
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanoscale and chiral metal–organic frameworks for asymmetric reactions in water: bridging Lewis acid catalysis and biological systems†
Watchara
Srimontree
,
Taku
Kitanosono
 *,
Yasuhiro
Yamashita
and
Shū
Kobayashi
*
*,
Yasuhiro
Yamashita
and
Shū
Kobayashi
*
Department of Chemistry, School of Science, The University of Tokyo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: tkitanosono@chem.s.u-tokyo.ac.jp; shu-kobayashi@chem.s.u-tokyo.ac.jp
First published on 6th May 2024
Abstract
Nowadays, stereoselective control over the sheer variety of chemical transformations benefits from the multipotency of chiral Lewis acids. Their use under biocompatible conditions has long posed a challenge because profuse amounts of biogenic nucleophiles readily deactivate them. To bridge the gap between chiral Lewis acid catalysis and biocompatible chemistry, the conversion of UiO(BPY)-type nanosized metal–organic frameworks (NMOFs) into chiral variants was herein exemplified. The combination of an elongated 2,2′-bipyridyl linker and scandium salt with a hydrophobic anion proved essential to implement traits such as robustness, biocompatibility, and catalytic activity. The catalyst could construct sufficiently hydrophobic environments sequestered within the framework, catalyzing asymmetric ring-opening reactions of meso-epoxide with low catalyst loading to afford β-amino acid alcohols in high yield (up to >99%) with high enantioselectivity (up to 88%). Most impressively, it exhibited a tolerance to the ex vivo poisoning of chiral Lewis acid catalysis by biogenic nucleophiles in sharp contrast to conventional water-compatible Lewis acids.
Introduction
Lewis acid catalysts are of considerable importance in modern organic chemistry because of their unique privilege in controlling reactivity and selectivity.1 Numerous examples explored thus far have demonstrated that sufficient activation of substrates can not only accelerate reactions but also affect regio- and stereoselectivity. Although this arena has been expanded to encompass a diverse range of organic transformations, classical Lewis acids are moisture-sensitive and thus corrosive because of evolved acidic gases. In this context, the advent of water-compatible Lewis acids2 was the cornerstone of green organic synthesis, accentuating their power in organic synthesis thereafter. In the meantime, notwithstanding the conspicuous prosperity of bioorthogonal catalysis,3,4 creating biocompatible Lewis acidic analogues compatible within the sea of cellular nucleophiles remains arduous. Because of their high electrophilicity, Lewis acids may be poisoned by Lewis basic functionalities such as amines and thiols present in proteins or biogenic small molecules, glutathione (GSH) and carnosine. This dysfunction of Lewis acids in cellular environments downgrades their potential applicability. Although chiral Lewis acids are too vulnerable to Lewis basic environments to deliver deteriorated yield and selectivity, nature executes Lewis acid-catalyzed enantioselective synthesis to function appositely in a living cell. While heterogeneous platforms, such as hydrophobic polymer and surfactant assemblies, embedding chiral Lewis acid complexes have been adopted for enantioselective reactions in aqueous environments,5–8 they are inept for biomedical purposes because of their aggregation behavior. An aspirational goal would therefore be to set the stage for chiral Lewis acids that allow enantioselective reactions even in the presence of cellular debris. While there have been a handful of successes in precious metal-based enantioselective reactions within living cells,9 successful applications of chiral Lewis acid catalysis rely absolutely on robust copper(II) complexes.10,11We envisioned implementing nanoscale metal–organic frameworks (MOFs) as an alternative platform to this end. Owing to their high surface area, permanent porosity, highly tunable pore structures, and flexible functionality, MOFs have emerged as novel devices with a wide spectrum of potential applications: gas storage, chemical sensing, biomedical imaging, drug delivery, and catalysis.12,13 Notwithstanding the brilliant successes of MOFs in diverse chemical transformations, two potential challenges that may limit their versatility can be identified: application to their chiral variant is often bottlenecked by the limited number of available chiral linkers and the limited techniques of postsynthetic modification.14 In addition, the involvement of MOFs in organic synthesis in aqueous solutions has barely been reported.15 This is surprising because of the inherent stability of nano-MOFs (NMOFs) in water16 and their biomedical applications,17–20 including drug delivery, cancer therapy, and medical diagnosis. In addition, we conceived that the confinement effects of NMOFs were fit for providing compartmentalized environments to conserve Lewis acidic sites from the sea of cellular nucleophiles. We herein report our efforts to address ex vivo the poisoning of chiral Lewis acid catalysts by biogenic nucleophiles.
