
A straightforward and rapid synthesis of polydithioacetals in the presence of chlorodimethylsilane†
Serter
Luleburgaz
,
Emre
Akar
,
Umit
Tunca
 and
Hakan
Durmaz
*
and
Hakan
Durmaz
*
Istanbul Technical University, Department of Chemistry, Maslak 34469, Istanbul, Turkey. E-mail: durmazh@itu.edu.tr
First published on 18th December 2023
Abstract
We focused on the synthesis of polydithioacetals (PDTAs) using chlorodimethylsilane (CDMS). In one of our previous studies, thioacetal formation was observed when CDMS was used to perform the reductive etherification reaction (RER) on a pendant aldehyde polymer platform along with thiols. This result was not surprising since CDMS has both Lewis acid and reducing agent characteristics, yet it was strikingly unexpected. Inspired by this study, PDTA synthesis using CDMS was proposed. Optimization of polymerization conditions was first carried out by comparing traditional acid sources with CDMS. The progress of polymerizations was kinetically monitored, and CDMS was found to perform best. Various parameters, such as the equivalent of reactants, solvent, and polymerization time, were then examined. A library of PDTAs with moderate to high molecular weights was created using a variety of aldehydes and dithiols. A mechanistic approach for the polymerization was proposed, and finally, a model degradation study on a representative PDTA was performed in the presence of hydrogen peroxide.
Introduction
Click reactions coined by Sharpless1 have been simply categorized as reactions providing high yields in common organic solvents at mild temperatures. Moreover, click reactions should display tolerance to various functional groups without complicated purification methods to recover the main product. Therefore, in synthetic polymer chemistry, it is always desirable to synthesize polymers via the click chemistry methodology under mild conditions, with readily available starting materials, and with high yield and efficiency.2,3 Without a doubt, thiol-based reactions, such as radical thiol–ene4–7,10,23/–yne10,11,19 or nucleophile-based thiol-Michael,4,7–10,17–23 thiol-epoxy,13,14 thiol-bromo,16 thiol-p-fluoro etc.,12,15 have made tremendous contributions to polymer science, as they meet most of the aforementioned requirements not only for polymer synthesis but also for polymer–polymer5,8,9,23 and/or polymer–biomolecule conjugation,4,5,8,15,20,23 polymer modification,4–6,8–10,14,15,17,19,20,23 polymer-based material synthesis, etc.4–23On the other hand, thiol chemistry should not be restricted by the above-mentioned reactions at the macromolecular level. For instance, the reaction of thiols with the carbonyl group of aldehydes and ketones, triggered by an acid catalyst, is fundamental in organic chemistry and yields a compound called thioacetal, one of the renowned functional groups in organic chemistry.24 Polydithioacetals (PDTAs), the polymeric analogs of dithioacetal, on the other hand, can be simply synthesized by reacting an aldehyde or a ketone with a dithiol in the presence of an acid catalyst,25–27 indicating another practical applicability of thiol-based reactions at the macromolecular level. Historically, the first example of PDTA was achieved by the reaction of the tetramercaptan derived from pentaerythritol (2,2-bis(mercaptomethyl)propane-1,3-dithiol) and 1,4-cyclohexadione to yield a spiran type of polymer by Fisher and Wiley.28 Next, linear PDTA synthesis was employed by Marvel and co-workers, using various carbonyl compounds and dithiols in the presence of dry HCl(g).29,30 Horvath et al. prepared sugar PDTAs using sugar and dithiols in dioxane catalyzed by HF for various polymerization times. The PDTAs showed low inherent viscosities in the range of 0.03–0.24 dL g−1.31 In the following work carried out by Imai, aromatic PDTAs with inherent viscosities of 0.12–0.24 dL g−1 were achieved from solution polycondensation of 4,4′-oxydibenzenethiol with aromatic aldehydes at 40–80 °C for 6 h through polyphosphoric acid trimethylsilyl ester as a condensing agent.32 After these preliminary studies, PDTA synthesis has been overlooked for a long time, but it has started to attract attention again in the last decade in parallel with the developments in polymer science.33–38 More recently, Yang and coworkers reported monomer–polymer recycling of PDTA.39 The authors prepared PDTA from 3,4,5-trimethoxybenzaldehyde and a variety of alkyl dithiols in acetonitrile catalyzed by p-toluenesulfonic acid (PTSA) at 50 °C for 24 h. Next, the authors described that the PDTA showed depolymerizability via ring-closing depolymerization into macrocycles, followed by entropy-driven ring-opening polymerization to reassemble the pristine PDTAs. In the meantime, Xu and Yuan synthesized PDTAs from aldehyde functionalized bio-based phenols and dithiols (or polymercaptans) under trifluoroacetic acid (CF3COOH) or ZrCl4 catalysis in methanol (MeOH) or tetrahydrofuran (THF) at room temperature for 30 min.40 The produced phenolic polymers showed vitrimer-like properties, and the associative dynamic exchange was found to promote reprocessing. The reactions of disulfide and thioester dynamic sulfur-based bonds employed in dynamic covalent chemistry were comprehensively reviewed by Orillo and Furlan.41
Moreover, the dithioacetal bond is labile in the presence of reactive oxygen species (ROS), while robust against acidic and basic conditions.42,43 Therefore, PDTAs have found extensive applications in bio-related fields where controlled degradation is desired, such as targeted drug delivery and tissue engineering.26,37,44–50 In particular, all these recent studies indicate the huge potential and the increasing importance of PDTA in terms of synthetic polymer chemistry.
The reductive etherification reaction (RER) is a straightforward method to obtain symmetric and unsymmetric ethers in the presence of a silane-based reducing agent and an acid catalyst.51–57 After the pioneering studies of Doyle on the RER, this strategy has been successfully employed at the macromolecular level by successive studies of the Yokozawa group58–62 and the recently published studies by our group.63–67 Notably, our studies have shown that chlorodimethylsilane (CDMS), which has both reducing agent and Lewis acid characteristics, introduced by Lee and Morandi,68 is an effective silane compound for obtaining polyethers as well as alkoxy pendant polymers.63,64 More recently, we showed that when a polymer possessing pendant aldehyde units was subjected to a CDMS-mediated RER using thiols, thioacetal and thioether structures were obtained in the side chain of the polymer.65 Inspired by this study, we envisioned using CDMS for linear PDTA synthesis and innovating a new synthesis method for this polymer structure. Hence, this paper aims to provide an in-depth overview of CDMS-mediated PDTA synthesis. The polymerization was optimized in many aspects to reveal the ideal conditions. Subsequently, a wide range of aldehydes and dithiols were used to create a polymer library (Scheme 1). Additionally, a mechanistic approach is also highlighted for polymerization.
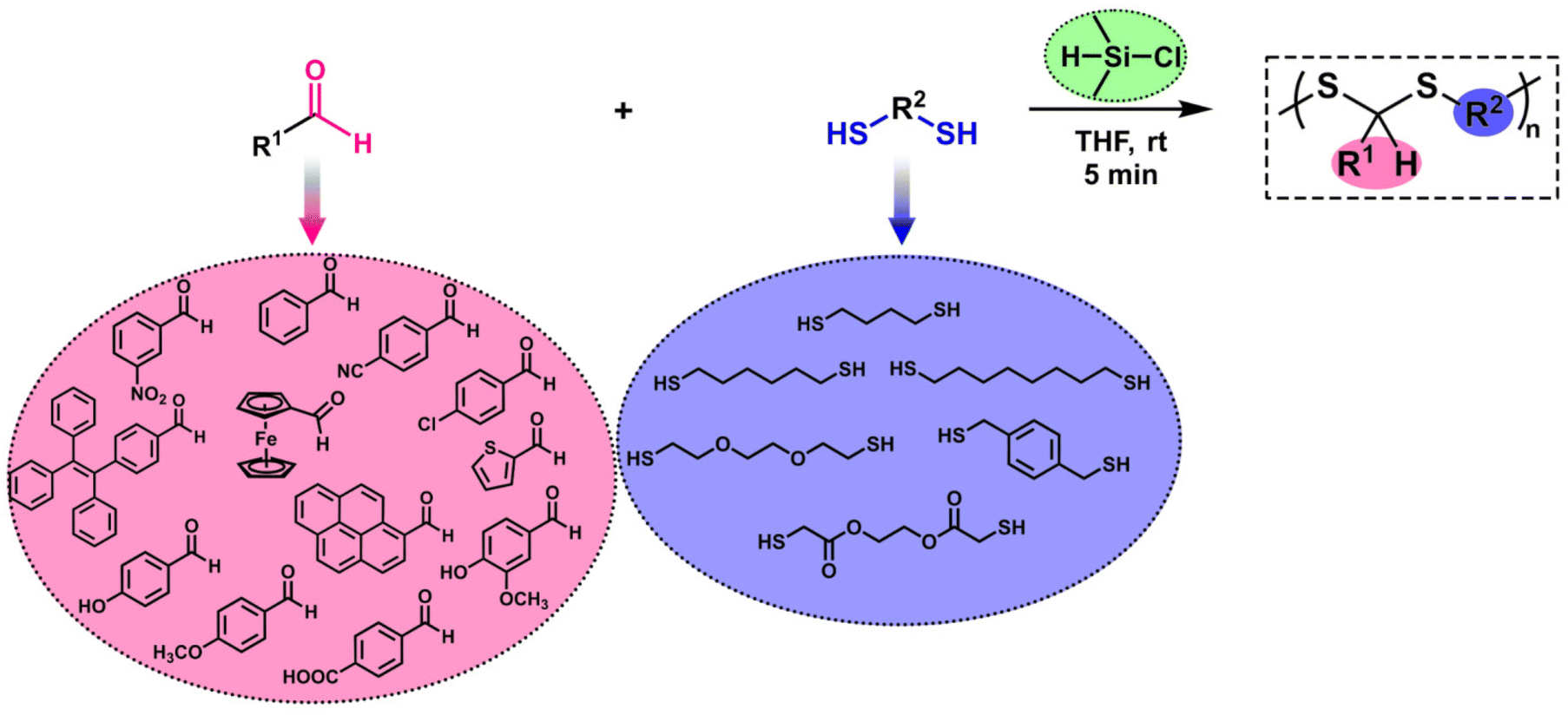 | ||
| Scheme 1 Synthetic route for the synthesis of PDTAs in the presence of CDMS. | ||
Experimental part
Materials
