
Emerging investigator series: mitigation of harmful algal blooms by electrochemical ozonation: from bench-scale studies to field applications†
Shasha
Yang
ab,
Luz Estefanny
Quispe Cardenas
ab,
Athkia
Fariha
a,
Nada
Shetewi
c,
Victor
Melgarejo Cazares
ab,
Nanyang
Yang
a,
Lewis
McCaffrey
 d,
Nicole
Wright
d,
Michael R.
Twiss
e,
Siwen
Wang
a,
Stefan J.
Grimberg
a and
Yang
Yang
*a
d,
Nicole
Wright
d,
Michael R.
Twiss
e,
Siwen
Wang
a,
Stefan J.
Grimberg
a and
Yang
Yang
*a
aDepartment of Civil and Environmental Engineering, Clarkson University, Potsdam, New York 13699, USA. E-mail: yanyang@clarkson.edu
bInstitute for a Sustainable Environment, Clarkson University, Potsdam, New York 13699, USA
cDepartment of Chemical Engineering, The Cooper Union, New York, New York 10003, USA
dNew York State Department of Environmental Conservation, 625 Broadway, Albany, New York 12333, USA
eFaculty of Science, Algoma University, Sault Ste. Marie, Ontario P6A 2G4, Canada
First published on 30th July 2024
Abstract
Harmful algal blooms (HABs) are an emerging threat to ecosystems, drinking water safety, and the recreational industry. As an environmental challenge intertwined with climate change and excessive nutrient discharge, HAB events occur more frequently and irregularly. This dilemma calls for fast-response treatment strategies. This study developed an electrochemical ozonation (ECO) process, which uses Ni–Sb–SnO2 anodes to produce locally concentrated ozone (O3) and hydroxyl radicals (·OH) to achieve ∼100% inactivation of cyanobacteria (indicated by chlorophyll-a degradation) and removal of microcystins within 120 seconds. More importantly, the proof-of-concept evolved into a full-scale boat-mounted completely mixed flow reactor for the treatment of HAB-impacted lake water. The single-pass treatment at a capacity of 544 m3 d−1 achieved 62% chlorophyll-a removal with an energy consumption of <1 Wh L−1. Byproducts (e.g., chlorate, bromate, trihalomethanes, and haloacetic acids) in the treated lake water were below the regulatory limits for drinking water. The whole effluent toxicity tests suggest that ECO treatment at 10 mA cm−2 posed certain chronic toxicity to the model crustacean invertebrate (Ceriodaphnia dubia). However, the treatment at 7 mA cm−2 (identified as the optimum condition) did not increase toxicity to model invertebrate and fish (Pimephales promelas) species. This study is a successful example of leveraging fundamental innovations in electrocatalysis to solve real-world problems.
Water impactThis study developed an innovative electrochemical ozonation (ECO) process to mitigate harmful algal blooms. The ECO process oxidizes water to generate ozone, which rapidly inactivates cyanobacteria and degrades toxins within minutes. Its high efficacy was demonstrated in both laboratory settings and pilot-scale real-world lake applications, showcasing low energy consumption and minimal byproduct formation. |
Introduction
Widespread harmful algal blooms (HABs) have become an emerging threat to ecosystems, recreational use of lakes, and drinking water supplies. Due to increased nutrient discharge and global climate change, the occurrence of HABs is expected to be more frequent.1,2 In the United States, the cyanobacterial HAB occurrence was 7 days per year per waterbody in 2017. The frequency was projected to be 18–39 days per year per water body by 2090.1 A recent study based on a 45-year record (1970–2015) revealed that the frequency of HABs occurring along the Chinese coast has increased by about 40% per decade.3 The surge production of hepatotoxic, cytotoxic, and neurotoxic microcystins can be expected in the HAB events.4Chemical oxidation (using chlorine, permanganate, H2O2, etc.)5–7 and advanced oxidation processes (UV/H2O2 and UV/chlorine)8 are promising for HAB mitigation at drinking water treatment works. However, they are less practical for on-site lake remediation due to the uncertain efficacy limited by hydrological conditions and concerns about the environmental impact of chemicals.
There is a critical need for in situ treatment technology that can proactively curb HABs at the early stage and precisely eliminate algae plumes and cyanotoxins in the impacted high-value areas (lake shores, public beaches, etc.). Since the bloom is usually concentrated in the top 0–1 m of the water column,9,10 we envision that mobile devices with pump-and-treat functionality could realize the precise treatment of the plume, instead of the whole water body, for maximum cost-effectiveness. Electrochemical oxidation (EO)-based technologies, with the advantages of small footprint, high efficiency, and additive-free operation, could be the best fit. Previously, the EO treatment of algae and microcystins equipped with anodes made of boron-doped diamond,11 metal oxide (RuO2 and IrO2),12,13 and graphite14 was investigated at the lab scale. We advanced the EO process by developing Ti4O7 filter anodes with pore sizes of 24–53 μm for the treatment of HAB-impacted lake water at both lab and full scales.15 The Ti4O7 filter anodes oxidize chloride in lake water to chlorine to inactivate algae cells and oxidize cyanotoxins. The flow-through operation (by pumping water out of the filter anodes immersed in the plume) significantly promoted convective mass transfer and thereby achieved higher treatment performance than homogenous chlorination. Though the scaled-up demonstration at a capacity of 110 m3 d−1 was successful, two limitations were identified: (1) the chlorine yield, which is associated with treatment performance, depends on chloride concentrations in lake water. (2) The micropores of the filter anode create large pressure drops ahead of the pump. The treatment capacity is limited by the pump suction head, which is significantly smaller than the lift head. The pumps were also vulnerable to damage by cavitation.
This study aims to address both engineering and fundamental challenges. The anode configuration was transformed from microporous cartridge filters to an array of mesh electrodes. Correspondingly, as will be discussed below, the treatment operation was changed from pumping water out of the submerged filter anode to pumping water through the mesh electrode array in a boat-mounted reactor to gain a larger treatment capacity (i.e., the flow rate is no longer limited by the pump suction head). The increase in design treatment capacity necessitates the use of more efficient electrodes that can produce various oxidants in addition to free chlorine, thereby removing the constraint of chloride concentration in lake water on treatment performance. In this regard, electrochemical ozone (O3) evolution (ECO) is a promising target reaction,16 as this reaction uses water molecules as the only feedstock (3H2O → O3(g) + 6H+ + 6e−, E0 = 1.51 VRHE).
Electrode materials made of Pt, PbO2, and boron-doped diamond (BDD) are able to produce O3.16 In addition to the cost concern, these electrodes have to be operated at high current densities (50-1000 mA cm−2) and acidic electrolyte (H2SO4 or HClO4) to achieve noticeable ozone production.17,18 The harsh conditions are less applicable to treating environmental water. Moreover, operations at high current density may promote the formation of halogenated byproducts.19 Recently, nickel-doped antimony tin oxide (NATO: Ni–Sb–SnO2) emerged as a more reactive electrode for O3 production. It could be operated at lower current densities (100–200 mA cm−2) for substantial current efficiencies (10–30%) in acidic electrolytes.20,21 Note that most of the studies on the ECO have been carried out in acidic electrolytes. This is because the apparent O3 evolution rate decreases with the increased pH,16 due to the promoted transformation of O3 to hydroxyl radical (·OH) and the decreased O3 solubility at elevated pH.22,23 Despite the potential compromise of O3 yield, recent environmental applications demonstrated that NATO could effectively produce O3 and the derivative ·OH in electrolytes and latrine wastewater at near-neutral pH, facilitating organic degradation and disinfection.24,25
This is the first study to explore the fundamentals, applications, and environmental implications of NATO electrodes in HAB mitigation. The significant advancement brought by NATO is the shift from using free chlorine to O3 and ·OH as the reactive species, enabling treatment performance to be independent of chloride concentration in lakes. In addition to process innovation and scaled-up demonstration, this study provides comprehensive evaluations of the environmental implications, covering process impacts on byproduct formation and effluent toxicity on an invertebrate and fish.
