
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnrevealing the potential of multicomponent metal-ion incorporation and sulfide modification in cobalt oxide for efficient water oxidation†
Muzzayab Masooda,
Muhammad Aamir *ac,
Muhammad Ejaz Khanb,
Muhammad Sherc,
Khush Bakhat Akramh,
Hafiz Zahid Shafid,
Hamad Almohamadi*e,
M. d. Akhtaruzzamanf and
M. d. Shahiduzzamang
*ac,
Muhammad Ejaz Khanb,
Muhammad Sherc,
Khush Bakhat Akramh,
Hafiz Zahid Shafid,
Hamad Almohamadi*e,
M. d. Akhtaruzzamanf and
M. d. Shahiduzzamang
aDepartment of Chemistry, Mirpur University of Science and Technology (MUST), Mirpur-10250 (AJK), Pakistan. E-mail: aamirorg@gmail.com
bDepartment of Computer Engineering, National University of Technology, Islamabad 44000, Pakistan
cDepartment of Chemistry, Allama Iqbal Open University, H-8, Islamabad, Pakistan
dMaterials Division, National Institute of Lasers & Optronics College (NILOP-C), Pakistan Institute of Engineering & Applied Science (PIEAS), P.O. Nilore 45650, Islamabad, Pakistan
eDepartment of Chemical Engineering, Faculty of Engineering, Islamic University of Madinah, Madinah, Abo Bakr Al Siddiq, Al Jamiah, Madinah 42351, Saudi Arabia
fThe Department of Chemistry, Faculty of Science, The Islamic University of Madinah, Madinah, Abo Bakr Al Siddiq, Al Jamiah, Madinah 42351, Saudi Arabia. E-mail: akhtar.brces@gmail.com
gNanomaterials Research Institute, Kanazawa University, Kakuma, Kanazawa 920-1192, Japan
hDepartment of Computer Science, National University of Technology, Islamabad 44000, Pakistan
First published on 1st July 2024
Abstract
The design and development of highly efficient electrocatalysts from transition metals have shown a great potential for substituting precious metal-based electrocatalysts in water-splitting processes. Cobalt oxide is one of the promising materials for oxygen evolution reaction (OER). Modifying the metal oxide by the incorporation of metal ions and substituting sulfides are effective but challenging strategies for achieving efficient OER activities. In the present work, we report the synthesis of CdCoO and CdCoS electrocatalysts deposited on the surface of nickel foam. These electrocatalysts and their composites CdCoO@CuCoO and CdCoS@CuCoS could deliver high catalytic activity for oxygen evolution reaction. The as-synthesized electrocatalysts were characterized using pXRD, FTIR spectroscopy, Raman spectroscopy, XPS, and SEM techniques. The CdCoS showed a lower OER overpotential of 199 mV at a current density of 10 mA cm−2 and 522 mV at 60 mA cm−2. The incorporation of Cd2+ ions in the cobalt oxides optimized the electronic states around the Co active sites, leading to improved catalytic activities and a lower overpotential compared to other reported cobalt oxides (such as oxyhydroxides). This work emphasizes the effect of metal-ion incorporation and sulfide modification on the OER activity of cobalt oxide for water splitting and provides a multicomponent engineering strategy for designing efficient electrocatalysts.
Introduction
The development of efficient and cost-effective electrocatalysts for oxygen-evolution reaction (OER) remains a serious challenge for realizing the full potential of electrochemical water splitting as a clean and sustainable energy source.1–3 This technology holds immense promise for generating hydrogen fuel, a clean-burning fuel with minimal environmental impact.4–7 Transition metal oxides, phosphides, and sulfides have emerged as promising OER electrocatalysts owing to their inherent abundance, good stability, and tunable physicochemical properties.8,9 The electrocatalytic activity of transition metal oxides can be tuned by the intentional incorporation of metal ions and the substitution of sulfur in the metal oxides, which can collectively enhance their OER activity.10,11 Thus, sulfur incorporation in metal oxides, particularly in cobalt oxide, produces the high spin state of the metal ions, e.g., Co3+ ions. Further, this high spin state has a higher electron population in the eg-orbitals of Co3+, improving the OER activity of cobalt oxide and its derivatives.12Metal sulfides have an enormous potential for development; however, their lower conductivity and scarcity of active sites are major challenges for their widespread applications.13 These challenges can be addressed by various strategies such as the combination of metal sulfides with oxides to create heterojunctions that subsequently improve the conductivity.11,14 Likewise, tuning the electronic properties of the metal oxides by introducing sulfur by a partial substitution results in lowering the covalency of the M–O bond through an inductive effect.15 Sulfur withdraws the electron density in M–O–S bonds and lowers the energy of antibonding orbitals, which promotes the formation of a high spin metal system with enhanced OER activities.16,17
Likewise, the substitution of metal ions in metal sulfides or metal incorporation in metal sulfides can optimize the local electronic structure of the active sites available for OER activities. Metal ion such as n-type or p-type incorporations in cobalt oxides and sulfides could be worthful exploring. For instance, Cd incorporation in cobalt oxide promotes electron transfer at the cobalt active sites. Moreover, the larger Cd2+ ion produces distortions in the cobalt oxide lattice, causing the exposure of cobalt sites for OER activities.18 The ionic radii of Cu2+ ions are comparable to Co3+ ions, And it has been reported that Cu-incorporated cobalt oxides with some optimum copper concentration can improve the OER activity of cobalt oxides for the OER.19 Moreover, the electronegativity of Cd2+ ions and Cu2+ is less than that of Co3+ ions, and CdO is an n-type and CuO is a p-type material, which affects the electronic properties of the active sites.20 Recently, the development of multi-elemental materials, also known as high entropy materials, having multiple elements in solid solutions and composites has been explored and these have shown excellent OER performances.21–23
In this work, we successfully synthesized Cd-incorporated cobalt oxide and a multicomponent composite of CdCoO with CuCoO. The sulfurized analogs were also synthesized through a deliberate design using sulfur precursors. Full characterizations were performed using pXRD, XPS, FTIR, Raman, and SEM techniques to analyze the as-synthesized materials. The electrochemical results showed that the OER potential was lower in Cd-incorporated cobalt oxide, while the overpotential was increased in the composites. The sulfide analog of the aforementioned electrocatalysts showed a lower overpotential than the oxides. The experimental results confirmed the influence of metal-ion substitution in the metal oxides and sulfides on the electronic properties around the active sites and the effect of the sulfide on the OER activity of the electrocatalysts. These findings add to the fundamental understanding needed to design new materials with improved OER activities.
Experimental
Chemicals
The commercially available analytical grade chemicals were used without further purification. These included cadmium sulfate hydrate (CdSO4·8/3H2O), cobalt(II) sulfate heptahydrate (CoSO4·7H2O), copper(II) sulfate pentahydrate (CuSO4·5H2O), thiourea (CH4N2S), ammonium dihydrogen phosphate (NH4H2PO4), polyvinylpyrrolidone (PVP, K-30, MW 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), 2-methylimidazole (C4H6N2), glycerol, isopropyl alcohol (IPA), N,N-dimethylformamide (DMF), ethanol, and deionized water. All the chemicals were purchased from Sigma Aldrich.
000), 2-methylimidazole (C4H6N2), glycerol, isopropyl alcohol (IPA), N,N-dimethylformamide (DMF), ethanol, and deionized water. All the chemicals were purchased from Sigma Aldrich.
