
Asymmetric α-oxyamination of aldehydes by synergistic catalysis of imidazolethiones and metal salts†
Xianrui Liang,
Na Li,
Xinlei Chen and
Weike Su*
Collaborative Innovation Center of Yangtze River Delta Region Green Pharmaceuticals, College of Pharmaceutical Sciences, Zhejiang University of Technology, Hangzhou 310014, China. E-mail: pharmlab@zjut.edu.cn; Tel: +86 57188320899
First published on 9th September 2014
Abstract
Novel and efficient imidazolethione catalysts combined with metal salts were successfully introduced to the asymmetric α-oxyamination of aldehydes. The desired products with high yields and good to excellent enantioselectivities were obtained via a one-pot oxidation–oxyamination reaction system.
Asymmetric organocatalysis play a pivotal role in the synthesis of a variety of chiral molecules. Among them, oxygen-bearing stereocenters, especially the enantioselective α-oxygenated carbonyl formations are one of the fundamental structural motifs in natural and synthetic products, such as taxol, bestai, and zaragozic acid.1 Many traditional methods including aminohydroxylations,2 olefin epoxydation3 and dihydroxylation4 exhibited good ability to provide these asymmetric oxygen-bearing stereocenters. Recently, the newly developed asymmetric α-oxidation of aldehydes as an important approach to form the enantioselective oxygen-bearing stereocenters has also been substantially studied in the presence of different catalytic systems.
MacMillan5 and Zhong6 reported the first direct α-oxidation of aldehydes using natural proline and nitrosobenzene in 2003. The catalytic enantioselective α-oxyamination of aldehydes using 2,2,6,6-tetramethylpiperidine-1-oxyl radical (TEMPO) as an electrophilic source of oxygen in the presence of imidazolidinone were reported by Sibi7 and MacMillan8 in their respective work in 2007 and 2010. In 2010, Kudo's group developed a bioinspired catalyst and used it to the asymmetric α-oxyamination of aldehydes via a tandem system with primary alcohol's oxidation.9 Maruoka used binaphthyl-based chiral amine catalysts and an oxoammonium salt instead of metal reagents to obtain stable α-oxy aldehydes with excellent enantioselectivity.10 Similar α-oxyamination reaction was also reported with using other catalyst systems, such as porous cross-linked polymers (PCPs) photocatalysts,11 TiO2 photocatalyst,12 or electrochemical oxidation.13 In 2012, MacMillan extended the scope of substrates and gained excellent enantioselectivity in low temperature using synergistic catalysis system of imidazolidinone and metal salts.14 Meanwhile, these asymmetric α-oxyamination aldehydes can be also obtained by many related multistep reactions15 and two of them were applied to the synthesis of oxylipins16 and pentoses17 successfully.
Although many of the reported organocatalysts show efficient in this asymmetric α-oxyamination of aldehydes, not all the substrates can be obtained with good enantioselectivity, and the yield still need to be improved via the one-pot reaction system. So new organocatalysts aimed to provide better yields and enantioselectivity with expanded substrate scopes are still worthwhile to investigate. In previous works,18 we had reported the successful application of newly developed chiral imidazolethiones catalysts (Fig. 1) in the enantioselective Friedel–Crafts reactions. The structure of thioamide was used to increase the rigidity of catalyst18 and the more ‘stiffer’ imidazolethiones were anticipated to help improve the stereoselectivity in reactions. Base on the good catalytic activity and applications of imidazolethiones, the synergistic catalysis of imidazolethiones and metal salts were introduced to the reactions of α-oxyamination of aldehydes in this work to provide another useful choice for asymmetric C–O bond formation. The possible synergistic catalytic mechanism was also discussed.
