
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReassessing anionic redox in conventional layered oxide cathodes for Li-ion batteries: ionic and covalent mechanisms†
Jianhua
Yin
a,
Zixin
Wu
*a,
Kai
Fang
a,
Yuanlong
Zhu
a,
Kang
Zhang
a,
Haitang
Zhang
a,
Yilong
Chen
a,
Li
Li
a,
Longlong
Fan
b,
Kang
Dong
 b,
Lirong
Zheng
b,
Qingsong
Wang
*c,
Huan
Huang
*b,
Jing
Zhang
*b,
Yu
Qiao
*a and
Shi-Gang
Sun
a
b,
Lirong
Zheng
b,
Qingsong
Wang
*c,
Huan
Huang
*b,
Jing
Zhang
*b,
Yu
Qiao
*a and
Shi-Gang
Sun
a
aState Key Laboratory of Physical Chemistry of Solid Surfaces, iChEM (Collaborative Innovation Center of Chemistry for Energy Materials), Department of Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, 361005, P. R. China. E-mail: zixinwu_gz@163.com; yuqiao@xmu.edu.cn
bBeijing Synchrotron Radiation Facility, Institute of High Energy Physics, Chinese Academy of Sciences, Beijing, 100049, P. R. China. E-mail: huanhuang@ihep.ac.cn; jzhang@ihep.ac.cn
cBavarian Center for Battery Technology (BayBatt), Department of Chemistry, University of Bayreuth, Universitätsstrasse 30, Bayreuth 95447, Germany. E-mail: qingsong.wang@uni-bayreuth.de
First published on 20th March 2025
Abstract
Efforts to improve the specific capacity and energy density of lithium nickel–cobalt–manganese oxide (NCM) cathodes focus on operating at high voltages or increasing nickel content. However, both approaches necessitate a thorough understanding of the charge compensation mechanism. Traditional ionic-bonding models which separate transition metal (TM) and oxygen redox processes prove inadequate as anionic redox becomes significant, ignoring crucial metal–oxygen interactions. In this study, we systematically investigate the charge compensation process in low-nickel and high-nickel NCMs under high-voltage conditions. Here, the involvement of oxygen is critical in redox, as it shares electrons with TM to form a strong TM–O covalent bond. Compared to low-Ni NCMs, high-Ni NCMs exhibit an oxygen dimerization stage with trapped O2, which leads to the aggregation of vacancies in the transition metal layer, thereby accelerating structural destabilization. This variation in oxygen dimerization behavior among NCMs is closely correlated with differences in elemental composition, spin states, and stacking faults. Our findings comprehensively reveal the redox behaviors of transition metals and oxygen, particularly highlighting oxygen behavior at each delithiation state, helping to optimize the utilization of oxygen redox reactions in commercial NCM compounds for high-capacity and high-energy-density lithium-ion batteries.
Introduction
Lithium nickel cobalt manganese oxide (LiNixCoyMnzO2, NCM) holds a competitive advantage in the high-end power battery market due to its high specific capacity, high energy density, and progressively optimized cost and safety.1–6 Generally, Ni serves as the active component, enhancing the specific capacity; Co maintains the stability of the layered cathode, thereby improving the rate capability; and Mn acts as an electrochemically inactive component, providing backbone support to preserve the crystal structure during lithium-ion (de)intercalation processes.2 Efforts to fully realize the benefits of NCM compounds for high-capacity, high-power batteries while reducing costs are focused on widening the operational voltage windows for low- to medium-Ni layered oxide electrodes and increasing the Ni content.1 However, these strategies are not straightforward, as increasing either the charging voltage or the nickel content can lead to degradation of the cathode material structure.1,2 This structural degradation is associated with the charge compensation process during delithiation, particularly at high levels of delithiation. Therefore, it is essential to accurately describe the charge compensation mechanism, to inform strategies that enhance electrochemical performance while maintaining stability.Operating the NCM cathodes at high voltage leads to oxygen involvement in the redox process, contributing to the increased instability of NCMs.7 Therefore, controlling the involvement of oxygen is a critical factor for achieving high voltage cycling while maintaining stability. Anion redox reactions have been extensively studied in lithium-rich layered oxide compounds, as they exhibit extra capacity beyond the cation redox reactions due to the ability to extract electrons from the non-bonding O 2p states along the Li–O–Li configuration.8 However, these compounds suffer from voltage hysteresis and significant stability issues, which remain unavoidable to date.9,10 Therefore, extending the concept of oxygen redox reactions to NCM cathodes presents a critical challenge and gives rise to several issues, such as the significance of anionic redox in NCM, its impact on the layered structure, and how to effectively regulate anionic redox, all of which remain ambiguous.
Generally, the charge compensation mechanism in layered oxide cathodes is typically described using a simple ionic-bonding model, which provides a straightforward description of the redox sequence.11 In this model, TM cations and O anions are considered separate from each other, each possessing discrete electronic bands. The energy of the TM 3d orbitals is higher than that of the O 2p orbitals, and thus, the redox bands are assumed to originate solely from the TM 3d states. However, due to the interaction between TM and O, particularly in compounds containing late-transition metals (e.g., Co, Ni), the activity of oxygen becomes strongly pronounced. This interaction is often overlooked when describing the charge compensation process in NCMs. Considering the electron interactions and charge-transfer effects, the ionic model can be improved using the Mott–Hubbard model, which incorporates the ligand-to-metal charge transfer (LMCT) effect, providing a more accurate representation of the interactions in correlated metal-oxide compounds.12,13 Charge fluctuations between TM 3d states can be suppressed due to the Coulomb interaction of the TM 3d electrons Udd, which describes the energy required to place two electrons into the same d site, thus introducing a band gap between unoccupied (upper Hubbard, UHB) and occupied (lower Hubbard, LHB) states.14 The ligand-to-metal charge transfer describes the process where the ligand shares electrons with the metal, creating electron holes on the ligand.15 The interactions between TM and O vary due to differences in the d-electron count, coordination environment, point group symmetries, and the effective charge of the central metal cation.15 The late transition metals exhibit negative charge transfer energy, causing the lower Hubbard band to merge with the anionic O 2p band, leading to the O sharing electron with metal.12 This implies that metal and oxygen form a correlated electronic system. As a result, the correlated electronic system complicates the assignment of oxidation states for both the TM and O. This complexity obscures the nature of the TM and O redox processes, underscoring the importance of using accurate and appropriate models when investigating charge compensation mechanisms. Furthermore, the precise mechanisms by which oxygen participates in charge compensation remain unclear, as does the connection to the O–O dimerization and oxygen evolution.
In this study, we systematically investigate the evolution of the electronic and geometric structures of two representative NCM cathodes, low-Ni and high-Ni, during high-voltage charging using various synchrotron X-ray spectroscopy techniques. As discussed in detail later, we refute the one-electron ionic approximation for describing the multi-electron system, as it fails to accurately interpret the redox reactions, particularly those involving oxygen. With the aid of various X-ray spectroscopy techniques, we have elucidated a comprehensive picture of the charge compensation process in NCM materials. We found that TM ionic redox reactions only occur at relatively low potentials. At a certain level of delithiation, the covalency of the system becomes strong enough to create electron holes in the oxygen, in other words, oxygen begins to participate in redox reactions through hybridization with TM before reaching the formal anion redox reaction region. In addition, pure oxygen oxidation occurs at a high delithiation state, providing extra capacity. This is achieved through cation disorder, resulting from Li/Ni mixing during synthesis or TM migration during the charging. Additionally, we further investigate the oxygen electronic structure, particularly the potential for dimerization, using high-resolution X-ray spectroscopy. The result elucidates the effects of elemental compositions on dimerization and how the oxygen redox reaction contributes to structural degradation. Our findings provide a more accurate description of the charge compensation mechanisms in NCMs, aligning more closely to the actual situations. This understanding enables more informed regulation of anion redox in NCMs, which will significantly contribute to the development of next-generation lithium-ion battery cathodes with enhanced capacity and stability.
Results and discussion
NCM structural information extracted from XAS characterization
LiNixCoyMnzO2 are conventional cathode materials that crystallize in a layer structure characterized by a hexagonal α-NaFeO2 structure with the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group (Fig. 1a). Operating these conventional compounds at high cut-off voltages, approaching their theoretical capacities, has emerged as a pivotal strategy to fully exploit the benefits of layered materials for high-capacity battery electrodes.1,2 Moreover, increasing the energy density of Li-ion battery cathodes through high-voltage operation necessitates a comprehensive understanding of the roles of transition metals and oxygen in the charge compensation process. The charge compensation mechanisms in these materials are determined by the characteristic features of valence electron states (VES).11 Consequently, investigating the VES is the main object of understanding the charge compensation mechanisms. X-ray absorption and emission spectroscopies serve as potent techniques to examine the VES, where a core-level electron is excited by the incident X-ray, and the VES are detected with the core electron state or core hole state as a probe.16,17 In this study, the roles of TMs and oxygen in the charge compensation process during delithiation were investigated using various X-ray spectroscopies (Fig. 1b). Typically, hard X-ray absorption spectroscopy, also known in nomenclature as X-ray Absorption Fine-Structure (XAFS) and beneficial for the TM K-edge, encompasses the X-ray Absorption Near-Edge Structure (XANES) region and the Extended X-ray Absorption Fine-Structure (EXAFS) region. The former is sensitive to the oxidation state of the absorber, and the shape of such spectra can be modulated by the average local coordination environment around the absorber.18 Meanwhile, the latter provides fine details of the partial pair distribution function of atoms surrounding the absorber, including distances, coordination numbers, and the Debye–Waller factor.19 Soft X-ray absorption spectroscopy, known as Near-Edge X-ray Absorption Fine Structure (NEXAFS), is particularly beneficial for the TM L-edge and 2p K-edge. This powerful and well-established technique provides crucial insights into the electronic structures of unoccupied states at the VES.20 In NEXAFS, the analysis of TM L-edges focuses on the spectral shifts, integrated intensity, branching ratio, and the crystal-field multiplet and charge-transfer multiplet calculations, which help determine the oxidation and spin states of TMs.18,20 By combining the soft X-ray TM L-edge and the hard X-ray TM K-edge, the TM 3d and 4p valence states can be probed, respectively. Resonant inelastic X-ray scattering (RIXS) is a powerful technique that combines X-ray absorption and X-ray emission to probe the electronic structure of materials.17 RIXS has recently been demonstrated to be an effective technique for fingerprinting the oxygen redox chemistry in TM oxide electrodes.9,21 In summary, the integrated application of XAS and RIXS to investigate the valence electron states of the TMs and oxygen affords a relatively comprehensive understanding of the charge compensation mechanism of NCM cathode materials during the Li-ion deintercalated process. Fig. 1a illustrates the first charge curves for LiNi1/3Co1/3Mn1/3O2 (NCM333) and LiNi0.8Co0.1Mn0.1O2 (NCM811), with each material charged to 4.8 V (vs. Li/Li+), respectively. To investigate the charge compensation mechanisms, X-ray spectroscopy measurements were conducted on a series of ex situ samples at different states of charge, as highlighted by the dot mark.