Synthesis of the electrocatalysts CdCoO and CdCoS
A solvothermal method was employed for the synthesis of CdCoO. Briefly, 0.125 g of CdSO4 and 0.25 g of CoSO4 were dissolved in 40 mL of isopropyl alcohol under magnetic stirring for 30 min. Subsequently, 8 mL of glycerol was added, and the solution was stirred for another hour to obtain a light pink homogeneous mixture. This mixture was then heated at 180 °C for 6 h in a sealed container. The resulting CdCo-glycolate precursor was cooled down, collected by centrifugation, and washed thoroughly with ethanol and deionized water. Finally, the dried CdCo-glycolate was calcined at 350 °C for 2 h to obtain CdCoO nanoparticles. For the synthesis of CdCoS, 0.2 g of thiourea was added to the reaction mixture alongside CdSO4 and CoSO4 using the same procedure described for CdCoO.Synthesis of CdCoO@CuCoO and CdCoS@CuCoS
The Cd–Co–O@Cu–Co–MOF composite was synthesized via a two-step method. First, 0.175 mg of PVP was dissolved in 10 mL of ethanol and stirred with 0.35 mg of presynthesized Cd–Co–O for 12 h. Subsequently, 10 mL each of deionized water and DMF were added along with 2 g of PVP, 0.25 mg of CoSO·46H2O, 0.75 mg of 2-methylimidazole, and 1.25 mg of CuSO4·H2O. The mixture was stirred for 30 min to achieve homogeneity and then heated at 150 °C for 10 h in a sealed container. The obtained product was cooled down, centrifuged at 6000 rpm for 15 min, washed repeatedly with appropriate solvents, and dried to obtain the Cd–Co–O@Cu–Co–MOF precursor. The synthesis of CdCoS@CuCo–MOF followed the same procedure, utilizing CdCoS instead of CdCoO in the initial step. CdCoO@CuCoO and CdCoS@CuCoS nanocomposites (NCs) were fabricated by thermal decomposition of the corresponding MOF precursors (M = Co or S). The calcination process was carried out at 450 °C for 2 h at a heating rate of 1 °C min−1 in air. Scheme 1 shows the steps involved in the synthesis of CdCoS and the CdCoS@CuCoS-CNs composite.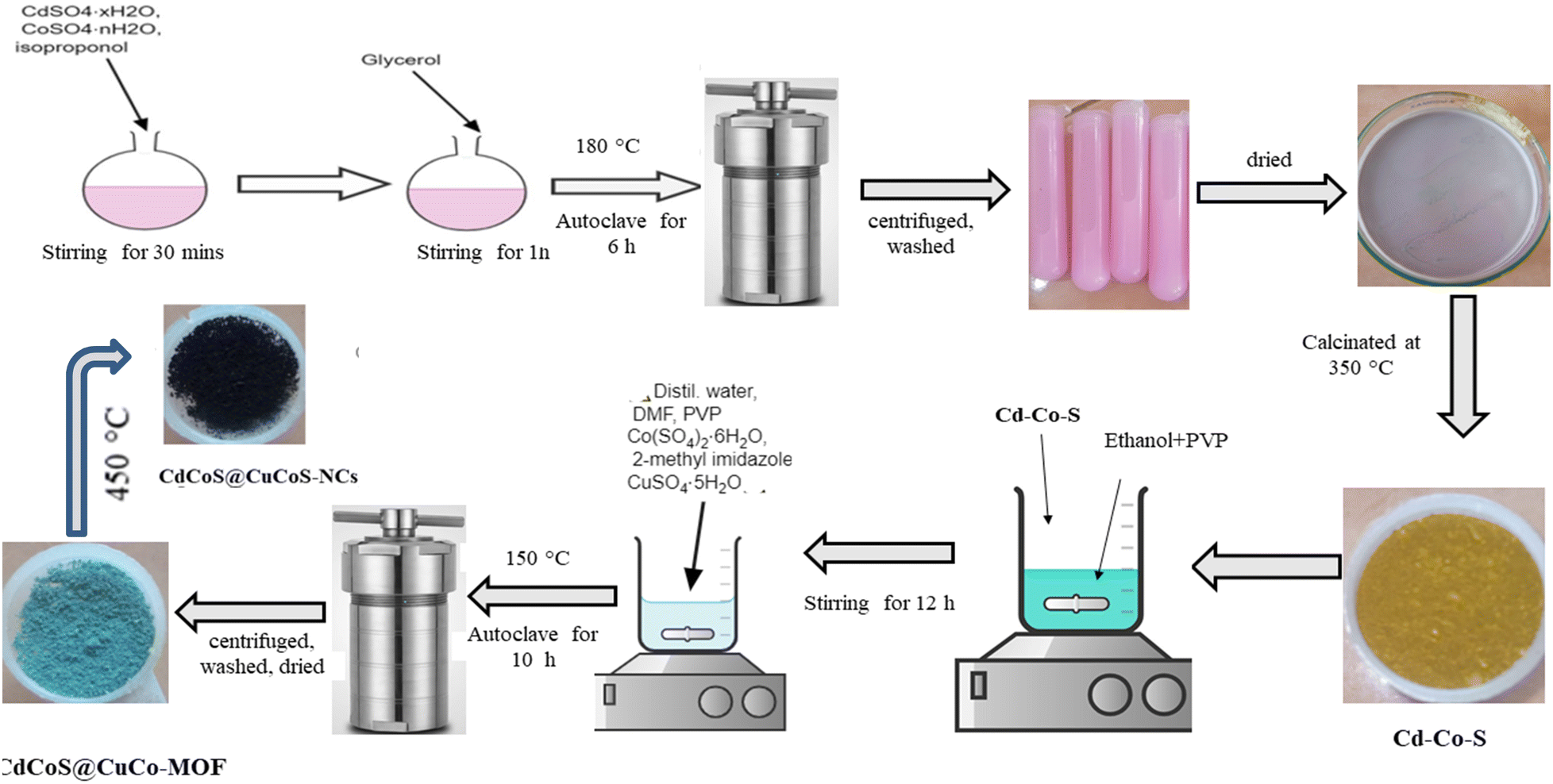 | ||
| Scheme 1 Schematic illustration for the synthesis of CdCoS and its composite CdCoS@CuCoS-NCs. | ||
Structural characterizations
The pXRD diffraction patterns were obtained using a Bruker-D8 DISCOVER system (Germany) under Cu Kα X-ray irradiation with λ = 1.5406 Å. Scanning electron microscopy (SEM) and energy dispersive X-ray (EDX) analysis were performed to obtain images and the elemental composition using the FESEM NOVA MIRA3XMM instrument. Fourier transform infrared spectroscopy (FT-IR) was performed to identify the functional groups using a SHIMADZU IRAffinity-1S system at room temperature. XPS was performed on a Kratos Axis Ultra DLD (Kratos Analytical, Japan)-based UHV spectrometer equipped with an Al Kα X-ray irradiation source (1486.6 eV).Electrochemical OER measurements
The electrochemical measurements were performed using an electrochemical workstation (Gamry Reference 3000) in a three-electrode setup with 15 mL aqueous KOH as an electrolyte, as-prepared samples on nickel foam as the working electrode, Pt as the counter electrode, and Ag/AgCl as the reference electrode. Electrochemical techniques, such as cyclic voltammetry (CV), linear sweep voltammetry (LSV), and electrochemical impedance spectroscopy (EIS), were employed to evaluate the performance of the electrocatalysts for OER activities.Linear sweep voltammetry (LSV) measurements
LSV was performed at a scan rate of 5 mV s−1 to get the precise overpotential. The conversion between the potentials versus Ag/AgCl and RHE was calculated using Nernst's equation, eqn (1), as follows.| ERHE = EAg/AgCl + (0.1976 + 0.059) V | (1) |
The overpotential of the electrocatalyst was determined by eqn (2).