 | ||
| Fig. 1 Chiral imidazolethione catalysts. | ||
To evaluate the efficiency of catalysts, the reaction of 3-phenylpropanal 3a and TEMPO was used as a model reaction based on the reaction conditions reported in literature.7 The 3a and TEMPO in DMF were treated with a stoichiometric amount of ferric chloride and a catalytic amount of different imidazolethione catalysts, respectively. Reduction of product α-aminoxy aldehyde 4a to the primary alcohol 5a was performed to aid analysis and the results were presented in Table 1. Compared with imidizolidinone 1a (Table 1, entry 1), a better stereoselectivity were obtained in the presence of imidazolethione 2a (Table 1, entry 2). The imidazolethione catalyst 2b with a larger cyclohexyl group was also evaluated for its catalytic activity, however, nearly no product was observed (Table 1, entry 3). The preliminary experiment results indicate that catalyst imidazolethione 2a has a better catalytic efficiency against 1a and 2b. The (S)-configurations were the main products according to the HPLC data reported in the literature.7
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Time (h) | Yieldb (%) | eec (%) |
| a Reaction condition: 3a (1 mmol), TEMPO (2 mmol), catalyst (20 mol%), HBF4 (20 mol%), FeCl3 (1 equiv.), DMF (2 mL), NaBH4 (2 equiv.).b Isolated yield.c Enantiomeric excess determined by HPLC analysis.d Not determined. | ||||
| 1 | 1a | 3 | 80 | 68 |
| 2 | 2a | 3 | 75 | 73 |
| 3 | 2b | 3 | Trace | n.d.d |

With the optimized catalyst imidazolethione 2a in hand, the reaction conditions including solvents, additives acids and synergistic metal salts were further examined with results summarized in Table 2. Among DMF, CH2Cl2, acetone, i-PrOH, H2O (Table 2, entries 1–5) and a series of mixed solvents (Table 2, entries 6–10), the proportion of H2O affecting the yield of the reaction significantly and higher yield and good selectivity were obtained in H2O at room temperature (Table 2, entry 5). Then additive acids that help to activate the substrates were evaluated and results showed that imidazolethione 2a with trifluoroacetic acid gave the product in high yield and enantioselectivity (Table 2, entries 5, 11, 12). To further improve the enantio-selectivity, the reaction was performed at 0 °C and −10 °C by adding a certain amount of acetone to H2O to lower its freezing point, but the ee value was still unsatisfactory (Table 2, entries 13 and 14). The synergistic metal salts, including FeCl3, FeCl2, CuCl2, CuSO4 and CuCl were chosen to facilitate the catalytic system. And better enantioselectivity was obtained when 10 mol% CuCl2 was used at −20 °C (Table 2, entry 23). At room temperature, both CuCl2 and CuCl gave good yields and ee values (Table 2, entries 17 and 19) with reaction time at 3 h. Lower reaction temperatures were also investigated to obtain even better enantioselectivity albeit with somewhat longer reaction time. Curiously, differences were observed between the synergistic metal salts of Cu(I) and Cu(II). The ee value decreased from 85% to 72% when the temperature was down to −20 °C in the presence of CuCl (Table 2, entry 21). However, the good ee value was obtained with CuCl2 at −20 °C (Table 2, entry 22). High yield and enantioselectivity could also be observed when the amount of CuCl2 and TEMPO were decreased to 10 mol% and 1.5 equiv. (Table 2, entry 23).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent (v/v) | Acid | Metal | T (°C) | Yieldb (%) | eec (%) |