Results and discussion
To exploit the chemical and thermal stability of Zr-based UiOs comprising robust zirconium–carboxylate bonds between oxocluster nodes and organic linkers,21 we intended to translate UiO-67 MOFs composed of a bipyridine linker to their chiral variant (Fig. 1a). 2,2′-Bipyridines have been extensively exploited for establishing numerous metal complexes in catalysis because of their robust redox stability, ease of functionalization, and entropically favored metal chelation.22 Inspired by the simplicity and the ease of access to water-stable multidentate coordination of chiral 2,2′-bipyridine bearing 6,6′-dicarbinol substituents 1,23 our study commenced with identifying the best 1-based linker for asymmetric catalysis. We opted for a divergent approach using an optically active intermediate 7 as a common intermediate to generate optically active NMOFs with UiO-67 topology (Fig. 1b). The length of candidate linkers is perceived to exhibit a sufficient size-exclusion effect to safeguard Lewis acidic metal ions encapsulated inside UiO-type NMOFs against nucleophilic residues of proteins. The scandium ion is a potent candidate to obtain proof-of-concept for the hypothesis due to its unique catalytic performance to other d-block elements. Ueno et al. leveraged the chiral environment of the β-helical tubular motif found in bacteriophage T4 for scandium catalysis. A covalent linkage of maleimide-substituted 2,2′-bipyridine to a cysteine residue and subsequent metalation led to the scandium enzyme, which catalyzed the asymmetric ring-opening reactions of meso-epoxides (Fig. 1c).24 The catalyst showed catalytic activity in the presence of nucleophilic residues, while the reaction suffered from low conversion and poor enantioselectivity even with the aid of organic cosolvent. In this work, we thus selected the asymmetric ring-opening reaction as a benchmark reaction to pursue both compatibility with biogenic nucleophiles and high catalytic performance.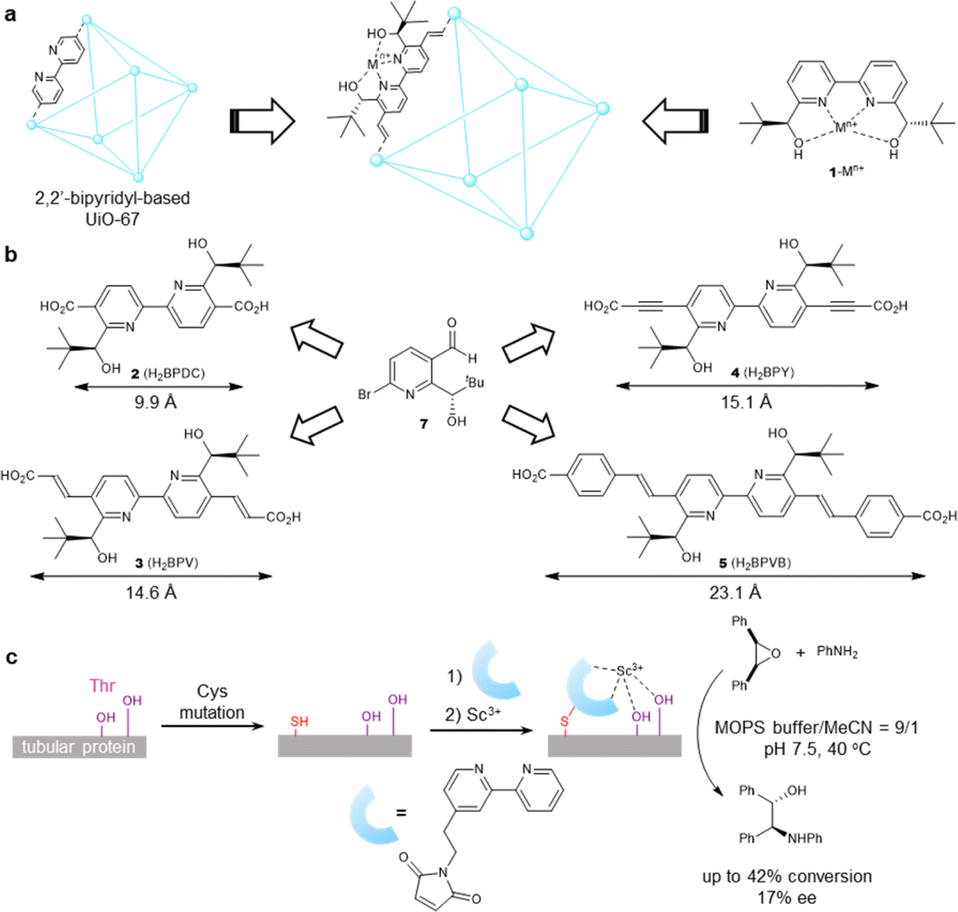 | ||
| Fig. 1 Design of biocompatible Lewis acid catalysts. (a) Concept of catalyst design. (b) Optically active 2,2′-bipyridyl linkers surveyed in this work. The values represent the length between carbon atoms of dicarboxylate. (c) Previous example of scandium-based Lewis acid metalloenzyme. | ||
The synthesis of optically active 2,2′-bipyridyl linkers is outlined in Fig. 2a. The synthesis of common intermediate 7 commenced with asymmetric transfer hydrogenation of compound 6 prepared from 2,6-dibromopyridine in 3 steps (Fig. 2a). Of note, adoption of type IIIb conditions25 (aqueous suspension) proved to be pivotal for asymmetric transfer hydrogenation because canonical conditions (formic acid–triethylamine mixture) failed to produce the product. Homocoupling of 8 and ensuing deprotection processes provided the chiral 5,5′-diformyl-2,2′-bipyridine 10. The chromium-based oxidation delivered 2,2′-bipyridine-5,5′-dicarboxylic acid (H2BPDC 2). Notwithstanding the successful assembly of the MOF upon solvothermal treatment with ZrCl4 and trifluoroacetic acid, this route faltered due to the severe degradation