m space group (Fig. 1a). Operating these conventional compounds at high cut-off voltages, approaching their theoretical capacities, has emerged as a pivotal strategy to fully exploit the benefits of layered materials for high-capacity battery electrodes.1,2 Moreover, increasing the energy density of Li-ion battery cathodes through high-voltage operation necessitates a comprehensive understanding of the roles of transition metals and oxygen in the charge compensation process. The charge compensation mechanisms in these materials are determined by the characteristic features of valence electron states (VES).11 Consequently, investigating the VES is the main object of understanding the charge compensation mechanisms. X-ray absorption and emission spectroscopies serve as potent techniques to examine the VES, where a core-level electron is excited by the incident X-ray, and the VES are detected with the core electron state or core hole state as a probe.16,17 In this study, the roles of TMs and oxygen in the charge compensation process during delithiation were investigated using various X-ray spectroscopies (Fig. 1b). Typically, hard X-ray absorption spectroscopy, also known in nomenclature as X-ray Absorption Fine-Structure (XAFS) and beneficial for the TM K-edge, encompasses the X-ray Absorption Near-Edge Structure (XANES) region and the Extended X-ray Absorption Fine-Structure (EXAFS) region. The former is sensitive to the oxidation state of the absorber, and the shape of such spectra can be modulated by the average local coordination environment around the absorber.18 Meanwhile, the latter provides fine details of the partial pair distribution function of atoms surrounding the absorber, including distances, coordination numbers, and the Debye–Waller factor.19 Soft X-ray absorption spectroscopy, known as Near-Edge X-ray Absorption Fine Structure (NEXAFS), is particularly beneficial for the TM L-edge and 2p K-edge. This powerful and well-established technique provides crucial insights into the electronic structures of unoccupied states at the VES.20 In NEXAFS, the analysis of TM L-edges focuses on the spectral shifts, integrated intensity, branching ratio, and the crystal-field multiplet and charge-transfer multiplet calculations, which help determine the oxidation and spin states of TMs.18,20 By combining the soft X-ray TM L-edge and the hard X-ray TM K-edge, the TM 3d and 4p valence states can be probed, respectively. Resonant inelastic X-ray scattering (RIXS) is a powerful technique that combines X-ray absorption and X-ray emission to probe the electronic structure of materials.17 RIXS has recently been demonstrated to be an effective technique for fingerprinting the oxygen redox chemistry in TM oxide electrodes.9,21 In summary, the integrated application of XAS and RIXS to investigate the valence electron states of the TMs and oxygen affords a relatively comprehensive understanding of the charge compensation mechanism of NCM cathode materials during the Li-ion deintercalated process. Fig. 1a illustrates the first charge curves for LiNi1/3Co1/3Mn1/3O2 (NCM333) and LiNi0.8Co0.1Mn0.1O2 (NCM811), with each material charged to 4.8 V (vs. Li/Li+), respectively. To investigate the charge compensation mechanisms, X-ray spectroscopy measurements were conducted on a series of ex situ samples at different states of charge, as highlighted by the dot mark.
 | ||
| Fig. 1 O3-type cathode and synchrotron spectroscopy methods. (a) First charge curves of NCM333 and NCM811 cathodes. The inset illustrates the O3-type structure, where dark cyan octahedrons represent TMO6 units, red spheres denote oxygen ions and green spheres indicate lithium ions. (b) Schematics of synchrotron-based X-ray absorption fine structure (XAFS), near-edge X-ray absorption fine structure (NEXAFS), and resonant inelastic X-ray scattering (RIXS) spectroscopy techniques, highlighting their application areas in characterizing cathode materials. | ||
Charge compensation from the aspects of TM
The normalized Ni, Co, and Mn K-edge transmission X-ray Absorption Fine Structure (XAFS) data for LiNi1/3Co1/3Mn1/3O2 (NCM333) and LiNi0.8Co0.1Mn0.1O2 (NCM811), specifically focusing on the X-ray Absorption Near-Edge Structure (XANES) region, are presented in Fig. 2a, respectively. In general, the threshold energy position of the absorption edge provides information about the state of the absorbing element while the line shape of the K-edge XANES offers unique insights into the coordination environment and site symmetry.22,23 For the fresh NCM333 cathode, XANES data indicate that the absorption edges for Ni, Co, and Mn closely align with those of NiO, LiCoO2, and MnO2, respectively, suggesting that the initial oxidation states are +2 for Ni, +3 for Co, and +4 for Mn. Meanwhile, the pristine NCM811 cathode displays initial oxidation states of +3 for Co, +4 for Mn, and Ni with a mixed state of Ni2+ and Ni3+ for Ni. As lithium ions are deintercalated, the Ni spectra of both NCM333 and NCM811 reveal that the entire absorption edge shifts to a higher energy region. Simultaneously, the line shape exhibits only a small shoulder peak at the edge that gradually washes out, which may correspond to the increasing hybridization between Ni and O. In contrast, none of the Co and Mn K-edge spectra show a systematic shift; instead, they exhibit complicated changes in the overall shape. It is well known that small changes in the coordination environment, including bond length, bond covalency, geometry, and site symmetry, resulting from delithiation, can significantly alter the XANES spectra.24,25 In the case of Co and Mn K-edge XANES results, the evolution of the spectral shape can be attributed to changes in bond length and covalency.26 Consequently, the influence of structural factors on the spectral shape complicates the interpretation of oxidation states via XANES. It is not feasible to ascertain changes in the oxidation state solely based on edge features, such as the half-height of the edge jump or peak position. In the face of such difficulties, we utilized an integration method to derive the average edge energy from each spectrum, which can assist in determining the formal oxidation states of each element (Fig. S1†). This integration method is reported to be insensitive to variations in the shape of the spectrum that occur without an overall shift of the edge position, thereby allowing for a more accurate determination of average edge energy.27 The edge energies derived from the integration of the Ni, Co, and Mn for the NCM333 and NCM811 cathodes are displayed in Fig. 2b and c, respectively. Co and Mn exhibit only minor changes in both NCM333 and NCM811, indicating their minimal involvement in charge compensation. Therefore, during the first charging process of NCMs, charge compensation at the metal sites is primarily achieved through the oxidation of Ni ions. Furthermore, the edge energy of Ni in NCM333 continues to increase up to 4.5 V (vs. Li/Li+) after which the edge position exhibits negligible changes. In contrast, for the NCM811 cathode, spectral changes in Ni become subtle after being charged to 4.2 V (vs. Li/Li+). Under high-voltage conditions, the TM K-edge spectra of all transition metals in NCMs converge to plateau-like features. Thus, an additional process must occur during the final stages of charging. | ||
| Fig. 2 Charge compensation from the perspective of transition metals revealed by TM K-edge XANES. (a) Normalized Mn, Co, and Ni K-edge XANES spectra of pristine and charged NCM333 and NCM811 cathodes charged to 4.2, 4.5, and 4.8 V (vs. Li/Li+). (b and c) Edge positions of the Ni, Co, and Mn K-edges for NCM333 (b) and NCM811 (c) cathodes at the pristine state and various states of charge. | ||
Hard X-ray extended absorption fine structure quantitative analysis provides insights into the local environment changes of TMO6, particularly TM–O bond lengths. EXAFS parameters were extracted from the data of samples at various states of charge. The Fourier transform magnitudes of the EXAFS spectra are shown in Fig. S5–S7,† and their fits are displayed in Fig. 3a, S8 and S9.† Bond distances for each element in NCMs are plotted in Fig. 3b with detailed analyses presented in Tables S1–S6.† The most significant change during charging is observed in the Ni–O coordination shell of both NCM333 and NCM811, indicating that charge compensation mainly occurs at Ni sites. In contrast, Co–O and Mn–O bond lengths decrease only slightly, suggesting their minor contributions to electrochemical reactions. Moreover, changes in Ni–TM, Co–TM, and Mn–TM bond lengths are attributed to variations in TM–O bonding during the delithiation due to the edge-shared octahedral layered structure. Therefore, XAFS analysis of the TM K-edge indicates that the oxidation states of TMs remain nearly constant beyond a certain degree of delithiation (4.5 V for NCM333 and 4.2 V for NCM811), where the evolution of the TM K-edge ceases and the TM–O bond lengths plateau. Interestingly, we find that the Ni–O bond length is larger than the Co–O and Mn–O bond lengths in the pristine state, resulting in the NiO6 octahedron being larger than the CoO6 and MnO6 (Fig. 3c). Consequently, in the layered structure, the different-sized octahedra must share edges to form continuous TMO2 slabs. This edge-sharing forces the ideal layered structure to distort, ultimately resulting in a “zigzag”-like structure. However, with delithiation, the Ni–O, Co–O, and Mn–O bond lengths become more uniform, resulting in a “ruler”-like oxygen layer. We hypothesize that this “zigzag”-like structure hinders slab gliding, as the uneven distribution of bond lengths creates structural hindrance. In contrast, the smooth “ruler”-like structure, with more uniform bond lengths, facilitates slab gliding, as it allows for easier movement.
