
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fuzziness endows viral motif-mimicry†
Norbert
Duro
,
Marton
Miskei
and
Monika
Fuxreiter
*
MTA-DE Momentum, Laboratory of Protein Dynamics, Department of Biochemistry and Molecular Biology, University of Debrecen, Hungary. E-mail: fmoni@med.unideb.hu
First published on 3rd August 2015
Abstract
Motif-mimicry is exploited by viruses to interfere with host regulatory networks and has also been suggested as a prevalent strategy for eukaryotic and prokaryotic pathogens. Using the same peptide motif however does not guarantee more effective interactions with the host. Motif-mediated interactions require a flexible or disordered environment, with structural and dynamic features that should differ between the competing host and viral proteins. Using the eukaryotic linear motif (ELM) database we analyzed the protein regions which contained the eukaryotic and viral motifs, including human and human virus ELMs with common target sites. We found that although the eukaryotic motifs are associated with a lack of structure, they are more stable than their flanking regions and can serve as molecular recognition elements. In contrast, eukaryotic viral motifs are often located in more ordered regions, but have increased local flexibility or disorder compared to their embedding environment. Most viral ELMs are devoid of stable binding elements and remain fuzzy after binding. Fuzziness reduces the entropic cost of binding and imparts versatile interaction modes to increase binding promiscuity and to compete with multiple host peptides. Fuzzy interactions confer further functional benefits such as the combinatorial usage of motifs, and a fine-tuning affinity via post-translational modifications.
Introduction
The functional diversity and adaptability of viruses is intriguing considering their small genome size. Viruses are proposed to have unique biophysical properties to cope with these evolutionary constraints: they possess loosely packed cores and a high propensity of non-regular secondary structure elements.1 Accordingly, viral protein segments often lack a stable tertiary structure, i.e. they are intrinsically disordered (ID) in the absence of a partner.2 This architecture can tolerate high mutation rates without the loss of the structural framework, which is required for function.3 Viral proteins were also experimentally demonstrated to maintain their intrinsically disordered state, even upon interacting with other components of the replication machinery.4 This phenomenon is termed as fuzziness.5,6 Nucleoprotein–phosphoprotein complexes in the measles,7 Hendra8 and Nipah9 viruses, for example, are characterized by a significant degree of conformational heterogeneity, which imparts dynamism on the recognition process. Fuzzy regions, for example, contribute to the organization of the nucleocapsid10 and also facilitate access to viral RNA.4Viruses invade their hosts via exploiting their regulatory networks. To this end, pathogens employ molecular mimicry and interact via short motifs that resemble those of the host system.11 Viral motifs are versatile: they can interfere with signaling pathways; control target protein levels or perturb post-translational modifications of host proteins. Viral motifs can also tune the cooperativity of host proteins and allosterically modulate the signaling outputs, as has recently been characterized in detail in the case of the E1A oncoprotein in a complex with the CREB binding protein and retinoblastoma protein.12,13 Motif-mimicry of host–peptide interactions is a powerful strategy, which is likely to be exploited by a large set of viral genomes (>2000)14 and is also employed by eukaryotic and prokaryotic pathogens.15 The robustness of short linear motifs (SLiMs) was also proposed to contribute to the adaptation and rapid evolution of viruses.11,14
Motif-mimicry does not necessarily mean that all viral motifs compete with the host motifs for the same site. For example, ubiquitin ligase recruitment16 or masking destruction motifs17 influence the host protein concentration or turnover, which might be regulated via different pathways in the host. Here we focus on cases where the viral and host motifs target the same host site. Experimental evidence demonstrates that the affinities of the viral motifs are higher than any of the host motifs, e.g. in the case of the PxxP (x could be any residue) SH3 binding motif of HIV Nef18 or the PTAG TSG101 binding motif of GAG-p6.19 As both the host and the pathogen utilize the same motif pattern (e.g. specificity determining residues) the higher affinities of the viral motifs likely rely on factors which are located outside the motif. These could provide additional contacts with the target protein or modulate the structural or dynamic properties of the motifs.
The eukaryotic linear motif (ELM) database is an excellent resource to analyze these features and compare the properties of the virus and host proteins.20 Previously it has been shown that ELMs tend to be located in protein regions that lack well-defined tertiary structures.21 The plasticity of ELM environments can impart versatility on binding modes and also enhance their adaptability. It is reasonable to assume that viral motifs employ a similar strategy. A local structural analysis on the protein segments that contain the viral motifs has not been carried out yet.
Here we aimed to reveal the molecular mechanisms of how viral motifs recognize their targets and those molecular factors that enable them to outperform the host motifs. The following scenarios were considered (Fig. 1): (i) protein regions containing viral motifs have increased plasticity as compared to those with eukaryotic motifs; (ii) viral peptide motifs are flanked by longer disordered regions, which can hamper the access of the competing host proteins to the target site by steric exclusion; and (iii) environments of the viral motifs provide further binding sites, which anchor the viral protein to the target and increase affinity. To investigate these mechanisms, the structural and dynamic features of the host and viral ELMs and their respective flanking regions were analyzed using two datasets (Fig. S1, ESI†): (i) experimentally verified viral and eukaryotic motifs in the ELM database; and (ii) human and human virus ELMs with common host target sites based on the VirusMentha22 and ELM interaction databases23 (Table S1, ESI†). Similar to previous studies,24 we found that the structural properties of viral motifs and their flanking regions vary over a wide range, but none of the proposed strategies were applicable. The environments of the eukaryotic motifs were found to be more flexible and include longer ID regions than those of the eukaryotic viral motifs. Flanking regions of eukaryotic ELMs contain more ID binding sites, which could establish buttressing interactions with the target proteins. Although these factors were comparable for human and human virus ELMs with common target sites, none of the scenarios could explain how viral motifs outcompete the host proteins. However, the local flexibility/disorder of the host and viral motifs relative to their embedding regions showed a significant difference. While the eukaryotic motifs are more stable and likely fold upon binding, the viral motifs have increased flexibility or disorder when compared to their flanking segments. Static or dynamic disorder, i.e. the fuzziness of the viral ELMs and their neighboring residues, is also observed upon interacting with the host target. Fuzziness of the motifs and their short flanking regions decreases the entropic cost of binding and improved affinity. The fuzzy virus–host interactions are amenable to fine-tuning via post-translational modifications or the combinatorial usage of motifs, which increases the binding versatility to interfere with host regulatory networks.
