
Biomaterials for mRNA delivery
Mohammad Ariful
Islam
a,
Emma K. G.
Reesor†
a,
Yingjie
Xu
b,
Harshal R.
Zope
a,
Bruce R.
Zetter
*b and
Jinjun
Shi
*a
aLaboratory for Nanoengineering & Drug Delivery, Brigham and Women's Hospital, Harvard Medical School, Boston, MA 02115, USA. E-mail: jinjun.shi@zeus.bwh.harvard.edu
bVascular Biology Program, Boston Children's Hospital, Harvard Medical School, Boston, MA 02115, USA. E-mail: bruce.zetter@childrens.harvard.edu
First published on 17th August 2015
Abstract
Messenger RNA (mRNA) has recently emerged with remarkable potential as an effective alternative to DNA-based therapies because of several unique advantages. mRNA does not require nuclear entry for transfection activity and has a negligible chance of integrating into the host genome which excludes the possibility of potentially detrimental genomic alternations. Chemical modification of mRNA has further enhanced its stability and decreased its activation of innate immune responses. Additionally, mRNA has been found to have rapid expression and predictable kinetics. Nevertheless, the ubiquitous application of mRNA remains challenging given its unfavorable attributes, such as large size, negative charge and susceptibility to enzymatic degradation. Further refinement of mRNA delivery modalities is therefore essential for its development as a therapeutic tool. This review provides an exclusive overview of current state-of-the-art biomaterials and nanotechnology platforms for mRNA delivery, and discusses future prospects to bring these exciting technologies into clinical practice.
 Mohammad Ariful Islam | Dr Mohammad Ariful Islam obtained his Bachelor in Biotechnology and Genetic Engineering from the University of Development Alternative, Dhaka, Bangladesh in 2007. He earned his Master (Feb 2010) and Doctoral degree (Feb 2013) from Seoul National University, South Korea under Prof. Chong-Su Cho and Prof. Cheol-Heui Yun. He developed several smart polymeric carriers to deliver gene (DNA, siRNA, microRNA) for cancer therapy and subunit antigens for vaccine therapy to prevent infectious diseases. He achieved several research awards for outstanding research performances include “Brain Korea 21 (BK21) Award” in 2012, the “Best Young Scientist Award” in 2013 from Seoul National University and some others in various scientific meetings. He has authored about 25 peer-reviewed articles and 3 of his research works have been issued/pending national and international patents. Currently, he is a Postdoctoral Fellow under Prof. Omid Farokhzad and Prof. Jinjun Shi at Harvard Medical School and Brigham & Women's Hospital (HMS/BWH), USA since Feb 2014, and working on developing mRNA nanotherapeutics for cancer immunotherapy & gene therapy, immunosenescence and asthma nanotherapeutics. Dr. Islam is a member of National Postdoctoral Association of USA and an Association of Young Biotechnologist of Bangladesh. He is also serving as a Co-Chair in the Communication Executive Committee of Postdoctoral Leadership Council of BWH at HMS. |
 Emma K. G. Reesor | Emma K. G. Reesor worked as a research assistant under the mentorship of Dr Mohammad Ariful Islam in the Omid Farokhzad and Jinjun Shi Laboratory at Harvard Medical School as a part of her Bachelor of Applied Sciences degree from the University of Waterloo, Canada. She is specialized in Nanotechnology Engineering, in the Departments of Chemistry, Chemical Engineering and Electrical Engineering of University of Waterloo, with a focus on Nanoscale Biosystems. |
 Yingjie Xu | Dr Yingjie Xu obtained her doctoral degree in the Department of Experimental Medicine at McGill University, under the supervision of Dr Moulay Alaoui-Jamali in 2011. After that, she joined Prof. Bruce Zetter's Laboratory as a Postdoctoral research fellow in the Vascular Biology Program at Boston Children's Hospital and Harvard Medical School. Yingjie's studies are focused on identifying the diagnostic and prognostic markers for cancer progression and chemoresistance and dissecting the underlying molecular mechanisms. |
 Harshal R. Zope | Dr Harshal R. Zope was born in Bhusawal, India on 26th Jan 1986. He pursued his interest in research by joining Institute of Biotechnology and Bioinformatics, University of Pune, India. In Pune University, he obtained a 5 years integrated M.Sc. degree in Biotechnology (2008) as well as M. Tech degree in Biotechnology (2009). Next, he pursued his interest by joining Leiden University for his PhD research under the supervision of Dr Alexander Kros (Feb 2010). During his PhD, he worked on peptide based functional biomaterials for various applications such as understanding membrane fusion, drug delivery and vaccine development. Currently, Harshal Zope is a Postdoctoral fellow at the Laboratory of Nanomedicine, Harvard Medical School/Brigham and Women's Hospital (HMS/BWH), Boston, USA. In the group of Prof. Omid Farokhzad and Prof. Jinjun Shi, he is developing a novel nanotechnology platform for HIV vaccine and cancer therapy. |
 Bruce R. Zetter | Dr Bruce Zetter is the Charles Nowiszewski Professor of Cancer Biology at Boston Children's Hospital and Harvard Medical School. He is highly regarded nationally and internationally as a leader in the research of tumor progression, cancer diagnosis, cancer metastasis, and tumor angiogenesis. His current research interests are focused on tumor metastasis, novel cancer treatments and on tests that predict future outcomes for cancer patients. Dr Zetter serves on numerous editorial boards of scientific journals, grant review panels for federal agencies and private foundations and scientific advisory boards of biotechnology companies. |
 Jinjun Shi | Dr Jinjun Shi holds an academic appointment as Assistant Professor at Harvard Medical School and directs the Laboratory for Nanoengineering & Drug Delivery in the Department of Anesthesiology at Brigham and Women's Hospital. He is also a research affiliate at Harvard-MIT Division of Health Sciences & Technology. His research involves an interdisciplinary combination of biomaterials, drug delivery, nanomedicine, and immunotherapy. Dr Shi has developed many multifunctional nanoparticle systems for the delivery of chemotherapeutics, proteins, vaccines, and nucleic acids, one of which has led to the first-in-human clinical trial of a synthetic nanoparticle vaccine for smoking cessation and relapse prevention. He has authored more than 30 papers, and holds over 25 issued/pending national and international patents. He has also received many awards, such as the NIH K99/R00 Career Development Award, the AACR Scholar-in-Training Award, the Movember-PCF Challenge Award, and the PCF Young Investigator Award. |
Introduction
Messenger RNA (mRNA), a natural biomolecule, is a transient entity that mediates the translation of genetic information from genes encoded in DNA to proteins located throughout the cell. The physical and temporal qualities of mRNA have allowed its use as a safe genetic material for gene-based therapy which does not require genomic integration.1,2 It is suitable for this use because of its potential to avoid nuclear localization and also because it allows rapid protein expression even in non-dividing and hard-to-transfect cells (e.g. dendritic cells and macrophages). These properties make mRNA an attractive molecule for immunotherapy.3 Another advantage of mRNA as a genetic element is its predictable, consistent protein expression kinetics, especially compared to DNA transfection which follows random onset time courses.4–7 However, relative to the technologies developed for DNA delivery, mRNA delivery strategies still require much more improvement and testing. Fortunately some of the carriers and biomaterials established for DNA and small interfering RNA (siRNA) have also been exhibiting a promising foundation for the development of mRNA delivery technologies.mRNA has been investigated for more than half a century, but its widespread applications in medical research and in the development of novel therapeutic modalities have been limited due to its perceived instability, susceptibility to degradation, insufficient translatability and immunostimulatory effects.8–10 Such challenges have partially been resolved thanks to an improved understanding of the structure of mRNA and its relationship to mRNA stability, as well as the development of a variety of chemical modification methods.10–15 These breakthroughs have subsequently facilitated the synthesis of mRNA with many different structural modifications (e.g., anti-reverse cap analogues (ARCA), 3′-globin UTR and poly-A tail), which still possess functional activity for immunotherapy and gene-based therapy. With these advances in stability and functionality, the use of mRNA as a therapeutic tool is now becoming a reality.14
Nevertheless, like other nucleic acids (e.g., DNA and siRNA), naked mRNA cannot readily cross the cell membrane on its own and thus requires delivery systems to enhance its cell permeation.16 Viral vectors have been used as mRNA carriers but may suffer from their potential immunologic side effects and toxicity as well as the vector-size limitations.1,17 Non-viral strategies such as electroporation, gene gun and sonoporation have been more thoroughly investigated as mRNA delivery systems.16,18,19 However, the ex vivo manipulation of cells with mRNA transfection using such approaches, while feasible, is highly laborious, expensive and overall ill-suited for extensive applications.20–22
Biomaterials represent an important step forward from the aforementioned non-viral strategies and have demonstrated promising potential for the delivery of various biomacromolecules such as DNA and siRNA.1,23 Compared to viral vectors and ex vivo technologies mentioned above, biomaterials are more biocompatible and diversified, and can be easily formulated for effective in vivo delivery and controlled release of therapeutics. Recently, biomaterials have also attracted considerable attention for mRNA delivery.5,24,25 For example, protamine has demonstrated remarkable abilities to ameliorate the transfection capabilities of mRNA and several protamine–mRNA complexes are now under clinical trials in cancer patients.26,27 Moreover, biomaterials-based nanoparticle platforms have in recent years been gradually applied to mRNA vaccine development and mRNA-based gene therapy.1,28,29
In this review, we summarize the strategies for chemical modification of mRNA, provide an overview of the currently available biomaterials and nanotechnology platforms for mRNA delivery and a critical analysis of how these mRNA delivery systems may be further improved to potentiate their therapeutic utility and discuss the challenges and opportunities in this exciting field.
