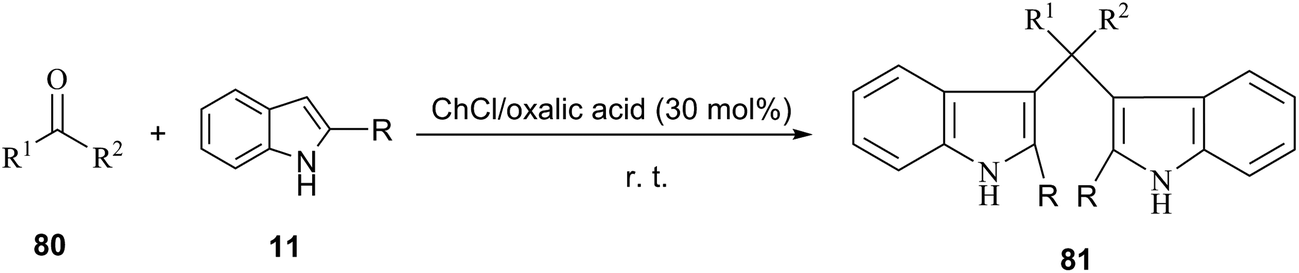 | ||
| Scheme 31 Synthesis of bis(indolyl)methanes catalyzed by ChCl/oxalic acid. | ||
In 2014, Handy and Westbrook have also reported that this reaction could efficient proceed well in ChCl/urea, providing bis(indolyl)methanes in high yields. Despite promising results, this process required high temperature (80 °C) and needed longer reaction time (4 h). In addition, the carbonyl compound as both ketones and aliphatic aldehydes failed to afford the desired products.66 Azizi et al. found that aliphatic aldehydes as well as simple ketones were good electrophilic partners when the reaction was carried out at room temperature in polyethylene glycol in the presence of ChCl/SnCl2.67 Various bis(indolyl)methane alkaloids could also synthesized when the reaction was performed in low melting mixture TA/DMU at 70 °C.68
Even more recently, oxalic acid/proline DES was reported to be capable of promoting the electrophilic reaction of indole and isatin for the synthesis of 3,3-di(indolyl)indolin-2-ones (83) at room temperature (Scheme 32).69 The reaction was performed in various solvents and yield obtained in oxalic acid/proline was much higher than those obtained in ethylene glycol, polyethylene glycol, trifluoroethanol, oxalic acid/glycine as well as guanidine hydrochloride/urea. A broad range of substrates were eligible for indole (11) and isatin derivatives (82) in this reaction, providing ready access to 3,3-di(indolyl)indolin-2-ones in high yields. The DES can be recycled and reused several times without appreciable loss of its activities.
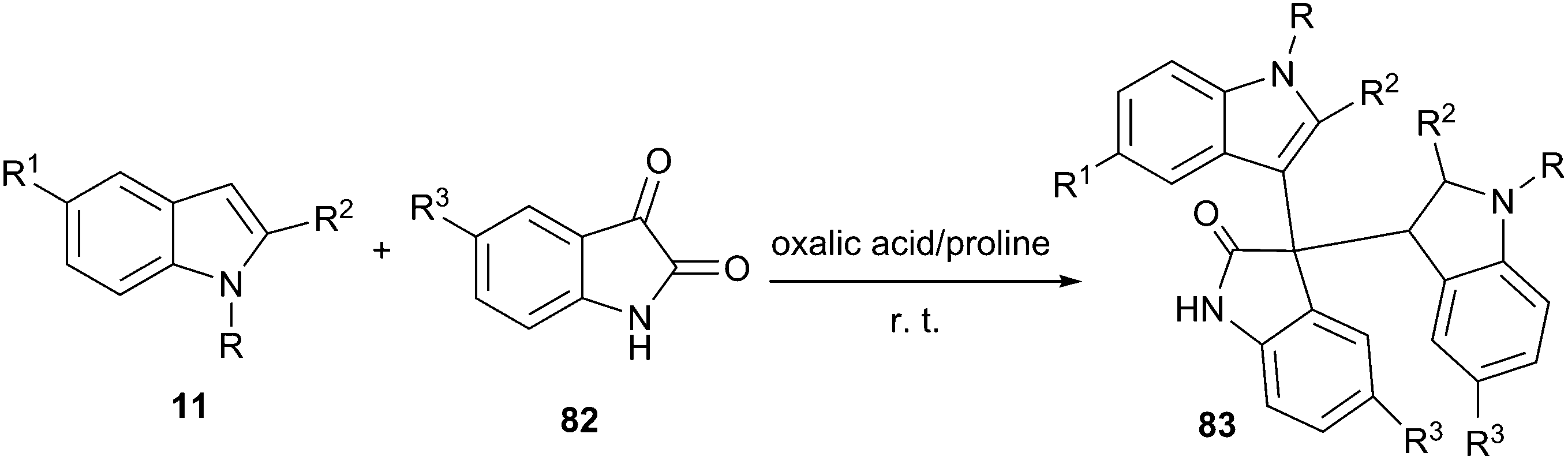 | ||
| Scheme 32 Synthesis of 3,3-diaryloxindoles in oxalic acid/proline. | ||
2.4 Multicomponent reactions
Multicomponent reactions (MCRs) are becoming powerful and valuable synthetic strategy for the construction of structurally complex molecules with a minimum of reaction steps and simple workup procedures from simple and readily accessible starting materials. They have a wide range of applications in synthetic, combinatorial and medicinal chemistry.70The Kabachnik–Fields reaction represents one of the most direct and appealing approaches for the preparation of α-aminophosphonates (85).71 A striking example of DES accelerated Kabachnik–Fields reaction was described by Disale and co-workers (Scheme 33).72 This one pot three-component synthesis of α-aminophosphonates from aryl aldehyde, aniline, and diethyl phosphite (84) usually required prolonged reaction times and heating when carried out in an organic solvent, water or under neat conditions.73 In contrast, it can proceed at room temperature using 15 mol% of choline based Zn deep eutectic mixture (ChCl/ZnCl2).
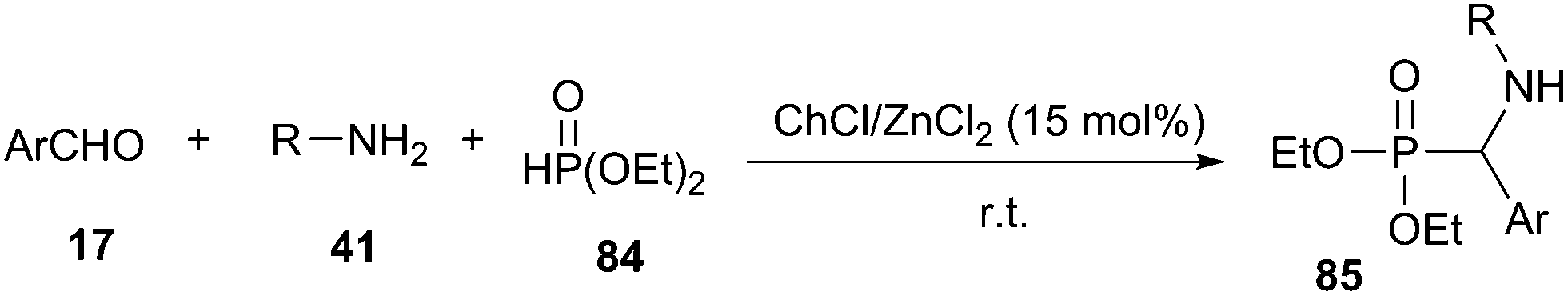 | ||
| Scheme 33 Kabachnik–Fields reaction catalyzed by ChCl/ZnCl2. | ||
The Mannich-type reaction is one of the most widely utilized chemical transformations for constructing β-amino carbonyl compounds. Mannich-type three-component reaction of aldehyde, aniline and ketone has been extensively investigated. Various Lewis acids, Lewis bases and metal salts have reported as catalysts for this reaction.74 However, difficulty in recycling the expensive catalysts restricts their application in practical synthesis. Recently, this three-component reaction has been investigated by Keshavarzipour et al. using DES. The reaction could also be performed using 5 mol% of ChCl/ZnCl2 at room temperature to give β-amino carbonyls (86) in good yields (52–98%).75 A proposed mechanism for this reaction was shown in Scheme 34. In these cases, actual species presented in ChCl/ZnCl2 may be [ZnCl3]−, [ZnCl5]−, [ZnCl7]− and [ZnCl3]−. These species may act as Lewis bases to adsorb a proton of ketone to give anionic species. Moreover, ChCl/ZnCl2 can form an acceptor–donor complex with the aldehyde and the imine. Thus, addition of the enol (or enolate) form of acetophenone could be facilitated.
 | ||
| Scheme 34 Proposed mechanism for Mannich reaction catalyzed by ChCl/ZnCl2. | ||
Undoubtedly, the Ugi reaction is the most studied and widely used MCR reaction, which has proven to be a powerful tool for rapid synthesis of lead compounds in drug discovery. The Ugi four-component reaction (U-4CR) involves the condensation of carbonyl derivatives, amines, carboxylic acids and isocyanides to afford α-amino acid derivatives. The classical Ugi reaction is carried out in organic solvents, such as methanol and dichloromethane.76 In 2013, Azizi et al. investigated this U-4CR in various solvents under catalyst-free conditions. ChCl/urea deep eutectic solvent led to much better results than conventional organic solvents, such as methanol, acetonitrile and dichloromethane. Furthermore, the experimental procedures were easy and the reactions went to completion at room temperature within 2–5 h. The products were easily separated by extraction of the deep eutectic solvent with water, and were usually obtained in high purity (Scheme 35).77
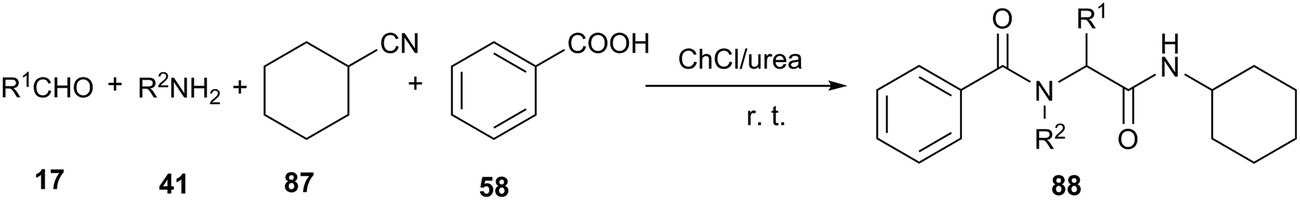 | ||
| Scheme 35 Ugi reaction in ChCl/urea. | ||
In the field of copper-catalyzed alkyne azide 1,3-dipolar cycloaddition (CuAAC), König and co-workers reported that 1,4-substituted 1,2,3-triazoles (92) were obtained by a one-pot process (from alkyne, benzyl bromide and sodium azide) in D-sorbitol/urea/NH4Cl in the presence of 5 mol% of CuI (Scheme 36).40
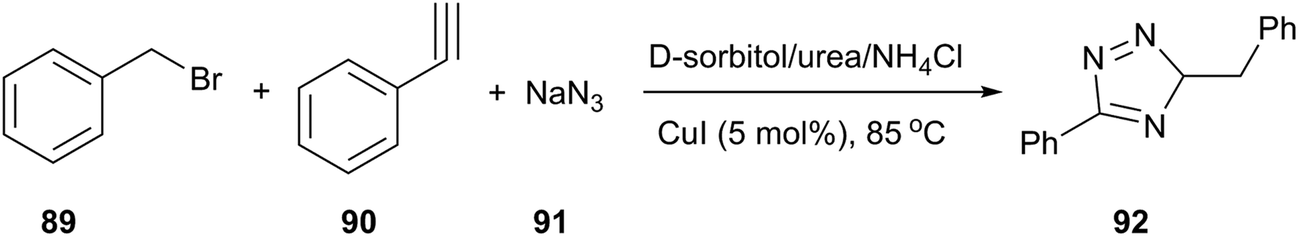 | ||
| Scheme 36 One-pot synthesis of 1,4-substituted 1,2,3-triazoles in D-sorbitol/urea/NH4Cl. | ||
In 2013, Mobinikhaledi and co-workers reported that ChCl/urea DES could be used as both catalyst and solvent for one-pot synthesis of 5-arylidene-2-imino-4-thiazolidinones (95) by condensation of the thioureas (93) with chloroacetyl chloride (94) and an aldehyde. This methodology was tolerant to a wide range of substrates since various thioureas and alkyl aldehydes were successfully reacted at 80 °C, affording the corresponding 5-arylidene-2-imino-4-thiazolidinones in 67–94% yields (Scheme 37).78
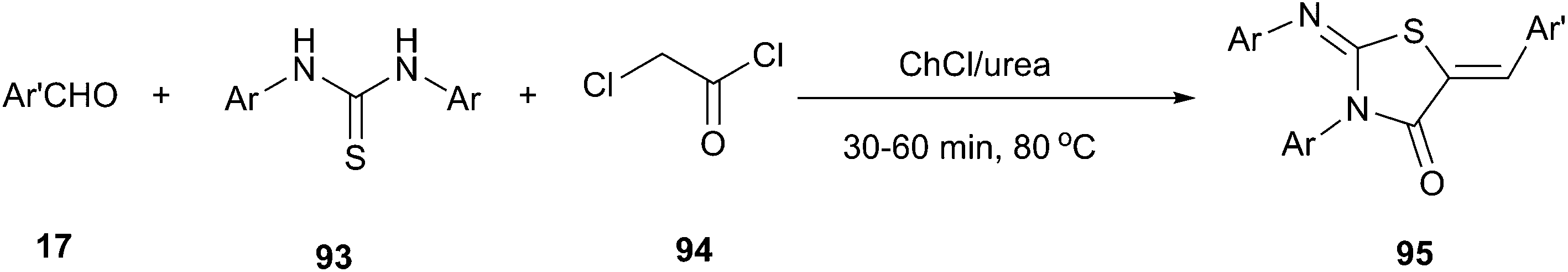 | ||
| Scheme 37 Synthesis of 5-arylidene-2-imino-4-thiazolidinones in ChCl/urea. | ||
A simple and efficient method has been described for the use of ChCl/urea deep eutectic solvent in one-pot reaction of alkyl halides, epoxides and thiourea. The corresponding β-hydroxy sulfides (96) could be prepared from in situ generated S-alkylisothiouronium salts in good to excellent yields. The plausible mechanistic proposition for this reaction is outlined in Scheme 38. The authors think that DES played a triple role, as an ionic reaction medium for the fast generation of S-alkylisothiouronium salts (intermediate M), activating the epoxide by hydrogen bonding and capture of the in situ generated urea.79
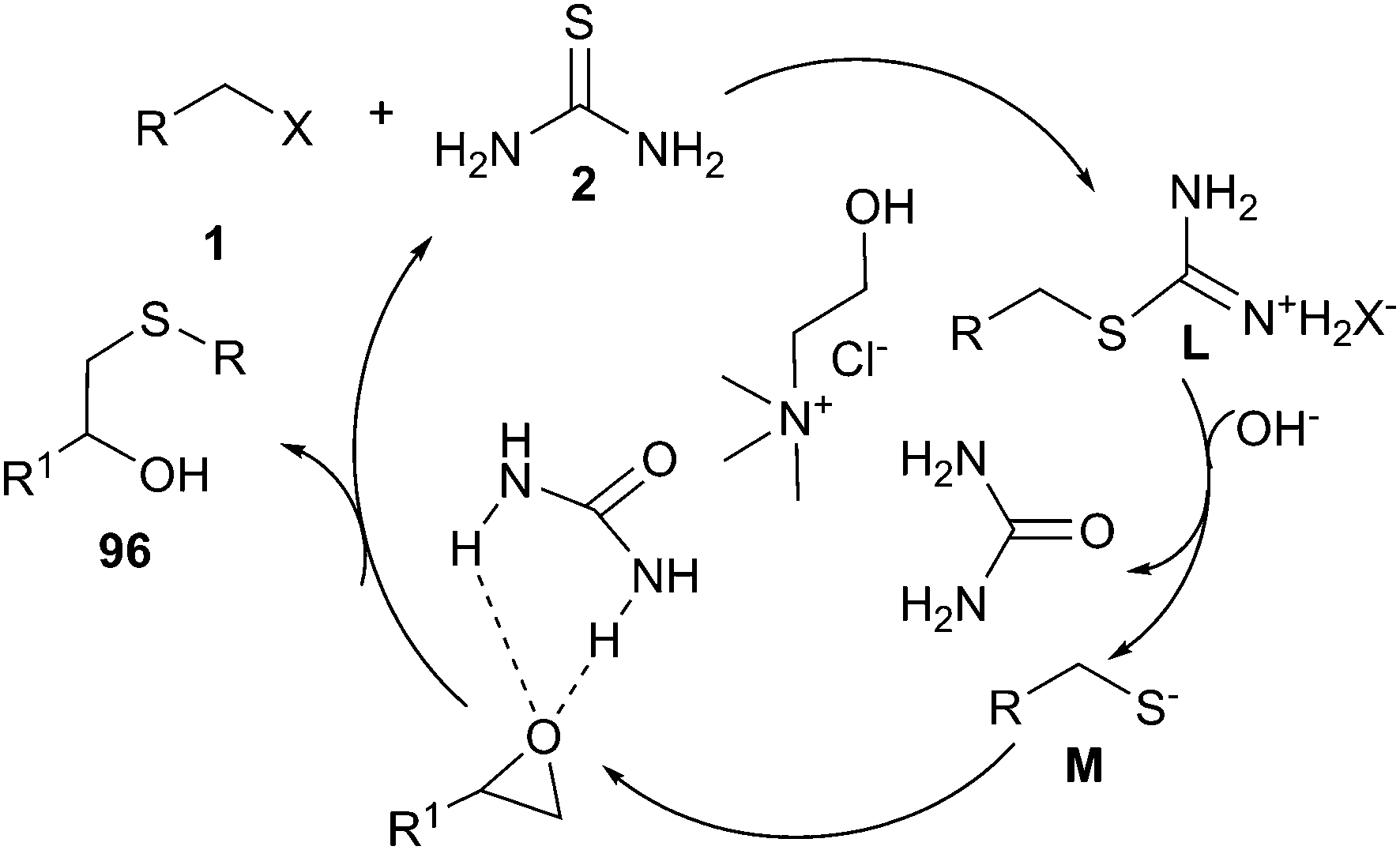 | ||
| Scheme 38 Postulated mechanism for one-pot reaction of alkyl halides, epoxides and thiourea in ChCl/urea. | ||
ChCl/urea promoted the environmentally friendly and fast synthesis of dithiocarbamate derivatives (99) via a one-pot, three-component condensation of an amine (97), carbon disulfide (98), and a variety of electrophilic reagents at ambient temperature has been reported by the same authors (Scheme 39).80 The same reaction could also be performed in polyethylene glycol (PEG). In this reaction, the DES and PEG could be recycled up to three runs without significant loss of their activity.
