DOI:
10.1039/C3RA41442F
(Review Article)
RSC Adv., 2013,
3, 14165-14182
Transition metal-catalyzed decarboxylative coupling reactions of alkynyl carboxylic acids
Received 24th March 2013, Accepted 31st May 2013
First published on 31st May 2013
Abstract
The decarboxylative coupling of alkynyl carboxylic acids is an attractive area of research in organic chemistry, because the structure of aryl alkyne is one of the important building blocks for the synthesis of p-conjugated compounds. The use of alkynyl carboxylic acid as an alkyne source has several advantages in handling and storage. As a catalyst, palladium, copper, nickel, and silver were employed in the decarboxylative coupling reactions. The formation of C–C (sp2–sp, sp–sp and sp3–sp), C–N, C–P and C–S bonds has been developed. This review aims to illustrate the development of the decarboxylative coupling reaction of alkynyl carboxylic acids.
 Kyungho Park | Kyungho Park was born in Jangheung, South Korea in 1984. He obtained his B. S. from Chonnam National University in 2009. After completed his M. S. (2011) from the Department of Chemistry at the same university, he commenced his Ph. D. under supervision of Prof. Sunwoo Lee in the same university. And he had short term research experience in Prof. Carsten Bolm group (RWTH Aachen University) for the NRF(Korea)-DAAD(German) summer research program (7th July–29th August 2011). His research interests include development of decarboxylative coupling reactions and their application in the synthesis of advanced functional materials. |
 Sunwoo Lee | Sunwoo Lee was born in Busan, South Korea in 1969. After his B.S. in Chemistry Education at Pusan National University (1987-94, including military service), he obtained his M.S. (1996) and Ph. D. (1999) at POSTECH under the supervision of Professor Jaiwook Park. He did his post-doctoral studies in the Department of Chemistry at Yale University (1999–2001) with Professor John F. Hartwig. He worked at LG Chem as a senior researcher (2001–2004). In 2004, he began his current position as a Professor in the Department of Chemistry at Chonnam National University. The main focus of his work is the development of catalytic transformations and their applications. |
1 Introduction
Palladium-catalyzed cross coupling reactions are some of the most useful tools in the synthesis of important molecules, such as natural products, bioactive compounds, polymers, and materials.1 Many types of reactions have been reported and widely used, such as the Kumada, Negishi, Stille, Suzuki, Hiyama, Heck, Sonogashira, and Buchwald–Hartwig coupling reactions.2 The first five of these reactions consist of the coupling reaction of aryl halides (or pseudo halides) and organometallic compounds bearing Mg, Al, Zn, Sn, B, and Si. These coupling partners have to be prepared from their corresponding simple arenes starting step processes for preparation,3 some of which may be hydrolytically sensitive and hence a limited range of starting materials can be used. Particularly, organometallic reagents require multi-functional group tolerance. To overcome this drawback, direct C–H activated coupling reactions have received much attention. Several catalysts have been used, including palladium, ruthenium, rhodium, copper, and others.4 However, these C–H activation methods have shown a narrow scope of substrates, having been limited to those which bear active protons by directing groups or low pKa values, despite the development of new methodology to address this issue. Compared with the traditional cross-couplings and C–H activation, decarboxylative coupling reactions using carboxylic acid derivatives have several advantages. Instead of metal waste from organometallic coupling reagents, less toxic carbon dioxide is released as a byproduct after the complete conversion, which reduces the cost of the process for the treatment of waste. In addition, carboxylic acid derivatives are easily available and stable for the handling and storage.Decarboxylative coupling was first reported by Nilsson in 1966.5 However, it had not been cited until the early 2000s, because it was not practical due to its low yield, limited scope, and requirement of large amounts of Cu2O. In 2002, Myers reported on palladium-catalyzed decarboxylative Heck reactions.6 Four years later, in 2006, Goossen developed efficient palladium-catalyzed decarboxylative coupling for the synthesis of biaryl from aryl carboxylic acids with aryl halides.7 Since then, a variety of related methodologies have been reported.8 In 2008, we first reported the decarboxylative coupling reaction of alkynyl carboxylic acid and aryl halides using a palladium catalyst, which afforded an efficient tool for the synthesis of symmetric and unsymmetric diarylalkynes.9 The use of alkynyl carboxylic acids as terminal alkyne surrogates provides a useful method for the handling of alkynes with a low boiling point.
Goossen8a and Liu8b independently reviewed the decarboxylative coupling reactions, including aryl carboxylic acids and some aryl alkynyl carboxylic acids. In this review, we focus on the decarboxylative coupling reactions of alkynyl carboxylic acid and recent related reports.
2 Palladium catalyzed-decarboxylative couplings
2.1. Coupling reactions with aryl halides and pseudo halides
The first decarboxylative coupling of alkynyl carboxylic acids was reported by S. Lee.9 They employed propiolic acid as an alkyne source for the coupling with aryl iodides and bromides with palladium catalyst in the presence of tetrabutylammonium fluoride (TBAF) (Scheme 1). While controlling the temperature, unsymmetric diarylalkynes were successfully obtained by one-pot reaction. However, this first report had some drawbacks, in that 6 equivalents of TBAF were required for high products yields, and 40 mol% Pt-Bu3, which is sensitive to air and moisture, was used for the synthesis of symmetric diarylalkynes from aryl bromides (Scheme 2).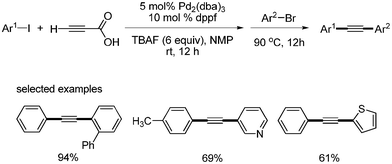 |
| | Scheme 1 Synthesis of unsymmetric diarylalkynes from propiolic acid and aryl halides. | |
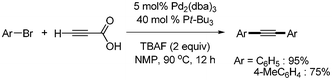 |
| | Scheme 2 Synthesis of symmetric diarylalkynes from propiolic acid and aryl halides. | |
One year later, S. Lee applied the decarboxylative coupling reactions toward 2-octynoic acid and phenylpropiolic acid.10 A variety of aryl bromides were coupled with alkynyl carboxylic acids, which afforded the desired aryl alkynes in good yields. In the case of 2-octynoic acid, Pd(PPh3)2Cl2 (1 mol%) and 1,4-bis(diphenylphosphino)butane (dppb) (2 mol%) showed good results in DMSO. Phenylpropiolic acid required Pd2(dba)3 (2.5 mol%) and Pt-Bu3 (10 mol%) for good yield. Aryl bromides bearing ester and ketone groups and heteroaromatic halides afforded the desired coupled products in good yields. In addition, chloro group could survive in these conditions (Scheme 3). However, both systems still required 2 equivalents of TBAF.
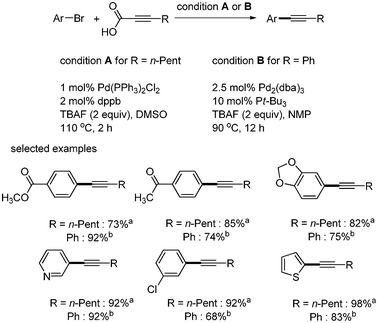 |
| | Scheme 3 Decarboxylative coupling with aryl bromides; acondition A; bcondition B. | |
In 2009, P. H. Lee reported the decarboxylative coupling reaction of phenylpropiolic acid with a variety of coupling partners, such as aryl iodides, aryl bromides, aryl triflates, vinyl bromides, and vinyl triflates.11 In the coupling with aryl iodides, the catalytic system of Pd2(dba)3·CHCl3 (2 mol%) and PPh3 (16 mol%) was used with Ag2O (1 equiv.) and LiI (3 equiv.) in DMF. Aryl iodides with aldehyde showed 85% yield in the coupling reaction. Sterically demanding ortho-substituted aryl iodides also provided good yields of the products (Scheme 4). In the case of aryl and vinyl bromides and triflates, Xantphos (8 mol%) and LiCl were employed instead of PPh3 and LiI, respectively. α- and β-bromo styrene afforded the desired coupled product in 89% and 92% yields, respectively (Scheme 5).