mRNA modification for clinical translation
Owing primarily to its instability, mRNA was originally considered to be unsuitable for use as a therapeutic molecule, despite the fact that research on mRNA delivery into cells was pioneered over three decades ago.30,31 Recent advances in molecular and structural biology, along with substantial progress in the understanding of mRNA biology and its degradation mechanism, have promoted the development of various chemical modification methods to improve mRNA stability and translation capacity.8,32 Below, we briefly outline the various modifications of mRNA's structure, including different capping techniques and elements that can be included to improve stability.In eukaryotic cells, mature mRNA is generally comprised of five distinct portions (Fig. 1a): (i) a cap structure, (ii) a 5′ untranslated region (5′ UTR), (iii) an open reading frame (ORF), (iv) a 3′ untranslated region (3′ UTR) and (v) a poly(A) tail (a tail of 100–250 adenosine residues).33,34 With recent progress in molecular biology techniques, in vitro transcription is commonly utilized to produce functional mRNA using a bacteriophage promoter.5 During in vitro mRNA transcription it has been observed that almost half of the caps are oriented in reverse which makes them unrecognizable to the cap-binding protein.35,36 Anti-reverse cap analogs (ARCAs) were developed to solve this problem. These are modifications in which OCH3 is used to replace or remove natural 3′ OH cap groups to avoid inappropriate cap orientation (Fig. 1b and c).11 It was found, moreover, that modification in the C2′ position, as well as the C3′ position, can also prevent inappropriate cap orientation.12 Additional modifications of ARCA structure have been reported for the purpose of improving the efficiency and stability of mRNA.5 For example, tetraphosphate ARCAs are reported to improve translation efficiency relative to that of other cap analogs,12 as well as phosphorothioate ARCAs, which confer hydrolysis resistance to mRNA, thus increasing translational stability.13
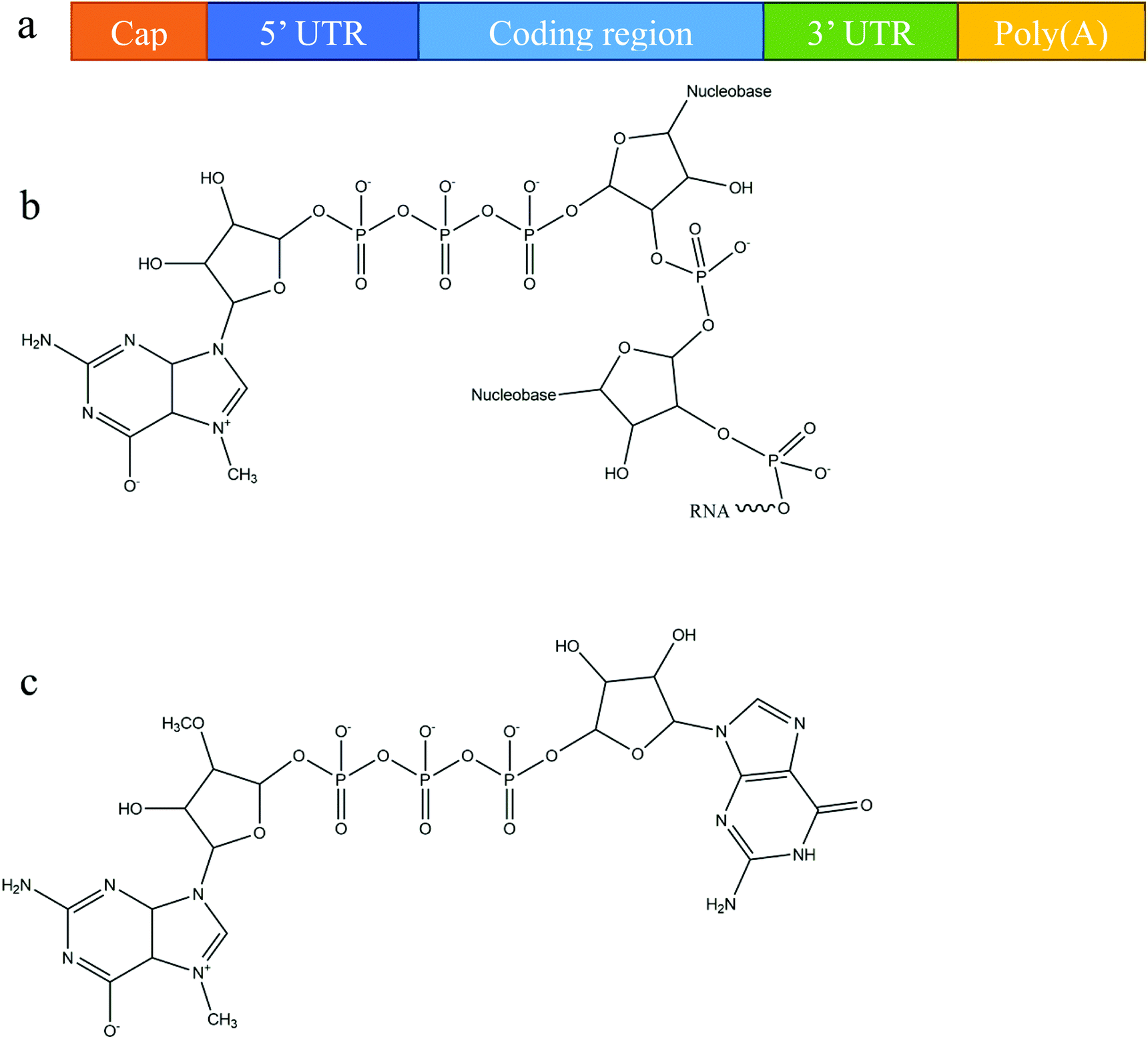 | ||
| Fig. 1 mRNA structural features: (a) a basic structure of a eukaryotic mRNA with five distinct components, (b) the cap structure of the 5′ end of eukaryotic mRNA, and (c) the structure of anti-reverse cap analogues (ARCA): 3′-O-Me-m7G(5′)ppp(5′)G. | ||
Polyadenylation, the addition of a poly adenine (A) tail to the 3′ end of mRNA, is catalyzed by poly(A) polymerase and is part of the process that produces mature mRNA in eukaryote cells. The poly(A) tail acts as the binding site for poly(A)-binding protein (PABP) which is exported with mRNA from nucleus to cytoplasm where it further binds to and recruit proteins that facilitate translation, such as translation initiation factor 4F (eIF4F).37 The poly(A) tail makes the mRNA molecule more stable as shortening or removal of the poly(A) tail accelerates mRNA degradation via enhanced exonucleotide digestion.38E. coli poly(A) polymerase (E-PAP) I has been optimized to add a poly(A) tail of at least 150 adenines to the 3′ terminal of in vitro transcribed mRNA. In some reports, instead of enzymatically attached poly(A) tail, in vitro transcribed RNA generated from plasmid templates consists of coding sequencing and a poly(A) tail with 30 to 120 nucleotides in length.39–42 The additional adenine residues confer stability to the in vitro transcribed capped mRNA and may increase its translational efficiency.43 Where direct comparisons have been made, longer poly(A) tails >100 adenines have generally been found to be preferable to smaller ones.40,41,44
The widely studied adenylate–uridylate rice elements (AREs) are important mRNA decay signals in the 3′ untranslated regions (3′ UTRs) of most eukaryotic mRNAs. mRNAs which contain AREs demonstrate reduced stability, perhaps due to the removal of the poly(A) tail. Stability is increased, however, when AREs are replaced with the 3′ UTR of a stable mRNA species such as β-globin mRNA.45 Iron responsive elements (IREs) represent an additional type of 3′ UTR which affect mRNA stability depending on their precise location in mRNA structure. In this case, the mRNA half-life increases when IREs occupy the 3′ UTR, whereas when that are located at the 5′ position, mRNA translational ability is improved.46
The multiple means of modulating structural elements of the mRNA, including the 5′ cap, 5′- and 3′-UTRs, the coding region, and the poly(A) tail, improve the intracellular stability and translational efficiency of in vitro transcribed mRNA. Although the degree of improvement for modulation of in vitro transcribed mRNA appears to depend on the means of modification, the cell type and cell differentiation state, reports range from modest levels of improvement to 2–3 orders of magnitude,12,40–42,47 which leads to the significantly higher protein expression and prolonged persistence of the protein from a range of a few minutes to longer than 1 week.40,47,48 Moreover, the incorporation of chemically modified nucleotides, such as substitution of cytidine triphosphate and uridine triphosphate with naturally occurring 5-methylcytidine and pseudouridine (ψ) triphosphate, respectively, into mRNA further suppresses the immune-stimulating property of in vitro transcribed mRNA.10
It is also noteworthy to clearly distinguish the different immune-stimulating effects that mRNA can have. First, there is an inherent response of the body to foreign, non-self mRNA. This is the type of response that the body uses to prevent threats such as RNA viruses, through the endosomal recognition.49 These RNA molecules can be encountered through immune sensors such as Toll-like receptors (TLRs).50 Some of the aforementioned mRNA modifications are specifically designed to reduce this innate immune response to minimize inherent host response. Second, the delivered therapeutic mRNA molecules can induce the desired immune response through stimulating appropriate immune cells and presentation of the specific antigen. This can also be referred to as the immune-stimulating effect of the mRNA, but in this case is a designed antigen-specific effect as opposed to an inherent reaction to foreign materials. It is also important to mention that the innate immunity is not preferable for mRNA-based gene therapy applications, where we only need to transfer mRNA into the cells and facilitate the expression of the protein of interest for therapeutic purpose. The results of these actions will be henceforth discussed with a variety of mRNA molecules and delivery methods.10,51–53
Naked mRNA, in different modified forms, has already been tested in clinical settings. The direct intradermal injections of naked mRNA were applied as a phase I/II non-randomized clinical trial in patients with stage IV renal cell cancer.54 In this study, granulocyte-macrophage colony stimulating factor (GM-CSF) was used as an adjuvant with mRNA encoding several tumor-associated antigens including mucin 1, carcinoembryonic, human epidermal growth factor receptor 2, telomerase, survivin and melanoma-associated antigen 1. Results revealed that vaccinations were well tolerated with no severe side effects and induced clinical responses.54 Recently, CureVac has developed self-adjuvanted two-component mRNA vaccines comprised of both free mRNA and protamine-complexed mRNA for the treatment of castration-resistant prostate cancer and stage IIIB/IV non-small cell lung cancer. They have demonstrated excellent safety as well as high levels of cellular immunity with T and B cell responses in several clinical trials.55–59 Moderna is another company that has been investigating mRNA as a vaccine and therapeutic technology, with two ventures Onkaido Therapeutics and Valera actively exploring the discovery and development of mRNA-based treatments in oncology and infectious diseases, respectively. Some representative studies using modified naked mRNA are summarized in Table 1.
| Category | Stage | mRNA | Activity and efficiency | Ref. |
|---|---|---|---|---|
| Naked mRNA | C57BL/6 mice | Luciferase and human CEA | After intramuscular injection mice demonstrated anti-CEA antibody response | 119 |
| CAF1 (H-2a) mice | S1509a | Intradermal injection induced immunity in tumors | 120 | |
| Mice | Chloramphenicol acetyltransferase and luciferase | After intramuscular injection CAT activity was detected and, in a separate experiment, a dose–response effect with luciferase was determined | 121 | |
| Genetically engineered mice (C57BL6/CFW background) | Human VEGF-A | After intramyocardial injection there was improved heart function and long-term survival because of the directed differentiation of endogenous heart progenitors | 122 | |
| Patients with stage IIIB/IV non-small cell lung cancer | MAGE-C1, MAGE-C2, NY-ESO-1, BIRC5, 5T4 | No dose limiting toxicity and good safety profile, along with significant induction of T and B cell responses in 65% of patients. | 59 | |
| Naked mRNA with GM-CSF | Patients with stage IV renal cell cancer | MUC1, CEA, Her-2/neu, telomerase, survivin and MAGE-A1 | The intradermal mRNA injections resulted in clinical response without any severe side effects and induced CD4+ and CD8+ T cell responses for the specific antigens. | 54 |
| Patients with stage III and IV metastatic melanoma | Autologous mRNA | Intradermal injection; safe and feasible; increase in antitumor humoral immune response in some patients; | 24 |
Whereas naked modified mRNA shows feasibility in their application as vaccines, several challenging issues remain elusive, such as enzymatic degradation, rapid elimination by renal excretion or by the mononuclear phagocyte system (MPS), as well as poor cellular uptake and endosomal escape, in particular for systemic mRNA delivery. Hence, to achieve more efficient mRNA delivery, a great number of biomaterials and nanoparticle platforms have been developed to protect mRNA from nuclease degradation and facilitate its cytosolic delivery for improved protein expression (Fig. 2 and 3). In the remainder of this review, we focus on recent advances in the delivery of mRNA to cells and tissues by means of contemporary biomaterials and nanotechnologies, including protamine complexes, lipid nanoparticles, polymeric nanoparticles, lipid–polymer hybrid nanoparticles, and gold nanoparticles.