in the postsynthetic deacetylation as well as unsuccessful deprotection before assembly. The use of 4 proved unfeasible due to the deprotection matter as well, despite the successful installation of an alkyne functionality through the Corey–Fuchs homologation of 10 and subsequent reaction with CO2 with alcohols protected by TBS. In the third plan, the Verley–Dobner modification of the Knoevenagel condensation and the subsequent deacetylation converted 10 to H2BPV 3. The solvothermal synthesis of a Zr-based MOF from 3 became deadlocked by the unwanted intramolecular 1,4-addition of an alcohol group to a cinnamic acid moiety that was deduced from the 1H NMR spectra of the digested sample. Thus, linker 5 emerged as an alternative way to bypass the issue. The formyl groups of 7 were elongated through Wittig olefination, which underwent a reductive homocoupling reaction and subsequent deprotection to afford H2BPVB 5 with perfect E configuration. A solvothermal reaction of 5 with ZrCl4, acetic acid, and H2O in N,N′-dimethylformamide (DMF) at 40 °C for 5 days afforded the targeted chiral BPVB-NMOF with a UiO framework as a pale yellow solid (Fig. 2b), despite the length of the organic linker being much larger than ever reported for Zr-MOFs.26,27 In the structure, the 2,2′-bipyridyl moiety was deemed to adopt cis conformation considering the conformational preference of 2,2′-bipyridyl under moderately acidic conditions.28,29 The presence of 6,6′-carbinols may restrict the inner rotational freedom after assembly, conferring benefits for postsynthetic metalation. The powder X-ray diffraction (PXRD) patterns of the as-synthesized MOF include the UiO-type structural fingerprint conformable with the reported pattern30 with line broadening due to the small size of the nanoparticles31 (Fig. 3c). The predominance of the (002) diffraction peak over (111) may originate from the richness of a hexagonal centered planar (hcp) structure over the usual face centered cubic (fcc) structure, indicating the presence of missing linker defects.32,33 Because the increased activity in ring-opening reactions of epoxides was reported for the hcp UiO-66,34 the presence of sufficient linker defects bodes well for exploration of chiral Lewis acid catalysis. The scanning electron microscopy (SEM) image also confirmed the nanostructure construction with an average diameter of 125 nm (Fig. 3a). Thermogravimetric analysis (TGA) revealed the missing linker defect to be 45% for a fcc structure and 25% for a hcp structure from the mass at 200 °C wherein the MOF is fully activated to the dehydroxylated compositions to the point of complete linker degradation (Fig. 3d). The open hysteresis found in the N2 adsorption–desorption isotherm may be attributed to irregular mesoporosity derived from such a high concentration of defects (Fig. S24†). To configure inner hydrophobic environments with increasing stability, we envisioned using hydrophobic anion, dodecyl sulfate (DS), for scandium installation. Because the length of dodecyl sulfate is 20.8 Å, dodecyl sulfate anions would interact with one another to form a “pseudomicelle” aggregate within a confined space in the UiO framework. This “pseudomicelle” is expected not to form a larger aggregate due to the confinement effect. Incubation with scandium tris(dodecyl sulfate) (ScDS3) in aqueous acetone solution led to the formation of a yellow precipitate. The collected powder was dried under vacuum at room temperature to afford BPVB-NMOF-Sc–DS.
