Monodisperse functional microspheres from step-growth “click” polymerizations: preparation, functionalization and implementation†
Chen
Wang
a,
Maciej
Podgórski
b and
Christopher N.
Bowman
*a
aDepartment of Chemical and Biological Engineering, University of Colorado, UCB 596, Boulder, Colorado 80303, US. E-mail: Christopher.bowman@colorado.edu
bFaculty of Chemistry, Department of Polymer Chemistry, MCS University, pl. Marii Curie Skłodowskiej 5, 20-031 Lublin, Poland
First published on 18th June 2014
Abstract
Herein, we demonstrate that, monodisperse, cross-linked clickable microspheres are made via step-growth thiol–acrylate Michael addition polymerization. The diameter of the microsphere is varied from 1–10 μm, depending on the cross-link density and the reaction conditions. Implementations of these microspheres including functionalized microspheres for “click” chemistry, polymeric composites, fluorescent labeling and polymer degradation are discussed and/or demonstrated.
Conceptual insightsStep-growth polymerizations have been extensively used for producing resins, films, fibers and adhesives, but are difficult to use in the preparation of microspheres. Herein, we demonstrate that click reactions enable just such an implementation due to the nature of these reactions. Further, this approach enables a new paradigm in micro particles, where “click” chemistry enables the fabrication of functional monodisperse microspheres from step-growth polymerization at ambient conditions that then preserve all the capabilities of being readily clickable after synthesis. |
Microspheres are materials with small size, high specific surface area and high diffusivity that are ubiquitous in coatings, composites, drug delivery, separations, and many other applications.1–3 Further, monodisperse microspheres have unique applications in chromatography, photonic crystals, micro-devices and biomedical analysis.3–5 Polymers prepared by free-radical chain growth polymerization have been extensively studied and primarily used in this area, including polymers such as polystyrene and poly(methyl methacrylate).6,7 On the contrary, industrial step-growth polymers, such as polyamides and polycarbonates, are rarely used in microsphere preparation.
As in chain-growth polymerization, no polymerization will happen until initiation occurs. In the case of step-growth reactions, polymerization continues in the presence of the reactive species, often a catalytic base, nucleophile or metal ion. However, in step-growth polymerizations the rate decelerates dramatically with increasing reactant consumption, generally compromising the final conversion and preventing the formation of appropriately reacted and stable particles. The presence of an uncontrolled amount of residual, unreacted functional groups within the microspheres is highly disfavored, leading to aggregation and numerous other problems.
Landfester and coworkers pioneered making microspheres from step-growth polymers such as polyurethanes by miniemulsion polymerization.8,9 Recently, other examples have been reported on making step-growth microspheres by implementing thiol-“click” chemistries in various approaches, including as both a polymerization strategy and as a crosslinking reaction of prepolymers.10–13 Shipp et al. recently reported preparing microbeads from thiol–ene radical suspension photo-polymerization.12 As one reaction in the “click” chemistry family, thiol-“click” chemistries have been widely used in dendrimer synthesis, polymer coupling, polymer functionalization and network formation due to their rapid kinetics and high product yields.14 The attributes of “click” chemistries render these reactions ideal candidates to address the issues of preparing microspheres by step-growth mechanism, because the reaction is able to achieve full conversion rapidly in benign conditions.
In chain-growth linear polymerizations, aliphatic main chains are typically generated from the reaction of a carbon–carbon double bond. On the other hand, the polymer backbone in step-growth systems incorporates various linking groups. In cross-linking polymerizations, structurally uniform networks are generated from step-growth mechanisms as opposed to the heterogeneous networks that result from traditional chain-growth polymerizations.14,15 Moreover, in step-growth polymerizations, a specific, controlled amount of functional groups remain available for further intrinsic functionalization after off-stoichiometric polymerizations. Herein, we demonstrate a versatile method for forming micron-size monodisperse microspheres by step-growth polymerization with all of the advantages of thiol-click processes used to make and subsequently modify the microspheres.