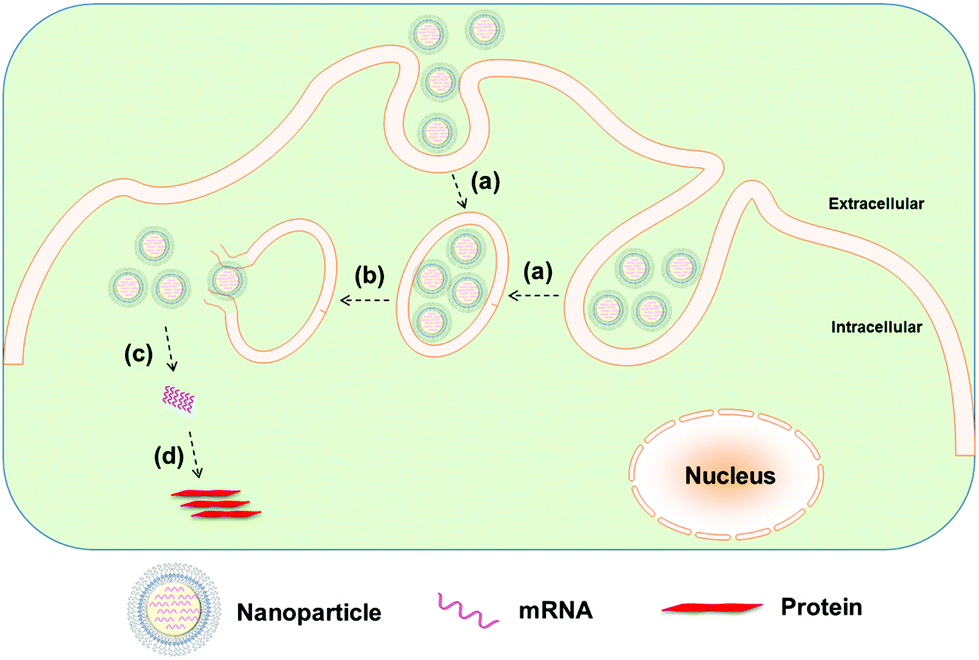 | ||
| Fig. 2 Nanoparticle-mediated delivery of mRNA: (a) cellular internalization of nanoparticles into endosome, (b) endosomal escape, (c) release of mRNA from nanoparticles, and (d) mRNA translation to protein without genomic integration. | ||
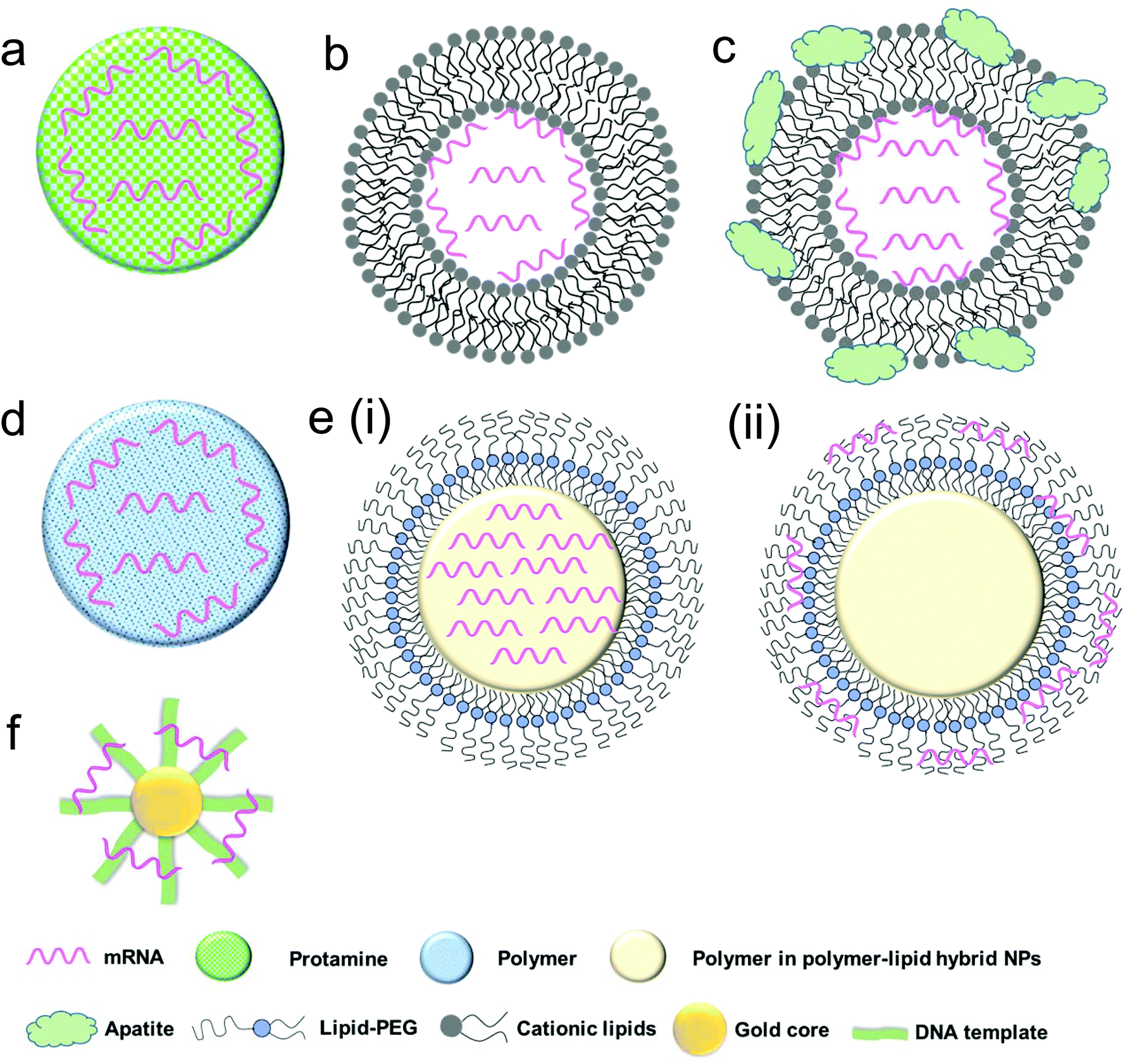 | ||
| Fig. 3 Schematic representation of various biomaterial-based systems for mRNA delivery: (a) protamine–mRNA complex; (b) lipid nanoparticle; (c) lipid nanoparticle with inorganic compounds (e.g. apatite); (d) cationic polymeric nanoparticle; (e) lipid–polymer hybrid nanoparticles including (i) mRNA–polymer complex core surrounded by a lipid shell and (ii) polymer core surrounded by a lipid shell with mRNA absorbed onto the surface; and (f) gold nanoparticle. | ||
Protamine–mRNA complexes
Protamine is a natural cationic protein, which gives it an excellent ability to complex nucleic acid including mRNA (Fig. 3a) and provides increased uptake of mRNA and transfection capabilities.60 It has been shown that protamine can effectively complex with mRNA and these complexes can act as a danger signal that activates murine cells through a MyD88-dependent pathway involving TLR7 and TLR8.60 Hoerr et al. demonstrated that these complexes degrade within 2 h when incubated in serum, which limits their abilities for survival in the circulation in vivo.53 Nevertheless, they later showed that even partially degraded mRNA–protamine complexes can still exhibit immunostimulatory activity for over 100 h.60 Moreover, it was found that protamine–mRNA complexes strongly activated a variety of white blood cells (such as granulocytes, B cells and NK cells) and overall significantly stimulated immune response compared to that of protamine–DNA systems.53,60 In a clinical study by Weide et al. the efficacy of this system was demonstrated by testing intradermally administered protamine-complexed mRNAs coding for Melan-A (a melanoma antigen), Tyrosinase (an enzyme which catalyzes the production of melanin), gp100 (a protein involved in melanosome maturation), Mage-A1 (a melanoma antigen), Mage-A3 (a melanoma antigen) and Survivin (a protein involved in the regulation of apoptosis) metastatic melanoma patients.26 No side effects greater than grade II (mild to moderate) were observed and overall there was a complete clinical response, including significant effect on the frequency of immunosuppressive cells and increase of antigen-specific T cells.26Recently, CureVac has also explored the use of protamine-complexed mRNA. It was found in an in vivo study that a two-component mRNA vaccine composed of both free and protamine-complexed mRNA resulted in successful antigen expression and immune stimulation, which were mediated by Toll-like receptor 7.51 A balanced adaptive immunity with both cellular (T cell-mediated) and humoral immunity was achieved which not only showed prophylactic activity but also exhibited therapeutic efficacy against tumor. CureVac has also put forward a clinical trial for patients with castrate-resistant prostate cancer using mRNA-encoding for prostate-specific antigen (PSA), prostate-specific membrane antigen (PSMA), prostate stem cell antigen (PSCA) and six transmembrane epithelial antigen of the prostate 1 (STEAP1).27 After intradermal vaccination, 80% of the subjects demonstrated immune response to the delivered mRNA antigen and 60% against multiple antigens, which ultimately correlated with longer survival.27 The currently investigated protamine–complexed mRNA systems and their therapeutic efficacy are summarized in Table 2.