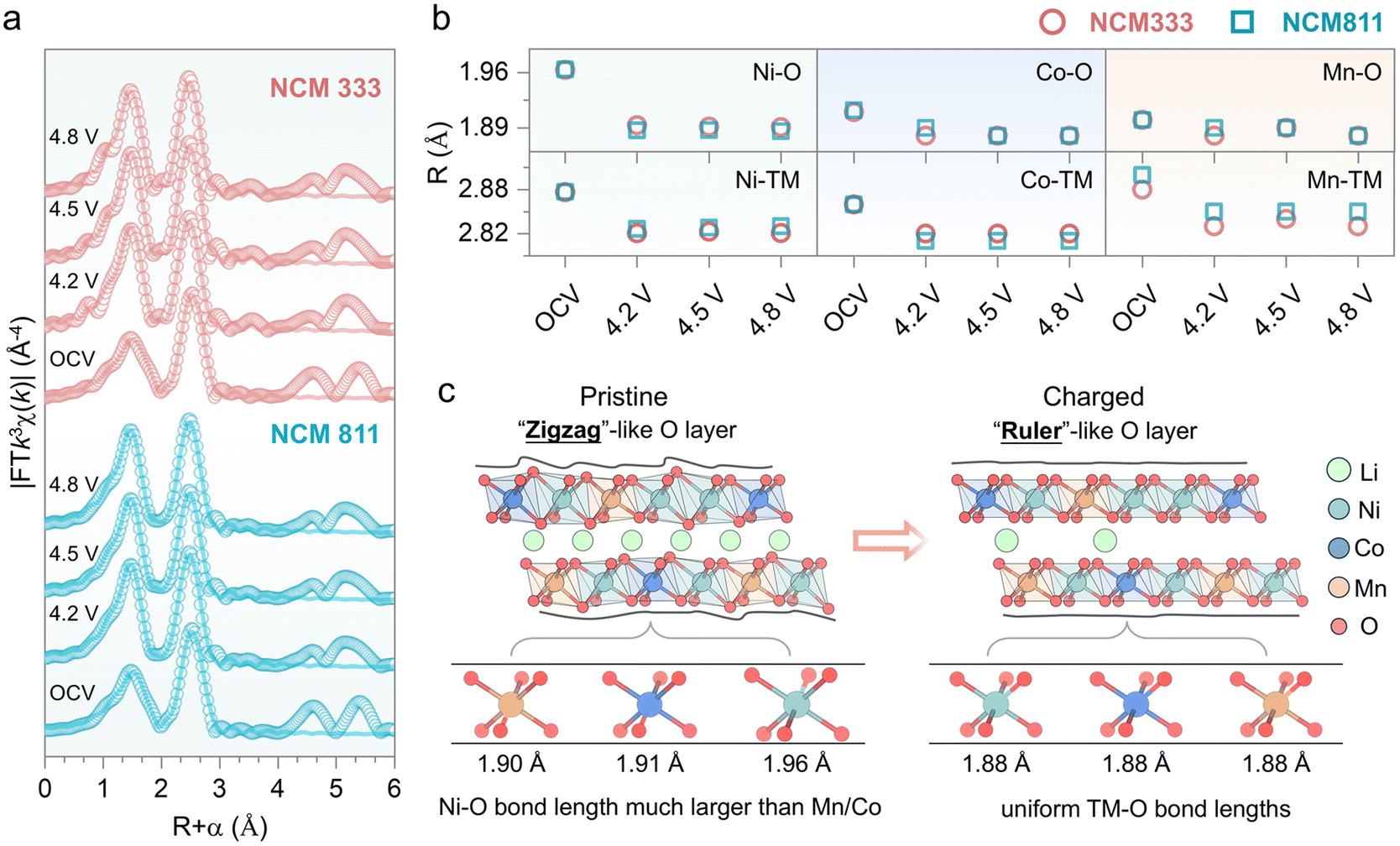 | ||
| Fig. 3 Charge compensation of transition metals revealed through local structure evolution. (a) Ni K-edge EXAFS fitting results of NCM333 and NCM811. (b) XAFS-derived bond lengths for TM–O (Ni–O, Co–O, Mn–O) and TM–TM (Ni–TM, Co–TM, Mn–TM) in NCM333 and NCM811 at the pristine state and various delithiation states. (c) Schematic illustration of the nearest coordination local structural changes and their effects on the layered structure during delithiation. | ||
Charge compensation achieved by TM–O hybridization and O-holes
As mentioned above, the L-edge NEXAFS of TMs is particularly useful in correlated electronic systems and provides insightful information about the electronic structure, including oxidation states, spin states, and bond covalency.18 Ni, Co, and Mn L2,3-edge NEXAFS data were collected in total electron yield mode from pristine and charged NCM333 and NCM811 cathodes (Fig. 4a, b and S10†). Due to splitting by the TM 2p spin–orbit interaction, the feature in the lower energy region, referred to as the L3 edge, corresponds to the transition from 2p3/2 to unoccupied d-character states, while the higher energy feature, the L2 edge, corresponds to the transition from 2p1/2. The Ni L3-edge was observed to have a dominant peak at approximately 853 eV (peak A), typically indicative of the Ni2+ state, while the feature around 855 eV (peak B) can be attributed to a Ni3+ state.28,29 As lithium ions are deintercalated from NCM333 and NCM811 cathodes, the higher energy peak increases at the expense of the lower energy peak, indicating a change in the effective nuclear charge of Ni ions, which is formally manifested as the oxidation of Ni (Fig. 4c). During the deintercalation of lithium ions from the NCM333 cathode, the peaks of Co L2,3-edge shift and broaden toward the higher energy region, with an increase in peak intensity observed before reaching 4.5 V (Fig. 4b). Subsequently, the spectra show only subtle changes. These phenomena are attributed to an increase in Co–O bond covalency resulting from local structure distortions of CoO6 octahedra as well as a partial increase in the effective nuclear charge of the Co ion.24 Therefore, the evolution of these spectral features suggests that Co in the NCM333 may participates to some extent in the charge compensation process. In contrast, the Co L2,3-edge of NCM811 remains largely unchanged with lithium-ion removal, indicating that Co ions remain mostly unchanged during charging (Fig. 4d). The difference in Co behavior between NCM333 and NCM811 may be attributed to the difficulty in detecting a small amount of oxidation in Co within NCM811 during charging. Nevertheless, the oxidation of Co in NCM333 is evidenced by changes in peak position and peak intensities. On the other hand, the Mn L2,3-edge spectra of NCM333 and NCM811 do not reveal differences in shape or energy position during charging, implying that Mn contributes minimally to the charge compensation (Fig. S10†). Fig. 4e and f illustrate the relationship between the states of charge and the peak ratio of peak B to peak A in the Ni L3-edge, as well as the peak position of the Co L3-edge. Overall, results from the TM L-edge NEXAFS also indicate that Ni is the primary oxidized metal element in NCM cathodes. | ||
| Fig. 4 Oxidation-state changes of transition metals upon delithiation revealed by TM L-edge NEXAFS. (a) Ni L2,3-edge NEXAFS spectra of pristine and charged NCM333 and NCM811 cathodes charged to 4.2, 4.5, and 4.8 V (vs. Li/Li+). (b) Co L2,3-edge NEXAFS spectra of pristine and charged NCM333 and NCM811 cathodes at various states. Reference spectra: Ni2+O, Ni23+O3, Co2+O, and Co23+O3. NEXAFS data were collected in total electron yield (TEY) mode. (c) Differential Ni L2,3-edge spectra of NCM333 and NCM811 cathodes at various states. (d) Differential Co L2,3-edge spectra of NCM333 and NCM811 cathodes at various states. (e) Quantification of the oxidation states from Ni and Co L2,3-edge NEXAFS of the NCM333 at various states. (f) Quantification of the oxidation states from Ni and Co L2,3-edge NEXAFS of the NCM811 at various states. | ||
Through the XAFS and NEXAFS analyses described above, the role of Ni in NCM cathodes has been identified as crucial in the charge compensation process. Moreover, the appearance of a plateau was observed in both metal-related spectroscopic techniques. In general, this plateau-like phenomenon is regarded as the endpoint of TM participation in charge compensation and the beginning of oxygen ion oxidation.30,31 This hypothesis posits that TMs oxidize independently of lattice oxygen; in other words, it suggests the separate oxidation of TM and O. The ionic-bonding model, incorporating the crystal field theory suggests that TM ions and oxygen ions possess discrete electronic bands, and an element becomes the redox center only when the higher-lying valence bands are empty (Fig. 5a). However, such an ionic model often downplays the role of covalency and the interactions between TM 3d and O 2p states are neglected in the data analysis. XAFS results reveal that it is difficult to oxidize Co and Mn in the presence of Ni, which contradicts predictions from the ionic-bonding model. Additionally, before Co and Mn contributed to the electrochemical reactions in NCM cathodes, electrolyte decomposition reactions, transition metal dissolution, oxygen release, and other side reactions had already occurred.8,32,33 Therefore, such an ionic description does not accurately represent the electronic structure in NCM cathodes. Pioneering work has demonstrated that the electronic structure of layered transition metal oxides can be interpreted using the qualitative Zaanen–Sawatzky–Allen picture of charge-transfer insulators.12,34 The band structure of insertion compounds can be inferred from the relative values of U and Δ, where U represents the Coulomb interactions in TM 3d states, and Δ is the ligand-to-metal charge transfer (LMCT) parameter (Fig. 5b).34 In such charge transfer model, U >> Δ indicates that the electron density is shifted from O 2p states to TM 3d states, forming covalent bonds with strong hybridization. Consequently, the oxidation of TMs and oxygen cannot be regarded as independent.
