
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and derivatization of epoxy-functional sterically-stabilized diblock copolymer spheres in non-polar media: does the spatial location of the epoxy groups matter?†
Csilla
György
a,
Timothy
Smith
b,
David J.
Growney
b and
Steven P.
Armes
 *a
*a
aDainton Building, Department of Chemistry, The University of Sheffield, Brook Hill, Sheffield, South Yorkshire S3 7HF, UK. E-mail: s.p.armes@shef.ac.uk
bLubrizol Ltd, Nether Lane, Hazelwood, Derbyshire DE56 4AN, UK
First published on 10th June 2022
Abstract
Epoxy-functional sterically-stabilized diblock copolymer spherical nanoparticles were synthesized via polymerization-induced self-assembly (PISA) in mineral oil. Epoxy groups were located either (i) in the nanoparticle cores or (ii) within the steric stabilizer chains. For the first system, reversible addition–fragmentation chain transfer (RAFT) dispersion polymerization of glycidyl methacrylate (GlyMA) was conducted using a poly(lauryl methacrylate) (PLMA63) precursor. The second system involved statistical copolymerization of GlyMA with lauryl methacrylate to produce a P(LMA50-stat-GlyMA9) precursor followed by chain extension using methyl methacrylate (MMA). 1H NMR studies and THF GPC analysis indicated that high monomer conversions (≥ 95%) and narrow molecular weight distributions (Mw/Mn ≤ 1.17) were obtained for both formulations. Dynamic light scattering indicated hydrodynamic diameters of 26 nm and 28 nm for P(LMA50-stat-GlyMA9)-PMMA67 and PLMA63-PGlyMA89 spheres, respectively. Transmission electron microscopy studies confirmed a well-defined spherical morphology in each case. Post-polymerization modification of these spherical nanoparticles was examined by reacting the epoxy groups with benzylamine. For the PLMA63-PGlyMA89 spheres, an [amine]/[epoxy] molar ratio of unity was sufficient to react all the epoxy groups. In contrast, the P(LMA50-stat-GlyMA9)-PMMA67 spheres required a fifty-fold excess of benzylamine for complete reaction. Furthermore, epoxy ring-opening reactions were conducted using either a trace amount of water or 50% v/v aqueous acetic acid at 110 °C. The extent of reaction was assessed using 1H NMR spectroscopy and THF GPC for the P(LMA50-stat-GlyMA9)-PMMA67 spheres and by FT-IR spectroscopy for the core-crosslinked PLMA63-PGlyMA89 spheres.
Introduction
Over the past two decades or so, reversible addition–fragmentation chain transfer (RAFT) polymerization has proven to be a powerful radical-based technique for the synthesis of a wide range of functional vinyl polymers.1–4 It is particularly useful in the context of polymerization-induced self-assembly (PISA), which is a powerful platform technology for the rational design of a wide range of diblock copolymer nano-objects.5–10 PISA typically involves growing a second block from one end of a soluble block in a suitable solvent such that the growing second block becomes insoluble in situ. Initially, this leads to micellar nucleation and eventually sterically-stabilized nano-objects are obtained. PISA is remarkably generic: robust formulations have