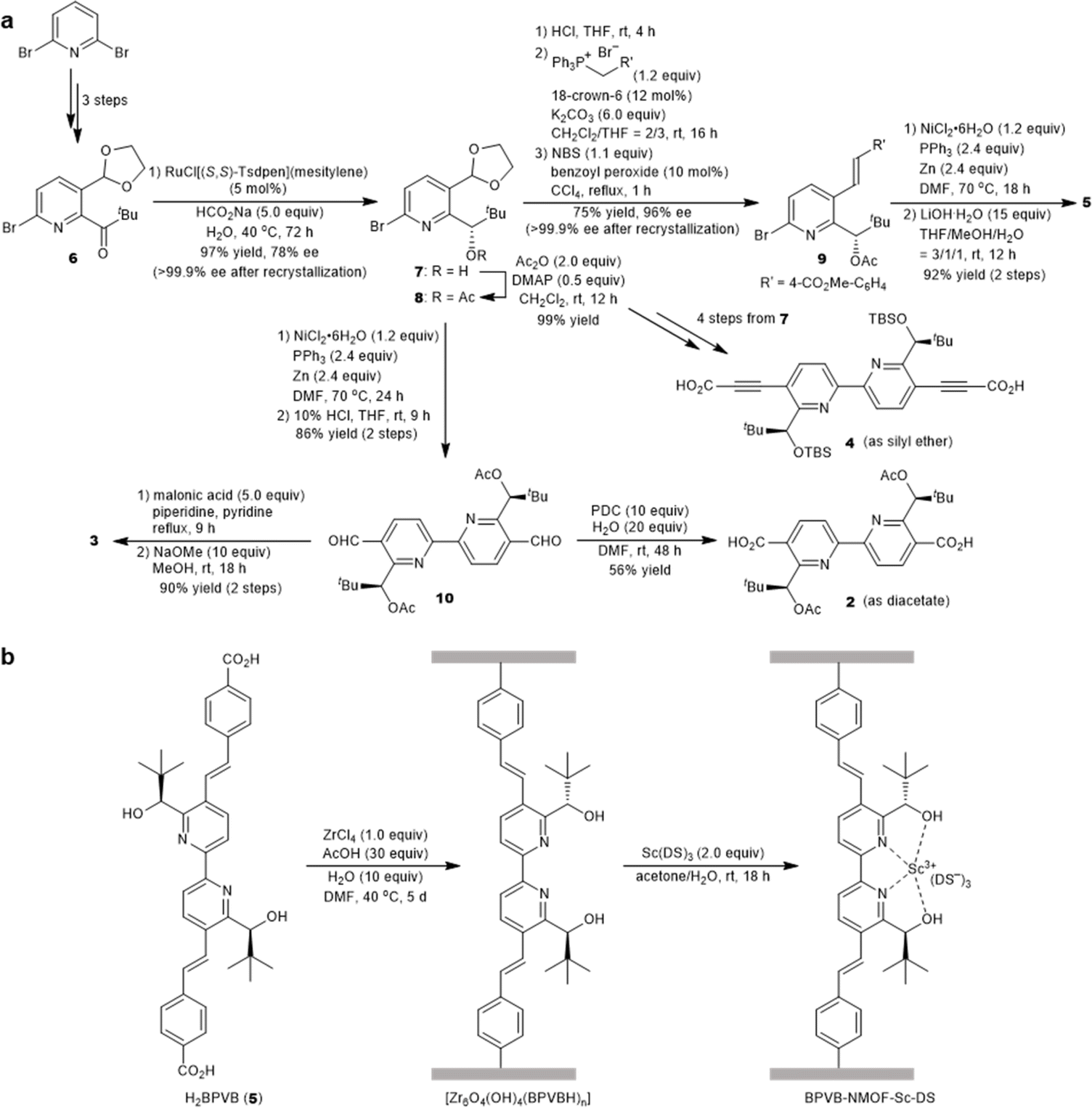 | ||
| Fig. 2 Design and synthesis of chiral BPVB-NMOF-Sc–DS. (a) Divergent synthesis of optically active 2,2′-bipyridyl linkers from common intermediate 7. (b) Preparation of BPVB-NMOF and postsynthetic metalation. DS stands for dodecyl sulfate. See the ESI† for details. | ||
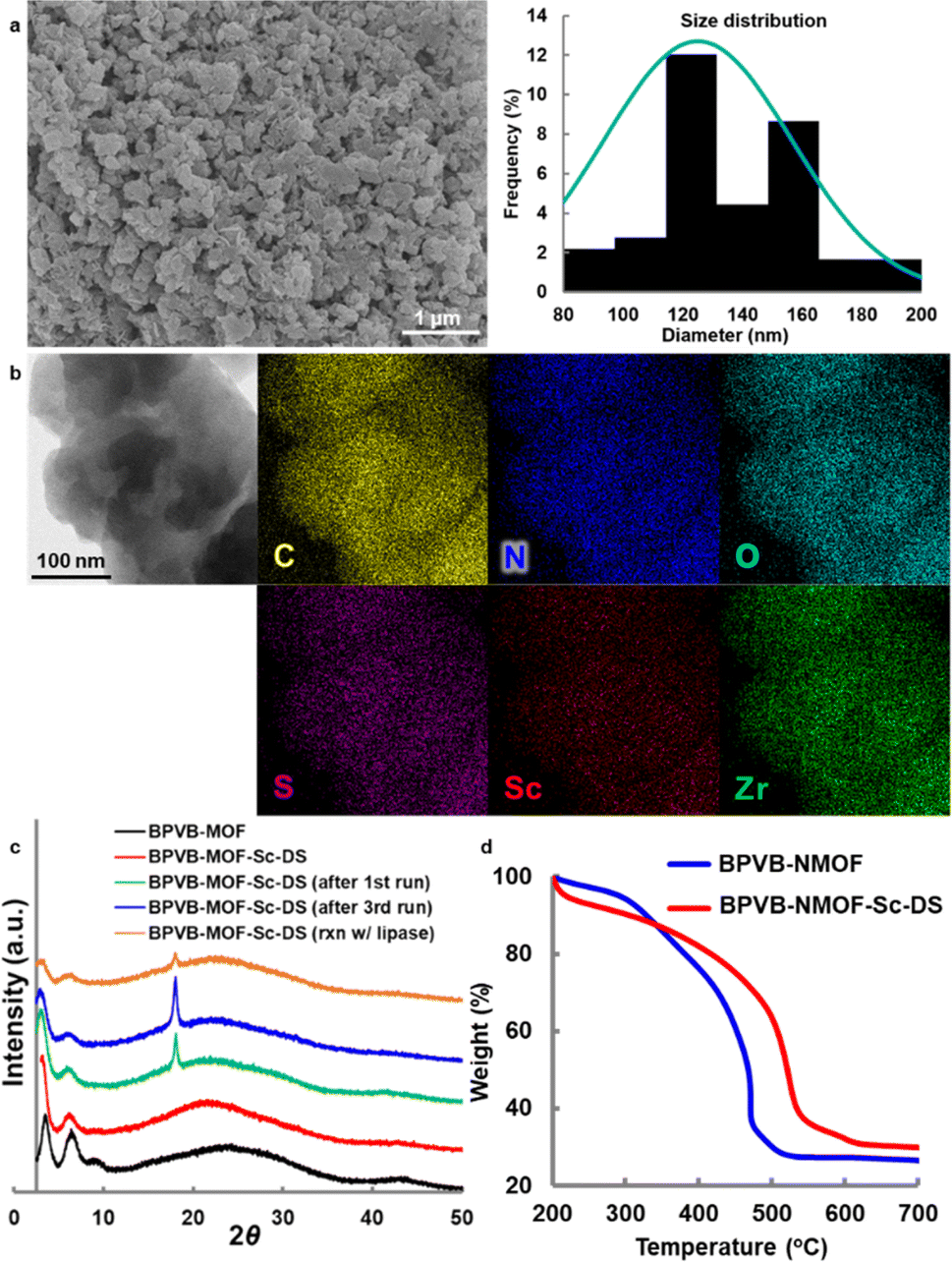 | ||
| Fig. 3 Characterization of BPVB-NMOFs. (a) SEM image of BPVB-NMOF and size distribution histogram. (b) STEM and EDS analysis of BPVB-NMOF-Sc–DS. (c) PXRD patterns of BPVB-NMOFs. (d) TGA plot. See the ESI† for details. | ||
Inductively coupled plasma (ICP) analysis revealed the average Sc content to be 0.5 mmol g−1. The N loading is twice as high as the Sc loading and the average Sc/Zr ratio was 55%, suggesting quantitative installation of the scandium ion into the UiO framework whether it is fcc or hcp. A blue shift of the interfacial N 1s peak by 1.6 eV in X-ray photoelectron spectroscopy (XPS) analysis also confirmed the coordination of the scandium ion to 2,2′-bipyridyl moieties (Fig. S26 and S27†). Because the tetradentate coordination of Sc3+ with two nitrogen atoms of 2,2′-bipyridine and two oxygen atoms of alcohols has been reported to be important for sufficient reaction efficiency24 and enantioselection,35 we anticipated analogous coordination environments, as corroborated by relatively high stability in water, activity comparable to Ueno's result (Fig. S8†), and high enantioselectivity. Energy dispersive X-ray spectroscopy (EDS) tomography confirmed the uniform distribution of all elements in BPVB-NMOF-Sc–DS particles (Fig. 3b). The coordination of scandium lengthened the linker length by ∼0.2 Å due to an increased torsion angle, thereby provoking subtle changes in morphology, especially a relative decrease in the fcc structure, as evidenced by the PXRD pattern (Fig. 3c). The significant isotherm transition from type II to type III after uptake of Sc(DS)3 suggested the occupancy of the interior space of a NMOF with dodecyl sulfate (Fig. S21†). The increased stability of BPVB-NMOF-Sc–DS particles was confirmed by TGA (Fig. 3d). This may be triggered by a loss of a certain amount of a linker, as shown in variability in Sc loadings. The scanning transmission electron microscopy (STEM) image of trifluoromethanesulfonate variant BPVB-NMOF-Sc–OTf manifested the presence of aggregates presumably derived from Sc(OTf)3 (Fig. S19†), bespeaking the importance of dodecyl sulfate on the precise coordination with 2,2′-bipyridyl moieties within the framework. This is also corroborated by scandium loading that is more than 1.2 times higher than that of BPVB-NMOF-Sc–DS. The further area EDS analysis revealed the formation of scandium oxide or hydroxide clusters after hydrolysis (Fig. S23†).