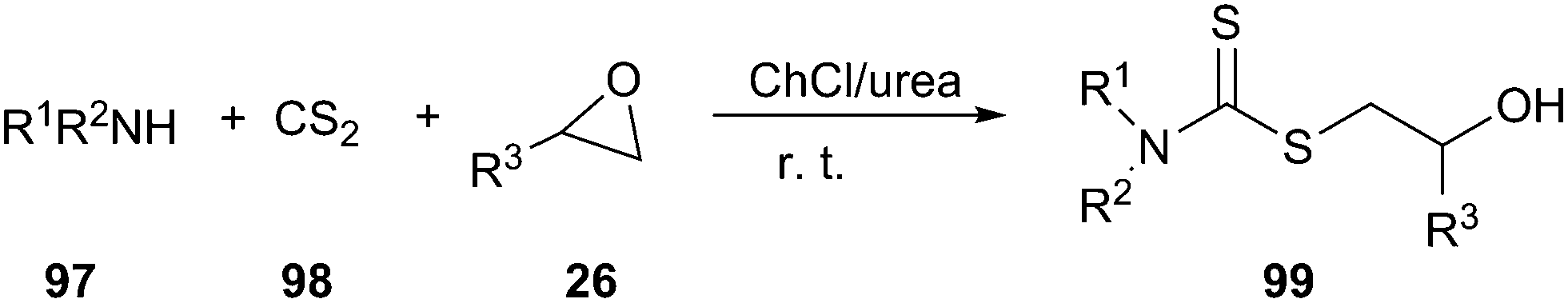 | ||
| Scheme 39 Synthesis of dithiocarbamates in ChCl/urea. | ||
As a type of multi-component reactions (MCRs), namely the Hantzsch reaction, one-pot condensation of aldehyde, dimedone, ethyl acetoacetate and ammonium acetate is one of the most straightforward procedures for the preparation of 1,4-dihydropyridine derivatives (103 and 104), a class of drugs possess a wide variety of biological and pharmacological actions.81 Various catalysts are known to affect this condensation.82 Though this reaction may proceed in ionic liquids, serious limitations have emerged for the industrial scale application of ionic liquids due to high cost, environmental toxicity and demand for high purity. In 2013, Pednekar et al. found that this reaction can be carried out in ChCl/urea deep eutectic solvent (Scheme 40). In such media, excellent yields of the resultant polyhydroacridines (PHA) have been obtained.83 Similarly, polyhydroquinolines (PHQ) have also been prepared in the one-pot condensation of dimedone, aldehydes and ammonium acetate. Other DESs such as ChCl/p-TsOH (1:1), ChCl/malonic acid (1:2), ChCl/glycerol (1:2), ChCl/HFIP (1:1.5) and ChCl/ZnCl2 (1:2) were also investigated by Wang et al., low to moderate yields of the products were obtained.84
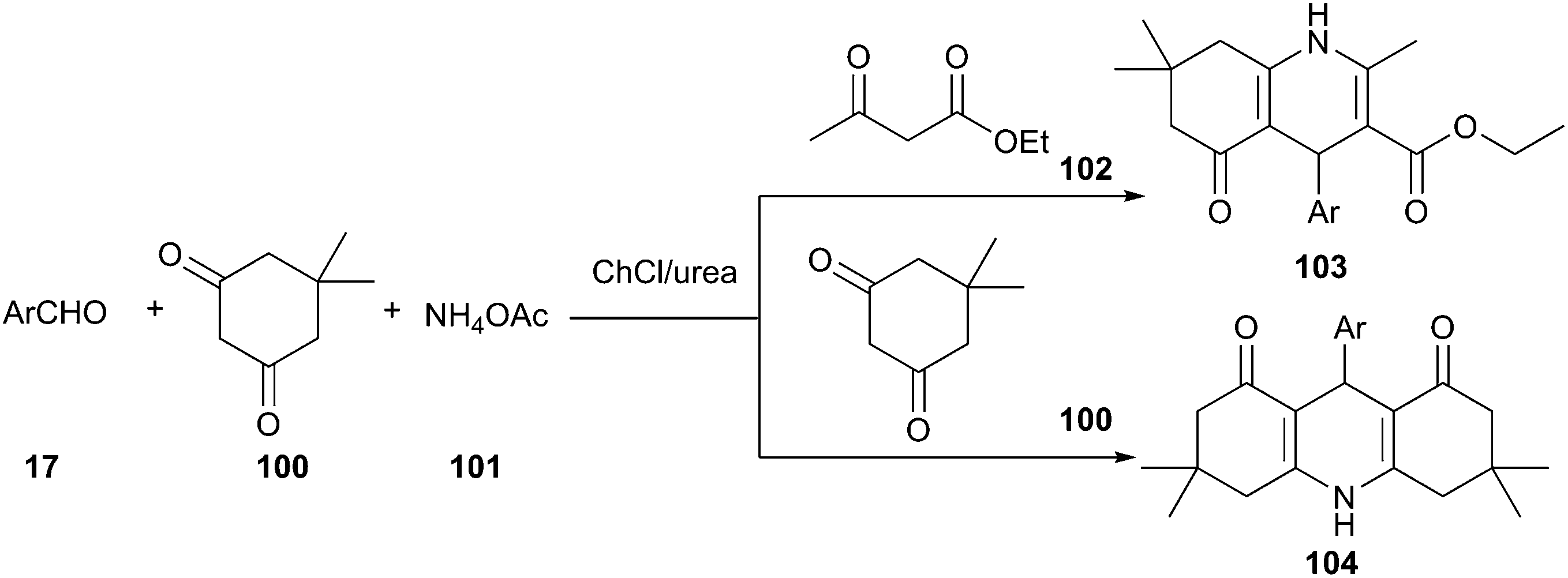 | ||
| Scheme 40 Synthesis of 1,4-dihydropyridine derivatives in ChCl/urea. | ||
2-Amino-4H-chromene derivatives are also useful intermediates for the synthesis of various compounds such as 1,4-dihydropyridines, pyranopyrazoles, pyridones, lactones, imidoesters as well as aminopyrimidines. The most straightforward synthesis of this heterocyclic system involves a three-component coupling of an aldehyde with alkylmalonates and diverse enolizable C–H activated acidic compounds in the presence of various acid or base catalysts.85 In 2014, Chaskar reported ChCl/urea (20 mol%) could be used as catalyst in one-pot three-component reaction of aldehydes dimedone/4-hydroxy coumarin, and active methylene nitriles (105) in aqueous medium at room temperature (Scheme 41).86 In this synthesis, the benzylidenemalononitrile firstly formed by Knoevenagel condensation of aldehyde and malononitrile eventually underwent Michael addition–cyclization with dimedone/4-hydroxy coumarin and afforded the chromenes (107, 108) in good to excellent yields. Azizi et al. also have demonstrated its high activity in this reaction.87
 | ||
| Scheme 41 Synthesis of 2-amino-4H-chromene derivatives catalyzed by ChCl/urea. | ||
Another interesting example of the use of ChCl/urea as the catalyst–solvent system is the one described by Azizi and coworkers in one-pot multicomponent reaction of isatin, or acenaphthoquinone, and malononitrile or cyanoacetic ester with 1,3-dicarbonyl compounds, naphtol and 4-hydroxycumarin (Scheme 42).88
 | ||
| Scheme 42 Synthesis of spirooxindole in ChCl/urea. | ||
A similar example can be found in one-pot reaction of salicylaldehyde (115) and malononitrile with various nucleophiles, including indoles, thiols, secondary amines, cyanides and azides in ChCl/urea. In such media, a broad range of salicylaldehydes and various nucleophiles were successfully converted to the desired 4H-chromenes (117) with high to excellent yields (Scheme 43).89 After completion, water was added to the reaction mixture, solid or viscous liquid was separated by filtration. The deep eutectic solvent was recovered from filtrate by evaporating the water. The recovered DES was then reused for three runs without obvious loss of activity (95%, 95%, 90% yields respectively).
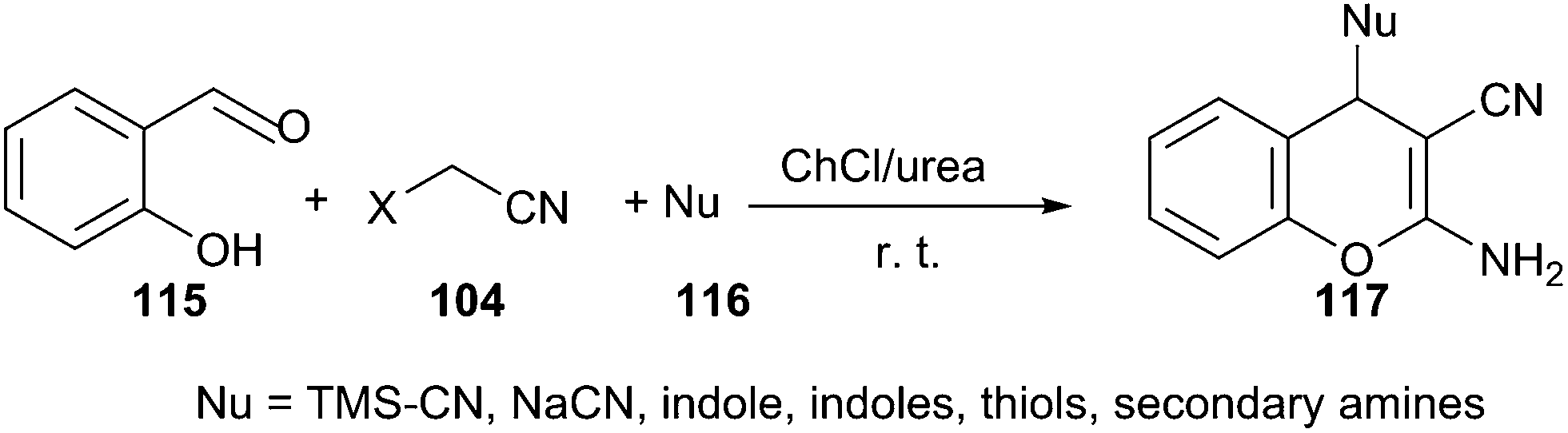 | ||
| Scheme 43 Multicomponent reactions of salicyladehydes and malononitrile with various nucleophiles in ChCl/urea. | ||
Spirooxindole is one of prevalent heterocycles found in numerous natural and synthetic products along with useful bio-, physio-, and pharmaceutical activities.90 The four-component reaction of phthalic anhydride (118), hydrazine hydrate (119), isatins and cyclic diketones/diamides (120) constitutes one of the most efficient manifolds for the synthesis of structurally diverse spirooxindoles spiroannulated (121) with pyrazolopyrimidophthalazines, indenopyrazolophthalazines, chromenopyrazolophthalazines and indazolophthalazines (Scheme 44).91 The reaction proceeded smoothly at 80 °C in ChCl/urea to give the corresponding products in good yields. ChCl/urea DES plays an important role in this reaction because of the fact that low yields were obtained in other solvent systems such as triethylamine–C2H5OH and piperidine–C2H5OH. The reaction mechanism may involve DES-catalyzed Knoevenagel condensation, Michael-type addition reaction and intramolecular dehydrative cyclization.
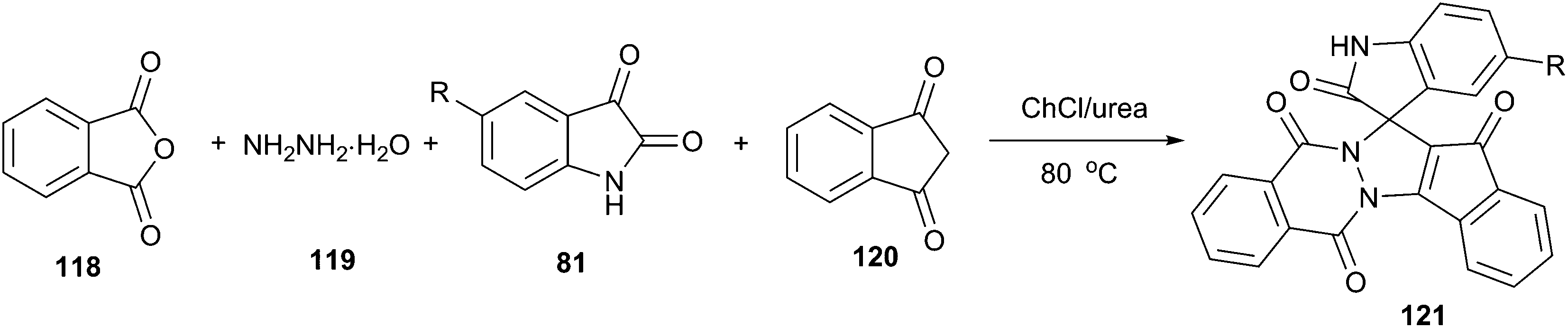 | ||
| Scheme 44 Synthesis of spirooxindoles in ChCl/urea. | ||
The same authors also reported a three-component reaction of aminouracils, isatins and cyclic carbonyl compounds for the synthesis of spirooxindoles (124) spiroannulated with pyranopyridopyrimidines, indenopyridopyrimidines, and chromenopyridopyrimidines using acid DES as a reaction medium.92 The three-component reaction of 6-amino-2-thiouracil (122), 4-hydroxy-6-methylpyran-2-one (123) and isatin as a simple model reaction to establish the feasibility of synthetic strategy (Scheme 45). The results clearly indicated that ChCl/oxalic acid showed superiority over the other systems such as ChCl/adipic acid, ChCl/succinic acid and ChCl/malonic acid. This reaction exhibited a broad substrate scope in such DES and the targeted spirooxindole derivatives were obtained with fair to excellent yields.
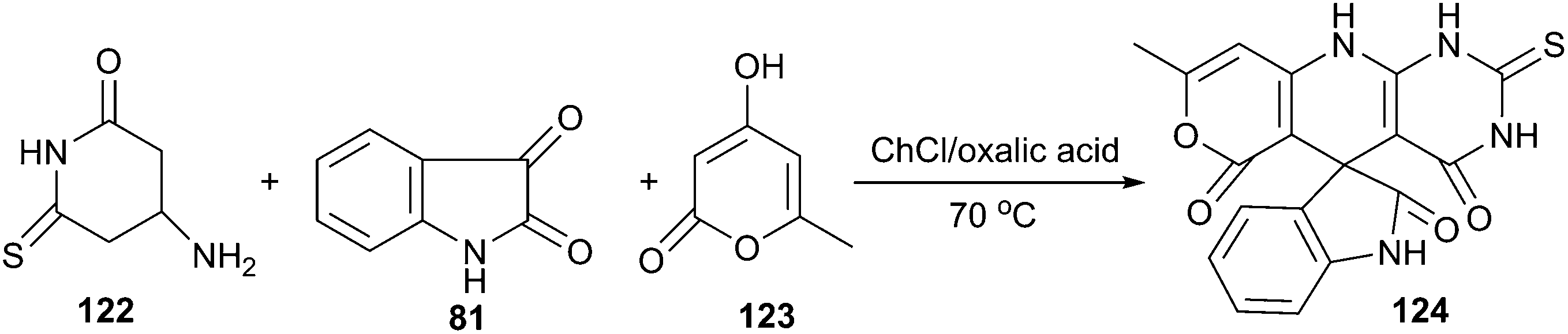 | ||
| Scheme 45 Synthesis of structurally diverse spirooxindoles in ChCl/oxalic acid. | ||
In 2015, Yan and co-workers reported that a series of novel spirooxindole derivatives (126) could be obtained by three-component domino Knoevenagel–Michael reaction of aldehydes, indole and 6,10-dioxaspiro[4.5]decane-7,9-dione (125) using choline chloride/oxalic acid as a catalyst (Scheme 46). The reactions were carried out at room temperature to give the spirooxindole derivatives in high yields. The reaction conditions allowed for the recycling of DES during five consecutive cycles, with no significant observation of loss of activity.93
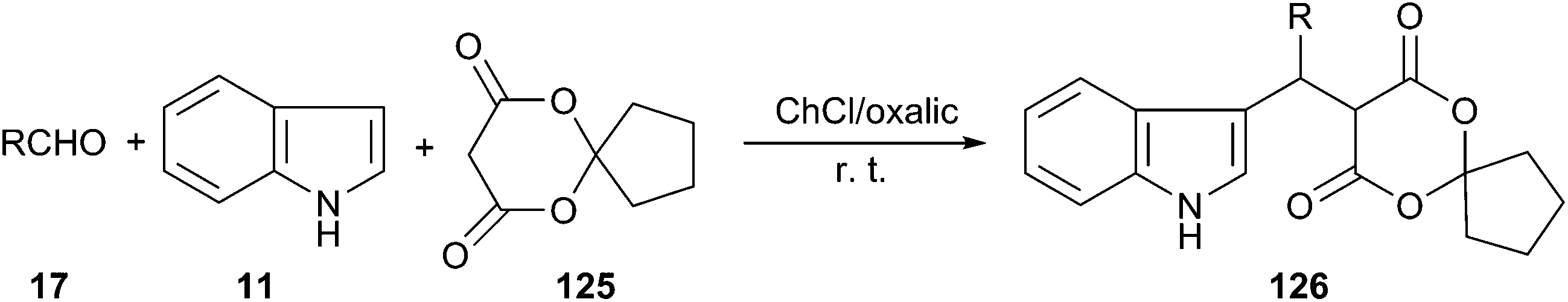 | ||
| Scheme 46 Synthesis of novel spirooxindoles in ChCl/oxalic acid. | ||
Heterocycles containing the phthalazine ring are important targets in synthetic and medicinal chemistry.94 He and his co-workers described an effective method for synthesis of 2H-indazolo[2,1-b]phthalazinetriones (128) by means of a three-component reaction of phthalhydrazide (127), an aldehyde, and dimedone using ChCl/p-TsOH DES as a catalyst (Scheme 47).95 The synthetic reaction was performed in a one-pot manner in the presence of 15 mol% of ChCl/p-TsOH. Under the same condition, methanol was found to be more effective than other organic solvents, such as EtOH, H2O, EtOH/H2O, MeOH/H2O, CH3CN, EtOAc, and toluene. At the end of the reaction, the product was easily and selectively extracted from the DES using ethyl acetate as an extraction solvent, thereby allowing the DES to successfully recycle at least 5 times without obvious loss in activity (86, 87, 82, 82 and 80% yield, respectively).
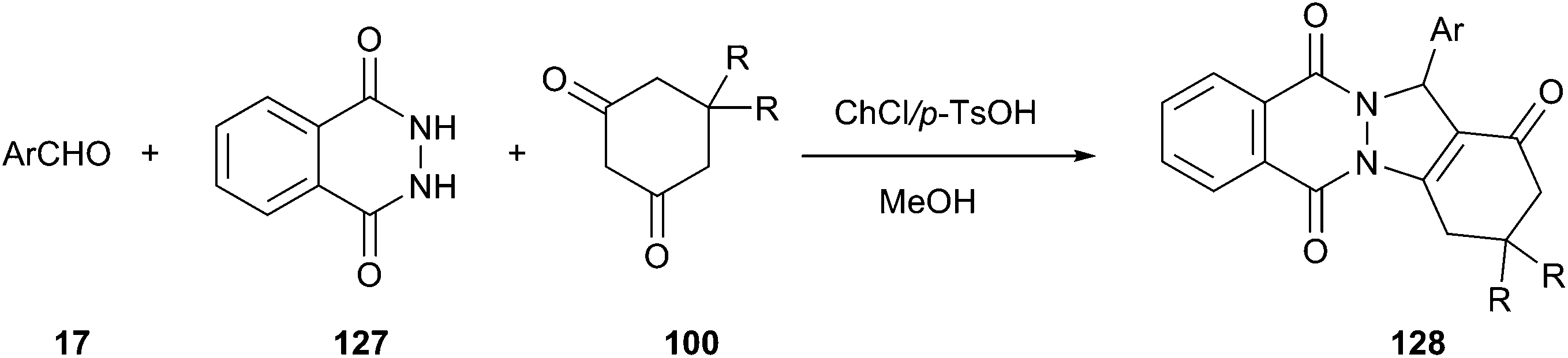 | ||
| Scheme 47 Synthesis of 2H-indazolo[2,1-b]phthalazine-triones catalyzed by ChCl/p-TsOH. | ||
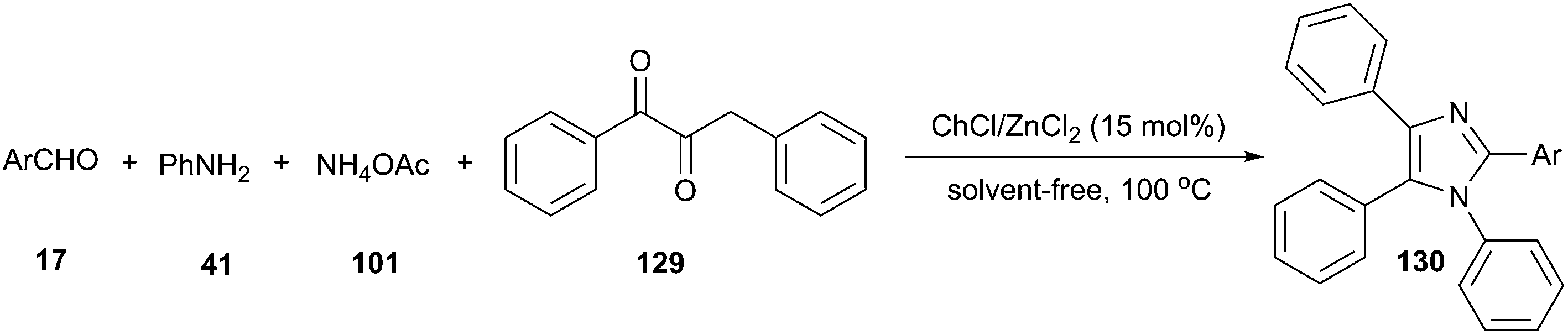 | ||
| Scheme 48 Synthesis of imidazoles catalyzed by ChCl/ZnCl2. | ||
In 2015, Mobinikhaledi and co-workers have investigated a four-component reaction of benzil, aldehyde, ammonium acetate and aniline in ChCl/MCl2 (M = Zn or Sn). The Lewis acid nature of these DES significantly enhanced the reaction rate. A broad range of aldehyde and aniline derivatives were successfully converted to the desired tetrasubstituted imidazoles (130) in high to excellent yields under solvent-free condition at 100 °C in the presence of 15 mol% of ChCl/ZnCl2 (Scheme 48).96
The same reaction can also be performed in ChCl/urea by using magnetically recoverable Fe3O4 nanoparticles as catalyst at 60 °C. The physical and chemical properties of the Fe3O4 in DES especially their Lewis acid sites had a significant influence on the catalytic activity. In this process, DES plays a triple role: as reaction medium, as hydrogen bond catalysis and stabilization of Fe3O4 nanoparticle.97
A Biginelli three-component reaction of aldehydes, 1,3-dicarbonyl compounds, and urea (131) has been developed by using ChCl/SnCl2 as a dual catalyst and environmentally benign reaction medium, solvent-free conditions, which generated various 3,4-dihydropyrimidin-2(1H)-one derivatives (132) in good to excellent yields (Scheme 49).98 In this MCR, ChCl/SnCl2 was proved to be a superior reaction medium when compared with other DESs, such as ChCl/urea (1:2), ChCl/ZnCl2 (1:2), and ChCl/ZnCl2/SnCl2 (1:1:1). In generally, the reactions were very clean, and no side products were obtained. The products obtained were of high purity and did not require any chromatographic separation in most cases.
 | ||
| Scheme 49 Synthesis of 3,4-dihydropyrimidin-2(1H)-ones in by ChCl/SnCl2. | ||
Azizi and his co-workers have demonstrated that ChCl/urea is an efficient promoting medium for synthesis of amino acid-based dithiocarbamates (135–137) by one-pot three-component reaction of the electrophilic reagent, carbon disulfide and α-amino acids (133). In situ preparation of dithiocarbamates by the reaction of different amino acids and carbon disulfide, followed by addition reaction with epoxides, alkyl halides and α,β-unsaturated enones at room temperature, gave the corresponding products in high yield with a short reaction time (Scheme 50).99 Interestingly, while the reaction proceeded very well in ChCl/urea, poor yields were obtained in other solvents such as tetrahydrofuran, acetonitrile, dichloromethane, toluene and ethyl acetate under the identical conditions, indicating that DES played an indispensable role in this reaction. In addition, DES could be recycled without activity or yields decrease.