been developed for various media, including water,5,11–19 polar solvents7,20–30 and non-polar solvents.31–59 In the case of dispersion polymerization formulations, using a relatively long stabilizer block typically produces kinetically-trapped spheres, whereas a relatively short stabilizer block combined with a relatively long insoluble block leads to the formation of vesicles. Highly anisotropic worm-like particles can also be obtained but this elusive morphology typically occupies relatively narrow phase space.Given the exceptional tolerance of RAFT polymerization towards monomer functionality, many examples of functional nanoparticles have been prepared using PISA.60–69 Of particular relevance to the present work is the use of glycidyl methacrylate (GlyMA) to design epoxy-functional nanoparticles.37,42,70–86 In principle, such nanoparticles can be derivatized by ring-opening the epoxy groups using nucleophiles such as water,87–89 amines,76,82,86,89–92 thiols72,76,82,89,92,93 carboxylic acids,94,95 azides86,89,96 or organosilyl reagents.97
The preparation of epoxy-functional spheres, worms or vesicles via RAFT aqueous emulsion polymerization of GlyMA has been reported by both Armes and co-workers and Tan et al.71,73,75,77,78,86,98 For such PISA formulations, premature loss of the epoxy groups via ring-opening attack by water can be minimized by choosing appropriate mild reaction conditions (e.g. neutral pH, moderate reaction temperatures of 20–50 °C) and ensuring short reaction times. Similarly, we have reported the facile synthesis of epoxy-functional diblock copolymer nano-objects directly in mineral oil.37,42 In this case, the survival of the epoxy groups is much less sensitive to the reaction conditions. Nevertheless, trace amounts of protic impurities can lead to intermolecular branching/crosslinking during the long-term storage of such copolymer dispersions at ambient temperature.37 Typically, the epoxy groups are located within the nanoparticle cores but an alternative strategy is to incorporate the epoxy groups within the steric stabilizer chains. In principle, this can be achieved either by using an appropriate epoxy-functional RAFT agent72 or by statistical copolymerization of a small amount of GlyMA with a suitable oil-soluble methacrylic comonomer.72,82,83
Herein we use PISA to prepare epoxy-functional sterically-stabilized diblock copolymer spheres directly in mineral oil whereby the epoxy groups are located either (i) within the nanoparticle cores or (ii) within the steric stabilizer chains, see Scheme 1. Both types of nanoparticles are characterized by 1H NMR spectroscopy, THF GPC, TEM and DLS. Subsequently, epoxy ring-opening reactions are conducted using either benzylamine or water. In particular, we demonstrate that the reaction conditions required to achieve high extents of reaction are highly dependent on the spatial location of the epoxy groups.
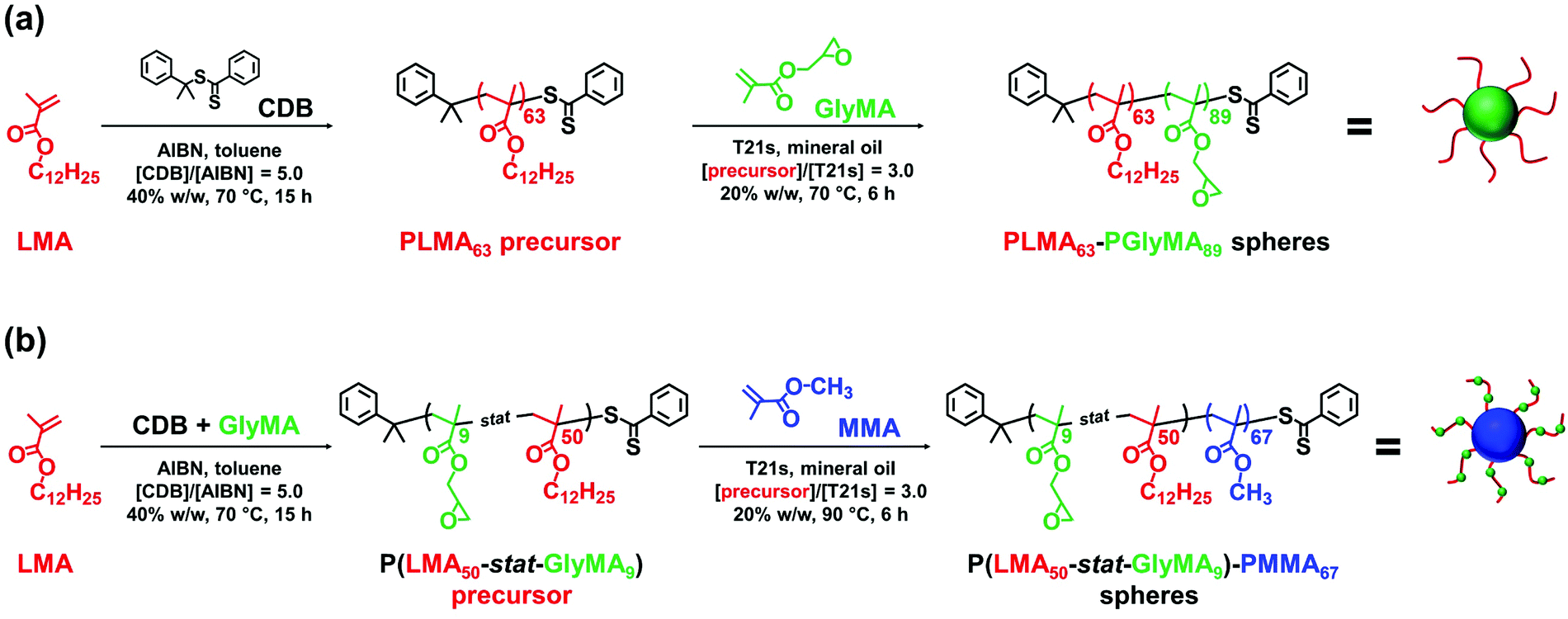 | ||