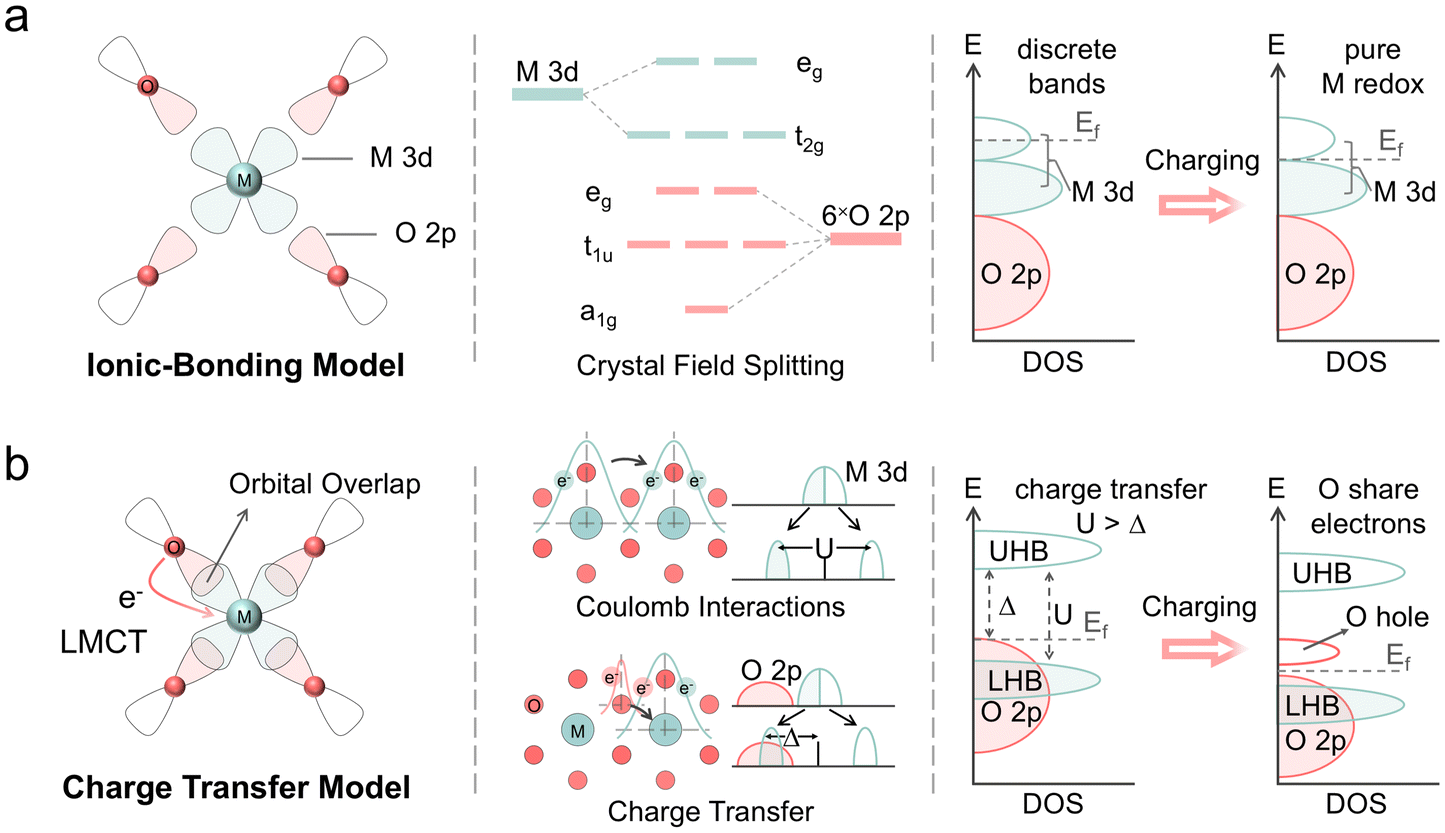 | ||
| Fig. 5 Illustrations of different electronic structure models. (a) Schematic representation of the ionic-bonding model, where TM cations and O anions are considered to possess discrete electronic bands. (b) Schematic illustrations represent the Mott–Hubbard model with charge transfer, depicting the d Coulomb interactions U and charge transfer Δ. These diagrams also demonstrate how delithiation affects the electronic structure as interpreted by each model. | ||
Thus, to further interpret the experimental data, Ni L2,3-edge NEXAFS spectra were simulated using a charge-transfer multiplet (CTM) approach combined with crystal field effects, employing a NiO6 octahedron cluster model, based on the number of electrons in the 3d state (Fig. 6a). The simulation can help us interpret the fine structure in the Ni L2,3-edge more comprehensively, including ligand field effects, charge transfer, spin state, Coulomb interactions, Jahn–Teller distortion, and other electronic-structure-related information.29,35,36 The simulations utilized the software package CTM4XAS, developed by de Groot et al.,37 which has been proven to successfully describe the TM 2p spectra in such correlated electronic systems of NCMs.29,38 The simulated 3d8, 3d8L, 3d7 and 3d7L spectra do not align with experimental data. However, a convolution of multiple configurations, including those with oxygen holes, is required to accurately reproduce the experimental spectrum (Fig. 6a and b). No characteristic features of 3d6 are observed in these spectra. This absence is ascribed to the stabilization of 3d7 through LMCT with a negative charge transfer character, (resulting in the formation of 3d7L) thereby making the formation of 3d6 practically impossible. Kleiner et al. have also observed this absence of 3d6-related features in NCM and NCA cathodes using NEXAFS.29,38 However, the CTM simulation, while providing an estimate of the number of d-electrons, cannot quantify the holes in the O 2p band. Nonetheless, these results demonstrate that during the charging process, electron density shifts from the oxygen 2p band to the nickel 3d band, leading to a transition of the Ni–O bond from ionic to covalent. Notably, O holes are already present in pristine NCM811, a finding comparable to previous studies of high-Ni layered cathodes and LiNiO2,39,40 which outlines the ligand-dominant ground state with increasing nickel content in NCM cathodes. Compared with NCM333, increasing the Ni content leads to a rise in the 3d7 (Ni3+) configuration, thereby allowing us to infer that the Ni3+ configuration is the driving force behind hole formation in the O 2p band. Additionally, it should be recognized that the TM 2p spectra collected in total electron yield (TEY) mode are subject to substantial surface signal contributions. As a result, the TEY data show a more reduced surface, indicating the inherent thermodynamic instability of the NiO6 octahedra with ligand holes. Overall, it can be concluded that oxygen contributes to the charge compensation through the formation of O 2p band holes by hybridizing with metals in NCM cathodes before the plateau in TM oxidation state evolution.
 | ||
| Fig. 6 O-hole formation induced by charge transfer. (a) Simulation of Ni L2,3-edge spectra of a NiO6 octahedron, comparing experimental NMC data with simulations for the 3d8, 3d8L, 3d7, and 3d7L electronic states. The simulations were conducted using the CTM4XAS software package. (b) Ni L2,3-edge spectra of charge of NCM333 and NCM811 at various charge states, fitted with simulated spectra in an Oh ligand field. | ||
Now, we can explore the contradiction that after a certain amount of delithiation, the TMs should continue to oxidize to maintain charge neutrality; yet, XANES analysis reveals the relative silence of TMs, or in other words, no formal TM oxidation will be observed. As discussed in previous sections, the Ni ions are the primary contributor to the charge compensation process. Upon charging, the Ni in the NCM is oxidized from the original ionic Ni–O bond to the covalent Ni–O bond with ligand holes, accompanied by hybridization of the Ni 3d and O 2p orbitals, which leads to the formation of covalent bonds. We emphasize a progressive shift from ionic-dominant to covalent-dominant interactions, without a specific turning point in this transition. High valence nickel ions, characterized by negative LMCT, are stabilized by electrons shared from oxygen, leading to hole formation in the oxygen.15,38 This ligand-hole stabilized configuration may prevent an increase in the effective nuclear charge of Ni. Thus, the K-edge XANES no longer shows blue shifts after the ionic Ni–O bond completely vanishes in the bulk, indicating that the predominant Ni–O bond is now covalent. Moreover, in the presence of Ni ions, it is difficult to observe a substantial O contribution through Co hybridization with O.38 This is because owing to the Co cation with a lower effective nuclear charge, the LMCT for the Co–O bond is weaker than that for the Ni cation. Although Co can also act as an electron transfer bridge in oxygen redox, its contribution is overshadowed by the stronger Ni–O covalency in Ni-rich systems. If only Co is present in a layered material, such as LiCoO2, electron–hole formation on O occurs through hybridizing with Co to respond to charge compensation.41 Ceder et al.42 and Tarascon et al.43 also emphasized that in late-transition-metal oxides (e.g., Co, Ni), it is the oxygen that is in large part responsible for the electron exchange, which can be explained based on the elegant electron–hole chemistry. Therefore, in NCM cathodes, oxygen is involved in charge compensation primarily through Ni–O hybridization, with minimal contribution from Co–O hybridization, across the entire electrochemical range. This involvement leaves no doubt about its substantial contribution to capacity throughout the entire delithiation process. However, this raises the question of whether oxygen redox, stimulated by interaction with transition metals, is necessary, and whether such oxygen redox would contribute to extra capacity, as observed in Li-rich cathodes.