The performance of the NMOF-Sc catalyst was evaluated in the asymmetric ring-opening reactions of epoxides with aniline in water (Fig. 4). A Sc loading of 1 mol% proved sufficient for the efficient reaction, delivering the desired product 13a quantitatively with 85% ee (Fig. 4a, entry 1). The reaction outcome proved not dependent on the Sc loading (Table S11†). No leaching of Sc3+ and Zr4+ both in the aqueous and organic phases is noteworthy (Table S10†). It is also worth pointing out that the reaction was highly enantioselective, notwithstanding the decent level of activity of BPVB-NMOF (entry 3). The activity of UiO series can be explained using the Lewis acidity of zirconium36,37 or the Brønsted acidity of zirconium-bound hydroxide,38 which is supposed to affect the enantioselectivity when the NMOF-Sc catalyst is used. A summary of control experiments is outlined in entries 2–9. The significant decrease in both yield and enantioselectivity of the reaction using linkers H2BPVB 5 and Sc(DS)3 supported the desired catalytic performance inside the framework (entry 2). The catalyst incubated with Sc(OTf)3, BPVB-NMOF-Sc–OTf, displayed poor activity along with moderate enantioselectivity (entry 4). This denotes that the framework alone fails to offer sufficient hydrophobicity for the reaction environments as well as that superfluous mounting of Sc3+ inside the framework is detrimental. This is consistent with our “pseudomicelle” hypothesis. Carrying out the model reaction with shorter reaction times still provided high product yields (entries 5 and 6). Organic solvents such as THF, CH2Cl2, and MeCN proved inappropriate (entries 7–9). Besides, the solvent-free reaction suffered from low enantioselectivity (entry 10), manifesting the pivotal role of water in controlling the stereoselectivity. With the optimal conditions in hand, the scope of the substrate was displayed as depicted in Fig. 4b. A variety of anilines could be employed irrespective of the electronic nature of substituents to afford optically active β-amino alcohols 13b–h in excellent yields (>90%) with high selectivities (up to 88% ee). The reaction proved scalable without deterioration of the reaction outcome (13a). In addition, the range of applicable nucleophiles was expanded to include 1-methyl indole and thiophenol that underwent C–C and C–S bond formations (13i and 13j), respectively. BPVB-NMOF-Sc–DS could be recovered by a simple filtration with no leaching of Sc3+ and then subjected to the next runs, resulting in excellent yields and high ee over three cycles (Fig. 4c). A facile separation of the NMOF catalyst from a post-reaction mixture probed our hypothesis of confined micelle formation. The gradual decline in the PXRD pattern of the fcc structure over the cycles may be ascribed to physical damage to the NMOF during the filtration (Fig. 3c). The emerging peak may be the impurities derived from the reactants, judging from the inconsistency with the reported peaks of scandium oxides or zirconium oxide.
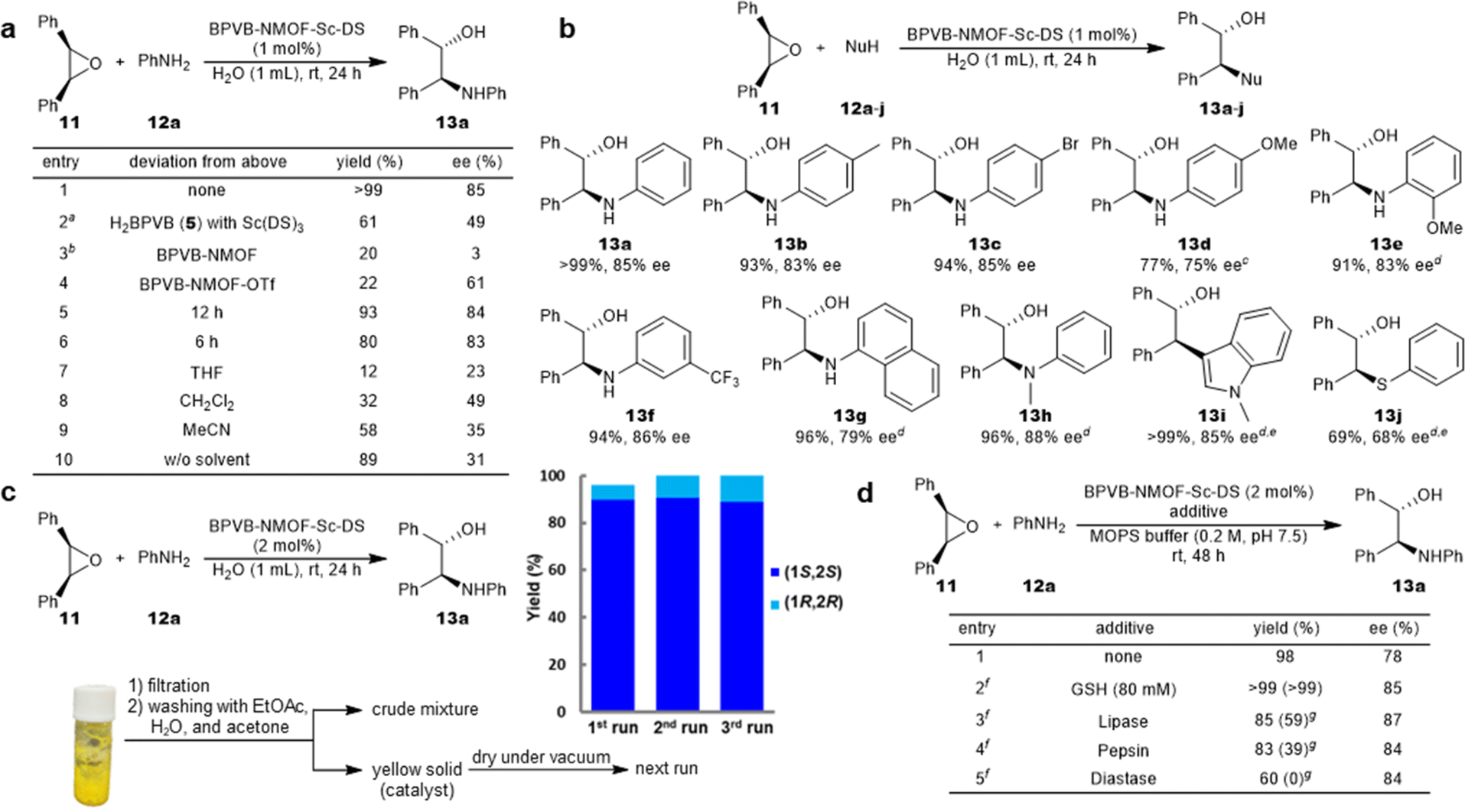 | ||
| Fig. 4 BPVB-NMOF-Sc–DS-catalyzed asymmetric ring-opening reactions of epoxides in aqueous media. (a) Control experiments. Standard reaction conditions: 11 (0.2 mmol), 12a (0.3 mmol), BPVB-NMOF-Sc–DS (1 mol%, 0.002 mmol), H2O (1 mL), rt, 24 h. Isolated yields after chromatography are given. aH2BPVB (1 mol%) and Sc(DS)3 (1 mol%) were premixed prior to the addition of reactants. bZr loading was controlled as the same as that under the standard conditions (2.8 mol% Zr). (b) Substrate scope: 11 (0.2 mmol), 12 (0.3 mmol), BPVB-NMOF-Sc–DS (1 mol%, 0.002 mmol), H2O (1 mL), rt, 24 h. Isolated yields after chromatography are given. cThree equivalents of nucleophile were used. dBPVB-NMOF-Sc–DS (2 mol%) was used. eRun for 48 h. (c) Reuse experiments: 11 (0.4 mmol), 12a (0.6 mmol), BPVB-NMOF-Sc–DS (2 mol%, 0.008 mmol), H2O (2 mL), rt, 24 h. The experimental procedure for catalyst recycling is briefly shown. The scale of the next run was calculated based on the Sc loading of the recovered catalyst. (d) Reaction in the presence of biogenic nucleophiles: 11 (0.2 mmol), 12a (0.3 mmol), BPVB-NMOF-Sc–DS (2 mol%, 0.004 mmol), MOPS buffer (2 mL), rt, 48 h. Isolated yields after chromatography are given. fThe additive (25 g L−1) was added. gRun with Sc(DS)3 (2 mol%, 0.004 mmol) and 1 (2.4 mol%, 0.0048 mmol) instead of BPVB-NMOF-Sc–DS. DS = dodecyl sulfate. MOPS = 3-morpholinopropane-1-sulfonic acid. See the ESI† for details. | ||