Benzaldehyde (BA, purified by redistillation, ≥99.5%, Sigma-Aldrich), 3-nitrobenzaldehyde (99%, Sigma-Aldrich), 4-cyanobenzaldehyde (95%, Sigma-Aldrich), 4-formylbenzoic acid (97%, Sigma-Aldrich), p-anisaldehyde (98%, Sigma-Aldrich), 4-hydroxybenzaldehyde (98%, Sigma-Aldrich), 4-chlorobenzaldehyde (98%, Sigma-Aldrich), 2-thiophenecarboxaldehyde (98%, Sigma-Aldrich), 4-hydroxy-3-methoxybenzaldehyde (vanillin, 99%, Sigma-Aldrich), 1-pyrenecarboxaldehyde (99%, Sigma-Aldrich), 4-(1,2,2-triphenylethenyl)benzaldehyde (Sigma-Aldrich), ferrocenecarboxaldehyde (98%, Sigma-Aldrich), 1,4-butanedithiol (98%, Sigma-Aldrich), 1,6-hexanedithiol (HDT, 96%, Sigma-Aldrich), 1,8-octanedithiol (97%, Sigma-Aldrich), ethylene glycol bismercaptoacetate (≥95.0%, Sigma-Aldrich), 2,2′-(ethylenedioxy) diethanethiol (95%, Sigma-Aldrich), sulfuric acid (H2SO4, 99.99%, Sigma-Aldrich), p-toluenesulfonic acid monohydrate (PTSA, ACS reagent, ≥98.5%, Sigma-Aldrich), methanesulfonic acid (MSA, ≥99.0%, Sigma-Aldrich), chlorodimethylsilane (CDMS, 98%, Aldrich), H2O2 (35%, Sigma-Aldrich), and trimethylsilyl chloride (≥98.0%, Sigma-Aldrich) were used as received. Tetrahydrofuran (THF, 99%, Sigma-Aldrich), chloroform (CHCl3, 99%, Sigma-Aldrich), 1,2-dichloroethane (DCE, 99.8%, Aldrich), 1,4-dioxane (for liquid chromatography LiChrosolv®, Supleco), 2-methyltetrahydrofuran (2-MeTHF, ≥99%, Sigma-Aldrich), N,N-dimethylformamide (DMF, 99.8%, Sigma-Aldrich), 1-methyl-2-pyrrolidinone (NMP, for liquid chromatography LiChrosolv®, Supleco) and N,N-dimethylacetamide (DMAc, 99.8%, Aldrich) were anhydrous and were of HPLC quality and used without further purification. Methanol (MeOH) was of reagent grade and used as received.Instrumentation
1H NMR (500 MHz) and 13C NMR (125 MHz) spectra were recorded using an Agilent VNMRS 500 instrument in CDCl3. Gel permeation chromatography (GPC) measurements were carried out with an Agilent instrument (series 1100) using a refractive index detector loaded with Waters Styragel columns (HR 5E, HR 4E, HR 3, HR 2, 4.6 mm internal diameter, 300 mm length, packed with 5 μm particles). The effective molecular weight ranges of the columns are 2000–4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000, 50–100000, 500–30000, and 500–20000 g mol−1, respectively. THF was used as an eluent at a flow rate of 0.3 mL min−1 at 30 °C, and 2,6-di-tert-butyl-4-methylphenol was used as an internal standard. The number-average molecular weight (Mn) and dispersity (Đ) of the polymers were calculated based on narrow linear polystyrene (PS) standards (Polymer Laboratories) ranging between 2300 and 3050000 g mol−1. FT-IR spectra were recorded on an Agilent Technologies Cary 630 FT-IR instrument over the range of 4000–400 cm−1. Differential scanning calorimetry (DSC) measurements were carried out on a TA DSC Q10 instrument under a nitrogen atmosphere in the temperature range from −50 °C to 150 °C at a scanning rate of 20 °C min−1. All data were collected from the second heating cycle, and the glass transition temperatures (Tg) were determined from the midpoint of calculated onset and endset temperatures of the glass transition region of the related DSC trace.
000000, 50–100000, 500–30000, and 500–20000 g mol−1, respectively. THF was used as an eluent at a flow rate of 0.3 mL min−1 at 30 °C, and 2,6-di-tert-butyl-4-methylphenol was used as an internal standard. The number-average molecular weight (Mn) and dispersity (Đ) of the polymers were calculated based on narrow linear polystyrene (PS) standards (Polymer Laboratories) ranging between 2300 and 3050000 g mol−1. FT-IR spectra were recorded on an Agilent Technologies Cary 630 FT-IR instrument over the range of 4000–400 cm−1. Differential scanning calorimetry (DSC) measurements were carried out on a TA DSC Q10 instrument under a nitrogen atmosphere in the temperature range from −50 °C to 150 °C at a scanning rate of 20 °C min−1. All data were collected from the second heating cycle, and the glass transition temperatures (Tg) were determined from the midpoint of calculated onset and endset temperatures of the glass transition region of the related DSC trace.
General procedure for the synthesis of linear polydithioacetal (synthesis of P1, run 6 in Table 2)
BA (224 μL, 2.20 mmol) was added to a 10 mL round-bottom flask and dissolved in 500 μL of THF. HDT (305 μL, 2.00 mmol) and CDMS (111 μL, 1.00 mmol) were then added to this solution, respectively, with continuous stirring at room temperature. Upon the addition of CDMS to the mixture, an exothermic reaction occurred, and the reaction medium turned into a slurry within 2 min. The reaction was allowed to stir for 5 min, then diluted with an additional 2 mL of THF and precipitated into 50 mL of MeOH. The dissolution–precipitation procedure (THF–MeOH) was repeated two times. Finally, the obtained polymer, P1, was dissolved in 5 mL of CHCl3 and transferred to a glass vial, and the solvent was evaporated to yield a white sticky solid (yield = 466 mg, 98%). 1H NMR (500 MHz, CDCl3): δ 7.41–7.25 (5H, ArH), 4.85 (1H, ArCHS), 2.49 (4H, SCH2(CH2)4CH2S), 1.50–1.29 (8H, SCH2(CH2)4CH2S); 13C NMR (125 MHz, CDCl3): 140.52, 128.52, 127.82, 127.68, 53.29, 32.16, 28.95, 28.38.Degradation of P1 in the presence of H2O2
P1 (run 6, Table 2) (200 mg) was dissolved in 10 mL of THF at room temperature. H2O2 (4 mL, 1 mL per 50 mg of P1) was added to the polymer solution. The heterogeneous mixture formed by the addition of H2O2 was stirred at 40 °C and aliquots of 50 μL were withdrawn from the reaction medium at different time intervals and precipitated in 1 mL of MeOH. The Mn values of the polymers were measured by GPC up to 8 h. After 24 h, the remaining solution was evaporated, and the crude product was dissolved in 10 mL of CHCl3 and extracted two times with water. Combined organic phases were dried over anhydrous Na2SO4, the solvent was evaporated, and the resulting crude product was analyzed by 1H NMR and GPC.Results and discussion
Optimization of the polymerization conditions for linear polydithioacetal PDTA (P1) synthesis was carried out in multiple aspects. For this purpose, benzaldehyde (BA) and 1,6-hexanedithiol (HDT) were chosen as model monomers, and the reactions were performed in THF at room temperature. It is well-known that PDTA synthesis can be carried out in the presence of Brønsted acids. Thus, the effect of traditional Brønsted acids, such as H2SO4, p-toluenesulfonic acid monohydrate (PTSA), and methanesulfonic acid (MSA), and Lewis acid chlorodimethylsilane (CDMS) on the polymerization was initially examined, and the obtained results are provided in Table 1. Polymerization reactions were carried out by using equimolar BA and HDT (2 mmol each) in the presence of 1 mol% of acid in 1 mL of THF at room temperature and monitored for 72 h. In all cases, PDTAs having low number-average molecular weights (Mns) were obtained in 1 h, and no distinct change was observed over 72 h for all the studied acids (runs 1, 4, 7, and 10 in Table 1). When the acid ratios were increased to 10 mol%, an improvement in the Mns of P1 (runs 2, 5, 8, and 11 in Table 1) was observed over time, especially in the presence of PTSA, MSA, and CDMS, for which the Mns increased to 11, 12, and 13 kDa, respectively, in 72 h. Finally, when the ratio of the acids was further increased to 25 mol%, low to high molecular weight polymers were obtained throughout the reactions (runs 3, 6, 9, and 12 in Table 1). Among them, the results obtained from CDMS were significantly higher than those from other acids; an Mn of 22 kDa was obtained after 8 h, which increased to 45 kDa at 72 h (run 12 in Table 1). Given the results obtained, CDMS was found to be the better alternative as an acid source compared to other traditional Brønsted acids, and thus, its performance was particularly examined in this study. It should also be noted here that with the addition of acid catalysts into the reactions, an exothermic reaction occurred (except for 1 mol% catalyst experiments), and the reaction media turned into a turbid solution during the polymerizations.|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Run | Acid catalyst | Equiv. | M n (kDa)b | |||||
| 1 h | 4 h | 8 h | 24 h | 48 h | 72 h | |||
|
a All reactions were carried out using BA:HDT at a molar ratio of 1:1 in 1 mL of THF at room temperature.