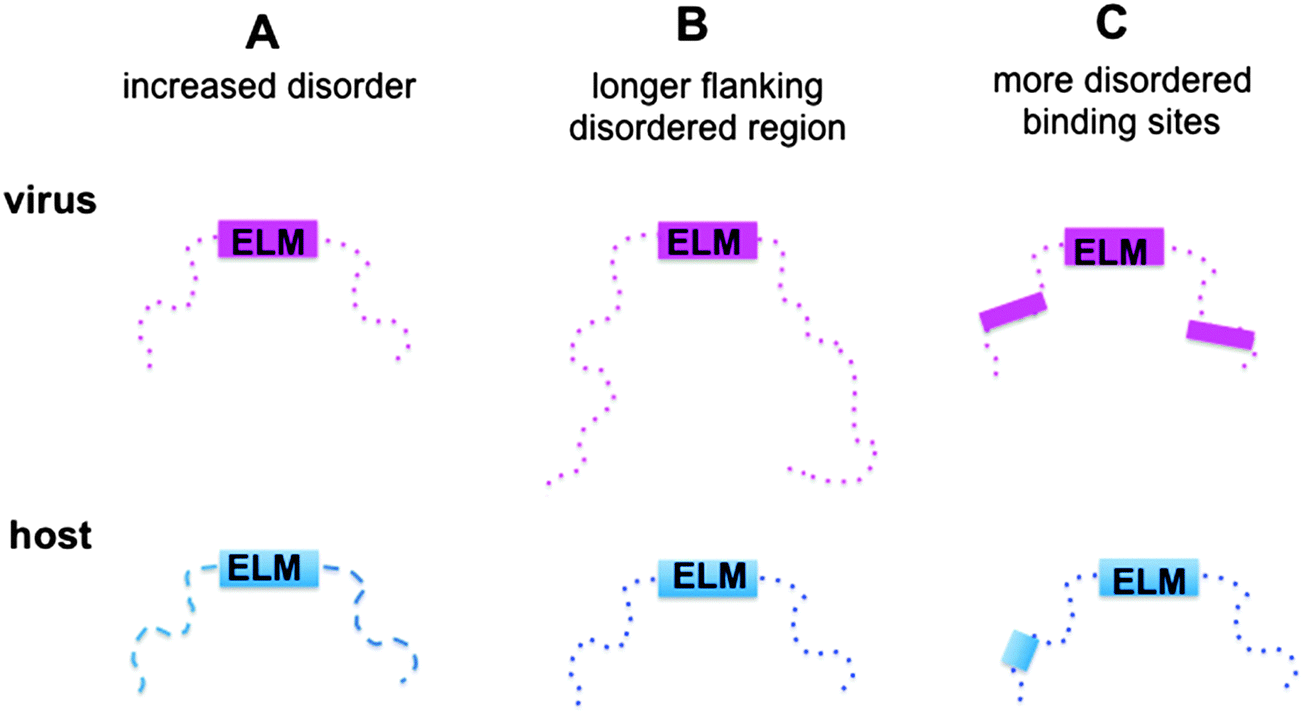 | ||
| Fig. 1 Schematic representation of the plausible scenarios of how viral ELMs (magenta) can outcompete host ELMs (blue). (A) Protein regions embedded with viral motifs have increased plasticity and a higher level of disorder as compared to those with eukaryotic motifs. (B) The viral motifs are flanked by longer disordered regions, which can hamper the access of the competing host proteins to the target site. (C) The environments of the viral motifs provide further binding sites, which can anchor the viral proteins to the target and increase their binding affinity. The ELMs are represented by solid labeled boxes. The disordered regions are displayed by dashed lines or dotted lines, the latter designates a higher level of disorder. The intrinsically disordered binding sites are represented by solid boxes. | ||
Results and discussion
ELM-containing viral proteins are more ordered than eukaryotic proteins
Increasing awareness of the structural flexibility of viral proteins gives the misleading impression that global disorder is a key feature for virus adaptability, survival and function. Recently it was shown, however, that the disorder of viral proteomes varies extensively and is not correlated to genome size.24 Comparing viral proteins to mesophilic eukaryotic proteins, no significant differences in contact densities, i.e. the tightness of the packing, were observed.1 Furthermore, viral proteins contain less regular secondary structure elements and more coils, but are equipped with fewer disordered regions. A recent comparative analysis between animal viruses and human proteins corroborated these results.14We focused on eukaryotic and virus proteins with experimentally verified ELMs (Table S2, ESI†) and compared their preference for a well-defined tertiary structure versus a disordered state. A significant difference between the average degree of disorder was observed (Fig. 2A): ELM-containing eukaryotic proteins are on the borderline between globular and disordered proteins (with a median value of 0.42; the average degree of disorder of the ID segments in the Disprot database v6.0225 is 0.44 using the IUpred program26), while the ELM-containing viral proteins are mostly structured (with a median value of 0.24). The fraction of intrinsically disordered residues in the eukaryotic ELM proteins is also significantly higher than in virus ELM proteins (Fig. 2B). This trend is in accord with the higher flexibility of the eukaryotic proteomes as compared to eukaryotic virus proteins, including all proteins, not only those with ELMs (Fig. S2, ESI†).
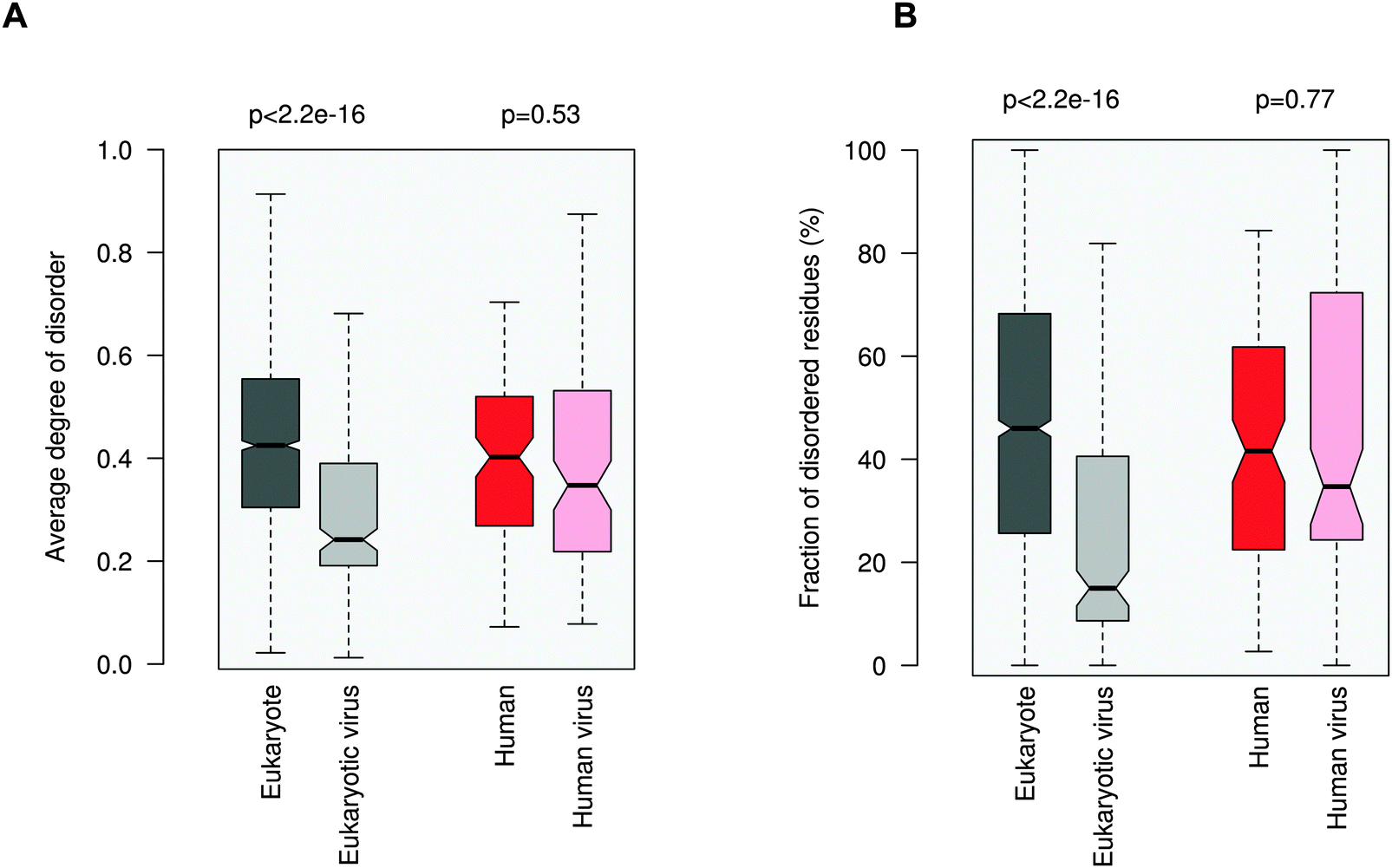 | ||
| Fig. 2 Average degree of disorder (A) and the fraction of disordered residues (B) of ELM-containing eukaryotic (dark gray), eukaryotic virus (light gray), human (dark red) and human virus (light red) proteins. The human and human virus ELMs target common host motif-binding domains. The p values were computed using the Wilcoxon test. | ||
Then we focused on human and human virus ELMs, which belonged to the same ELM classes (Table S3, ESI†). Human ELM proteins are also more pliable than human virus ELM proteins and have a higher fraction of ID residues (not shown). Finally we compared the subset of human and human virus ELMs, where the common motif-binding domains have been experimentally demonstrated (Tables S1 and S4, ESI†). Here no significant difference in the average degree of disorder or in the propensity of ID residues was seen between human and human virus ELM proteins. Comparing the disorder of different viral families using all proteins, excludes that this is owing to a biased selection of ELMs from given families (Fig. S3, ESI†). These results were consistent with the disorder predictions using the PONDR VSL1 algorithm27 (Fig. S4, ESI†).