The compatibility of the NMOF catalyst in the presence of biogenic nucleophiles was evaluated as shown in Fig. 4d. The reactions were run in MOPS buffer with comparison to micellar catalysis (type IIb conditions)25 using Sc(DS)3 and 1. When GSH was loaded into the system, the reaction was promoted efficiently to provide excellent yield and high enantioselectivity in both cases (entry 2). The absence of negative effects can be explained by the absence of uptake of GSH in the interior of the micelle due to the small size of GSH. The reaction was therefore carried out in the presence of proteins to understand the tolerance to biogenic nucleophiles because the formation of SDS–protein composites was reported.39 As a result, the activity of BPVB-NMOF-Sc–DS proved to be higher than that of the micellar catalysis in the presence of proteins. Of note, the activity of Sc(DS)3 was completely inhibited in the presence of diastase, underpinning the proof of concept in this study. Because diastase is stable in acidic or neutral solutions,40 the severe poisoning effects may have been exerted for the micellar catalysis. The confined environments of the NMOF are suited for the exclusion of these proteins: e.g. pepsin (48 Å × 64 Å,41 the smallest among three).
Conclusions
In summary, we have successfully developed a new UiO-67(BPY) MOF composed of a chiral 2,2′bipyridyl-functionalized dicarboxylate linker (H2BPVB). The limited free rotation of 2,2′-bipyridyl by introduction of a 6,6′-carbinol moiety and fabrication of a defect-rich structure due to longer linker length resulted in a quantitative postsynthetic uptake of scandium salt. The use of Sc(DS)3 was of vital importance for our design to precisely emplace Sc3+ and to forge sufficiently hydrophobic “pseudomicelle” environments sequestered inside the framework. Such a concept of the confined micelle is, to our knowledge, without precedent. The as-prepared BPVB-NMOF-Sc–DS efficiently catalyzed the asymmetric ring-opening reactions of epoxides in water without any organic cosolvent to deliver high yields and enantioselectivities with a broad range of nucleophiles. The impaired performance in pure organic solvents or under solvent-free conditions adorned this catalysis. The reusability of the catalyst by expedient filtration underscores its relative robustness even in water. Remarkably, this NMOF catalyst showed superior catalytic performance to the usual micellar catalysis in the presence of proteins, demonstrating the ability to prevent active Lewis acid sites from mortiferous deactivation. The ways of designing and preparing water-stable chiral NMOFs represent a unique direction for synthetic organic chemistry to build an abiological path without impairing homeostasis. As such, it warrants further study to build a bridge between organic chemistry and biochemistry.Data availability
All the data supporting this study are included in the main text and the ESI.†Author contributions
T. K. and S. K. conceived and directed the study. S. W. performed the experiments. T. K. and S. W. analyzed the data. T. K. performed DFT calculations to optimize the structure of chiral linkers. All the authors prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Grants-in-Aid for Science Research (JP19H05288 to TK and JP22H04972 to TK, YY, and SK) from the Japan Society for the Promotion of Science (JSPS). We are grateful to Dr Woogyum Kim (The University of Tokyo) for his contribution to the early stage of this work. We also thank Dr Tei Maki (The University of Tokyo and JEOL Ltd.) for STEM analyses.References
- A. Corma and H. García, Lewis acids: from conventional homogeneous to green homogeneous and heterogeneous catalysis, Chem. Rev., 2003, 103, 4307–4366 Search PubMed.