| Category | Platform | Stage | mRNA | Activity and efficiency | Ref. |
|---|---|---|---|---|---|
| Abbreviations: (N,N-[bis(2-hydroxyethyl)]-N-[2,3-bis(tetradecanoyloxy) propyl] ammonium chloride (DMDHP); 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP); 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE); 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[biotinyl(polyethylene glycol)-2000] (ammonium salt); 1,2-distearoy-phosphatidylethanolamine-polyethylene glycol (DSPE-PEG); 1,2-distearoy-phosphatidylethanolamine-polyethylene glycol-anisamide (DSPE-PEG-AA); Antigen-presenting cell (APC); Anti-Reverse Cap Analog (ARCA); Bone-marrow-derived dendritic cell (BM-DC); Carcinoembryonic antigen (CEA); Cluster of differentiation 70 (CD70); Cluster of differentiation 86 (CD86); C–X–C chemokine receptor type 4 (CXCR-4); Cytomegalovirus (CMV); Cytotoxic T cell (CTL); Dendritic cell (DC); Diethylaminoethyl methacrylate (DEAEMA) and butyl methacrylate (BMA); Diethylaminoethyl methacrylate (DMAEMA); Enhanced green fluorescent protein (EGFP); Granulocyte-macrophage colony stimulating factor (GM-CSF); Green fluorescent protein (GFP); Human bronchial epithelia cell line (BEAS-2B); Human epidermal growth factor receptor 2 (Her-2/neu); Interleukin 12 (IL-12); Interleukin 6 (IL-6); Melanoma-associated antigen 1 (MAGE-A1); Melanoma-associated antigen family C1 (MAGE-C1); Melanoma-associated antigen family C2 (MAGE-C2); Mucin 1 (MUC1); New York esophageal squamous cell carcinoma 1 (NY-ESO-1); N-[1-(2,3-dioleyloxy)propyl]-N,N,N-trimethylammonium chloride (DOTMA); Oligo(ethylene glycol) methyl ether methacrylate (OEGMA); Ovalbumin (OVA); Peripheral blood mononuclear cell (PBMC); Poly(ethylene glycol) methyl ether methacrylate (PEGMA); Polyethylenimine (PEI); Prostate-specific antigen (PSA); Prostate-specific membrane antigen (PSMA); Prostate stem cell antigen (PSCA); Six transmembrane epithelial antigen of the prostate 1 (STEAP1); Toll-like receptor (TLR); Tumor necrosis factor alpha (TNF-α); Tumor-associated antigen (TAA); Vascular endothelial growth factor-A (VEGF-A); β-Galactosidase (β-gal); Trophoblast glycoprotein (5T4). | |||||
| Protamine–mRNA complex | Protamine complexed β-gal mRNA | HeLa-Kb cells injected into B6 (H2b) mice and BALB/c mice | β-Gal and GFP | Successful CTL response, dependent on injection site | 53 |
| Human PBMC | β-Gal, EGFP or CMV pp65 | Complexes triggered strong IL-6 and TNF-α release, activation of innate immune system and other APCs | 60 | ||
| Murine BM-DC | β-Gal or CMV pp65 | Stimulated mouse BM-DC: release of IL-6 and IL-12 and up-regulation of CD86 | 123 | ||
| Protamine complexed Melan-A, Tyrosinase, gp100, Mage-A1, Mage-A3, and Survivin mRNA | Patients with metastatic melanoma | Melan-A, Tyrosinase, gp100, Mage-A1, Mage-A3, and Survivin | Increased frequency of immunosuppressive and vaccine-directed T cells | 26 | |
| CureVac®: A two-component mRNA vaccine composed of free mRNA and mRNA complexed with protamine | BALB/c and C57BL/6 mice | OVA (GgOVA), control (Ecβ-gal sh), PSMA (HsPSMA) and STEAP (HsSTEAP) | Demonstrated antitumor effects after intravenous injection; activated the adaptive and innate immune systems | 51 | |
| Ability to inhibit established tumors | |||||
| CureVac® (CV9201) | Patients with stage IIIB/IV non-small cell lung cancer | MAGE-C1, MAGE-C2, NY-ESO-1, survivin and 5T4 | This is a phase I/IIa clinical trial | 58 | |
| CureVac® (CV9202) | Patients with stage IV non-small cell lung cancer | NY-ESO-1, MAGEC1, MAGEC2, 5 T4, Survivin, and MUC1 | mRNA vaccines potentially induced immune response to antigens expressed in tumors | 57 | |
| CureVac® (CV9202) in combination with radiotherapy | Patients with stage IV non-small cell lung cancer | NY-ESO-1, MAGEC1, MAGEC2, 5 T4, Survivin, and MUC1 | 19 patients have been recruited thus far, recruitment finished by the end of 2014 | 57 | |
| CureVac® (CV9103) | Patients with castrate-resistant prostate cancer | PSA, PSCA, PSMA and STEAP1 | Intradermal injections; maximum tolerated dose not defined; activation of TLR7 | 27,55 | |
| Antigen-specific T-cells were detected in 79% of patients and 58% of the immunological responders reacted against multiple antigens; | |||||
| Increased antigen-specific T-cells and antigen-unspecific B cells in 74% of patients | |||||
| Safe, well-tolerated and induces high levels of cellular immunogenicity | |||||
| CureVac® (CV9104) | Patients with metastatic castrate-refractory prostate cancer | mRNA-encoding six antigens (this is an advancement of CV9103 encoding two more antigens) | This is a randomised, double-blind, placebo-controlled, Phase I/II trial scheduled to be finished in 2016. | 27,124 | |
| Lipid nanoparticle | DOTAP liposomes covered with apatite nanoparticles | HeLa | Luciferase | Together with ARCA (over cap) had over 100-fold improvement compare to DOTAP, percentage not determined | 78 |
| HeLa | Luciferase | 9–14 better than mRNA liposome alone, percentage not determined | 79 | ||
| NIH 3T3 | |||||
| HeLa | Luciferase | DOTAP-apatite outperformed DOTAP alone and Lipofectamine 2000, percentage not determined | 80 | ||
| HUVEC | |||||
| Fibronectin complexed to DOTAP liposomes covered with apatite nanoparticles | HeLa | Luciferase | Fn-DOTAP-apatite more than 50-times better than DOTAP, percentage not determined | 61 | |
| DOTAP/DOPE/DSPE-PEG-2000-biotin | Primary murine bone marrow-derived DC from C57BL/6 mice | TriMix mRNA encoding CD40-ligand,TLR4 and CD70 | 19% | 19 | |
| DOTAP/DOPE/DSPE-PEG-2000-biotin lipoplex loaded microbubbles | DC primary cultures from the bone marrow of C57BL/6 mice | Luciferase | 24% | 21 | |
| DOTMA | NIH 3T3 | Luciferase | Percentage not determined, at least 20–30% RNase resistant | 31 | |
| Lipofectamine 2000 and TransIT | Neurospheres from subventricular zone of adult | EGFP | 40–50% | 125 | |
| C57BL/6 mice | |||||
| MLRI/DOPE and TransFast | CHO | GFP and luciferase | >50% | 126 | |
| NIH 3T3 | >45% | ||||
| Novel cationic lipids: X2, S1, S2, S3, 2X3 and 2D3 with DOPE (helper lipid) | DC cells cultured from the bone marrow of C57BL/6 mice | EGFP and B-16 | Up to 47% of DC progenitors | 127 | |
| Up to 57% of immature DCs | |||||
| DOTAP/cholesterol liposome with DSPE-PEG and DSPE-PEG-AA, encapsulating protamine/mRNA cores | NCI-H460 xenograft | Herpes simplex virus 1-thymidine kinase (HSV1-tk) | 68–78% | 10 | |
| DOTAP/DOPE | HeLa | Luciferase, GFP and CXCR4 | ∼80% | 92 | |
| Stemfect | JAWS II | Luciferase and GFP | 80% | 128 | |
| DC2.4 | Luciferase and GFP | >97% | |||
| Human primary DCs | Luciferase and GFP | >50% | |||
| Murine primary DCs | Luciferase and GFP | >60% | |||
| Polymeric nanoparticle | Linear PEI | BEAS-2B | Luciferase | 5-Fold higher than Lipo 2000 | 70 |
| Poly(DMAEMA-co-OEGMA) | BEAS-2B | Luciferase | Same level as Lipo 2000 | ||
| Poly(DMAEMA) with PEG | BEAS-2B | Luciferase | 3-Fold improvement after PEGylation | ||
| Branched PEI (25 kDa) | PC3 | GFP | ∼30% | 93 | |
| Linear PEI | HeLa | Luciferase, GFP and CXCR4 | 40% | 92 | |
| Branched PEI (2 kDa) conjugated to melittin, with chloroquine | HeLa | GFP | 52.2% | 91 | |
| HUVEC | 71.6% | ||||
| Triblock copolymer (comprising DMAEMA, PEGMA, DEAEMA and BMA) | DC2.4 | EGFP and OVA | 50% | 28 | |
| RAW264.7 | 77% | ||||
| Polymer-lipid hybrid nanoparticle | Poly-(β-amino ester) core, phospholipid bilayer shell | DC2.4 | Luciferase and GFP | 30% | 106 |
| Mannosylated histidylated lipopolyplexes | DC2.4 | EGFP and MART-1 | 60% | 71 | |
| DMDHP | CHO | Luciferase and GFP | 70–80% | 129 | |
| Gold nanoparticle | DNA oligonucleotide-conjugated gold nanoparticle | HeLa | BAX | Fluorescent signal uniformly detected, percentage not determined. | 29 |
| HepG2 | |||||
Lipid nanoparticles
The field of lipid nanoparticles for nucleic acid delivery is relatively mature compared to other nanotechnologies. As such, quite a variety of different lipid formulations have been tested for mRNA delivery. Cationic lipids are used ubiquitously owing to their favourable electrostatic interactions with negatively charged mRNA to form nanoparticles (Fig. 3b). This field was pioneered in 1989 with a study of the use of DOTMA (N-[1-(2,3-dioleyloxy)propyl]-N,N,N-trimethylammonium chloride) to transfect human, rat, mouse, xenopus (frog) and drosophila cells with luciferase mRNA.31 The clinical development of such cationic lipid tools has however been hampered by their toxicity.31 Despite these concerns, DOTMA remains a commonly used material, along with DOTAP (1,2-dioleoyloxy-3-trimethylammonium propane chloride).61 An important note about these cationic lipids is that while positively charged lipid nanoparticles can be effective in vitro, in vivo results are less promising because cationic liposomes can be quickly eliminated by the mononuclear phagocyte system (MPS).The coating of poly(ethylene) glycol (PEG) layer on lipid carriers has been widely used for the delivery of nucleic acid payloads, such as siRNA62 and DNA,63 to improve the formulation process, reduce aggregation and increase the blood circulation time.64–67 However, the surface PEGylation has also been shown to decrease cellular uptake, an effect which may be minimized by optimizing PEG size and content.63,68,69 PEG modification has also been applied to nanoparticle-based mRNA delivery, particularly to polymer nanoparticles70 and lipid–polymer hybrid nanoparticles,71,72 both of which will be discussed in later sections. The application of PEGylation to lipid-based mRNA delivery systems to improve their in vivo efficacy remains elusive. Another strategy is the incorporation of a helper lipid such as 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) to reduce aggregation of the lipid systems (as well as to improve endosomal escape).73–76
To further enhance mRNA transfection potency in both mitotic and non-mitotic cells, Akaike and co-workers developed a different approach by coating inorganic carbonate apatite nanoparticles on liposomal carriers (Fig. 3c). It was evident that these inorganic additives helped to increase mRNA uptake through effective endocytosis. They also demonstrated that decorating mRNA-containing DOTAP-apatite particles with RGD, which is known for its ability to bind integrins,77 enhanced cytoplasmic expression of delivered mRNA. This method is versatile and holds a great potential due to its abilities for targeting and efficient delivery of mRNA.61,78–80
More recently, a novel lipid/protamine/mRNA nanoparticle platform was proposed and comprehensively explored for systemic delivery to tumors.10 In this study DOTAP liposomes were used to encapsulate mRNA–protamine complexes and then coated with DSPE-PEG and DSPE-PEG-anisamide.10 These particles demonstrated stability against degradation in serum, high in vitro transfection ability in NCI-H460 cells, low cytotoxicity (even at a concentration 50 times higher than the dose they found to induce effective transfection), accumulation in tumor site and anticancer action in vivo.10 Together this approach presents a very promising hybrid system for the systemic delivery of mRNA. Currently, Tekmira Pharmaceuticals is also investigating lipid nanoparticles for systemic mRNA delivery to the liver.