Flanking regions of competing human and human virus ELMs have similar levels of disorder
ELMs in general tend to be located in disordered regions, which impart plasticity on linear motifs.21,28 This feature is also exploited for ELM discovery.29 In accordance with previous observations, 20AA regions flanking the eukaryotic ELMs have higher disorder scores than the average values for the corresponding proteins (0.58 vs. 0.42, Fig. 3A). Similarly, regions containing eukaryotic virus ELMs are also more flexible than the other protein regions (0.37 versus 0.24). These results were also corroborated by comparing the flanking environments of eukaryotic and eukaryotic virus ELMs to randomly chosen segments of the corresponding proteins with the same length (Fig. S5, ESI†). In line with their higher level of disorder, ID regions neighboring eukaryotic ELMs are significantly longer than those flanking eukaryotic virus ELMs, likely owing to the smaller genome size (Fig. 3B).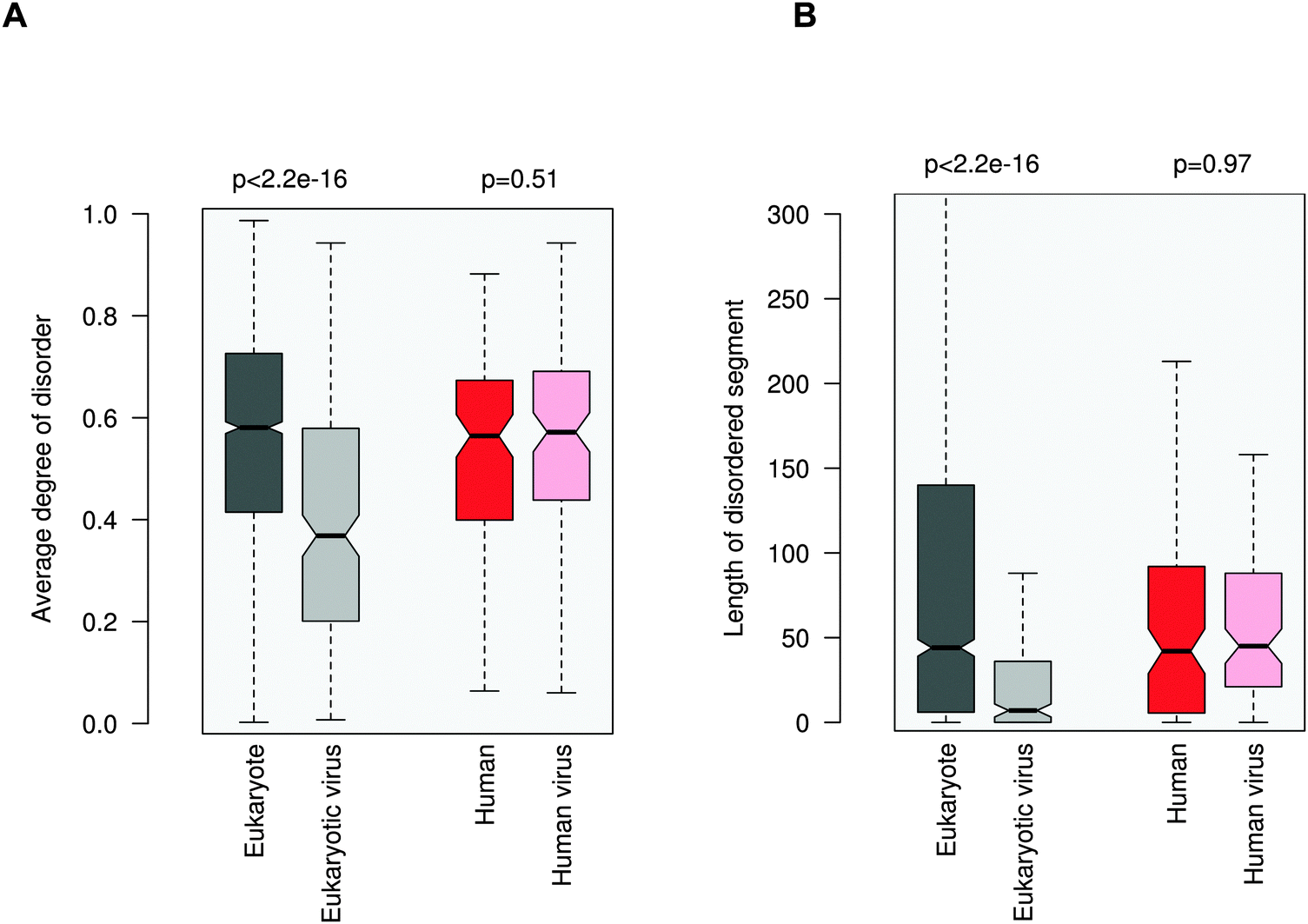 | ||
| Fig. 3 Disorder properties of the protein regions flanking the eukaryotic (dark gray), eukaryotic virus (light gray), human (dark red) and human virus (light red) ELMs. The human and human virus ELMs target common host motif-binding domains. (A) The average degree of disorder of the 20AA flanking region. (B) The length of the disordered segment flanking the motif. The p values were computed using the Wilcoxon test. | ||
In contrast to the marked difference between the disorder properties of the eukaryotic and eukaryotic virus ELM environments, the regions embedded with the human and human virus ELMs are rather similar. Neither the degree of disorder nor the length of the motif-flanking ID segments exhibit a significant difference between the human and human virus ELMs (Fig. 3). These results indicate some sort of constraint on the disorder properties of the competing motifs. This conclusion was supported by comparing the flanking regions to randomly chosen segments of the corresponding proteins (Fig. S6, ESI†) as well as by using a different disorder prediction method, PONDR VSL1 (Fig. S7, ESI†).