| Overpotential = ERHE − 1.23 V | (2) |
Tafel plots
The polarization curves obtained from the LSV measurements were plotted as overpotential versus the log current. The Tafel slope was obtained by taking the slope of the fitted linear portion of the Tafel plot according to the eqn (3):| η = b log[J] + a | (3) |
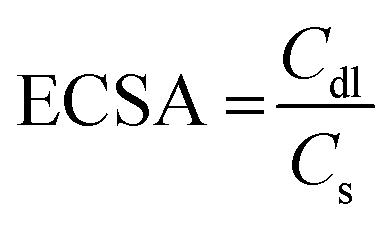 | (4) |
iR corrections
The solution resistance was determined by impedance spectroscopy. The potential was corrected by subtraction of 100% of the solution resistance according to eqn (5).| EiR corrected = E − iR | (5) |
Results and discussion
FTIR spectra of the as-synthesized CdCoO, CdCoO@CuCoO/C–NC, CdCoS, and CdCoS@CuCoS/C–NC are presented in Fig. 1a. The stretching vibrational bands at 1635 and 3266 cm−1 were credited to the –OH group present on the surface of CdCoO and its derivatives.24 The presence of H2O and CO2 was indicated by the symmetric and asymmetric stretching of C–O vibrations observed at 1000–1500 cm−1.25 The band located at 2358 cm−1 corresponded to the stretching vibrations of C–H bonds.26 The sharp bands at 592 and 675 cm−1 indicated the presence of spinel structure vibrations of the M–O bonds in tetrahedral and octahedral fields.27,28 In CdCoO@CuCoO/C–NC, the M–O vibrational bands became weaker and broader due to the formation of M–O, M–N, and M–C bonds due to the metal oxides and oxygen and nitrogen-functionalized carbons.29 Likewise, the band at 1618 cm−1 indicated the presence of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C linkage in CdCoO@CuCoO/C–NC.25 The symmetric stretching of C–O appeared at 1024 cm−1. In CdCoS, the vibrational bands at 592 and 671 cm−1 belonged to Co–S and Cd–S, respectively.26 Similar to the oxide-based samples, the weak vibrational band of M–S in CdCoS@CuCoS/C–NC also became broader and weaker.
C linkage in CdCoO@CuCoO/C–NC.25 The symmetric stretching of C–O appeared at 1024 cm−1. In CdCoS, the vibrational bands at 592 and 671 cm−1 belonged to Co–S and Cd–S, respectively.26 Similar to the oxide-based samples, the weak vibrational band of M–S in CdCoS@CuCoS/C–NC also became broader and weaker.
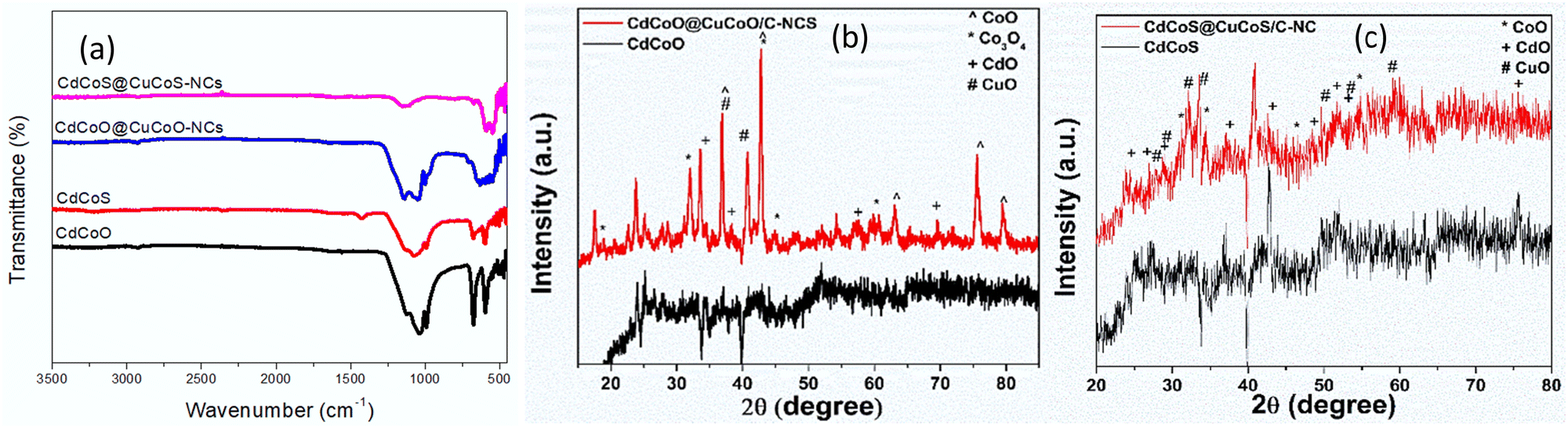 | ||
| Fig. 1 (a) FTIR spectra of CdCoO, CdCoS, CdCoO@CuCoO-NCs, and CdCoS@CuCoS-NCs. pXRD patterns of (b) CdCoO, CdCoO@CuCoO-NCs (c) CdCoS, and CdCoS@CuCoS-NCs. | ||
The crystal structure of the as-synthesized samples was determined by X-ray diffraction (XRD) analysis. Fig. 1b shows the XRD patterns of CdCoO and CdCoO@CuCoO/C–NCs. The diffraction peaks at 2θ degrees of 18.9, 32, 36.9, 42.7, 44.9, and 60.77 degrees could be indexed to the (111), (220), (311), (222), (400), (422), (511), and (440) planes of Co3O4 (JCPD Card: 75-2480).30 According to JCPD file No. 01-074-2391, the presence of CoO was confirmed by the peaks diffracted at 2θ degrees of 36.9, 42.7, 63.05, 75.7, and 79.2 that could be indexed to the (111), (200), (220), (311), and (222) planes.31 The presence of Cd–O was confirmed by the appearance of diffraction peaks for (111), (200), (220), (311), and (222) at degree angles of 33.6, 38.2, 57.1, and 69.5 (JCPD card no 75-0592).32 Two diffraction peaks for (200) and (111) at 36.9 and 40.8 degrees appeared from the incorporation of CuO (JCPD card 45-0937) in CdCoO@CuCoO/C–NCS.33 Similarly, in CdCoS and CdCoS@CuCoS/C–NCs, the diffraction peaks at degree angles of 31.2, 34.5, 46.6, and 54.7 were consistent with JCPD card # 19-0366, corresponding to the (204), (220), (306), and (330) planes of CoS (Fig. 1c).34 The diffraction peaks of CdS appeared at 2θ degrees of 23.9, 27, 28.5, 37.09, 42.8, 51.9, and 53.6, belonging to the (100), (002), (101), (102), (110), (103), (200), (112), and (201) planes respectively.35 The diffraction peaks observed at degree angles of 27.8, 29.1, 32.1, 33.4, 49.9, 53.5, and 59 were found to have characteristics of CuS and were assigned to the indices (101), (102), (103), (006), (110), (108), and (116), respectively (JCPD no. 01-079-2321).36