b Determined by GPC, calibrated based on linear PS standards in THF.
|
||||||||
| 1 | H2SO4 | 0.01 | 0.9 | 1.1 | 1.0 | 1.4 | 1.3 | 1.4 |
| 2 | 0.10 | 0.9 | 1.1 | 1.5 | 2.0 | 2.6 | 3.3 | |
| 3 | 0.25 | 1.8 | 4.5 | 5.3 | 9.2 | 14 | 17 | |
| 4 | PTSA | 0.01 | 0.8 | 1.0 | 1.1 | 1.2 | 1.1 | 1.3 |
| 5 | 0.10 | 1.1 | 1.4 | 3.0 | 5.7 | 11 | 11 | |
| 6 | 0.25 | 2.2 | 4.7 | 14 | 20 | 25 | 24 | |
| 7 | MSA | 0.01 | 0.6 | 0.7 | 0.8 | 0.9 | 1.2 | 1.3 |
| 8 | 0.10 | 0.9 | 1.3 | 2.6 | 5.0 | 8.5 | 12 | |
| 9 | 0.25 | 1.3 | 2.1 | 3.8 | 6.1 | 5.5 | 6.4 | |
| 10 | CDMS | 0.01 | 1.4 | 1.2 | 1.7 | 2.3 | 2.2 | 2.8 |
| 11 | 0.10 | 1.4 | 1.2 | 1.6 | 4.6 | 10 | 13 | |
| 12 | 0.25 | 5.2 | 13 | 22 | 29 | 38 | 45 | |
First, the effects of BA, HDT, and CDMS molar concentrations on the polymerization were kinetically monitored by taking samples from the reaction medium at regular intervals to determine the optimum polymerization conditions, and the resulting data are given in Table 2. As shown in Table 2, when 25 mol% of CDMS (run 1, Table 2) was used, the Mn of PDTA attained was 3.2 kDa within 2 min, and it gradually increased to 29 kDa over 24 h. When the CDMS amount was increased to 50 mol% (run 2, Table 2), a distinct increment was observed in the obtained Mns: 7 kDa in 2 min and 43 kDa in 24 h. A further increase in CDMS (100 mol%) showed a similar trend to the 50 mol% case, yet the Mn showed a little decline after 24 h compared to the 50 mol% case (run 3, Table 2). This result can be attributed to the increased acidity of the reaction medium due to the high loading of CDMS, leading to chain scission at longer durations in the resulting PDTA. As a result, 50 mol% of CDMS was chosen as the ideal equivalent (based on monomers) for the acid source and used in the subsequent experiments. Next, the effect of monomer concentration on the polymerization was examined. When the monomer concentration increased to 4 M based on HDT, the Mn of PDTA was found to be 14 kDa in 2 min and it increased to 45 kDa over 24 h (run 4, Table 2). On the other hand, reducing the monomer concentration to 1 M resulted in a dramatic decrease in the Mn of the resulting polymer: 2.3 kDa in 2 min and 4.1 kDa in 24 h (run 5, Table 2). It is worth noting here that, as highlighted in our previous studies, CDMS can directly reduce the aldehyde carbonyl to the corresponding alcohol, leading to an equivalent imbalance within the reactants.63,64 For this purpose, another iteration was made by increasing the BA amount by 10%, which significantly increased the Mn of P1. Strikingly, the Mn of the polymer was found to be 44 kDa in 2 min, which increased to 64 kDa in 5 min and somewhat slightly increased to 78 kDa in 24 h (run 6, Table 2). Yet, further increment in the amount of BA resulted in a lower molecular weight over 24 h (Mn = 56 kDa), which can be attributed to a new equivalent imbalance between BA and HDT monomers (run 7, Table 2). Notably, satisfactory Mn values were also obtained when CDMS was replaced with trimethylsilyl chloride under the conditions of run 6 (Table 2); Mn = 6 kDa was obtained in 2 min, increasing to 38 kDa in 1 h and 48 kDa in 24 h (run 8, Table 2). The results of this experiment show that trimethylsilyl chloride is also a suitable acid source to proceed with the proposed polymerization system; however, the Mn values obtained are still lower than those from CDMS, which might be attributed to the sterically hindered structure of trimethylsilyl chloride, and therefore, it might slow down the polymerization (see the polymerization mechanism in Fig. 5A).
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Run | BA:HDT:CDMS (equiv.) |
HDT [M] |
M
nb (kDa) |
|||||||
| 2 min | 5 min | 15 min | 30 min | 60 min | 240 min | 480 min | 1440 min | |||
| a All reactions were carried out in THF at room temperature. b Determined by GPC, calibrated based on linear PS standards in THF. c Trimethylsilyl chloride was used instead of CDMS. d Reactions were performed at 0 °C. | ||||||||||
| 1 | 1:1:0.25 |
2 | 3.2 | 5.3 | 5.6 | 6.6 | 5.2 | 13 | 22 | 29 |
| 2 | 1:1:0.50 |
2 | 7.0 | 18 | 19 | 23 | 24 | 25 | 26 | 43 |
| 3 | 1:1:1 |
2 | 11 | 15 | 24 | 27 | 32 | 34 | 32 | 39 |
| 4 | 1:1:0.50 |
4 | 14 | 33 | 34 | 34 | 38 | 38 | 40 | 45 |
| 5 | 1:1:0.50 |
1 | 2.3 | 2.8 | 2.4 | 3.5 | 3.5 | 3.7 | 3.8 | 4.1 |
| 6 |
1.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1:0.50 :1:0.50
|
4 | 44 | 64 | 64 | 65 | 66 | 72 | 80 | 78 |
| 7 | 1.2:1:0.50 |
4 | 43 | 48 | 52 | 52 | 55 | 52 | 56 | 56 |
| 8c | 1.1:1:0.50 |
4 | 6.0 | 11 | 24 | 35 | 38 | 39 | 44 | 48 |
| 9d | 1:1:0.50 |
4 | 16 | 28 | 38 | 41 | 46 | 54 | 57 | 65 |
| 10d | 1.1:1:0.50 |
4 | 25 | 34 | 35 | 39 | 43 | 45 | 44 | 45 |
On the other hand, as discussed above, since the reaction is exothermic at room temperature, it was thought that this might be the reason for the equivalent imbalance that facilitates the conversion of aldehyde to alcohol. As such, the model monomers were reacted at 0 °C under the same conditions described in run 4 (Table 2) to examine the role of temperature in polymerization. Here, a gradual increase in Mn values was observed; for instance, the Mn of the polymer was found to be 16 kDa in 2 min, which increased to 46 kDa in 1 h and continued to increase, reaching 65 kDa in 24 h (run 9, Table 2). It should be noted here that these results are higher than the values obtained at room temperature (run 4, Table 2). In addition, when the same reaction was repeated under the conditions of run 6 (Table 2) (i.e., BA equivalent was increased to 1.1), the Mn values were higher than those for the 1:1 case in the first 5 min (Mn = 25 kDa in 2 min and 34 kDa in 5 min), then remained below them and were almost constant over time (Mn = 43 kDa in 1 h and 45 kDa in 24 h) (run 10, Table 2). These results highlight that a better end-group control for BA and, thus, a stoichiometric balance between the reactants could be achieved at 0 °C. However, the Mn values obtained under the conditions of 1.1:1:0.5 (BA:HDT:CDMS) at room temperature (run 6, Table 2) are still higher than the values at 0 °C, implying that room temperature is still required to promote the proposed polymerization system to reach higher molecular weights in shorter durations, although a slight excess of BA needs to be used due to some loss in the polymerization. Given these results, it was decided that the ideal equivalents for BA:HDT:CDMS were 1.1:1:0.5, the optimum monomer concentration was 4 M (based on HDT), and the polymerization temperature was room temperature. In addition, since no dramatic increase in the Mn of the resulting polymer was observed after 5 min, the polymerization time was set to be 5 min and used in the next experiments with the above-mentioned conditions.
Finally, we also examined the effect of solvent on polymerization. Among the solvents tested for polymerization, THF afforded PDTA with high Mn and yield (run 1, Table 3). When chlorinated solvents, such as CHCl3 and DCE, were used in the polymerizations, relatively high Mns of 43 and 54 kDa and high yields of 89 and 90%, respectively, were achieved (runs 2 and 3, Table 3). In addition, different etheric solvents, such as 1,4-dioxane and 2-MeTHF, were also found to be as effective as THF, leading to very similar results in terms of both Mns (60 and 62 kDa) and yields (runs 4 and 5, Table 3). However, the solvents containing carbonyl units such as DMF, NMP, and DMAc were found to be not an ideal choice for the synthesis of PDTA. When DMF and NMP (runs 6 and 7, Table 3) were used, the molecular weights were found to be quite low (Mn = 1.4 and 1.8 kDa) compared to those when using the aforementioned solvents, and even no polymerization was observed in the presence of DMAc (run 8, Table 3). These results could be attributed to the affinity of CDMS towards carbonyl units in these solvents, inhibiting the evolution of polymerizations. As a result, THF was still a better choice for the proposed strategy and was used in conjunction with the above-mentioned conditions in the rest of this study.
| Run | Solvent |
M
n
b (kDa) |
Đ | Yieldc (%) |
|---|---|---|---|---|
|
a All reactions were carried out using BA:HDT:CDMS at a molar ratio of 1.1:1:0.5 in 500 μL of solvent at room temperature for 5 min.
b Determined by GPC, calibrated based on linear PS standards in THF.
c Isolated yields.
|
||||
| 1 | THF | 64 | 1.53 | 98 |
| 2 | CHCl3 | 43 | 1.74 | 89 |
| 3 | DCE | 54 | 1.50 | 90 |
| 4 | 1,4-Dioxane | 60 | 1.46 | 90 |
| 5 | 2-MeTHF | 62 | 1.54 | 96 |
| 6 | DMF | 1.4 | 3.64 | 54 |
| 7 | NMP | 1.8 | 3.32 | 52 |
| 8 | DMAc | No reaction | ||
Before examining the scope of reactants, P1 was characterized in detail by various spectroscopic measurements. Here, when 1H NMR of P1 is examined, the characteristic backbone methine proton (ArCHS) of PDTA can be seen at 4.85 ppm. In addition, the signals between 7.41 and 7.25 ppm can be assigned as the aromatic protons of BA, and the signals that appear at 2.49, 1.50, and 1.29 ppm are the methylene protons of HDT (Fig. 1A). From the 13C NMR spectrum of P1, it was detected that signals at 53.29 and 140–128 ppm could be assigned as the methine carbon (ArCHS) and aromatic carbons of the PDTA backbone, respectively (Fig. 1B). Furthermore, from the FT-IR spectrum of P1, a strong C–S thioether stretching peak of the PDTA backbone was detected at around 750 cm−1 (Fig. 1C). Finally, the GPC chromatogram of P1 showed a monomodal distribution trace with a Đ of 1.53, indicating smooth polymerization (Fig. 1D).