An analogous system to typical free-radical dispersion polymerization is designed for step-growth thiol-Michael addition polymerization. In a dispersion polymerization system, the reaction starts from a homogeneous mixture comprising monomer, initiator, surfactant and organic solvent. The reaction media is subtly chosen to dissolve monomer but not polymer, so that the polymer could precipitate and would be stabilized by surfactant to generate microspheres. As one typical system, we selected a tetra-thiol, pentaerythritol tetrakis(3-mercaptopropionate) (PETMP) and a tri-acrylate, trimethylolpropane triacrylate (TMPTA) as monomers. The monomer mixture is dissolved in methanol in the presence of polyvinylpyrrolidone (PVP) as a surfactant. The mixture was a clear solution until the reaction was triggered by adding hexylamine, which is an efficient catalyst for thiol-Michael addition.16 Then, the mixture quickly became a white, colloidal dispersion. The polymerization proceeded rapidly and quantitatively with no observable monomer in the solution just 5 min after the reaction was triggered (1H NMR, Fig. S1†). The mixture was left stirring for 2 h prior to any additional steps to assure complete conversion.
Images from scanning electron microscopy show that the product consisted of relatively monodisperse, spherical particles (Fig. 1). The product can be re-dispersed in methanol by sonication and re-form a stable colloid, indicating that the microspheres are well isolated from each other. Isolated microspheres indicate that full conversion was achieved, and further reaction was not observed by any means.
 | ||
| Fig. 1 SEM images of microspheres prepared by thiol-Michael addition dispersion polymerization of stoichiometric PETMP and TMPTA in methanol, with various amount of hexylamine as the initiator: (a) 1.6 wt% (b) 4.0 wt% (c) 10.0 wt% hexylamine with respect to the mass of monomers. Polymerization conditions: PETMP (3.66 g, 7.5 mmol), TMPTA (3.0 g, 10 mmol), PVP (1.5 g) and methanol (150 mL), the mixture was stirred at 400 RPM for 2 h after the addition of hexylamine. The scale bar length is 10 μm. | ||
Microspheres with varied diameter are achieved by altering the catalyst loading. Fig. 1a–c show polymerizations initiated with 1.6 wt%, 4.0 wt% and 10.0 wt% hexylamine, resulting in microspheres with diameters of 3.57 μm, 2.10 μm and 1.39 μm, respectively. The coefficients of variance (CV) of the microspheres shown in Fig. 1 are all lower than 5%, indicating monodispersity. Previously, it was reported that particle size increases with higher loading of initiator in the dispersion polymerization of styrene.17 However, in our studies, the diameter of the microspheres decreased with increasing catalyst concentration in step-growth dispersion polymerization, indicating a fundamentally different particle formation mechanism from the one accepted for free-radical chain growth dispersion polymerizations.
In cross-linking step-growth polymerization, the functionality of monomers determines the degree of cross-linking and the physical properties of the polymer network, such as the rubbery modulus and glass transition temperature (Tg). We selected a series of monomers of various functionalities to investigate the effects of varied cross-linking densities, as listed in Scheme 1. EGBMP (1), TMPTMP (2) and PETMP (3) are di-, tri- and tetra-functional thiol monomers; TMPTA (4) and DTPTA (5) are tri- and tetra-functional acrylate monomers. The microspheres prepared by different combinations of these monomers are listed in Table 1 (entries 1–4). All the experiments were conducted under the same levels of functional group concentration (30 mmol for both thiol and acrylate), solvent (150 mL methanol), surfactant (1.5 g PVP), catalyst loading (60 mg hexylamine, 2 mol% to each reactive species) and mechanical stirring (400 rpm). Interestingly, the size and size distribution of the microspheres was found to be strongly dependent on the monomer functionalities. As the number of functional groups increased, the microsphere diameter decreased from 9.87 μm to 2.79 μm (Table 1, entry 1–4, SEM images shown in Fig. S2†). It is reported that, for dispersion polymerization, polymer precipitates when it reaches a critical length/size and forms nuclei. Subsequently, we hypothesize that the nuclei react with growing polymeric chains and increase in size uniformly, leading to the desired microspheres.18 When the degree of functionality increases, the polymer has poorer solubility at a similar conversion. As a result, nucleation starts at lower conversions, and more nuclei are formed. Given that full conversion is still achieved, each nucleus grows with the addition of fewer monomer units with the end result being smaller microspheres. We also observed that the polymerization mixture became turbid much more quickly for more cross-linked (i.e., higher functionality) systems. For example, the tetrathiol–tetraacrylate system (entry 4) mixture became turbid so quickly that the catalyst was not able to disperse uniformly. As a result, the coefficient of variance increased to 20%, disturbing the falling trend. We believe that the monodispersity is achievable for each of these step-growth systems, when polymerization conditions are optimized.