Until now, lipid nanoparticles have been widely utilized, particularly to introduce mRNA to immune cells for vaccine purposes. The detailed overview of currently available lipid-based nanoparticles for mRNA delivery was presented in several recent reports,3,16,25,81 and a summary of these advances is presented in Table 2. As the lipid nanoparticles represent a more thoroughly explored system, their further development and optimization may open up the foundation for the creation of more effective mRNA delivery systems.
Polymeric nanoparticles
Polymeric nanoparticles have emerged as effective delivery vehicles for a variety of payloads, such as DNA,82 siRNA,83 mRNA,28 proteins,84 chemotherapeutic agents85 and others. To date, cationic polymers have been used primarily because of their favourable electrostatic interactions with negatively charged nucleic acids (Fig. 3d) and cell membranes. As compared with other delivery vehicles such as liposomes, multiple parameters must be considered in their design. Pack et al. enumerated some of these in their recent report in which they emphasized optimization with respect to synthesis of mRNA carriers, transfection and endosomal escape capabilities.86 Additionally, polymers to be used for pharmaceutical purposes should be biocompatible, non-toxic and non-immunogenic.Generally, polymeric nanoparticles are prepared by nanoprecipitation87,88 or emulsion techniques.89,90 When properly optimized, these polymeric carriers may serve as a highly functional delivery vehicle for mRNA. The surfaces can be easily modified with other materials such as ligands to confer targeting abilities and other polymers to afford properties such as pH-responsive release for tuning the release profile.91 The particles themselves must overcome extra- and intracellular barriers to ensure that the mRNA is available for protein expression (Fig. 2). Additionally, specific to polymeric particles, the charge and size of the polymers used must be carefully considered since the binding strength between the mRNA and polymer is a key factor in mRNA expression efficiency.91
Among various polymeric carriers for gene delivery, polyethylenimine (PEI) (Fig. 4a) is a classic material and is always used as a standard control in nucleic acid transfection. Rejman et al. explored the use of linear PEI polyplexes in HeLa cells and found moderate mRNA transfection efficiency of approximately 40% using C–X–C chemokine receptor (CXCR) type 4. This was exceeded by a cationic lipid formulation (DOTAP/DOPE) (Fig. 4b and c).92 However, concerns regarding PEI include non-degradability and potential toxicity (depending on the molecular weight and type of PEI). Recently, a few different studies have investigated the efficiency of modified PEI as an mRNA delivery system with high transfection efficiency and reduced toxicity. Read et al. explored the use of reducible polycations with histidine and polylysine residues (HIS-RPCs) and, with their optimised formulations, found transfection efficiencies of over 90% in human prostate cancer cells (PC3).93 In contrast, they found that nanoparticles made of unmodified branched PEI (25 kDa) were only able to achieve efficiencies of about 30%.93 Bettinger et al. also investigated the transfection activity of PEI (branched but with low molecular weight of 2 kDa) along with another well-known cationic polymer, poly(L-lysine) (Fig. 4d). In this study, they included additive compounds to increase transfection capability of PEI and, here, they found that the polymer in the presence of chloroquine (an agent to aid endosomolysis) and in conjugation to melittin (a membrane-active peptide) significantly increased the transfection efficiency.91 With these additives, transfection efficiencies of 52.2% and 71.6% were achieved in HeLa and HUVEC cells, respectively, and were greater than with the cationic lipid (DOTAP) alone.91 The effect of molecular weight of PEI was also investigated with the conclusion that overall higher MW polymers had better endosomolytic activity but bound mRNA too tightly for it to be released.91 These studies and their conflicting results also bring forward the important note that the efficacy and utility of a polymer candidate can be markedly improved through the use of additives and tuning of the MW and size of the particle.
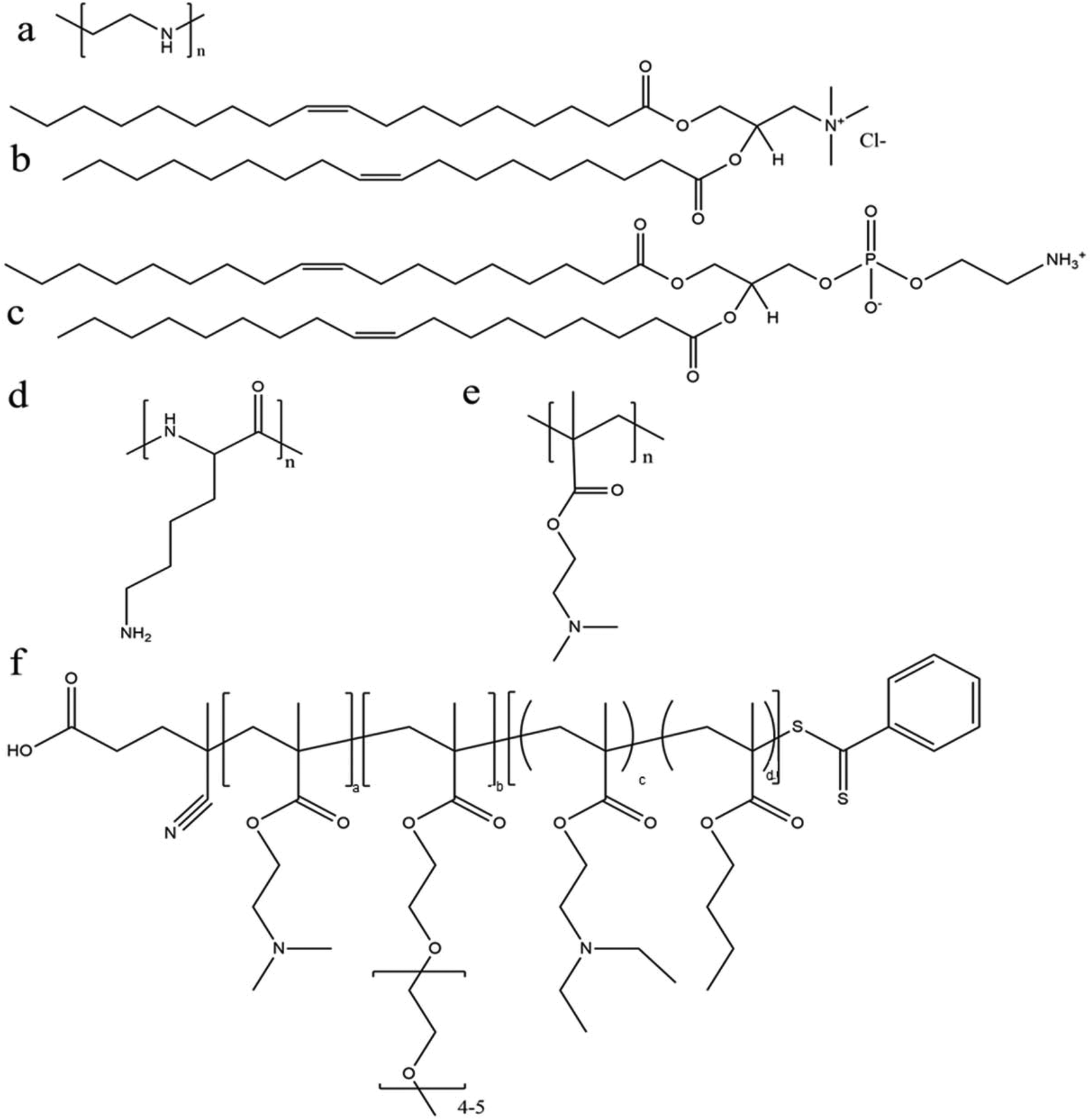 | ||
| Fig. 4 Chemical structure of some representative polymers and lipids used for mRNA delivery: (a) polyethylenimine (PEI); (b) 1,2-dioleoyl-3-trimethylammonium-propane (chloride salt) (DOTAP); (c) 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE); (d) poly(L-lysine) (PLL); (e) poly(2-(dimethylamino)ethyl methacrylate) (p(DMAEMA)); and (f) poly(DMAEMA-PEGMA-DEAEMA-co-BMA). | ||
The effect of PEGylation on the polymer–mRNA binding efficacy and mRNA transfection abilities was also investigated. It was demonstrated that adding PEG side chains to poly(N,N-diethylaminoethyl methacrylate) (p(DMAEMA)) (Fig. 4e) increased its ability to complex mRNA and tendency to form monodispersed particles, as compared to their unmodified variants as well as linear PEI (used as a positive control).70 Nonetheless, linear PEI displayed the highest mRNA transfection capabilities and the PEGylated mRNA nanoparticle exhibited significantly higher transfection capability over its non-PEGylated counterpart.70 Moreover, through testing with an influenza-peptide 7 (an endolysosomal release peptide), it was found that the PEGylated copolymer further supported the endosomal release of their mRNA payload and improved transfection efficiency,70 suggesting further investigation into this polymer chemistry may be fruitful.
With rapid advances in polymer chemistry, a vast variety of other cationic polymers have been synthesized and applied to mRNA delivery. Recently a novel triblock polymer has been developed (Fig. 4f) using: (i) diethylaminoethyl methacrylate (DMAEMA) to promote mRNA condensation, (ii) poly(ethylene glycol) methyl ether methacrylate (PEGMA) to enhance stability and biocompatibility and (iii) a copolymer of diethylaminoethyl methacrylate (DEAEMA) and butyl methacrylate (BMA) to facilitate cytosolic entry.28 The triblock polymer nanoparticles were ranged in size from 86 to 216 nm after complexation with mRNA and achieved transfection efficiency of up to 77% in RAW264.7 macrophage cells and 50% in DC2.4 dendritic cells, compared with approximately 30% in both cell lines where the commercial transfection agent Lipofectamine 2000 was used.28 A further study was performed using a number of differing formulations made of the similar three polymers and found that the optimal arrangement was DMAEMA-PEGMA-DEAEMA-co-BMA, which provided an ideal balance between stability and charge shielding.28 With this arrangement, the copolymers showed decreased cytotoxicity in vitro with the decreasing of molecular weights for the second (PEGMA) block.28
The use of polymeric nanoparticles for mRNA delivery is still in its infancy but has shown great potential. The field of polymeric mRNA delivery began with the use of diethylaminoethyl-dextran30 and has now evolved to rival the abilities of many well-established lipid systems. Various novel polymeric carriers as mRNA delivery systems are summarized in Table 2. The future of this field largely depends on the further discovery and optimization of more efficient polymers to improve mRNA transfection efficiency, as well as safety and biocompatibility.