The vibrations of metal–oxygen bonds, nearby surface imperfections, and the coordinating environment were next investigated using Raman spectroscopy. The Raman spectra of the prepared oxide nanocomposites are shown in Fig. 2a. All the characteristic vibration peaks of CdCoO could be seen in the Raman spectra. The vibrational peaks at 184.8, 487.92, 524.6, 636.01, and 713 cm−1 were assigned to asymmetric and bending vibrations of Co2O3.37 CdO exhibited Raman modes at 281, 376, 599.7, and 801 cm−1.38 In CdCoO@CuCoO/C–NCs, the peaks at 287.18, 328, and 615 cm−1 were assigned to the vibrations of Cu–O.39 In the case of CdCoS, the vibrations at 477.6, 494, and 677.3 cm−1 and peaks at 298.9, 392.2, and 586.7 cm−1 were assigned to the Raman modes of CoS and CdS, respectively (Fig. 2b).35,40 Likewise, in CdCoS@CuCoS/C–NCS, the vibrational modes for CuS at 285.1 and 485.2 cm−1 were the characteristics peaks for Cu–S bond (Fig. 2b).41
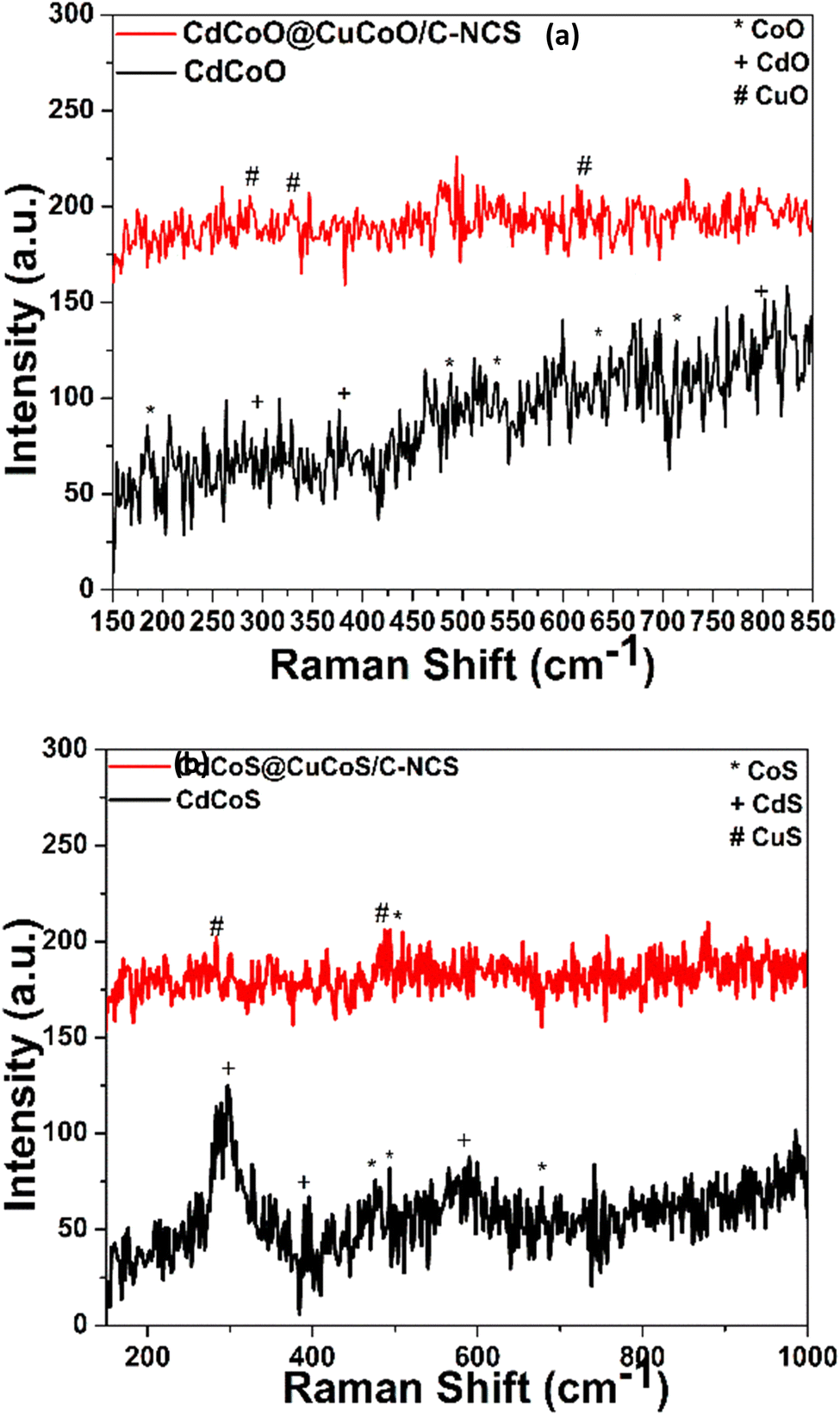 | ||
| Fig. 2 Raman spectra of (a) CdCoO and CdCoO@CuCoO/C–NCs, and (b) CdCoS and CdCoS@CuCoS/C–NCs. | ||
The X-ray photoelectron spectra were recorded on the surface of the as-synthesized materials to examine their valence shell states. The XPS survey spectra of CdCoO, and CdCoO@CuCoO/C–NCs (CdCoO@CuCoO) are shown in Fig. 3a. The presence of Cd, Co, C, and O at the surface of the synthesized materials could be observed in the survey spectrum of CdCoO. The only difference in the XPS survey spectra of CdCoO@CuCoO and CdCoO was the presence of Cu-related peaks, indicating the successful formation of the required materials. Fig. 3b shows the comparative survey XPS spectra of CdCoS and CdCoS@CuCoS/C–NCs (CdCoS@CuCoS). The presence of S 2p peaks in the survey spectra showed the formation of the respective sulfides. Fig. 3c shows major peaks at 406 and 412 eV corresponding to Cd 3d3/2 and Cd 3d5/2, respectively, confirming the presence of Cd2+.42,43 Whereas, the deconvoluted spectrum of Co 2p with spin–orbit bimodal pairs showed peaks for Co 2p1/2 and Co 2p3/2, which appeared at 798.6 and 783. 6 eV, respectively, with satellite peaks at 804 and 786 eV, confirming the presence of CoO (Fig. 3c).44,45 The oxidation state of the cobalt could not be determined based purely on the main oxidation peaks.46 The satellite peak intensity and their binding energy positions were used to identify the Co valence state.47 Cobalt showed 2+ and 3+ ionic states, in which Co2+ displayed two satellite peaks due to the crystal field splitting in the tetrahedral crystal field environment. Therefore, it is obvious from the spectra that the cobalt was present in mixed valence states in the as-synthesized materials.46,48
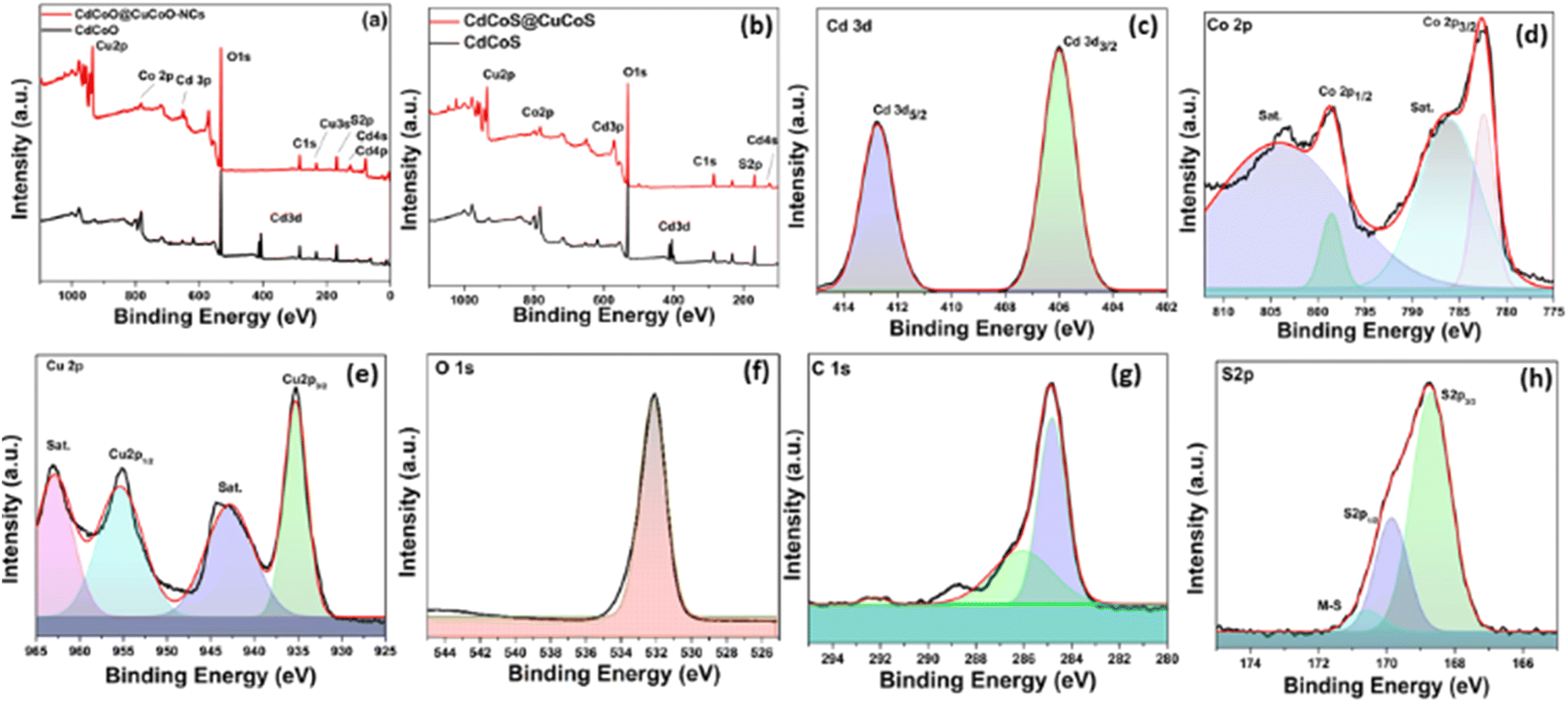 | ||
| Fig. 3 XPS survey spectra of (a) CdCoO, CdCoO@CuCoO/CNs, and (b) CdCoS and CdCoS@CuCO@/CNs. XPS deconvoluted spectra for (c) Cd, (d) Co, (e) Cu, (f) O,(g) C, and (h) S in the as-synthesized samples. | ||
Fig. 3d displays the deconvoluted spectra of Cu 2p into Cu 2p3/2 and Cu 2p1/2 at 935.1 and 955.1 eV, respectively. The energy difference between the two peaks was 20 eV, which corresponded to the previous CuO spectra.49 The appearance of two satellite peaks at 943 and 962.6 eV with the main peaks also confirmed the synthesis of CuO.50,51 The core level spectrum of O 1s is illustrated in Fig. 3(d). A distinct peak at 532 eV was observed, attributed to the O−2.52 Similarly, spectral peaks for C 1s appeared at 292.1, 287.5, and 284.9 eV, which were attributed to the presence of C–O and C–C linkages due to the oxidation of imidazole to form carbon species (Fig. 3e).53 Similarly, in CdCoS@CdCuS, peaks for S−2 were observed at 168 eV and 170 eV for S 2p3/2 and S 2p1/2, respectively (Fig. 3g).54,55 A peak observed at 171.2 eV corresponding to the presence of metal-sulfur linkages.56 The presence of S 2p and O 1s characteristic peaks in the sulfide-based samples indicated the formation of partial sulfide and oxide-based systems (Fig. 3h).