Methods
Cyanobacteria cultivation and materials
Two cyanobacteria strains, Synechococcus elongatus (UTEX 3055) and Microcystis aeruginosa (UTEX LB 3037), were incubated in a shaker incubator (Innova S44i, Eppendorf) with 10–30% photosynthetic light following our previous study.15 The bench-scale tests involved mesh-type (5 cm × 5 cm with double-sided coating; 0.5 × 0.8 mm rhombus openings) anode with either NATO or antimony tin oxide (ATO: Sb–SnO2) coatings. The NATO and ATO anodes were prepared by dip-coating titanium mesh into sol–gel solutions of metal-citrate complexes, followed by calcination.24 The precursor solution for ATO contained 360 mM SnCl4·5H2O (98%, Aldrich) and 40 mM SbCl3 (>99.0%, Aldrich), while that for NATO contained 360 mM SnCl4·5H2O, 15 mM SbCl3, and 4 mM Ni(OCOCH3)2·4H2O (98%, Aldrich). Metal salts were mixed with 1.2 mol L−1 of citric acid in ethylene glycol at 90 °C for 30 min to form stable sol–gels.The NATO anode comprises NATO out-layer coating (1 mg cm−2 projected area) and ATO base coating (1 mg cm−2 projected area) on Ti mesh, while the ATO anode only contains the ATO base coating. The dual-layer design of the NATO anode bestows the electrode with superior durability and O3 yield. Details were provided in our previous publication.24 The full-scale electrode array contains 20 pieces of mesh NATO anode (50 × 50 cm; 0.5 × 0.8 mm rhombus openings) sandwiched by 21 pieces of perforated stainless steel sheet cathode (50 × 50 cm; round opening at Ø 3 mm) at an interspace of 0.5 cm. The full-scale mesh NATO anodes were manufactured by Square One Coating Systems following the same sol–gel coating + calcination procedure described above.
Testing conditions
All tests were conducted in 95 mL phosphate buffer solution (PBS; pH = 7.7, conductivity = 329 μS cm−1) or lake water collected from Lake Neatahwanta, NY. The synthetic water or lake water was spiked with cyanobacteria culture to reach an initial concentration of 100 μg L−1 as chlorophyll-a (Chl-a). In bench-sale electrochemical treatment, a mesh anode (NATO or ATO; 5 × 5 cm) was coupled with a stainless steel cathode (5 × 5 cm) at a distance of 0.5 cm and operated at current densities of 7 or 10 mA cm−2. For the homogeneous ozonation treatment, O3 gas (generated from an A2Z ozone generator) was purged into 95 mL PBS solution till target concentrations (0.8–2.5 mg L−1) were reached. The O3 bubbling was stopped, and Synechococcus elongatus culture was spiked into the PBS solution to reach 100 μg Chl-a L−1. The reactor was then sealed from the air. Samples were taken within 300 s. The loss of dissolved O3 after the 300 s reaction was less than 20%.Analytical approaches
For the Chl-a measurement, water samples (5 mL) were passed through a 0.2 μm polycarbonate membrane filter. The filter was then extracted in 10 mL of 90% acetone. After placing in the dark at 4 °C for 8 h, the Chl-a extract was quantified by a fluorometer (TD-700, Turner Designs). The MC-LR in water was quantified by a UPLC system coupled with a triple-stage quadrupole mass spectrometer (MS/MS; Thermo Scientific, Vanquish-TSQ ALTIS) equipped with a Phenomenex Luna Omega PS C18 column (1.6 μm, 100 mm × 2.1 mm). The MS/MS was operated in electrospray in negative ion mode. In the tests involving Microcystis aeruginosa (an MC-LR toxin-producing strain), MC-LR in the bulk solution was defined as extracellular MC-LR. Intracellular MC-LR within the Microcystis aeruginosa cells was extracted by acetone after retaining cells by filtering the water sample through a polycarbonate membrane with a pore size of 0.2 μm.Free chlorine was measured by a portable Hach DR900 colorimeter (HACH, CO) using a DPD (N,N-diethyl-p-phenylenediamine) reagent. Dissolved O3 concentrations were determined by the indigo method on a NanoDrop OneC spectrophotometer (Thermo Fisher Scientific).26 When measuring electrochemical O3 evolution in the presence of chloride, the anodically generated free chlorine was masked by 1 M malonic acid.24 Trihalomethanes (THMs) and haloacetic acids (HAAs) were measured by gas chromatography/mass spectrometry (GC/MS) following EPA Method 624.1. Inorganic anions were analyzed by ion chromatography (Thermo Dionex Integrion HPIC).27 The whole effluent toxicity tests on Pimephales promelas and Ceriodaphnia dubia were independently conducted by AquaTOX Research, Inc., following EPA Methods 2000.0 and 2002.0, respectively (EPA-821-R-02-013).
Results and discussion
Electrode characterization
Fig. 1a shows the cyclic voltammetric (CV) profiles of NATO and ATO. The anodic potential (Ea) was corrected for the ohmic drop between the reference electrode and anode by considering the product of current (i) and uncompensated resistance (Ru). The measured value was converted to the reversed hydrogen electrode scale (RHE) to account for the pH effect using the equation below.28| ERHE = Emeasured + 0.0591 × pH | (1) |
 | ||
| Fig. 1 (a) Cyclic voltammetry of ATO and NATO anodes measured in 100 mM NaClO4 electrolyte (pH = 7). The intercept of the dashed lines with the x-axis represents the onset potential. (b) Dissolved O3 produced by NATO in 95 mL PBS electrolyte (pH 7.7), PBS electrolyte amended 1 mM Cl− or Synechococcus elongatus culture (initial Chl-a concentration of 100 μg L−1), and lake water (pH 7.8) collected from Lake Neatawantah. Data in Fig. 1b are presented as the mean value of triplicate ± standard deviation. | ||
We adopted the Ni dopant level of 1 at% (Ni/(Ni + Sb + Sn)) as optimized in previous studies.29,30 The Ni doping shifts the CV hysteresis toward lower potentials. The shift of onset potentials from 2.48 V for ATO to 2.38 V for NATO and the rise of the current response can be attributed to the enhanced evolution of oxygen and O3.20 In the lake water treatment, the mesh electrodes were operated at current densities between 6–10 mA cm−2 projected area, which leads to anodic potentials around 2.6–2.7 VRHE (Fig. 1a), surpassing the thermodynamic criteria for ·OH radical evolution (·OH/H2O: 2.7 VNHE).31
Electrolysis using ATO could not generate O3. In contrast, the NATO generated dissolved O3 in PBS electrolyte in the presence or absence of Cl− (Fig. 1b). The evolution rate and current efficiency in PBS electrolyte at 10 mA cm−2 are 0.0078 mmol m−2 s−1 and 4.5%, respectively. Chlorine evolution capability was tested in PBS electrolyte amended with 1 mM Cl−, a typical Cl− concentration found in lake waters treated in this study (Table S1†). NATO and ATO demonstrated comparable chlorine evolution rates of 0.022 and 0.024 mmol m−2 s−1, respectively, at 10 mA cm−2 (Fig. S1†). The electrochemical reactive surface area (ECSA) of the NATO mesh anode was measured by the double-layer capacitance method.32 The capacitance is measured as 16.64 mF, corresponding to an ECSA of 443.6 cm2 (Fig. S2†). The estimated ECSA is 8.9 times larger than the projected surface area (50 cm2).