Lipid–polymer hybrid nanoparticles
The lipid–polymer hybrid nanoparticle platform has several potential advantages when compared with either lipid- or polymer-based nanoparticles for the delivery of therapeutic compounds including small molecules, peptides, proteins and oligonucleotides, and has gained significant attention in last decade.94–98 Structurally, the core of the nanoparticle is made up of a polymeric material coated with lipids and/or lipid–PEGs which can enhance the nanoparticle stability and pharmacokinetics, and confer surface tunable properties.94,99–101 Alternatively, the properties of the core can be tuned in order to respond to external stimuli which can help to facilitate endosomal escape.71,102,103 We have also recently reported the development of several such systems which exhibited sustained delivery of siRNA and enhanced gene silencing.104,105 Lipid–polymer hybrid nanoparticle systems have also been employed as delivery vehicles for mRNA.71,72,102There are two structural strategies which have been employed to design lipid–polymer hybrid nanoparticles for mRNA delivery. In the first method, negatively charged mRNA is complexed with the cationic polymer and then the surface can be decorated with lipids or lipid–PEG conjugates (Fig. 3e (i)).10,106 In the second method, mRNA is adsorbed to the surface of the cationic nanoparticle core (Fig. 3e (ii)).107 Using both methods, mRNA delivery can be achieved; however release profiles may vary and it has been hypothesized by Su et al.102 that surface adsorbed mRNA has a faster release profile. They also showed that adsorbed mRNA has increased stability compared to naked mRNA which suggests that even if the mRNA is on the surface of nanoparticle, degradation can still be reduced due to the complexation.
To increase endosomal mRNA delivery, various pH-responsive polymers have been used. Upon endocytosis, changes in pH cause a conformational change in the polymer which results in osmotic shock followed by endosome disruption. For example, a PEGylated derivative of histidylated polylysine and L-histidine-(N,N-di-n-hexadecylamine)ethylamide liposomes (termed as ‘histidylated lipopolyplexes’) was prepared for an mRNA based cancer vaccine in which polylysine was used to complex mRNA, and histidylation contributed in charge switching at endosomal pH condition due to the imidazole group with pI of 6. Additionally, they found that mannosylation of this delivery system significantly enabled targeted mRNA delivery to dendritic cells through interaction with the mannose receptor.71 The PEGylated histidylated mRNA lipopolyplexes were also efficient in inducing an anti-B16 specific cellular immune response and conferring protection against B16F10 melanoma in mice.72 In another report, poly-β-amino ester (PBAE), developed by Su et al., also used pH-responsiveness to disrupt endosomal membrane.106 The PBAE polymer was coated with lipids containing DOTAP which was used to adsorb mRNA on the particle as described earlier. Both of the pH-responsive systems have great potential for developing an effective delivery system for mRNA.102
A summary of the advances on lipid–polymer hybrid nanoparticles is presented in Table 2. As of now it is difficult to postulate which is the most efficient method for mRNA delivery since there is a lack of sufficient studies to compare different mRNA delivery systems and determine the optimum one. Evidently, there is a compelling need to investigate various delivery options and establish quick optimization methods to screen a large number of mRNA delivery systems at once. This would accelerate the developmental progress of this highly promising biomedical research field.
Gold nanoparticles
Another type of nanoparticle delivery system for mRNA is gold nanoparticle-DNA oligonucleotide (AuNP–DNA) conjugates (Fig. 3f).29 This system has demonstrated significant transfection activity in HeLa (cervical carcinoma) and HepG2 (hepatocyte carcinoma) cells (detected using confocal fluorescent microscopy). The nanoparticles were tested in vivo using direct injection into xenograft tumors and the results showed that the (AuNP–DNA conjugate)–mRNA nanoparticles were able to induce the production of biologically functionalized Bcl-2-associated X (BAX) protein,29 suggesting a promising start for this mRNA nanoparticle formulation. While gold nanoparticles have not been extensively studied for mRNA delivery, their promising results with other nucleic acids (e.g., siRNA and DNA) indicate that they have significant potential for the delivery of mRNA.Indeed, the gold nanoparticle-oligonucleotide system has been more widely studied with DNA and siRNA.108–110 This system has demonstrated significant cellular uptake in a number of cell lines.109,111–113 Recently, the mechanism behind the uptake of gold oligonucleotide particles was further elucidated. It has been shown that their uptake is highest in serum-free conditions and the membrane proteins responsible for the uptake of these particles are the scavenger receptors.111 Other delivery systems based upon gold nanoparticles include gold nanoparticles passivated with BSA-SV40 large T antigen conjugates,114 layer-by-layer gold nanoparticles with polymers for the delivery of siRNA115,116 and plasmid DNA delivery using carbon dot-gold nanoparticles conjugated with PEI.117 These exciting techniques and technologies all may have the potential to be applied to mRNA delivery, demonstrating that the field of gold nanoparticle-mediated mRNA transport is rich with new possibilities.
Conclusion
mRNA-based vaccine and gene therapy technologies are innovative, promising and rapidly emerging strategies in biomedical research for the purpose of treating acquired and congenital diseases. Although naked mRNA has prospective clinical efficacy, this field faces various challenges such as the delivery issues, targeting administration and short-term gene expression, all of which are critical drawbacks and could be the major limitations to translate this field to widespread clinical application. The rapid advancements in biomaterials and nanotechnology could significantly help overcome these obstacles. This review has receptively presented the progress of cutting-edge biomaterials and nanotechnology platforms to improve the efficacy of mRNA delivery. While further improvements are necessary, it is encouraging to see that some of the mRNA-based treatment strategies24,26,118 have already entered the clinical trial stage within a short time following their development. More efforts are still required to find novel biomaterials/nanoparticles and optimized formulations which can provide high transfection activity and biocompatibility with minimal carrier-specific toxicity/immunostimulation, high selectivity and specificity, and effective systemic in vivo delivery and prolonged protein expression (particularly for gene therapy). With the advancement and proper design of biomaterials, it is expected that mRNA technology will be of high interest in clinical applications for years to come.Acknowledgements
This work was supported by the National Institutes of Health (NIH) grants R00CA160350 (J. S.) and R01CA37393 (B. R. Z.), and the Movember-PCF Challenge Award (J. S.) and PCF Young Investigator Award (J. S.). Y. X. received DoD PCRP Postdoctoral Training Award (W81XWH-14-1-0268).References
- H. Yin, R. L. Kanasty, A. A. Eltoukhy, A. J. Vegas, J. R. Dorkin and D. G. Anderson, Nat. Rev. Genet., 2014, 15, 541–555 CrossRef CAS PubMed.
- E. S. Quabius and G. Krupp, Nat. Biotechnol., 2015, 32, 229–235 CAS.
- R. P. Deering, S. Kommareddy, J. B. Ulmer, L. A. Brito and A. J. Geall, Expert Opin Drug Deliv., 2014, 11, 885–899 CrossRef CAS PubMed.
- J. Lee, D. Boczkowski and S. Nair, Methods Mol. Biol., 2013, 969, 111–125 CAS.
- A. Yamamoto, M. Kormann, J. Rosenecker and C. Rudolph, Eur. J. Pharm. Biopharm., 2009, 71, 484–489 CrossRef CAS PubMed.
- C. Leonhardt, G. Schwake, T. R. Stogbauer, S. Rappl, J. T. Kuhr, T. S. Ligon and J. O. Radler, Nanomedicine, 2014, 10, 679–688 CrossRef CAS PubMed.
- T. S. Ligon, C. Leonhardt and J. O. Radler, PLoS One, 2014, 9, e107148 Search PubMed.
- X. Wu and G. Brewer, Gene, 2012, 500, 10–21 CrossRef CAS PubMed.
- E. Grudzien-Nogalska, J. Kowalska, W. Su, A. N. Kuhn, S. V. Slepenkov, E. Darzynkiewicz, U. Sahin, J. Jemielity and R. E. Rhoads, Methods Mol. Biol., 2013, 969, 55–72 CAS.
- Y. Wang, H. H. Su, Y. Yang, Y. Hu, L. Zhang, P. Blancafort and L. Huang, Mol. Ther., 2013, 21, 358–367 CrossRef CAS PubMed.
- J. Stepinski, C. Waddell, R. Stolarski, E. Darzynkiewicz and R. E. Rhoads, RNA, 2001, 7, 1486–1495 CAS.
- J. Jemielity, T. Fowler, J. Zuberek, J. Stepinski, M. Lewdorowicz, A. Niedzwiecka, R. Stolarski, E. Darzynkiewicz and R. E. Rhoads, RNA, 2003, 9, 1108–1122 CrossRef CAS.
- E. Grudzien-Nogalska, J. Jemielity, J. Kowalska, E. Darzynkiewicz and R. E. Rhoads, RNA, 2007, 13, 1745–1755 CrossRef CAS PubMed.
- J. Peng, E. L. Murray and D. R. Schoenberg, Methods Mol. Biol., 2008, 419, 215–230 CAS.
- M. S. Kormann, G. Hasenpusch, M. K. Aneja, G. Nica, A. W. Flemmer, S. Herber-Jonat, M. Huppmann, L. E. Mays, M. Illenyi, A. Schams, M. Griese, I. Bittmann, R. Handgretinger, D. Hartl, J. Rosenecker and C. Rudolph, Nat. Biotechnol., 2011, 29, 154–157 CrossRef CAS PubMed.
- G. Tavernier, O. Andries, J. Demeester, N. N. Sanders, S. C. De Smedt and J. Rejman, J. Controlled Release, 2011, 150, 238–247 CrossRef CAS PubMed.
- C. E. Thomas, A. Ehrhardt and M. A. Kay, Nat. Rev. Genet., 2003, 4, 346–358 CrossRef CAS PubMed.
- M. D. Mattozzi, M. J. Voges, P. A. Silver and J. C. Way, J. Vis. Exp., 2014, 86, e51234 Search PubMed.
- H. Dewitte, S. Van Lint, C. Heirman, K. Thielemans, S. C. De Smedt, K. Breckpot and I. Lentacker, J. Controlled Release, 2014, 194, 28–36 CrossRef CAS PubMed.
- S. Van Meirvenne, L. Straetman, C. Heirman, M. Dullaers, C. De Greef, V. Van Tendeloo and K. Thielemans, Cancer Gene Ther., 2002, 9, 787–797 CrossRef CAS PubMed.
- M. L. De Temmerman, H. Dewitte, R. E. Vandenbroucke, B. Lucas, C. Libert, J. Demeester, S. C. De Smedt, I. Lentacker and J. Rejman, Biomaterials, 2011, 32, 9128–9135 CrossRef CAS PubMed.
- H. Dewitte, S. Van Lint, C. Heirman, K. Thielemans, S. C. De Smedt, K. Breckpot and I. Lentacker, J. Controlled Release, 2014, 194c, 28–36 CrossRef PubMed.
- R. Kanasty, J. R. Dorkin, A. Vegas and D. Anderson, Nat. Mater., 2013, 12, 967–977 CrossRef CAS PubMed.
- B. Weide, J. P. Carralot, A. Reese, B. Scheel, T. K. Eigentler, I. Hoerr, H. G. Rammensee, C. Garbe and S. Pascolo, J. Immunother., 2008, 31, 180–188 CrossRef CAS PubMed.