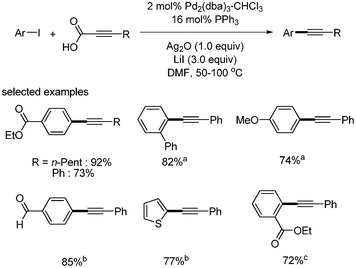 |
| | Scheme 4 Decarboxylative coupling with aryl iodides, a2 equiv. of Ag2O, 2 equiv. of phenylpropiolic acid and 6 equiv. of LiI were used, b8 mol% of Xantphos as a ligand, c1.5 equiv. of Ag2O and phenylpropiolic acid were used. | |
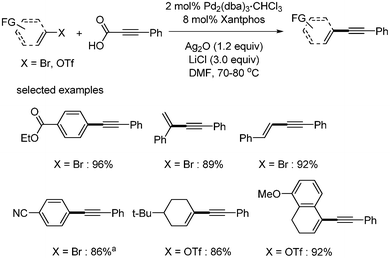 |
| | Scheme 5 Decarboxylative coupling with aryl and vinyl bromides and triflates. aLiI was used. | |
As shown in Scheme 6, lithium phenylacetylide afforded the desired product in 35% yield, and silver phenylacetylide showed 51% yield. On the basis of these results, it was suggested that silver phenylacetylide might be involved in the transmetallation step rather than lithium phenylacetylide.
 |
| | Scheme 6 Decarboxylative coupling with metal phenylacetylides. aTHF as solvent; babsence of LiI. | |
Li expanded the scope of substrates in palladium-catalyzed decarboxylative coupling reactions.12 They developed a catalytic system consisting of Pd(OAc)2 and XPhos, and ran the coupling reaction in the presence of Cs2CO3 in THF. A variety of benzyl chlorides, bromides, and acetate afforded the desired benzyl alkynes in good yields (Scheme 7). They first reported the decarboxylative coupling with aryl chlorides, although only three examples were shown. Chlorobenzene and 4-chloroacetophenone were coupled with phenylpropiolic acid, giving the corresponding aryl alkynes in 76% and 75% yields, respectively. However, 4-chloroanisole produced the desired product in 19% yield (Scheme 8).
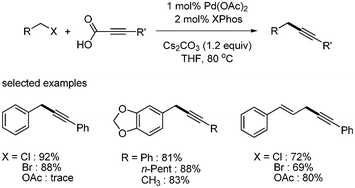 |
| | Scheme 7 Decarboxylative coupling with benzyl halides and acetates. | |
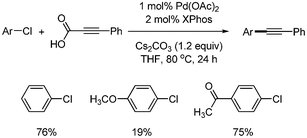 |
| | Scheme 8 Decarboxylative coupling with aryl chlorides. | |
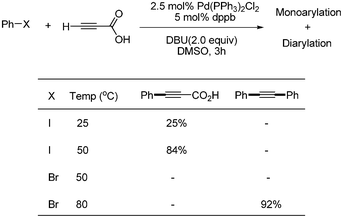
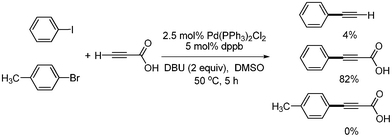
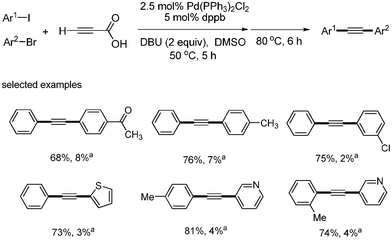
S. Lee developed a catalytic system of Pd(PPh3)2Cl2 (5 mol%), dppb (10 mol%), DBU (2 equiv.), and DMSO for the synthesis of symmetric and unsymmetric diarylalkynes in decarboxylative coupling reactions.13 This system was an improved method meant to solve the previous problems associated with the necessity of excess of TBAF and air sensitive Pt-Bu3. It was known that symmetric diaryalkynes were obtained in high yields when 1 equivalent of propiolic acid and 2 equivalents of aryl bromides were reacted at 80 °C in this reaction system. Similar results were obtained when 2-butynedioic acid was reacted at 110 °C instead of propiolic acid (Scheme 9). Diarylated alkyne was found to form as a major product at 80 °C, even though 1 equivalent of phenyl bromide was reacted with 1 equivalent of propiolic acid. However, among the monoarylated products that were formed predominantly in the coupling reaction with aryl iodides at low temperatures, such as 25 and 50 °C, more Sonogashira coupling products (aryl alkynyl carboxylic acids) was formed than the decarboxylative coupling products (aryl acetylenes). However, phenyl bromide did not couple with propiolic acid at 50 °C (Scheme 10). When phenyl iodide and p-tolyl bromide were reacted with propiolic acid in the same reaction vessel under the optimized conditions, only phenyl iodide coupled product was formed as a major product (Scheme 11). Unsymmetric diarylalkynes were successfully synthesized by controlling temperature. All reagents, including aryl iodides, bromides, and propiolic acid, were added at the beginning of the reaction. When the reaction first proceeded at 50 °C for 5 h and then ran at 80 °C for 6 h, the desired unsymmetric diarylalkynes were obtained in good yields. However, the symmetrical diarylalkynes were formed as byproduct in 2–8% yields (Scheme 12).
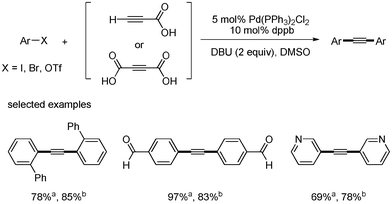 |
| | Scheme 9 Synthesis of symmetric diarylalkynes. aYield from propiolic acid; byield from 2-butynedioic acid. | |
 |
| | Scheme 10 Coupling of propiolic acid with phenyl iodide and bromide. | |
 |
| | Scheme 11 Comparative experiment of aryl iodide and bromide in the coupling reaction. | |
 |
| | Scheme 12 Synthesis of unsymmetric diarylalkynes. aYield of symmetric diarylalkyne. | |
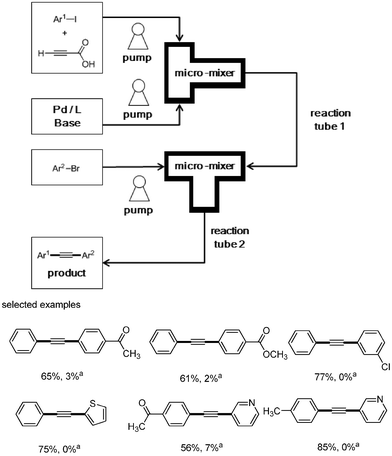
To solve this problem, a continuous microflow reaction system was employed in the synthesis of unsymmetrical diarylalkynes (Scheme 13).14 The catalytic system is almost the same as that in the batch reaction. However, it is most important to find a system for all reagents to be in solution in order to apply the batch reaction methodology to flow chemistry. Because organic reactions that require insoluble reagents cannot be applied in flow chemistry. The optimized condition was first investigated under which all reagents exist in solution at room temperature in the reservoir. The optimized condition found involved one reservoir containing aryl iodide and propiolic acid in DMSO, and another reservoir containing palladium, ligand, and DBU in DMSO. These two reservoirs were connected to a micromixer, and all reagents in two reservoirs were mixed and reacted in the microtube at 50 °C for 4 h. Then, the resulting mixture was mixed with aryl bromide in the second micromixer, and reacted in the microtube at 120 °C for 4 h. This continuous flow reaction system afforded unsymmetric diarylalkynes in high yields with a trace amount of symmetric diarylalkynes as byproducts, and showed high selectivity compared to the batch reaction system.
 |
| | Scheme 13 Microflow reaction of decarboxylative coupling reaction, aYield of symmetric diarylalkynes. | |
Two different aryl bromides were employed instead of aryl iodides and bromides for the synthesis of unsymmetric diarylalkynes from the decarboxylative coupling of propiolic acid.15 Goossen reported that [allylPdCl]2 (2.5 mol%), SPhos (7.5 mol%), 3 equiv. of TBAF·3H2O showed good yields in the synthesis of unsymmetric diarylalkynes. This selective reaction was also controlled by temperature. The first step consists of a Sonogashira coupling of the first aryl bromides with propiolic acid to afford aryl alkynyl carboxyl acid at 50 °C. The second step was run at 80 °C after the other aryl bromide was added. Good yields were shown in a wide range of unsymmetric diarylalkynes. Aryl bromides bearing hydroxyl and amine groups afforded the desired coupled product in good yields. However, there were no examples of one-pot reaction, in which all reagents are added at the beginning (Scheme 14).
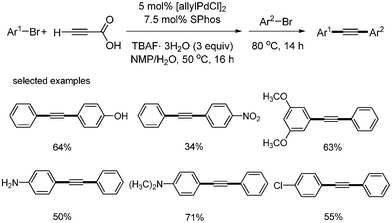 |
| | Scheme 14 Synthesis of unsymmetric diarylalkynes from two different aryl bromides. | |
S. Lee reported an aqueous system for the synthesis of symmetrical diarylalkynes from palladium-catalyzed decarboxylative coupling of propiolic acid and aryl bromides (Scheme 15).16 They were focused on propiolic acid as a suitable reagent for water-based solvent systems in the coupling reaction of alkyne due to its good solubility in water. Pd(TPPMS)2Cl2, a water-soluble palladium source, and TPPMS, a water-soluble ligand, showed good reactivity in the presence of octadecyl trimethyl ammonium chloride (OTAC) as a phase transfer surfactant (PTS). This method can be used under both microwave and thermal heating conditions. The microwave system produced the desired product in 15 min, and the thermal heating produced the product in 3–20 h. However, they did not succeed in the decarboxylative coupling with aryl iodides in the aqueous medium.