Additional disordered binding sites facilitate the binding of both host and virus ELMs
ID regions in general are reminiscent of multi-partite interactions, which are mediated either by the linear motifs or ID binding sites that gain structure upon binding. The latter can be transient secondary structure conformations, which are biased toward their bound state,30,31 or hydrophobic patches, which are stabilized by intermolecular interactions.32 ID binding sites have a variety of names, their definitions and relationships are detailed elsewhere.33 In general they have lower disorder scores than the embedding disordered protein regions34 and can also mediate non-specific interactions by anchoring the short linear motifs.32 This suggests a plausible scenario to increase the affinity of motif binding via engaging additional binding regions for partner interactions. We compared the number of ID binding sites which tend to fold upon binding in the 100AA flanking regions of the eukaryotic and eukaryotic virus ELMs using the Anchor program.32More ID binding sites were observed in the protein segments neighboring the eukaryotic ELMs than in the regions flanking the eukaryotic viral ELMs (Fig. 4). This is however not merely a consequence of the higher disorder of the eukaryotic proteins or the longer ID regions flanking the motif (Fig. S8, ESI†). The difference between the number of ID binding sites in the respective 100AA motif-flanking segments of eukaryotic and eukaryotic virus proteins is higher than the differences when comparing other 100AA regions of the corresponding eukaryotic and viral proteins. We should also note that ID binding sites are enriched in the environments of both eukaryotic and eukaryotic virus ELMs as compared to the corresponding proteins on average.
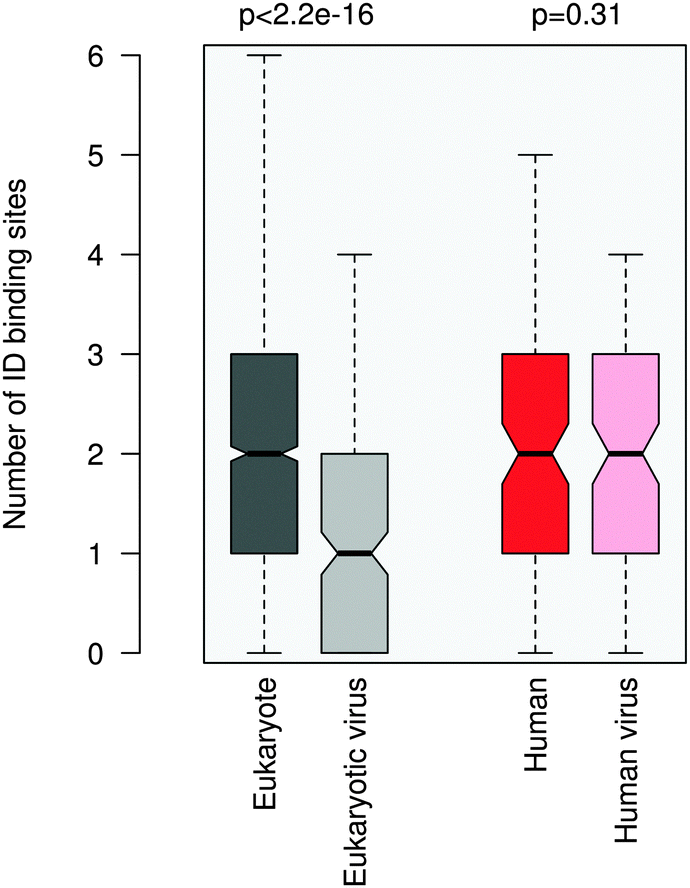 | ||
| Fig. 4 Number of intrinsically disordered binding sites within the 100AA flanking regions of the eukaryotic (dark gray), eukaryotic virus (light gray), human (dark red) and human virus (light red) ELMs. The human and human virus ELMs target common host motif-binding domains. The p value was computed using the Wilcoxon test. | ||
The flanking regions of human and human virus ELMs comprise a comparable number of ID binding sites, while this markedly deviates in the corresponding proteins. For this dataset we also repeated the ID binding site calculations using the Disopred3 algorithm.35 Disopred3 employs a support vector machine approach, which predicts more irregular structural elements to bind than Anchor and excludes potential transient contacts. For these reasons Disopred3 predicts more ID binding sites in the 100AA regions flanking the human virus ELMs than in those neighboring the human ELMs (Fig. S9, ESI†). This indicates that buttressing contacts by ID binding sites may contribute to the higher efficiency of the human viral motifs. The DGR motif of the capsid protein VP1 of adeno-associated virus 2, for example, competes with the four NGR integrin binding sites of fibronectin (LIG_Integrin_isoDGR_1 motif in ELM; see Table S1, ESI†) for host entry. The 100AA flanking region of the viral DGR motif contains more ID binding sites than the neighboring segments of the fibronectin NGR motifs of the same size, in accordance with the critical role of the flanking residues in facilitating the adhesion of the cell surface and integrin receptor switching.36
Eukaryotic ELMs tend to fold, while viral motifs remain fuzzy
Probing the three proposed scenarios (Fig. 1) did not provide a conclusive answer as to how viral motifs compete with their host counterparts. Hence we assumed that the structural or dynamic properties of the viral ELMs themselves are responsible for more efficient partner recognition. By comparing the disorder properties of the human and human virus ELMs with common host motif-binding domains we found that the human virus ELMs are significantly more flexible or disordered than their human counterparts (Fig. 5A) despite the comparable level of disorder in the embedding regions (Fig. 3A). This suggests that human and human virus motifs have different characteristics as compared to their flanking protein regions. Thus we computed the