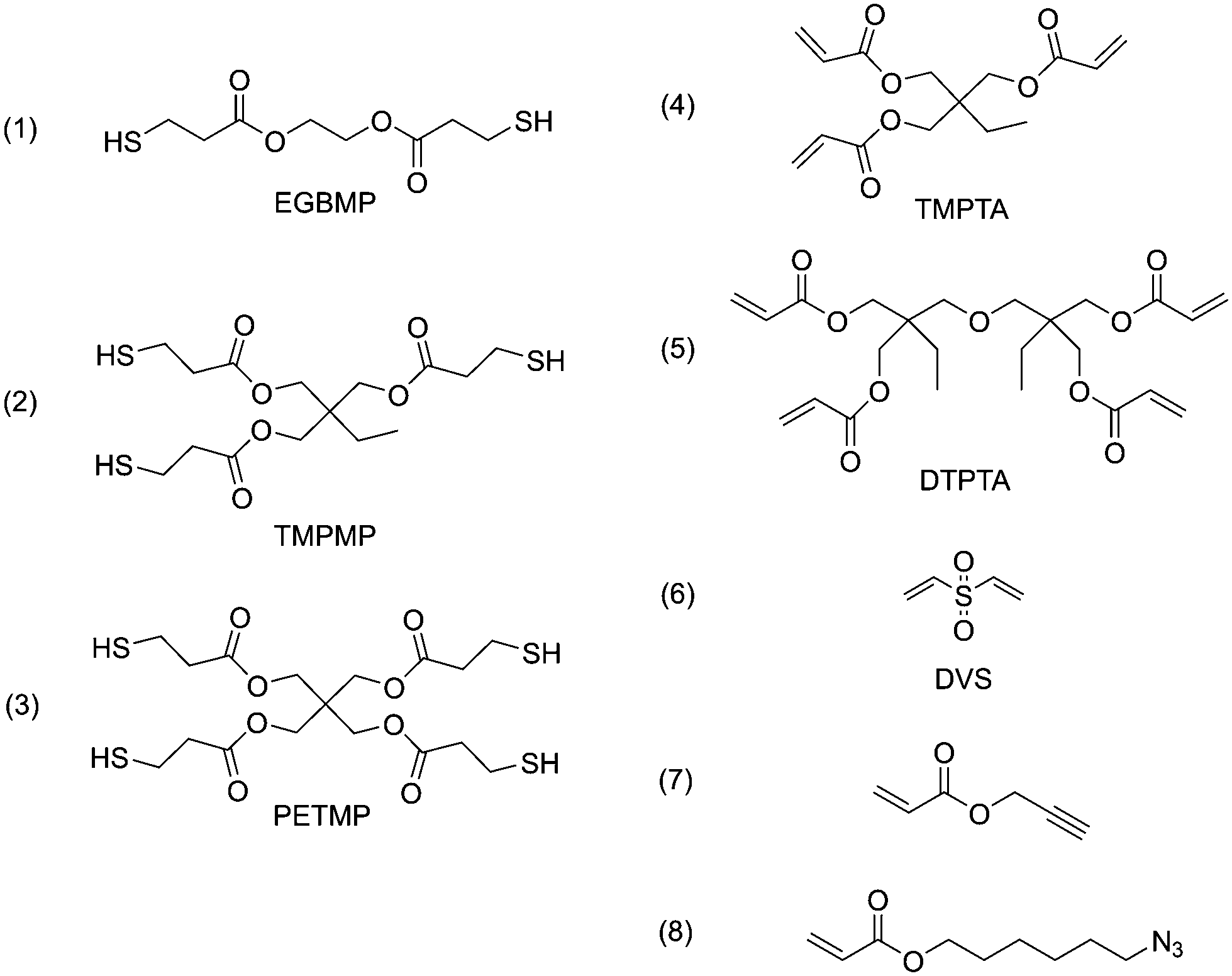 | ||
| Scheme 1 Monomers used in this work: (1) ethylene glycol bis(3-mercaptopropionate) (EGBMP); (2) trimethylolpropane tris(3-mercaptopropionate) (TMPMP); (3) pentaerythritol tetrakis(3-mercaptopropionate) (PETMP); (4) trimethylolpropane triacrylate (TMPTA); (5) di(trimethylolpropane) tetraacrylate (DTPTA); (6) divinyl sulfone (DVS); (7) propargyl acrylate; (8) 6-azidohexyl acrylate. | ||
| Entry | Thiol/ene monomer functionalityb | D n/μm | CV/% | Yield/% | T g /°C |
|---|---|---|---|---|---|
| a Polymerization conditions: 30 mmol of both thiol and acrylate functional groups, 150 mL MeOH, 1.5 g PVP and 60 mg hexylamine. b Monomers of varying functionality are shown in Scheme 1. c The glass transition temperature was measured by DSC. d Divinylsulfone is used as the ene monomer instead of the acrylate. | |||||
| 1 | Di/tri | 9.9 | 36 | 72 | −24 |
| 2 | Tri/tri | 6.1 | 11 | 90 | −6 |
| 3 | Tetra/tri | 3.6 | 3.7 | 94 | 8 |
| 4 | Tetra/tetra | 2.8 | 20 | 90 | 16 |
| 5d | Tetra/di | 1.1 | 4.0 | 95 | 13 |
It is also shown in Table 1 that the Tg is readily manipulated by changing the degree of cross-linking. As the crosslink density increases from the dithiol–triene system to the tetrathiol–tetraene resin (entries 1–4 in Table 1), the Tg increases from −24 °C to 16 °C, as measured by differential scanning calorimetry (DSC, Fig. S3†). More interestingly, as the structure of network linking chains is easily tuned in step-growth polymerizations by using different types of vinyl monomer, microspheres with significantly different properties were obtained. For the tetrathiol–diene (entry 5), divinyl sulfone (DVS, Scheme 1(6)) was used as a Michael acceptor. The polymerization conditions for the PETMP–DVS system were analogous to all the other thiol–acrylate systems. However, not only did smaller microspheres result from the PETMP–DVS polymerization, but also their Tg was higher than in similarly cross-linked thiol–acrylate network microspheres. The bulky sulfone groups combined with the low molecular weight DVS structure lead to a stiffer backbone polymer chain, which affected the material's Tg. It is indicated that this polymerization technique is potentially useful for all kinds of monomers that are applicable to thiol-Michael reactions, and the corresponding properties of such polymer microspheres can be facilely adjusted.