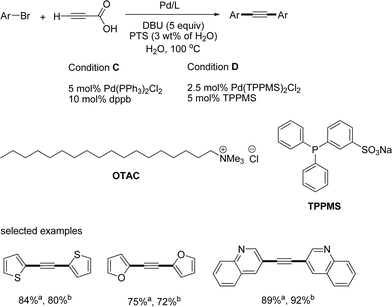 |
| | Scheme 15 Decarboxylative couplings in aqueous conditions; acondition C; bcondition D. | |
S. Lee and coworkers prepared Pd–CNT nanocomposite from a composite of thiolated MWCNT and Na2PdCl4.17 This palladium nanocomposite has an average diameter of 2.9 nm with a narrow size distribution for the Pd–CNT. This Pd–CNT was applied as a catalyst (5 mol% Pd) in the decarboxylative coupling of aryl alkynyl carboxylic acid and aryl iodides. Aryl iodides bearing cyano, bromo, and ketone groups produced the corresponding products in 58%, 87%, and 92%, respectively. This reaction system does not require an additional ligand, and showed good activities in a variety of aryl iodides as well as aryl alkynyl carboxylic acids (Scheme 16).
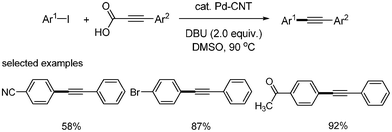 |
| | Scheme 16 Pd–CNT-catalyzed decarboxylative coupling. | |
Very recently, Kantam reported a phosphine-free catalytic system for the decarboxylative coupling reaction of aryl halides.18 They synthesized a carboxyamido/carbene ligand and applied its palladium complex as shown in Scheme 17 in the decarboxylative coupling reaction of alkynyl carboxylic acid and aryl bromides. Only 0.5 mol% palladium catalyst was required for the synthesis of aryl alkynes. However, the reaction temperature was high, and only two examples of aryl chlorides were reported.
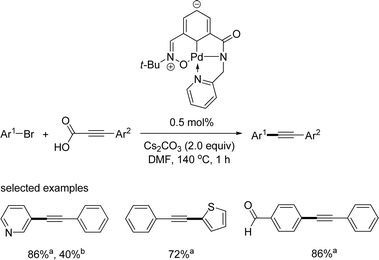 |
| | Scheme 17 Phosphine-free decarboxylative coupling reaction. aYield from ArBr; byield from ArCl. | |
Wu and coworkers reported decarboxylative coupling reaction of aryl chlorides by using cyclopalladated ferrocenylimine catalyst.19 The coupling of aryl chlorides with alkynyl carboxylic acids afforded the desired product in good yield as shown in Scheme 18. They showed that aryl chlorides bearing electron-poor, electron-neutral and even inactive sterically hindered electron-rich substituents afforded the desired decarboxylative coupled products in good yields.
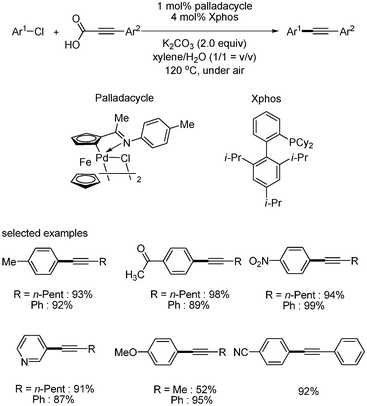 |
| | Scheme 18 Cyclopalladated ferrocenylimine-catalyzed decarboxylative couplings with aryl chlorides. | |
To reveal the real ligand and the role of the air, Wu and coworkers conducted the controlling experiments as shown in Scheme 19. They found that phosphine ligand L1 showed activity. However, phosphine oxide L2, which is easily formed from the oxidation of L1, did not give the coupled product. Based on these results, they suggested that the real ligand must be the alkyl phosphine ligand L1.
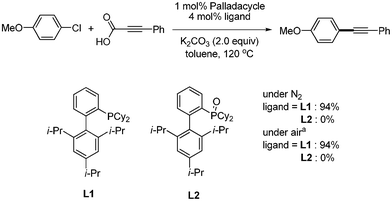 |
| | Scheme 19 Controlling experiments to define the real ligand under air, aToluene/H2O (1/1) as solvent. | |
S. Lee and coworkers also studied the mechanism of the decarboxylative coupling using gas chromatography and FT-IR spectroscopy.20 They found that the decarboxylation of phenyl propiolic acid occurred at 80 °C in the presence of DBU without any metal. Pathways A and B were suggested (Scheme 20). In path A, the decarboxylation step proceeds first, and then the decarboxylated alkyne reacts with aryl palladium(II) iodide complex. In path B, phenylpropiolic acid reacts with aryl palladium(II) iodide complex to produce the phenylpropiolate palladium complex before the decarboxylation step took place. They found that the yield of decarboxylative coupling is higher than that of the decarboxylation in 2 h at 80 °C. This result and the additional experimental data suggested that path B was suggested as the predominant pathway.
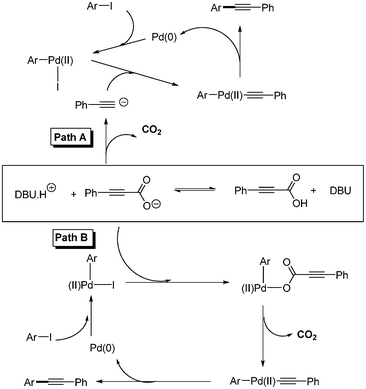 |
| | Scheme 20 Two proposed possible pathways in the decarboxylative coupling reaction. | |
2.2. Coupling reactions with aryl boronic acids
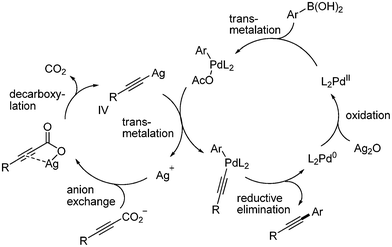
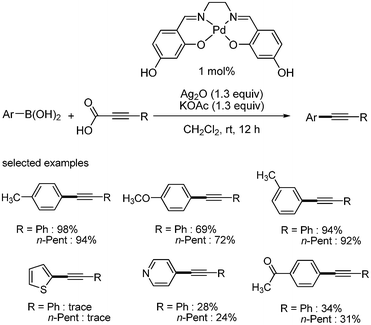
Loh developed the decarboxylative coupling reaction with aryl boronic acid instead of aryl halides.21 They employed Pd(OAc)2 (2 mol%) as a catalyst, Ag2O as an oxidant, and KOAc as a base. The desired coupled product was obtained in good yields at room temperature. As shown in Scheme 21, heteroaromatic boronic acid provided good yield of the desired product. Aryl boronic acids possessing nitro group afforded the desired coupled product without the reduction of nitro group. With the 4-bromophenyl boronic acid, the aromatic halide survived in this coupling reaction. Alkyl alkynyl carboxylic acids showed good yields in this coupling reaction. The following mechanism was proposed: aryl palladium species was formed from the transmetallation of aryl boronic acid and palladium, and alkynyl silver complex was formed from the decarboxylation of aryl alkynyl carboxylic acid. The transmetallation between these two metal complexes produced palladium complex bearing two organic ligands, aryl and alkynyl group. Reductive elimination afforded the desired product and palladium(0) species, which is then oxidized by silver to regenerate the palladium(II). It was suggested that silver oxide works as an oxidant to oxidize the palladium(0), and as a decarboxylative agent to generate the desired alkynyl metal species (Scheme 22).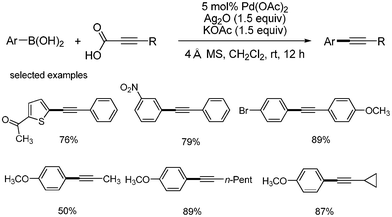 |
| | Scheme 21 Decarboxylative coupling with aryl boronic acids. | |
 |
| | Scheme 22 Proposed mechanism of decarboxylative coupling with aryl boronic acids. | |
 |
| | Scheme 23 Pd–hydroxysalen-catalyzed decarboxylative coupling with aryl boronic acids. | |
S. Lee and coworkers also reported decarboxylative coupling with aryl boronic acid using palladium hydroxysalen complex as a catalyst.22 The reaction showed good reactivity in the presence of 1 mol% of palladium hydroxyl salene complex at room temperature (Scheme 23). However, Loh and Lee had never reported that the decarboxylative coupling with propiolic acid and aryl boronic acid to give the corresponding aryl alkynes.