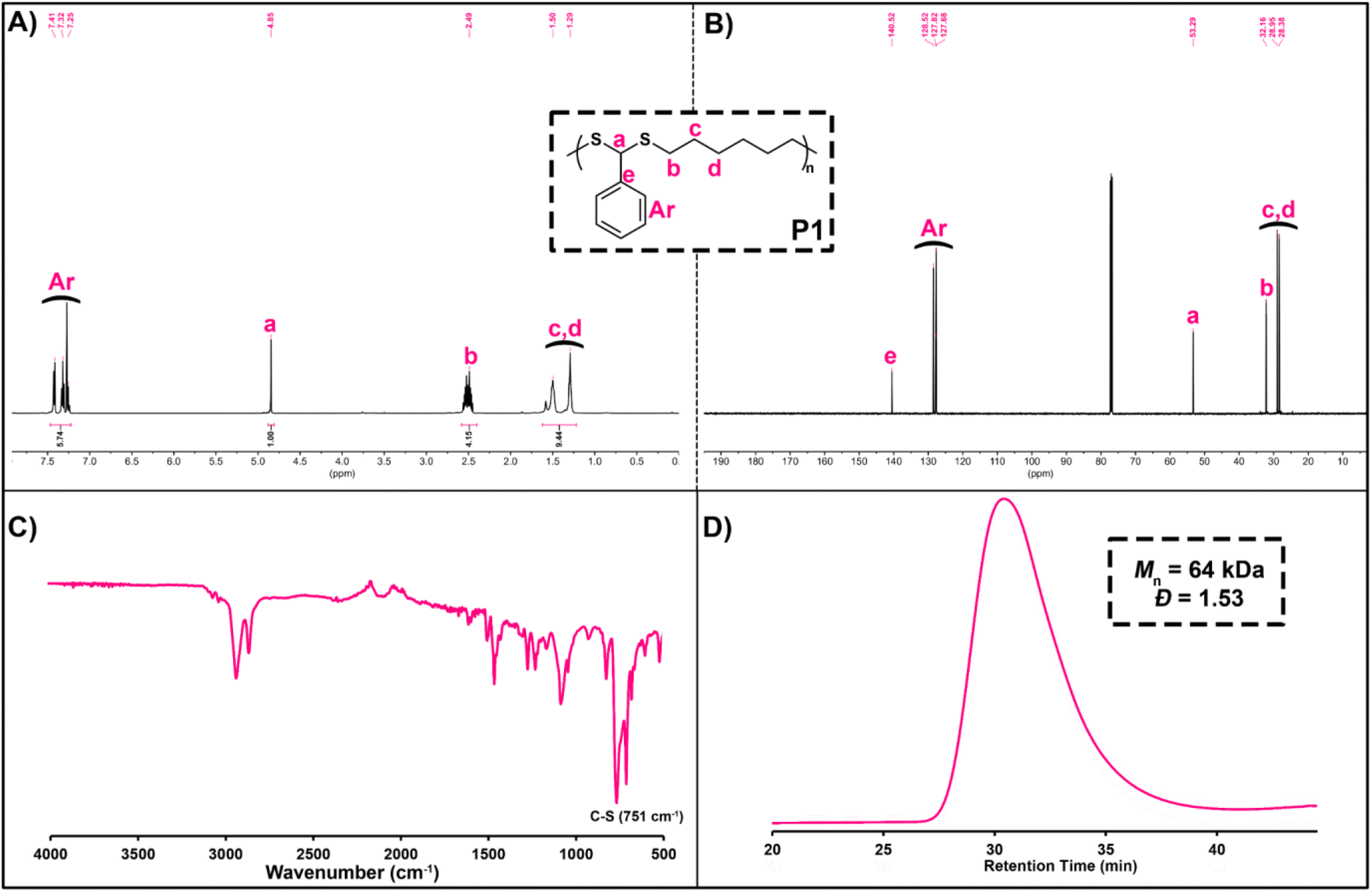 | ||
| Fig. 1 1H NMR spectrum (A) in CDCl3 (500 MHz), 13C NMR spectrum (B) in CDCl3 (125 MHz), FT-IR spectrum (C), and GPC chromatogram (D) of P1. | ||
With the optimized conditions in hand, various aldehydes and dithiols were then reacted to generate a PDTA library, and the results are shown in Table 4. The final structures of the obtained polymers and the 1H NMR spectra of some selected polymers confirming the chemical structure of precursor monomers that are used in polymerizations can be seen in Fig. 2 and 3 (relevant data for the remaining polymers are provided in the ESI†). While examining the aldehyde scope, HDT was used as the model dithiol. First, the electronic effects of different substituents on the aldehydes were examined in the polymerization. When electron-withdrawing-group-containing aldehydes were used, PDTAs were achieved with high yields and low to moderate Mns, such as for 3-nitrobenzaldehyde (P2, Mn = 18 kDa), 4-cyanobenzaldehyde (P3, Mn = 9 kDa), and 4-formylbenzoic acid (P4, Mn = 9 kDa). PDTAs with low to moderate Mns were again obtained when aldehydes containing electron-donating substituents such as p-anisaldehyde, 4-hydroxybenzaldehyde, and 4-chlorobenzaldehyde were utilized: P5 (Mn = 15 kDa), P6 (Mn = 8 kDa), and P7 (Mn = 6 kDa), respectively. These results suggest that substituents on the phenyl ring have no distinct effect on the proposed polymerization system and, thus, on the obtained Mns of PDTAs. Likewise, a natural aldehyde, namely vanillin, yielded a PDTA with moderate Mn in good yield (P8, Mn = 16 kDa, 87%). Notably, the Mns of PDTA P6 and P8 were comparable to those obtained from the recent work of Xu and Yuan.40 However, they prepared these phenolic PDTAs from corresponding aldehydes and dithiols within 30 min catalyzed by CF3COOH in MeOH at room temperature.