difference between the degree of disorder of the human and human virus ELMs and their flanking regions (ΔID = IDELM − IDFlanking20AA). We found that the human ELMs have a lower degree of disorder than their environment, while the human virus ELMs have an elevated level of disorder as compared to their embedding environments (Fig. 5B). Pair-wise differences of disorder scores between the human and human virus ELMs and their 20AA flanking regions underscore this observation (p = 4.7 × 10−5 with the Wilcoxon test, p = 1.5 × 10−4 with the Kolmogorov–Smirnov test). Taken together the human ELMs appear to be more stable, while the human virus ELMs are more flexible or dynamic than their 20AA flanking regions.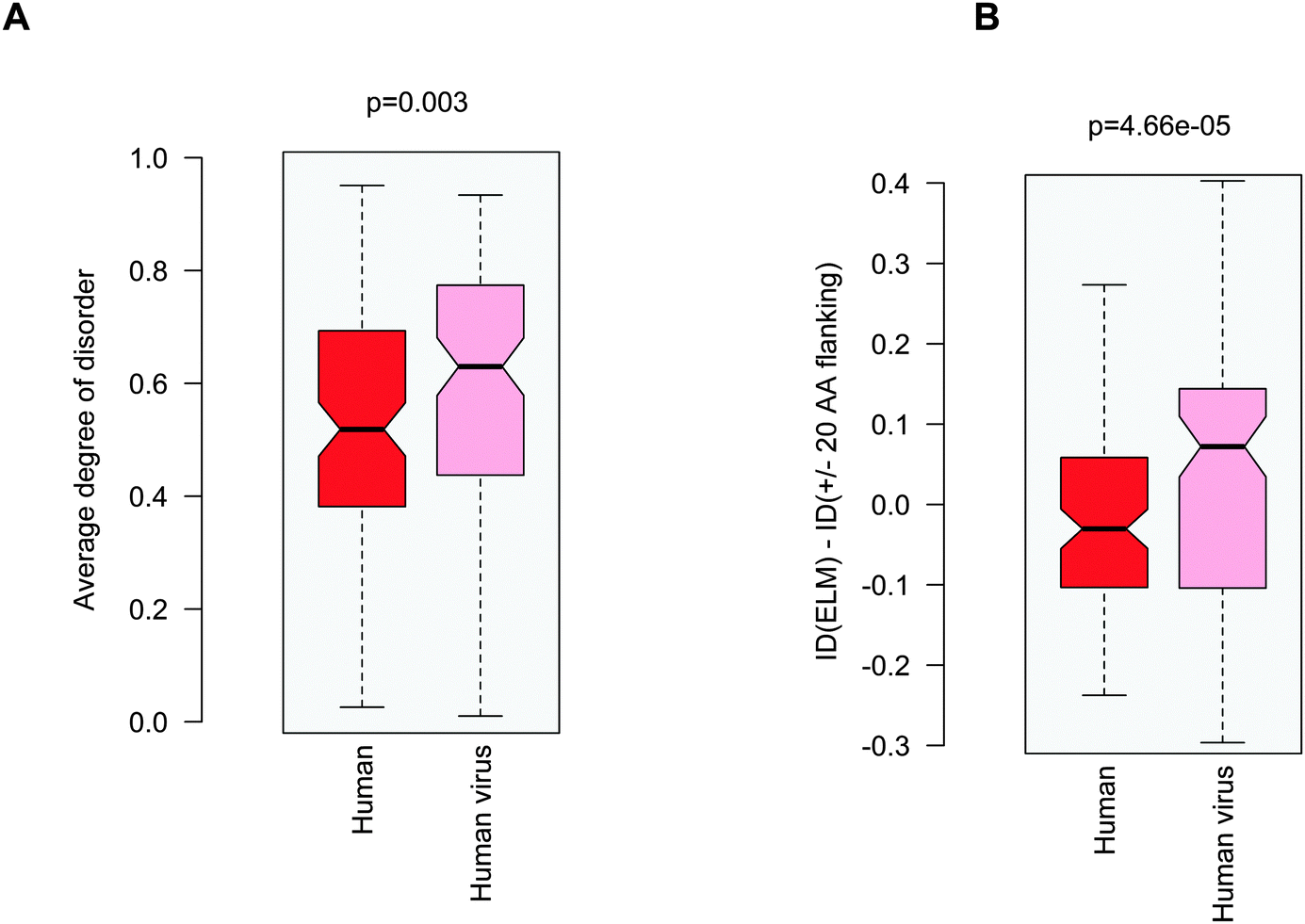 | ||
| Fig. 5 (A) Average degree of disorder of the human and human virus ELMs that target common host motif-binding domains. (B) Difference in the degree of disorder between the human and human virus ELMs and their respective 20AA flanking regions. The p values were computed using the Wilcoxon test. | ||
The decreased disorder of human ELMs as compared to their flanking regions indicates that they can serve as preformed30 or molecular recognition elements,31 which exhibit transient secondary structures biased toward their bound conformation. These binding sites could fold upon interacting with their partner. Accordingly, 68% of human ELMs are associated with non-regular secondary elements (NORS).37 Along these lines, 54% of the disordered human and 28% of the disordered viral ELMs are predicted to fold upon binding,32 irrespective of their secondary structure preferences. Taken together, the human virus ELMs follow a different strategy for partner recognition than the corresponding host motifs, which is driven by increased local flexibility or disorder. To corroborate this observation, 87% of the disordered viral motifs are flanked by short (at least 5AA) fuzzy regions, which remain dynamic even when bound to their partners.
Experimental evidence for fuzziness in virus–host interactions
Despite the experimental difficulties in characterizing fuzzy protein regions,38 growing evidence supports the existence of the disordered state, i.e. fuzziness, in viral complexes.2 Two examples of how fuzziness contributes to motif-mimicry are detailed below.Nonstructural protein 5A (NS5A) of the hepatitis C virus (HCV) has two PxxP motifs (PP2.1 and PP2.2), out of which the PP2.2 motif can interact with a variety of SH3 domains of the Src kinase family. NMR results reveal two additional PxxP motifs, serving as low-affinity sites for noncanonical SH3 binding.39 All NS5A binding motifs compete for the same pocket on the SH3 domain via mutually exclusive binding modes. Although the noncanonical sites are embedded in transiently structured α-helical regions, the population of helical conformations decreases upon binding. The heteronuclear Overhauser effect (hetNOE) values of the NS5A residues at the binding interface also decrease upon interacting with SH3 domains, indicating increased conformational flexibility and more heterogeneous structural ensemble. The fuzzy nature of the complex provides a favorable entropic contribution to the binding free energy and results in a 2–3 fold increase in Kd values.
The hepatitis B virus (HBV) preS1 domain contains multiple motifs, which resemble the recognition sites of the cell–surface receptor γ2-adaptin. preS1 does not exhibit a preformed secondary structure, and interactions with the γ2-adaptin EAR domain do not induce structure-formation.40 NOE enhancements show that the binding motifs, which are flanked by proline residues, have a distinct dynamic character and remain fuzzy in the context of the binding partner. Deletion experiments demonstrate that the flanking residues also contribute to the binding affinity of preS1 to the γ2-adaptin EAR domain. In preS1, fuzziness enables combinatorial usage of the motifs, which increases the functional versatility of the viral protein.
A further series of experimental evidence supports that fuzziness is present in paramyxovirus complexes3 upon interactions of the NTAIL of a nucleoprotein with a phosphoprotein in Measles, Nipah and Hendra viruses.