2.3. Decarboxylative coupling for the synthesis of diynes
Jiao and coworkers reported the synthesis of unsymmetrical diynes by the copper catalyzed decarboxylative sp–sp carbon bond formation of aryl alkynyl carboxylic acids and terminal alkynes.23 The yields of the desired products was moderate under air, but it was trace under N2. The optimized condition was that 10 mol% CuI and 10 mol% 1,10-phenanthroline was employed with Et3N (2.0 equiv.). With the optimal condition, they showed the broad scope of the substrates (Scheme 24). But 3-(furan-2-yl)propiolic acid and alkyl substituted terminal alkynes showed lower yields than those from any others.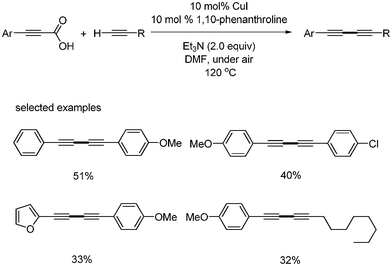 |
| | Scheme 24 Copper-catalyzed decarboxylative coupling with aryl propiolic acid with terminal alkynes. | |
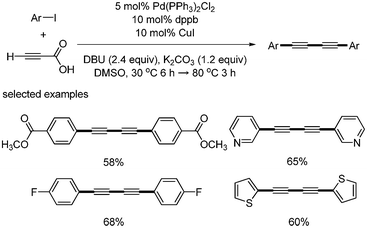
The direct synthesis of 1,4-diarylsubstituted 1,3-diynes from aryl iodides and propiolic acid was independently developed by S. Lee24 and Kim.25 They used Pd(PPh3)2Cl2 and CuI as catalysts, but different bases and solvents were employed. S. Lee used DBU for the coupling reaction of propiolic acid and aryl iodides, and used K2CO3 for the homocoupling of aryl alkynyl carboxylic acid in DMSO solvent (Scheme 25). Kim used Et3N and Ag2CO3 in DMF (Scheme 26). The major difference is that S. Lee conducted a one-pot reaction by controlling temperature, whereas Kim performed two-step reactions by the sequential addition of reagent. However, the formation of diarylalkynes as a byproduct was inevitable in both cases.
 |
| | Scheme 25 Synthesis of diynes in the presence of K2CO3. | |
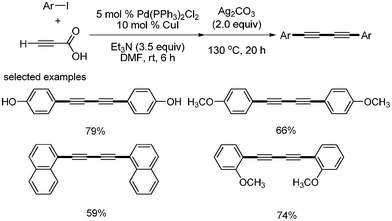 |
| | Scheme 26 Synthesis of diynes in the presence of Ag2CO3. | |
Fu reported the copper-catalyzed decarboxylative coupling reaction of potassium alkynyl carboxylates with 1,1-dibromo-1-alkenes for the synthesis of unsymmetrical diynes.26 They found that neocuproine was the best ligand than any others phosphine and amine based ligands (Scheme 27).
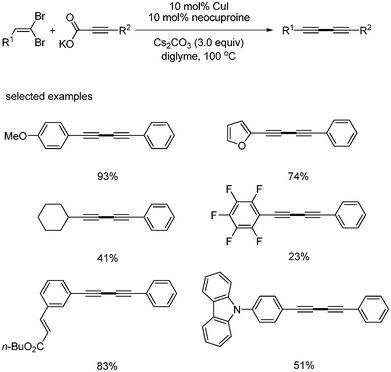 |
| | Scheme 27 Synthesis of diynes from 1,1-dibromo-1-alkenes and alkynyl carboxylic acids. | |
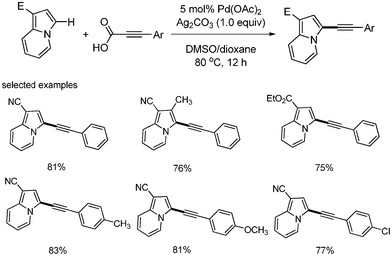 |
| | Scheme 28 C–H activation with decarboxylative coupling. | |
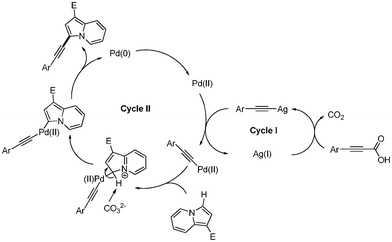 |
| | Scheme 29 Proposed mechanism of C–H activation with decarboxylative coupling. | |
2.4. Decarboxylative coupling with C–H activation
Zhao reported the synthesis of C-3 alkynylated indolizines via the C–H activation of indolizine and decarboxylation of alkynyl carboxylic acid.27 The palladium-catalyzed direct arylation of heteroaromatic compounds such as indolizines with aryl halides has been studied. This C–H activation toward decarboxylative coupling reaction was expanded. Substituted indolizines provided C–H activated coupled products in good yields in the presence of Pd(OAc)2 (5 mol%) and Ag2CO3 (1.0 equiv.) (Scheme 28). The proposed mechanism was as follows: the palladium(II) was reacted with alkynylsilver, which was formed from decarboxylation of phenyl propiolic acid, affording alkynyl palladium complex, followed by electrophilic attack of the generated Pd(II) species to indolizine group to produce alkynyl indolizinyl palladium complex. Reductive elimination of palladium complex releases coupled product and Pd(0) species, which are regenerated and oxidized to Pd(II) by silver (Scheme 29).2.5. Decarboxylative carbonylations
Sonogashira-type decarboxylative carbonylation was first reported by S. Lee.28 Alkynyl carboxylic acid was decarboxylated in a carbon monoxide atmosphere, affording carbonylative coupled product. 0.5 mol% Pd(PPh3)2Cl2 was employed without any additional ligand. When Et3N was used as a base instead of DBU, the product yields showed good. Under these optimized conditions, a variety of aryl iodides produced the desired α,β-alkynyl aryl ketones in good yields (Scheme 30).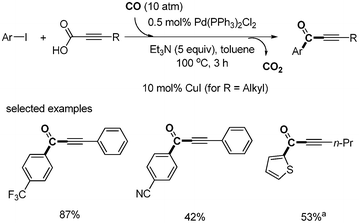 |
| | Scheme 30 Decarboxylative carbonylation, a10 mol% of CuI were used. | |
Non-carbonylative coupling product was formed as a byproduct in the presence of CuI. However, in the absence of CuI, the desired carbonylative coupling product was formed without any formation of non-carbonylative coupling product (Scheme 31).
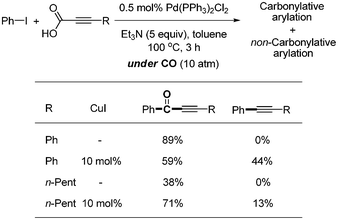 |
| | Scheme 31 Copper effect in the decarboxylative carbonylation. | |
As an expansion of decarboxylative carbonylation, S. Lee reported the synthesis of aryl alkynones from the palladium-catalyzed selective carbonylative and non-carbonylative coupling reaction of propiolic acid.29 When aryl iodide (2 equiv.) and propiolic acid (1 equiv.) were reacted under CO (8 atm) with Pd(PPh3)2Cl2 (5 mol%), CuI (10 mol%), and Et3N (6 equiv.), aryl iodide proceeded in selectively non-carbonylative and carbonylative coupling toward propiolic acid (Scheme 32). They found that alkynyl carboxylic acid prefers carbonylative coupling in both the presence and absence of CuI. However, terminal alkyne afforded carbonylative product in the presence of CuCl and non-carbonylative product in the absence of CuCl.
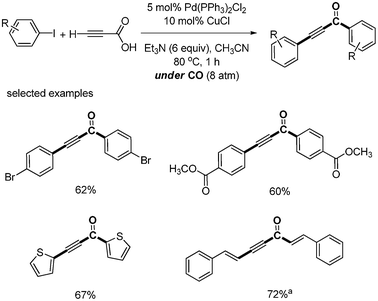 |
| | Scheme 32 One-pot synthesis of diaryl alkynones. aβ-Bromostyrene was used. | |
3. Copper-catalyzed decarboxylative couplings
Xue and coworkers first reported the copper-catalyzed decarboxylative coupling of aryl alkynyl carboxylic acid with aryl iodides and bromides.30 The catalytic system of CuI (10 mol%) and phenanthroline (10 mol%) afforded the desired product in good yields in the presence of Cs2CO3. Although the reaction temperature was high (130 °C), this reaction method showed good tolerance toward functional groups such as alcohols, amines, ketones, esters, nitros, and carboxylic acids (Scheme 33). In addition, they provided the one-pot synthesis of benzofuran from the domino reaction of 2-iodophenyl and alkynyl carboxylic acid (Scheme 34). Based on computational investigation, they suggested that the oxidative addition of aryl iodides to copper(I) first occurred and produced copper(III) complex, which subsequently reacted with alkynyl carboxylic acid to give the desired coupled product through decarboxylation and reductive elimination (Scheme 35).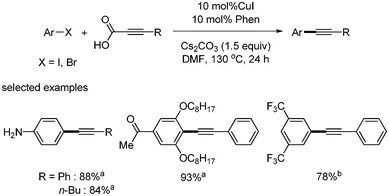 |
| | Scheme 33 Decarboxylative coupling by copper catalyst. aYield from aryl iodide; byield from aryl bromide. | |
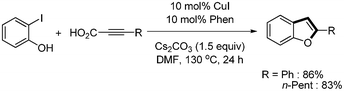 |
| | Scheme 34 Synthesis of benzofuran from the domino reaction. | |
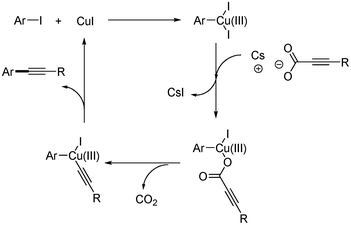 |
| | Scheme 35 Proposed mechanism of the copper-catalyzed decarboxylative coupling reaction. | |
Mao reported an improved method (lower catalyst loading) for copper-catalyzed decarboxylative coupling reactions (Scheme 36).31 They used Fe(acac)3 as a co-catalyst and reduced the amount of copper catalyst to 0.5 mol%. 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 or 1:2 Fe catalyst to Cu catalyst ratios had better results than the 1:1 ratio. The reaction was carried out with K3PO4 at 140 °C under argon. In their reaction system, the addition of palladium catalyst did not improve the yield of product (Scheme 37). Based on this result, contamination by residual transition metals in the catalytic system of Cu/Fe was ruled out.