The stability of the NATO electrode was previously investigated by the accelerated lifetime test.24 According to the established lifetime prediction model, the NATO anode operated at the optimum current density of 7 mA cm−2 (see below) has an expected lifetime of 9800 h. The leaching of metal ions, for example, Sb, is only significant (<0.05% loss of the mass loading) after 90 min electrolysis.24 This explains the results observed in this study that Ni, Sb, and Sn were not detected (by inductively coupled plasma mass spectrometry with a detection limit of 1 μg L−1) after 3 min electrolysis when >99% Chl-a was achieved (to be discussed in the next section and Fig. 2).
 | ||
| Fig. 2 Inactivation of Synechococcus elongatus by electrolysis using (a) ATO and NATO anodes in PBS electrolyte, (b) NATO anode in different electrolytes. (c) Comparison of Chl-a destruction kinetics of NATO and ATO in the presence of 1 mM Cl−. (d) Comparison of Chl-a destruction by ECO and homogeneous ozonation with various initial O3 dosages (0.8 and 1.5 mg L−1 dissolved O3). All tests were conducted in 95 mL PBS (pH 7.7) amended with Synechococcus elongatus at an initial Chl-a concentration of 100 μg L−1 at 10 mA cm−2. Throughout the electrolysis, pH was not changed. Data are presented as the mean value of triplicate ± standard deviation. | ||
Bench-scale investigation
The treatment performance of mesh-type ATO and NATO was evaluated based on the inactivation efficiency of Synechococcus elongatus and Microcystis aeruginosa, two strains of common cyanobacteria in algal blooms.33,34 These two strains have similar Gram-negative cell wall structures.35Synechococcus elongatus does not produce toxins. It was used in most bench-scale tests to study the inactivation kinetics without the potential interference of MC-LR. The optimized reaction conditions were then applied to treat electrolyte spiked with Microcystis aeruginosa culture with co-existing MC-LR.This study uses extracted chlorophyll-a (Chl-a) fluorescence as an indicator of cyanobacteria viability. Chl-a cannot maintain its structural integrity and light-adsorbing properties after cell damage. It is commonly used as an indicator to quantify the viable cell concentration.36–39 We demonstrated clear linear correlations between optical density (a standard index for estimating the concentration of cells in a suspension) and Chl-a concentrations in Synechococcus elongatus and Microcystis aeruginosa cultures (Fig. S3†). These data support the feasibility of using Chl-a concentration to represent viable cell density. More importantly, under the context of lake water remediation, Chl-a concentration is a straightforward algae proxy adopted by the World Health Organization and regulators. For example, the New York State Department of Environmental Conservation (NYSDEC) uses pigment concentration >25 μg L−1 as one of the criteria of HAB rather than cell density.40 Therefore, we use the degradation of Chl-a as the performance metric for HAB mitigation.
The tests of inactivation of Synechococcus elongatus were first performed in PBS electrolyte (pH = 7.7 throughout the reactions) at a current density of 10 mA cm−2. Note that both ATO and NATO have sufficiently high anodic potentials of ∼2.7 VRHE to produce ·OH (Fig. 1a). It was found that ATO showed negligible reactivity in Chl-a removal, and NATO noticeably outperformed ATO (Fig. 2a). The poor performance of ATO suggests that surface-bound ·OH generated from the Sb–SnO2 sites is less efficient for algae inactivation. Given that both NATO and ATO share Sb–SnO2 as the primary catalyst component, except NATO containing an additional 1 mol% Ni dopant, the yield of surface-bound ·OH on the Sb–SnO2 sites of NATO should be comparable to that of ATO. However, NATO possesses the additional ability to produce O3, which can be partially transformed into ·OH in the water phase. Consequently, the superior performance of NATO should be attributed to the combined effects of O3 and its derivative ·OH.
O3 played a key role in algae inactivation, evidenced by the consumption of O3 (i.e., lower residual O3 concentrations) during the electrolysis in PBS electrolyte spiked with Synechococcus elongatus (Fig. 1b). We believe that both O3 and derivative ·OH contributed to algae inactivation, but it is challenging to quantify their contribution. This is because the conversion of O3 to ·OH is only pronounced when pH > 9,23 which means O3 and ·OH co-exist in most of the treatment scenarios (pH 7–8) demonstrated in this study. Moreover, the ECO process employs a distinct O3 dosing mechanism compared to ozonation. Thus, the strategy of adding a radical quencher to investigate direct oxidation by O3 alone is not suitable for investigating the ECO process. Specifically, the O3 evolution on NATO requires the combination of surface-bound ·OH with O2 to form ·HO3, then O3.20,41 Therefore, the addition of tert-butyl alcohol (TBA) to scavenge ·OH also halted the O3 production (below the detection limit of 0.1 mg L−1) and consequently Chl-a removal by NATO was retarded in the presence of 100 mM TBA (Fig. 2b). Though we were unable to determine whether O3 and secondary product ·OH contributed the most, the electrochemical production of O3 is the driving force of the treatment process. Therefore, in the following content, we will continue to use the term ECO to distinguish this process from other radical-mediated advanced oxidation processes.
In the presence of Cl−, NATO could produce free chlorine and O3. If chlorine made a significant contribution to Chl-a removal, then the Chl-a removal kinetics in PBS with Cl− should be faster than PBS without Cl−. However, our observation suggests otherwise. The presence of up to 3 mM Cl− (a high record in lakes of the United States42) did not further accelerate Chl-a removal (Fig. 2b; w/ Cl−vs. PBS). Previously, we determined the pseudo-first-order rate constant of Chl-a in Synechococcus elongatus and free chlorine as 0.027 L (mg−1 min−1).43 With the presence of 1 mM Cl−, the time-average free chlorine concentrations produced by NATO at 10 mA cm−2 within 120 s electrolysis should be ∼7 mg L−1 (Fig. S1†). If free chlorine is responsible for Chl-a removal, the half-life of the reaction (t1/2) should be 220 s instead of the 48 s shown in Fig. 2b. Therefore, we conclude that even in the presence of chloride, O3 and derivative ·OH are the two primary reactive species for cyanobacteria inactivation by NATO. The treatment performance of ATO can be enhanced in the presence of Cl− due to the production of free chlorine. However, as shown in Fig. 2c, a comparison of Chl-a removal kinetics in PBS with 1 mM Cl− at 10 mA cm−2 indicates that NATO (t1/2 ≈ 60 s) still significantly outperforms ATO (t1/2 ≈ 160 s).