In terms of physical properties, homogeneous networks fabricated by step-growth polymerization have narrow Tg's and robust response to heating. To explore such behavior in microspheres, we prepared a microparticle-filled polymeric composite material by dispersing PETMP–DVS microspheres (40 wt%) well into a mixture of TMPMP–TMPTA monomers (60 wt%), which were then polymerized. The composite material was an opaque elastomer at ambient conditions. Its dynamic mechanical analysis (DMA) is shown in Fig. 2. The material exhibits two distinct Tg's: one near 0 °C distinctive of the TMPMP–TMPTA matrix and another near 37 °C from PETMP–DVS microspheres. Both Tg's are higher than those from DSC. The storage modulus drops from 1400 MPa in the glassy state to 80 MPa after the first transition, and then drops to 10 MPa after the second one. It is worth noting that both phases in the composite are composed of highly cross-linked polymers. Two distinct Tg's within 40 °C confirm the homogeneity of step-growth networks and result in multiple thermal responses, which could readily be engineered into functional materials such as triple shape memory polymers.19
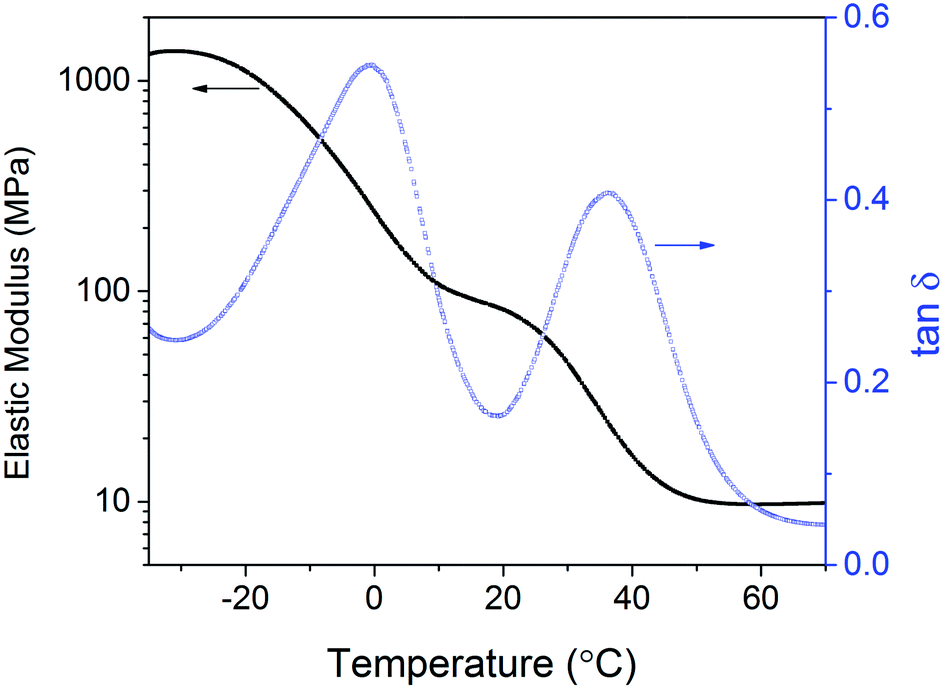 | ||
| Fig. 2 Dynamic mechanical analysis of polymeric composite of PETMP–DVS microspheres in TMPTMP–TMPTA matrix. The composite is composed of 0.5 g of PETMP–DVS microspheres (42 wt%) embedded in 0.7 g of TMPTMP–TMPTA matrix (58 wt%). | ||
In terms of formation of functional polymer microspheres, for example, polystyrene based microspheres, there are usually two ways: post-polymerization reactions that immobilize functional groups onto the surface of microspheres, or copolymerization of functional monomers. The former method involves multiple steps whereas the latter is only applicable to functional groups that are inert to radicals. However many useful functionalities are vulnerable to radical reactions. In contrast, the thiol-Michael addition is a stoichiometric addition reaction between a thiol and an electron deficient double bond, which enables the