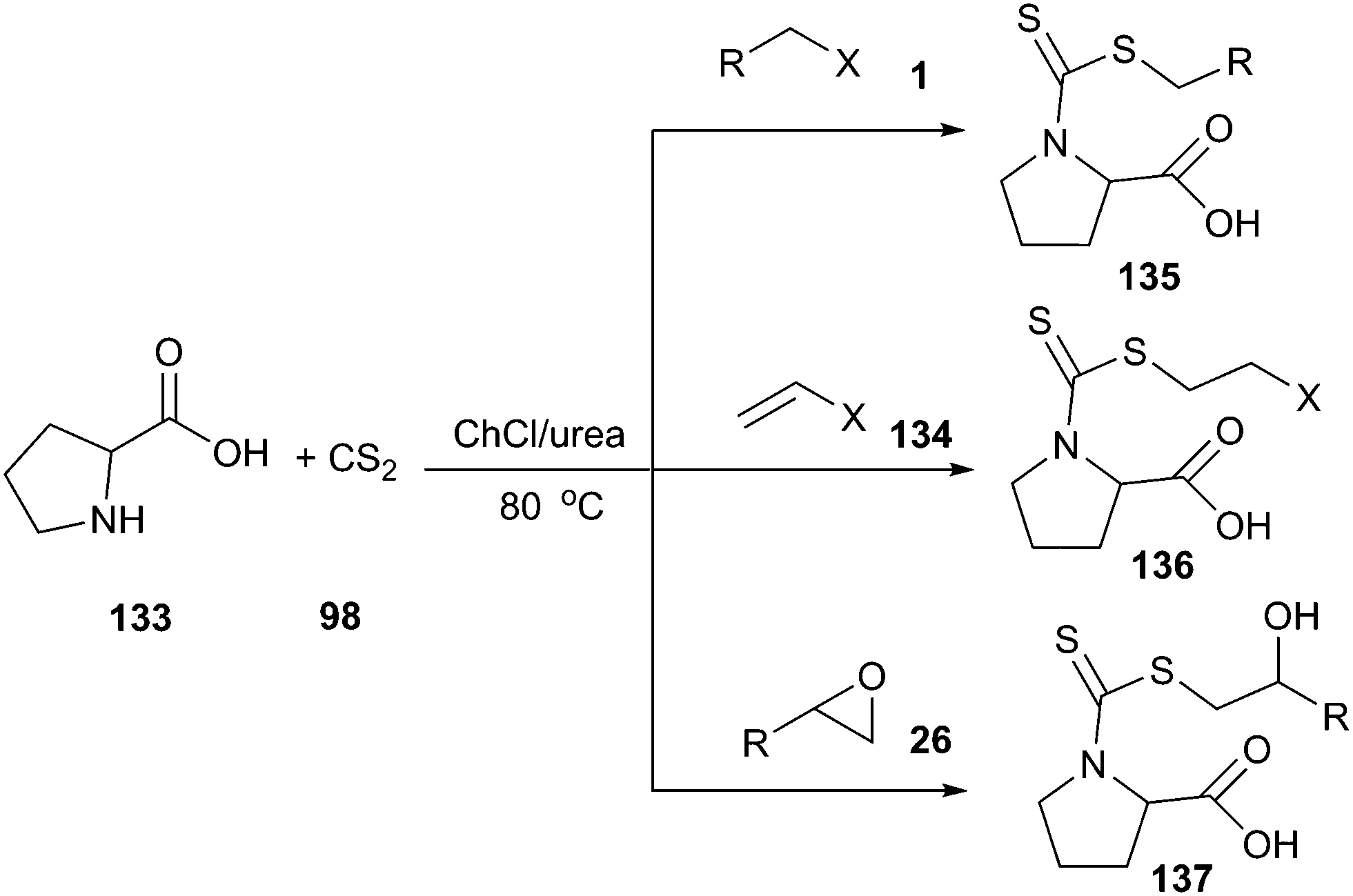 | ||
| Scheme 50 One-pot synthesis of proline dithiocarbamate in ChCl/urea. | ||
Another interesting example of the use of DES as the catalyst–solvent system was described by above authors for four-component synthesis of dithiocarbamate derivatives. The one-pot reaction of various aromatic aldehydes, ketones, aliphatic amines, and carbon disulfide, in the presence of potassium hydroxide in ChCl/urea at room temperature, led to the corresponding dithiocarbamates (138) in good to excellent yields (Scheme 51).100 In this MCR, ChCl/urea showed a significant beneficial effect on the reaction yields compared to classical organic solvents (THF or CH2Cl2) and water.
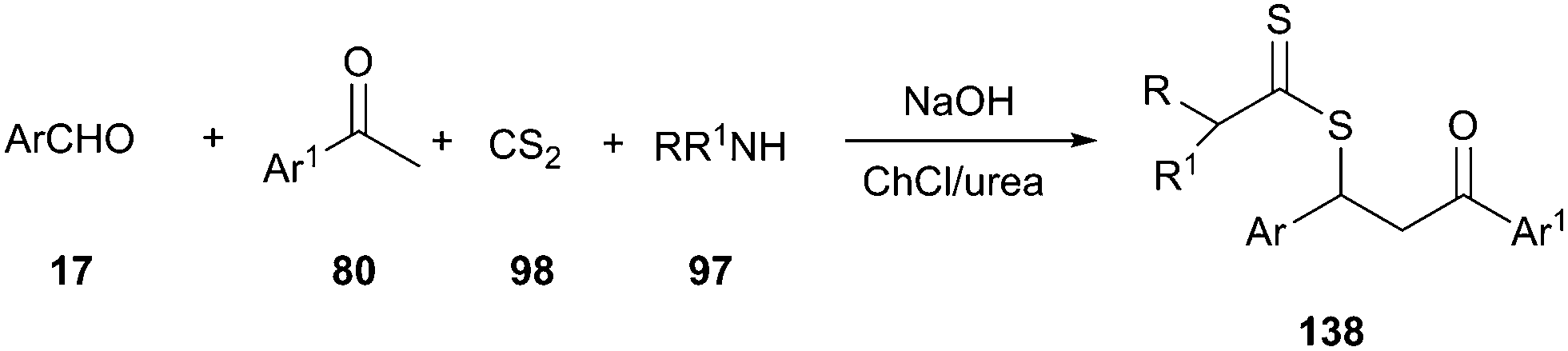 | ||
| Scheme 51 Four-component synthesis of dithiocarbamates in ChCl/urea. | ||
More recently, the above group has been found that ChCl/urea is a highly effective catalyst and reaction medium for one-pot synthesis of 2-aminothiazole and 2-aminoxazole derivatives. Three-component reactions of active methylene compounds, urea or thiourea (139) and N-bromosuccinimide (NBS) (140) proceeded without organic solvent and in the absence of basic or acidic catalyst at 60 °C to furnish structurally diverse 2-aminoxazoles and 2-aminothiazoles (141) in good to excellent yields with short reaction times (Scheme 52).101
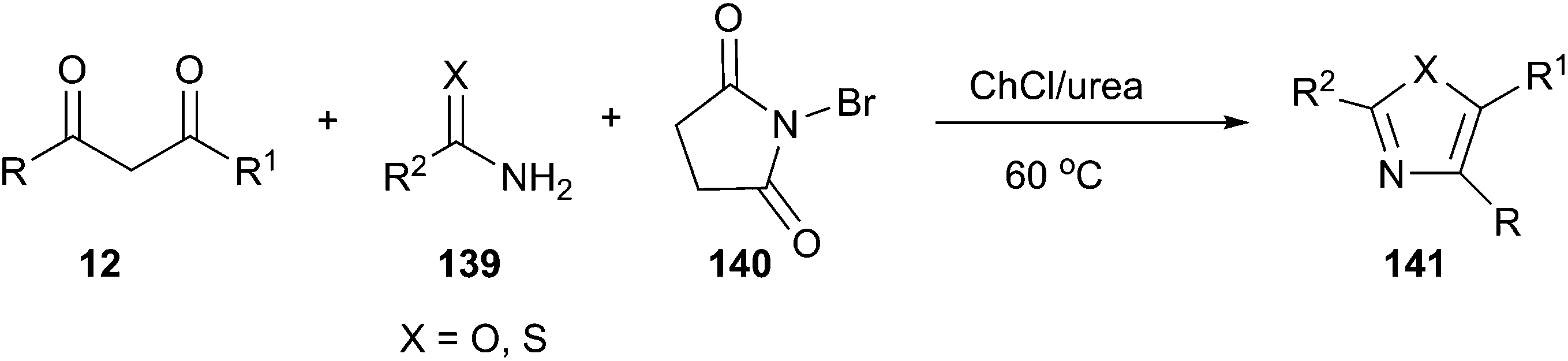 | ||
| Scheme 52 Synthesis of substituted 2-aminothiazoles and 2-aminoxazole derivatives in ChCl/urea. | ||
In 2012, we reported an efficient three-component reaction of 2-aminoaryl ketones (142), aldehyde, and ammonium acetate under aerobic oxidation conditions using a low melting mixture of maltose/dimethylurea/NH4Cl as reaction medium, which generated quinazoline derivatives (143) in high yields (Scheme 53).102 For comparative purposes, the suitability of other organic solvents, such as CH3CN, DMF, DMSO or toluene as well as various melt mixtures, including citric acid/DMU, D-(−)/fructose/DMU, L-(+)-tartaric acid/DMU, L-(+)-tartaric acid/choline chloride, mannose/DMU/NH4Cl, lactose/DMU/NH4Cl, and maltose/DMU/NH4Cl, in this catalyst-free MCR was evaluated under the same experimental conditions. In all cases, the desired products were obtained either in a trace amount or in a significantly lower yield than in maltose/DMU/NH4Cl. The low melting mixture could be recovered easily after the reaction, and reused up to three cycles as a reaction medium with a slight decrease in yields.
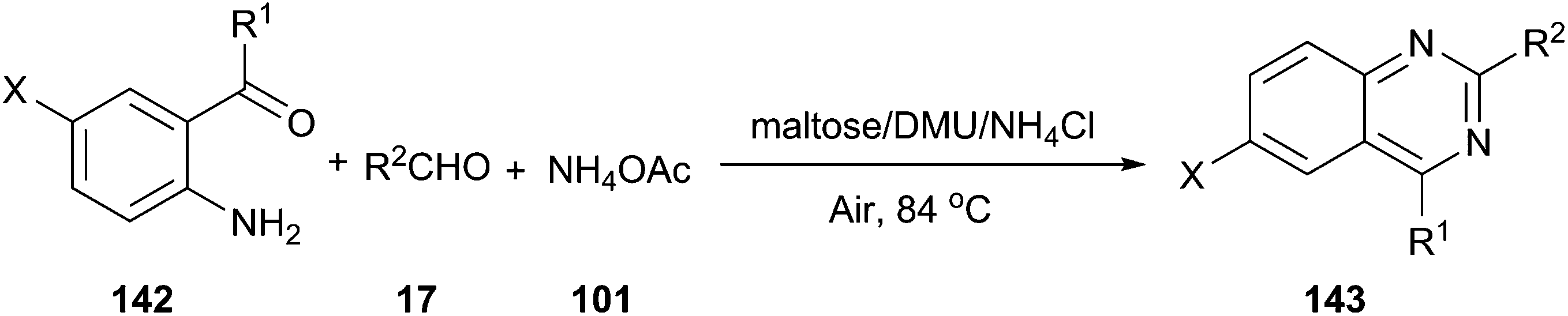 | ||
| Scheme 53 Synthesis of quinazoline derivatives in maltose/DMU/NH4Cl. | ||
MCRs have emerged as vital tools for the preparation of substituted and functionalized pyrrole derivatives (145) from simple materials.103 In 2015, we explored a one-pot, four-component reaction of amines, aldehydes, 1,3-dicarbonyl compounds and nitromethane (144) using a DES as reaction medium and catalyst. In order to optimize the reaction condition, the model reaction of aniline, 4-chlorobenzaldehyde, acetylacetone and nitromethane was investigated (Scheme 54).104 Among all screened DESs such as ChCl/FeCl3, ChCl/ZnBr2, ChCl/itaconic acid, ChCl/fumaric acid, ChCl/malic acid, ChCl/succinic acid, ChCl/malonic acid led to the highest yield. It was found that 12% yield of product was obtained when only malonic acid was used. And, no reaction was observed in the case of ChCl in the absence of malonic acid, which indicated that neither ChCl nor malonic acid promote the reaction alone. The co-existence of ChCl and malonic acid had shown the synergetic effect on the reaction, which was perhaps the main reason for the high catalytic activity of the DES. This work exhibited a broad substrate scope and products of the reaction were conveniently isolated from the DES by extraction with ethyl acetate, allowing the DES to be recycled. This reaction protocol provides a very efficient and straightforward route to construct various functionalized pyrrole without using an acidic condition or metal catalysts. It is worth mentioning that the reaction time is shortened obviously compared with previous reported method.105
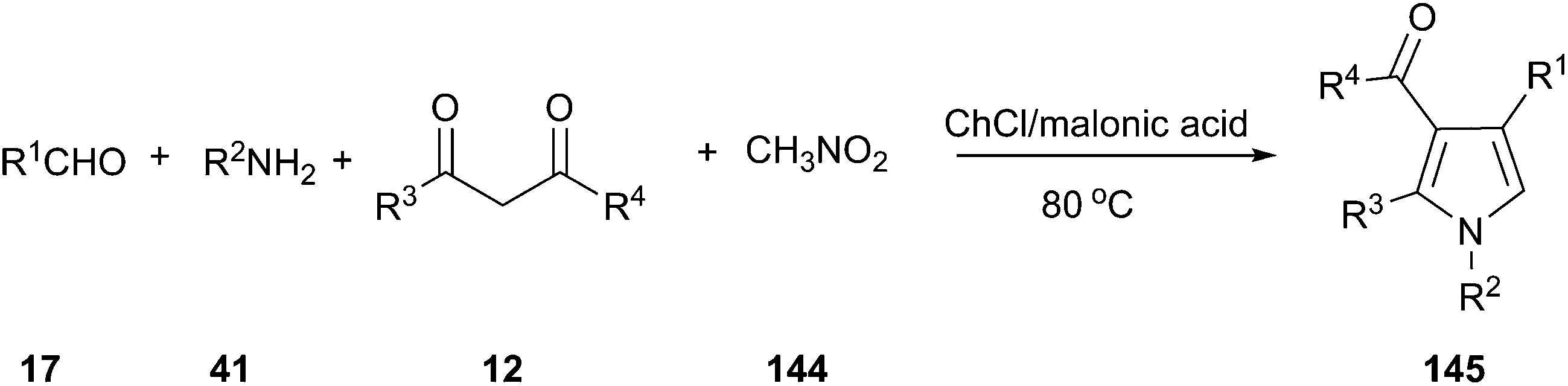 | ||
| Scheme 54 Synthesis of functionalized pyrroles in ChCl/malonic acid. | ||
Another example for synthesis of annulated pyrroles (148) by three-component reaction between free sugars (146), amines and 1,3-dicarbonyl compounds (147) has been reported by Rokade and co-workers using ChCl/urea (Scheme 55).106 The reaction may proceed via formation of intermediate enamines, generated in situ from amines and 1,3-dicarbonyl compounds. And condensation of the enamines with free sugars is facilitated by hydrogen bonding of urea with the anomeric hydroxyl group. The resulting aldol product on subsequent cyclodehydration followed by aromatization afforded the annulated pyrrole. Ease of recovery and reusability of DES with high activity make this protocol more practical for preparation of annulated pyrroles in large scale.
 | ||
| Scheme 55 Synthesis of annulated pyrroles in ChCl/urea. | ||
The combination of DES and magnetic nanoparticles as catalyst was used to synthesise imidazo[1,2-a]pyridines (151) by one-pot, three-component reaction of 2-aminopyridines (149), aldehydes, and alkynes (150) (Scheme 56).107 By using magnetic CuFeO2 as catalyst, the synthesis was examined in many organic solvents and DESs such as methanol, ethanol, toluene, acetonitrile, N,N-dimethylformamide, polyethylene glycol D-(−)-fructose/DMU, mannose/DMU/NH4Cl and lactose/DMU/NH4Cl, and it was found that the reaction in citric acid/DMU gave the best result. After reaction, citric acid/DMU solvent as well as CuFeO2 catalyst can be recovered and reused, thus making this protocol economically and potentially viable.
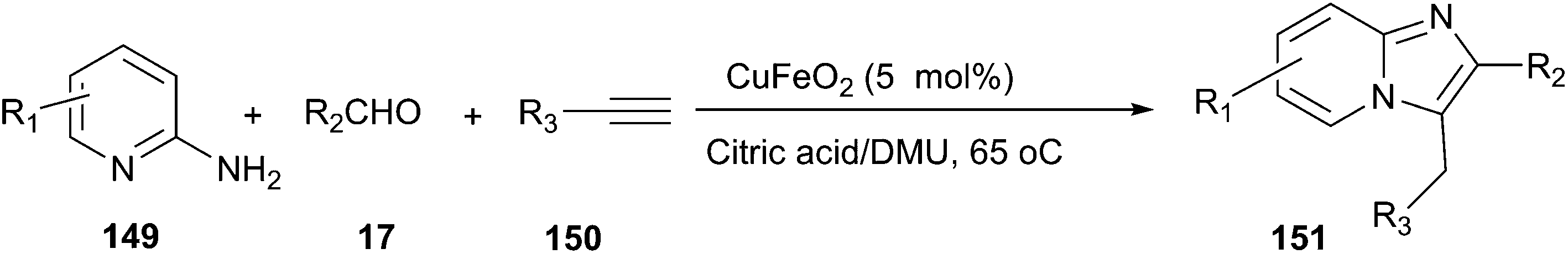 | ||
| Scheme 56 Synthesis of imidazo[1,2-a]pyridines using CuFeO2 in citric acid/DMU. | ||
In 2013, Siddalingamurthy et al. have developed a versatile and general method using ChCl/urea as a catalyst for the three-component coupling of N-methyl indole, Meldrum's acid (152) and aldehydes to get Meldrum's adduct (153) followed by reaction of amines with this Meldrum's adduct to get indole-3-propanamides (154) (Scheme 57).108
 | ||
| Scheme 57 Synthesis of indole-3-propanamides using ChCl/urea. | ||
2.5 DES in condensation reactions
ChCl/urea has also been described as catalyst and reaction medium by Pawar and co-workers in 2011 in Perkin reaction of aldehydes with anhydrides for the synthesis of cinnamic acid and its derivatives (155) (Scheme 58).109 This method eliminates the use of hazardous organic solvents and toxic catalysts, and the reaction time and temperature can be remarkably reduced. Furthermore, the DES could be reused up to three times without any loss in activity. | ||
| Scheme 58 Perkin reaction using ChCl/urea. | ||
Another interesting example of the use of ChCl/urea as the catalyst–solvent system is described by Liu and coworkers in Knoevenagel condensation of aromatic aldehydes with active methylene compounds (156) such as malononitrile, ethyl cyanoacetate, benzimidazole-2-acetonitrile and benzothiazole-2-acetonitrile (Scheme 59).110 In order to optimize the reaction condition, the different molar ratios of ChCl and urea and the different amount of DES had been studied by the authors. The control experiments demonstrated that 20% ChCl/urea (1:2) under solvent-free conditions gave best yield (92%). Sonawane et al. reported that a series of symmetrical and unsymmetrical chromophores can be synthesized by Knoevenagel condensation of 4,4′-hexyliminobisbenzaldehyde and different active methylene compounds such as 2-(3,5,5-trimethylcyclohex-2-enylidene)malononitrile, 2-(4-methoxy phenyl)acetonitrile, 2-phenylacetonitrile, ethyl 2-cyanoacetate, 2-(thiophene-2-yl)acetonitrile, (2E)-2-cyano-2-(3,5,5-trimethylcyclohex-2-enylidene)acetate, 2-(4-nitrophenyl) acetonitrile, and malononitrile in ChCl/urea.111
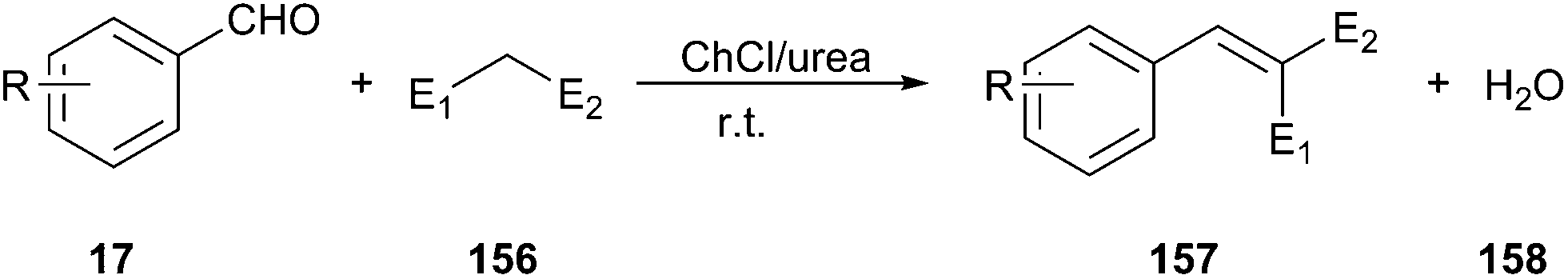 | ||
| Scheme 59 Knoevenagel condensation using ChCl/urea. | ||
Coumaranones were also synthesized by a Knoevenagel condensation of coumaranones with aldehydes in short reaction times and high yields in ChCl/urea.112 Harishkumar et al. applied this method to prepare coumarins (161) via condensation starting from salicyl aldehydes (159) and active methylene compounds (160) such as meldrum's acid, diethylmalonate, ethyl cyanoacetate, dimethylmalonate (Scheme 60).113 More recently, this work was extended to synthesize a series novel coumarin styryl dyes containing the 2-(1-(7-(diethylamino)-2-oxo-2H-chromen-3-yl)ethylidene)malononitrile moiety.114 In these process, DES played the dual role of catalyst as well as solvent, thereby negating the use of any additional catalyst.