The efficacy of ECO was compared with homogeneous ozonation (Fig. 2d). The homogeneous ozonation at 1.5 mg L−1 O3 achieved Chl-a removal kinetics similar to ECO. In contrast, ECO generated 0.48 mg L−1 O3 at 120 s, corresponding to a time-average [O3] of 0.24 mg L−1. ECO produced less residual bulk [O3] (0.24 vs. 1.5 mg L−1) to realize faster Chl-a removal kinetics than homogeneous ozonation. Therefore, the faster cyanobacteria inactivation could be attributed to the locally concentrated O3 produced at the vicinity of the NATO anode, and the regional O3 concentration is equivalent to ∼1.5 mg L−1.
The ECO treatment of two cyanobacteria strains individually spiked in PBS electrolyte with 1 mM Cl− shows that Microcystin aeruginosa and Synechococcus elongatus have similar degradation kinetics (Fig. 3a). This similarity is attributed to their identical Gram-negative cell wall structures,35 suggesting that the inactivation mechanism is likely due to cell wall damage. After elucidating the reaction mechanism, the treatment tests were operated in more field-like scenarios. In most of the field tests, the ECO full-scale system was operated at 7 mA cm−2 (see discussion below). ECO at 7 mA cm−2 is equally efficient for the inactivation of Microcystis aeruginosa (Fig. 3b) and Synechococcus elongatus (Fig. S4†) in PBS amended with 1 mM Cl−. The treatment was further operated in Lake Neatahwanta water (collected before the HAB season and filtered to exclude background cyanobacteria and MC-LR) spiked with Microcystis aeruginosa. The Chl-a removal kinetics were slightly retarded by the water matrices, but the >90% removal efficiency can still be achieved after 180 s electrolysis.
 | ||
| Fig. 3 (a) Comparison of degradation kinetics of Microcystis aeruginosa and Synechococcus elongatus in PBS amended with 1 mM Cl− at 10 mA cm−2. (b) Inactivation of Microcystis aeruginosa and (c) destruction of intracellular (In) and extracellular (Ex) MC-LR by electrolysis using NATO anode in 95 mL PBS amended with 1 mM Cl− or 95 mL Lake Neatahwanta (Lake-N) water at 7 mA cm−2. The initial Chl-a concentration for both Microcystis aeruginosa and Synechococcus elongatus is 100 μg L−1. The initial concentrations of intracellular and extracellular MC-LR in PBS electrolyte are 1 and 3 μg L−1, respectively. The PBS electrolyte has a pH reading of 7.7 throughout the electrolysis. The pH values of Lake-N before and after treatment are 7.8 and 7.5, respectively. Data are presented as the mean value of triplicate ± standard deviation. | ||
The lake water and PBS electrolyte spiked with Microcystis aeruginosa culture contains both intra- and extracellular MC-LR. In both media, intra-cellular MC-LR was degraded slower than extra-cellular MC-LR (Fig. 3c). This is because the destruction of intra-cellular MC-LR requires the cell structure to be broken down first.
The bench-scale treatability study concluded that the ECO process is efficient for the removal of cyanobacteria and MC-LR at minute-level retention time. The remaining roadblock toward field development is understanding the environmental implications of the ECO technology. Given the fast Chl-a degradation kinetics, we hypothesize that significant cyanobacteria inactivation could be achieved before the yield of byproducts. Concern about the formation of inorganic byproducts was first addressed in the bench-scale study under field-like conditions. Other environmental aspects (organic byproducts and eco-toxicity) were investigated in the field application (see the following content). Chlorate (ClO3−) was identified as the product stemming from the electrochemical oxidation of Cl−. Fig. S5† shows the evolution of ClO3− in PBS electrolyte amended with 1 mM Cl− and lake water containing ∼1 mM Cl− at 10 mA cm−2. The results indicate that the evolution of ClO3− was suppressed in the lake water compared with electrolysis in PBS electrolytes, possibly due to the depletion of free chlorine by reacting with matrice components. As will be discussed below, the full-scale ECO reactor was operated at a Chl-a removal efficiency of ∼40%, corresponding to a treatment duration of 60 s in batch mode. In this scenario, the [ClO3−] was below the detection limit (5 μg L−1) and the World Health Organization (WHO) guideline of 0.7 mg L−1.44
Bromate is a byproduct derived from the ozonation of bromide (Br−).45 We do not believe this is a concern in the lake water treatment in this study because Br− was not detected in the lake water of two test sites (Table S1†). To address the concern about the future applications in treating Br− containing water, we investigated the formation BrO3− in the ECO treatment of PBS electrolyte containing 0.2 mg L−1 Br−, a concentration reported in some surface water.46 The results show that, throughout the 180 s ECO treatment process at 7 mA cm−2, the dominant product is HOBr. Formation of BrO3− was not detected (Fig. S6†). This finding highlights the advantage of ECO over homogeneous ozonation as the former yields less bulk O3 to form BrO3−.
The bench-scale investigation concludes that (1) ECO effectively removes cyanobacteria and cyanotoxins at short, minute-level retention times; (2) the reaction is built upon the locally concentrated O3 produced at the NATO anode. The efficacy is independent of chloride concentration, which broadens the treatment scenarios in fresh waters.