Scanning electron microscopy (SEM) was used to explore the morphology of the as-synthesized materials, as shown in Fig. 4. Fig. 4a shows that CdCoO comprised flake-like aggregated microstructures. The CdCoO@CuCoO composite showed flower-like morphologies due to the presence of carbon materials (Fig. 4b). Whereas, CdCoS exhibited smaller fused spherical morphologies due to agglomeration, as displayed in Fig. 4c. In comparison to CdCoO, CdCoS comprised smaller particles. Like CdCoO@CuCoO, the CdCoS@CuCoS composites displayed similar kinds of flake-like structures due to the carbon materials (Fig. 4d).
 | ||
| Fig. 4 SEM images of the as-synthesized (a) CdCoO, (b) CdCoO@CuCoO, (c) CdCoS, and (d) CdCoS@CuCoS. | ||
Electrochemical OER activity
The as-synthesized materials showed typical cyclic voltammetry (CV) behaviors in 1.0 M KOH, as shown in Fig. 5a–d. The oxidation peak between 1.42–1.66 V (vs. RHE) corresponded to the Co2+/Co3+ redox couple, which is an active electrocatalyst for OER activity. The plot between the anodic peak current and the square root of the scan rate exhibited a linear relationship (Fig. 5e). The slope values for CdCoS, CdCoS@CuCoS, CdCoO, and CdCoO@CuCoO were 33.1784, 27.6949, 16.8722, and 17.6006, respectively. The highest slope value was observed for CdCoS, indicating the enhanced diffusion properties of OH−. Therefore, more electroactive supplies, such as Co–SOOH, can be formed on the surface of CdCoS. Likewise, the CV profiles were also used to determine the double-layer capacitance (Cdl) of the as-synthesized catalysts, as presented in Fig. 5f.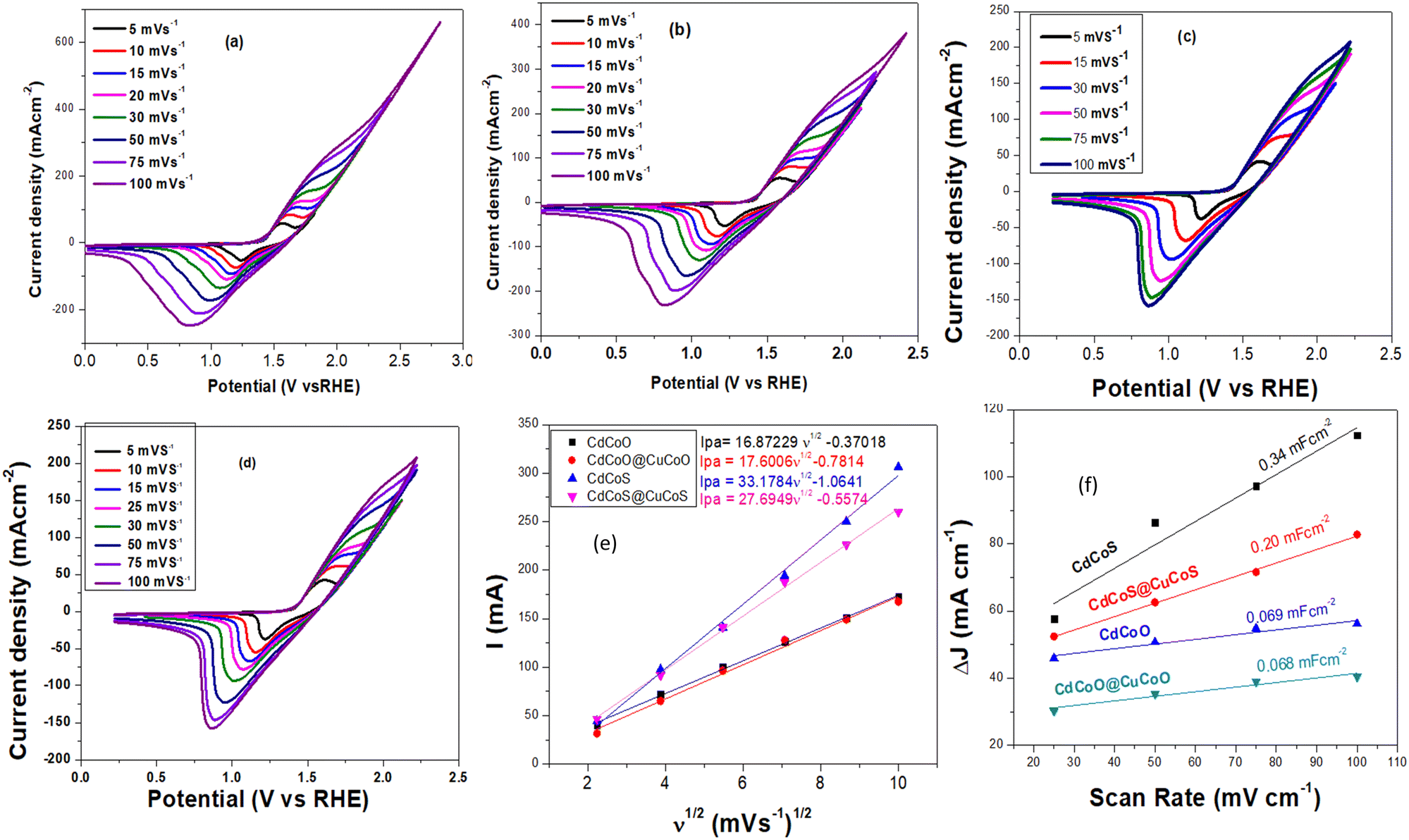 | ||
| Fig. 5 Cyclic voltammograms (CV) of the catalysts (a) CdCoS, (b) CdCoS@CuCoS, (c) CdCoO, and CdCoO@CuCoO. The plots are between the anodic peak current (Ipa) of cyclic voltammograms in 1.0 M KOH and the square root of the scan rates (v1/2) for all the as-synthesized catalysts. | ||
The average Cdl values of CdCoS, CdCuS@CuCoS, CdCoO, and CdCoO@CuCoO were found to be 0.34, 0.20, 0.069, and 0.068 mF cm−2, respectively (Table 1). The larger Cdl value of CdCoS also suggested it had more active sites for OER activity. The Cdl results supported the findings of the anodic peak current and the square root of the scan rate plot. The ECSA is another parameter to correlate the catalytic efficiency of materials and was calculated using the average Cdl values presented in Table 1.
| Catalysts | Average Cdl (mF cm−2) | ECSA (cm−2) | Rct (Ω) |
|---|---|---|---|
| CdCoS | 0.34 | 15.22 | 3.2 |
| CdCoS@CuCoS | 0.20 | 8.95 | 8 |
| CdCoO | 0.068 | 3.045 | 10.78 |
| CdCoO@CuCoO | 0.069 | 3.09 | 23.2 |
CdCoS showed an ECSA value of 15.22 cm−2, whereas the ESCA values for CdCuS@CuCoS, CdCoO, and CdCoO@CuCoO were 8.95, 3.045, and 3.09 cm−2 respectively (Table 1). The greater ESCA value for CdCoS showed that it offered a larger surface area and more active sites for the intrinsic electrocatalytic activity of the catalyst compared to the other as-synthesized materials. Based on these results, it could be determined that CdCoS and CdCoO can show a greater electrocatalytic performance than the composites since the composites comprised large-sized particles with a reduced surface area; moreover, the surface of the catalyst was covered by amorphous carbon, which blocks the active sites.