In the field of charge compensation mechanisms, it is widely recognized that redox mechanisms are categorized into TM redox and O redox.44 The former refers to any redox process involving TM 3d orbitals, classified as TM redox regardless of potential contributions from hybridized anion ligands, while the latter describes a redox process that only involves O 2p orbitals, termed intrinsic anion redox.44,45 This concept of redox mechanism, with an ionic charge compensation perspective, is simple yet powerful, as it reveals the electron count, and the contribution from component atoms and provides insights into or predictions of structural or chemical properties, such as the anomalous first charge capacity of layered Li-rich oxide cathodes.46,47 Nevertheless, it is worthwhile to reconsider the appropriateness of depicting the redox process as two separate events, TM and O oxidation, in terms of formal oxidation states or valence, rather than focusing on changes in charge density, which refers to the distribution of electrons in chemical systems. In addition, higher oxidation states of oxygen can be observed before the anion redox regime, a phenomenon that raises the conclusion that the lattice oxygen oxidation occurs in the so-called TM oxidation regime.48 Thus, because the formal oxidation starts to blur in correlated systems and ploy-ion compounds, it is not quite wise to utilize the formal oxidation state from an ionic perspective to divide the charge compensation process into separated entities and ignore the interactions between ions, particularly the strong interplay between metal ions and ligand ions. There are certain ambiguities and uncertainties regarding the oxidation state, ionicity, covalency, and charge distributions, along with their interrelationships. Although there is a lack of advanced theoretical methods to detail the electronic structure in solids, we stress here that a complete understanding of the involvement of oxygen is critical in the battery process. During the conventional nomenclature TM oxidation regime, oxygen plays a role as important as TMs in charge compensation, facilitated by strong TM–O hybridization. Moreover, the formation of oxygen holes, accompanied by an increase in TM–O hybridization, significantly affects electrochemical properties and structural degeneration. The formation of the rock-salt phase and surface reduction can be attributed to reactions involving these oxygen holes.15,38 Oxygen participation in the charge compensation process through electron sharing with the metal does not contribute to extra capacity (Fig. 7a). Even if the metal does not induce oxygen electron sharing, it must still be oxidized to a high oxidation state to achieve such capacity. Furthermore, this oxygen oxidation process complicates electron donation, as the overlap between the metal and oxygen bands increases with greater delithiation. Oxygen can be involved in redox reactions independently of hybridization with TMs, a process denoted as intrinsic oxygen redox. This process occurs in the O 2p non-bonding states, where electrons are removed directly from this unhybridized state.8 This process is independent of the transition metal in terms of the redox process and has the potential to contribute extra capacity (Fig. 7b). In NCM cathodes, this unhybridized oxygen 2p state is formed by Li/Ni mixing49,50 and TM migration.51 Accordingly, we identify two pathways through which oxygen contributes to charge compensation: hybridization with transition metals and involvement in non-bonding O 2p states.
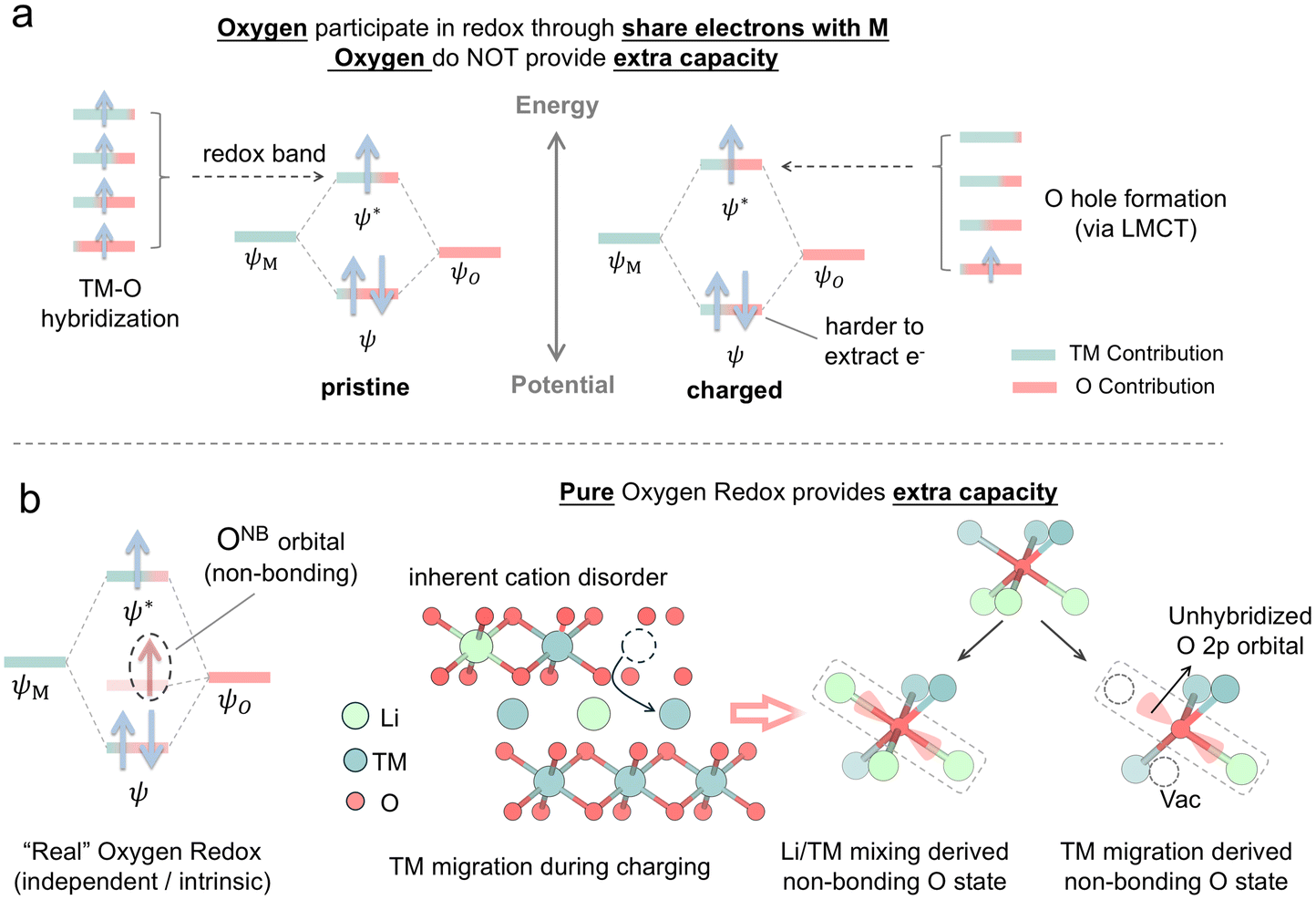 | ||
| Fig. 7 Relationship between the covalent electron structure and extra capacity. (a) Schematic illustration of the covalent model and its influence on the charging process. Oxygen contributes to capacity through hybridization with metal but does not provide extra capacity. (b) Schematic illustration of the non-bonding O 2p states formed by TM migrations or Li/TM mixing and their effect on the charge compensation. | ||
Intrinsic oxygen redox reaction induced by structure degeneration
O K-edge RIXS measurements were conducted to further investigate the oxygen redox process.52 The O K-edge RIXS spectrum of these layered cathode materials reveals three primary features (Fig. 8a). There is a peak with a broad energy loss width ranging from −5 eV to −10 eV, attributed to TM–O hybridization.53 The peak centered around −8 eV is regarded as the main peak of the oxidized oxygen state, indicating the intrinsic oxygen redox.7,53 Additionally, RIXS has been proven to be the most reliable tool to detect oxygen dimerization, through a fingerprint feature characterized by a progression of energy-loss peaks at −2–0 eV, associated with vibrations of the O–O bond and a fundamental vibrational frequency of about 1600 cm−1, which is the vibrational frequency of molecular O2.54 The spectral feature at −5 eV, also observed in LiNiO2 and other Ni-rich cathodes, has not yet been fully understood but is believed to be linked to an increase in Ni–O hybridization.53 NCM cathodes demonstrate different behaviors of intrinsic oxygen redox spectral features in the RIXS data, depending on the Ni content and the state of charge. Intrinsic oxygen redox is observed only at 4.8 V in NCM333, with no trapped O2 features, while both are present in NCM811 at 4.5 V (Fig. 8b). The formation mechanism of these trapped O2 molecules, widely discussed in the literature, is acknowledged to be related to the interlayer migration of TM ions from the TM layer to the Li layer, which is accompanied by intralayer migration of TMs to form the TM vacancy clusters where molecular O2 can reside within the lattice (Fig. 8c).54,55 TM migration induces the reorganization of the electronic structure and shifts the O 2p states to higher energy relative to the hybridized TM–O state, thus prioritizing the depopulation of the unhybridized O 2p state.51 In the early stages of charging, oxygen primarily participates in redox through the LMCT process, resulting in the formation of oxygen holes (Fig. 8d). The accumulation of oxygen holes may promote the formation of oxygen dimers (O2). Li/Ni mixing in NCM333 primarily contributes oxygen with the non-bonded states, followed by oxygen loss associated with TM migration.7 The presence of trapped O2 in NCM811 suggests more pronounced TM migration, including interlayer migration that induces cation disorder and intralayer migration leading to vacancy clusters. TM migration in NCM811 is one of the sources of O non-bond states.51 With further delithiation, the intensified TM migration brings about oxygen loss, while the trapped O2 also escapes from the lattice. Accordingly, the oxygen redox behavior in NCM is governed by TM migration and cation disorder. Consequently, it is essential to understand how electronic and geometric structures, as well as the elemental composition, influence these factors to clarify the underlying differences in oxygen redox behaviors. | ||
| Fig. 8 Oxygen redox behavior and its interaction with the local structure. (a) O K-edge RIXS spectra of a Li-rich (Li1.2Ni0.2Mn0.6O2) cathode in the pristine state and charged to 4.8 V (vs. Li/Li+). (b) O K-edge RIXS spectra of NCM333 and NCM811 cathodes at pristine, 4.2 V, 4.5 V, and 4.8 V (vs. Li/Li+). (c) Illustration of trapped-O2 formation, highlighting the migration and aggregation of vacancies leading to O–O dimerization ends with oxygen molecules within the lattice. (d) Schematic illustration of anion redox behavior in NCM333 and NCM811. Before oxygen evolution, NCM811 undergoes the trapped-O2 stage with a TM vacancy cluster, whereas NCM333 does not. | ||