Static fuzziness and binding promiscuity of the human virus ELMs
Owing to the genetic compaction of viruses, the encoded proteins should be involved in multiple functions, e.g. capable of establishing interactions with different partners. This could be facilitated by the underlying conformational heterogeneity of the viral proteins, which allows the ensemble to shift between different conformational states upon responding to different signals. Our results indicate that viral motifs have increased local flexibility or disorder relative to their 20AA flanking regions. We discussed examples for dynamic fuzziness, when the protein interconverts between multiple conformations in the bound state.5,6 In the case of static fuzziness however, the ID protein folds upon interacting with the partner, but adopts alternative conformations.5,6 17% of the human virus ELMs are located in ordered regions; these were analyzed for static fuzziness. To this end, we collected the structures of the host–virus complexes of these ELMs and different secondary structure conformations upon targeting the same host protein were identified (Table S5, ESI†). The adenovirus E1A protein interacts with a retinoblastoma protein in multiple locations via short peptides, which despite being the same sequence adopt different secondary structures in the complex (Fig. 6).41 The E2F transcription factor uses the same contacts to inactivate the viral oncoprotein. Another example is the YNSTFF motif (MOD_N-GLC_1) of the SARS coronavirus spike receptor-binding domain (SRBD), which interacts with its receptor using α-helix, turn or β-bridge structural elements (PDB codes: 2ajf, 3scj).42 In addition, the FNATKF motif of the SARS SRBD adopts 3 different conformations upon binding to the receptor, increasing the structural and functional versatility of the virus–host interaction. This illustrates that static fuzziness can also increase binding promiscuity and enable viral ELMs to compete with various human motifs (Table S1, ESI†) for their respective targets.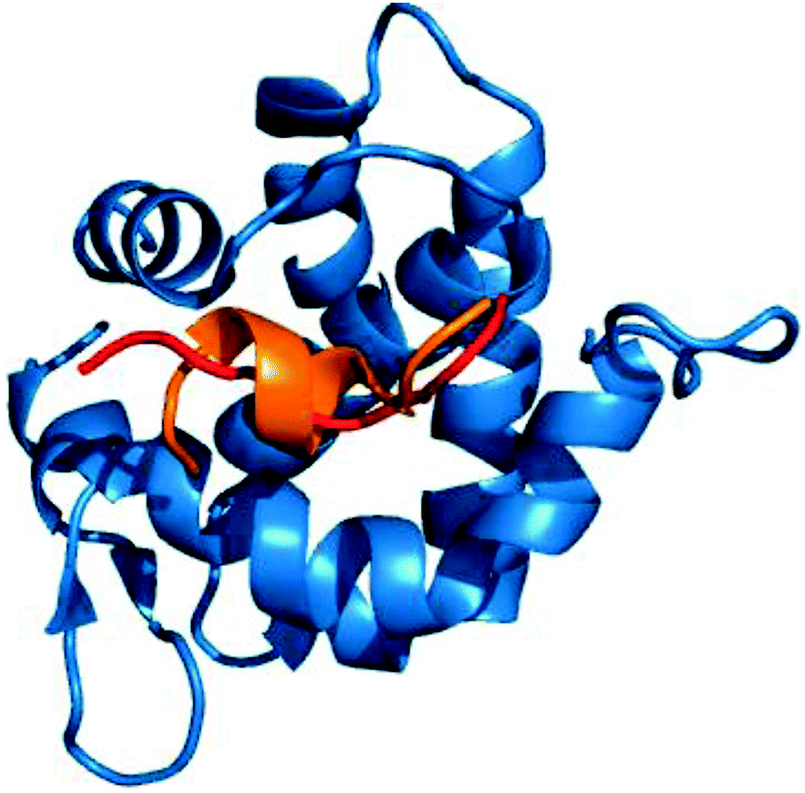 | ||
| Fig. 6 The interaction of the E1A viral oncoprotein with a retinoblastoma protein is realized via an α-helix and a turn secondary structure element (PDB code: 2r7g). | ||
Implications of viral motif fuzziness for antiviral strategies
Disrupting virus–host interactions is an exciting challenge to develop antiviral therapeutics. As motif-mimicry is exploited by numerous viral genomes, a possible strategy is to design a motif-mimetic, which targets the same binding site of the host and outcompetes the viral motif.43The evolutionary plasticity of viral ELMs however, could be a bottleneck for this approach. Another disadvantage is the low selectivity/binding promiscuity of the motif, which can lead to side-effects via binding to undesired host proteins.44 One can attempt to block the viral motif directly by specific antibodies and overcome the problem of resistance.45 Our results indicate that fuzziness or increased local flexibility of the viral motifs is critical for their interactions. Consequently, rigidifying viral motifs should hamper their binding to the host proteins, and host peptides should become more effective at targeting the same site. This offers an alternative strategy for developing motif-mimetic drugs.
Experimental and bioinformatics results indicate that the disorder properties of a given site could be modulated by a longer protein sequence (approx. 100AA).26,32 Binding a small-molecule compound into this region could thus decrease the local dynamics of the viral motif, which would stabilize the structural elements and reduce the conformational heterogeneity. Targeting the more ordered flanking regions of the viral motifs could be more feasible than the dynamic viral ELM itself, yet could impact its function. Rigidifying the viral motifs and their proximal residues might also impair capsid formation and disfavor replication. As the embedding regions for the viral ELMs are more structured, they are likely less mutation-prone than the motif itself, and thereby could be more suitable drug targets.
Conclusion
Viral motif-mimicry is a successful strategy to invade the host and reprogram its regulatory networks. While eukaryotic ELMs serve as molecular recognition elements for host–peptide interactions, viral ELMs have increased local flexibility compared to their environments. Experimental evidence supports that the fuzziness of the viral ELMs improves binding affinity and promiscuity. Perturbing dynamic properties of viral ELMs via small-molecule binding at the flanking regions could offer an alternative approach to direct motif-based drug development.We must note that viral proteins use versatile mechanisms to reprogram the host system by having an alternative set of motifs to the host proteins, or to alter the turnover owing to the presence or absence of degradation signals. This analysis focused on only a subset of viral motifs, those which use the same functional classes as host peptide motifs.
Experimental procedures
Eukaryotic and virus ELM datasets
2585 ELMs were downloaded from the ELM database (http://elm.eu.org).20 We ignored all cases where the evidence class was ‘predicted’ or ELMs with ‘no further instance evidence’, discarding in total 556 ELMs. All of the remaining 2029 ELMs have been corroborated by experimental evidence. This dataset contained 1801 eukaryotic, 220 virus and 8 prokaryotic ELMs. Owing to their small number and the absence of the corresponding prokaryotic viral motifs, the prokaryotic ELMs were not analyzed. Out of the 1801 eukaryotic ELMs, 1148 were human ELMs and out of the 220 eukaryotic viral ELMs, 160 were human virus ELMs. As one protein can contain multiple ELMs, the analyzed ELMs belonged to 1182 eukaryotic, 698 human, 123 eukaryotic virus and 82 human virus proteins.The distribution of eukaryotic and eukaryotic virus ELMs amongst different ELM classes is displayed in Table S2 (ESI†). Then we paired the human and human virus ELMs based on their ELM classes. First, all possible pairs of human and human virus ELMs with identical ELM classes (e.g. integrin_isoDGR_1) were created, resulting in 1159 pairs (from 31 ELM classes, Table S3, ESI†). Obviously, the same ELM type does not guarantee that the human and human virus ELMs will have identical host domain targets. Hence using two resources, the iELM database23 and the VirusMentha database,22 we collected experimental evidence to filter out those pairs where the human and human virus motifs target the same host protein domain. The selection process is displayed in Fig. S1 (ESI†). iELM provided 32 examples with identical motif-binding domains for human and human virus ELMs. The VirusMentha database provided 202 cases where the human virus ELM and the human ELM targeted the same protein. The VirusMentha database however does not provide information on the target binding domains. Therefore, the human–human virus ELM pair examples, which were filtered based on the VirusMentha database, were cross-validated using the iELM database interaction domains. Here, domains which bind a given motif were collected. Cross-validation could assign a motif-binding domain to 90 out of those 202 human–human virus ELM pairs found using the VirusMentha database. 14 pairs out of the 90 hits overlapped with the examples selected from the iELM interactions database. Thus 76 human–human virus ELM pair examples were found using the VirusMentha database with interacting motif binding domains and were validated against the iELM interacting domains. In total, the two databases resulted in 108 (32 + 76) human and human virus ELM pairs with identical host binding domains. As in the VirusMentha database, we applied the binary interaction filter, and in the 32 iELM cases where the experimental binding affinities are provided, we excluded that the 108 motif-domain associations are resulted by indirect interactions. The distribution of the 108 human and human virus pairs amongst the different ELM classes is displayed in Table S4 (ESI†).