Field applications
A boat-mount ECO system was developed for the scaled-up treatment of HAB-impacted lake water (Fig. 4a). The full-scale electrode array contains 20 pieces of mesh NATO anode (50 × 50 cm; 0.5 × 0.8 mm rhombus openings) sandwiched by 21 pieces of perforated stainless steel sheet cathode (50 × 50 cm; round opening at Ø 3 mm) at an interspace of 0.5 cm (Fig. 4b). The polycarbonate reactor has a 190 L effective liquid volume. Other on-board components include a custom-made DC power supply, generators, and a water pump. Algae plumes were captured by an intake pipe at an adjustable depth (0–1 m) underwater. An intake screen with a mesh opening size of 1 mm was installed on the intake port to prevent the entry of fish and other large aquatic life (Fig. S7†). Lake water was transferred from the pipe to the pump and pushed through the 190 L reactor at a flow rate of 378 L min−1 (i.e., treatment capacity of 544 m3 d−1), corresponding to a hydraulic retention time of 30 seconds. Lake water was treated as is without chemical addition.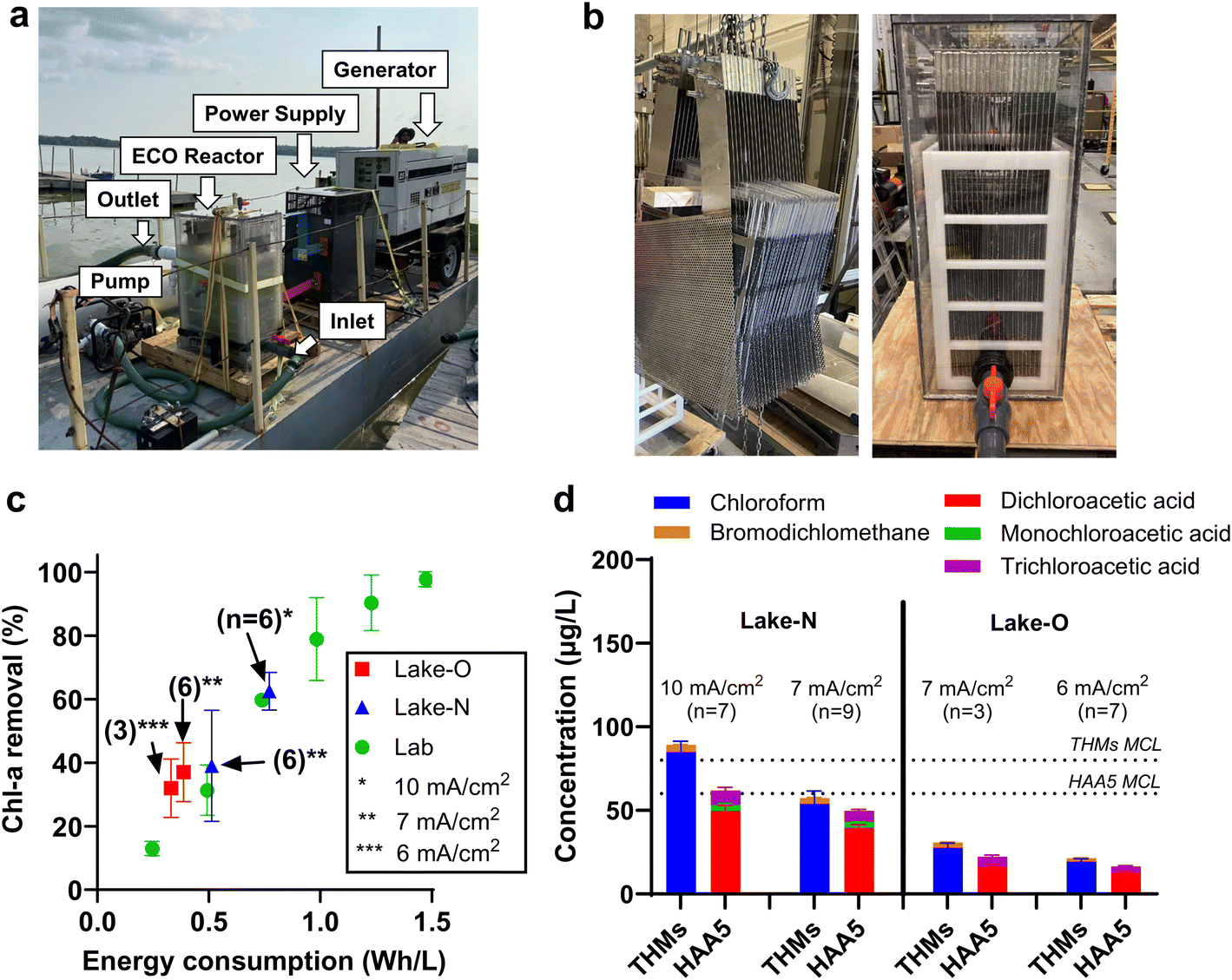 | ||
| Fig. 4 (a) Layout of the boat-mount ECO system. (b) Side view of ECO reactor with mesh electrodes installed. (c) Chl-a removal efficiencies benchmarked by energy consumption. Data were collected from lab-scale investigation and field tests performed in Lake Neatahwanta (Lake-N) and Oneida Lake Oneida (Lake-O). n is the number of pairs of inlet and outlet samples taken. The pH values of Lake-N (7.8) and Lake-O (8.3) did not change after treatment. (d) Trifluoromethanes (THMs) and five haloacetic acids (HAA5) in the treated Lake Neatahwanta effluent when the reactor was operated at 7 mA cm−2. MCL is the maximum contaminant level regulated by the U.S. EPA. | ||
The electrode array has a total projected anode area of 10 m2. Applying 600–1000 A total current on the electrode array resulted in current densities of 6–10 mA cm−2 with a cell voltage ranging from 12–20 V. In operation, water passing through the electrode mesh incurred efficient mass transfer. We have conducted preliminary tests in the field to show that the Chl-a concentrations in samples collected from the mid-height sampling port (representing bulk concentration) and outlet were the same (Fig. S8†). Therefore, the reactor can be considered a completely mixed-flow reactor (CMFR).
Based on the bench-scale batch reactions (data set of “PBS w/ 1 mM Cl−” in Fig. 2b and S4†), the pseudo-first-order rate constants (kECO's) at 7 and 10 mA cm−2 are 0.026 and 0.061 s−1, respectively. When transforming the reaction from batch to CMFR, the Chl-a removal efficiency can be estimated by
| Removal efficiency (%) = (1 − C/C0) × 100% = 1 − 1/(1 + τkECO) | (2) |
The performance of the full-scale system was validated in the treatment of algal blooms that occurred in Lake Neatahwanta (43°18′30′′N 76°26′14′′W) and Oneida Lake (43°10′25′′N 75°55′50′′W) in New York state. Lake Neatahwanta was treated in the midst of an algal bloom with 105 μg L−1 Chl-a and 3 μg L−1 MC-LR, while tests on Oneida Lake happened in the early blooming stage with Chl-a concentration of 5.4 μg L−1 (Table S1†). Each field deployment lasted for one day, with 9 to 12 pairs of influent and effluent samples taken. Tests in Oneida Lake were performed at current densities of 6–7 mA cm−2, while those in Lake Neatahwanta were conducted at 7–10 mA cm−2. As shown in Fig. 4c, treatment operated at 7 and 10 mA cm−2 achieved Chl-a removal efficiency of 39% and 62%, respectively, approaching the predicted values based on the CMFR model. More importantly, plotting removal efficiency data against specific energy consumption (Wh L−1) shows that the field test performances align well with the bench-scale data set. The convergence of laboratory results and full-scale performance on Chl-a removal demonstrates the promising linear scalability of the ECO process in cyanobacteria inactivation using specific energy consumption as the key design parameter, given that the same anodes (i.e., NATO) are used.
Intracellular MC-LR was not found in the Lake Neatahwanta water samples. The treatment at 7 mA cm−2 led to an effluent extracellular MC-LR concentration (i.e., MC-LR resides in lake water) of 0.41 μg L−1 (vs. C0 = 3 μg L−1), corresponding to a destruction efficiency of 86%. Note that it is challenging to study the extracellular MC-LR degradation kinetics due to the rapid degradation in ECO within a minute. Nonetheless, the 86% destruction of MC-LR when 40% Chl-a degradation was obtained in the full-scale CMFR reactor is comparable with the results observed in the lab-scale batch reactor (>95% destruction of MC-LR when ∼40% Chl-a degradation was achieved, as shown in Fig. 3).
For all the field tests, the residual chlorine and O3 in the treated effluent are below the detection limits (both at 0.1 mg L−1). This is likely due to the short retention time of 30 seconds, which did not allow for the accumulation of residual oxidants in the continuous flow reactor. Additionally, the loss of oxidants during sample shipment is possible. Samples were transported from the boat to the shore for water analysis, and oxidant decay may have occurred during this transfer. To verify the generation of oxidants, we conducted supplementary tests at bench-scale by electrolyzing Lake Neatahwanta water at 7 and 10 mA cm−2 in batch mode (Fig. 1b). At a retention time of 30 seconds (equivalent to the hydraulic retention time of the field-scale CMFR), the O3 concentrations were measured to be 0.2 mg L−1 under 7 mA cm−2 and 0.4 mg L−1 under 10 mA cm−2. Chlorine concentrations were below the detection limit of 0.1 mg L−1 (Fig. S1b†). The O3 concentration range of 0.2–0.4 mg L−1 likely mirrors the residual O3 concentrations in the effluent immediately after exiting the reactor.