From an electronic structure perspective, the tendency of TM migration is influenced by the number of valence electrons (3d electrons) and the spin state. Based on octahedral site stabilization energy, TMs in high-spin states exhibit a greater tendency to migrate, whereas those in low-spin states show a reduced tendency to migrate (Fig. 9a).56 Ni2+ exists in a high-spin state and possesses a radius like that of Li+, facilitating migration and Li/Ni mixing. In contrast, neither Ni3+ nor Ni4+ is prone to Li/Ni mixing. Furthermore, high-spin Ni3+ is more susceptible to migration, while Ni4+ is likely to adopt a low-spin state. The branching ratio, defined as the ratio of integrated L3 intensity to the overall L2,3-edge intensity, is higher for high-spin states than for low-spin states (Fig. 9b).57 Therefore, it can be used as a diagnostic tool for the metal spin state. From the branching ratio at the TM L2,3-edge, it is observed that Ni adopts a low-spin configuration during the charging process in both NCM333 and NCM811 cathodes. Therefore, the low-spin electronic configuration renders Ni migration in these cathodes electronically structurally unfavorable. NCM811, however, exhibits a RIXS feature corresponding to trapped O2, suggesting that other factors enhance the probability of TM migration. Therefore, we present an in situ X-ray powder diffraction analysis conducted on NCM333 and NCM811 (Fig. S12 and S13†), to examine the phase transitions of NCM333 and NCM811 cathodes during the charging process. As indicated in Fig. S12,† NCM333 undergoes a series of phase transitions during Li extraction, transitioning from the original layered structure (H1) to the monoclinic phase (M).58 In contrast, NCM811 exhibits two additional hexagonal phases (H2 and H3), undergoing an H2–H3 phase transition at excessive delithiation.58 Yu et al. discussed that the structural essence of the H3 phase is an O3–O1 intergrowth structure, in which the O1 slabs originate from the gliding of original O3 slabs in high-nickel layered oxide cathodes.59 During the transformation from the O3 phase to the O1 phase, the oxygen stacking sequence changes from cubic close packing (ABCABC) in the O3 phase to hexagonal close packing (ABAB) in the O1 phase, accompanied by pronounced TM migration.59,60 This is due to the different migration pathways in the O1 and O3 phases, resulting in a lower energy barrier for Ni ion migration into the Li layer in the O1 phase.60 For the NCM333 cathode, Ni2+ in the pristine state is prone to mixing and migration, while in NCM811, Ni is also easy to migrate, but both without vacancies within the Li layer. Upon delithiation, both transition to a low spin state which inhibits migration; however, the formation of the O1 slab in NCM811 enhances the migration tendency (Fig. 9d). Additionally, Co and Mn, acting as “nail” elements, do not migrate. Thus, vacancies in NCM333 are separated by these immobile barriers, resulting in dispersed vacancies. In contrast, in NCM811, due to its higher Ni content, the TM vacancy clusters are formed by intralayer migration. Overall, while the electronic structure discourages TM migration, the geometric structure encourages it. Furthermore, the elemental composition influences the aggregation of vacancies.
 | ||
| Fig. 9 Effects of electronic and geometric structures on anion redox behavior. (a) Schematic diagram illustrating the relationship of spin states with TM migration. TM ions in high spin states are prone to migration, whereas TM ions with ionic radii close to that of Li+ (0.76 Å) are susceptible to cation mixing. (b) Illustration of the branching ratio concept and its relationship with spin states. (c) Branching ratios of Ni L2,3-edge spectra of NCM333 and NCM811 at various states indicate that Ni in the charged NCMs exists in a low spin state. (d) The scheme shows the influence of spin states, stacking faults, and elemental composition on cation mixing and TM migration. | ||
Conclusions
To improve specific capacity and energy density while maintaining structural stability during high states of delithiation in lithium nickel cobalt manganese oxide (NCM) cathodes, we have investigated the charge compensation process across a wide voltage range in both low-Ni and high-Ni content NCM materials using state-of-the-art synchrotron X-ray spectroscopy to characterize both cation and anion states. The results provide detailed information about the contributions of TMs and oxygen to redox processes in NCMs at various states of delithiation. Moreover, this study has comprehensively elucidated several related innovative insights and conclusions:(I) At relatively lower depths of charging, the system exhibits more ionic electronic structures, and thus the process primarily involves pure TM redox. Beyond a certain level of delithiation, the higher TM oxidation states cause electrons around the oxygen to transfer to TM, creating holes in the oxygen ligand and forming strong covalent TM–O bonds. In other words, oxygen participates in redox through strong TM–O hybridization and ligand-to-metal electron sharing (regarded as Ligand to Metal Charge Transfer, LMCT), even before the “formal” pure anionic redox reaction region. Additionally, in the Ni-rich material, NCM811, oxygen holes can be observed at any state of delithiation, including in the pristine state, indicating that the Ni3+ content is crucial for promoting electron holes on oxygen.
(II) Compared to NCM333, the NCM811 material undergoes a process involving trapped O2 before oxygen loss, which leads to the formation of vacancy clusters in the transition metal layer. This process is partially irreversible, and the aggregated vacancies from repeated cycling eventually destroy the layered structure. This variation in oxygen dimerization behavior is closely correlated with elemental composition, spin states, and the presence of stacking faults. Specifically, in NCM materials, Co and Mn are “nail” elements that do not migrate; thus, Ni is the only atom that can migrate within these materials. During the charging process, Ni is continually oxidized to high oxidation states and low spin states. However, these low-spin states, due to their stronger interaction with oxygen, are less likely to migrate. NCM811 is more susceptible to stacking faults in the O1 layer than NCM333, which geometrically increases the likelihood of Ni interlayer migration and favors the formation of vacancies in the TM layer. Compared to NCM333, where vacancies in the TM layer are distributed in such a way that intralayer migration to form vacancy clusters (a relatively aggregative region of vacancies) is challenging, the higher mobility of the active element Ni in NCM811 increases the probability of forming these clusters.
(III) Nickel is the metallic element most predominantly involved in charge compensation, with the Ni–O bond contracting significantly during charging. Notably, in the pristine state, the unequal metal–oxygen bond lengths (with the Ni–O bond being longer than others) cause the layered structure to adopt a “zigzag” configuration. Upon delithiation, the metal–oxygen bond lengths become more uniform, resulting in a “ruler” configuration. We hypothesize that the changes in the layered structure induced by local structural evolution have significant implications for phase transitions and the formation of stacking faults.
We have elucidated the charge compensation mechanisms in low- and high-nickel NCMs during high-voltage charging. Specifically, our analysis focused on the behavior of oxygen and the associated structure evolution to enhance our understanding of redox chemistry. This insight provides a broader context for discussions on anion redox reactions and informs strategies to improve performance. We expect that our findings will inspire numerous future studies, leading to a deeper fundamental understanding of the reaction mechanisms and material optimization, ultimately maximizing the energy density of NCM compounds.
Data availability
The data that support the findings of this study are available within the article and its ESI.†Author contributions
J. Y.: investigation, data curation, formal analysis, visualization, writing the original draft, manuscript review and editing. Z. W.: investigation, visualization, manuscript review and editing. K. F.: data curation. Y. Z.: data curation. K. Z.: data curation. H. Z.: data curation. Y. C.: data curation. L. L.: data curation. L. F.: data curation, formal analysis. K. D.: data curation, formal analysis. L. Z.: data curation, formal analysis. Q. W.: conceptualization, supervision, data curation, formal analysis, manuscript review. H. H.: conceptualization, investigation, supervision, formal analysis, manuscript review. J. Z.: conceptualization, investigation, supervision, formal analysis, manuscript review. Y. Q.: conceptualization, investigation, supervision, writing the original manuscript, manuscript review and editing. S. S.: supervision, manuscript review.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was partially supported by the Natural Science Foundation of China (grant no. 22179111), the Ministry of Science and Technology of China (grant no. 2023YFB2406200), the Fundamental Research Funds for the Central Universities (grant no. 20720220010), and the Principal Fund from Xiamen University (grant no. 20720210015). The authors are thankful to the Beijing Synchrotron Radiation Facility (BSRF) for providing support with characterization at stations 1W1B and 4B9B. The authors appreciate the help from D. Wong, C. Schulz, and M. Bartkowiak for the RIXS characterization (proposal-Info: 221-11099-ST) at beamline U41-PEAXIS of Bessy II, Helmholtz-Zentrum Berlin für Materialien und Energie (HZB), Berlin, Germany.Notes and references
- W. Li, E. M. Erickson and A. Manthiram, High-nickel layered oxide cathodes for lithium-based automotive batteries, Nat. Energy, 2020, 5, 26–34 CrossRef CAS.
- M. M. Thackeray and K. Amine, Layered Li–Ni–Mn–Co oxide cathodes, Nat. Energy, 2021, 6, 933 Search PubMed.
- C. Zhang, X. Zhao, R. Sacchi and F. You, Trade-off between critical metal requirement and transportation decarbonization in automotive electrification, Nat. Commun., 2023, 14, 1616 Search PubMed.