Analysis of the disorder
The preference for a well-defined structure or the lack of a stable structure was estimated based on low-resolution pair-wise potentials by the IUPred program.26 The results were corroborated using the PONDR VSL1 and VSL2 neural network algorithms,46 which provided very similar results. Hence we only display the data computed by VSL1 in the ESI.† The average degree of disorder was computed by averaging the disorder score of all residues. In the IUPred ‘long’ algorithm we applied a 0.44 threshold to discriminate between ordered and disordered residues. Using this binary classification, we determined the propensity of the disordered residues (NID/NAA, where NID is the number of disordered residues, and NAA is the length of the sequence). The 0.44 limit is based on the average disorder score of the disordered residues in the 6.02 version of the Disprot database.25 To analyze the local disorder properties, the 20AA ELM-flanking regions were considered to make the results comparable to the previous analysis on the eukaryotic ELMs.21Analysis of the disordered binding sites and fuzzy regions
Intrinsically disordered binding regions, which fold upon binding, were computed using the Anchor program32 and were defined as continuous stretches of at least 5 residues with Anchor scores > 0.5. The dynamic fuzzy regions do not adopt a well-defined structure upon interacting with their partners. The fuzzy regions were defined as disordered regions that do not overlap with the intrinsically disordered binding sites. The fuzzy regions were identified applying two conditions: IUPred score > 0.44 (to define an ID region) and Anchor score < 0.5 (to exclude the formation of a stable structure).Non-regular secondary structure (NORS) elements were identified using the PredictProtein server.47
Statistical analysis
All statistics were performed using the R program (http://www.r-project.org/). Both the Wilcoxon rank sum test (Mann–Whitney) and the Wilcoxon signed rank test were computed.Acknowledgements
The support of the Hungarian Science Fund program (OTKA NN 106562) and the Momentum program (LP2012-41) of the Hungarian Academy of Sciences is gratefully acknowledged (M.F.). We thank Sonia Longhi for fruitful discussions.References
- N. Tokuriki, C. J. Oldfield, V. N. Uversky, I. N. Berezovsky and D. S. Tawfik, Do viral proteins possess unique biophysical features?, Trends Biochem. Sci., 2009, 34(2), 53–59 CrossRef CAS PubMed.
- B. Xue, D. Blocquel, J. Habchi, A. V. Uversky, L. Kurgan, V. N. Uversky and S. Longhi, Structural disorder in viral proteins, Chem. Rev., 2014, 114(13), 6880–6911 CrossRef CAS PubMed.
- J. Habchi and S. Longhi, Structural disorder within paramyxovirus nucleoproteins and phosphoproteins, Mol. BioSyst., 2012, 8(1), 69–81 RSC.
- M. R. Jensen, G. Communie, E. A. J. Ribeiro, N. Martinez, A. Desfosses, L. Salmon, L. Mollica, F. Gabel, M. Jamin, S. Longhi, R. W. Ruigrok and M. Blackledge, Intrinsic disorder in measles virus nucleocapsids, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(24), 9839–9844 CrossRef CAS PubMed.
- P. Tompa and M. Fuxreiter, Fuzzy complexes: polymorphism and structural disorder in protein–protein interactions, Trends Biochem. Sci., 2008, 33(1), 2–8 CrossRef CAS PubMed.
- M. Fuxreiter, Fuzziness: linking regulation to protein dynamics, Mol. BioSyst., 2012, 8(1), 168–177 RSC.
- A. D'Urzo, A. Konijnenberg, G. Rossetti, J. Habchi, J. Li, P. Carloni, F. Sobott, S. Longhi and R. Grandori, Molecular Basis for Structural Heterogeneity of an Intrinsically Disordered Protein Bound to a Partner by Combined ESI-IM-MS and Modeling, J. Am. Soc. Mass Spectrom., 2015, 26(3), 472–481 CrossRef PubMed.
- G. Communie, J. Habchi, F. Yabukarski, D. Blocquel, R. Schneider, N. Tarbouriech, N. Papageorgiou, R. W. Ruigrok, M. Jamin, M. R. Jensen, S. Longhi and M. Blackledge, Atomic resolution description of the interaction between the nucleoprotein and phosphoprotein of Hendra virus, PLoS Pathog., 2013, 9(9), e1003631 CAS.
- J. Habchi, S. Blangy, L. Mamelli, M. R. Jensen, M. Blackledge, H. Darbon, M. Oglesbee, Y. Shu and S. Longhi, Characterization of the interactions between the nucleoprotein and the phosphoprotein of Henipavirus, J. Biol. Chem., 2011, 286(15), 13583–13602 CrossRef CAS PubMed.
- R. Ivanyi-Nagy and J. L. Darlix, Fuzziness in the core of the human pathogenic viruses HCV and HIV, Adv. Exp. Med. Biol., 2012, 725, 142–158 CrossRef CAS.
- N. E. Davey, G. Trave and T. J. Gibson, How viruses hijack cell regulation, Trends Biochem. Sci., 2011, 36(3), 159–169 CrossRef CAS PubMed.
- J. C. Ferreon, M. A. Martinez-Yamout, H. J. Dyson and P. E. Wright, Structural basis for subversion of cellular control mechanisms by the adenoviral E1A oncoprotein, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(32), 13260–13265 CrossRef CAS PubMed.
- A. C. Ferreon, J. C. Ferreon, P. E. Wright and A. A. Deniz, Modulation of allostery by protein intrinsic disorder, Nature, 2013, 498(7454), 390–394 CrossRef CAS PubMed.
- T. Hagai, A. Azia, M. M. Babu and R. Andino, Use of host-like peptide motifs in viral proteins is a prevalent strategy in host–virus interactions, Cell Rep., 2014, 7(5), 1729–1739 CrossRef CAS PubMed.
- A. Via, B. Uyar, C. Brun and A. Zanzoni, How pathogens use linear motifs to perturb host cell networks, Trends Biochem. Sci., 2015, 40(1), 36–48 CrossRef CAS PubMed.
- B. J. Stanley, E. S. Ehrlich, L. Short, Y. Yu, Z. Xiao, X. F. Yu and Y. Xiong, Structural insight into the human immunodeficiency virus Vif SOCS box and its role in human E3 ubiquitin ligase assembly, J. Virol., 2008, 82(17), 8656–8663 CrossRef CAS PubMed.
- M. Welcker and B. E. Clurman, The SV40 large T antigen contains a decoy phosphodegron that mediates its interactions with Fbw7/hCdc4, J. Biol. Chem., 2005, 280(9), 7654–7658 CrossRef CAS PubMed.
- T. Stangler, T. Tran, S. Hoffmann, H. Schmidt, E. Jonas and D. Willbold, Competitive displacement of full-length HIV-1 Nef from the Hck SH3 domain by a high-affinity artificial peptide, Biol. Chem., 2007, 388(6), 611–615 CrossRef CAS PubMed.
- A. Schlundt, J. Sticht, K. Piotukh, D. Kosslick, N. Jahnke, S. Keller, M. Schuemann, E. Krause and C. Freund, Proline-rich sequence recognition: II. Proteomics analysis of Tsg101 ubiquitin-E2-like variant (UEV) interactions, Mol. Cell. Proteomics, 2009, 8(11), 2474–2486 CAS.
- H. Dinkel, K. Van Roey, S. Michael, N. E. Davey, R. J. Weatheritt, D. Born, T. Speck, D. Kruger, G. Grebnev, M. Kuban, M. Strumillo, B. Uyar, A. Budd, B. Altenberg, M. Seiler, L. B. Chemes, J. Glavina, I. E. Sanchez, F. Diella and T. J. Gibson, The eukaryotic linear motif resource ELM: 10 years and counting, Nucleic Acids Res., 2014, 42(Database issue), D259–D266 CrossRef CAS PubMed.