preservation of functional groups for off-stoichiometric polymerization. Nair et al. reported a dual-cure polymerization system that exploited this self-limiting nature of the reaction.20,21 In their work, the acrylate was used both in a cross-linking thiol-Michael addition polymerization (to form an initial, stable polymer), and then the remaining excess acrylate was homo-polymerized radically to form the final polymer. Herein, our method provides a facile route to prepare monodisperse functional microspheres where an excess of either functional group in the reaction is subsequently useful in facilitating functionalization and additional reactions.
By off-stoichiometric thiol–acrylate polymerization, microspheres with excess thiol (or acrylate) are made in a single, simple step. The residual functionality from the self-limiting reaction is confirmed by FT-IR, as shown in Fig. 3 where the peaks around 2570 cm−1 and 810 cm−1 are used to identify the thiol and acrylate groups, respectively (Fig. 3a(1) and (2)). It is noted that only the excess functionality can be seen on FT-IR, indicating the reaction is indeed stoichiometric, self-limiting and that the polymerization achieves complete conversion of the limiting reactant. By copolymerizing functional monomers, we introduce a variety of functionalities into our model thiol–acrylate microspheres. We selected alkyne and azide groups as examples, since they are the building blocks for the copper catalyzed alkyne–azide cycloaddition reaction (CuAAC), which has been widely used in bioconjugation and bio-detection analysis. Propargyl acrylate, which is commercially available and 6-azidohexyl acrylate, which is synthesized as reported, are copolymerized into PETMP–TMPTA based model microspheres (SEM images see Fig. S4†).22Fig. 3a(3) and (4) shows the confirmation of the azide and alkyne moieties as observed by FT-IR where peaks around 2100 cm−1 and 3260 cm−1 were used to identify the azide and alkyne, respectively. We expect that these “clickable” microspheres will find uses in applications where particles need to be tuned, particularly through reactive functionalization and in composite structures where excellent integration of the microparticles is necessary.