- A. L. Cheng, E. R. H. Fuchs, V. J. Karplus and J. J. Michalek, Electric vehicle battery chemistry affects supply chain disruption vulnerabilities, Nat. Commun., 2024, 15, 2143 CrossRef CAS PubMed.
- Z. Xue, F. Wu, M. Ge, X. Huang, Y. S. Chu, P. Pianetta and Y. Liu, Mesoscale interplay among composition heterogeneity, lattice deformation, and redox stratification in single-crystalline layered oxide cathode, eScience, 2024, 4, 100251 CrossRef.
- S. N. Lauro, J. N. Burrow and C. B. Mullins, Restructuring the lithium-ion battery: a perspective on electrode architectures, eScience, 2023, 3, 100152 Search PubMed.
- G. H. Lee, J. Wu, D. Kim, K. Cho, M. Cho, W. Yang and Y. M. Kang, Reversible Anionic Redox Activities in Conventional LiNi1/3 Co1/3Mn1/3O2 Cathodes, Angew Chem. Int. Ed. Engl., 2020, 59, 8681–8688 CrossRef CAS PubMed.
- K. Luo, M. R. Roberts, R. Hao, N. Guerrini, D. M. Pickup, Y. S. Liu, K. Edstrom, J. Guo, A. V. Chadwick, L. C. Duda and P. G. Bruce, Charge-compensation in 3d-transition-metal-oxide intercalation cathodes through the generation of localized electron holes on oxygen, Nat. Chem., 2016, 8, 684–691 CrossRef CAS PubMed.
- J. J. Marie, R. A. House, G. J. Rees, A. W. Robertson, M. Jenkins, J. Chen, S. Agrestini, M. Garcia-Fernandez, K. J. Zhou and P. G. Bruce, Trapped O2 and the origin of voltage fade in layered Li-rich cathodes, Nat. Mater., 2024, 23, 818–825 CrossRef CAS.
- P. M. Csernica, K. McColl, G. M. Busse, K. Lim, D. F. Rivera, D. A. Shapiro, M. S. Islam and W. C. Chueh, Substantial oxygen loss and chemical expansion in lithium-rich layered oxides at moderate delithiation, Nat. Mater., 2025, 24, 92–100 CrossRef CAS.
- A. Manthiram, A reflection on lithium-ion battery cathode chemistry, Nat. Commun., 2020, 11, 1550 CrossRef CAS PubMed.
- J. Zaanen, G. Sawatzky and J. Allen, Band gaps and electronic structure of transition-metal compounds, Phys. Rev. Lett., 1985, 55, 418 Search PubMed.
- P. Olalde-Velasco, J. Jiménez-Mier, J. Denlinger, Z. Hussain and W. Yang, Direct probe of Mott-Hubbard to charge-transfer insulator transition and electronic structure evolution in transition-metal systems, Phys. Rev. B:Condens. Matter Mater. Phys., 2011, 83, 241102 Search PubMed.
- G. Sawatzky and J. Allen, Magnitude and origin of the band gap in NiO, Phys. Rev. Lett., 1984, 53, 2339 CrossRef CAS.
- M. J. W. Ogley, A. S. Menon, G. C. Pandey, G. J. Páez Fajardo, B. J. Johnston, I. McClelland, V. Majherova, S. Huband, D. Tripathy, I. Temprano, S. Agrestini, V. Celorrio, G. E. Pérez, S. G. Booth, C. P. Grey, S. A. Cussen and L. F. J. Piper, Metal-ligand redox in layered oxide cathodes for Li-ion batteries, Joule, 2025, 9, 101775 CrossRef CAS.
- F. de Groot, High-Resolution X-ray Emission and X-ray Absorption Spectroscopy, Chem. Rev., 2001, 101, 1779–1808 CrossRef CAS PubMed.
- F. M. F. de Groot, M. W. Haverkort, H. Elnaggar, A. Juhin, K.-J. Zhou and P. Glatzel, Resonant inelastic X-ray scattering, Nat. Rev. Methods Primers, 2024, 4, 45 Search PubMed.
- G. S. Henderson, F. M. De Groot and B. J. Moulton, X-ray absorption near-edge structure (XANES) spectroscopy, Rev. Mineral. Geochem., 2014, 78, 75–138 CrossRef CAS.
- M. Newville, Fundamentals of XAFS, Rev. Mineral. Geochem., 2014, 78, 33–74 CrossRef CAS.
- F. M. de Groot, J. Fuggle, B. Thole and G. Sawatzky, 2p X-ray absorption of 3d transition-metal compounds: an atomic multiplet description including the crystal field, Phys. Rev. B:Condens. Matter Mater. Phys., 1990, 42, 5459 Search PubMed.
- K. Dai, J. Wu, Z. Zhuo, Q. Li, S. Sallis, J. Mao, G. Ai, C. Sun, Z. Li, W. E. Gent, W. C. Chueh, Y.-d. Chuang, R. Zeng, Z.-x. Shen, F. Pan, S. Yan, L. F. J. Piper, Z. Hussain, G. Liu and W. Yang, High Reversibility of Lattice Oxygen Redox Quantified by Direct Bulk Probes of Both Anionic and Cationic Redox Reactions, Joule, 2019, 3, 518–541 CrossRef CAS.
- K. Fang, J. Yin, G. Zeng, Z. Wu, Y. Tang, D. Yu, H. Luo, Q. Liu, Q. Zhang, T. Qiu, H. Huang, Z. Ning, C. Ouyang, L. Gu, Y. Qiao and S.-G. Sun, Elucidating the Structural Evolution of O3-type NaNi1/3Fe1/3Mn1/3O2: A Prototype Cathode for Na-Ion Battery, J. Am. Chem. Soc., 2024, 146, 31860–31872 CrossRef CAS.
- M. L. Baker, M. W. Mara, J. J. Yan, K. O. Hodgson, B. Hedman and E. I. Solomon, K- and L-edge X-ray Absorption Spectroscopy (XAS) and Resonant Inelastic X-ray Scattering (RIXS) Determination of Differential Orbital Covalency (DOC) of Transition Metal Sites, Coord. Chem. Rev., 2017, 345, 182–208 CrossRef CAS PubMed.
- W.-S. Yoon, M. Balasubramanian, K. Y. Chung, X.-Q. Yang, J. McBreen, C. P. Grey and D. A. Fischer, Investigation of the charge compensation mechanism on the electrochemically Li-Ion deintercalated Li1-xCo1/3Ni1/3Mn1/3O2 electrode system by combination of soft and hard X-ray absorption spectroscopy, J. Am. Chem. Soc., 2005, 127, 17479–17487 CrossRef CAS PubMed.
- J. R. Croy, J. S. Park, F. Dogan, C. S. Johnson, B. Key and M. Balasubramanian, First-Cycle Evolution of Local Structure in Electrochemically Activated Li2MnO3, Chem. Mater., 2014, 26, 7091–7098 Search PubMed.
- A. Deb, U. Bergmann, S. P. Cramer and E. J. Cairns, In situ X-ray absorption spectroscopic study of the Li[Ni1/3Co1/3Mn1/3]O2 cathode material, J. Appl. Phys., 2005, 97, 113523 Search PubMed.
- H. Dau, P. Liebisch and M. Haumann, X-ray absorption spectroscopy to analyze nuclear geometry and electronic structure of biological metal centers-potential and questions examined with special focus on the tetra-nuclear manganese complex of oxygenic photosynthesis, Anal. Bioanal. Chem., 2003, 376, 562–583 CrossRef CAS PubMed.
- Y. Yu, P. Karayaylali, L. Giordano, J. Corchado-Garcia, J. Hwang, D. Sokaras, F. Maglia, R. Jung, F. S. Gittleson and Y. Shao-Horn, Probing Depth-Dependent Transition-Metal Redox of Lithium Nickel, Manganese, and Cobalt Oxides in Li-Ion Batteries, ACS Appl. Mater. Interfaces, 2020, 12, 55865–55875 CrossRef CAS.
- K. Kleiner, J. Melke, M. Merz, P. Jakes, P. Nagel, S. Schuppler, V. Liebau and H. Ehrenberg, Unraveling the Degradation Process of LiNi0.8Co0.15Al0.05O2 Electrodes in Commercial Lithium Ion Batteries by Electronic Structure Investigations, ACS Appl. Mater. Interfaces, 2015, 7, 19589–19600 CrossRef CAS.
- E. Hu, X. Yu, R. Lin, X. Bi, J. Lu, S. Bak, K.-W. Nam, H. L. Xin, C. Jaye, D. A. Fischer, K. Amine and X.-Q. Yang, Evolution of redox couples in Li- and Mn-rich cathode materials and mitigation of voltage fade by reducing oxygen release, Nat. Energy, 2018, 3, 690–698 CrossRef CAS.
- B. Li, Z. Zhuo, L. Zhang, A. Iadecola, X. Gao, J. Guo, W. Yang, A. V. Morozov, A. M. Abakumov and J.-M. Tarascon, Decoupling the roles of Ni and Co in anionic redox activity of Li-rich NMC cathodes, Nat. Mater., 2023, 22, 1370–1379 CrossRef CAS PubMed.
- N. Laszczynski, S. Solchenbach, H. A. Gasteiger and B. L. Lucht, Understanding electrolyte decomposition of graphite/NCM811 cells at elevated operating voltage, J. Electrochem. Soc., 2019, 166, A1853–A1859 CrossRef CAS.
- B. Strehle, K. Kleiner, R. Jung, F. Chesneau, M. Mendez, H. A. Gasteiger and M. Piana, The role of oxygen release from Li- and Mn-rich layered oxides during the first cycles investigated by on-line electrochemical mass spectrometry, J. Electrochem. Soc., 2017, 164, A400 CrossRef CAS.