:1 or 1:2 Fe catalyst to Cu catalyst ratios had better results than the 1:1 ratio. The reaction was carried out with K3PO4 at 140 °C under argon. In their reaction system, the addition of palladium catalyst did not improve the yield of product (Scheme 37). Based on this result, contamination by residual transition metals in the catalytic system of Cu/Fe was ruled out.
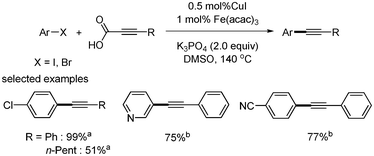 |
| | Scheme 36 Copper–iron-catalyzed decarboxylative coupling. aAryl iodide was used; baryl bromide was used. | |
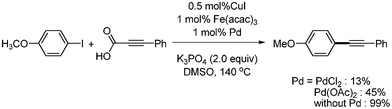 |
| | Scheme 37 Palladium effects of copper–iron-catalyzed decarboxylative coupling. | |
Two years later, a catalytic system of CuI/PPh3 without iron in the decarboxylative coupling of alkynyl carboxylic acids with aryl iodides and bromides was reported.32 Compared with the previous report, the amount of copper catalyst was increased to 2 mol%, and the reaction temperature was decreased to 90 °C. However, the reaction was still conducted under argon. Among the phosphines and amines ligands tested, PPh3 showed the best yield (Scheme 38). This catalytic system was employed for decarboxylative coupling in water.33 The reaction conditions were almost the same as those in the previous reported system. Aryl iodides were reacted in the presence of CuI (2 mol%), PPh3 (4 mol%), and K2CO3 (3 equiv.), affording the desired product in good yields (Scheme 39). In the case of aryl bromides, TBAB (tetrabutyl ammonium bromide) as the phase transfer catalyst and 2 equiv. of NaI were required for higher yields. The suggested mechanism was that copper(I) first reacts with alkynyl carboxylic acid and produces the alkynyl copper complex through decarboxylation. The alkynyl copper complex reacts with aryl halides and forms aryl alkynyl copper complex, which produces the desired product after reductive elimination. (Scheme 40). Their suggested mechanism was opposite to the previous report by Xue and coworker, in which the oxidative addition of aryl halide occurred on Cu(I) before decarboxylation.
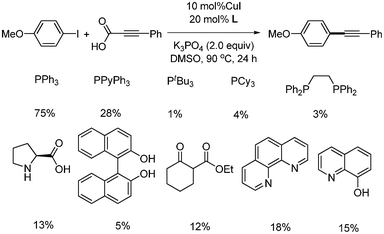 |
| | Scheme 38 Screening of ligands in the copper-catalyzed decarboxylative coupling reaction. | |
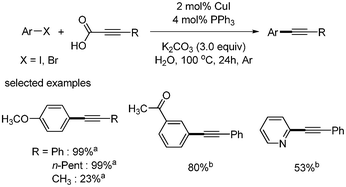 |
| | Scheme 39 Cu/PPh3-catalyzed decarboxylative coupling reactions in water. aAryl iodide was used; baryl bromide, 2 equiv. of NaI and 1 equiv. of TBAB were used. | |
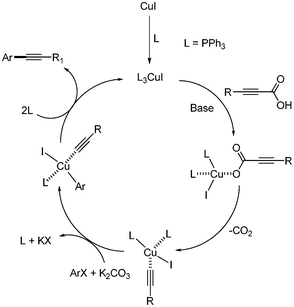 |
| | Scheme 40 Proposed mechanism of the copper-catalyzed decarboxylative coupling reaction in aqueous condition. | |
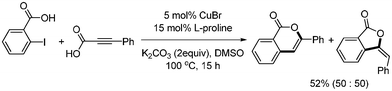
Muthusubramanian reported the copper-catalyzed synthesis of 2-arylindoles from domino decarboxylative coupling and cyclization.34 They prepared aryl alkynyl carboxylic acids from the coupling reaction of aryl iodides and propiolic acid in the presence of Pd(PPh3)2Cl2 (2 mol%), CuI (4 mol%), and iPr2NH (2.5 equiv.). When 2-iodotrifluoroacetanilide and aryl alkynyl carboxylic acids were reacted with CuBr (5 mol%), L-proline (15 mol%), and K2CO3 (2 equiv.), the desired 2-arylindole was formed in good yields (Scheme 41). When 2-iodobenzoic acid and phenyl propiolic acid were reacted under the same conditions in DMF, a mixture of isocoumarin and phthalide was formed in equal amounts with an overall yield of 52% (Scheme 42).
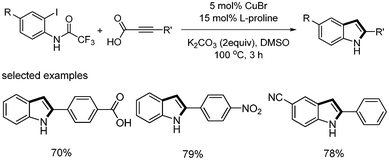 |
| | Scheme 41 Synthesis of 2-arylindoles. | |
 |
| | Scheme 42 Synthesis of isocoumarin and phthalide. | |
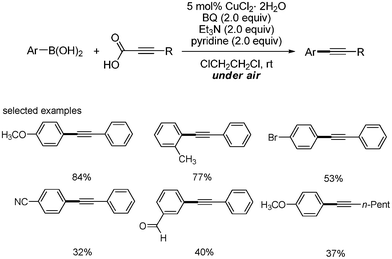
Jiao and coworkers reported the coupling reaction of aryl boronic acids instead of aryl halides by the copper catalyst.35 When benzoquinone, Et3N and pyridine (2 equivalents) and under an air atmosphere with CuCl2·2H2O showed good catalytic activity for the coupling reaction. However, the usage of the electron deficient aryl boronic acids or alkyl alkyne carboxylic acid for the reaction resulted in the low yields of the coupled products (Scheme 43).
 |
| | Scheme 43 Copper-catalyzed decarboxylative coupling of aryl boronic acids. | |
4. Nickel-catalyzed coupling reaction
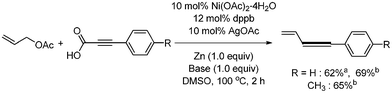
Nickel-catalyzed decarboxylative coupling reaction of alkynyl carboxylic acids were first reported by S. Lee36 Allyl acetate was employed as a coupling partner. Ni(OAc)2·4H2O (10 mol%) and dppb (12 mol%) were used as a catalyst and ligand, respectively (Scheme 44). 10 mol% AgOAc and 1 equivalent of Zn were respectively employed as an additive and reducing agent of Ni(II). The reactions were carried out at 100 °C for 0.5 h. A variety of the substituted aryl alkynylic carboxylic acid was coupled with allyl acetate, affording the desired products in good yields. Interestingly, the allenes were obtained in the presence of bases such as Cs2CO3 and DBU (Scheme 45).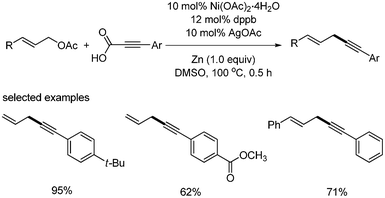 |
| | Scheme 44 Nickel-catalyzed decarboxylative coupling. | |
 |
| | Scheme 45 Synthesis of allenes, aCs2CO3 was used, bDBU was used. | |
5. Silver catalyzed-coupling reaction
The decarboxylative addition of aryl propiolic acid and NaN3 was reported by Choi and S. Lee37 Silver-decorated graphene oxide catalyst (GOSH–Ag) was prepared by depositing silver nanoparticles on thiolated grapheme oxide (GOSH) surfaces. When 1 mol% of GOSH–Ag was used in the reaction of phenyl propiolic acid and NaN3, the desired 1,2,3-triazole was formed in good yields (Scheme 46). This catalyst could easily be separated and recovered from the reaction mixture and reused several times.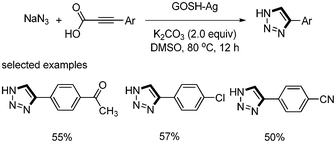 |
| | Scheme 46 Silver-catalyzed decarboxylative cyclization reactions leading to triazoles. | |
6. Synthesis of aryl alkynyl carboxylic acids
With increasing attention being paid to the decarboxylative coupling of alkynyl carboxylic acid, a variety of synthetic methods have been developed for their synthesis. There are a number of methods to synthesize alkynecarboxylic acids, including the hydrolysis of alkynyl carboxylates,38a the elimination of bromo-substituted carboxylic acids,38b the oxidation of alkynyl alcohols and aldehydes,38c and the carboxylation of alkynes.38d,38e Among these, C–H carboxylation of terminal alkynes with carbon dioxide has been studied more often and more widely used. However, this approach has several drawbacks, such as the preparation of the terminal alkyne as a starting material from the Sonogashira coupling, and the employment of carbon dioxide, which necessitates special equipment. To overcome these problems, the direct synthesis of aryl alkynyl carboxylic acid from the site-selective Sonogashira coupling reaction of propiolic acid and aryl halides has been developed.The Sonogashira coupling reaction of propioic acid and aryl bromides was first reported by Buchwald39 The reaction was carried out with Pd(CH3CN)2Cl2 and sulfonated XPhos, a water-soluble phosphine ligand (Scheme 47). The reaction was conducted in a mixture solvent of water and CH3CN. However, the desired product aryl alkynyl carboxylic acid was converted to methyl ester using trimethylsilyldiazomethane to ease purification. For this reaction to proceed the reaction temperature was set at 60 °C, and the ligand has to be prepared (Scheme 48).