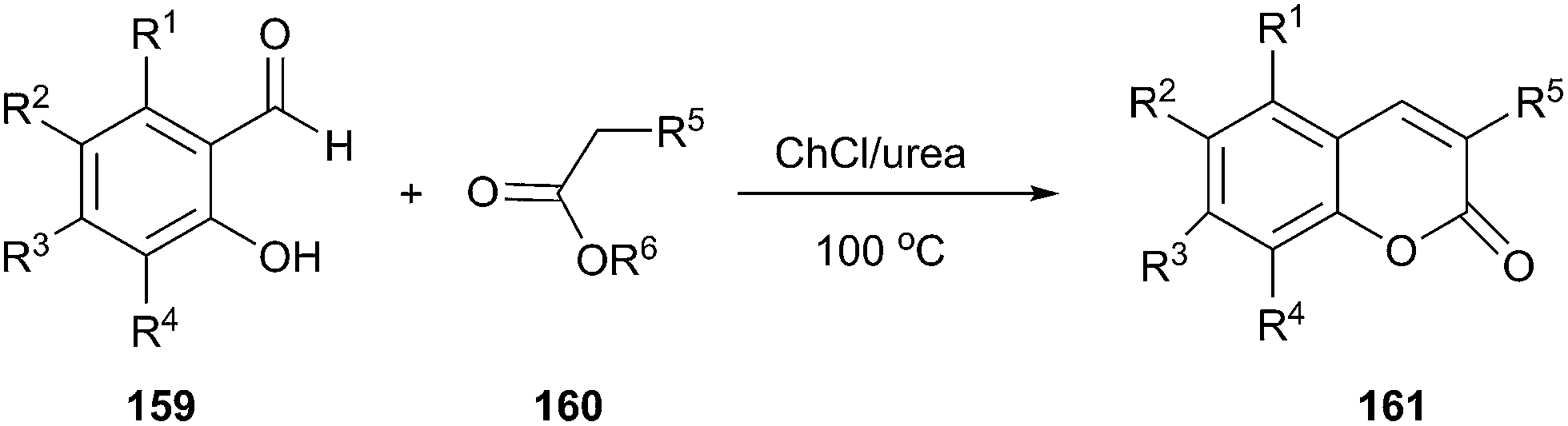 | ||
| Scheme 60 Synthesis of coumarins in ChCl/urea. | ||
Acid-catalyzed condensation of aldehydes with 1,3-dicarbobonyl compounds to xanthene derivatives has gained considerable attention in recent years since xanthenes are one of the important classes of heterocyclic compounds and are known to possess a wide range of pharmacological activities and used as valuable synthetic precursors for many organic compounds.115 In 2013, we have found that condensation of aromatic aldehydes and cyclic 1,3-dicarbobonyl compounds could also be conveniently performed in an acidic DES composed of citric acid and DMU. This reaction exhibited a broad substrate scope in such DES and the targeted xanthene derivatives were obtained with high to excellent yields (Scheme 61).116 The use of other low melting mixture such as glucose/urea/CaCl2, glucose/urea/NaCl, sucrose/ChCl, glucose/guanidinium HCl, L-(+)-tartaric acid/ChCl, and L-(+)-tartaric acid/DMU in this reaction led to decrease of yields.
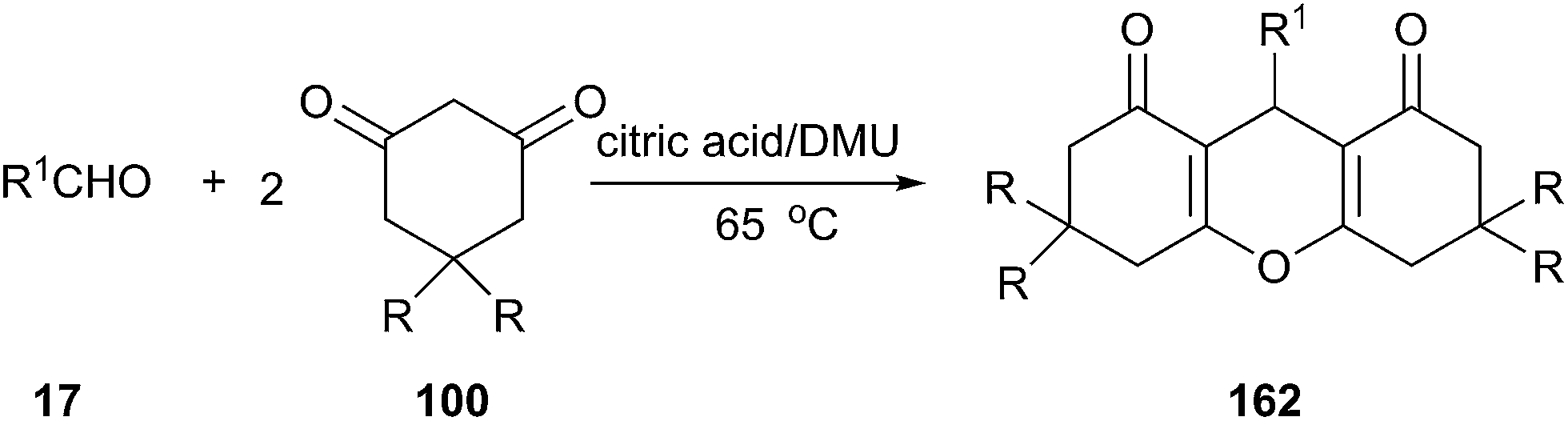 | ||
| Scheme 61 Synthesize of xanthene derivatives using citric acid/DMU. | ||
In 2006, Duan and co-workers reported that ChCl/ZnCl2 DES has been used as an efficient and moisture-stable Lewis acidic catalyst for protection of carbonyls to 1,3-dioxanes at room temperature under solvent-free conditions (Scheme 62). Various aldehydes could be successfully converted to corresponding acetals (164) using neopentyl glycol (163) as protection reagent. The catalytic system can be reused up to five times without appreciable loss of activity.117
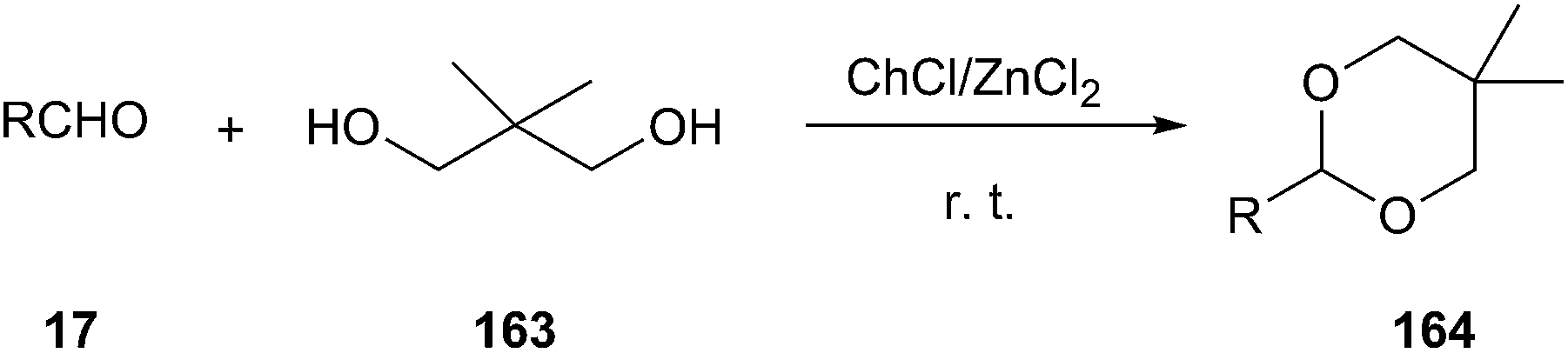 | ||
| Scheme 62 Synthesize of 1,3-dioxanes using ChCl/ZnCl2. | ||
An interesting iodine-catalyzed selective synthesis of monoarylureas (165) was reported by Mahajan et al. The reactions were carried out in citric acid/urea/mannitol mixture (30:20:5) at 80 °C in the presence of catalytic amount of iodine, various monoarylureas were produced from anilines in good yields (Scheme 63).118
 | ||
| Scheme 63 Selective synthesis of monoarylureas in citric acid/urea/mannitol mixture. | ||
A facile and efficient approach for synthesis of gem-bisamides (166) starting from aldehydes using ChCl/urea has been developed by Azizi group (Scheme 64).119 Various arylaldehydes including some heteroaryl derivatives such as 2-furaldehyde, thiophene-2-carbaldehyde and pyridine-carbaldehyde as well as aliphatic aldehydes could undertake this reaction in good yields. In this process, ChCl/urea plays the triple role of being solvent, catalyst and reagent.
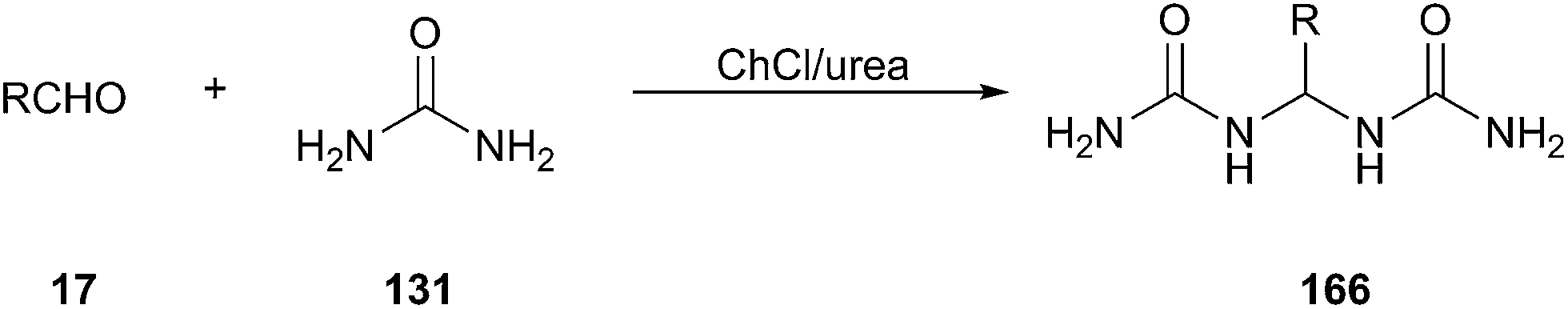 | ||
| Scheme 64 Synthesis of bisamides using ChCl/urea. | ||
More recently, Azizi et al. reported a fast and green method for the formation of carbon–nitrogen bond using ChCl/urea as a cost effective and eco-friendly solvent.120 The N,N′-bis(arylmethylidene)arylmethane diamines and imines from the reaction of aromatic aldehydes with ammonia and a variety of amines were prepared in ChCl/urea at 60 °C (Scheme 65). The reaction was found to work well with a variety of aromatic aldehydes bearing electrondonating as well as electron-withdrawing groups. However, aliphatic aldehydes did not participate in the reaction.
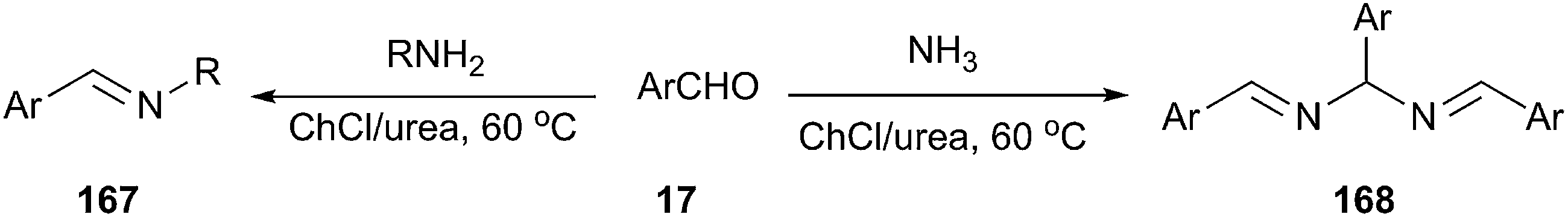 | ||
| Scheme 65 Synthesis of bisamides using ChCl/urea. | ||
In another study, it was reported by Lobo et al. that the DES from choline chloride and malonic acid is an efficient catalyst for synthesis of N-aryl phthalimide derivatives (169) by condensation of phthalic anhydride and primary aromatic amines (Scheme 66). The reaction proceeded smoothly at 65 °C in methanol to give the corresponding product in good yield.121
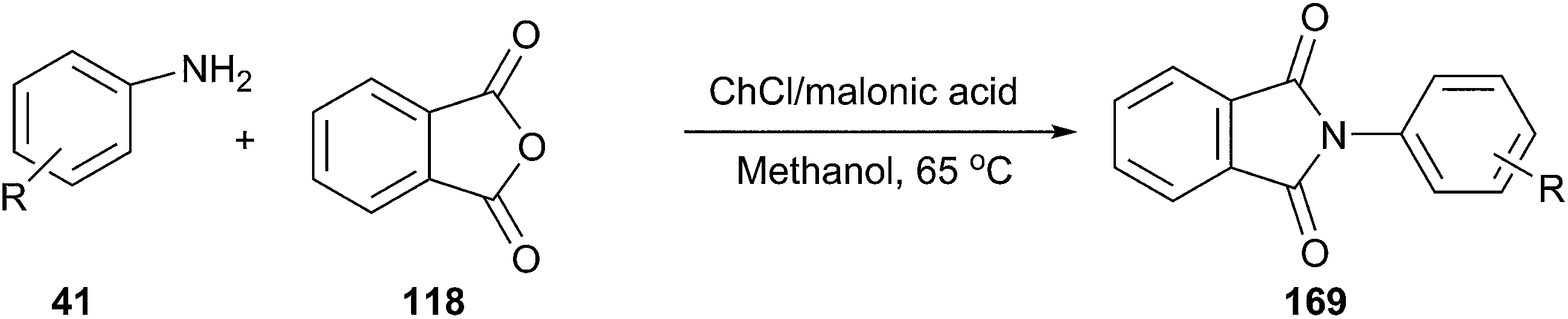 | ||
| Scheme 66 Synthesis of N-aryl phthalimide derivatives using ChCl/malonic acid. | ||
One-pot synthesis of nitriles from aldehydes and hydroxylamine hydrochloride (157) has also been carried out efficiently using ChCl/urea as a catalyst. The corresponding nitriles were obtained in good to excellent yields (68–92%) in short reaction times (10 min) with the aid of microwave irradiation.122 When the reaction was performed in ChCl/ZnCl2 at 100 °C, the corresponding primary amides were obtained in high yields (Scheme 67).123
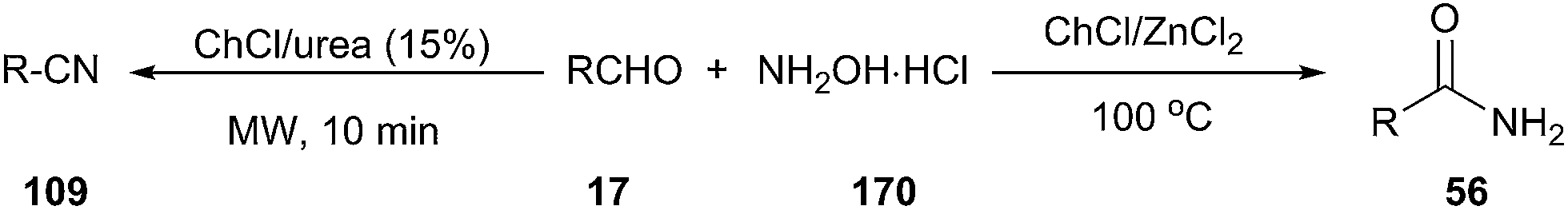 | ||
| Scheme 67 Synthesis of nitriles using ChCl/urea. | ||
In another study, Singh and coworkers explored the efficacy of the DES for benzylation of phenols (171) with benzyl bromide (Scheme 68).124 It was found that ChCl/urea was promising solvent and phase-transfer-medium for benzylation of phenols. Lower yields were obtained in other media such as ChCl/glycerol and ChCl/ZnCl2. Various bases such as NaOH, K2CO3, and K3PO4 were also investigated. Among all the bases, KOH gave the highest yield of the desired product. No benzylation of phenol was observed when the reaction was carried out in the absence of DES, demonstrating the necessity of DES. Structurally diverse phenols with different functional groups such as methyl, methoxy, chloro, bromo, tertiary butyl, acyl, and trifluoromethyl groups underwent benzylation with various aromatic bromides and the products were obtained in good to excellent yields.
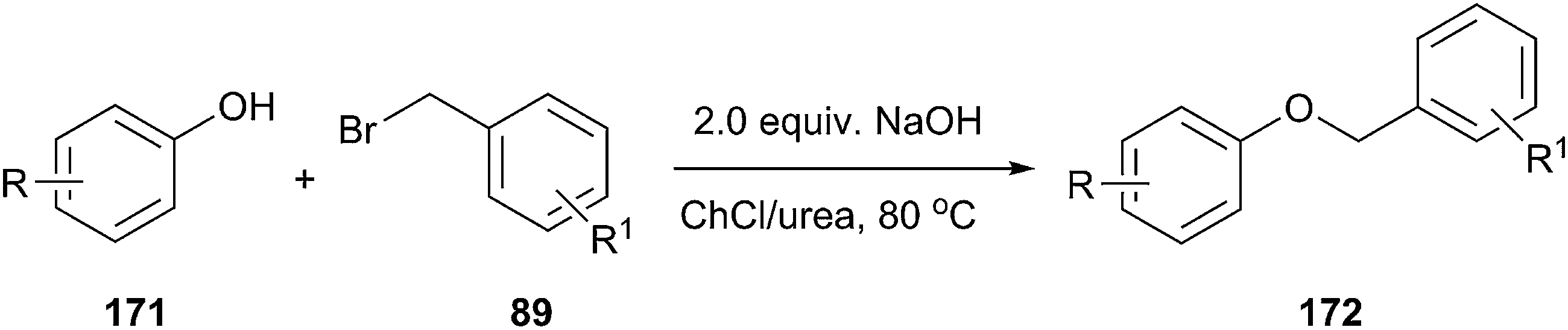 | ||
| Scheme 68 The benzylation of phenols using ChCl/urea. | ||
In 2012, Azizi employed ChCl/SnCl2 deep eutectic solvent as a green catalyst and reaction medium for the efficient N-formylation of aromatic amines under mild reaction conditions (Scheme 69).125 A series of deep eutectic solvents, such as ChCl/SnCl2, ChCl/urea, ChCl/ZnCl2, ChCl/ZnCl2/SnCl2 and ChCl/Gly were tested, and ChCl/SnCl2 showed better activity in terms of the yields of the products. The corresponding N-formyl derivatives were obtained in good to excellent yields by the reaction of aromatic amines with formic acid in ChCl/SnCl2 at 70 °C. In the presence of ChCl/SnCl2, the reaction of aniline with trimethyl orthoformate, depending on the reaction conditions, provided either product 173 or 175 as the major product. Treatment of aromatic amines with trimethyl orthoformate at 70 °C in the presence of water (0.5 mL) gave the corresponding N-formyl derivatives. When the same reaction condition was carried out under anhydrous conditions at 90 °C, N,N′-diarylamidines (175) was obtained.
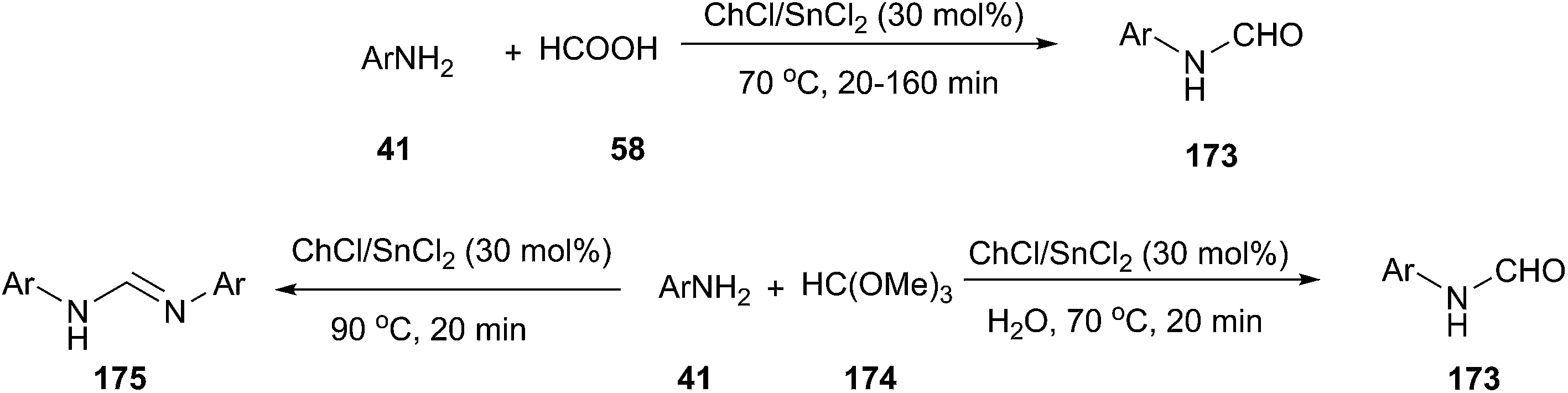 | ||
| Scheme 69 Synthesis of formamides and N,N′-diarylamidines using ChCl/SnCl2. | ||
2.6 DES in oxidation reactions
Oxidation of alcohols to the corresponding carbonyl compounds is considered a most important transformation in organic synthesis. In 2014, Azizi found that ChCl/urea was a highly efficient catalyst and reaction medium for rapid oxidation of alcohols (176) to aldehydes and ketones using N-bromosuccinimide (NBS) as the oxidant (Scheme 70).126 In this method they tried different DESs and organic solvents such as ChCl/urea, ChCl/SnCl2, ChCl/ZnCl2, ChCl/LaCl3, ChCl/p-TSA, dichloromethane, ethanol, methanol, acetonitrile and ethyl acetate, but found ChCl/urea appeared to be the best reaction medium for this reaction, showing the highest yield. This mild procedure can make selective oxidation of secondary alcohols in the presence of primary alcohols. Furthermore, the synthesis of 2-bromoacetophenone was achieved through ChCl/urea-mediated tandem oxidation and bromination of alcohol.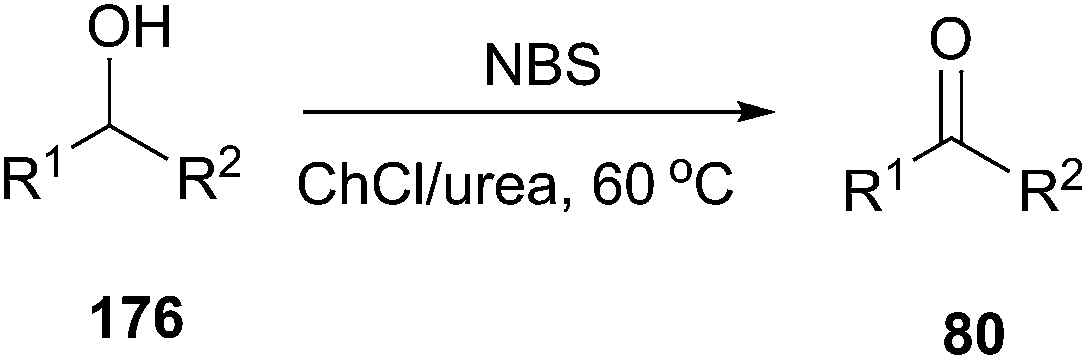 | ||
| Scheme 70 Oxidation of alcohols in ChCl/urea. | ||
In a recent report, Gadilohar and coworkers prepared choline peroxydisulfate (ChPS) based on peroxydisulfate anion and choline cation (Scheme 71) and investigated its catalytic performance in the above reaction.127 The prepared oxidant exhibited a superior oxidant activity in the synthesis of aldehydes and ketones under mild solvent-free reaction conditions with high yields and excellent selectivities. This ChPS is cheap, easy to prepare, and easily biodegradable.