- P. Midoux and C. Pichon, Expert Rev. Vaccines, 2015, 14, 221–234 CrossRef CAS PubMed.
- B. Weide, S. Pascolo, B. Scheel, E. Derhovanessian, A. Pflugfelder, T. K. Eigentler, G. Pawelec, I. Hoerr, H. G. Rammensee and C. Garbe, J. Immunother., 2009, 32, 498–507 CrossRef CAS PubMed.
- K. J. Kallen, U. Gnad-Vogt, B. Scheel, G. Rippin and A. Stenzl, J. Immunother. Cancer, 2013, 1, 1–1 CrossRef.
- C. Cheng, A. J. Convertine, P. S. Stayton and J. D. Bryers, Biomaterials, 2012, 33, 6868–6876 CrossRef CAS PubMed.
- J. H. Yeom, S. M. Ryou, M. Won, M. Park, J. Bae and K. Lee, PLoS One, 2013, 8, e75369 CAS.
- G. Koch, Curr. Top. Microbiol. Immunol., 1973, 62, 89–138 CAS.
- R. W. Malone, P. L. Felgner and I. M. Verma, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 6077–6081 CrossRef CAS.
- M. Strenkowska, J. Kowalska, M. Lukaszewicz, J. Zuberek, W. Su, R. E. Rhoads, E. Darzynkiewicz and J. Jemielity, New J. Chem., 2010, 34, 993–1007 RSC.
- A. K. Banerjee, Microbiol. Rev., 1980, 44, 175–205 CAS.
- R. J. Jackson, Cell, 1993, 74, 9–14 CrossRef CAS.
- A. E. Pasquinelli, J. E. Dahlberg and E. Lund, RNA, 1995, 1, 957–967 CAS.
- A. Cai, M. Jankowska-Anyszka, A. Centers, L. Chlebicka, J. Stepinski, R. Stolarski, E. Darzynkiewicz and R. E. Rhoads, Biochemistry, 1999, 38, 8538–8547 CrossRef CAS PubMed.
- L. Weill, E. Belloc, F. A. Bava and R. Mendez, Nat. Struct. Mol. Biol., 2012, 19, 577–585 CAS.
- D. Schwartz, C. J. Decker and R. Parker, RNA, 2003, 9, 239–251 CrossRef CAS.
- V. F. Van Tendeloo, P. Ponsaerts, F. Lardon, G. Nijs, M. Lenjou, C. Van Broeckhoven, D. R. Van Bockstaele and Z. N. Berneman, Blood, 2001, 98, 49–56 CrossRef CAS PubMed.
- S. Holtkamp, S. Kreiter, A. Selmi, P. Simon, M. Koslowski, C. Huber, O. Tureci and U. Sahin, Blood, 2006, 108, 4009–4017 CrossRef CAS PubMed.
- M. Mockey, C. Goncalves, F. P. Dupuy, F. M. Lemoine, C. Pichon and P. Midoux, Biochem. Biophys. Res. Commun., 2006, 340, 1062–1068 CrossRef CAS PubMed.
- U. Sahin, K. Kariko and O. Tureci, Nat. Rev. Drug Discovery, 2014, 13, 759–780 CrossRef CAS PubMed.
- G. J. Cao and N. Sarkar, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 10380–10384 CrossRef CAS.
- A. C. Beckel-Mitchener, A. Miera, R. Keller and N. I. Perrone-Bizzozero, J. Biol. Chem., 2002, 277, 27996–28002 CrossRef CAS PubMed.
- G. Shaw and R. Kamen, Cell, 1986, 46, 659–667 CrossRef CAS.
- R. D. Klausner, T. A. Rouault and J. B. Harford, Cell, 1993, 72, 19–28 CrossRef CAS.
- K. J. Kallen and A. Thess, Therapeutic Advances in Vaccines, 2014, vol. 2, pp. 10–31 Search PubMed.
- K. Kariko, A. Kuo and E. Barnathan, Gene Ther., 1999, 6, 1092–1100 CrossRef CAS PubMed.
- S. S. Diebold, T. Kaisho, H. Hemmi, S. Akira and C. Reis e Sousa, Science, 2004, 303, 1529–1531 CrossRef CAS PubMed.
- S. R. Nallagatla, R. Toroney and P. C. Bevilacqua, RNA Biol., 2008, 5, 140–144 CrossRef CAS.
- M. Fotin-Mleczek, K. M. Duchardt, C. Lorenz, R. Pfeiffer, S. Ojkic-Zrna, J. Probst and K. J. Kallen, J. Immunother., 2011, 34, 1–15 CrossRef CAS PubMed.
- M. Fotin-Mleczek, K. Zanzinger, R. Heidenreich, C. Lorenz, A. Thess, K. M. Duchardt and K. J. Kallen, J. Gene Med., 2012, 14, 428–439 CrossRef CAS PubMed.
- I. Hoerr, R. Obst, H. G. Rammensee and G. Jung, Eur. J. Immunol., 2000, 30, 1–7 CrossRef CAS.
- S. M. Rittig, M. Haentschel, K. J. Weimer, A. Heine, M. R. Muller, W. Brugger, M. S. Horger, O. Maksimovic, A. Stenzl, I. Hoerr, H. G. Rammensee, T. A. Holderried, L. Kanz, S. Pascolo and P. Brossart, Mol. Ther., 2011, 19, 990–999 CrossRef CAS PubMed.
- H. Kubler, T. Maurer, A. Stenzl, S. Feyerabend, U. Steiner, M. Schostak, W. Schultze-Seemann, F. Vom Dorp, L. Pilla and G. Viatali, J. Clin. Oncol., 2011, 29, 4535 Search PubMed.
- S. Rausch, C. Schwentner, A. Stenzl and J. Bedke, Hum. Vaccin. Immunother., 2014, 10, 3146–3152 CrossRef PubMed.
- M. Sebastian, A. Papachristofilou, C. Weiss, M. Fruh, R. Cathomas, W. Hilbe, T. Wehler, G. Rippin, S. D. Koch, B. Scheel, M. Fotin-Mleczek, R. Heidenreich, K. J. Kallen, U. Gnad-Vogt and A. Zippelius, BMC Cancer, 2014, 14, 748 CrossRef PubMed.
- ClinicalTrials.gov, Trial of an RNActive®-Derived Cancer Vaccine in Stage IIIB/IV Non Small Cell Lung Cancer (NSCLC), https://clinicaltrials.gov/ct2/show/NCT00923312?term=cV9201&rank=1, (accessed 06 April, 2015).
- M. Sebastian, L. V. Boehmer, A. Zippelius, F. Mayer, M. Reck, D. Atanackovic, M. Thomas, F. Schneller, S. W. Jan, E. Goekkurt, H. Bernhard, A. Groeschel, B. Scheel, S. D. Koch, T. Lander, G. Rippin, V. Wiegand, U. S. Gnad-Vogt, K. J. Kallen and A. Knuth, J. Clin. Oncol., 2012, 2012, 30 Search PubMed , Abstract: 2573.
- B. Scheel, R. Teufel, J. Probst, J. P. Carralot, J. Geginat, M. Radsak, D. Jarrossay, H. Wagner, G. Jung, H. G. Rammensee, I. Hoerr and S. Pascolo, Eur. J. Immunol., 2005, 35, 1557–1566 CrossRef CAS PubMed.
- F. T. Zohra, Y. Maitani and T. Akaike, Biol. Pharm. Bull., 2012, 35, 111–115 CAS.
- Y. Bao, Y. Jin, P. Chivukula, J. Zhang, Y. Liu, J. Liu, J. P. Clamme, R. I. Mahato, D. Ng, W. Ying, Y. Wang and L. Yu, Pharm. Res., 2013, 30, 342–351 CrossRef CAS PubMed.
- P. Harvie, F. M. Wong and M. B. Bally, J. Pharm. Sci., 2000, 89, 652–663 CrossRef CAS.
- J. Heyes, K. Hall, V. Tailor, R. Lenz and I. MacLachlan, J. Controlled Release, 2006, 112, 280–290 CrossRef CAS PubMed.
- M. C. Woodle, Adv. Drug Delivery Rev., 1998, 32, 139–152 CrossRef CAS.
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2005, 4, 145–160 CrossRef CAS PubMed.
- M. L. Immordino, F. Dosio and L. Cattel, Int. J. Nanomedicine, 2006, 1, 297–315 CrossRef CAS PubMed.
- H. Hatakeyama, H. Akita, K. Kogure, M. Oishi, Y. Nagasaki, Y. Kihira, M. Ueno, H. Kobayashi, H. Kikuchi and H. Harashima, Gene Ther., 2007, 14, 68–77 CrossRef CAS PubMed.
- L. Y. Song, Q. F. Ahkong, Q. Rong, Z. Wang, S. Ansell, M. J. Hope and B. Mui, Biochim. Biophys. Acta, 2002, 1558, 1–13 CrossRef CAS.
- S. Uzgun, G. Nica, C. Pfeifer, M. Bosinco, K. Michaelis, J. F. Lutz, M. Schneider, J. Rosenecker and C. Rudolph, Pharm. Res., 2011, 28, 2223–2232 CrossRef PubMed.
- F. Perche, T. Benvegnu, M. Berchel, L. Lebegue, C. Pichon, P. A. Jaffres and P. Midoux, Nanomedicine, 2011, 7, 445–453 CrossRef CAS PubMed.
- M. Mockey, E. Bourseau, V. Chandrashekhar, A. Chaudhuri, S. Lafosse, E. Le Cam, V. F. Quesniaux, B. Ryffel, C. Pichon and P. Midoux, Cancer Gene Ther., 2007, 14, 802–814 CrossRef CAS PubMed.
- D. Hirsch-Lerner, M. Zhang, H. Eliyahu, M. E. Ferrari, C. J. Wheeler and Y. Barenholz, Biochim. Biophys. Acta, 2005, 1714, 71–84 CrossRef CAS PubMed.
- L. Wasungu, M. C. Stuart, M. Scarzello, J. B. Engberts and D. Hoekstra, Biochim. Biophys. Acta, 2006, 1758, 1677–1684 CrossRef CAS PubMed.
- H. Farhood, N. Serbina and L. Huang, Biochim. Biophys. Acta, 1995, 1235, 289–295 CrossRef.
- L. Wasungu and D. Hoekstra, J. Controlled Release, 2006, 116, 255–264 CrossRef CAS PubMed.
- S. E. D'Souza, M. H. Ginsberg and E. F. Plow, Trends Biochem. Sci., 1991, 16, 246–250 CrossRef.
- F. T. Zohra, E. H. Chowdhury, S. Tada, T. Hoshiba and T. Akaike, Biochem. Biophys. Res. Commun., 2007, 358, 373–378 CrossRef CAS PubMed.
- F. T. Zohra, E. H. Chowdhury, M. Nagaoka and T. Akaike, Anal. Biochem., 2005, 345, 164–166 CrossRef CAS PubMed.
- F. T. Zohra, E. H. Chowdhury and T. Akaike, Biomaterials, 2009, 30, 4006–4013 CrossRef CAS PubMed.