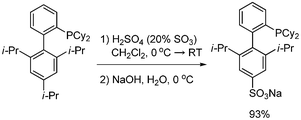 |
| | Scheme 47 Preparation of sulfonated XPhos. | |
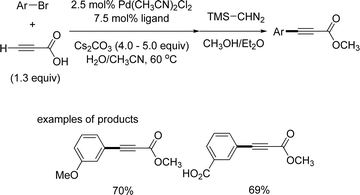 |
| | Scheme 48 Coupling reaction of propiolic acid and aryl bromides. | |
S. Lee developed the direct synthesis of aryl alkynyl carboxylic acid from the site-selective reaction of propiolic acid with aryl iodides.40 The reaction was carried out with Pd(PPh3)2Cl2 (2.5 mol%), dppb (5 mol%), and DBU (2 equiv.) at 50 °C. When Cu(acac)2 was added to the reaction mixture, the desired aryl alkynyl carboxylic acid was formed at 25 °C, and the resulting mixture afforded decarboxylated product, aryl acetylene, at 60 °C. This reaction method provided a simple and efficient tool for the synthesis of aryl alkyne carboxylic acid and aryl acetylene from aryl iodides. Under these reaction conditions, only aryl iodides coupled with propiolic acid, and produced the corresponding aryl alkynyl carboxylic acid and terminal alkynes (Scheme 49).
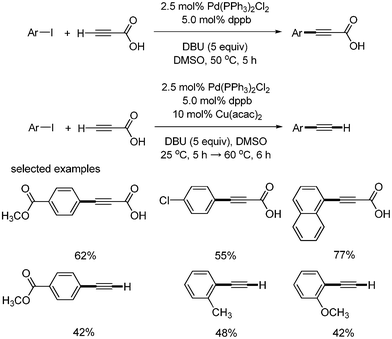 |
| | Scheme 49 Synthesis of aryl alkynyl carboxylic acids and aryl alkynes. | |
S. Lee and coworkers reported the synthesis of aryl alkynyl carboxylic acid from the Sonogashira coupling of propiolic acid and aryl bromides or aryl triflates using Pd(PPh3)4.41 The reaction temperature was 25 °C for aryl bromides bearing electron withdrawing groups, and 35 °C for those bearing electron donating groups. Their method showed good tolerance toward functional groups such as alkoxy, ketone, ester, aldehydes, cyano, nitro, and hydroxyl (Scheme 50). In addition, they found that propiolic acid showed higher reactivity than other terminal alkynes. Among terminal alkynes, only propiolic acid coupled with 4-tert-butylbromobenzene to produce the desired product in 83% yield (Scheme 51). They found that the oxidative adduct palladium complex reacted with propiolic acid to produce the coupled product, even in the presence of phenyl acetylene (Scheme 52). Based on mechanistic studies using chronoamperometric analysis,41 it was suggested that the key reaction step for the high reactivity of propiolic acid might be ligand exchange between the acetylide and bromide at palladium, and/or reductive elimination, but not the oxidative addition step.
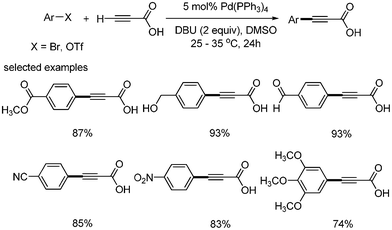 |
| | Scheme 50 Synthesis of aryl alkynyl carboxylic acids from aryl bromides. | |
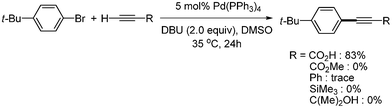 |
| | Scheme 51 Sonogashira reaction with terminal alkynes. | |
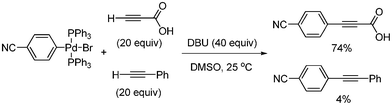 |
| | Scheme 52 Comparative coupling reaction of phenyl acetylene and propiolic acid. | |
7. Decarboxylation
Kolarovic first reported the copper-catalyzed decarboxylation of alkynyl carboxylic acids.42 The catalytic systems of Pd(OH)2/C did not give satisfactory results in the decarboxylations (Scheme 53). The best result was obtained when CuCl (5 mol%) was employed in CH3CN at 60 °C. It was found that the presence of 3 equivalents of Et3N accelerated the reaction rate in some cases (Scheme 54).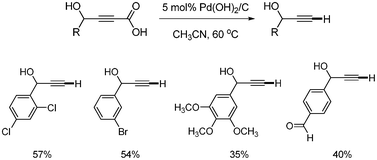 |
| | Scheme 53 Decarboxylation using by Pd(OH)2/C. | |
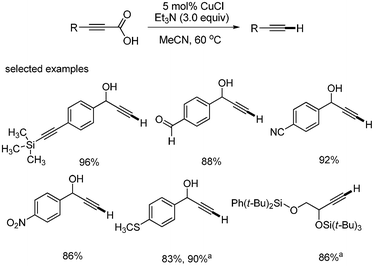 |
| | Scheme 54 Copper-catalyzed decarboxylation. aEt3N 3.0 equiv. was used. | |
8. Coupling reactions for the formation of C–N C−1–P/C–S bonds
Decarboxylative carbon–nitrogen bond formation was reported by Jiao43 The coupling reaction of alkynyl carboxylic acids and amides afforded the desired ynamides via copper-catalyzed oxidative amidation. The optimized conditions involved CuCl2·2H2O (10 mol%) as a catalyst and Na2CO3 as a base. In addition, air was needed as an oxidant to complete the catalytic cycle. A variety of substituted aryl alkynyl carboxylic acids showed good yields, but only secondary amines bearing electron withdrawing groups such as carbonyl and sulfonyl produced the desired C–N bonded products. Tolerance toward functional groups such as ester, aldehyde, and sulfone was demonstrated (Scheme 55). The mechanism shown in Scheme 56 was suggested showing that alkynyl copper(II) was formed via the decarboxylation of copper(II) intermediate. Nucleophilic attack by the amide compound affords the Cu(II)(alkynyl)(amidate) intermediate, followed by reductive elimination leading to the desired product. Cu(0) was reoxidized by air to fulfill the catalytic cycle (Scheme 56).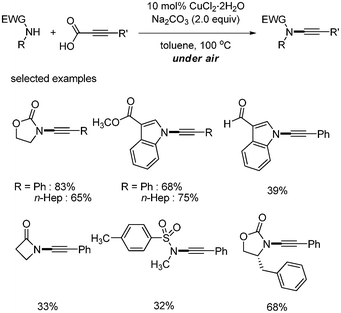 |
| | Scheme 55 Decarboxylative C–N bond formation. | |
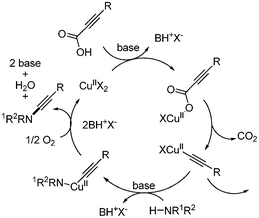 |
| | Scheme 56 Proposed mechanism of decarboxylative C–N bond formation. | |
Yang and coworkers have developed the C–P bond formation of aryl alkynyl carboxylic acids with HP(O)R2 through the catalytic combined efforts of both palladium and copper (Scheme 57).44 The usage of 1,10-phenanthroline and PPh3 as ligands, and AgOAc as oxidant in NMP provided good yields of the coupling reaction products.
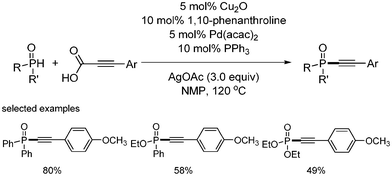 |
| | Scheme 57 Decarboxylative C–P bond formation. | |
Liu reported copper-catalyzed decarboxylative C–S cross-coupling.45 When alkynyl carboxylic acid and thiol were reacted in the presence of CuI (4 mol%) and Cs2CO3 (1.2 equiv.), vinyl sulfides were formed, not the alkynyl sulfides. All thiols afforded anti-Markovnikov coupling products in good to excellent yields with high stereoselectivity for Z-isomers. Importantly, this method was tolerant of a broad range of functional groups, including amines, alcohols, halides, and nitrogen-containing heterocycles. The tolerance of halides enables further derivatization through cross-coupling reactions (Scheme 58).