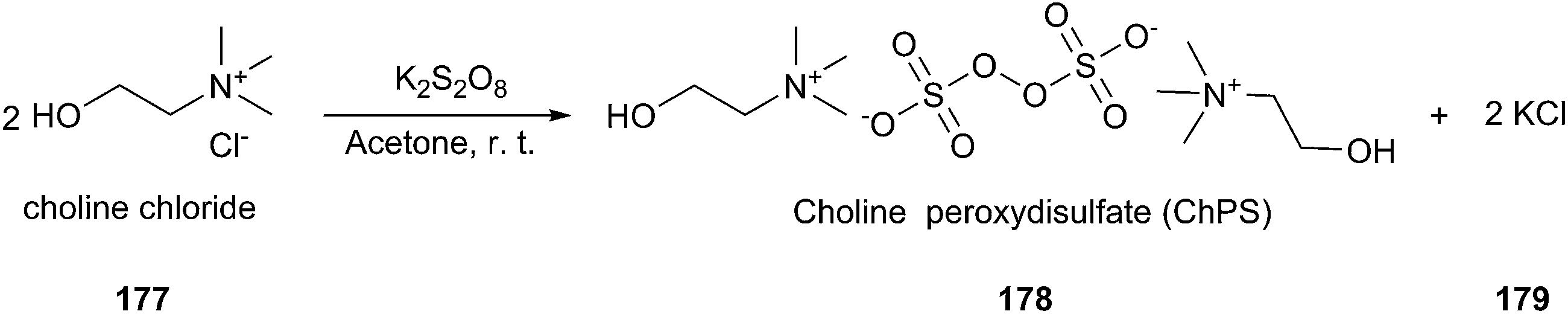 | ||
| Scheme 71 Synthesis of ChPS. | ||
More recently, Zhang et al. designed a DES supported TEMPO (DES–TEMPO) composed of N,N-dimethyl-(4-(2,2,6,6-tetramethyl-1-oxyl-4-piperidoxyl)butyl)dodecyl ammonium salt ([Quaternium-TEMPO]+Br−) and urea. TEMPO in combination with Fe(NO3)3 as co-catalyst showed excellent performances on the oxidation of various alcohols to their corresponding carbonyl compounds with molecular oxygen as oxidant under solvent-free conditions. The DES–TEMPO could be easily recovered from the reaction mixture and reused up to five times with no significant loss of its catalytic activity.128
Sulfoxides (180) are important synthetic intermediates for the synthesis of various biologically active molecules. Meanwhile, the use of hydrogen peroxide has attracted much attention in organic synthesis since it is readily available, cheap and environmentally benign, with water as the only byproduct.129 In 2014, Dai et al. found that ChCl/p-TsOH can be used an efficient catalyst for the selective oxidation of sulfides (181) with H2O2 as the oxidant (Scheme 72).130 Moreover, the use of ChCl/p-TsOH, instead of neat p-TsOH, can greatly reduce the acid sewage especially in large-scale synthetic processes.
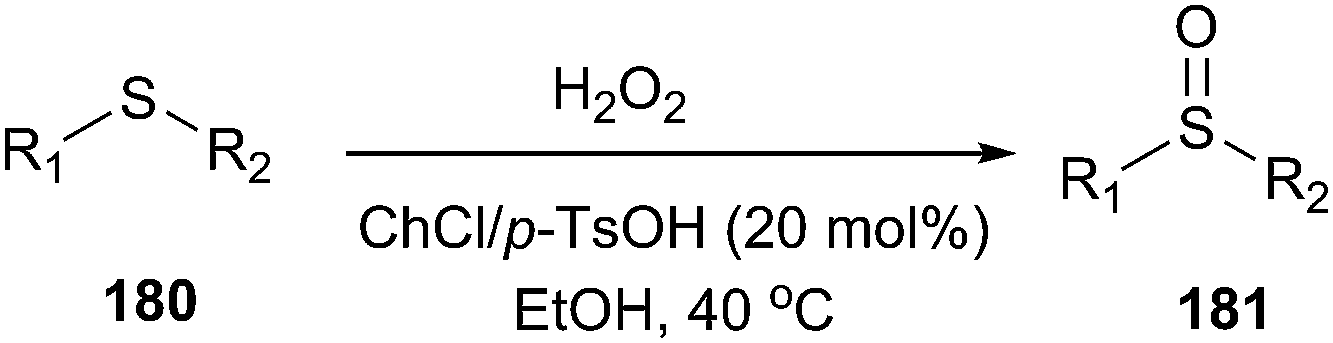 | ||
| Scheme 72 Selective oxidation of sulfides using ChCl/p-TsOH. | ||
The use of a new deep eutectic mixture based on ChCl and 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) as a catalyst in transformation of aryl/heteroaryl boronic acids (169) to the corresponding phenols (63) in water using hydrogen peroxide as the oxidant have been developed by Wang et al. (Scheme 73).131 Broad substrate compatibility, metal- and additive-free conditions as well as reusability of the catalyst made this procedure more environmentally benign.
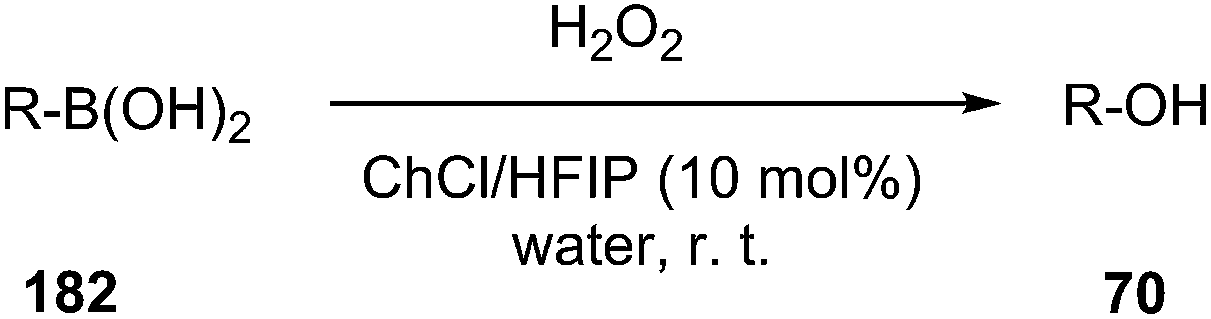 | ||
| Scheme 73 Oxidative hydroxylation of arylboronic acid using ChCl/HFIP. | ||
2.7 DES in the reduction reactions
The reduction of carbonyl compounds to the corresponding alcohols is an important transformation in organic synthesis. The combination of ChCl/urea with sodium borohydride has been successfully applied in reduction of carbonyl compounds.132 Thus, a variety of functionalized aldehydes and ketones as well as α,β-unsaturated carbonyl compounds in ChCl/urea in the presence of NaBH4 at room temperature were reduced to the corresponding alcohols in high yields. The chemoselective reduction of epoxide compounds by the nucleophilic attack by NaBH4 resulted in the formation of the corresponding alcohols in ChCl/urea (Scheme 74).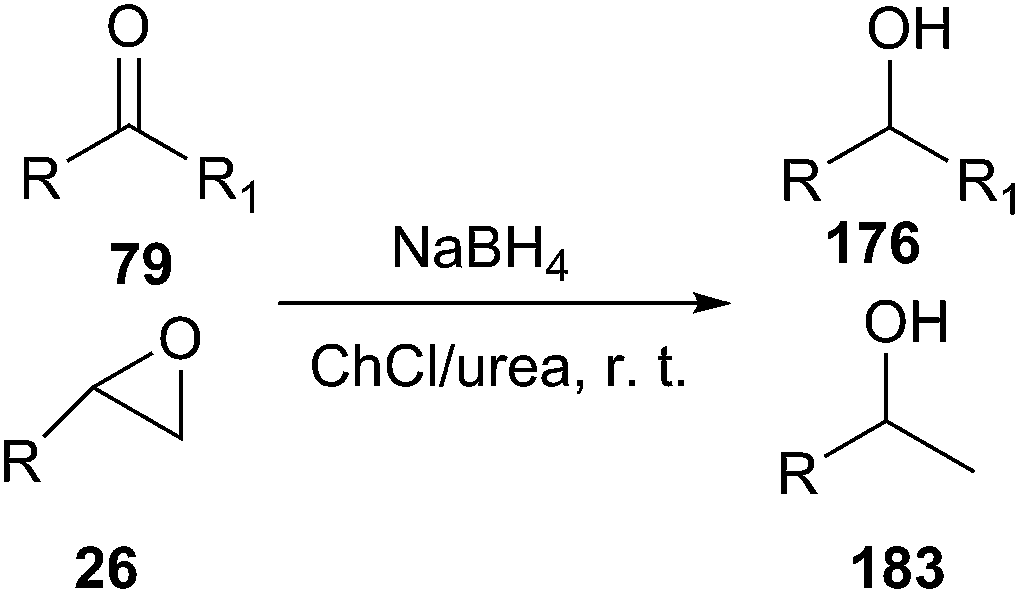 | ||
| Scheme 74 Reduction of carbonyl compounds and epoxides in ChCl/urea. | ||
In a related study, Xu and coworkers have also used ChCl/urea to improve biocatalytic asymmetric reduction of 3-chloropropiophenone (CPE) (184)to (S)-3-chloro-1-phenylpropanol (CPL) (185) catalyzed by immobilized Acetobacter sp. CCTCC M209061 (Scheme 75). In this process, the preparation of (S)-CPL on a large scale was feasible in the ChCl/urea-containing system, in which the biocatalyst exhibited much higher operational stability.133
 | ||
| Scheme 75 Asymmetric reduction of CPE in ChCl/urea-containing aqueous system. | ||
It is noteworthy that in 2014, Maugeri et al. reported that whole-cell biocatalysis with the use of baker's yeast in different mixtures of water with deep eutectic solvent ChCl/glycerol for enantioselective ketone reduction (Scheme 76).134 It was observed that the enantioselective reduction of ethyl acetoacetate needed long reaction times (>200 h), which suggests that the whole cells remain stable in these neoteric solvents. By changing the proportion of the DES added in water, a complete inversion of enantioselectivity was observed, from approximately 95% enantiomeric excess (ee) (S) in pure water to approximately 95% ee (R) in the pure ChCl/glycerol. The authors thinks that some (S)-oxidoreductases present in baker's yeast are inhibited by DESs, whereas R-selective oxidoreductases would stay as active biocatalysts within the whole cell. It is our opinion that this work demonstrates that DESs may actually become promising unconventional reaction media for whole-cell biocatalysis.
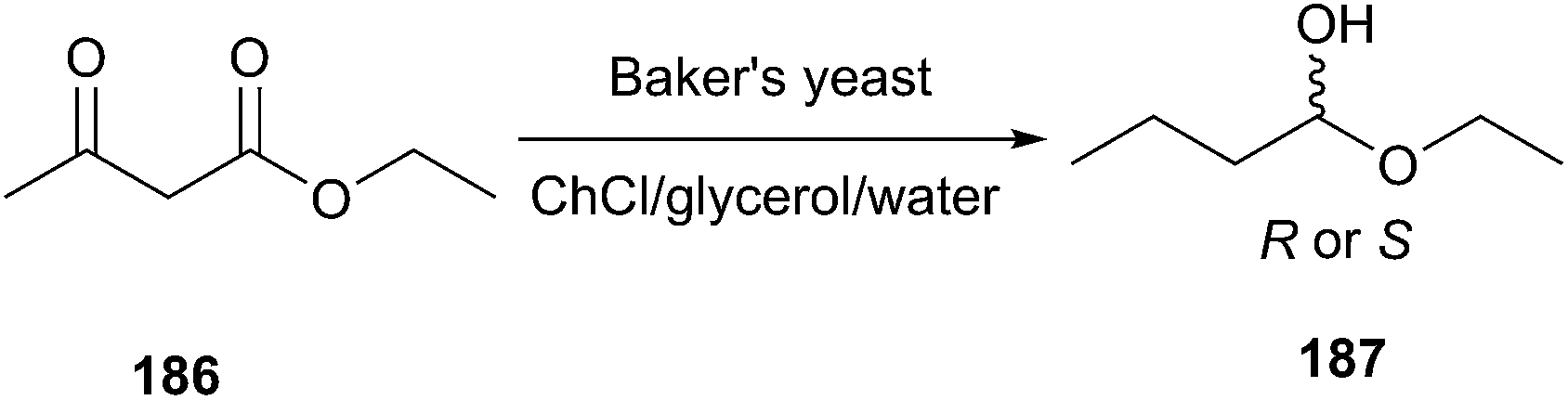 | ||
| Scheme 76 Enantioselective reduction of ethyl acetoacetate in ChCl/glycerol-containing aqueous system. | ||
Metal-catalyzed hydrogenation reaction has captured considerable attention, because it provides environmentally benign approaches to synthetically a large variety of commodity and fine chemicals, as well as pharmaceutical substances. In 2006, König and co-workers reported the hydrogenation of methyl α-cinnamide (188) catalyzed by the Wilkinson's complex [RhCl(PPh3)3] in the carboxylic acid-based eutectic mixture, at 90 °C and 1 atm of dihydrogen (Scheme 77).9b It was found that the reaction proceeded rapidly, cleanly and quantitatively in citric acid/DMU melt. Notably, even using chiral eutectic mixtures such as sorbitol/urea/NH4Cl and manniyol/DMU/NH4Cl as reaction media, no enantioselectivity was obtained in all cases.
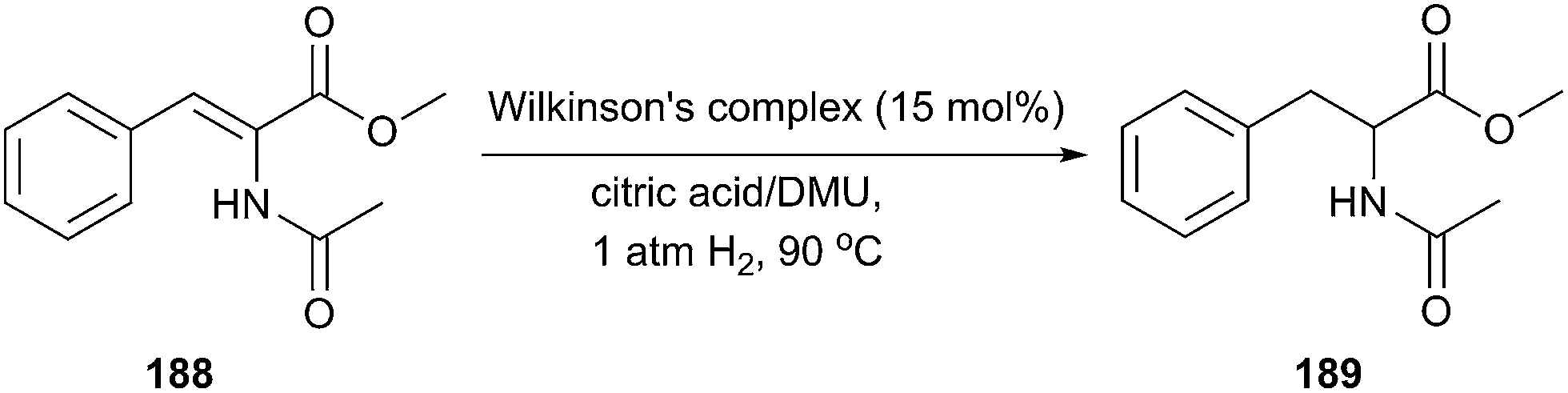 | ||
| Scheme 77 Rh(I)-catalyzed hydrogenation of methyl α-cinnamide in citric acid/DMU. | ||
Amine formation is one of most important transformations in organic synthesis. The direct reductive amination (DRA) is one of the most powerful and widely used synthetic strategies, which allows creation of a new C–N bond by coupling diverse carbonyl compounds with amines.135 In 2014, Saberi and co-workers have developed a highly efficient and ecofriendly method for one-pot reductive amination of carbonyl compounds using ChCl/urea as the catalyst in the presence of sodium borohydride under mild conditions (Scheme 78).136 The reactions were carried out in MeOH at room temperature and the desired secondary amines were obtained in high yields.
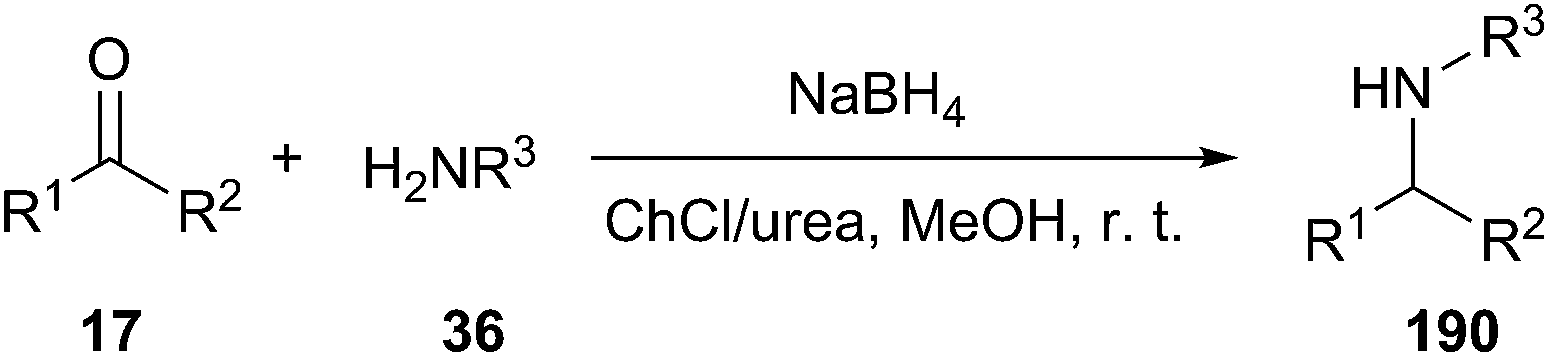 | ||
| Scheme 78 Reductive amination of carbonyl compounds using ChCl/urea. | ||
2.8 Other reactions
Earlier work in the utilization of low melting mixtures for transition-metal-catalyzed reactions such as Suzuki, Heck, and Sonogashira reactions as well as the Huisgen 1,3-dipolar cycloaddition, have been extensively investigated by the research group from König in 2009.40 In these kinds of reactions, most of catalyst–solvent systems could be reused without significant loss of their catalytic activity.Recently, Zhao et al. improved Suzuki–Miyaura and Heck reactions by using low-melting β-cyclodextrin (CD)/NMU mixture as reaction media in the presence of β-CD supported Pd nanoparticles (Scheme 79).137 The reactions could be performed in air without any ligand at 85 °C and afforded the corresponding cross-coupled products in good to excellent isolated yields after a simple workup under every low Pd loading (0.05 mol%). The catalytic system can be recycled and reused four times without loss of catalytic activity.
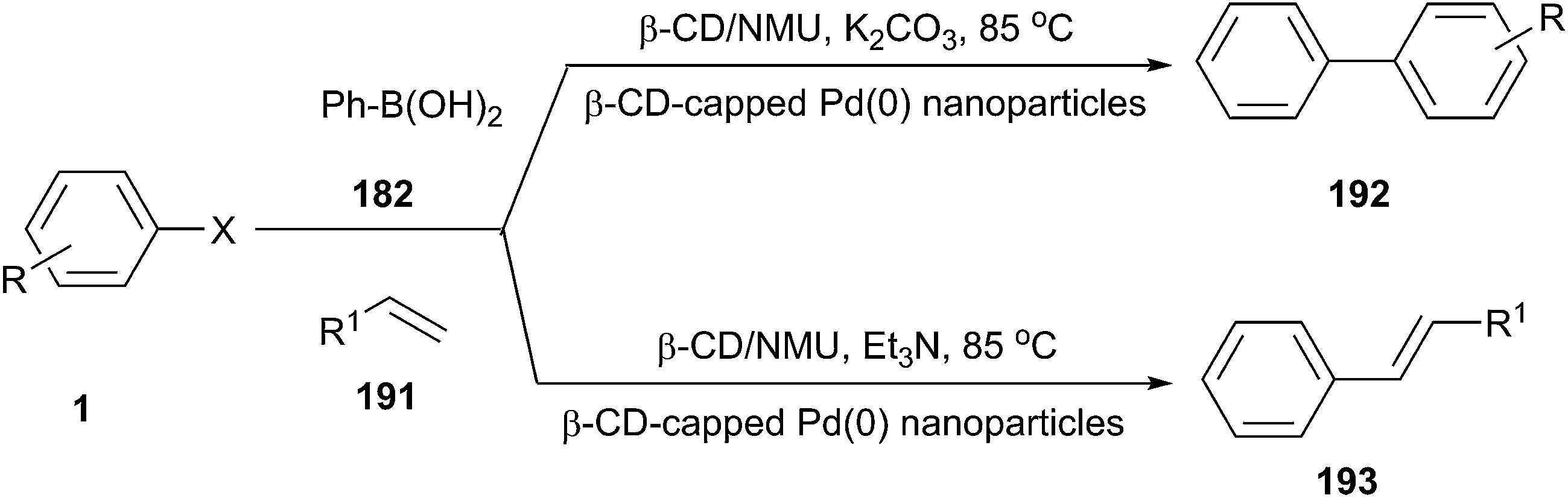 | ||
| Scheme 79 Suzuki–Miyaura and Heck reactions in β-CD/NMU. | ||
In 2014, Hajipour and co-workers prepared ChCl/CuCl via the reaction of choline chloride with CuCl in a 1:1 ratio. It has been employed as an efficient catalytic system in the palladium free Sonogashira-type cross-coupling reactions of phenylacetylene with a variety of aryl halides (Scheme 80).138 Choline chloride, as an OH-functionalized ligand and also as a quaternary ammonium salt support, protects the Cu(I) species against oxidation by air during the reaction. The reaction was found to be versatile, affording good to high yields for various aryl iodides with both electron-rich and electron-deficient functional groups in DMF in the presence of 20 mol% of ChCl/CuCl at 140 °C. It should be noted that the reactions of aryl iodides with phenylacetylene in this catalytic system were fast, and quantitative coupling yields were obtained. However, aryl bromide substrates required longer reaction times and aryl chlorides were far less reactive.