- M. Fuxreiter, P. Tompa and I. Simon, Local structural disorder imparts plasticity on linear motifs, Bioinformatics, 2007, 23(8), 950–956 CrossRef CAS PubMed.
- A. Calderone, L. Licata and G. Cesareni, VirusMentha: a new resource for virus–host protein interactions, Nucleic Acids Res., 2015, 43(Database issue), D588–D592 CrossRef PubMed.
- R. J. Weatheritt, P. Jehl, H. Dinkel and T. J. Gibson, iELM – a web server to explore short linear motif-mediated interactions, Nucleic Acids Res., 2012, 40(Web Server issue), W364–W369 CrossRef CAS PubMed.
- R. Pushker, C. Mooney, N. E. Davey, J. M. Jacque and D. C. Shields, Marked variability in the extent of protein disorder within and between viral families, PLoS One, 2013, 8(4), e60724 CAS.
- M. Sickmeier, J. A. Hamilton, T. LeGall, V. Vacic, M. S. Cortese, A. Tantos, B. Szabo, P. Tompa, J. Chen, V. N. Uversky, Z. Obradovic and A. K. Dunker, DisProt: The Database of Disordered Proteins, Nucleic Acids Res., 2007, 35(Database issue), D786–D793 CrossRef CAS PubMed.
- Z. Dosztanyi, V. Csizmok, P. Tompa and I. Simon, The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins, J. Mol. Biol., 2005, 347(4), 827–839 CrossRef CAS PubMed.
- K. Peng, P. Radivojac, S. Vucetic, A. K. Dunker and Z. Obradovic, Length-dependent prediction of protein intrinsic disorder, BMC Bioinf., 2006, 7, 208 CrossRef PubMed.
- N. E. Davey, K. Van Roey, R. J. Weatheritt, G. Toedt, B. Uyar, B. Altenberg, A. Budd, F. Diella, H. Dinkel and T. J. Gibson, Attributes of short linear motifs, Mol. BioSyst., 2012, 8(1), 268–281 RSC.
- N. E. Davey, D. C. Shields and R. J. Edwards, SLiMDisc: short, linear motif discovery, correcting for common evolutionary descent, Nucleic Acids Res., 2006, 34(12), 3546–3554 CrossRef CAS PubMed.
- M. Fuxreiter, I. Simon, P. Friedrich and P. Tompa, Preformed structural elements feature in partner recognition by intrinsically unstructured proteins, J. Mol. Biol., 2004, 338(5), 1015–1026 CrossRef CAS PubMed.
- C. J. Oldfield, Y. Cheng, M. S. Cortese, P. Romero, V. N. Uversky and A. K. Dunker, Coupled folding and binding with alpha-helix-forming molecular recognition elements, Biochemistry, 2005, 44(37), 12454–12470 CrossRef CAS PubMed.
- B. Meszaros, I. Simon and Z. Dosztanyi, Prediction of protein binding regions in disordered proteins, PLoS Comput. Biol., 2009, 5(5), e1000376 Search PubMed.
- R. Pancsa and M. Fuxreiter, Interactions via intrinsically disordered regions: what kind of motifs?, IUBMB Life, 2012, 64(6), 513–520 CrossRef CAS PubMed.
- A. Mohan, C. J. Oldfield, P. Radivojac, V. Vacic, M. S. Cortese, A. K. Dunker and V. N. Uversky, Analysis of molecular recognition features (MoRFs), J. Mol. Biol., 2006, 362(5), 1043–1059 CrossRef CAS PubMed.
- D. T. Jones and D. Cozzetto, DISOPRED3: precise disordered region predictions with annotated protein-binding activity, Bioinformatics, 2015, 31(6), 857–863 CrossRef PubMed.
- F. Curnis, A. Cattaneo, R. Longhi, A. Sacchi, A. M. Gasparri, F. Pastorino, P. Di Matteo, C. Traversari, A. Bachi, M. Ponzoni, G. P. Rizzardi and A. Corti, Critical role of flanking residues in NGR-to-isoDGR transition and CD13/integrin receptor switching, J. Biol. Chem., 2010, 285(12), 9114–9123 CrossRef CAS PubMed.
- J. Liu, H. Tan and B. Rost, Loopy proteins appear conserved in evolution, J. Mol. Biol., 2002, 322, 53–64 CrossRef CAS.
- N. Rezaei-Ghaleh, M. Blackledge and M. Zweckstetter, Intrinsically disordered proteins: from sequence and conformational properties toward drug discovery, ChemBioChem, 2012, 13(7), 930–950 CrossRef CAS PubMed.
- M. Schwarten, Z. Solyom, S. Feuerstein, A. Aladag, S. Hoffmann, D. Willbold and B. Brutscher, Interaction of nonstructural protein 5A of the hepatitis C virus with Src homology 3 domains using noncanonical binding sites, Biochemistry, 2013, 52(36), 6160–6168 CrossRef CAS PubMed.
- M. C. Jurgens, J. Voros, G. J. Rautureau, D. A. Shepherd, V. E. Pye, J. Muldoon, C. M. Johnson, A. E. Ashcroft, S. M. Freund and N. Ferguson, The hepatitis B virus preS1 domain hijacks host trafficking proteins by motif mimicry, Nat. Chem. Biol., 2013, 9(9), 540–547 CrossRef PubMed.
- X. Liu and R. Marmorstein, Structure of the retinoblastoma protein bound to adenovirus E1A reveals the molecular basis for viral oncoprotein inactivation of a tumor suppressor, Genes Dev., 2007, 21(21), 2711–2716 CrossRef CAS PubMed.
- W. Kabsch and C. Sander, Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features, Biopolymers, 1983, 22(12), 2577–2637 CrossRef CAS PubMed.
- J. A. Wells and C. L. McClendon, Reaching for high-hanging fruit in drug discovery at protein–protein interfaces, Nature, 2007, 450(7172), 1001–1009 CrossRef CAS PubMed.
- T. Vavouri, J. Semple, R. Garcia-Verdugo and B. Lehner, Intrinsic Protein Disorder and Interaction promiscuity are widely associated with Dosage Sensitivity, Cell, 2009, 138, 198–208 CrossRef CAS PubMed.
- P. Chames, M. Van Regenmortel, E. Weiss and D. Baty, Therapeutic antibodies: successes, limitations and hopes for the future, Br. J. Pharmacol., 2009, 157(2), 220–233 CrossRef CAS PubMed.
- Z. Obradovic, K. Peng, S. Vucetic, P. Radivojac, C. J. Brown and A. K. Dunker, Predicting intrinsic disorder from amino acid sequence, Proteins, 2003, 53(suppl 6), 566–572 CrossRef CAS PubMed.
- G. Yachdav, E. Kloppmann, L. Kajan, M. Hecht, T. Goldberg, T. Hamp, P. Honigschmid, A. Schafferhans, M. Roos, M. Bernhofer, L. Richter, H. Ashkenazy, M. Punta, A. Schlessinger, Y. Bromberg, R. Schneider, G. Vriend, C. Sander, N. Ben-Tal and B. Rost, PredictProtein – an open resource for online prediction of protein structural and functional features, Nucleic Acids Res., 2014, 42(Web Server issue), W337–W343 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5mb00301f |
| This journal is © The Royal Society of Chemistry 2015 |