| a Reaction condition: 3a (1 mmol), TEMPO (2 mmol), 2a (20 mol%), HX (20 mol%), metal (1 equiv.), solvent (2 mL), NaBH4 (2 equiv.).b Isolated yield.c Enantiomeric excess determined by HPLC analysis.d 12 h.e 24 h.f 48 h.g CuCl2 (10 mol%), TEMPO (1.5 equiv.), 48 h. | ||||||
| 1 | DMF | TFA | FeCl3 | 25 | 78 | 76 |
| 2 | CH2Cl2 | TFA | FeCl3 | 25 | Trace | n.d. |
| 3 | Acetone | TFA | FeCl3 | 25 | 23 | n.d. |
| 4 | i-PrOH | TFA | FeCl3 | 25 | 15 | n.d. |
| 5 | H2O | TFA | FeCl3 | 25 | 85 | 75 |
| 6 | H2O–CH2Cl2 (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
TFA | FeCl3 | 25 | 85 | 74 |
| 7 | H2O–CH2Cl2 (1:1) |
TFA | FeCl3 | 25 | 53 | 65 |
| 8 | H2O–CH2Cl2 (1:4) |
TFA | FeCl3 | 25 | 8 | n.d. |
| 9 | H2O–acetone (4:1) |
TFA | FeCl3 | 25 | 83 | 70 |
| 10 | H2O–DMF (4:1) |
TFA | FeCl3 | 25 | 73 | 67 |
| 11 | H2O | HBF4 | FeCl3 | 25 | 80 | 73 |
| 12 | H2O | p-TSA | FeCl3 | 25 | 57 | 64 |
| 13d | H2O | TFA | FeCl3 | 0 | 80 | 79 |
| 14e | H2O–acetone (4:1) |
TFA | FeCl3 | −10 | 76 | 82 |
| 15 | H2O | TFA | FeCl2 | 25 | 83 | 72 |
| 16 | H2O | TFA | FeSO4 | 25 | 77 | 72 |
| 17 | H2O | TFA | CuCl2 | 25 | 89 | 80 |
| 18 | H2O | TFA | CuSO4 | 25 | 77 | 73 |
| 19 | H2O | TFA | CuCl | 25 | 83 | 85 |
| 20e | H2O–acetone (4:1) |
TFA | CuCl | −10 | 74 | 78 |
| 21f | H2O–acetone (2:1) |
TFA | CuCl | −20 | 65 | 72 |
| 22f | H2O–acetone (2:1) |
TFA | CuCl2 | −20 | 83 | 89 |
| 23g | H2O–acetone (2:1) |
TFA | CuCl2 | −20 | 80 | 90 |
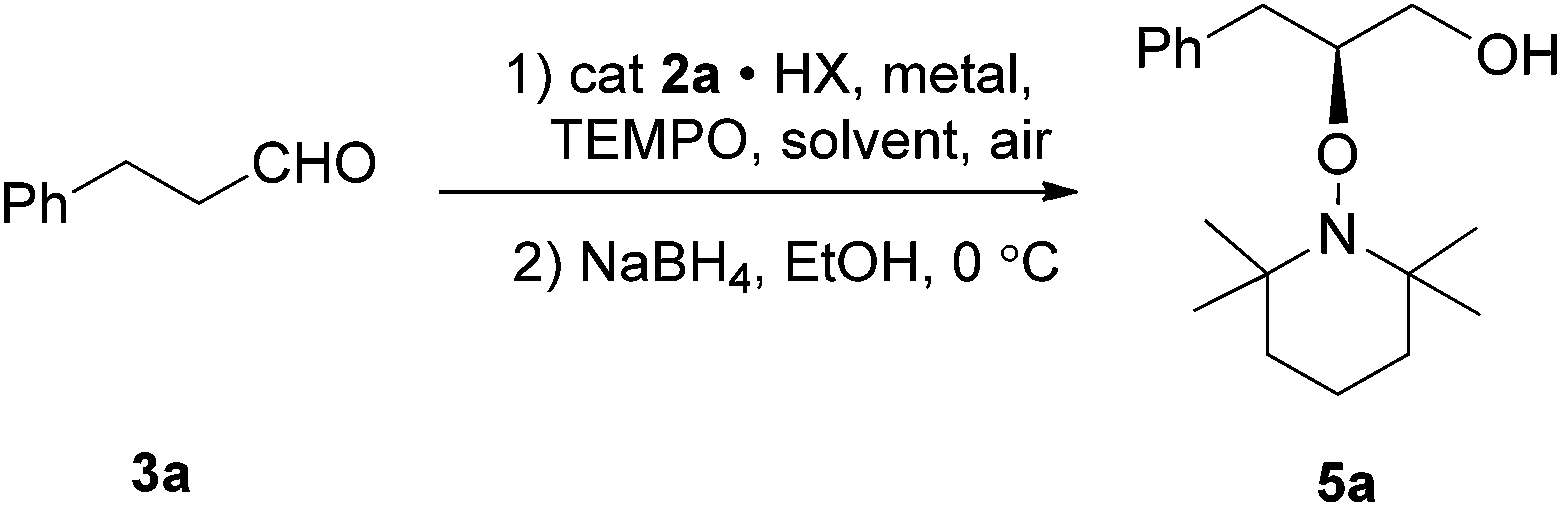
Allowed for the unstable properties of aldehydes and observed good yield and ee value in H2O–CH2Cl2 (Table 2, entry 6). We developed a one-pot reaction system under the optimal reaction conditions (Table 2, entry 23) including oxidation of primary alcohols, followed by the asymmetric α-oxyamination of aldehydes, and then the NaBH4 reduction of aldehydes to corresponding alcohols as shown in Table 3. The TEMPO and NaClO system was chosen as oxidizing agent in the first step due to its mild reaction condition and high conversion rates. The oxidation–oxyamination and reduction reactions of alcohols 3 provided products 5 in good yields and with high to excellent enantioselectivities except 3b. It was observed that the reaction of 2-phenylethanol 3b provided the product in very high yield but showed very low enantioselectivity with 4% ee, which might have resulted from the rapid racemization of phenylacetaldehyde. Both aromatic and aliphatic alcohols were applied in these sequential reaction systems. When 4-phenyl-1-butanol 3c was used as substrate, the product 5c can be obtained in good yield and enantioselectivity. This was the case with several long-chain and short-chain aliphatic alcohols 3d, 3e and 3f. It was worth noting that the yields of product 5g and 5h were decreased when the γ-position substituted alcohols were used. It might be due to the presence of the steric groups hinder the attack of TEMPO. Olefin, cyclohexyl, ester containing alcohols 3h, 3i and 3j were also confirmed to be the competent substrates, which yielded the corresponding products in good yields and excellent enantioselectivity. Overall, the above results demonstrate that this one-pot sequential reaction system with optimized reaction conditions catalyzed by imidazolethione 2a can be applied to a broad substrate scope.