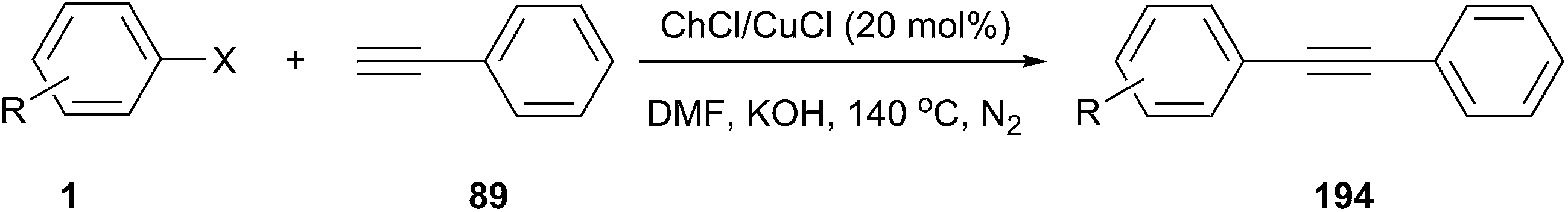 | ||
| Scheme 80 Sonogashira cross-coupling reactions catalyzed by ChCl/CuCl. | ||
In a recent report, García-Álvarez et al. found that bis-allyl Ru(IV) complex [Ru(η3:η3-C10H16)Cl2(benzimidazole)] (196) is an active catalyst in the redox isomerization of allylic alcohols in ChCl/glycerol (Gly) (Scheme 81).139 In this process, a series of primary and secondary allylic alcohols (195) could be isomerized into the corresponding carbonyl compounds (197) in the absence of base at 70 °C. The authors demonstrated that only 0.2 mol% of catalyst for monosubstituted was necessary to obtain good product yields. However, high catalyst loading and longer reaction times for their disubstituted counterparts were always required.
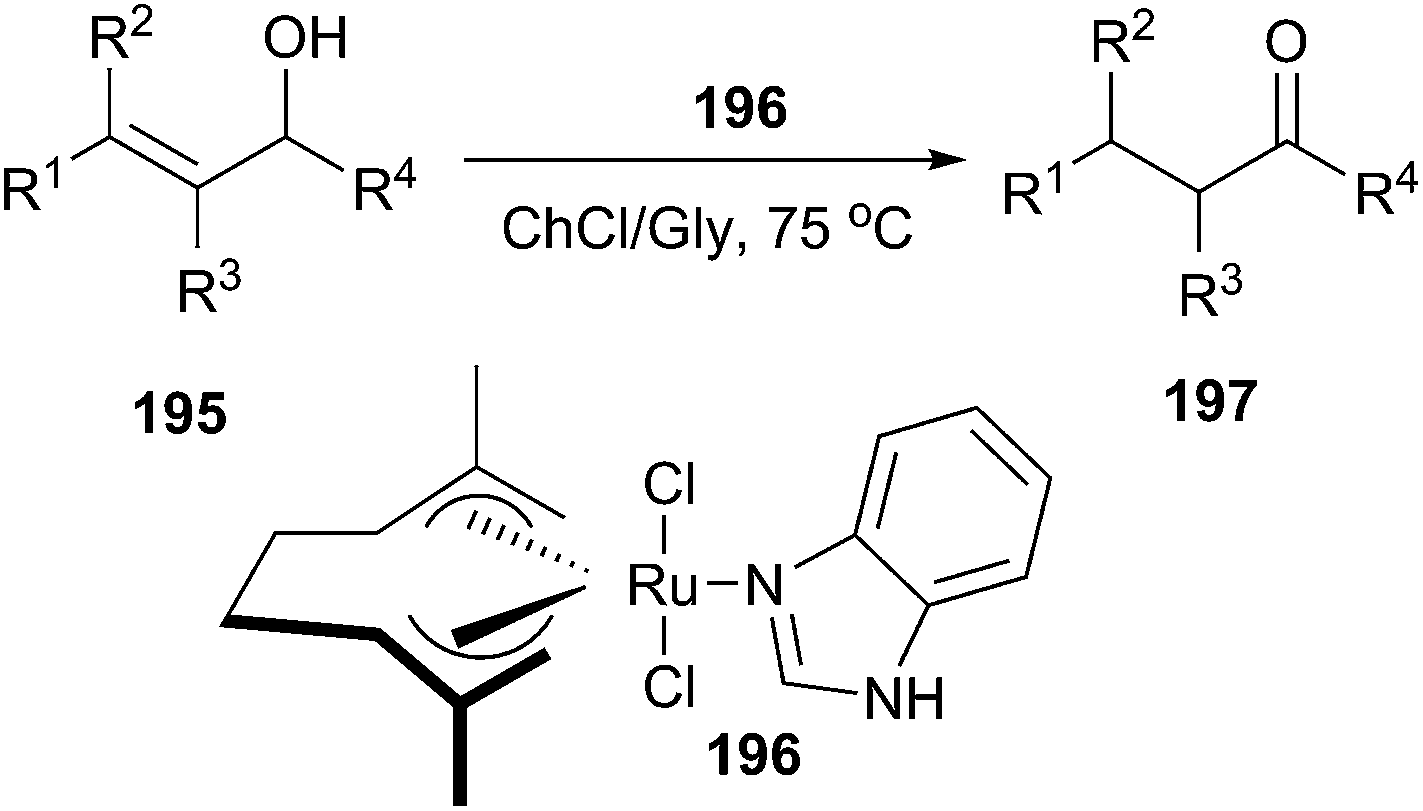 | ||
| Scheme 81 Ru(IV)-catalyzed redox isomerization of allylic alcohols in ChCl/Gly. | ||
In 2014, low melting mixtures (LMM) based on β-CD derivatives and DMU have also been prepared and applied as reaction media for Tsuji–Trost reactions and hydroformylation.140 In the case of the palladium-catalyzed cleavage of allyloctylcarbonate (Tsuji–Trost reaction), rapid conversion of allyloctylcarbonate was attained (5 min) at 90 °C in randomly methylated-β-CD (RAME-β-CD)/DMU under nitrogen with diethylamine as an allyl scavenger (Scheme 82).
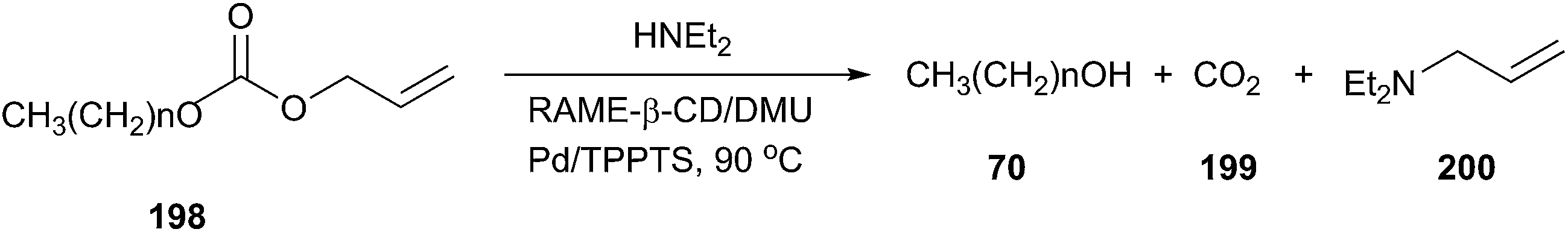 | ||
| Scheme 82 Palladium catalyzed cleavage of allyloctylcarbonate (n = 7) or allyloctadecylcarbonate (n = 17) in RAME-β-CD/DMU. | ||
In hydroformylation reaction, tris-(m-sulfonatophenyl)phosphine trisodium salt (TPPTS) was used as a polar ligand. It was found that the catalytic activity of Rh(acac)(CO)2 in the mixture between DMU and RAME-β-CD is higher than those reported for classical systems based on CD in water. The best result was obtained in the case of RAME-β-CD (30/70) (Scheme 83).141 It was also established that the conversion increased with the 1-decene solubility and decreased with the LMM viscosity.
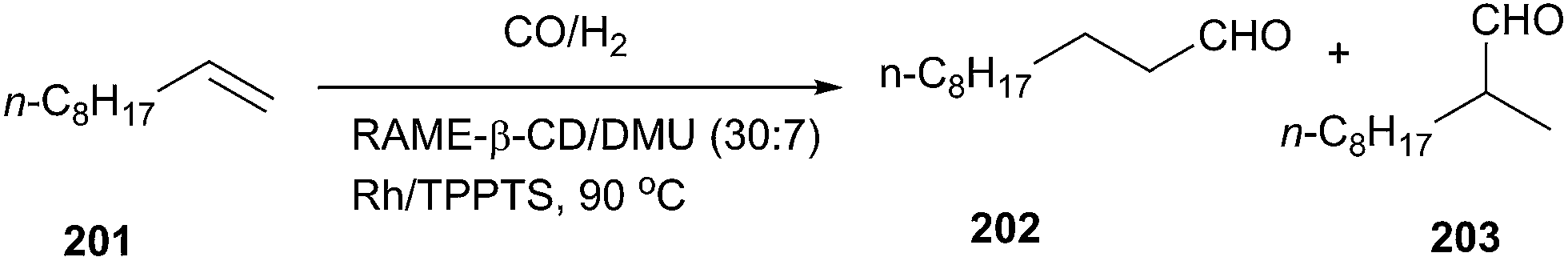 | ||
| Scheme 83 Rhodium-catalyzed hydroformylation of 1-decene in RAME-β-CD/DMU. | ||
Efficient conversion of biorenewable carbohydrates such as glucose, sucrose or fructose to 5-hydroxymethyl-furfural (HMF) is a key step for using these carbohydrates to produce liquid fuels and value molecule platform, especially for the fabrication of safer polymers and fuel additives. The most convenient and efficient method for the preparation of HMF is the acid catalyzed dehydration of D-fructose. For fructose conversion, a variety of reaction systems such as high boiling organic solvents like dimethylsulfoxide, water, ionic liquids or biphasic systems using both homogeneous and heterogeneous catalysts are used. However, these methods suffer from some drawbacks, such as the difficulty in separation of the 5-HMF product from the high boiling point solvents or the use of a large amount of expensive ionic liquid solvents. To date, much effort has been paid to find economically and environmentally feasible processes for HMF production, especially using cheap and safe DESs.
In 2008, Han and co-workers were the first to describe the catalytic activity of several Lewis acids (ZnCl2 or CrCl3) in the aforementioned dehydration reaction of fructose to HMF in various ChCl-based eutectic mixtures. However, this catalytic system displayed only moderate conversions and selectivities.142 In order to overcome these problems, König and co-workers have next investigated the production of HMF in a eutectic mixture composed of choline chloride and carbohydrates (Scheme 84). These high concentrated melts exhibit a low melting point, a low viscosity and a low toxicity. Under this catalytic condition, apart from the monosaccharides D-fructose, other monosaccharide glucose, disaccharide sucrose and the polyfructan inulin can also be successfully converted into HMF.143 Later, Ruß et al. extended the scope of this process for dehydration of L-sorbose in choline chloride melts to give HMF in moderate yields.144
 | ||
| Scheme 84 Acid-catalyzed dehydration of carbohydrates to HMF in low melting mixture. | ||
Han and co-workers have reported that HMF can also be produced by one-pot tandem depolymerization/dehydration of inulin, a biopolymer of fructose, in ChCl-derived DES without assistance of metal chloride-based catalysts.145 HMF was produced with 56% and 51% yield in ChCl/oxalic acid and ChCl/citric acid at 80 °C, respectively. In a biphasic system (ChCl/oxalic acid/ethyl acetate), the yield of HMF can be slightly improved up to 64%. After separation of the ethyl acetate phase containing HMF, the ChCl/oxalic acid was recycled at least 6 times without appreciable loss of HMF yield. In a similar way, De Oliveira Vigier and coworker have demonstrated that betaine hydrochloride (BHC), a co-product of the sugar beet industry, can be used as a renewably sourced Brönsted acid in combination with ChCl, glycerol or water for the production of HMF from fructose and inulin. In a ternary mixture ChCl/BHC/water (10/0.5/2), HMF was produced with 75% yield at 90 °C from 40 wt% of fructose. This system is also capable of promoting the tandem hydrolysis/dehydration of inulin to HMF in 52% yield.146
In 2013, Domínguez de María and co-workers reported that the immo-CAL-B could be used as a biocatalyst for the (trans)esterification of HMF with different acyl donors such as carboxylic acids, natural oils, carbonates, methyl- and ethyl esters. At the end of the reaction, the separation of unreacted HMF and HMF esters is performed by using DES such as ChCl/Gly or ChCl/urea as separation agents. These DESs are able to dissolve hydrogen bond donors (e.g., HMF), whereas non-hydrogen-bond donors (in this case HMF esters) form a second phase. By using this approach, high ester purities (>99%) and efficiencies (up to >90% HMF ester recovery) in separations were obtained.147
From above results, these DESs may are considered as ideal solvents for the catalytic conversion of valuable renewable raw materials such as starch, lignin and cellulose to HMF because (1) most of DESs are able to dissolve enormous amounts of saccharides; (2) HMF can be conveniently isolated from the reaction media by liquid–liquid extraction; (3) the eutectic mixture is able to dilute the water generated in the dehydration process, thus avoiding the rehydration of HMF to form undesired side products levulinic and formic acid.
Furfural, derived from lignocellulosic biomass, is a very versatile and key bio-based platform chemical used for the production of important non-petroleum-derived chemicals. Adopting the concept of dehydration of carbohydrates in choline chloride melts, Ruß et al. reported that the dehydration of isomaltulose (206) to 5-(α-D-glucosyloxymethyl)furfural (GMF) (207) could also conveniently take place in an isomaltulose/choline chloride melt by acidic catalysts. The greatest yield (52%) was obtained by using ZnCl2 as catalyst (Scheme 85).148
 | ||
| Scheme 85 Acid-catalyzed dehydration of isomaltulose to GMF in low melting mixture. | ||
More recently, Pöhnlein and co-workers reported that transesterification between glucose and vinyl hexanoate catalyzed by lipase (Novozyme 435) could be achieved in two deep eutectic solvents consisting of choline chloride and urea and choline chloride and glucose (CC/Glc), which led to the formation of a glucose-6-O-hexanoate (Scheme 86). These findings demonstrate that for the enzymatic synthesis of glycolipids it not only was possible to replace the use of organic solvents with DES, it also was shown that the alternative use of DES provides further beneficial aspects to this synthesis, e.g. the good solubility of both substrates in DESs and the ability of one DES (CC/Glc) to act as substrate and solvent at the same time.149
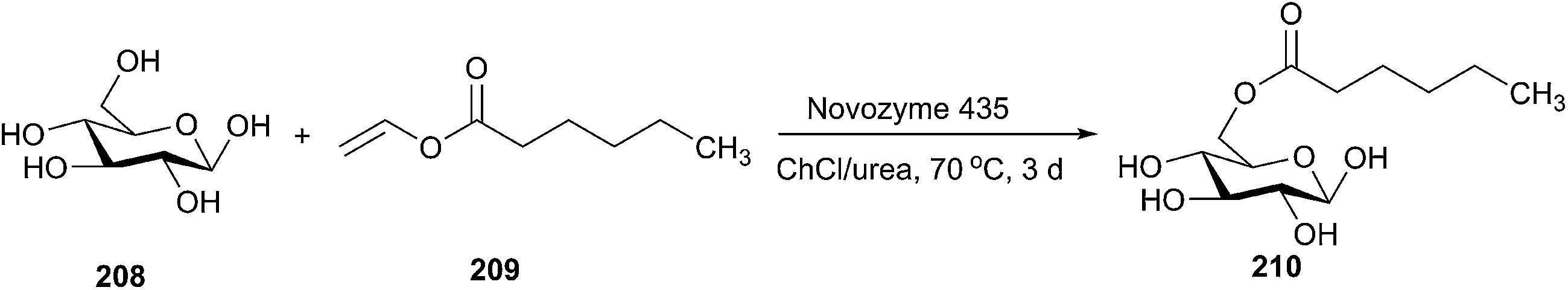 | ||
| Scheme 86 Lipase-catalyzed transesterification between glucose and vinyl hexanoate in ChCl/urea. | ||
Additionally, in 2014, the combination of bio- and organocatalysis in DES is reported by the group of Domínguez de María and coworkers for the first time. To this end, immobilized lipase B from Candida antarctica (CAL-B) was used as the biocatalyst for in situ acetaldehyde formation. In the same DES media (ChCl/glycerol), proline-based organocatalyst performed the crossed aldehyde–acetaldehyde aldol reaction to yield the desired chiral β-hydroxyacetaldehyde (212) in a one-pot process. The subsequent addition of ethyl acetate enables the efficient extraction of the product by the formation of a second phase. After phase decantation, both catalysts and DES can be reused. Sodium borohydride is added to the ethyl acetate phase to reduce the β-hydroxyacetaldehyde to the corresponding 1,3-diol (213) (Scheme 87).150 This leads to an integrative concept with straightforward product recovery and promising catalyst recycling.
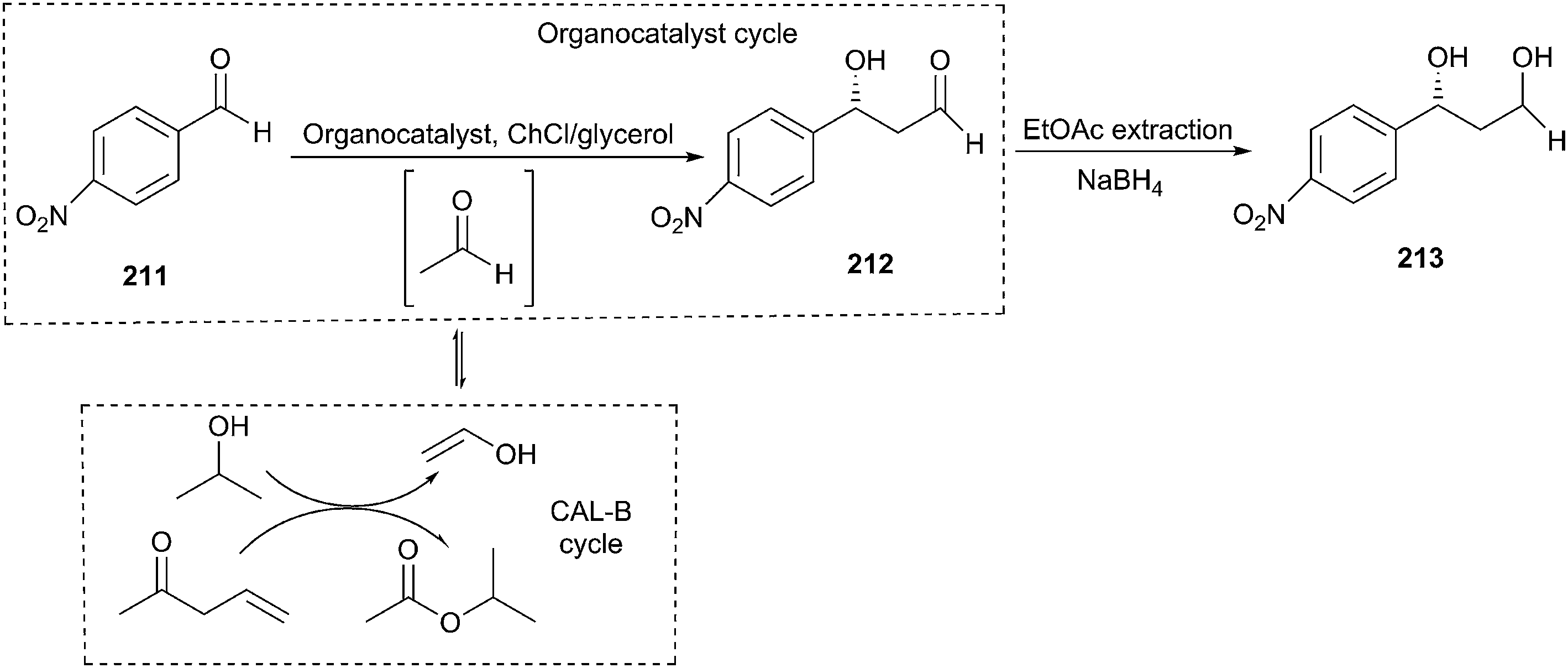 | ||
| Scheme 87 Tandem enzyme-organocatalyst crossed aldol reactions in ChCl/glycerol. | ||
Other particularly interesting aspect for using EDSs is the resolution of racemic compounds. In 2015, the group of Xu used deep eutectic solvent as solvent to improve the performance of immobilized Acetobacter sp. CCTCC M209061 cells for asymmetric oxidation of racemic 1-(4-methoxyphenyl)ethanol (MOPE, 214).151 The ChCl/glycerol enhanced the efficiency of resolution of racemic MOPE and improved the stability of the biocatalyst owing to its excellent solvent property for MOPE and its benign biocompatibility with Acetobacter sp. CCTCC M209061 cells. In this process, MOPE was oxidized to 4′-methoxyacetophenone (MOAP, 215) and gave enantiopure (S)-MOPE (216). Meanwhile, NAD(P)+ is converted to NAD(P)H, and the co-substrate, acetone, is simultaneously reduced, driving the asymmetric oxidation by regenerating NAD(P)+ from NAD(P)H (Scheme 88).