| Scheme 1 (a) Synthesis of poly(lauryl methacrylate) (PLMA63) via RAFT solution polymerization of LMA in anhydrous toluene at 40% w/w solids using cumyl dithiobenzoate (CDB) RAFT agent and 2,2′-azoisobutyronitrile (AIBN) initiator at 70 °C. This precursor was then chain-extended by RAFT dispersion polymerization of glycidyl methacrylate (GlyMA) using a tert-butyl peroxy-2-ethylhexanoate (T21s) initiator at 70 °C in mineral oil and targeting 20% w/w solids. (b) Synthesis of a poly(lauryl methacrylate-stat-glycidyl methacrylate) [P(LMA50-stat-GlyMA9)] precursor via RAFT solution copolymerization of LMA with GlyMA in anhydrous toluene at 40% w/w solids using a CDB RAFT agent and AIBN initiator at 70 °C, followed by the RAFT dispersion polymerization of methyl methacrylate (MMA) at 90 °C in mineral oil using T21s initiator and targeting 20% w/w solids. | ||
Experimental
Materials
Methyl methacrylate (MMA, 99%) was purchased from Alfa Aesar (Germany), passed through basic alumina to remove its inhibitor and then stored at −20 °C prior to use. Lauryl methacrylate (LMA, 96%), glycidyl methacrylate (GlyMA, 97%), cumyl dithiobenzoate (CDB), benzylamine (99%), N-methylaniline (99%), 2-(dimethylamino)propylamine (99%), triethylenetetramine (≥ 97%), 1-butanethiol (99%), CDCl3 and n-dodecane were purchased from Merck (UK) and used as received. 2,2′-Azoisobutyronitrile (AIBN) was obtained from Molekula (UK) and tert-butyl peroxy-2-ethylhexanoate (T21s) was purchased from AkzoNobel (The Netherlands). CD2Cl2 was purchased from Goss Scientific (UK). Tetrahydrofuran was obtained from VWR Chemicals (UK). Methanol, acetic acid (99%) and toluene were purchased from Fisher Scientific (UK). Group III hydroisomerized mineral oil (viscosity = 4.3 cSt at 100 °C) was kindly provided by Lubrizol Ltd (Hazelwood, Derbyshire, UK).Synthesis of PLMA63 precursor via RAFT solution polymerization in toluene
A PLMA63 precursor was synthesized at 40% w/w solids according to a previously reported synthesis protocol.43 Briefly, this synthesis was conducted as follows. LMA (77.0 g; 302.8 mmol), CDB (1.50 g; 5.5 mmol; target DP = 55), AIBN (181 mg; 1.10 mmol; CDB/AIBN molar ratio = 5.0) and anhydrous toluene (118.1 g) were weighed into a 250 mL round-bottomed flask. The sealed flask was purged with nitrogen for 30 min and immersed in a preheated oil bath at 70 °C. The reaction solution was stirred continuously, and the ensuing polymerization was quenched after 15 h by exposing the reaction solution to air while cooling the flask to room temperature. A final LMA conversion of 90% was determined by 1H NMR spectroscopy. The crude polymer was purified by three consecutive precipitations into a ten-fold excess of methanol (with redissolution in THF after each precipitation). The mean DP of this PLMA precursor was calculated to be 63 using 1H NMR spectroscopy by comparing the ten aromatic protons assigned to the cumyl and dithiobenzoate end-groups at 7.10–8.00 ppm to the two oxymethylene protons attributed to PLMA at 3.75–4.20 ppm. THF GPC analysis using a refractive index detector and a series of near-monodisperse poly(methyl methacrylate) calibration standards indicated an Mn of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 g mol−1 and an Mw/Mn of 1.19.