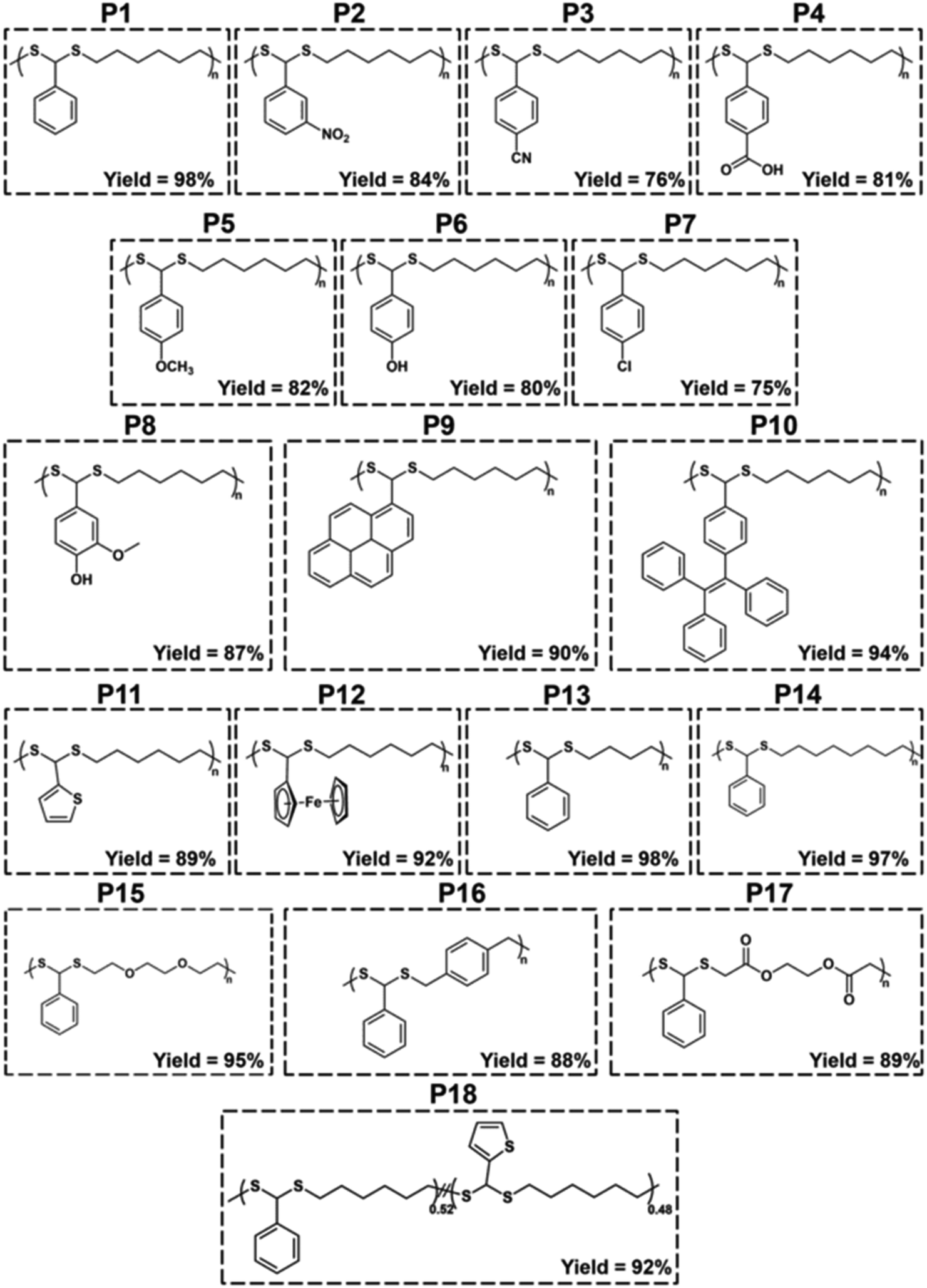 | ||
| Fig. 2 Final structures of the resulting polymers. | ||
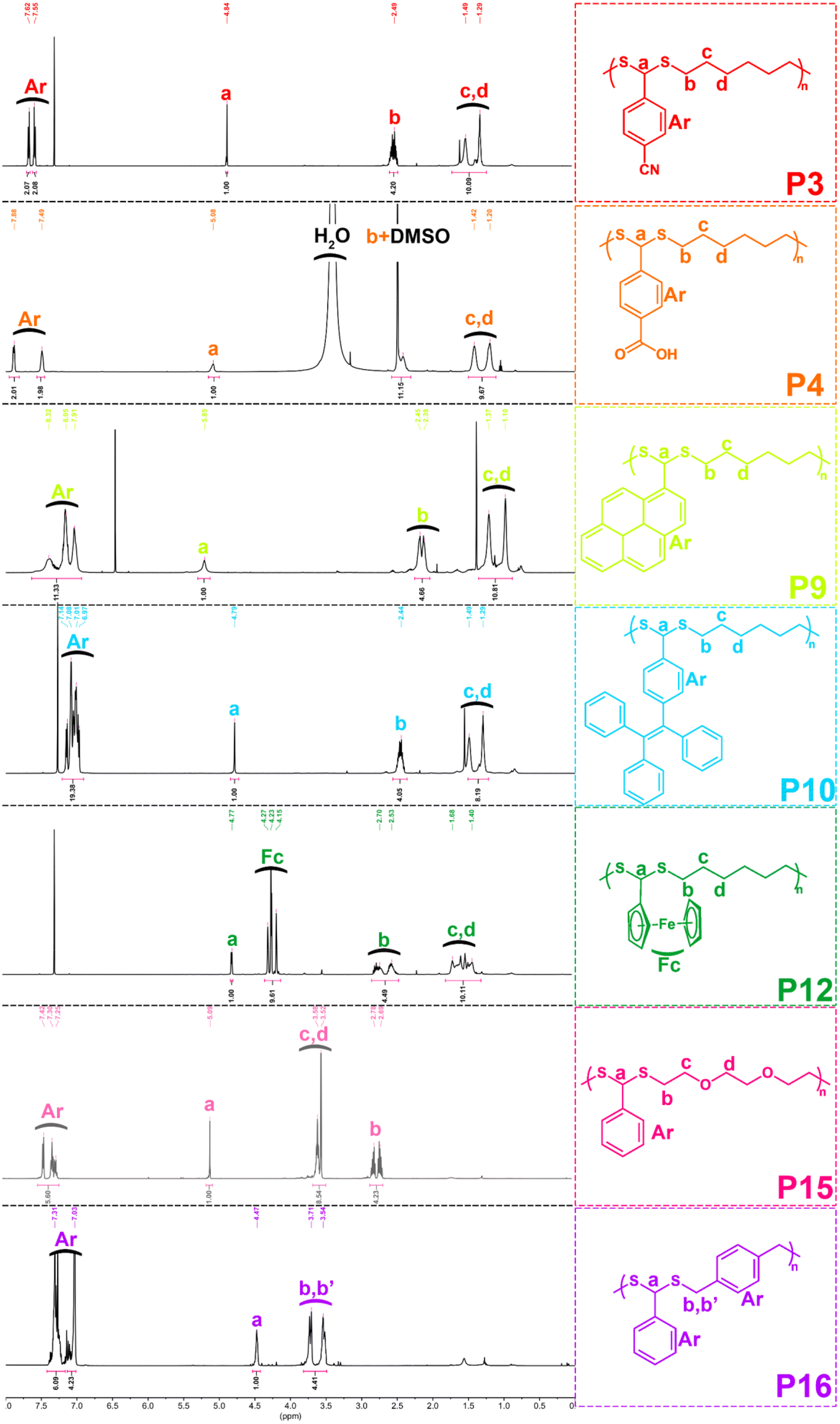 | ||
| Fig. 3 1H NMR spectra of representative polymers in CDCl3 (500 MHz). | ||
| Polymer | Aldehyde | Dithiol |
M
nb (kDa) |
Đ |
T
gc (°C) |
Yieldd (%) |
|---|---|---|---|---|---|---|
|
a All reactions were carried out using aldehyde:dithiol:CDMS at a molar ratio of 1.1:1:0.5 in 500 μL of THF at room temperature for 5 min.
b Determined by GPC, calibrated based on linear PS standards in THF.
c
T
g values were determined by DSC from the second heating cycle.
d Isolated yields.
e Equal moles (1.10 mmol each) of aldehydes were used.
f Not detected.
|
||||||
| P1 | BA | HDT | 64 | 1.53 | −29.6 | 98 |
| P2 | 3-Nitrobenzaldehyde | HDT | 18 | 2.00 | −26.8 | 84 |
| P3 | 4-Cyanobenzaldehyde | HDT | 9 | 2.16 | −0.2 | 76 |
| P4 | 4-Formylbenzoic acid | HDT | 9 | 3.17 | 64.6 | 81 |
| P5 | p-Anisaldehyde | HDT | 15 | 1.58 | −24.2 | 82 |
| P6 | 4-Hydroxybenzaldehyde | HDT | 8 | 2.57 | 6.1 | 80 |
| P7 | 4-Chlorobenzaldehyde | HDT | 6 | 2.07 | −15.3 | 75 |
| P8 | Vanillin | HDT | 16 | 1.71 | 7.9 | 87 |
| P9 | 1-Pyrenecarboxaldehyde | HDT | 16 | 1.97 | 54.3 | 90 |
| P10 | 4-(1,2,2-Triphenylethenyl)benzaldehyde | HDT | 47 | 2.09 | 56.5 | 94 |
| P11 | 2-Thiophenecarboxaldehyde | HDT | 30 | 1.62 | ndf | 89 |
| P12 | Ferrocenecarboxaldehyde | HDT | 14 | 1.62 | −0.6 | 92 |
| P13 | BA | 1,4-Butanedithiol | 66 | 1.69 | −17.3 | 98 |
| P14 | BA | 1,8-Octanedithiol | 58 | 1.88 | −35.0 | 97 |
| P15 | BA | 2,2′-(Ethylenedioxy)diethanethiol | 59 | 1.65 | −28.7 | 95 |
| P16 | BA | 1,4-Benzenedimethanethiol | 14 | 1.67 | 45.5 | 88 |
| P17 | BA | Ethylene glycol bis-mercaptoacetate | 14 | 2.25 | 0.9 | 89 |
| P18e | BA/2-Thiophenecarboxyaldehyde | HDT | 49 | 1.63 | −32.8 | 92 |
Different functionalities were also introduced into the PDTA backbone as part of the aldehyde library. For instance, a PDTA with a relatively high Mn was obtained when 1-pyrenecarboxyaldehyde was used in polymerization (P9, Mn = 16 kDa). A sterically congested aldehyde, 4-(1,2,2-triphenylethenyl)benzaldehyde, afforded the corresponding PDTA (P10) with high Mn = 47 kDa in high yield (94%). This result was particularly striking since lower activity was expected from this aldehyde. In addition, this protocol could be a useful tool to achieve luminogenic materials starting from tetraphenylethene building blocks since there is an increasing interest in two-photon absorption and aggregation-induced emission.69 A heteroatom-containing aromatic aldehyde 2-thiophenecarboxyaldehyde resulted in the corresponding PDTA (P11) with high Mn = 30 kDa in good yield (89%). In this study, ferrocenecarboxyaldehyde was also examined and it yielded the corresponding PDTA (P12) with moderate Mn = 14 kDa. It should be noted that this approach is believed to be promising as it brings a new perspective to the synthesis of organometallic polymers.70
The dithiol toolbox was also examined to explore the scope of the polymerization, and BA was used as the model aldehyde in these studies. Similar to P1, PDTAs of high Mns were observed when linear aliphatic dithiols such as 1,4-butanedithiol (P13, Mn = 66 kDa) and 1,8-octanedithiol (P14, Mn = 58 kDa) were used in the polymerization reactions. Moreover, 2,2′-(ethylenedioxy)diethanethiol gave the corresponding PDTA (P15) with high Mn = 59 kDa. However, when an aromatic dithiol, 1,4-benzenedimethanethiol, was used to perform the polymerization, the resulting PDTA (P16) was found to have moderate Mn = 14 kDa. Similarly, ethylene glycol bis-mercaptoacetate resulted in a PDTA (P17) with moderate Mn = 14 kDa.