 | ||
| Fig. 3 (A) FT-IR spectra of functionalized PETMP–TMPTA microspheres: (1) thiol-excess (33 mol%); (2) acrylate-excess (20 mol%); (3) azide-functionalized (with 10 mol% azide with respect to thiol functionality); (4) alkyne-functionalized (with 15 mol% alkyne with respect to thiol functionality). Images for fluorescently-labeled PETMP–TMPTA microspheres (60× objective): (B) rhodamine B labeled thiol-excess microspheres; (C) rhodamine 110 labeled alkyne-functionalized microspheres. | ||
Functionalized microspheres are utilized widely in fluorescent labeling. To demonstrate the utility of these functionalizable microspheres, thiol-excess microspheres were formed and then reacted with a fluorescent acrylic monomer (acryloxyethyl thiocarbamoyl rhodamine B) by a second thiol-Michael reaction. In addition, alkyne-functionalized microspheres were reacted with a fluorescent azide (rhodamine 110 conjugated PEG azide) by the CuAAC reaction in methanol. The fluorescence of both thiol-excess and alkyne-functionalized microspheres was confirmed by optical microscopy, as shown in Fig. 3b and c, respectively. We expect these microspheres will serve as a strong tool for fluorescence-related applications, especially because of the capability for preparing such microspheres.
An additional use of microspheres involves their controlled degradation for one of a variety of purposes. In particular, esters are subject to hydrolysis in acidic and basic environments, and polymers with ester groups in the backbone have been studied for biodegradation and controlled release.23 Our thiol–acrylate model microspheres contain thio-ether esters as building blocks, which have been found to be particularly susceptible to hydrolysis.24 We dispersed such microspheres in 1 mol L−1 NaOH (aq.), and the mixture turned from turbid to clear in just 30 min, indicative of particle degradation. More interestingly, fluorescent dye was released during the degradation of microspheres. Fig. 4 shows the amount of fluorophore released as a function of the degradation time for rhodamine B labeled thiol-excess microspheres, as monitored by UV-vis spectroscopy. The amount of dye released from the microspheres increases continually with degradation time. Moreover, no dye was released from these same microspheres dispersed in water overnight since the dye was covalently couple through a thio-ether ester functionality. Subsequent exposure of these particles to a strongly basic solution caused degradation and led to an identical amount of dye release as particles that were immediately placed in basic solution. Clearly, one of the most powerful aspects of the thiol-Michael reaction is the ability to vary the chemistry of these microparticles, including their degradation and release characteristics. While relatively harsh conditions were used here to achieve rapid degradation and release, it is readily feasible to degrade these same esters under much more mild conditions, to incorporate more rapidly cleavable esters (or other hydrolytically labile species), or even to include moieties that utilize a different stimulus, e.g., light or heat, to trigger degradation and release.
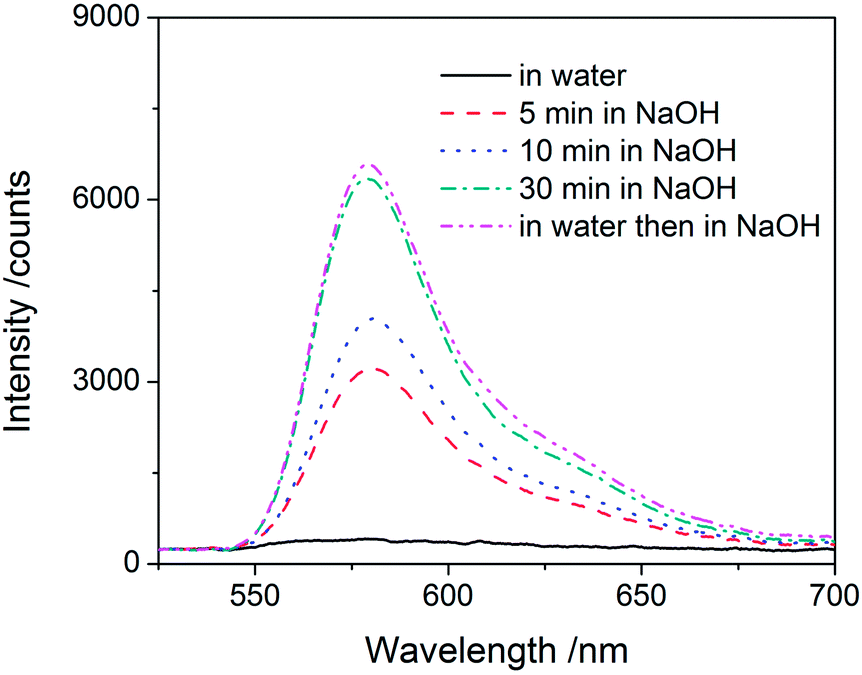 | ||
| Fig. 4 UV-vis spectra of the solution from the degradation of rhodamine B labeled PETMP–TMPTA microspheres in various conditions. | ||
Conclusions
Monodispersed microspheres were successfully prepared by step-growth thiol-Michael addition polymerization. Compared with traditional polymeric microspheres, the nature of step-growth polymerization endorses such microspheres with facilely tunable backbone and intrinsic functionalization. Polymeric composite, fluorescent labeled microspheres and degradable microspheres are cases shown here as implementations for this method. We expect the method developed here to be a strong tool for research area that requires monodisperse functional microspheres, such as photonic crystal, colloidal molecule, chromatography and bio-analysis.Acknowledgements
We would like to thank the National Science Foundation (CHE 1214109) for providing financial support to this work.Notes and references
- H. Kawaguchi, Prog. Polym. Sci., 2000, 25, 1171–1210 CrossRef CAS.