- S. Kobayashi, Lanthanide trifluoromethanesulfonates as stable Lewis acids in aqueous media. Yb(OTf)3 catalyzed hydroxymethylation reaction of silyl enol ethers with commercial formaldehyde solution, Chem. Lett., 1991, 2187–2190 CrossRef CAS.
- J. A. Prescher and C. R. Bertozzi, Chemistry in living systems, Nat. Chem. Biol., 2005, 1, 13–21 Search PubMed.
- E. M. Sletten and C. R. Bertozzi, Bioorthogonal chemistry: fishing for selectivity in a sea of functionality, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 Search PubMed.
- T. Kitanosono, F. Lu, K. Masuda, Y. Yamashita and S. Kobayashi, Efficient recycling of catalyst-solvent couples from Lewis acid catalyzed asymmetric reactions in water, Angew. Chem., Int. Ed., 2022, 61, e202202335 Search PubMed.
- S. Azoulay, K. Manabe and S. Kobayashi, Catalytic asymmetric ring opening of meso-epoxides with aromatic amines in water, Org. Lett., 2005, 7, 4593–4595 CrossRef CAS PubMed.
- M. Kokubo, C. Ogawa and S. Kobayashi, Lewis acid catalysis in water with a hydrophilic substrate: scandium-catalyzed hydroxymethylation with aqueous formaldehyde in water, Angew. Chem., Int. Ed., 2008, 47, 6909–6911 CrossRef CAS PubMed.
- M. Ohara, Y. Hara, T. Ohnuki and S. Nakamura, Direct enantioselective three-component synthesis of optically active propargylamines in water, Chem.–Eur. J., 2014, 20, 8848–8851 CrossRef CAS PubMed.
- M. Wittwer, U. Markel, J. Schiffels, J. Okuda, D. F. Sauer and U. Schwaneberg, Engineering and emerging applications of artificial metalloenzymes with whole cells, Nat. Catal., 2021, 4, 814–827 CrossRef CAS.
- W. Ghattas, V. Dubosclard, A. Wick, A. Bendelac, R. Guillot, R. Ricoux and J.-P. Mahy, Receptor-based artificial metalloenzymes on living human cells, J. Am. Chem. Soc., 2018, 140, 8756–8762 Search PubMed.
- S. Chordia, S. Narasimhan, A. L. Paioni, M. Baldus and G. Roelfes, In vivo assembly of artificial metalloenzymes and application in whole-cell biocatalysis, Angew. Chem., Int. Ed., 2021, 60, 5913–5920 Search PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, The chemistry and applications of metal–organic frameworks, Science, 2013, 341, 1230444 Search PubMed.
- G. Lan, K. Ni and W. Lin, Nanoscale metal–organic frameworks for phototherapy of cancer, Coord. Chem. Rev., 2019, 379, 65–81 Search PubMed.
- W. Gong, Z. Chen, J. Dong, Y. Liu and Y. Cui, Chiral metal–organic frameworks, Chem. Rev., 2022, 122, 9078–9144 Search PubMed.
- A. Dhakshinamoorthy, A. M. Asiric and H. Garcia, Catalysis by metal–organic frameworks in water, Chem. Commun., 2014, 50, 12800–12814 Search PubMed.
- X. Zhang, B. Wang, A. Alsalme, S. Xiang, Z. Zhang and B. Chen, Design and applications of water-stable metal–organic frameworks: status and challenges, Coord. Chem. Rev., 2020, 423, 213507 Search PubMed.
- F. Wang, Y. Zhang, Z. Liu, Z. Du, L. Zhang, J. Ren and X. A. Qu, Biocompatible heterogeneous MOF-Cu catalyst for in vivo drug synthesis in targeted subcellular organelles, Angew. Chem., Int. Ed., 2019, 58, 6987–6992 CrossRef CAS PubMed.
- N. Singh, S. Qutub and N. M. Khashab, Biocompatibility and biodegradability of metal organic frameworks for biomedical applications, J. Mater. Chem. B, 2021, 9, 5925–5934 RSC.
- X. Cai, Z. Xie, D. Li, M. Kassymova, S.-Q. Zang and H.-L. Jiang, Nano-sized metal–organic frameworks: synthesis and applications, Coord. Chem. Rev., 2020, 417, 213366 CrossRef CAS.
- P. Behera, S. Subudhi, S. P. Tripathy and K. Parida, MOF derived nano-materials: a recent progress in strategic fabrication, characterization and mechanistic insight towards divergent photocatalytic applications, Coord. Chem. Rev., 2022, 456, 214392 CrossRef CAS.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, A new zirconium inorganic building brick forming metal organic frameworks with exceptional stability, J. Am. Chem. Soc., 2008, 130(42), 13850–13851 CrossRef PubMed.