600 g mol−1 and an Mw/Mn of 1.19.
Synthesis of P(LMA50-stat-GlyMA9) precursor via RAFT solution polymerization in toluene
The P(LMA50-stat-GlyMA9) precursor was synthesized at 40% w/w solids as follows. LMA (63.0 g; 247.8 mmol), GlyMA (5.5 g; 38.5 mmol), CDB (1.50 g; 5.50 mmol; target DP = 52), AIBN (181 mg; 1.10 mmol; CDB/AIBN molar ratio = 5.0) and anhydrous toluene (105.3 g) were weighed into a 250 mL round-bottomed flask. Anhydrous toluene was used to minimize the potential loss of epoxy groups via ring-opening with water. The sealed flask was purged with nitrogen for 30 min and immersed in a preheated oil bath at 70 °C. The reaction solution was stirred continuously, and the ensuing polymerization was quenched after 15 h by exposing the reaction solution to air while cooling the flask to room temperature. An overall comonomer conversion of 90% was determined by 1H NMR spectroscopy. The crude copolymer was purified by three consecutive precipitations into a ten-fold excess of methanol (with redissolution in THF after each precipitation). The overall mean DP of this P(LMA-stat-GlyMA) precursor was calculated to be 59 (with 50 LMA units and 9 GlyMA units per copolymer chain), by using 1H NMR spectroscopy to compare the ten aromatic protons assigned to the cumyl and dithiobenzoate end-groups at 7.10–8.00 ppm to the two oxymethylene protons attributed to the LMA repeat units at 3.85–4.20 ppm and the methine proton corresponding to the epoxide ring at 3.10–3.30 ppm, respectively. THF GPC analysis using a refractive index detector and a series of near-monodisperse poly(methyl methacrylate) calibration standards indicated an Mn of 12300 g mol−1 and an Mw/Mn of 1.19.
Synthesis of poly(lauryl methacrylate)-poly(glycidyl methacrylate) (PLMA63-PGlyMA89) spherical nanoparticles via RAFT dispersion polymerization of GlyMA in mineral oil
PLMA63-PGlyMA89 spherical nanoparticles were synthesized at 20% w/w solids using the following protocol. PLMA63 precursor (1.50 g; 92.0 μmol), GlyMA (1.18 g; 8.28 mmol; target DP = 90), T21s initiator (6.64 mg; 30.7 μmol; precursor/T21s molar ratio = 3.0; 10.0% v/v in mineral oil) and mineral oil (10.74 g) were weighed into a glass vial and purged with nitrogen for 30 min. The sealed vial was immersed in a preheated oil bath at 70 °C and the reaction mixture was magnetically stirred for 6 h. 1H NMR analysis indicated 99% GlyMA conversion by comparing the integrated monomer vinyl signal at 6.17 ppm to the integrated epoxy methine signals corresponding to both PGlyMA and GlyMA at 3.20–3.33 ppm. THF GPC analysis using a refractive index detector and a series of near-monodisperse poly(methyl methacrylate) calibration standards indicated an Mn of 22000 g mol−1 and an Mw/Mn of 1.18.
Synthesis of poly(lauryl methacrylate-stat-glycidyl methacrylate)-poly(methyl methacrylate) (P(LMA50-stat-GlyMA9)-PMMA67) spherical nanoparticles via RAFT dispersion polymerization of MMA in mineral oil
P(LMA50-stat-GlyMA9)-PMMA67 spherical nanoparticles were synthesized at 20% w/w solids as follows. P(LMA50-stat-GlyMA9) precursor (2.00 g; 140.13 μmol), T21s initiator (10.10 mg; 46.71 μmol; precursor/T21s molar ratio = 3.0; 10.0% v/v in mineral oil) and mineral oil (11.97 g) were weighed into a glass vial and purged with nitrogen for 30 min. MMA monomer (1.04 mL; 9.81 mmol; target DP = 70) was degassed separately then added to the reaction mixture via syringe. The sealed vial was immersed in a preheated oil bath at 90 °C and the reaction mixture was magnetically stirred for 6 h. 1H NMR analysis indicated 95% MMA conversion by comparing the integrated methyl signal assigned to MMA at 3.77 ppm to the integrated methyl signal corresponding to PMMA at 3.55–3.72 ppm. THF GPC analysis using a refractive index detector and a series of near-monodisperse poly(methyl methacrylate) calibration standards indicated an Mn of 21100 g mol−1 and an Mw/Mn of 1.17.