- K. K. Phua, S. K. Nair and K. W. Leong, Nanoscale, 2014, 6, 7715–7729 RSC.
- J. U. Menon, P. Ravikumar, A. Pise, D. Gyawali, C. C. Hsia and K. T. Nguyen, Acta Biomater., 2014, 10, 2643–2652 CrossRef CAS PubMed.
- H. Gul-Uludag, J. Valencia-Serna, C. Kucharski, L. A. Marquez-Curtis, X. Jiang, L. Larratt, A. Janowska-Wieczorek and H. Uludag, Leuk. Res., 2014, 38, 1299–1308 CrossRef CAS PubMed.
- M. C. Chen, K. Sonaje, K. J. Chen and H. W. Sung, Biomaterials, 2011, 32, 9826–9838 CrossRef CAS PubMed.
- A. A. Ranade, P. P. Bapsy, S. Nag, D. Raghunadharao, V. Raina, S. H. Advani, S. Patil, A. Maru, V. P. Gangadharan, C. Goswami, J. S. Sekhon, K. Sambasivaiah, P. Parikh, A. Bakshi and R. Mohapatra, Asia Pac. J. Clin. Oncol., 2013, 9, 176–181 CrossRef PubMed.
- D. W. Pack, A. S. Hoffman, S. Pun and P. S. Stayton, Nat. Rev. Drug Discovery, 2005, 4, 581–593 CrossRef CAS PubMed.
- L. Zhang, J. M. Chan, F. X. Gu, J. W. Rhee, A. Z. Wang, A. F. Radovic-Moreno, F. Alexis, R. Langer and O. C. Farokhzad, ACS Nano, 2008, 2, 1696–1702 CrossRef CAS PubMed.
- J. Wu, N. Kamaly, J. Shi, L. Zhao, Z. Xiao, G. Hollett, R. John, S. Ray, X. Xu, X. Zhang, P. W. Kantoff and O. C. Farokhzad, Angew. Chem., Int. Ed., 2014, 53, 8975–8979 CrossRef CAS PubMed.
- M. G. Nava-Arzaluz, E. Pinon-Segundo, A. Ganem-Rondero and D. Lechuga-Ballesteros, Recent Pat. Drug Delivery Formulation, 2012, 6, 209–223 CrossRef CAS.
- E. Pinon-Segundo, M. G. Nava-Arzaluz and D. Lechuga-Ballesteros, Recent Pat. Drug Delivery Formulation, 2012, 6, 224–235 CrossRef CAS.
- T. Bettinger, R. C. Carlisle, M. L. Read, M. Ogris and L. W. Seymour, Nucleic Acids Res., 2001, 29, 3882–3891 CrossRef CAS PubMed.
- J. Rejman, G. Tavernier, N. Bavarsad, J. Demeester and S. C. De Smedt, J. Controlled Release, 2010, 147, 385–391 CrossRef CAS PubMed.
- M. L. Read, S. Singh, Z. Ahmed, M. Stevenson, S. S. Briggs, D. Oupicky, L. B. Barrett, R. Spice, M. Kendall, M. Berry, J. A. Preece, A. Logan and L. W. Seymour, Nucleic Acids Res., 2005, 33, e86 CrossRef PubMed.
- L. Zhang, J. M. Chan, F. X. Gu, J.-W. Rhee, A. Z. Wang, A. F. Radovic-Moreno, F. Alexis, R. Langer and O. C. Farokhzad, ACS Nano, 2008, 2, 1696–1702 CrossRef CAS PubMed.
- S. Krishnamurthy, R. Vaiyapuri, L. Zhang and J. M. Chan, Biomater. Sci., 2015, 3, 923–936 RSC.
- C. Salvador-Morales, L. Zhang, R. Langer and O. C. Farokhzad, Biomaterials, 2009, 30, 2231–2240 CrossRef CAS PubMed.
- B. Mandal, H. Bhattacharjee, N. Mittal, H. Sah, P. Balabathula, L. A. Thoma and G. C. Wood, Nanomedicine, 2013, 9, 474–491 CrossRef CAS PubMed.
- S. D. Kong, M. Sartor, C. M. Hu, W. Zhang, L. Zhang and S. Jin, Acta Biomater., 2013, 9, 5447–5452 CrossRef PubMed.
- C. Salvador-Morales, L. Zhang, R. Langer and O. C. Farokhzad, Biomaterials, 2009, 30, 2231–2240 CrossRef CAS PubMed.
- D. E. Owens Iii and N. A. Peppas, Int. J. Pharm., 2006, 307, 93–102 CrossRef PubMed.
- B. T. Luk, R. H. Fang and L. Zhang, Theranostics, 2012, 2, 1117–1126 CrossRef CAS PubMed.
- X. Su, J. Fricke, D. G. Kavanagh and D. J. Irvine, Mol. Pharm., 2011, 8, 774–787 CrossRef CAS PubMed.
- C. Clawson, L. Ton, S. Aryal, V. Fu, S. Esener and L. Zhang, Langmuir, 2011, 27, 10556–10561 CrossRef CAS PubMed.
- J. Shi, Z. Xiao, A. R. Votruba, C. Vilos and O. C. Farokhzad, Angew. Chem., Int. Ed., 2011, 123, 7165–7169 CrossRef PubMed.
- J. Shi, Y. Xu, X. Xu, X. Zhu, E. Pridgen, J. Wu, A. R. Votruba, A. Swami, B. R. Zetter and O. C. Farokhzad, Nanomedicine, 2014, 10, 897–900 CrossRef CAS PubMed.
- X. Su, J. Fricke, D. G. Kavanagh and D. J. Irvine, Mol. Pharm., 2011, 8, 774–787 CrossRef CAS PubMed.
- D. M. Anderson, L. L. Hall, A. R. Ayyalapu, V. R. Irion, M. H. Nantz and J. G. Hecker, Hum. Gene Ther., 2003, 14, 191–202 CrossRef CAS PubMed.
- S. M. Ryou, S. Kim, H. H. Jang, J. H. Kim, J. H. Yeom, M. S. Eom, J. Bae, M. S. Han and K. Lee, Biochem. Biophys. Res. Commun., 2010, 398, 542–546 CrossRef CAS PubMed.
- N. L. Rosi, D. A. Giljohann, C. S. Thaxton, A. K. Lytton-Jean, M. S. Han and C. A. Mirkin, Science, 2006, 312, 1027–1030 CrossRef CAS PubMed.
- J. H. Kim, J. H. Yeom, J. J. Ko, M. S. Han, K. Lee, S. Y. Na and J. Bae, J. Biotechnol., 2011, 155, 287–292 CrossRef CAS PubMed.
- P. C. Patel, D. A. Giljohann, W. L. Daniel, D. Zheng, A. E. Prigodich and C. A. Mirkin, Bioconjugate Chem., 2010, 21, 2250–2256 CrossRef CAS PubMed.
- D. A. Giljohann, D. S. Seferos, W. L. Daniel, M. D. Massich, P. C. Patel and C. A. Mirkin, Angew. Chem., Int. Ed., 2010, 49, 3280–3294 CrossRef CAS PubMed.
- D. A. Giljohann, D. S. Seferos, P. C. Patel, J. E. Millstone, N. L. Rosi and C. A. Mirkin, Nano Lett., 2007, 7, 3818–3821 CrossRef CAS PubMed.
- J. A. Ryan, K. W. Overton, M. E. Speight, C. N. Oldenburg, L. Loo, W. Robarge, S. Franzen and D. L. Feldheim, Anal. Chem., 2007, 79, 9150–9159 CrossRef CAS PubMed.
- A. Elbakry, A. Zaky, R. Liebl, R. Rachel, A. Goepferich and M. Breunig, Nano Lett., 2009, 9, 2059–2064 CrossRef CAS PubMed.
- E. Zhao, Z. Zhao, J. Wang, C. Yang, C. Chen, L. Gao, Q. Feng, W. Hou, M. Gao and Q. Zhang, Nanoscale, 2012, 4, 5102–5109 RSC.
- J. Kim, J. Park, H. Kim, K. Singha and W. J. Kim, Biomaterials, 2013, 34, 7168–7180 CrossRef CAS PubMed.
- H. Kübier, A. Stenzl, W. Schultze-Seemann, F. vom Dorp, L. Pilla, C. Hampel, D. Jocham and K. Miller, Eur. J. Cancer, 2011, 47, S498–S499 CrossRef.
- R. M. Conry, A. F. LoBuglio, M. Wright, L. Sumerel, M. J. Pike, F. Johanning, R. Benjamin, D. Lu and D. T. Curiel, Cancer Res., 1995, 55, 1397–1400 CAS.
- R. D. Granstein, W. Ding and H. Ozawa, J. Invest. Dermatol., 2000, 114, 632–636 CrossRef CAS PubMed.
- J. A. Wolff, R. W. Malone, P. Williams, W. Chong, G. Acsadi, A. Jani and P. L. Felgner, Science, 1990, 247, 1465–1468 CAS.
- L. Zangi, K. O. Lui, A. von Gise, Q. Ma, W. Ebina, L. M. Ptaszek, D. Spater, H. Xu, M. Tabebordbar, R. Gorbatov, B. Sena, M. Nahrendorf, D. M. Briscoe, R. A. Li, A. J. Wagers, D. J. Rossi, W. T. Pu and K. R. Chien, Nat. Biotechnol., 2013, 31, 898–907 CrossRef CAS PubMed.
- B. Scheel, S. Braedel, J. Probst, J. P. Carralot, H. Wagner, H. Schild, G. Jung, H. G. Rammensee and S. Pascolo, Eur. J. Immunol., 2004, 34, 537–547 CrossRef CAS PubMed.
- ClinicalTrials.gov, Trial of RNActive®-Derived Prostate Cancer Vaccine in Metastatic Castrate-refractory Prostate Cancer, https://www.clinicaltrials.gov/ct2/show/NCT01817738?term=curevac&rank=2, (accessed 12 May, 2015).
- S. McLenachan, D. Zhang, A. B. Palomo, M. J. Edel and F. K. Chen, PLoS One, 2013, 8, e83596 Search PubMed.
- S. Zou, K. Scarfo, M. H. Nantz and J. G. Hecker, Int. J. Pharm., 2010, 389, 232–243 CrossRef CAS PubMed.
- O. O. Markov, N. L. Mironova, M. A. Maslov, I. A. Petukhov, N. G. Morozova, V. V. Vlassov and M. A. Zenkova, J. Controlled Release, 2012, 160, 200–210 CrossRef CAS PubMed.
- K. K. Phua, K. W. Leong and S. K. Nair, J. Controlled Release, 2013, 166, 227–233 CrossRef CAS PubMed.
- D. M. Anderson, L. L. Hall, A. R. Ayyalapu, V. R. Irion, M. H. Nantz and J. G. Hecker, Hum. Gene Ther., 2003, 14, 191–202 CrossRef CAS PubMed.
Footnote |
| † Present address: Nanotechnology Engineering Program, University of Waterloo, Waterloo, ON N2L 3G1, Canada. |
| This journal is © The Royal Society of Chemistry 2015 |