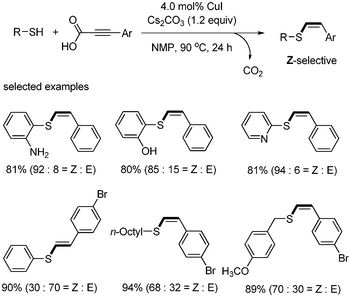 |
| | Scheme 58 Decarboxylative C–S bond formation. | |
9. Multicomponent coupling reactions
Van der Eycken and coworkers reported the synthesis of 1,4-diamino-2-butynes using copper-catalyzed one-pot coupling reaction of propiolic acid, aldehyde, and amine.46 Symmetrical substituted diamino alkynes were obtained when the reaction was conducted under microwave conditions. Alkyl aldehydes showed good yields, but benzaldehyde gave only 28% yield. Only secondary amines afforded the desired product. The primary amine produced only trace amounts of the desired product (Scheme 59).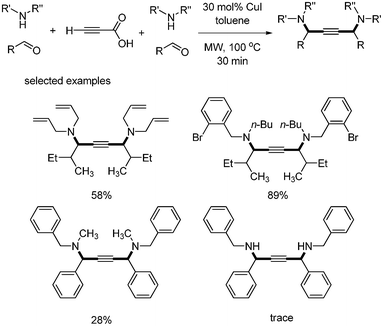 |
| | Scheme 59 Synthesis of 1,4-diamino-2-butynes from three-component reaction. | |
Three-component click-chemistry coupling reaction was reported by Kolarovic.47 Aryl iodides, NaN3, and alkynyl carboxylic acid were reacted in the presence of CuSO4·5H2O (10 mol%), sodium ascorbate (20 mol%), L-proline (20 mol%), and K2CO3 (1.2 equiv.) to afford the desired 1,2,3-triazoles in good yields. This method avoids the usage of volatile low-molecular-weight terminal alkynes, reduces the handling of potentially unstable and explosive azides, and provides the desired product with good purity without any additional purification steps. In addition, alternative reaction conditions were provided to allow for both non-decarboxylative and decarboxylative coupling product (Scheme 60). Similar decarboxylative click-chemistry reaction was reported by Schoffstall48 They employed alpha-bromoacetophenone instead of aryl halides (Scheme 61).
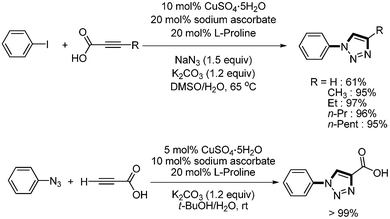 |
| | Scheme 60 Synthesis of 1,2,3,-triazole from alkynyl carboxylic acid. | |
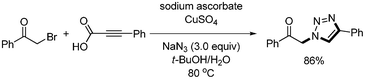 |
| | Scheme 61 Three-component reaction for the synthesis of 1,2,3-triazole. | |
S. Lee reported the copper-catalyzed one-pot synthesis of imidazo[1,2-a]pyridines from the three-component reaction of 2-aminopyridine, aldehyde, and alkynyl carboxylic acids.49 The catalysts of CuI and Cu(OTf)2 afforded the desired imidazo[1,2-a]pyridines in good yields. Good yields were obtained with propiolic acid (Scheme 62). Aryl alkynyl carboxylic acids were prepared in situ, and used for the coupling reaction with 2-aminopyridine and benzaldehyde. Based on this concept, S. Lee also reported the four-component reactions of 2-aminopyridine, aldehyde, aryl iodide and propiolic acid to produce the desired imidazo[1,2-a]pyridine in good yields. (Scheme 63). In the reaction with propiolic acid, two pathways were proposed to form 3-methyl-2-phenylimidazo[1,2-a]pyridine: path A, in which the decarboxylative coupling reaction occurs first, and path B, in which the reaction at the terminal alkynyl carbon occurs first. Deuterium exchange experiments showed that dideuterated product was formed much more than trideuterated product. This result suggested that two alternative pathways, C and D, are both involved in the reaction, but that path C dominates over path D (Scheme 64).
![Three-component reaction for the synthesis of imidazo[1,2-a]pyridines. a2-Pentynoic acid was used; bpropiolic acid was used.](/image/article/2013/RA/c3ra41442f/c3ra41442f-s62.gif) |
| | Scheme 62 Three-component reaction for the synthesis of imidazo[1,2-a]pyridines. a2-Pentynoic acid was used; bpropiolic acid was used. | |
![Four-component reaction for the synthesis of imidazo[1,2-a]pyridines.](/image/article/2013/RA/c3ra41442f/c3ra41442f-s63.gif) |
| | Scheme 63 Four-component reaction for the synthesis of imidazo[1,2-a]pyridines. | |
![Proposed mechanism of the decarboxylative coupling reaction in the synthesis of imidazo[1,2-a]pyridines.](/image/article/2013/RA/c3ra41442f/c3ra41442f-s64.gif) |
| | Scheme 64 Proposed mechanism of the decarboxylative coupling reaction in the synthesis of imidazo[1,2-a]pyridines. | |
10. Applications
T. C. Lin and coworkers synthesized a series of star-shaped multipolar chromophores containing functionalized quinoxaline and quinoxalinoid units that showed two-photon absorption properties, both in the femtosecond and the nanosecond time domain.50 For the synthesis of diaryl alkyne precursor, 2-butynedioic acid was employed as an alkyne source, which was developed by S. Lee, instead of using their previous methodology.13 The desired diaryl alkyne was successfully obtained in 85% yield from the coupling reaction of aryl bromides and 2-butynedioic acid (Scheme 65).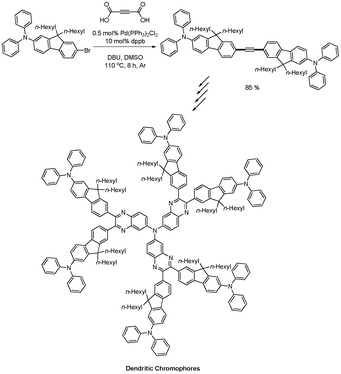 |
| | Scheme 65 Synthesis of star-shaped multipolar chromophores. | |
Loye synthesized unsymmetrically substituted heterocyclic alkyne 5-(pyridin-3-ylethynyl) picolinonitrile via one-pot Sonogashira and decarboxylative coupling reaction, which was developed in 2008 by S. Lee.9 3-Iodopyridine was employed for the Sonogashira coupling with propiolic acid, and 5-bromo-2-cyanopyridine was employed for the decarboxylative coupling.51 The coupled product was transformed into the corresponding acid and employed as a ligand for the construction of two-dimensional lead(II) coordination polymer (Scheme 66).
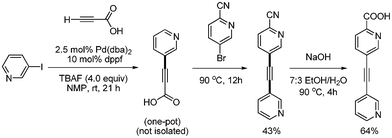 |
| | Scheme 66 Synthesis of unsymmetrically substituted heterocyclic alkyne. | |
Skrydstrup reported the synthesis of two isotopically labeled variants of the β-amyloid binding compound FSB.52 In the course of the synthesis of the final products, one-pot Sonogashira and decarboxylative coupling reaction were used instead of the previous three-step synthetic procedure which was Sonogashira coupling, deprotection and another Sonogashira coupling. The desired coupled product was obtained in 43% yield from 1,4-dibromo-2-fluorobenzene, 2-chloro-4-iodoaniline, and propiolic acid under a catalytic system of Pd(PPh3)2Cl2 and dppf. The yield of this reaction is an impressive 81% per C–C bond formation for the four generated C–C bonds. Aryl bromide was used as the first substrate, and aryl iodide was used as the second substrate, which was opposite to the method9,13 employed by S. Lee (Scheme 67).
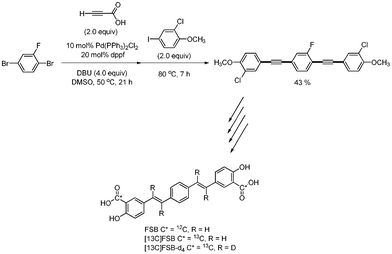 |
| | Scheme 67 Synthesis of the fibril binding compound FSB. | |
11. Conclusion
Over the past five years, great advances have been made in the decarboxylative coupling reaction of alkynyl carboxylic acids by transition-metal catalysts. Despite the many significant developments in this field, there remain a number of unsolved problems. Until now, only one example of C–H activation has been reported. In addition, no coupling with alkyl group has been developed. Therefore, further studies are needed to achieve high activity and versatility in these areas. It has been known that the decarboxylation of alkynyl carboxylic acid occurs in the absence of transition-metal catalyst.20 Based on this concept, it is expected that transition-metal-free decarboxylative coupling reactions will be developed.Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2012R1A1B3000871)References
- E. I. Negishi, Handbook of Organopalladium Chemistry for Organic Synthesis, Wiley-Interscience, New York, 2002 Search PubMed.
- A. de Meijere and F. Diederich, in Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH, Germany, 2nd edn, 2004 Search PubMed.