LSV was performed using a standard three-electrode system in 1.0 M KOH alkaline medium in the potential window of 1.2–2.3 V (vs. RHE) to explore the electrochemical OER performance of the as-synthesized catalysts. As shown in Fig. 6a, the onset potentials for CdCoS, CdCuS@CuCoS, CdCoO, and CdCoO@CuCoO were observed at 1.42, 1.435, 1.438, and 1.446 V (vs. RHE), respectively.
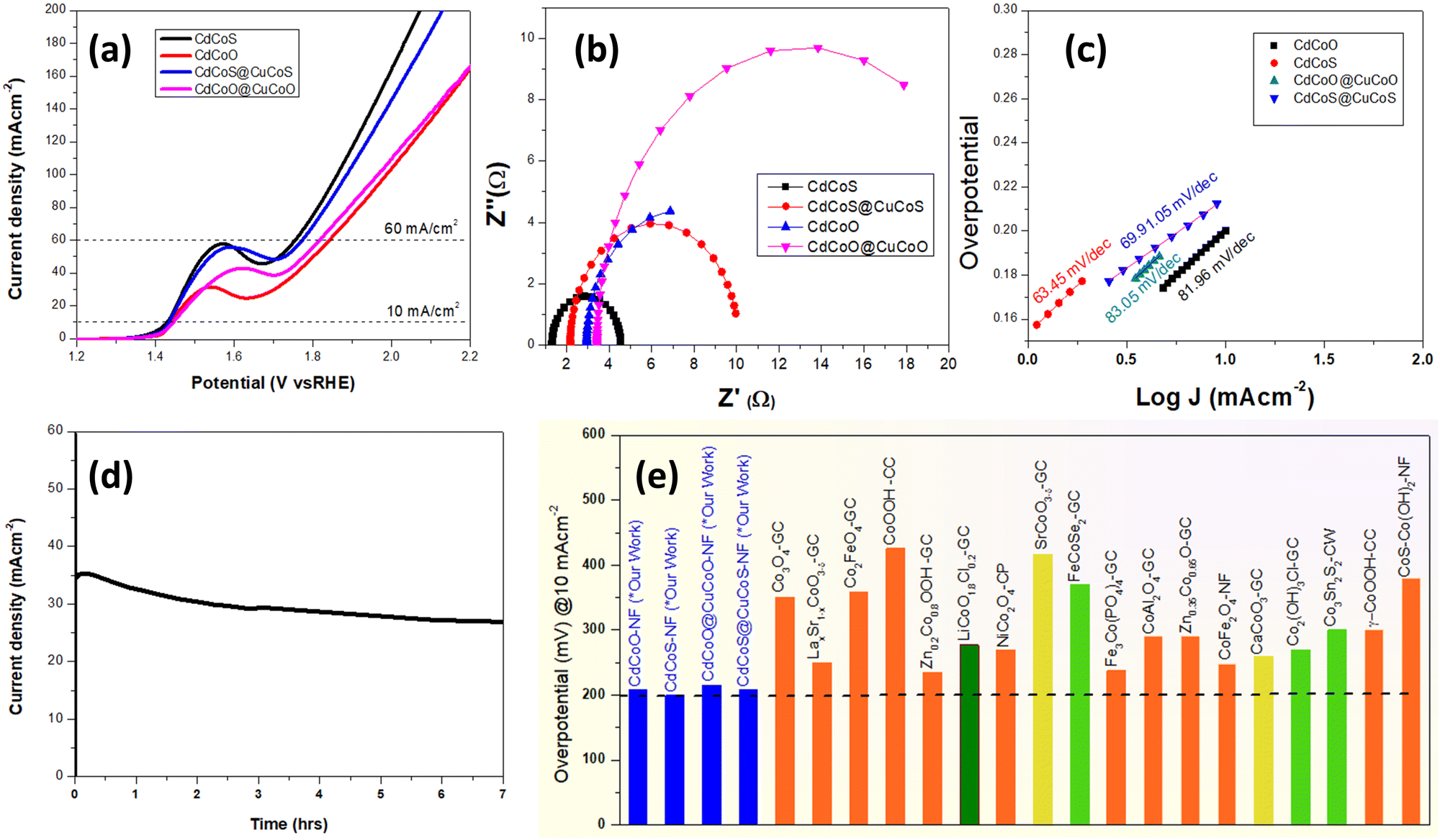 | ||
| Fig. 6 (a) iR uncorrected LSV plots (vs. RHE), (b) EIS Nyquist plots to determine the charge-transfer resistance and solution resistance, (c) Tafel plots to determine the kinetics parameters of the as-synthesized CdCoS, CdCoS@CuCoS, CdCoO, and CdCoO@CuCoO. (d) Chronoamperometric stability tests of the best performing CdCoS electrocatalysts, and (e) overpotential at 10 mA cm−2 of the various cobalt-based electrocatalysts for diverse material classes reported in the literature (appropriate references are given in Table S1, ESI†); GC = glassy carbon, NF = nickel foam, CC = carbon cloth, CW = copper wire, CP = carbon paper. | ||
Whereas the values of the overpotential at 10 mA cm−2 were 199, 205, 208, and 215 mV for CdCoS, CdCuS@CuCoS, CdCoO, and CdCoO@CuCoO, respectively. It could thus be observed that CdCoS had a lower overpotential than all the other as-synthesized catalysts. However, these overpotential values at 10 mA cm−2 do not truly represent the overpotential for the OER. There was also a contribution from the oxidation peak current of materials in the same potential range. Thus, we also measured the overpotential at 60 mA cm−2 to eliminate the oxidation peak current factor. At 60 mA cm−2, there was no involvement of oxidation peaks of the as-synthesized catalysts. The overpotential values showed the same trend as that observed at 10 mA cm−2; however, the value of the overpotential was high, as expected. The overpotential values for CdCoS, CdCuS@CuCoS, CdCoO, and CdCoO@CuCoO were 522, 588, 543, and 616 mV at 60 mA cm−2, respectively. It could be observed that CdCoS and CdCoO had superior performance compared to their respective composites CdCoS@CuCoS and CdCoO@CuCoO. These results indicate that the poor charge transfer between the heterostructures, increased particle size after the formation of composite, and blockage of active sites by the carbon were the major contributors.
Likewise, we calculated the TOF values of the as-synthesized electrocatalysts to determine the intrinsic activity of the catalysts, by assuming that the Co2+ is the only active sites and is oxidized just before the onset of the OER. The TOF values were calculated at 10 mA cm−2 and 60 mA cm−2. CdCoS showed values of 0.673 s−1 (10 mA cm−2) and 4.04 s−1 (60 mA cm−2) while, TOF values of 1.04 s−1 (10 mA cm−2) and 6.26 s−1 (60 mA cm−2) were observed for CdCoO. On the other hand, for CdCuS@CuCoS, the TOF values were 1.1 s−1 (10 mA cm−2) and 6.59 s−1 (60 mA cm−2) and for CdCoO@CuCoO, TOF values of 1.32 s−1 (10 mA cm−2) and 7.93 s−1 (60 mA cm−2) were observed.