It is important to emphasize that the inactivation of algae should be achieved after a single pass through the CMFR due to the exposure of cells to locally concentrated O3 and ·OH around NATO anodes, as discussed above. The decay of 0.2–0.4 mg L−1 residual O3 during sample transportation is unlikely to contribute further to the treatment as our findings demonstrated that ozonation with O3 up to 0.8 mg L−1 did not noticeably remove Chl-a (Fig. 2d).
The current density of 7 mA cm−2 (total current of 700 A and cell voltage of 14 V in treating Lake Neatahwanta) was identified as the optimum condition, balancing the significant algae removal, low byproduct formation, and insignificant toxicity (discussed below). It is important to note that the ∼40% Chl-a removal at a short retention time of 30 s already incurred instant improvement in water clarity (Fig. S9†). Improvement in local water quality can be expected by the circulative treatment of water. The energy consumption of 0.5 Wh L−1 is also lower than the chlorine-based electrochemical HAB mitigation system (1.1 Wh L−1) reported in our previous study.43
Environmental implications
The formation of halogenated byproducts, including trihalomethanes (THMs) and five haloacetate acids (HAA5), was investigated in the treatment of Lake Neatahwanta and Oneida Lake. THMs and HAA5 were not detected in the influent samples. In the effluent samples, chloroform and dichloroacetic acid were the dominant THMs and HAAs, respectively. For treatment operated at current densities of 6 and 7 mA cm−2, the total concentrations of THMs and HAA5 were below the maximum contaminant level (MCL) values regulated by the U.S. EPA (Fig. 4d).Whole effluent toxicity tests were conducted to evaluate whether the effluent from the ECO process would negatively impact the aquatic life in the receiving water. Samples of influent (INF) and treated effluent (EFF) were collected during the operation in Lake Neatahwanta (Table S2†) and Oneida Lake (Table S3†) at 6–10 mA cm−2. In the toxicity tests, model freshwater invertebrate (Ceriodaphnia dubia) and fish (Pimephales promelas) species were exposed to INF samples, EFF samples, and lab control water (CON). Ten replicates were used for Ceriodaphnia dubia, and four replicates for Pimephales promelas for each INF, EFF, and CON exposure. Comparing the toxicity test results of samples obtained at 10 mA cm−2 (Table S2†), although Ceriodaphnia dubia survival was 100% at both 48 h (acute) and 6–7 days (chronic), reproduction is lower in EFF than INF, suggesting the treatment induced toxicity for the invertebrate species. Conversely, no effects were observed on the survival and growth of Pimephales promelas. The analysis of samples derived from operation at 6 and 7 mA cm−2 show that there were no significant survival impacts at either 48 h (acute) or 6–7 days (chronic) for either the invertebrate or fish species, with survival rates of 100%. Similarly, there were no significant effects on the reproduction of invertebrates or the growth of fish in EFF compared to INF. The whole effluent toxicity tests confirmed that the ECO process operated at 6 and 7 mA cm−2 did not induce acute or chronic toxicity in the treated effluent, echoing our conclusion that 7 mA cm−2 is the optimum current density. Lastly, it is important to note that the ECO process alone cannot remove nutrients. We are currently exploring the integration of separation processes, such as coagulation and filtration, as pretreatment steps to the ECO process. Incorporating these algae harvest processes may enable the treatment train to remove intracellular nutrients and reduce the recurrence of algal blooms.
Conclusions
This study developed an ECO process for the rapid inactivation of cyanobacteria and the destruction of MC-LR. The process used NATO anodes to generate locally concentrated O3 as the major reactive species. Compared with homogeneous ozonation, the ECO process achieved the same degree of treatment but yielded lower bulk [O3]. Note that a majority of electrochemical water treatment processes rely on the electrolysis of chloride to produce free chlorine as oxidants, and the treatment performance depends on the chloride concentration.47,48 The shift of the dominant oxidant from chlorine to O3 provides a new strategy for sustaining high treatment efficiency in freshwater with a limited chloride source and suppressing the formation of byproducts.The significant merit of the engineering of this study is the scaled-up application of the ECO process to solve real-world problems. The performance of batch mode lab-scale reactor was replicated in a boat-mounted CMFR reactor using specific energy consumption as a benchmark. In addition to the successful demonstration of HAB mitigation, we also evaluated whole effluent toxicity to show that the ECO process operated at the optimized current density of 7 mA cm−2 did not increase the effluent toxicity. This case study sets an example for the scaled-up application of electrified water treatment technology in environmental remediation.
Data availability
Source data for all graphs are available from the corresponding author upon request.Author contributions
S. Y.: data curation, formal analysis, investigation, visualization, original draft preparation. L. E. Q. C. and A. F.: formal analysis, investigation, visualization. N. S., V. M. C., and N. Y.: formal analysis, investigation. L. M.: funding acquisition, methodology, resources, review & editing. N. W.: methodology, review & editing. M. R. T.: methodology, resources, review & editing. S. W.: data curation, investigation, methodology, resources, supervision, review & editing. S. J. G.: funding acquisition, investigation, project administration, resources, supervision, review & editing. Y. Y.: conceptualization, funding acquisition, formal analysis, investigation, project administration, resources, supervision, review & editing.Conflicts of interest
The authors declare no competing interest.Acknowledgements
This research was supported by the New York State Center of Excellence in Healthy Water Solutions (C190175), New York State Department of Environmental Conservation (MOU# AM10700, ESF-PRJ-19-07) and Innovation Fund Manufacturing Grant (FuzeHub CON0002479). Opinions, results, findings and/or interpretations of data contained therein are the responsibility of the co-authors and do not necessarily represent the opinions, interpretations or policy of the State New York State. The New York State Department of Environmental Conservation funding does not imply endorsement of commercial products, currently or in the future. The lab-scale investigation was partially supported by the National Science Foundation (STTR Phase I-2321315). We thank Scott Hodge and Jacob Weller of the Clarkson University Machine Shop for assembling and modifying the boat-mounted ECO system. We thank Clarkson University graduate students Bryan Dwyer and Philip Hekeler for their assistance in field operations.References
- S. C. Chapra, B. Boehlert, C. Fant, V. J. Bierman, J. Henderson, D. Mills, D. M. L. Mas, L. Rennels, L. Jantarasami, J. Martinich, K. M. Strzepek and H. W. Paerl, Climate Change Impacts on Harmful Algal Blooms in U.S. Freshwaters: A Screening-Level Assessment, Environ. Sci. Technol., 2017, 51(16), 8933–8943, DOI:10.1021/acs.est.7b01498.
- H. W. Paerl and J. T. Scott, Throwing Fuel on the Fire: Synergistic Effects of Excessive Nitrogen Inputs and Global Warming on Harmful Algal Blooms, Environ. Sci. Technol., 2010, 44(20), 7756–7758, DOI:10.1021/es102665e.