There existed two mainly differential opinions about the mechanism of asymmetric α-oxyamination of aldehydes. In 2007, Sibi7 proposed a singly occupied molecular orbital (SOMO) activation mechanism. In 2010, MacMillan and his co-workers8 revealed the reaction proceeded via a more traditional enamine addition pathway. Based on the enamine addition mechanism8,19 and experimental results in this work, we proposed a synergistic catalysis mechanism (Scheme 1). The catalyst imidazolethione coupled with Cu(II) activated aldehydes and TEMPO in their own catalytic cycles, respectively. The two activated intermediates narrowed the energy gap and facilitated reaction. Firstly, aldehyde was activated by chiral imidazolidinone catalysts and formed the enamine intermediate I. Simultaneously, TEMPO as another substrate was activated by Cu(II) and formed the Cu(II)–TEMPO complex II. Then, the complex as electrophile attacked enamine intermediate to produce the iminium ion III. The hydrolysis of intermediate III released imidazolethiones and Cu(I). The Cu(I) was reoxidized by ambient oxygen to regenerate the Cu(II) salt and worked in the cycle again.
 | ||
| Scheme 1 The possible synergistic catalytic mechanism. | ||
Conclusions
In summary, the imidazolethione catalyst 2a is successfully applied to the asymmetric α-oxyamination of aldehydes via the efficient one-pot sequential reaction system. The corresponding products are obtained in good yields and high to excellent enantioselectivities. Applications of this synergistic catalysis system to α-functionalization of carbonyl compounds are underway in our laboratory.Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant no. 21206148).References
- E. Dinca, P. Hartmann, J. Smrček, I. Dix, P. G. Jones and U. Jahn, Eur. J. Org. Chem., 2012, 24, 4461–4482 CrossRef
.
-
(a) G. De Faveri, G. Ilyashenko and M. Watkinson, Chem. Soc. Rev., 2011, 40, 1722–1760 RSC
-
(a) T. J. Donohoe, C. J. R. Bataille and P. Innocenti, Org. React., 2012, 76, 1–48 CAS
-
(a) C. E. I. Knappke and A. J. Von Wangelin, ChemCatChem, 2010, 2, 1381–1383 CrossRef CAS
- S. P. Brown, M. P. Brochu, C. J. Sinz and D. W. C. MacMillan, J. Am. Chem. Soc., 2003, 125, 10808–10809 CrossRef CAS PubMed
- G. F. Zhong, Angew. Chem., Int. Ed., 2003, 42, 4247–4250 CrossRef CAS PubMed
- M. P. Sibi and M. Hasegawa, J. Am. Chem. Soc., 2007, 129, 4124–4125 CrossRef CAS PubMed
- J. F. Van Humbeck, S. P. Simonovich, R. R. Knowles and D. W. C. MacMillan, J. Am. Chem. Soc., 2010, 132, 10012–10014 CrossRef CAS PubMed
-
(a) K. Akagawa, T. Fujiwara, S. Sakamoto and K. Kudo, Org. Lett., 2010, 15, 1804–1807 CrossRef PubMed
- T. Kano, H. Mii and K. Maruoka, Angew. Chem., Int. Ed., 2010, 49, 6638–6641 CrossRef CAS PubMed
- Z. Xie, C. Wang, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 2056–2059 CrossRef CAS PubMed
- X. H. Ho, M. J. Kang, S. J. Kim, E. D. Park and H. Y. Jang, Catal. Sci. Technol., 2011, 1, 923–926 CAS
- N. N. Bui, X. H. Ho, S. i. Mho and H. Y. Jang, Eur. J. Org. Chem., 2009, 31, 5309–5312 CrossRef
- S. P. Simonovich, J. F. Van Humbeck and D. W. C. MacMillan, Chem. Sci., 2012, 3, 58–61 RSC
-
(a) H. S. Yoon, X. H. Ho, J. Jang, H. J. Lee, S. J. Kim and H. Y. Jang, Org. Lett., 2012, 14, 3272–3275 CrossRef CAS PubMed
- G. A. Abeykoon, S. Chatterjee and J. S. Chen, Org. Lett., 2014, 16, 3248–3251 CrossRef CAS PubMed
- M. Peifer, R. Berger, V. W. Shurtle, J. C. Conrad and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 5900–5903 CrossRef CAS PubMed
-
(a) X. R. Liang, S. M. Li and W. K. Su, Tetrahedron Lett., 2012, 53, 289–291 CrossRef CAS PubMed
- A. E. Allen and D. W. C. MacMillan, Chem. Sci., 2012, 3, 633–658 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization data. See DOI: 10.1039/c4ra08556f |
| This journal is © The Royal Society of Chemistry 2014 |