- B. Jeong, Y. H. Bae, D. S. Lee and S. W. Kim, Nature, 1997, 388, 860–862 CrossRef CAS PubMed.
- J. Wang, P. A. G. Cormack, D. C. Sherrington and E. Khoshdel, Angew. Chem., Int. Ed., 2003, 42, 5336–5338 CrossRef CAS PubMed.
- M. Deutsch, Y. A. Vlasov and D. J. Norris, Adv. Mater., 2000, 12, 1176–1180 CrossRef CAS.
- A. Terray, J. Oakey and D. W. M. Marr, Science, 2002, 296, 1841–1844 CrossRef CAS PubMed.
- W. Ming, J. Zhao, X. Lu, C. Wang and S. Fu, Macromolecules, 1996, 29, 7678–7682 CrossRef CAS.
- M. Sivakumar and K. P. Rao, React. Funct. Polym., 2000, 46, 29–37 CrossRef CAS.
- F. Tiarks, K. Landfester and M. Antonietti, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2520–2524 CrossRef CAS.
- M. Antonietti and K. Landfester, Prog. Polym. Sci., 2002, 27, 689–757 CrossRef CAS.
- R. A. Prasath, M. T. Gokmen, P. Espeel and F. E. Du Prez, Polym. Chem., 2010, 1, 685–692 RSC.
- C. O. Bounds, R. Goetter, J. A. Pojman and M. Vandersall, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 409–422 CrossRef CAS.
- O. Z. Durham, S. Krishnan and D. A. Shipp, ACS Macro Lett., 2012, 1, 1134–1137 CrossRef CAS.
- J. Zou, C. C. Hew, E. Themistou, Y. Li, C. K. Chen, P. Alexandridis and C. Cheng, Adv. Mater., 2011, 23, 4274–4277 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- T. Gong, B. J. Adzima, N. H. Baker and C. N. Bowman, Adv. Mater., 2013, 14, 2024–2028 CrossRef PubMed.
- W. Xi, C. Wang, C. J. Kloxin and C. N. Bowman, ACS Macro Lett., 2012, 1, 811–814 CrossRef CAS.
- C. K. Ober and M. L. Hair, J. Polym. Sci., Part A: Polym. Chem., 1987, 25, 1395–1407 CrossRef CAS.
- J. S. Downey, R. S. Frank, W.-H. Li and H. D. H. Stöver, Macromolecules, 1999, 32, 2838–2844 CrossRef CAS.
- J. Zhao, M. Chen, X. Wang, X. Zhao, Z. Wang, Z.-M. Dang, L. Ma, G.-H. Hu and F. Chen, ACS Appl. Mater. Interfaces, 2013, 5, 5550–5556 CAS.
- D. P. Nair, N. B. Cramer, J. C. Gaipa, M. K. McBride, E. M. Matherly, R. R. McLeod, R. Shandas and C. N. Bowman, Adv. Funct. Mater., 2012, 22, 1502–1510 CrossRef CAS.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2013, 26, 724–744 CrossRef.
- A.-M. Zorn, M. Malkoch, A. Carlmark and C. Barner-Kowollik, Polym. Chem., 2011, 2, 1163–1173 RSC.
- M. A. Tracy, K. L. Ward, L. Firouzabadian, Y. Wang, N. Dong, R. Qian and Y. Zhang, Biomaterials, 1999, 20, 1057–1062 CrossRef CAS.
- A. E. Rydholm, S. K. Reddy, K. S. Anseth and C. N. Bowman, Biomacromolecules, 2006, 7, 2827–2836 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization methods and supplemental SEM images are included. See DOI: 10.1039/c4mh00082j |
| This journal is © The Royal Society of Chemistry 2014 |