- C. Kaes, A. Katz and M. W. Hosseini, Bipyridine: the most widely used ligand. A review of molecules comprising at least two 2,2′-bipyridine units, Chem. Rev., 2000, 100, 3553–3590 CrossRef CAS PubMed.
- C. Bolm, M. Zehnder and D. Bur, Optically active bipyridines in asymmetric catalysis, Angew. Chem., Int. Ed., 1990, 29, 205–207 CrossRef.
- H. Inaba, S. Kanamaru, F. Arisaka, S. Kitagawa and T. Ueno, Semi-synthesis of an artificial scandium(III) enzyme with a β-helical bio-nanotube, Dalton Trans., 2012, 41, 11424–11427 RSC.
- T. Kitanosono and S. Kobayashi, Reactions in water involving the “on-water” mechanism, Chem.–Eur. J., 2020, 26, 9408–9429 CrossRef CAS PubMed.
- K. Manna, P. Ji, F. X. Greene and W. Lin, Metal-organic framework nodes support single-site magnesium-alkyl catalysts for hydroboration and hydroamination reactions, J. Am. Chem. Soc., 2016, 138, 7488–7491 CrossRef CAS PubMed.
- L. Feng, S. Yuan, J.-S. Qin, Y. Wang, A. Kirchon, D. Qiu, L. Cheng, S. T. Madrahimov and H.-C. Zhou, Lattice expansion and contraction in metal-organic frameworks by sequential linker reinstallation, Matter, 2019, 1, 156–167 CrossRef.
- S. T. Howard, Conformers, energetics, and basicity of 2,2’-bipyridine, J. Am. Chem. Soc., 1996, 118, 10269–10274 CrossRef CAS.
- I. Alkorta, J. Elguero and C. Roussel, A theoretical study of the conformation, basicity and NMR properties of 2,2’-, 3,3’- and 4,4’-bipyridines and their conjugated acids, Comput. Theor. Chem., 2011, 966, 334–339 CrossRef CAS.
- F. Dong, X. Wang, Z. Zhao, S. Gao, G. Wang, K. Li and Y. Lin, Ligand “lock and fix” strategy provides stability to metal-organic frameworks for photodegradation processes, Cell Rep. Phys. Sci., 2023, 4, 101545 CrossRef CAS.
- A. Demessence, P. Horcajada, C. Serre, C. Boissière, D. Grosso, C. Sanchez and G. Férey, Elaboration and properties of hierarchically structured optical thin films of MIL-101(Cr), Chem. Commun., 2009, 7149–7151 RSC.
- M. J. Cliffe, E. C. Martínez, Y. Wu, J. Lee, A. C. Forse, F. C. N. Firth, P. Z. Moghadam, D. F. -Jimenez, M. W. Gaultois, J. A. Hill, O. V. Magdysyuk, B. Slater, A. L. Goodwin and C. P. Grey, Metal-organic nanosheets formed via defect-mediated transformation of a hafnium metal-organic framework, J. Am. Chem. Soc., 2017, 139, 5397–5404 CrossRef CAS PubMed.
- T. Bailey, L. Yang, E. Humphreys, F. Esat, B. Douglas and N. Hondow, The controlled microfluidic formation of stable mixed phase HCP/FCC-UiO-67(Zr)-benzoic acid through modification of water concentration, J. Porous Mater., 2024, 31, 267–279 CrossRef CAS.
- X. Chen, Y. Lyu, Z. Wang, X. Qiao, B. C. Gates and D. Yang, Tuning Zr12O22 node defects as catalytic sites in the metal-organic framework hcp UiO-66, ACS Catal., 2020, 10, 2906–2914 CrossRef CAS.
- C. Schneider, A. R. Sreekanth and E. M. Dipl.-Chem., Scandium–bipyridine-catalyzed enantioselective addition of alcohols and amines to meso-epoxides, Angew. Chem., Int. Ed., 2004, 43, 5691–5694 CrossRef CAS PubMed.
- F. Vermoortele, B. Bueken, G. L. Bars, B. V. de Voorde, M. Vandichel, K. Houthoofd, A. Vimont, M. Daturi, M. Waroquier, V. V. Speybroeck, C. Kirschhock and D. E. De Vos, Synthesis modulation as a tool to increase the catalytic activity of metal-organic frameworks: the unique case of UiO-66(Zr), J. Am. Chem. Soc., 2013, 135, 11465–11468 CrossRef CAS PubMed.
- A. Das, N. Anbu, H. Reinsch, A. Dhakshinamoorthy and S. Biswas, A thiophene-2-carboxamide-functionalized Zr(IV) organic framework as a prolific and recyclable heterogeneous catalyst for regioselective ring opening of epoxides, Inorg. Chem., 2019, 58, 16581–16591 CrossRef CAS PubMed.
- Y. Liu, R. C. Klet, J. T. Hupp and O. Farha, Probing the correlations between the defects in metal-organic frameworks and their catalytic activity by an epoxide ring-opening reactions, Chem. Commun., 2016, 52, 7806–7809 RSC.
- K. Ibel, R. P. May, K. Kirschner, H. Szadkowski, E. Mascher and P. Lundahl, Protein-decorated micelle structure of sodium-dodecyl-sulfate–protein complexes as determined by neutron scattering, Eur. J. Biochem., 1990, 190, 311–318 CrossRef CAS PubMed.
- M. Carlsen, J. Nielsen and J. Villadsen, Kinetic studies of acid-inactivation of α-amylase from Aspergillus oryzae, Chem. Eng. Sci., 1996, 51, 37–43 CrossRef CAS.
- Y. Chen, P. Li, J. A. Modica, R. J. Drout and O. K. Farha, Acid-resistant mesoporous metal−organic framework toward oral insulin delivery: protein encapsulation, protection, and release, J. Am. Chem. Soc., 2018, 140, 5678–5681 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc01343c |
| This journal is © The Royal Society of Chemistry 2024 |