Derivatization of PLMA63-PGlyMA89 spherical nanoparticles with benzylamine
A typical protocol for functionalization of PLMA63-PGlyMA89 spheres with benzylamine was conducted as follows: benzylamine (67.6 μL, 0.619 mmol; [amine]/[epoxy] molar ratio = 1.0) was added to 1.0 g of a 20% w/w dispersion of PLMA63-PGlyMA89 spheres in mineral oil via micropipet. The sealed vial was immersed in a preheated oil bath at 70 °C and the reaction mixture was magnetically stirred for 17 h. Unfortunately, the final copolymer proved to be insoluble in various deuterated solvents, which prevented its analysis by 1H NMR spectroscopy. Thus the extent of functionalization was assessed by FT-IR spectroscopy. The core-crosslinked copolymer nanoparticles were purified by three consecutive precipitations into a ten-fold excess of methanol (with redispersion in THF after each precipitation) and then dried under vacuum for 24 h prior to analysis.Derivatization of P(LMA50-stat-GlyMA9)-PMMA67 spherical nanoparticles using various amines
A typical protocol for functionalization of P(LMA50-stat-GlyMA9)-PMMA67 spheres with benzylamine was conducted as follows: benzylamine (9.2 μL, 84.58 μmol; [amine]/[epoxy] molar ratio = 1.0) was added to 1.0 g of a 20% w/w dispersion of P(LMA50-stat-GlyMA9)-PMMA67 spheres in mineral oil via micropipet. The sealed vial was immersed in a preheated oil bath at 70 °C and the reaction mixture was magnetically stirred for 17 h. The reduction in epoxy groups (Y%) was determined using 1H NMR spectroscopy by comparing the satellite signal of the PMMA backbone at 3.43–3.47 ppm to the integrated epoxy methine signal at 3.12–3.30 ppm. The same reaction was also studied using a 2, 5, 10, 20 or 50-fold excess of benzylamine under otherwise identical conditions. In addition, when using an equimolar amount of amine, the reaction time was varied from 17 h to 88 h, the solids content was adjusted from 20 to 35% w/w solids and the reaction temperature ranged from 20 to 90 °C. Furthermore, alternative nucleophiles such as N-methylaniline, 2-(dimethylamino)propylamine or triethylenetetramine (using an [amine]/[epoxy] molar ratio = 1.0 in each case) were also examined at 20% w/w solids in mineral oil for 17 h at 70 °C. Finally, 1-butanethiol ([thiol]/[epoxy] molar ratio = 50) was also examined at 20% w/w solids in mineral oil for 17 h at 70 °C.Epoxy ring-opening reactions of PLMA63-PGlyMA89 and P(LMA50-stat-GlyMA9)-PMMA67 spherical nanoparticles using water
A typical protocol for reacting the epoxy groups of PLMA63-PGlyMA89 and P(LMA50-stat-GlyMA9)-PMMA67 spheres with water was conducted as follows. A 20% w/w nanoparticle dispersion in mineral oil (1.0 g) was weighed into a glass vial. The sealed vial was immersed in a preheated oil bath at 110 °C and the reaction mixture was magnetically stirred for 17 h. The reduction in epoxy groups (Y%) for the P(LMA50-stat-GlyMA9)-PMMA67 spheres was determined by using 1H NMR spectroscopy to compare the satellite signal of the PMMA backbone at 3.43–3.47 ppm to the integrated epoxy methine signal at 3.12–3.30 ppm. The extent of epoxy ring-opening for the PLMA63-PGlyMA89 spheres was assessed by FT-IR spectroscopy. The core-crosslinked copolymer nanoparticles were purified by three consecutive precipitations into a ten-fold excess of methanol (with redispersion in THF after each precipitation) and then dried under vacuum for 24 h prior to analysis.Epoxy ring-opening reactions of PLMA63-PGlyMA89 spherical nanoparticles using 50% v/v acetic acid