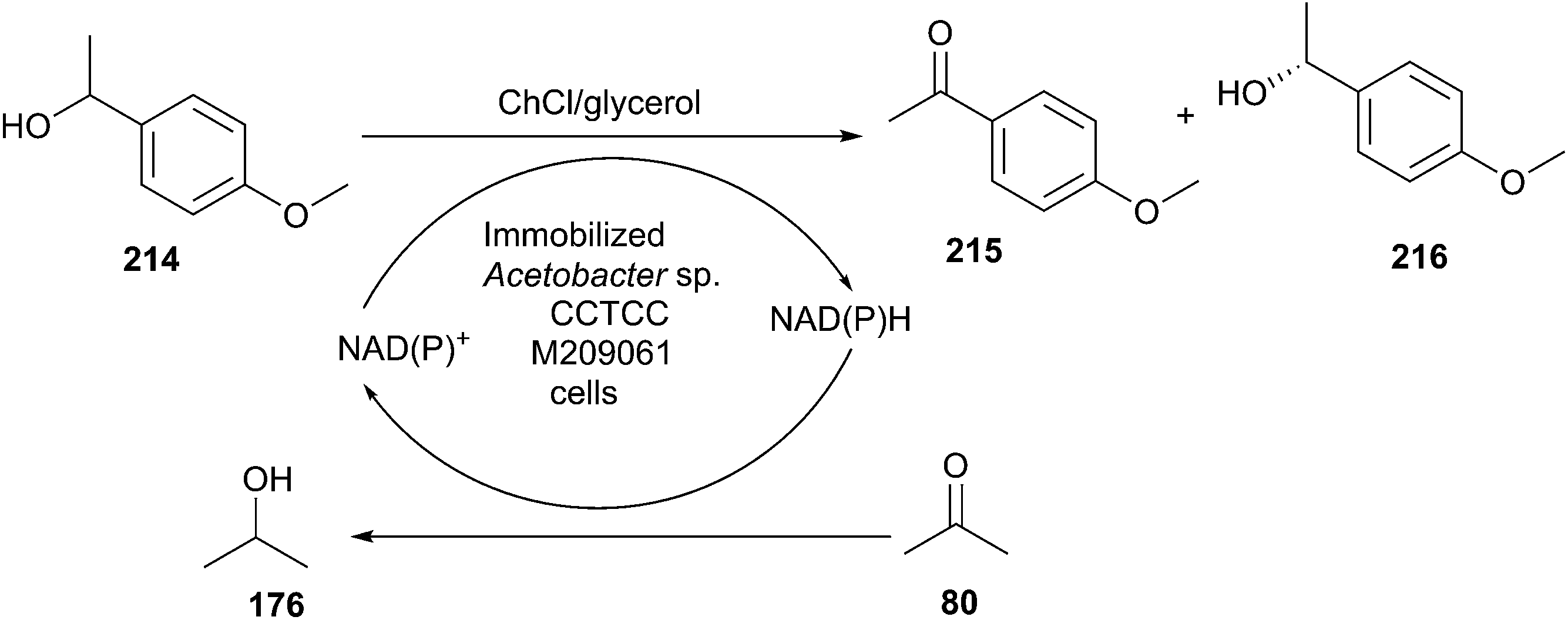 | ||
| Scheme 88 Asymmetric oxidation of racemic MOPE in ChCl/glycerol. | ||
Finally, in 2014, the group of Cao investigated the reaction of 1,4-dialkoxybenzene and paraformaldehyde catalyzed by [ChCl]/[MCl]2 (M = Fe, Zn or Sn) and [ChCl/[MCl·H2O]2 (M = Mg, Al, Cr, Mn, Co, Ni and Cu). The Lewis acid nature of these eutectic mixtures significantly enhanced the reaction rate. It was found that ChCl/FeCl3 (15 mol%) is the most effective catalyst than others. Under the optimal conditions, the pillar[5]arene (219) and pillar[6]arene (220) can be obtained in CH2Cl2 at room temperature with the yield of 35% and 53% respectively (Scheme 89).152
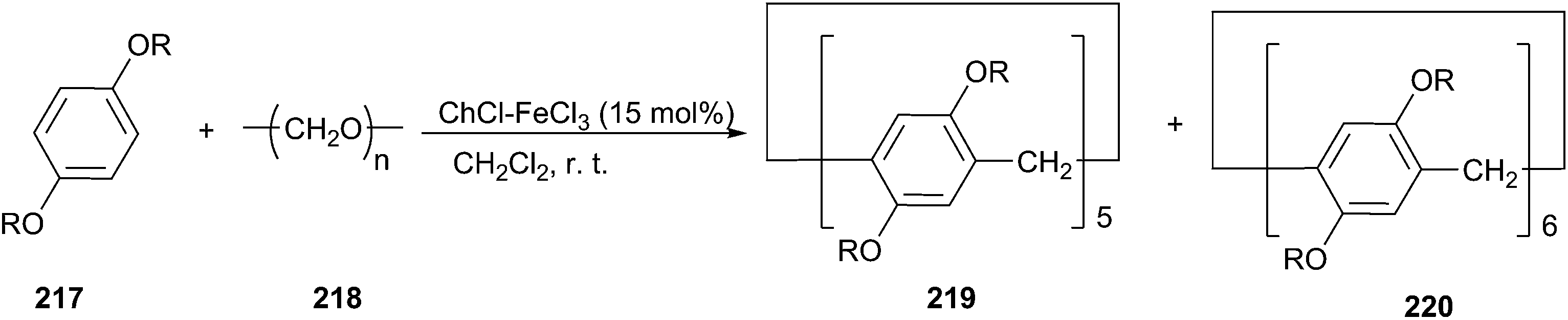 | ||
| Scheme 89 Synthesis of pillar[n]arenes (n = 5 and 6) using ChCl/FeCl3. | ||
3. Conclusions and perspectives
Green chemistry emphasizes that the solvent should be a safe, non-toxic, green, readily available, cheap, recyclable and biodegradable. Deep eutectic solvents' properties, such as low price, chemical inertness with water, easy to prepare, tuneable physicochemical properties, high thermal stability, and biocompatible, meet these requirements. In this review, we highlight the up-to-date progress in employing DESs as green solvent and/or catalyst in organic reactions. The examples complied in this review clearly demonstrate the importance and advantages of DESs in several important organic transformations. In many cases, large increases in reactivity and selectivity have been achieved using these media. Despite all these promising advances, only a narrow range of DESs have been utilized in organic synthesis, both opportunities and challenges still remain in this field.Based on the results in this review, more attention should be paid to design novel specific-task DESs from available natural materials and hence further increase the applications of these solvents in organic reactions. There is a large potential using the combination of DESs and enzyme catalysts in organic reactions based on the unique solvent properties of DESs. DESs have obvious potential as reaction media for biotransformations of highly polar substrates, such as (poly)-saccharides, which cannot be performed in water. Such replacement of volatile media by nonvolatile DESs in this field will doubtlessly continue. Furthermore, it is to be expected that DESs-based solvent systems will have enormous potential in metallic-catalyzed organic transformations. To date, an understanding as to how DESs affect organic reactions is limited. It can be expected that more theoretical calculation should be devoted to better understanding the reaction mechanism and the development of new DES catalysts. In addition, from an application point of view, the development of less expensive DESs as media or catalysts in the synthesis of important commercial compounds and drugs on an industrial scale replacing the current media or catalysts is particularly appealing. Finally, the toxicity of the DESs should be investigated, in order to make sure that they are less toxic than the original components.
It is believed that this review will be helpful for the researchers in the area of DESs to stimulate critical discussions and kindle interest. These economical DESs will provide new opportunities for the study of the green organic synthesis, environmentally friendly media and catalysts in particular, and aid in the clean and efficient cycle of chemical materials. With the fast development of green chemistry and our unremitting efforts, it is expected that the DESs will play a considerably important role in the organic synthesis in the near future.
Acknowledgements
The authors gratefully acknowledge the financial support by the National Natural Science Foundation of China (no. 21272053) and the Nature Science Foundation of Hebei Province (no. B2015205182).References
- (a) Y. L. Gu, Green Chem., 2012, 14, 2091–2128 RSC; (b) M. C. Bubalo, S. Vidović, I. R. Redovniković and S. Jokić, J. Chem. Technol. Biotechnol., 2015, 90 DOI:10.1002/jctb.4668.
- (a) T. Y. Cheng, D. C. Zhang, H. X. Li and G. H. Liu, Green Chem., 2014, 16, 3401–3427 RSC; (b) A. K. Rathi, M. B. Gawande, R. Zboril and R. S. Varma, Coord. Chem. Rev., 2015, 291, 68–94 CrossRef CAS PubMed.
- Y. Medina-Gonzalez, S. Camy and J. S. Condoret, ACS Sustainable Chem. Eng., 2014, 2, 2623–2636 CrossRef CAS.
- W. Zhang, Green Chem., 2009, 11, 911–920 RSC.
- (a) F. He, P. Li, Y. L. Gu and G. X. Li, Green Chem., 2009, 11, 1767–1773 RSC; (b) Y. L. Gu and F. Jerome, Green Chem., 2010, 12, 1127–1138 RSC; (c) A. E. Diaz-Alvarez, J. Francos, B. Lastra-Barreira, P. Crochet and V. Cadierno, Chem. Commun., 2011, 47, 6208–6227 RSC; (d) Y. L. Liu, X. P. Zeng and J. Zhou, Chem.–Asian. J., 2012, 7, 1759–1763 CrossRef CAS PubMed; (e) J. I. Garcia, H. Garcia-Marin and E. Pires, Green Chem., 2014, 16, 1007–1103 RSC; (f) P. Cintas, S. Tagliapietra, E. C. Gaudino, G. Palmisano and G. Cravotto, Green Chem., 2014, 16, 1056–1065 RSC.
- (a) Y. L. Gu and F. Jerome, Chem. Soc. Rev., 2013, 42, 9550–9570 RSC; (b) J. Yang, J. N. Tan and Y. L. Gu, Green Chem., 2012, 14, 3304–3317 RSC; (c) B. L. Li, P. H. Li, X. N. Fang, C. X. Li, J. L. Sun, L. P. Mo and Z. H. Zhang, Tetrahedron, 2013, 69, 7011–7018 CrossRef CAS PubMed; (d) R. Y. Guo, P. Wang, G. D. Wang, L. P. Mo and Z. H. Zhang, Tetrahedron, 2013, 69, 2056–2061 CrossRef CAS PubMed.
- (a) J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed; (b) M. Petkovic, K. R. Seddon, L. P. N. Rebelo and C. S. Pereira, Chem. Soc. Rev., 2011, 40, 1383–1403 RSC; (c) N. Isambert, M. D. S. Duque, J. C. Plaquevent, Y. Genisson, J. Rodriguez and T. Constantieux, Chem. Soc. Rev., 2011, 40, 1347–1357 RSC; (d) A. Taheri, X. J. Pan, C. H. Liu and Y. L. Gu, ChemSusChem, 2014, 7, 2094–2098 CrossRef CAS PubMed; (e) A. Taheri, C. H. Liu, B. B. Lai, C. Cheng, X. J. Pan and Y. L. Gu, Green Chem., 2014, 16, 3715–4371 RSC; (f) B. S. Wang, S. J. Yang, L. J. Min, Y. L. Gu, Y. Y. Zhang, X. P. Wu, L. F. Zhang, E. H. M. Elageed, S. Wu and G. H. Gao, Adv. Synth. Catal., 2014, 356, 3125–3134 CrossRef CAS PubMed; (g) A. Taheri, B. B. Lai, C. G. Cheng and Y. L. Gu, Green Chem., 2015, 17, 812–816 RSC; (h) P. Domínguez de María and Z. Maugeri, Curr. Opin. Chem. Biol., 2011, 15, 220–225 CrossRef PubMed.
- (a) A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2003, 70–71 RSC; (b) A. P. Abbott, D. Boothby, G. Capper, D. L. Davies and R. K. Rasheed, J. Am. Chem. Soc., 2004, 126, 9142–9147 CrossRef CAS PubMed; (c) E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed.
- (a) G. Imperato, E. Eibler, J. Niedermaier and B. König, Chem. Commun., 2005, 1170–1172 RSC; (b) G. Imperato, S. Hoger, D. Lenoir and B. König, Green Chem., 2006, 8, 1051–1055 RSC.
- Y. T. Dai, J. van Spronsen, G. J. Witkamp, R. Verpoorte and Y. H. Choi, Anal. Chim. Acta, 2013, 766, 61–68 CrossRef CAS PubMed.
- M. Francisco, A. van den Bruinhorst and M. C. Kroon, Angew. Chem., Int. Ed., 2013, 52, 3074–3085 CrossRef CAS PubMed.
- M. A. Skopek, M. A. Mohamoud, K. S. Ryder and A. R. Hillman, Chem. Commun., 2009, 935–937 RSC.
- Q. H. Zhang, K. De Oliveira Vigier, S. Royer and F. Jérôme, Chem. Soc. Rev., 2012, 41, 7108–7146 RSC.
- B. K. Tang and K. H. Row, Monatsh. Chem., 2013, 144, 1427–1454 CrossRef CAS PubMed.
- Y. Y. Zhang, X. H. Lu, X. Feng, Y. J. Shi and X. Y. Ji, Prog. Chem., 2013, 25, 881–892 CAS.
- Z. Maugeri and P. Domínguez de María, RSC Adv., 2012, 2, 421–425 RSC.
- F. Cardellini, M. Tiecco, R. Germani, G. Cardinali, L. Corte, L. Roscini and N. Spreti, RSC Adv., 2014, 4, 55990–56002 RSC.
- F. del Monte, D. Carriazo, M. C. Serrano, M. C. Gutierrez and M. L. Ferrer, ChemSusChem, 2014, 7, 999–1009 CrossRef CAS PubMed.
- (a) M. Francisco, A. van den Bruinhorst and M. C. Kroon, Green Chem., 2012, 14, 2153–2157 RSC; (b) S. Q. Xia, G. A. Baker, H. Li, S. Ravula and H. Zhao, RSC Adv., 2014, 4, 10586–10596 RSC.
- (a) D. V. Wagle, H. Zhao and G. A. Baker, Acc. Chem. Res., 2014, 47, 2299–2308 CrossRef CAS PubMed; (b) D. Carriazo, M. C. Serrano, M. C. Gutierrez, M. L. Ferrer and F. del Monte, Chem. Soc. Rev., 2012, 41, 4996–5014 RSC.
- (a) H. Zhao and G. A. Baker, J. Chem. Technol. Biotechnol., 2013, 88, 3–12 CrossRef CAS PubMed; (b) S. K. Tang, G. A. Baker and H. Zhao, Chem. Soc. Rev., 2012, 41, 4030–4066 RSC; (c) H. Zhao, G. A. Baker and S. Holmes, J. Mol. Catal. B: Enzym., 2011, 72, 163–167 CrossRef CAS PubMed; (d) H. Zhao, G. A. Baker and S. Holmes, Org. Biomol. Chem., 2011, 9, 1908–1916 RSC; (e) L. Gu, W. Huang, S. K. Tang, S. J. Tian and X. W. Zhang, Chem. Eng. J., 2015, 259, 647–652 CrossRef CAS PubMed.
- (a) E. Durand, J. Lecomte and P. Villeneuve, Eur. J. Lipid Sci. Technol., 2013, 115, 379–385 CrossRef CAS PubMed; (b) Z.-L. Huang, B.-P. Wu, Q. Wen, T.-X. Yang and Z. Yang, J. Chem. Technol. Biotechnol., 2014, 89, 1975–1981 CrossRef CAS PubMed; (c) M. C. Gutierrez, M. L. Ferrer, L. Yuste, F. Rojo and F. del Monte, Angew. Chem., Int. Ed., 2010, 49, 2158–2162 CrossRef CAS PubMed.
- (a) L. L. Sze, S. Pandey, S. Ravula, S. Pandey, H. Zhao, G. A. Baker and S. N. Baker, ACS Sustainable Chem. Eng., 2014, 2, 2117–2123 CrossRef CAS; (b) N. R. Rodriguez, A. S. B. Gonzalez, P. M. A. Tijssen and M. C. Kroon, Fluid Phase Equilib., 2015, 385, 72–78 CrossRef CAS PubMed.
- (a) A. R. Hillman, K. S. Ryder, C. J. Zaleski, V. Ferreira, C. A. Beasley and E. Vieil, Electrochim. Acta, 2014, 135, 42–51 CrossRef CAS PubMed; (b) P. Sebastian, E. Valles and E. Gomez, Electrochim. Acta, 2014, 123, 285–295 CrossRef CAS PubMed.
- (a) F. S. Oliveira, A. B. Pereiro, L. P. N. Rebelo and I. M. Marrucho, Green Chem., 2013, 15, 1326–1330 RSC; (b) F. Pena-Pereira and J. Namiesnik, ChemSusChem, 2014, 7, 1784–1800 CrossRef CAS PubMed; (c) T. N. Gu, M. L. Zhang, T. Tan, J. Chen, Z. Li, Q. H. Zhang and H. D. Qiu, Chem. Commun., 2014, 50, 11749–11752 RSC; (d) B. Tang, H. Zhang and K. H. Row, J. Sep. Sci., 2015, 38, 1053–1064 CrossRef CAS PubMed.
- A. Abo-Hamad, M. Hayyan, M. A. AlSaadi and M. A. Hashim, Chem. Eng. J., 2015, 273, 551–567 CrossRef CAS PubMed.
- A. L. Wang, X. L. Zheng, Z. Z. Zhao, C. P. Li and X. F. Zheng, Prog. Chem., 2014, 26, 784–795 Search PubMed.
- (a) Z. F. Li, H. L. Hou, A. G. Ying and S. L. Xu, Chin. J. Org. Chem., 2014, 34, 1074–1091 CrossRef CAS; (b) C. Huang, Y. Q. Yin, J. H. Guo, J. Wang, B. M. Fan and L. J. Yang, RSC Adv., 2014, 4, 10188–10195 RSC.
- N. Azizi, Z. Yadollahy and A. Rahimzadeh-Oskooee, Tetrahedron Lett., 2014, 55, 1722–1725 CrossRef CAS PubMed.
- V. Krishnakumar, N. G. Vindhya, B. K. Mandal and F. R. N. Khan, Ind. Eng. Chem. Res., 2014, 53, 10814–10819 CrossRef CAS.
- U. N. Yadav and G. S. Shankarling, J. Mol. Liq., 2014, 195, 188–193 CrossRef CAS PubMed.
- A. K. Sanap and G. S. Shankarling, RSC Adv., 2014, 4, 34938–34943 RSC.
- Z. Maugeri and P. Domínguez de María, J. Mol. Catal. B: Enzym., 2014, 107, 120–123 CrossRef CAS PubMed.
- A. K. Sanap and G. S. Shankarling, Catal. Commun., 2014, 49, 58–62 CrossRef CAS PubMed.
- B. S. Singh, H. R. Lobo and G. S. Shankarling, Catal. Commun., 2012, 24, 70–74 CrossRef CAS PubMed.
- N. Azizi and E. Batebi, Catal. Sci. Technol., 2012, 2, 2445–2448 CAS.
- G. Imperato, E. Eibler, J. Niedermaier and B. König, Chem. Commun., 2005, 1170–1172 RSC.
- F. Ilgen and B. König, Green Chem., 2009, 11, 848–854 RSC.
- C. Vidal, J. García-Álvarez, A. Hernán-Gómez, A. R. Kennedy and E. Hevia, Angew. Chem., Int. Ed., 2014, 53, 5969–5973 CrossRef CAS PubMed.
- S. Gore, S. Baskaran and B. König, Green Chem., 2011, 13, 1009–1013 RSC.
- S. Gore, S. Baskaran and B. Koenig, Adv. Synth. Catal., 2012, 354, 2368–2372 CrossRef CAS PubMed.
- (a) Z. H. Zhang, J. J. Li and T. S. Li, Ultrason. Sonochem., 2008, 15, 673–676 CrossRef CAS PubMed; (b) S. D. Joshi, U. A. More, V. H. Kulkarni and T. M. Aminabhavi, Curr. Org. Chem., 2013, 17, 2279–2304 CrossRef CAS.
- S. Handy and K. Lavender, Tetrahedron Lett., 2013, 54, 4377–4379 CrossRef CAS PubMed.
- J. Stockigt, A. P. Antonchick, F. R. Wu and H. Waldmann, Angew. Chem., Int. Ed., 2011, 50, 8538–8564 CrossRef CAS PubMed.
- S. Handy and M. Wright, Tetrahedron Lett., 2014, 55, 3440–3442 CrossRef CAS PubMed.
- (a) J. Tummatorn, M. P. Gleeson, S. Krajangsri, C. Thongsornkleeb and S. Ruchirawat, RSC Adv., 2014, 4, 20048–20052 RSC; (b) A. Porcheddu, M. G. Mura, L. De Luca, M. Pizzetti and M. Taddei, Org. Lett., 2012, 14, 6112–6115 CrossRef CAS PubMed; (c) E. Siddalingamurthy, K. M. Mahadevan, J. N. Masagalli and H. N. Harishkumar, Tetrahedron Lett., 2013, 54, 5591–5596 CrossRef CAS PubMed.