- X. Xiao, S. Agustí, Y. Pan, Y. Yu, K. Li, J. Wu and C. M. Duarte, Warming Amplifies the Frequency of Harmful Algal Blooms with Eutrophication in Chinese Coastal Waters, Environ. Sci. Technol., 2019, 53(22), 13031–13041, DOI:10.1021/acs.est.9b03726.
- H. Dong, Md. T. Aziz and S. D. Richardson, Transformation of Algal Toxins during the Oxidation/Disinfection Processes of Drinking Water: From Structure to Toxicity, Environ. Sci. Technol., 2023, 57(35), 12944–12957, DOI:10.1021/acs.est.3c01912.
- P. Xie, J. Ma, J. Fang, Y. Guan, S. Yue, X. Li and L. Chen, Comparison of Permanganate Preoxidation and Preozonation on Algae Containing Water: Cell Integrity, Characteristics, and Chlorinated Disinfection Byproduct Formation, Environ. Sci. Technol., 2013, 47(24), 14051–14061, DOI:10.1021/es4027024.
- A. Zamyadi, Y. Fan, R. I. Daly and M. Prévost, Chlorination of Microcystis Aeruginosa: Toxin Release and Oxidation, Cellular Chlorine Demand and Disinfection by-Products Formation, Water Res., 2013, 47(3), 1080–1090, DOI:10.1016/j.watres.2012.11.031.
- L. Spoof, S. Jaakkola, T. Važić, K. Häggqvist, T. Kirkkala, A.-M. Ventelä, T. Kirkkala, Z. Svirčev and J. Meriluoto, Elimination of Cyanobacteria and Microcystins in Irrigation Water—Effects of Hydrogen Peroxide Treatment, Environ. Sci. Pollut. Res., 2020, 27(8), 8638–8652, DOI:10.1007/s11356-019-07476-x.
- B. Yu, Y. Zhang, H. Wu, W. Yan, Y. Meng, C. Hu, Z. Liu, J. Ding and H. Zhang, Advanced Oxidation Processes for Synchronizing Harmful Microcystis Blooms Control with Algal Metabolites Removal: From the Laboratory to Practical Applications, Sci. Total Environ., 2024, 906, 167650, DOI:10.1016/j.scitotenv.2023.167650.
- T. Kutser, Quantitative Detection of Chlorophyll in Cyanobacterial Blooms by Satellite Remote Sensing, Limnol. Oceanogr., 2004, 49(6), 2179–2189, DOI:10.4319/lo.2004.49.6.2179.
- T. S. Moore, J. H. Churnside, J. M. Sullivan, M. S. Twardowski, A. R. Nayak, M. N. McFarland, N. D. Stockley, R. W. Gould, T. H. Johengen and S. A. Ruberg, Vertical Distributions of Blooming Cyanobacteria Populations in a Freshwater Lake from LIDAR Observations, Remote Sens. Environ., 2019, 225, 347–367, DOI:10.1016/j.rse.2019.02.025.
- W. Liao, M. Murugananthan and Y. Zhang, Electrochemical Degradation and Mechanistic Analysis of Microcystin-LR at Boron-Doped Diamond Electrode, Chem. Eng. J., 2014, 243, 117–126, DOI:10.1016/j.cej.2013.12.091.
- W. Liang, J. Qu, L. Chen, H. Liu and P. Lei, Inactivation of Microcystis Aeruginosa by Continuous Electrochemical Cycling Process in Tube Using Ti/RuO2 Electrodes, Environ. Sci. Technol., 2005, 39(12), 4633–4639, DOI:10.1021/es048382m.
- Y. Zhou, H. Peng, L. Jiang, X. Wang, Y. Tang and L. Xiao, Control of Cyanobacterial Bloom and Purification of Bloom-Laden Water by Sequential Electro-Oxidation and Electro-Oxidation-Coagulation, J. Hazard. Mater., 2024, 462, 132729, DOI:10.1016/j.jhazmat.2023.132729.
- C. Huang, W. Huang, J. Xiong and S. Wang, Mechanism and Excellent Performance of Graphite Felt as Anodes in Electrochemical System for Microcystis Aeruginosa and Microcystin-LR Removal with No pH Limitation nor Chemical Addition, Sep. Purif. Technol., 2021, 277, 119502, DOI:10.1016/j.seppur.2021.119502.
- S. Yang, M. R. Twiss, S. Fernando, S. J. Grimberg and Y. Yang, Mitigation of Cyanobacterial Harmful Algal Blooms (cHABs) and Cyanotoxins by Electrochemical Oxidation: From a Bench-Scale Study to Field Application, ACS ES&T Eng., 2022, 2(7), 1160–1168, DOI:10.1021/acsestengg.1c00344.
- P. A. Christensen, T. Yonar and K. Zakaria, The Electrochemical Generation of Ozone: A Review, Ozone: Sci. Eng., 2013, 35(3), 149–167, DOI:10.1080/01919512.2013.761564.
- L. G. De Sousa, D. V. Franco and L. M. Da Silva, Electrochemical Ozone Production Using Electrolyte-Free Water for Environmental Applications, J. Environ. Chem. Eng., 2016, 4(1), 418–427, DOI:10.1016/j.jece.2015.11.042.
- K. Arihara, C. Terashima and A. Fujishima, Electrochemical Production of High-Concentration Ozone-Water Using Freestanding Perforated Diamond Electrodes, J. Electrochem. Soc., 2007, 154(4), E71, DOI:10.1149/1.2509385.
- J. T. Jasper, Y. Yang and M. R. Hoffmann, Toxic Byproduct Formation during Electrochemical Treatment of Latrine Wastewater, Environ. Sci. Technol., 2017, 51(12), 7111–7119, DOI:10.1021/acs.est.7b01002.
- P. A. Christensen, P. S. Attidekou, R. G. Egdell, S. Maneelok, D. A. C. Manning and R. Palgrave, Identification of the Mechanism of Electrocatalytic Ozone Generation on Ni/Sb-SnO2, J. Phys. Chem. C, 2017, 121(2), 1188–1199, DOI:10.1021/acs.jpcc.6b10521.
- P. A. Christensen, K. Zakaria and T. P. Curtis, Structure and Activity of Ni- and Sb–Doped SnO2 Ozone Anodes, Ozone: Sci. Eng., 2012, 34(1), 49–56, DOI:10.1080/01919512.2012.639687.
- J. A. Roth and D. E. Sullivan, Solubility of Ozone in Water, Ind. Eng. Chem. Fundam., 1981, 20(2), 137–140, DOI:10.1021/i100002a004.
- J. Staehelin and J. Hoigne, Decomposition of Ozone in Water: Rate of Initiation by Hydroxide Ions and Hydrogen Peroxide, Environ. Sci. Technol., 1982, 16(10), 676–681, DOI:10.1021/es00104a009.
- Y. Zhang, Y. Yang, S. Yang, E. Quispe-Cardenas and M. R. Hoffmann, Application of Heterojunction Ni–Sb–SnO2 Anodes for Electrochemical Water Treatment, ACS ES&T Eng., 2021, 1(8), 1236–1245, DOI:10.1021/acsestengg.1c00122.