-
(a) A. K. Steib, T. Thaler, K. Komeyama, P. Mayer and P. Knochel, Angew. Chem., Int. Ed., 2011, 50, 3303 CrossRef CAS;
(b) K.-H. Wu and H.-M. Gau, J. Am. Chem. Soc., 2006, 128, 14808 CrossRef CAS;
(c) G. Manolikakes, C. M. Hernandez, M. A. Schade, A. Metzger and P. Knochel, J. Org. Chem., 2008, 73, 8422 CrossRef CAS;
(d) M. E. Doster, J. A. Hatnean, T. Jeftic, S. Modi and S. A. Johnson, J. Am. Chem. Soc., 2010, 132, 11923 CrossRef CAS;
(e) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS;
(f) A. Postigo and R. A. Rossi, Org. Lett., 2001, 3, 1197 CrossRef CAS.
-
(a) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS;
(b) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068 RSC;
(c) J. Wencel-Delord, T. Droge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC;
(d) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 Search PubMed;
(e) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC;
(f) T. C. Boorman and I. Larrosa, Chem. Soc. Rev., 2011, 40, 1910 RSC;
(g) J. Q. Yu and Z. J. Shi, C–H Activation, Springer, Berlin, Germany, 2010.
- M. Nilsson, Acta Chem. Scand., 1966, 20, 423 CrossRef CAS.
- A. G. Myers, D. Tanaka and M. R. Mannion, J. Am. Chem. Soc., 2002, 124, 11250 CrossRef CAS.
- L. J. Goossen, G. Deng and L. M. Levy, Science, 2006, 313, 662 CrossRef.
-
(a) N. Rodriguez and L. J. Goossen, Chem. Soc. Rev., 2011, 40, 5030 RSC;
(b) R. Shang and L. Liu, Sci. China: Chem., 2011, 54, 1670 Search PubMed.
- J. Moon, M. Jeong, H. Nam, J. Ju, J. H. Moon, H. M. Jung and S. Lee, Org. Lett., 2008, 10, 945 CrossRef CAS.
- J. Moon, M. Jang and S. Lee, J. Org. Chem., 2009, 74, 1403 CrossRef CAS.
- H. Kim and P. H. Lee, Adv. Synth. Catal., 2009, 351, 2827 CrossRef CAS.
- W.-W. Zhang, X.-G. Zhang and J.-H. Li, J. Org. Chem., 2010, 75, 5259 CrossRef CAS.
- K. Park, G. Bae, J. Moon, J. Choe, K. H. Song and S. Lee, J. Org. Chem., 2010, 75, 6244 CrossRef CAS.
- H. J. Lee, K. Park, G. Bae, J. Choe, K. H. Song and S. Lee, Tetrahedron Lett., 2011, 52, 5064 Search PubMed.
- S. Tartaggia, O. D. Lucchi and L. J. Goossen, Eur. J. Org. Chem., 2012, 1431 Search PubMed.
- K. Park, G. Bae, A. Park, Y. Kim, J. Choe, K. H. Song and S. Lee, Tetrahedron Lett., 2011, 52, 576 CrossRef CAS.
- A. Pyo, J. D. Kim, H. C. Choi and S. Lee, J. Organomet. Chem., 2013, 724, 271 Search PubMed.
- P. V. Reddy, P. Srinivas, M. Annapurna, S. Bhargava, J. Wagler, N. Mirzadeh and M. L. Kantam, Adv. Synth. Catal., 2013, 355, 705 Search PubMed.
- X. Li, F. Yang and Y. Wu, J. Org. Chem., 2013, 78, 4543 Search PubMed.
- A. Pyo, Y. H. Kim, K. Park, G. C. Kim, H. C. Choi and S. Lee, Appl. Organomet. Chem., 2012, 26, 650 Search PubMed.
- C. Feng and T.-P. Loh, Chem. Commun., 2010, 46, 4779 RSC.
- Y. Heo, Y. Y. Kang, T. Palani, J. Lee and S. Lee, Inorg. Chem. Commun., 2012, 23, 1 Search PubMed.
- M. Yu, D. Pan, W. Jia, W. Chen and N. Jiao, Tetrahedron Lett., 2010, 51, 1287 CrossRef CAS.
- Y. Kim, A. Park, K. Park and S. Lee, Tetrahedron Lett., 2011, 52, 1766 Search PubMed.
- J. Park, E. Park, A. Kim, S.-A. Park, Y. Lee, K.-W. Chi, Y. H. Jung and I. S. Kim, J. Org. Chem., 2011, 76, 2214 CrossRef CAS.
- Z. Huang, R. Shang, Z.-R. Zhang, X.-D. Tan, X. Xiao and Y. Fu, J. Org. Chem., 2013, 78, 4551 Search PubMed.
- B. Zhao, Org. Biomol. Chem., 2012, 10, 7108 RSC.
- A. Park, K. Park, Y. Kim and S. Lee, Org. Lett., 2011, 13, 944 CrossRef CAS.
- W. Kim, K. Park, A. Park, J. Choe and S. Lee, Org. Lett., 2013, 14, 1654 Search PubMed.
- D. Zhao, C. Gao, X. Su, Y. He, J. You and Y. Xue, Chem. Commun., 2010, 46, 9049 RSC.
- J. Mao, M. Wu, G. Xie and S. Ji, Adv. Synth. Catal., 2009, 351, 2101 CrossRef CAS.
- X. Qu, T. Li, P. Sun, Y. Zhu, H. Yang and J. Mao, Org. Biomol. Chem., 2011, 9, 6938 RSC.
- T. Li, P. Sun, H. Yang, Y. Zhu, H. Yan, L. Lu and J. Mao, Tetrahedron, 2012, 68, 6413 Search PubMed.
- T. Ponpandian and S. Muthusubramanian, Tetrahedron Lett., 2012, 53, 4248 Search PubMed.
- L. Shi, W. Jia, X. Li and N. Jiao, Tetrahedron Lett., 2013, 54, 1951 Search PubMed.
- J. Choe, J. Yang, K. Park, T. Palani and S. Lee, Tetrahedron Lett., 2012, 53, 6908 Search PubMed.
- J. D. Kim, T. Palani, M. R. Kumar, S. Lee and H. C. Choi, J. Mater. Chem., 2012, 22, 20665 RSC.
-
(a) A. S.-Y. Lee, Y.-J. Hu and S.-F. Chu, Tetrahedron, 2001, 57, 2121 CrossRef CAS;
(b) A. Katritzky, S. Ozcan and E. Todadze, Org. Biomol. Chem., 2010, 8, 1296 RSC;
(c) E. Dalcanale and F. Montanari, J. Org. Chem., 1986, 51, 567 CrossRef;
(d) X. Zhang, W.-Z. Zhang, X. Ren, L.-L. Zhang and X.-B. Lu, Org. Lett., 2011, 13, 2402 CrossRef CAS;
(e) X. Wang, Y. N. Lim, C. Lee, H.-Y. Jang and B. Y. Lee, Eur. J. Org. Chem., 2013, 1867 Search PubMed.
- K. W. Anderson and S. L. Buchwald, Angew. Chem., 2005, 117, 6329 Search PubMed.
- K. Park, T. Palani, A. Pyo and S. Lee, Tetrahedron Lett., 2012, 53, 733 CrossRef CAS.
- K. Park, J.-M. You, S. Jeon and S. Lee, Eur. J. Org. Chem., 2013, 1973 Search PubMed.
- A. Kolarovic and Z. Faberova, J. Org. Chem., 2009, 74, 7199 CrossRef CAS.
- W. Jia and J. Jiao, Org. Lett., 2010, 12, 2000 CrossRef CAS.
- J. Hu, N. Zhao, B. Yang, G. Wang, L.-N. Guo, Y.-M. Liang and S.-D. Yang, Chem.–Eur. J., 2011, 17, 5516 CrossRef CAS.
- S. Ranjit, Z. Duan, P. Zhang and X. Liu, Org. Lett., 2010, 12, 4134 CrossRef CAS.
- H. Feng, D. S. Ermolat'ev, G. Song and E. V. Van der Eycken, J. Org. Chem., 2012, 77, 5149 Search PubMed.
- A. Kolarovic, M. Schnurch and M. D. Mihovilovic, J. Org. Chem., 2011, 76, 2613 CrossRef CAS.
- D. E. Mendes and A. M. Schoffstall, J. Chem. Educ., 2011, 88, 1582 Search PubMed.
- T. Palani, K. Park, M. R. Kumar, H. M. Jung and S. Lee, Eur. J. Org. Chem., 2012, 5038 Search PubMed.
- T.-C. Lin, Y.-H. Lee, C.-Y. Liu, B.-R. Huang, M.-Y. Tsai, Y.-J. Huang, J.-H. Lin, Y.-K. Shen and C.-Y. Wu, Chem.–Eur. J., 2013, 19, 749 Search PubMed.
- R. C. Severance, E. S. Rountree, M. D. Smith and H.-C. zur Loye, Solid State Sci., 2012, 14, 1512 Search PubMed.
- M. N. Burhardt, R. Taaning, N. C. Nielsen and T. Skrydstrup, J. Org. Chem., 2012, 77, 5357 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.