- R. C. Morales, V. Tambyrajah, P. R. Jenkins, D. L. Davies and A. P. Abbott, Chem. Commun., 2004, 158–159 RSC.
- S. Gore, S. Baskaran and B. König, Org. Lett., 2012, 14, 4568–4571 CrossRef CAS PubMed.
- F. P. Ma, G. T. Cheng, Z. G. He and Z. H. Zhang, Aust. J. Chem., 2012, 65, 409–416 CAS.
- B. Bafti and H. Khabazzadeh, J. Chem. Sci., 2014, 126, 881–887 CrossRef CAS.
- B. S. Singh, H. R. Lobo, D. V. Pinjari, K. J. Jarag, A. B. Pandit and G. S. Shankarling, Ultrason. Sonochem., 2013, 20, 287–293 CrossRef CAS PubMed.
- P. A. More, B. L. Gadilohar and G. S. Shankarling, Catal. Lett., 2014, 144, 1393–1398 CrossRef CAS PubMed.
- S. Gore, K. Chinthapally, S. Baskaran and B. König, Chem. Commun., 2013, 49, 5052–5054 RSC.
- M. J. Rodríguez-Álvarez, C. Vidal, J. Díez and J. García-Álvarez, Chem. Commun., 2014, 50, 12927–12929 RSC.
- (a) F. P. Ma, P. H. Li, B. L. Li, L. P. Mo, N. Liu, H. J. Kang, Y. N. Liu and Z. H. Zhang, Appl. Catal., A, 2013, 457, 34–41 CrossRef CAS PubMed; (b) B. Zuo, J. X. Chen, M. C. Liu, J. C. Ding, H. Y. Wu and W. Su, J. Chem. Res., 2009, 14–16 CrossRef CAS; (c) H. J. Deng, Y. J. Fang, G. W. Chen, M. C. Liu, H. Y. Wu and J. X. Chen, Appl. Organomet. Chem., 2012, 26, 164–216 CrossRef CAS PubMed; (d) C. C. Silveira, M. P. Fortes and S. R. Mendes, Curr. Org. Chem., 2012, 16, 1540–1548 CrossRef CAS; (e) D. Akbaslar, O. Demirkol and S. Giray, Synth. Commun., 2014, 44, 1323–1332 CrossRef CAS PubMed.
- P. Wang, F. P. Ma and Z. H. Zhang, J. Mol. Liq., 2014, 198, 259–262 CrossRef CAS PubMed.
- G. Imperato, R. Vasold and B. König, Adv. Synth. Catal., 2006, 348, 2243–2247 CrossRef CAS PubMed.
- V. De Santi, F. Cardellini, L. Brinchi and R. Germani, Tetrahedron Lett., 2012, 53, 5151–5155 CrossRef CAS PubMed.
- L. Wang, K. Q. Zhu, Q. Chen and M. Y. He, Dyes Pigm., 2015, 112, 274–279 CrossRef CAS PubMed.
- Z. Z. Chen, W. Zhu, Z. B. Zheng and X. Z. Zou, J. Fluorine Chem., 2010, 131, 340–344 CrossRef CAS PubMed.
- Z. Z. Chen, B. Zhou, H. H. Cai, W. Zhu and X. Z. Zou, Green Chem., 2009, 11, 275–278 RSC.
- B. Singh, H. Lobo and G. Shankarling, Catal. Lett., 2011, 141, 178–182 CrossRef CAS.
- (a) Y. H. Liu, Q. S. Liu and Z. H. Zhang, Tetrahedron Lett., 2009, 50, 916–921 CrossRef CAS PubMed; (b) Z. H. Zhang and J. Lin, Synth. Commun., 2007, 37, 209–215 CrossRef CAS PubMed; (c) G. Bartoli, G. Bencivenni and R. Dalpozzo, Chem. Soc. Rev., 2010, 39, 4449–4465 RSC; (d) M. Shiri, Chem. Rev., 2012, 112, 3508–3549 CrossRef CAS PubMed; (e) R. Dalpozzo, Chem. Soc. Rev., 2015, 44, 742–778 RSC.
- (a) L. P. Mo, Z. C. Ma and Z. H. Zhang, Synth. Commun., 2005, 35, 1997–2004 CrossRef CAS; (b) S. A. R. Mulla, A. Sudalai, M. Y. Pathan, S. A. Siddique, S. M. Inamdar, S. S. Chavan and R. S. Reddy, RSC Adv., 2012, 2, 3525–3529 RSC; (c) D. Q. Liang, W. Z. Huang, L. Yuan, Y. H. Ma, J. M. Ma and D. M. Ning, Catal. Commun., 2014, 55, 11–14 CrossRef CAS PubMed.
- U. N. Yadav and G. S. Shankarling, J. Mol. Liq., 2014, 191, 137–141 CrossRef CAS PubMed.
- S. Handy and N. M. Westbrook, Tetrahedron Lett., 2014, 55, 4969–4971 CrossRef CAS PubMed.
- N. Azizi and Z. Manocheri, Res. Chem. Intermed., 2012, 38, 1495–1500 CrossRef CAS.
- R. R. Jella and R. Nagarajan, Tetrahedron, 2013, 69, 10249–11025 CrossRef CAS PubMed.
- D. Chandam, A. Mulik, P. Patil, S. Jagdale, D. Patil, S. Sankpal and M. Deshmukh, J. Mol. Liq., 2015, 207, 14–20 CrossRef CAS PubMed.
- (a) C. de Graaff, E. Ruijter and R. V. A. Orru, Chem. Soc. Rev., 2012, 41, 3969–4009 RSC; (b) Y. L. Gu, Green Chem., 2012, 14, 2091–2128 RSC; (c) H. G. O. Alvim, E. N. da Silva and B. A. D. Neto, RSC Adv., 2014, 4, 54282–54299 RSC; (d) R. Y. Guo, Z. M. An, L. P. Mo, S. T. Yang, H. X. Liu, S. X. Wang and Z. H. Zhang, Tetrahedron, 2013, 69, 9931–9938 CrossRef CAS PubMed; (e) X. T. Li, A. D. Zhao, L. P. Mo and Z. H. Zhang, RSC Adv., 2014, 4, 51580–51588 RSC; (f) X. T. Li, Y. H. Liu, X. Liu and Z. H. Zhang, RSC Adv., 2015, 5, 25625–25633 RSC; (g) M. H. Li, A. Taheri, M. Liu, S. H. Sun and Y. L. Gu, Adv. Synth. Catal., 2014, 356, 537–556 CrossRef CAS PubMed.
- G. Keglevich and E. Balint, Molecules, 2012, 17, 12821–12835 CrossRef CAS PubMed.
- S. T. Disale, S. R. Kale, S. S. Kahandal, T. G. Srinivasan and R. V. Jayaram, Tetrahedron Lett., 2012, 53, 2277–2279 CrossRef CAS PubMed.
- (a) G. Y. Sun, J. T. Hou, J. J. Dou, J. Lu, Y. J. Hou, T. Xue and Z. H. Zhang, J. Chin. Chem. Soc., 2010, 57, 1315–1320 CrossRef CAS PubMed; (b) J. T. Hou, J. W. Gao and Z. H. Zhang, Appl. Organomet. Chem., 2011, 25, 47–53 CrossRef CAS PubMed; (c) S. Sobhani, Z. M. Falatooni and M. Honarmand, RSC Adv., 2014, 4, 15797–15806 RSC.
- (a) R. G. Arrayas and J. C. Carretero, Chem. Soc. Rev., 2009, 38, 1940–1948 RSC; (b) B. Karimi, D. Enders and E. Jafari, Synthesis, 2013, 45, 2769–2812 CrossRef CAS PubMed.
- F. Keshavarzipour and H. Tavakol, Catal. Lett., 2015, 145, 1062–1066 CrossRef CAS PubMed.
- S. Brauch, S. S. van Berkel and B. Westermann, Chem. Soc. Rev., 2013, 42, 4948–4962 RSC.
- N. Azizi, S. Dezfooli and M. M. Hashemi, C. R. Chim., 2013, 16, 1098–1102 CrossRef CAS PubMed.
- A. Mobinikhaledi and A. K. Amiri, Res. Chem. Intermed., 2013, 39, 1491–1498 CrossRef CAS PubMed.
- N. Azizi, Z. Yadollahy and A. Rahimzadeh-oskooee, Synlett, 2014, 25, 1085–1088 CrossRef PubMed.
- N. Azizi and E. Gholibeglo, RSC Adv., 2012, 2, 7413–7416 RSC.
- S. A. Khedkar and P. B. Auti, Mini-Rev. Med. Chem., 2014, 14, 282–290 CrossRef CAS.
- J. P. Wan and Y. Y. Liu, RSC Adv., 2012, 2, 9763–9777 RSC.
- S. Pednekar, R. Bhalerao and N. Ghadge, J. Chem. Sci., 2013, 125, 615–621 CrossRef CAS PubMed.
- L. Wang, K.-Q. Zhu, Q. Chen and M.-Y. He, Green Process. Synth., 2014, 3, 457–461 CAS.
- (a) W. B. Sun, P. Zhang, J. Fan, S. H. Chen and Z. H. Zhang, Synth. Commun., 2010, 40, 587–594 CrossRef CAS PubMed; (b) R. Y. Guo, Z. M. An, L. P. Mo, R. Z. Wang, H. X. Liu, S. X. Wang and Z. H. Zhang, ACS Comb. Sci., 2013, 15, 557–563 CrossRef CAS PubMed.
- A. Chaskar, Lett. Org. Chem., 2014, 11, 480–486 CrossRef CAS.
- N. Azizi, S. Dezfooli, M. Khajeh and M. M. Hashemi, J. Mol. Liq., 2013, 186, 76–80 CrossRef CAS PubMed.
- N. Azizi, S. Dezfooli and M. M. Hashemi, J. Mol. Liq., 2014, 194, 62–67 CrossRef CAS PubMed.
- N. Azizi, M. Mariami and M. Edrisi, Dyes Pigm., 2014, 100, 215–221 CrossRef CAS PubMed.
- (a) X. N. Zhang, Y. X. Li and Z. H. Zhang, Tetrahedron, 2011, 67, 7426–7430 CrossRef CAS PubMed; (b) J. Deng, L. P. Mo, F. Y. Zhao, Z. H. Zhang and S. X. Liu, ACS Comb. Sci., 2012, 14, 335–341 CrossRef CAS PubMed; (c) R. Y. Guo, P. Wang, G. D. Wang, L. P. Mo and Z. H. Zhang, Tetrahedron, 2013, 69, 2056–2061 CrossRef CAS PubMed.
- A. Rajawat, S. Khandelwal and M. Kumar, RSC Adv., 2014, 4, 5105–5112 RSC.
- S. Khandelwal, A. Rajawat, Y. K. Tailor and M. Kumar, Comb. Chem. High Throughput Screening, 2014, 17, 763–769 CrossRef CAS.
- N. Yan, Y. K. Xiong, J. H. Xia, P. X. Rui, Z. W. Lei, W. L. Liao and B. Xiong, Chin. J. Org. Chem., 2015, 35, 384–438 CrossRef CAS.
- (a) H. J. Wang, X. N. Zhang and Z. H. Zhang, Monatsh. Chem., 2010, 141, 425–430 CrossRef CAS; (b) X. N. Zhao, G. F. Hu, M. Tang, T. T. Shi, X. L. Guo, T. T. Li and Z. H. Zhang, RSC Adv., 2014, 4, 51089–51097 RSC.
- L. Wang, M. Zhou, Q. Chen and M. Y. He, J. Chem. Res., 2013, 598–600 CrossRef CAS.
- A. Mobinikhaledi and A. Amiri, Res. Chem. Intermed., 2015, 41, 2063–2070 CrossRef CAS PubMed.
- N. Aziizi, Z. Manochehri, A. Nahayi and S. Torkashvand, J. Mol. Liq., 2014, 196, 153–158 CrossRef CAS PubMed.
- N. Azizi, S. Dezfuli and M. M. Hahsemi, Sci. World J., 2012, 908702 Search PubMed.
- N. Azizi and M. Marimi, Environ. Chem. Lett., 2013, 11, 371–376 CrossRef CAS PubMed.
- N. Azizi, M. Khajeh, M. Hasani and S. Dezfooli, Tetrahedron Lett., 2013, 54, 5407–5410 CrossRef CAS PubMed.
- N. Azizi, Z. Rahimi and M. Alipour, C. R. Chim., 2015, 18 DOI:10.1016/j.crci.2014.10.001.
- Z. H. Zhang, X. N. Zhang, L. P. Mo, Y. X. Li and F. P. Ma, Green Chem., 2012, 14, 1502–1506 RSC.
- (a) V. Estevez, M. Villacampa and J. C. Menendez, Chem. Soc. Rev., 2014, 43, 4633–4657 RSC; (b) B. L. Li, M. Zhang, H. C. Hu, X. Du and Z. H. Zhang, New J. Chem., 2014, 38, 2435–2442 RSC.
- H. C. Hu, Y. H. Liu, B. L. Li, Z. S. Cui and Z. H. Zhang, RSC Adv., 2015, 5, 7720–7728 RSC.
- (a) S. Maiti, S. Biswas and U. Jana, J. Org. Chem., 2010, 75, 1674–1683 CrossRef CAS PubMed; (b) A. T. Khan, M. Lal, P. R. Bagdi, R. S. Basha, P. Saravanan and S. Patra, Tetrahedron Lett., 2012, 53, 4145–4150 CrossRef CAS PubMed; (c) A. B. Atar and Y. T. Jeong, Tetrahedron Lett., 2013, 54, 5624–5628 CrossRef CAS PubMed; (d) J. B. Bharate, R. Sharma, S. Aravinda, V. K. Gupta, B. Singh, S. B. Bharate and R. A. Vishwakarma, RSC Adv., 2013, 3, 21736–21742 RSC.
- S. M. Rokade, A. M. Garande, N. A. A. Ahmad and P. M. Bhate, RSC Adv., 2015, 5, 2281–2284 RSC.
- J. Lu, X. T. Li, E. Q. Ma, L. P. Mo and Z. H. Zhang, ChemCatChem, 2014, 6, 2854–2859 CrossRef CAS PubMed.
- E. Siddalingamurthy, K. M. Mahadevan and T. O. S. Kumar, Synth. Commun., 2013, 43, 3153–3316 CrossRef CAS PubMed.
- P. M. Pawar, K. J. Jarag and G. S. Shankarling, Green Chem., 2011, 13, 2130–2134 RSC.
- S. Liu, Y. X. Ni, W. J. Wei, F. L. Qiu, S. L. Xu and A. G. Ying, J. Chem. Res., 2014, 186–188 CrossRef.
- Y. A. Sonawane, S. B. Phadtare, B. N. Borse, A. R. Jagtap and G. S. Shankarling, Org. Lett., 2010, 12, 1456–1459 CrossRef CAS PubMed.
- I. Hawkins and S. T. Handy, Tetrahedron, 2013, 69, 9200–9204 CrossRef CAS PubMed.
- H. N. Harishkumar, K. M. Mahadevan, K. H. C. Kiran and N. D. Satyanarayan, Org. Commun., 2011, 4, 26–32 CAS.
- S. B. Phadtare, K. J. Jarag and G. S. Shankarling, Dyes Pigm., 2013, 97, 105–112 CrossRef CAS PubMed.
- H. Y. Lu, J. J. Li and Z. H. Zhang, Appl. Organomet. Chem., 2009, 23, 165–169 CrossRef CAS PubMed.
- P. H. Li, F. P. Ma, P. Wang and Z. H. Zhang, Chin. J. Chem., 2013, 31, 757–763 CrossRef CAS PubMed.
- Z. Y. Duan, Y. L. Gu and Y. Q. Deng, Catal. Commun., 2006, 7, 651–656 CrossRef CAS PubMed.
- H. Mahajan, M. Bhardwaj and S. Paul, Org. Prep. Proced. Int., 2014, 46, 463–468 CrossRef CAS PubMed.
- N. Azizi and M. Alipour, J. Mol. Liq., 2015, 206, 268–271 CrossRef CAS PubMed.
- N. Azizi and M. Edrisi, Monatsh. Chem., 2015, 146 DOI:10.1007/s00706-015-1447-2.
- H. R. Lobo, B. S. Singh and G. S. Shankarling, Green Chem. Lett. Rev., 2012, 5, 487–533 CrossRef CAS PubMed.
- U. B. Patil, S. S. Shendage and J. M. Nagarkar, Synthesis, 2013, 45, 3295–3299 CrossRef CAS PubMed.
- U. B. Patil, A. S. Singh and J. M. Nagarkar, RSC Adv., 2014, 4, 1102–1106 RSC.
- A. S. Singh, S. S. Shendage and J. M. Nagarkar, Tetrahedron Lett., 2014, 55, 7243–7246 CrossRef CAS PubMed.
- N. Azizi, E. Gholibeglo, M. Babapour, H. Ghafuri and S. M. Bolourtchian, C. R. Chim., 2012, 15, 768–773 CrossRef CAS PubMed.
- N. Azizi, M. Khajeh and M. Alipour, Ind. Eng. Chem. Res., 2014, 53, 15561–15565 CrossRef CAS.
- B. L. Gadilohar, H. S. Kumbhar and G. S. Shankarling, Ind. Eng. Chem. Res., 2014, 53, 19010–19018 CrossRef CAS.
- Y. C. Zhang, F. L. Lu, X. H. Cao and J. Q. Zhao, RSC Adv., 2014, 4, 40161–40169 RSC.
- (a) D. A. G. Sanchez, Synlett, 2008, 1101–1110 CrossRef CAS PubMed; (b) Y. H. Liu, Z. H. Zhang and T. S. Li, Synthesis, 2008, 3314–3331 CAS; (c) Y. H. Liu, J. Deng, J. W. Gao and Z. H. Zhang, Adv. Synth. Catal., 2012, 354, 441–447 CrossRef CAS PubMed; (d) P. H. Li, B. L. Li, Z. M. An, L. P. Mo, Z. S. Cui and Z. H. Zhang, Adv. Synth. Catal., 2013, 355, 2952–2959 CrossRef CAS PubMed.
- D. Y. Dai, L. Wang, Q. Chen and M. Y. He, J. Chem. Res., 2014, 38, 183–185 CrossRef CAS.
- L. Wang, D. Y. Dai, Q. Chen and M. Y. He, J. Fluorine Chem., 2014, 158, 44–47 CrossRef CAS PubMed.
- N. Azizi, E. Batebi, S. Bagherpour and H. Ghafuri, RSC Adv., 2012, 2, 2289–2293 RSC.
- P. Xu, Y. Xu, X.-F. Li, B.-Y. Zhao, M.-H. Zong and W.-Y. Lou, ACS Sustainable Chem. Eng., 2015, 3, 718–724 CrossRef CAS.
- Z. Maugeri and P. Domínguez de María, ChemCatChem, 2014, 6, 1535–1537 CrossRef CAS PubMed.
- J. Deng, L. P. Mo, F. Y. Zhao, L. L. Hou, L. Yang and Z. H. Zhang, Green Chem., 2011, 13, 2576–2584 RSC.
- D. Saberi, J. Akbari, S. Mandudi and A. Heydari, J. Mol. Liq., 2014, 196, 208–210 CrossRef CAS PubMed.
- X. H. Zhao, X. Liu and M. Lu, Appl. Organomet. Chem., 2014, 28, 635–640 CrossRef CAS PubMed.
- A. R. Hajipour, S. H. i. Nazemzadeh and F. Mohammadsaleh, Tetrahedron Lett., 2014, 55, 654–656 CrossRef CAS PubMed.
- C. Vidal, F. J. Suárez and J. García-Álvarez, Catal. Commun., 2014, 44, 76–79 CrossRef CAS PubMed.
- F. Jérôme, M. Ferreira, H. Bricout, S. Menuel, E. Monflier and S. Tilloy, Green Chem., 2014, 16, 3876–3880 RSC.
- M. Ferreira, F. Jérôme, H. Bricout, S. Menuel, D. Landy, S. Fourmentin, S. Tilloy and E. Monflier, Catal. Commun., 2015, 63, 62–65 CrossRef CAS PubMed.
- S. Hu, Z. Zhang, Y. Zhou, B. Han, H. Fan, W. Li, J. Song and Y. Xie, Green Chem., 2008, 10, 1280–1283 RSC.
- F. Ilgen, D. Ott, D. Kralisch, C. Reil, A. Palmberger and B. König, Green Chem., 2009, 11, 1948–1954 RSC.
- C. Ruß, A. H. Begli and B. Koenig, Synth. Commun., 2013, 43, 2452–2456 CrossRef PubMed.
- S. Hu, Z. Zhang, Y. Zhou, J. Song, H. Fan and B. Han, Green Chem., 2009, 11, 873–877 RSC.
- K. De Oliveira Vigier, A. Benguerba, J. Barrault and F. Jérôme, Green Chem., 2012, 14, 285–289 RSC.
- M. Krystof, M. Pérez-Sánchez and P. Domínguez de María, ChemSusChem, 2013, 6, 630–634 CrossRef CAS PubMed.
- C. Ruß, C. Luff, A. H. Begli and B. Koenig, Synth. Commun., 2012, 42, 3112–3116 CrossRef PubMed.
- M. Pöehnlein, J. Ulrich, F. Kirschhöefer, M. Nusser, C. Muhle-Goll, B. Kannengiesser, G. Brenner-Weiβ, B. Luy, A. Liese, C. Syldatk and R. Hausmann, Eur. J. Lipid Sci. Technol., 2015, 117, 161–166 CrossRef PubMed.
- C. R. Müller, I. Meiners and P. Domínguez de María, RSC Adv., 2015, 4, 46097–46101 RSC.
- P. Xu, J. Cheng, W. Y. Lou and M. H. Zong, RSC Adv., 2015, 5, 6357–6364 RSC.
- J. Cao, Y. H. Shang, B. Qi, X. Z. Sun, L. Zhang, H. W. Liu, H. B. Zhang and X. H. Zhou, RSC Adv., 2015, 5, 9993–9996 RSC.
| This journal is © The Royal Society of Chemistry 2015 |