The thermal behavior of the resulting polymers was revealed by DSC analysis, and the collected data are given in Table 4. The table shows that most of the synthesized PDTAs exhibit low Tg values ranging from −35 to 7.9 °C due to the dithiols that impart flexibility to the polymer structures. Among them, P4 exhibited the highest Tg (64.6 °C); most likely, the intra and/or intermolecular H-bonding ability of the carboxylic acid units has a significant impact leading to the relatively high Tg. Moreover, PDTAs produced from sterically hindered aldehydes (P9 and P10) exhibited high Tg values, 54.3 and 56.5 °C, respectively. Therefore, these results can be attributed to the stiffness of the related polymers due to the pyrene and triphenylethenyl units that restrict chain movements. A relatively high Tg = 45.5 °C was also observed when 1,4-benzenedimethanethiol (P16) was used as a dithiol source, which is related to the decrease in main chain flexibility due to the replacement of aliphatic dithiols with an aromatic one.
A copolymerization study was also performed to extend the scope of polymerization, as described in Scheme 2. For this purpose, equal moles (1.10 mmol each) of BA and 2-thiophenecarboxyaldehyde were reacted with HDT under the same polymerization conditions to yield the corresponding copolymer PDTA (P18) with high Mn = 49 kDa (Fig. 4B). The 1H NMR spectrum of P18 showed that the ratio of integral areas of the CH methine protons resulted in a nearly equal distribution (0.52/0.48) regarding the feeding ratio of BA:2-thiophenecarboxyaldehyde (Fig. 4A). The data indicate that different copolymeric backbones can be achieved by employing this protocol and the desired feed ratio of the starting materials.
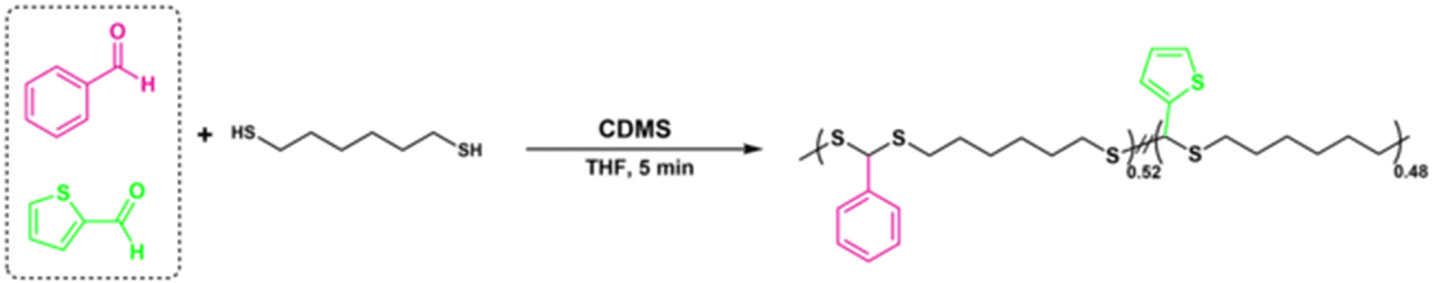 | ||
| Scheme 2 Synthetic route for copolymer PDTA (P18) synthesis. | ||
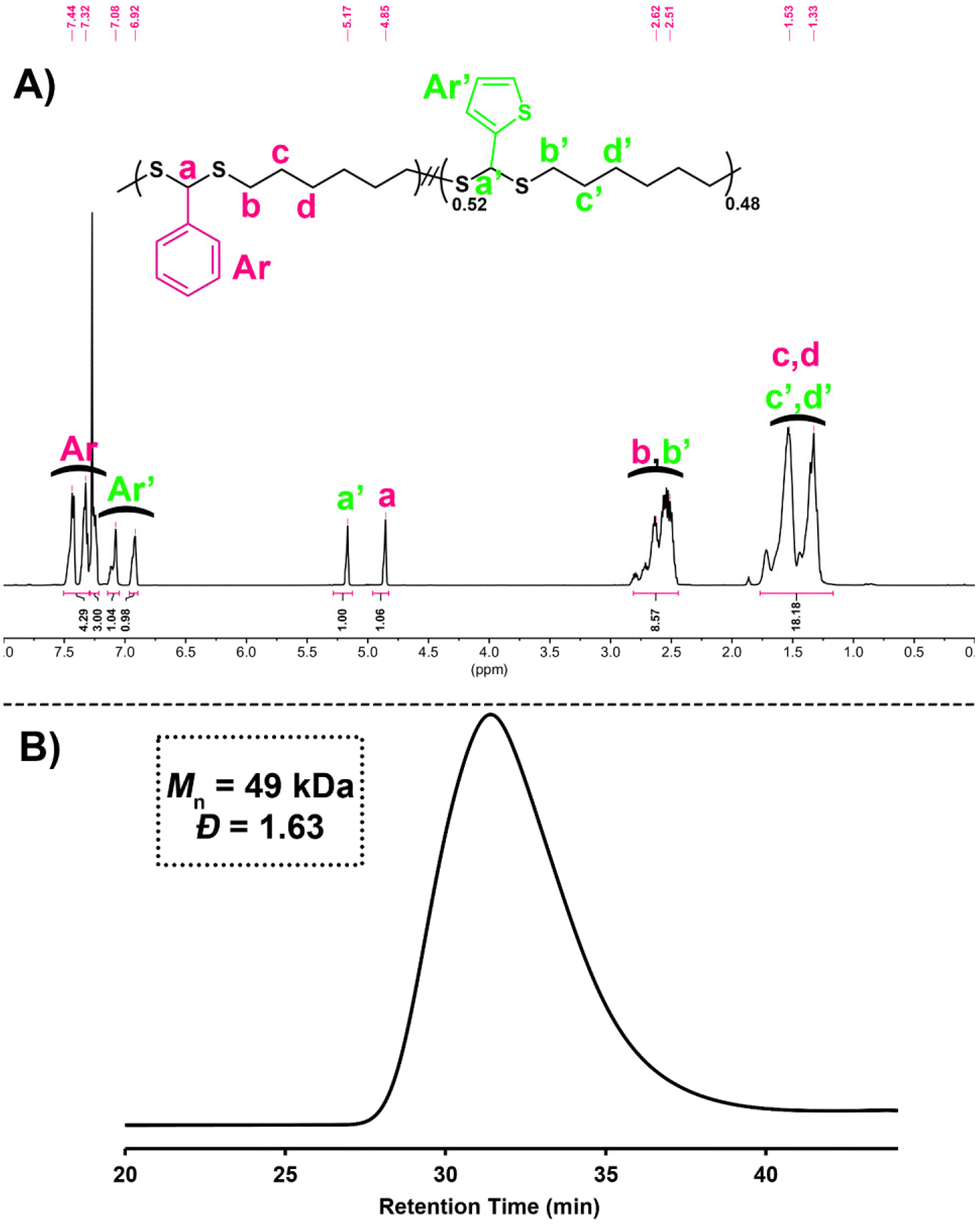 | ||
| Fig. 4 1H NMR spectrum (A) in CDCl3 (500 MHz) and GPC chromatogram (B) of P18. | ||
Next, the CDMS-mediated PDTA synthesis described in this study was examined in terms of mechanistic aspects. Fig. 5 shows the proposed mechanism for the polymerization protocol. Since CDMS acts as a Lewis acid but also generates HCl in situ during the reaction, a Brønsted acid character should also be expected. To this end, we envisioned two mechanisms working together during polymerization. For the Lewis acid route (Fig. 5A), as explained in our previous studies,63,64 the reaction starts with the silylation of the aldehyde unit in the presence of CDMS, where the chlorine ion acts as the leaving group in order to form the silylated aldehyde (I). Then, I is attacked by the dithiol, resulting in a hemithioacetal-type intermediate II, followed by a protonation step to yield III, and then silanol leaves the structure after the attack of another dithiol to give IV. Finally, polymerization takes place through a condensation reaction between III and IV.