As the electrocatalysis was performed in the solution phase, therefore, solution resistance plays a key role in the electrocatalysis. To determine the solution resistance, electrochemical impedance spectroscopy (EIS) was performed and the results are presented in Fig. 6b. EIS was performed in 1.0 M KOH solution, at an open circuit potential. Solution resistances of 1.27, 2.14, 2.94, and 3.41 Ω were observed for the CdCoS, CdCuS@CuCoS, CdCoO, and CdCoO@CuCoO catalysts, respectively. The Rct values for CdCoS, CdCuS@CuCoS, CdCoO, and CdCoO@CuCoO were 3.2, 8, 10.78, and 23.2 Ω respectively. The lowest Rct value was observed for CdCoS, indicating its faster electron transport compared to the other electrocatalysts.
The kinetics is another parameter to determine the performance of an electrocatalyst. Fig. 6c shows the Tafel plots for CdCoS, CdCuS@CuCoS, CdCoO, and CdCoO@CuCoO with values of 63.45, 69.91, 81.98, and 83.05 mV dec−1 respectively. CdCoS showed the lowest Tafel slope value, indicating its fast kinetics compared to all the other samples. Likewise, in comparison, CdCoS had a lower Tafel slope value compared to CdCoS@cuCoS whereas, that for CdCoO was lower than for the corresponding composite (Table 2). These results are in accordance with the LSV results. Next, the stability of the prepared catalyst was determined at a constant voltage of 0.6 V and the current was measured for 7 h, as shown in Fig. 6d. A slight decrease in current density was observed after a few hours, but overall the catalyst exhibited good stability for 7 h.
| Catalyst | Overpotential (mV) | Tafel slope (mV dec−1) | |
|---|---|---|---|
| At 10 mA cm−2 | At 60 mA cm−2 | ||
| CdCoS | 199 | 522 | 63.45 |
| CdCoO | 208 | 543 | 81.98 |
| CdCoS@CuCoS | 205 | 588 | 69.91 |
| CdCoO@CuCoO | 215 | 616 | 83.05 |
Activity enhancements in the as-synthesized electrocatalysts
Zheng et al.11 compared the OER activity of CuCoO and CuCoS by partially substituting the oxides with sulfides. It was revealed that the charge density of the Co active sites was increased due to the removal of the highly electronegative oxygen atoms, thus a low valence state of the central metal was observed. The increase in the electron density on the Co3+ state resulted in a change of the electron-filled configuration t62g e1g in the crystal field, which became closer to that of the Co2+ ions. According to the eg orbital theory, if the electrons in the eg orbital are greater than 1, the adsorption capacity for the OER intermediates becomes weak, and an appropriate adsorption energy for the binding of intermediates can be achieved.57,58 Likewise, the sulfides increased the covalency of the M–O bond, and the binding energy of the OER intermediates on the surface of electrocatalysts could be optimized to get improved OER performances.12 In the present work, the increase in OER activity of CdCoS compared to CdCoO was attributed to the substitution of sulfides.The incorporation of the Cd in the cobalt oxides/sulfide also influences the electron density of the metal active sites and exposes the active sites by creating defects in the cobalt oxide lattice. Here, Cd2+ has larger ionic radii compared to the Co2+/Co3+ ions and has filled 3d orbitals.59 The large ionic radii produce distortions in the crystal lattices, leading to exposure of the Co active sites.18 Moreover, like Zn2+,60 Cd2+ having filled 3d orbitals causes a weak binding with oxygen 2p orbitals, leading to a localized electron density around the oxygen in the Cd–O bonds. The electronegativity of the Cd2+ ions is less than Co2+/Co3+ ions, thus causing an increase in electron density in the Co2+/Co3+ active sites. Thus, excellent OER activity was observed in the CdCoO and CdCoS electrocatalysts compared to the various other cobalt oxides(hydroxides), and sulfides of various classes of materials (Fig. 6e and Table S1 of the ESI†).
However, interestingly the OER activity of the electrocatalysts decreased after the formation of the composite. The CdCoO@CuCoO-NCs (CdCoO@CuCoO) were MOF-derived and had two major contributing factors: copper and carbon. The Cu2+ ions have a d9 electronic configuration, have lower electronegativity than Co2+/Co3+, and have similar ionic radii.59 We propose that the Cu2+ incorporation and carbon collectively cover the surface of the materials and remove the surface defects, which we believe collectively lowers the catalytic activity of the electrocatalysts. Whereas, the sulfide-based CdCoS@CuCoS electrocatalysts showed greater OER activity than CdCoO@CuCoO due to the sulfide effect, as described above. The OER performances of CdCoS@CuCoS and CdCoO@CuCoO were still superior to the various reported cobalt-based materials as presented in Fig. 6e and Table S1 (ESI†).
Conclusions
In summary, the present work reports the rational design of multicomponent electrocatalysts by incorporating Cd in cobalt oxide to improve the oxidative water-splitting performance. Sulfide-modified CdCoS was also synthesized to explore the synergistic effect of Cd incorporation and the sulfide effect on the OER performance of electrocatalysts. The CdCoS exhibited an overpotential of 199 mV at 10 mA cm−2, which was superior to many of the cobalt-based various materials reported in the literature. The composite of CdCoO and CdCoS was also synthesized with CuCoO and CuCoS via a MOF-derived route. The resultant CdCoO@CuCoO and CdCoS@CuCoS exhibited good OER activity, but lower than that of CdCoO and CdCoS. The lower performance of the composites was attributed to the coating of the active sites needed for catalysis. Moreover, CdCoS also showed good durability after 7 h of stability testing. This work is expected to provide a new avenue for the development of efficient electrocatalysts by utilizing a multicomponent doping strategy.Data availability
No software or code have been included and no new data were generated or analysed as part of this study.Conflicts of interest
There are no conflicts to declare.References
- T. u Haq and Y. Haik, Electrochemical Water Splitting: Fundamentals, Challenges and Advances, Springer, 2024, pp. 1–19 Search PubMed.
- M.-I. Jamesh and X. Sun, J. Power Sources, 2018, 400, 31–68 Search PubMed.
- K. Li, Y. Tong, J. He, X.-Y. Liu and P. Chen, Mater. Horiz., 2023, 10, 5277–5287 Search PubMed.
- H. Wang and H. c D. Abruña, J. Phys. Chem. C, 2021, 125, 7188–7203 Search PubMed.
- X. Chen, H. Wang, R. Meng, B. Xia and Z. Ma, ACS Appl. Energy Mater., 2020, 3, 1305–1310 Search PubMed.
- H. Wang, Y. Tong, K. Li and P. Chen, J. Colloid Interface Sci., 2022, 628, 306–314 Search PubMed.
- Y. Tong, P. Chen, L. Chen and X. Cui, ChemSusChem, 2021, 14, 2576–2584 Search PubMed.
- L. Gong, X. Y. E. Chng, Y. Du, S. Xi and B. S. Yeo, ACS Catal., 2018, 8, 807–814 Search PubMed.
- X. Cheng and Y. Tong, ACS Appl. Energy Mater., 2023, 6, 9577–9584 Search PubMed.
- D. A. Kuznetsov, B. Han, Y. Yu, R. R. Rao, J. Hwang, Y. Román-Leshkov and Y. Shao-Horn, Joule, 2018, 2, 225–244 Search PubMed.
- J. Zheng, D. Meng, J. Guo and Z. Wang, J. Alloys Compd., 2023, 968, 172254 Search PubMed.
- X. Zhang, H. Zhong, Q. Zhang, Q. Zhang, C. Wu, J. Yu, Y. Ma, H. An, H. Wang and Y. Zou, Nat. Commun., 2024, 15, 1383 Search PubMed.
- G. Fu and J.-M. Lee, J. Mater. Chem. A, 2019, 7, 9386–9405 Search PubMed.
- Z. Shao, Q. Zhu, Y. Sun, Y. Zhang, Y. Jiang, S. Deng, W. Zhang, K. Huang and S. Feng, Adv. Mater., 2022, 34, 2110172 Search PubMed.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 Search PubMed.