- S. Y. Yang, D. Kim and H. Park, Shift of the Reactive Species in the Sb–SnO2-Electrocatalyzed Inactivation of E. Coli and Degradation of Phenol: Effects of Nickel Doping and Electrolytes, Environ. Sci. Technol., 2014, 48(5), 2877–2884, DOI:10.1021/es404688z.
- H. Bader and J. Hoigné, Determination of Ozone in Water by the Indigo Method, Water Res., 1981, 15(4), 449–456, DOI:10.1016/0043-1354(81)90054-3.
- S. Yang, S. Fernando, T. M. Holsen and Y. Yang, Inhibition of Perchlorate Formation during the Electrochemical Oxidation of Perfluoroalkyl Acid in Groundwater, Environ. Sci. Technol. Lett., 2019, 6(12), 775–780, DOI:10.1021/acs.estlett.9b00653.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, 2nd edn, Wiley, New York, 2000 Search PubMed.
- P. A. Christensen, K. Zakaria, H. Christensen and T. Yonar, The Effect of Ni and Sb Oxide Precursors, and of Ni Composition, Synthesis Conditions and Operating Parameters on the Activity, Selectivity and Durability of Sb-Doped SnO2 Anodes Modified with Ni, J. Electrochem. Soc., 2013, 160(8), H405, DOI:10.1149/2.023308jes.
- S. Y. Yang, Y. S. Choo, S. Kim, S. K. Lim, J. Lee and H. Park, Boosting the Electrocatalytic Activities of SnO2 Electrodes for Remediation of Aqueous Pollutants by Doping with Various Metals, Appl. Catal., B, 2012, 111–112, 317–325, DOI:10.1016/j.apcatb.2011.10.014.
- Y. Yang, Recent Advances in the Electrochemical Oxidation Water Treatment: Spotlight on Byproduct Control, Front. Environ. Sci. Eng., 2020, 14(5), 85, DOI:10.1007/s11783-020-1264-7.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, Benchmarking Heterogeneous Electrocatalysts for the Oxygen Evolution Reaction, J. Am. Chem. Soc., 2013, 135(45), 16977–16987, DOI:10.1021/ja407115p.
- J. Li, Z. Chen, Z. Jing, L. Zhou, G. Li, Z. Ke, X. Jiang, J. Liu, H. Liu and Y. Tan, Synechococcus Bloom in the Pearl River Estuary and Adjacent Coastal Area–With Special Focus on Flooding during Wet Seasons, Sci. Total Environ., 2019, 692, 769–783, DOI:10.1016/j.scitotenv.2019.07.088.
- A. D'ors, M. C. Bartolomé and S. Sánchez-Fortún, Toxic Risk Associated with Sporadic Occurrences of Microcystis Aeruginosa Blooms from Tidal Rivers in Marine and Estuarine Ecosystems and Its Impact on Artemia Franciscana Nauplii Populations, Chemosphere, 2013, 90(7), 2187–2192, DOI:10.1016/j.chemosphere.2012.11.029.
- J. Šmarda, D. Šmajs, J. Komrska and V. Krzyžánek, S-Layers on Cell Walls of Cyanobacteria, Micron, 2002, 33(3), 257–277, DOI:10.1016/S0968-4328(01)00031-2.
- K. Schulze, D. A. López, U. M. Tillich and M. Frohme, A Simple Viability Analysis for Unicellular Cyanobacteria Using a New Autofluorescence Assay, Automated Microscopy, and ImageJ, BMC Biotechnol., 2011, 11(1), 118, DOI:10.1186/1472-6750-11-118.
- F. Sun, H.-Y. Pei, W.-R. Hu and M.-M. Song, A Multi-Technique Approach for the Quantification of Microcystis Aeruginosa FACHB-905 Biomass during High Algae-Laden Periods, Environ. Technol., 2012, 33(15), 1773–1779, DOI:10.1080/09593330.2011.644868.
- L. J. Hazeem, G. Kuku, E. Dewailly, C. Slomianny, A. Barras, A. Hamdi, R. Boukherroub, M. Culha and M. Bououdina, Toxicity Effect of Silver Nanoparticles on Photosynthetic Pigment Content, Growth, ROS Production and Ultrastructural Changes of Microalgae Chlorella Vulgaris, Nanomaterials, 2019, 9(7), 914, DOI:10.3390/nano9070914.
- Y. Song, Z. Li, A. Feng, J. Zhang, Z. Liu and D. Li, Electrokinetic Detection and Separation of Living Algae in a Microfluidic Chip: Implication for Ship's Ballast Water Analysis, Environ. Sci. Pollut. Res., 2021, 28(18), 22853–22863, DOI:10.1007/s11356-020-12315-5.
- Harmful Algal Blooms (HABS) Program Guide, https://extapps.dec.ny.gov/docs/water_pdf/habsprogramguide.pdf, (accessed 2024-06-07) Search PubMed.
- Y.-H. Wang and Q.-Y. Chen, Anodic Materials for Electrocatalytic Ozone Generation, Int. J. Electrochem., 2013, 2013, 1–7, DOI:10.1155/2013/128248.
- H. A. Dugan, S. L. Bartlett, S. M. Burke, J. P. Doubek, F. E. Krivak-Tetley, N. K. Skaff, J. C. Summers, K. J. Farrell, I. M. McCullough, A. M. Morales-Williams, D. C. Roberts, Z. Ouyang, F. Scordo, P. C. Hanson and K. C. Weathers, Salting Our Freshwater Lakes, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(17), 4453–4458, DOI:10.1073/pnas.1620211114.
- S. Yang, M. R. Twiss, S. Fernando, S. J. Grimberg and Y. Yang, Mitigation of Cyanobacterial Harmful Algal Blooms (cHABs) and Cyanotoxins by Electrochemical Oxidation: From a Bench-Scale Study to Field Application, ACS ES&T Eng., 2022, 2(7), 1160–1168, DOI:10.1021/acsestengg.1c00344.
- World Health Organization, Chlorite and Chlorate in Drinkingwater, WHO Guidelines for Drinkingwater Quality; WHO/SDE/WSH/05.08/86, 2005 Search PubMed.
- B. Legube, B. Parinet, K. Gelinet, F. Berne and J.-P. Croue, Modeling of Bromate Formation by Ozonation of Surface Waters in Drinking Water Treatment, Water Res., 2004, 38(8), 2185–2195, DOI:10.1016/j.watres.2004.01.028.
- Bromide in Surface Waters, Capps Lab at UGA, http://cappslab.ecology.uga.edu/additional-info/bromide-in-surface-water/, (accessed 2023-10-13) Search PubMed.
- Y. Yang, J. Shin, J. T. Jasper and M. R. Hoffmann, Multilayer Heterojunction Anodes for Saline Wastewater Treatment: Design Strategies and Reactive Species Generation Mechanisms, Environ. Sci. Technol., 2016, 50(16), 8780–8787, DOI:10.1021/acs.est.6b00688.
- K. Cho, Y. Qu, D. Kwon, H. Zhang, C. A. Cid, A. Aryanfar and M. R. Hoffmann, Effects of Anodic Potential and Chloride Ion on Overall Reactivity in Electrochemical Reactors Designed for Solar-Powered Wastewater Treatment, Environ. Sci. Technol., 2014, 48(4), 2377–2384, DOI:10.1021/es404137u.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ew00490f |
| This journal is © The Royal Society of Chemistry 2024 |