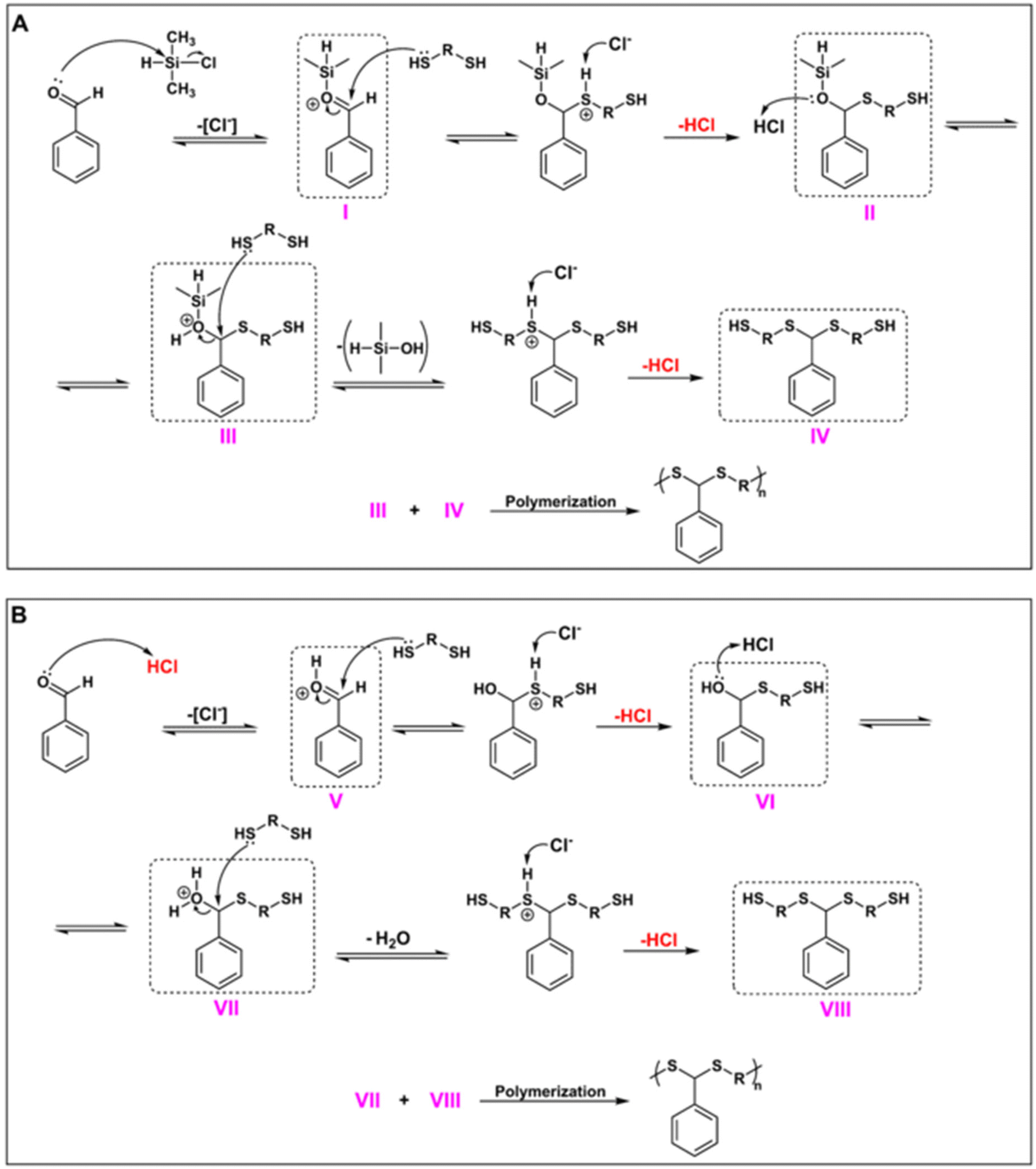 | ||
| Fig. 5 Proposed mechanism for CDMS-mediated PDTA synthesis: Lewis acid route (A) and Brønsted acid route (B). | ||
Moreover, since in situ HCl formation was observed in the Lewis acid route, it is also possible to expect the Brønsted acid route during the reaction (Fig. 5B). Hence, aldehyde can also be protonated with HCl to form a protonated aldehyde intermediate (V). From there, the reaction is expected to follow the traditional acid-catalyzed dithioacetal formation mechanism, where V is then attacked by a dithiol to form VI, followed by a protonation step to remove the water from this intermediate (VII) and attack from another dithiol to form VIII. Finally, the condensation reaction between VII and VIII will result in the formation of PDTA. Since polymerization takes place through two possible mechanisms, it is most likely to expect that CDMS resulted in the formation of PDTAs with much higher Mns in short durations when compared to other acids that were examined during this study.
As mentioned before, one of the most important features of dithioacetal bonds is their ROS-sensitive character. In this regard, P1 was subjected to degradation using H2O2 as the source of ROS, and the progress of the reaction was monitored kinetically by taking samples from the reaction medium at regular intervals and analyzing by GPC (Scheme 3). As seen in Fig. 6B, the degradation of the polymer started within 1 h and continued over time, and complete degradation was observed in 24 h. The degradation of P1 was confirmed by 1H NMR analysis. The CH methine signal of P1 at 4.85 ppm completely disappeared, while the aldehyde proton at 10.05 ppm reappeared, emphasizing the regeneration of the initial reactants (Fig. 6A).
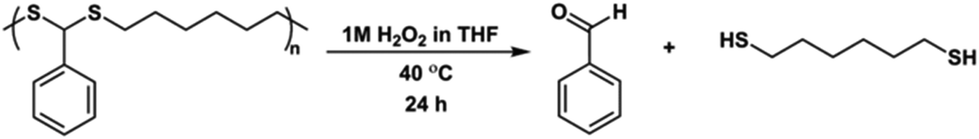 | ||
| Scheme 3 Degradation of P1 (run 6, Table 2) in the presence of H2O2. | ||
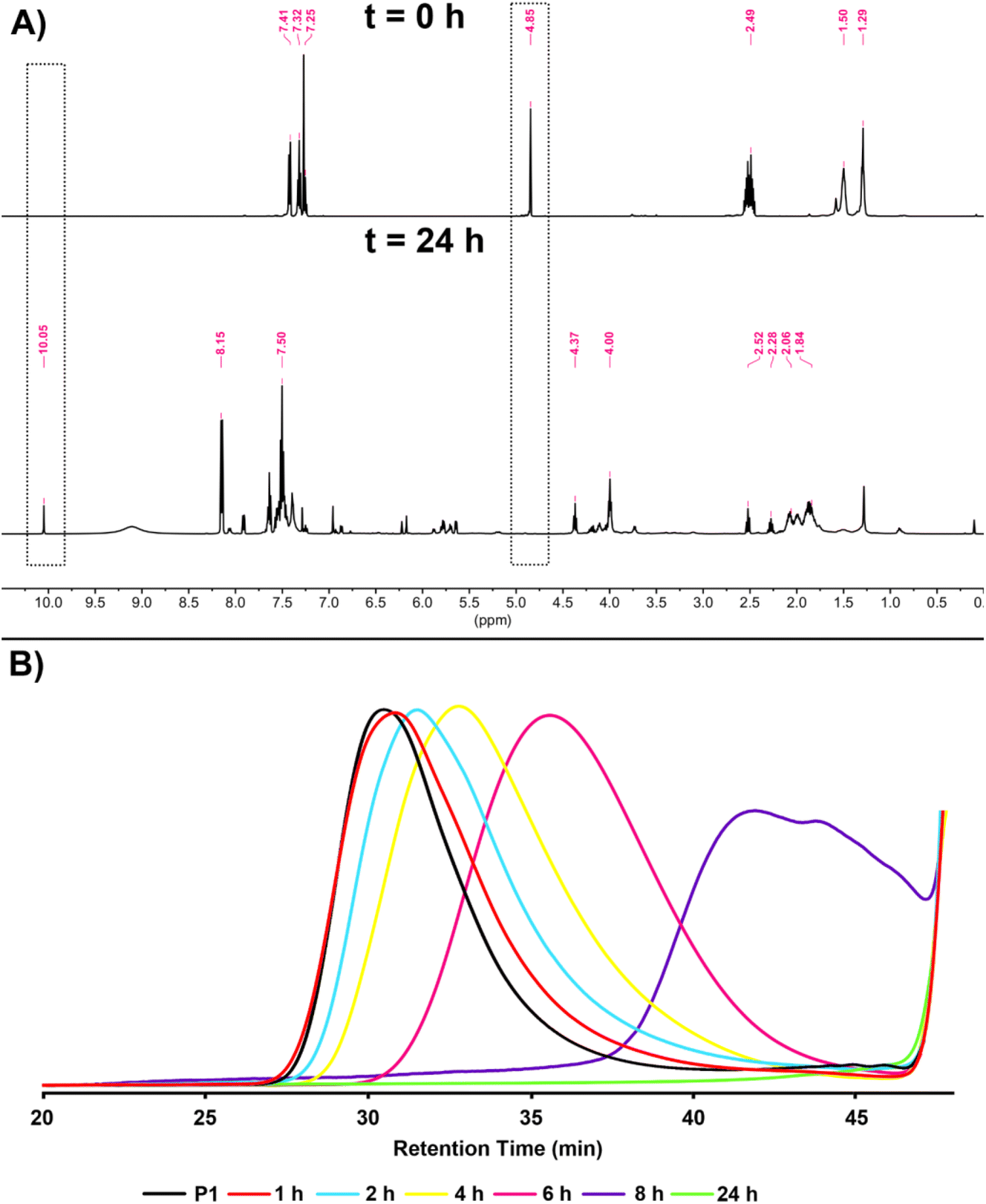 | ||
| Fig. 6 1H NMR spectra (A) at t = 0 and after t = 24 h in CDCl3 (500 MHz) and overlaid GPC traces (B) obtained at different time intervals during the degradation of P1. | ||
Conclusions
We have successfully prepared a wide variety of PDTAs in the presence of CDMS. Optimization studies performed between BA and HDT revealed that 0.50 equiv. of CDMS and slightly excess BA (1.1 equiv.) were required to obtain relatively high molecular weight polymers. The proposed protocol proceeded straightforwardly under mild conditions, resulting in PDTAs with high isolated yields in just 5 minutes. Although PDTA synthesis is known in the polymer chemistry literature, comprehensive work was performed to investigate various polymerization parameters. The PDTA backbone was tailored by selecting different aldehydes and dithiols, forming a huge polymer library with Mns ranging from 6 to 66 kDa. A degradation study on a model PDTA (P1) was also performed, and a complete degradation was observed in 24 h.Collectively, a straightforward and rapid PDTA synthesis method has been revealed in this study, which promises to circumvent the harsh conditions required in a classical PDTA synthesis. We believe that this work will pave a new way for PDTA synthesis, and we aim to inspire further research in this field and facilitate the development of PDTA-based materials with tailored properties for diverse applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Scientific and Technological Research Council of Turkey (TUBITAK) (Project Number: 119Z516) and the Scientific Research Projects Departments of Istanbul Technical University (ITU BAP Project Number: TDK-2021-43456).References
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- Z. Geng, J. J. Shin, Y. Xi and C. J. Hawker, J. Polym. Sci., 2021, 59, 963–1042 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743–750 CrossRef CAS.
- O. I. Kalaoglu-Altan, B. Verbraeken, K. Lava, T. N. Gevrek, R. Sanyal, T. Dargaville, K. D. Clerk, R. Hoogenboom and A. Sanyal, ACS Macro Lett., 2016, 5, 676–681 CrossRef CAS PubMed.