- Y. Xie, M. Saubanère and M.-L. Doublet, Requirements for reversible extra-capacity in Li-rich layered oxides for Li-ion batteries, Energy Environ. Sci., 2017, 10, 266–274 RSC.
- R. Parmar, S. J. Rezvani, F. Nobili, A. Di Cicco, A. Trapananti, M. Minicucci, S. Nannarone, A. Giglia, F. Maroni and R. Gunnella, Electrochemical Response and Structural Stability of the Li+ Ion Battery Cathode with Coated LiMn2O4 Nanoparticles, ACS Appl. Energy Mater., 2020, 3, 8356–8365 CrossRef CAS.
- E. Bjorklund, C. Xu, W. M. Dose, C. G. Sole, P. K. Thakur, T. L. Lee, M. F. L. De Volder, C. P. Grey and R. S. Weatherup, Cycle-Induced Interfacial Degradation and Transition-Metal Cross-Over in LiNi0.8Mn0.1Co0.1O2-Graphite Cells, Chem. Mater., 2022, 34, 2034–2048 CrossRef CAS PubMed.
- E. Stavitski and F. M. De Groot, The CTM4XAS program for EELS and XAS spectral shape analysis of transition metal L edges, Micron, 2010, 41, 687–694 CrossRef CAS PubMed.
- K. Kleiner, C. A. Murray, C. Grosu, B. Ying, M. Winter, P. Nagel, S. Schuppler and M. Merz, On the Origin of Reversible and Irreversible Reactions in LiNixCo(1−x)/2Mn(1−x)/2O2, J. Electrochem. Soc., 2021, 168(12), 120533 CrossRef CAS.
- A. R. Genreith-Schriever, H. Banerjee, A. S. Menon, E. N. Bassey, L. F. J. Piper, C. P. Grey and A. J. Morris, Oxygen hole formation controls stability in LiNiO2 cathodes, Joule, 2023, 7, 1623–1640 CrossRef CAS.
- V. N. Kothalawala, K. Suzuki, J. Nokelainen, A. Hyvönen, I. Makkonen, B. Barbiellini, H. Hafiz, P. Tynjälä, P. Laine, J. Välikangas, T. Hu, U. Lassi, K. Takano, N. Tsuji, Y. Amada, A. A. S. Devi, M. Alatalo, Y. Sakurai, H. Sakurai and A. Bansil, Compton scattering study of strong orbital delocalization in a LiNiO2 cathode, Phys. Rev. B, 2024, 109, 035139 CrossRef CAS.
- Z. Wu, G. Zeng, J. Yin, C.-L. Chiang, Q. Zhang, B. Zhang, J. Chen, Y. Yan, Y. Tang, H. Zhang, S. Zhou, Q. Wang, X. Kuai, Y.-G. Lin, L. Gu, Y. Qiao and S.-G. Sun, Unveiling the Evolution of LiCoO2 beyond 4.6 V, ACS Energy Lett., 2023, 8, 4806–4817 CrossRef CAS.
- G. Ceder, Y. M. Chiang, D. R. Sadoway, M. K. Aydinol, Y. I. Jang and B. Huang, Identification of cathode materials for lithium batteries guided by first-principles calculations, Nature, 1998, 392, 694–696 CrossRef CAS.
- J. Tarascon, G. Vaughan, Y. Chabre, L. Seguin, M. Anne, P. Strobel and G. Amatucci, In situ structural and electrochemical study of Ni1−xCoxO2 metastable oxides prepared by soft chemistry, J. Solid State Chem., 1999, 147, 410–420 CrossRef CAS.
- W. E. Gent, I. I. Abate, W. Yang, L. F. Nazar and W. C. Chueh, Design Rules for High-Valent Redox in Intercalation Electrodes, Joule, 2020, 4, 1369–1397 CrossRef CAS.
- J. Hong, W. E. Gent, P. Xiao, K. Lim, D.-H. Seo, J. Wu, P. M. Csernica, C. J. Takacs, D. Nordlund, C.-J. Sun, K. H. Stone, D. Passarello, W. Yang, D. Prendergast, G. Ceder, M. F. Toney and W. C. Chueh, Metal–oxygen decoordination stabilizes anion redox in Li-rich oxides, Nat. Mater., 2019, 18, 256–265 CrossRef CAS.
- K. Luo, M. R. Roberts, N. Guerrini, N. Tapia-Ruiz, R. Hao, F. Massel, D. M. Pickup, S. Ramos, Y.-S. Liu, J. Guo, A. V. Chadwick, L. C. Duda and P. G. Bruce, Anion Redox Chemistry in the Cobalt Free 3d Transition Metal Oxide Intercalation Electrode Li[Li0.2Ni0.2Mn0.6]O2, J. Am. Chem. Soc., 2016, 138, 11211–11218 CrossRef CAS.
- Z. Lu and J. R. Dahn, Understanding the Anomalous Capacity of Li/Li [NixLi(1/3−2x/3)Mn(2/3−x/3)]O2 Cells Using In Situ X-Ray Diffraction and Electrochemical Studies, J. Electrochem. Soc., 2002, 149, A815 CrossRef CAS.
- R. A. House, G. J. Rees, M. A. Pérez-Osorio, J.-J. Marie, E. Boivin, A. W. Robertson, A. Nag, M. Garcia-Fernandez, K.-J. Zhou and P. G. Bruce, First-cycle voltage hysteresis in Li-rich 3d cathodes associated with molecular O2 trapped in the bulk, Nat. Energy, 2020, 5, 777–785 CrossRef CAS.
- D.-H. Seo, A. Urban and G. Ceder, Calibrating transition-metal energy levels and oxygen bands in first-principles calculations: accurate prediction of redox potentials and charge transfer in lithium transition-metal oxides, Phys. Rev. B:Condens. Matter Mater. Phys., 2015, 92, 115118 CrossRef.
- D. H. Seo, J. Lee, A. Urban, R. Malik, S. Kang and G. Ceder, The structural and chemical origin of the oxygen redox activity in layered and cation-disordered Li-excess cathode materials, Nat. Chem., 2016, 8, 692–697 CrossRef CAS PubMed.
- A. Gao, X. Li, Q. Zhang, T. Lin, Y. Wang, Y. Chen, W. Lin, S. Wang, P. Ji, Z. Luo, J. Wang, Y. Guo and L. Gu, Dynamic Transition Metal Network via Orbital Population Design Stabilizes Lattice Oxygen Redox in Stoichiometric Layered Cathodes, Adv. Mater., 2025, 37, 2412673 CrossRef CAS PubMed.
- C. Schulz, K. Lieutenant, J. Xiao, T. Hofmann, D. Wong and K. Habicht, Characterization of the soft X-ray spectrometer PEAXIS at BESSY II, J. Synchrotron Radiat., 2020, 27, 238–249 CrossRef CAS PubMed.
- G.-H. Lee, S. Lee, J. Zhang, B. Rinkel, M. J. Crafton, Z. Zhuo, Y. Choi, J. Li, J. Yang, J. W. Heo, B.-C. Park, B. D. McCloskey, M. Avdeev, W. Yang and Y.-M. Kang, Oxygen Redox Activities Governing High-Voltage Charging Reversibility of Ni-Rich Layered Cathodes, Energy Environ. Sci., 2024, 17, 9154–9163 RSC.
- R. A. House, U. Maitra, M. A. Perez-Osorio, J. G. Lozano, L. Jin, J. W. Somerville, L. C. Duda, A. Nag, A. Walters, K. J. Zhou, M. R. Roberts and P. G. Bruce, Superstructure control of first-cycle voltage hysteresis in oxygen-redox cathodes, Nature, 2020, 577, 502–508 CrossRef CAS PubMed.
- R. A. House, J.-J. Marie, M. A. Pérez-Osorio, G. J. Rees, E. Boivin and P. G. Bruce, The role of O2 in O-redox cathodes for Li-ion batteries, Nat. Energy, 2021, 6, 781–789 Search PubMed.
- E. Hu, S. M. Bak, Y. Liu, J. Liu, X. Yu, Y. N. Zhou, J. Zhou, P. Khalifah, K. Ariyoshi and K. W. Nam, Utilizing environmental friendly iron as a substitution element in spinel structured cathode materials for safer high energy lithium-ion batteries, Adv. Energy Mater., 2016, 6, 1501662 CrossRef.
- B. Thole and G. Van der Laan, Branching ratio in X-ray absorption spectroscopy, Phys. Rev. B:Condens. Matter Mater. Phys., 1988, 38, 3158 CrossRef CAS PubMed.
- H.-H. Ryu, K.-J. Park, C. S. Yoon and Y.-K. Sun, Capacity Fading of Ni-Rich Li[NixCoyMn1−x−y]O2 (0.6 ≤ x ≤ 0.95) Cathodes for High-Energy-Density Lithium-Ion Batteries: Bulk or Surface Degradation?, Chem. Mater., 2018, 30, 1155–1163 Search PubMed.
- D. Yu, G. Zeng, D. Chen, Y. Yan, Y. Zou, Q. Liu, K. Zhang, K. Fang, J. Xu, W. Yin, Y.-H. Hong, T. Qiu, H.-G. Liao, X. Kuai, Y. Sun, Y. Qiao and S.-G. Sun, Revealing Gliding-Induced Structural Distortion in High-Nickel Layered Oxide Cathodes for Lithium-Ion Batteries, ACS Nano, 2024, 18, 27654–27664 CrossRef CAS PubMed.
- C. Wang, X. Wang, R. Zhang, T. Lei, K. Kisslinger and H. L. Xin, Resolving complex intralayer transition motifs in high-Ni-content layered cathode materials for lithium-ion batteries, Nat. Mater., 2023, 22, 235–241 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc00429b |
| This journal is © The Royal Society of Chemistry 2025 |