- V. V. Pavlishchuk and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97–102 Search PubMed.
- A. Padhi, K. Nanjundaswamy, C. Masquelier, S. Okada and J. Goodenough, J. Electrochem. Soc., 1997, 144, 1609 Search PubMed.
- H. Yan, R. Deng, S. Zhang, H. Yao, J. Duan, H. Bai, Y. Li, R. Liu, K. Shi and S. Ma, J. Alloys Compd., 2023, 954, 170072 Search PubMed.
- Q. Zhang, Z. D. Wei, C. Liu, X. Liu, X. Q. Qi, S. G. Chen, W. Ding, Y. Ma, F. Shi and Y. M. Zhou, Int. J. Hydrogen Energy, 2012, 37, 822–830 Search PubMed.
- K. Li and D. Xue, J. Phys. Chem. A, 2006, 110, 11332–11337 Search PubMed.
- K. Li, G. Zhou, Y. Tong, Y. Ye and P. Chen, ACS Sustainable Chem. Eng., 2023, 11, 14186–14196 Search PubMed.
- X. Ren and Y. Tong, Int. J. Hydrogen Energy, 2024, 49, 489–497 Search PubMed.
- K. Li, J. He, X. Guan, Y. Tong, Y. Ye, L. Chen and P. Chen, Small, 2023, 19, 2302130 Search PubMed.
- K. Kannan, D. Radhika, D. Gnanasangeetha, S. K. Lakkaboyana, K. K. Sadasivuni, K. Gurushankar and M. M. Hanafiah, Inorg. Chem. Commun., 2021, 125, 108429 Search PubMed.
- A. Q. Mugheri, A. Tahira, U. Aftab, M. I. Abro, A. B. Mallah, G. Z. Memon, H. Khan, M. A. Abbasi, I. A. Halepoto and S. R. Chaudhry, RSC Adv., 2019, 9, 34136–34143 Search PubMed.
- Z. K. Heiba, M. B. Mohamed, M. Abdellatief and A. Albassam, Appl. Phys. A: Mater. Sci. Process., 2020, 126, 1–10 Search PubMed.
- S. Farhadi, M. Javanmard and G. Nadri, Acta Chim. Slov., 2016, 63, 335–343 Search PubMed.
- V. Revathi and K. Karthik, J. Mater. Sci.: Mater. Electron., 2018, 29, 18519–18530 Search PubMed.
- K. Kannan, D. Radhika, M. P. Nikolova, V. Andal, K. K. Sadasivuni and L. S. Krishna, Optik, 2020, 218, 165112 Search PubMed.
- L. Belles, C. Moularas, S. Smykała and Y. Deligiannakis, Nanomaterials, 2021, 11, 925 Search PubMed.
- A. Jena, T. R. Penki, N. Munichandraiah and S. Shivashankar, J. Electroanal. Chem., 2016, 761, 21–27 Search PubMed.
- A. M. Mostafa, S. A. Yousef, W. H. Eisa, M. A. Ewaida and E. A. Al-Ashkar, Appl. Phys. A: Mater. Sci. Process., 2017, 123, 1–9 Search PubMed.
- L.-C. Jiang and W.-D. Zhang, Biosens. Bioelectron., 2010, 25, 1402–1407 Search PubMed.
- L.-L. Ren, L.-H. Wang, Y.-F. Qin and Q. Li, Front. Chem., 2022, 9, 818255 Search PubMed.
- S. K. Yadav, A. K. Vishwakarma and L. Yadava, Macromol. Symp., 2023, 407, 2100441 Search PubMed.
- C. V. Thulasi-Varma, S. S. Rao, C. S. S. P. Kumar, C. V. Gopi, I. K. Durga, S.-K. Kim, D. Punnoose and H.-J. Kim, Dalton Trans., 2015, 44, 19330–19343 Search PubMed.
- B. Rivas-Murias and V. Salgueiriño, J. Raman Spectrosc., 2017, 48, 837–841 Search PubMed.
- S. Jambure and C. Lokhande, Mater. Lett., 2013, 106, 133–136 Search PubMed.
- J. Xu, W. Ji, Z. Shen, W. Li, S. Tang, X. Ye, D. Jia and X. Xin, J. Raman Spectrosc., 1999, 30, 413–415 Search PubMed.
- S. Peng, X. Han, L. Li, Z. Zhu, F. Cheng, M. Srinivansan, S. Adams and S. Ramakrishna, Small, 2016, 12, 1359–1368 Search PubMed.
- H. Wu and W. Chen, Nanoscale, 2011, 3, 5096–5102 Search PubMed.
- S. Xie, X. Lu, T. Zhai, J. Gan, W. Li, M. Xu, M. Yu, Y.-M. Zhang and Y. Tong, Langmuir, 2012, 28, 10558–10564 Search PubMed.
- Q. Gu, H. Zhuang, J. Long, X. An, H. Lin, H. Lin and X. Wang, Int. J. Photoenergy, 2012, 2012, 857345 Search PubMed.
- Q. Cheng, C. Yang, K. Tao and L. Han, Electrochim. Acta, 2020, 341, 136042 Search PubMed.
- R. Samal, S. Mondal, A. S. Gangan, B. Chakraborty and C. S. Rout, Phys. Chem. Chem. Phys., 2020, 22, 7903–7911 Search PubMed.
- T. Mizokawa, Y. Morita, T. Sudayama, K. Takubo, I. Yamada, M. Azuma, M. Takano and Y. Shimakawa, Phys. Rev. B, 2009, 80, 125105 Search PubMed.
- H. Singh, A. Sinha, S. Gupta, M. Singh and H. Ghosh, arXiv preprint arXiv, 2015, 1507, 05190 Search PubMed.
- Y. Ikedo, J. Sugiyama, H. Nozaki, H. Itahara, J. Brewer, E. Ansaldo, G. Morris, D. Andreica and A. Amato, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 054424 Search PubMed.
- P. Kulkarni, S. Mahamuni, M. Chandrachood, I. Mulla, A. Sinha, A. S. Nigavekar and S. Kulkarni, J. Appl. Phys., 1990, 67, 3438–3442 Search PubMed.
- K. Gupta, M. Bersani and J. A. Darr, J. Mater. Chem. A, 2016, 4, 13786–13794 Search PubMed.
- J. Qiao, M. Fan, Y. Fu, Z. Bai, C. Ma, Y. Liu and X.-D. Zhou, Electrochim. Acta, 2015, 153, 559–565 Search PubMed.
- G. Wang, H. Wang, Y. Ling, Y. Tang, X. Yang, R. C. Fitzmorris, C. Wang, J. Z. Zhang and Y. Li, Nano Lett., 2011, 11, 3026–3033 Search PubMed.
- S. Zhang, D. Li, S. Chen, X. Yang, X. Zhao, Q. Zhao, S. Komarneni and D. Yang, J. Mater. Chem. A, 2017, 5, 12453–12461 Search PubMed.
- W. Teng, M. Huo, Z. Sun, W. Yang, X. Zheng, C. Ding and S. Zhang, Front. Chem., 2020, 8, 334 Search PubMed.
- Y. Song, Y. Guo, S. Qi, K. Zhang, J. Yang, B. Li, J. Chen, Y. Zhao and Y. Lou, J. Alloys Compd., 2021, 884, 161035 Search PubMed.
- Y. Ning, D. Ma, Y. Shen, F. Wang and X. Zhang, Electrochim. Acta, 2018, 265, 19–31 Search PubMed.
- Z. Wang, Y. You, J. Yuan, Y.-X. Yin, Y.-T. Li, S. Xin and D. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 6520–6528 Search PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 Search PubMed.
- R. t Shannon and C. Prewitt, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1970, 26, 1046–1048 Search PubMed.
- Z.-F. Huang, J. Song, Y. Du, S. Xi, S. Dou, J. M. V. Nsanzimana, C. Wang, Z. J. Xu and X. Wang, Nat. Energy, 2019, 4, 329–338 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ya00327f |
| This journal is © The Royal Society of Chemistry 2024 |