- D. P. Nair, M. Podgorski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- O. Daglar, S. Luleburgaz, E. Baysak, U. S. Gunay, G. Hizal, U. Tunca and H. Durmaz, Eur. Polym. J., 2020, 137, 109926 CrossRef CAS.
- H. Mutlu, E. B. Ceper, X. Li, J. Yang, W. Dong, M. M. Ozmen and P. Theato, Macromol. Rapid Commun., 2019, 40, 1800650 CrossRef PubMed.
- R. Hoogenboom, Angew. Chem., Int. Ed., 2010, 49, 3415–3417 CrossRef CAS PubMed.
- S. Aksakal, R. Aksakal and C. R. Becer, Polym. Chem., 2018, 9, 4507–4516 RSC.
- S. De and A. Khan, Chem. Commun., 2012, 48, 3130–3132 RSC.
- M. C. Stuparu and A. Khan, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3057–3070 CrossRef CAS.
- S. Agar, E. Baysak, G. Hizal, U. Tunca and H. Durmaz, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1181–1198 CrossRef CAS.
- B. M. Rosen, G. Lligadas, C. Hahn and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3931–3939 CrossRef CAS.
- U. S. Gunay, M. Cetin, O. Daglar, G. Hizal, U. Tunca and H. Durmaz, Polym. Chem., 2018, 9, 3037–3054 RSC.
- O. Daglar, U. S. Gunay, G. Hizal, U. Tunca and H. Durmaz, Macromolecules, 2019, 52, 3558–3572 CrossRef CAS.
- X. Fu, A. Qin and B. Z. Tang, Aggregate, 2023, e350, DOI:10.1002/agt2.350.
- J. C. Worch, C. J. Stubbs, M. J. Price and A. P. Dove, Chem. Rev., 2021, 121, 6744–6776 CrossRef CAS PubMed.
- O. Daglar, E. Cakmakci, U. S. Gunay, G. Hizal, U. Tunca and H. Durmaz, Macromolecules, 2020, 53, 2965–2975 CrossRef CAS.
- J. X. Lei, Q. Y. Wang, F. S. Du and Z. C. Li, Chin. J. Polym. Sci., 2021, 39, 1146–1154 CrossRef CAS.
- U. Tunca, Macromol. Chem. Phys., 2018, 219, 1800163 CrossRef.
- P. G. M. Wuts and T. W. Greene, Greene's Protective Groups in Organic Synthesis, John Wiley & Sons, 2006 Search PubMed.
- S. Sensfuss, Angew. Makromol. Chem., 1996, 234, 191–207 CrossRef CAS.
- B. Liu and S. Thayumanavan, Cell Rep. Phys. Sci., 2020, 1, 100271 CrossRef CAS.
- Y. Men, T. G. Breve, H. Liu, A. G. Denkova and R. Eelkema, Polym. Chem., 2021, 12, 3612–3618 RSC.
- N. G. Fisher and R. H. Wiley, US Patent, 2389662, 1945 Search PubMed.
- C. S. Marvel, E. H. H. Shen and R. R. Chambers, J. Am. Chem. Soc., 1950, 72, 2106–2109 CrossRef CAS.
- C. S. Marvel and R. C. Farrar Jr., J. Am. Chem. Soc., 1957, 79, 986–988 CrossRef CAS.
- E. G. Horvath, S. Gardlund, S. K. Sen, J. W. Berry and A. J. Deutschman Jr., J. Polym. Sci., Part A: Polym. Chem., 1965, 3, 1985–1992 CAS.
- M. Kakimoto, T. Seri and Y. Imai, Polym. J., 1987, 19, 1303–1307 CrossRef CAS.
- S. Hilf and A. F. M. Kilbinger, Macromolecules, 2009, 42, 4127–4133 CrossRef CAS.
- S. Chatterjee and S. Ramakrishnan, ACS Macro Lett., 2012, 1, 593–598 CrossRef CAS PubMed.
- S. Chatterjee and S. Ramakrishnan, ACS Macro Lett., 2014, 3, 953–957 CrossRef CAS PubMed.
- M. Uchiyama, M. Osumi, K. Satoh and M. Kamigaito, Angew. Chem., Int. Ed., 2020, 59, 6832–6838 CrossRef CAS PubMed.
- M. Uchiyama, M. Osumi, K. Satoh and M. Kamigaito, Macromol. Rapid Commun., 2021, 42, 2100192 CrossRef CAS PubMed.
- S. Kim, H. Park, F. Fuss and Y. Lee, Polym. Chem., 2023, 14, 2610–2616 RSC.
- L. S. Kariyawasam, J. Rolsma and Y. Yang, Angew. Chem., Int. Ed., 2023, 62, e202303039, DOI:10.1002/anie.202303039.
- Y. Jin, C. Hu, J. Wang, Y. Ding, J. Shi, Z. Wang, S. Xu and L. Yuan, Angew. Chem., Int. Ed., 2023, 62, e202305677, DOI:10.1002/ange.202305677.
- A. G. Orrillo and R. L. E. Furlan, Angew. Chem., Int. Ed., 2022, 61, e202201168, DOI:10.1002/anie.202201168.
- G. Sartori, R. Ballini, F. Bigi, G. Bosica, R. Maggi and P. Righi, Chem. Rev., 2004, 104, 199–250 CrossRef CAS PubMed.
- H. F. Yu, Synth. Commun., 2013, 43, 1280–1286 CrossRef CAS.
- D. S. Wilson, G. Dalmasso, L. Wang, S. V. Sitaraman, D. Merlin and N. Murthy, Nat. Mater., 2010, 9, 923–928 CrossRef CAS PubMed.
- C. Xu, R. Han, H. Liu, Y. Zhu, J. Zhang and L. Xu, ChemistrySelect, 2021, 6, 3277–3285 CrossRef CAS.
- L. Xu, M. Zhao, H. Zhang, W. Gao, Z. Guo, X. Zhang, J. Zhang, J. Cao, Y. Pu and B. He, Biomacromolecules, 2018, 19, 4658–4667 CrossRef CAS PubMed.
- Q. Zong, J. Li, X. Xiao, X. Du and Y. Yuan, Acta Biomater., 2022, 154, 97–107 CrossRef CAS PubMed.
- Y. Tu, X. Xiao, Y. Dong, J. Li, Y. Liu, Q. Zong and Y. Yuan, Biomaterials, 2022, 289, 121795 CrossRef CAS PubMed.
- A. Rinaldi, R. Caraffi, M. V. Grazioli, N. Oddone, L. Giardino, G. Tosi, M. A. Vandelli, L. Calzà, B. Ruozi and J. T. Duskey, Polymers, 2022, 14, 687 CrossRef CAS PubMed.
- Y. Lu, P. Shan, W. Lu, X. Yin, H. Liu, X. Lian, J. Jin, Y. Qi, Z. Li and Z. Li, Chem. Eng. J., 2023, 463, 142311 CrossRef CAS.
- M. P. Doyle, D. J. DeBruyn and D. A. Kooistra, J. Am. Chem. Soc., 1972, 94, 3659–3661 CrossRef CAS.
- C. T. West, S. J. Donnelly, D. A. Kooistra and M. P. Doyle, J. Org. Chem., 1973, 38, 2675–2681 CrossRef CAS.
- M. P. Doyle, D. J. DeBruyn, S. J. Donnelly, D. A. Kooistra, A. A. Odubela, C. T. West and S. M. Zonnebelt, J. Org. Chem., 1974, 39, 2740–2747 CrossRef CAS.
- M. P. Doyle and C. T. West, J. Org. Chem., 1975, 40, 3821–3829 CrossRef CAS.
- M. P. Doyle and C. T. West, J. Org. Chem., 1975, 40, 3829–3834 CrossRef CAS.
- M. P. Doyle and C. T. West, J. Org. Chem., 1975, 40, 3835–3838 CrossRef CAS.
- M. P. Doyle, C. T. West, S. J. Donnelly and C. C. McOsker, J. Organomet. Chem., 1976, 117, 129–140 CrossRef CAS.
- T. Yokozawa and F. Nakamura, Makromol. Chem., Rapid Commun, 1993, 14, 167–172 CrossRef CAS.
- T. Yokozawa and F. Nakamura, Macromolecules, 1995, 28, 4668–4674 CrossRef CAS.
- T. Yokozawa and K. Takenoya, React. Funct. Polym., 1996, 30, 251–260 CrossRef CAS.
- K. Takenoya and T. Yokozawa, Macromolecules, 1998, 31, 2906–2910 CrossRef CAS.
- L. Niimi, S. Hiraoka and T. Yokozawa, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5440–5448 CrossRef CAS.
- S. Luleburgaz, G. Hizal, U. Tunca and H. Durmaz, Macromolecules, 2021, 54, 5106–5116 CrossRef CAS.
- S. Luleburgaz, G. Hizal, U. Tunca and H. Durmaz, Macromolecules, 2022, 55, 1533–1543 CrossRef CAS.
- E. Akar, D. Kandemir, S. Luleburgaz, V. Kumbaraci and H. Durmaz, Eur. Polym. J., 2022, 177, 111440 CrossRef CAS.
- S. Luleburgaz, U. Tunca and H. Durmaz, Polym. Chem., 2023, 14, 2949–2957 RSC.
- S. Luleburgaz, U. Tunca and H. Durmaz, Macromol. Chem. Phys., 2023, 2300063, DOI:10.1002/macp.202300063.
- Y. H. Lee and B. Morandi, Synlett, 2017, 28, 2425–2428 CrossRef CAS.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- M. Serrano-Ruiz, F. Scalambra and A. Romerosa, in Advances in Organometallic Chemistry and Catalysis, ed. A. J. L. Pombeiro, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2013, pp. 379–405, DOI:10.1002/9781118742952.ch29.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py01128c |
| This journal is © The Royal Society of Chemistry 2024 |