A typical protocol for ring-opening the epoxy groups of PLMA63-PGlyMA89 spherical nanoparticles using 50% v/v acetic acid was conducted as follows: 4.0 g of a 20% w/w nanoparticle dispersion in mineral oil was weighed into a 10 mL round-bottomed flask. This flask was then immersed in a preheated oil bath at 110 °C and connected via plastic tubing to a second round-bottomed flask (also immersed in an oil bath set to 110 °C) containing 10 mL of 50% v/v aqueous acetic acid (see Fig. S1†). The latter flask was connected to a stream of N2 gas to generate acidic vapor. Both flasks were magnetically stirred for 17 h whereby essentially all of the 50% v/v aqueous acetic acid had evaporated. The epoxy ring-opening reaction for the PLMA63-PGlyMA89 spheres was analyzed by FT-IR spectroscopy. The copolymer was purified by three consecutive precipitations into a ten-fold excess of methanol (with redispersion in THF after each precipitation) then dried under vacuum for 24 h prior to analysis.1H NMR spectroscopy
1H NMR spectra were recorded in either CD2Cl2 or CDCl3 using a 400 MHz Bruker Avance spectrometer. Typically, 64 scans were averaged per spectrum.Gel permeation chromatography (GPC)
Molecular weight distributions (MWDs) were assessed by GPC using THF as an eluent. The GPC system was equipped with two 5 μm (30 cm) Mixed C columns and a WellChrom K-2301 refractive index detector operating at 950 ± 30 nm. The THF mobile phase contained 2.0% v/v triethylamine and 0.05% w/v butylhydroxytoluene (BHT) and the flow rate was fixed at 1.0 ml min−1. A series of twelve near-monodisperse poly(methyl methacrylate) standards (Mp values ranging from 800 to 2200000 g mol−1) were used for column calibration in combination with a refractive index detector.
Dynamic light scattering (DLS)
DLS studies were performed using a Zetasizer Nano ZS instrument (Malvern Instruments, UK) at a fixed scattering angle of 173°. Copolymer dispersions were diluted to 0.10% w/w solids using n-dodecane prior to analysis at 20 °C. The z-average diameter and polydispersity of the nanoparticles were calculated by cumulants analysis of the experimental correlation function using Dispersion Technology Software version 6.20. Data were averaged over ten runs each of thirty seconds duration.Transmission electron microscopy (TEM)
TEM studies were conducted using a FEI Tecnai G2 spirit instrument operating at 80 kV and equipped with a Gatan 1k CCD camera. A single droplet of a 0.10% w/w copolymer dispersion was placed onto a carbon-coated copper grid and allowed to dry, prior to exposure to ruthenium(VIII) oxide vapor for 7 min at 20 °C.99 This heavy metal compound acts as a positive stain for the core-forming PGlyMA or PMMA block to improve contrast. The ruthenium(VIII) oxide was prepared as follows: ruthenium(IV) oxide (0.30 g) was added to water (50 g) to form a black slurry; addition of sodium periodate (2.0 g) with continuous stirring produced a yellow solution of ruthenium(VIII) oxide within 1 min at 20 °C.Fourier transform infrared (FT-IR) spectroscopy
FT-IR spectra were recorded for the PLMA63-PGlyMA89 precursor and derivatized PLMA63-PGlyMA89 diblock copolymers at 20 °C using a Thermo-Scientific Nicolet iS10 FT-IR spectrometer equipped with a Golden Gate Diamond ATR accessory. The spectral resolution was 4 cm−1 and 96 scans were averaged per spectrum.Results and discussion
Synthesis of PLMA63-PGlyMA89 and P(LMA50-stat-GlyMA9)-PMMA67 spherical nanoparticles
A PLMA63 precursor was synthesized via RAFT solution polymerization of LMA in anhydrous toluene at 70 °C using CDB as a RAFT agent (see Scheme 1). A P(LMA50-stat-GlyMA9) precursor was prepared by copolymerization of LMA with GlyMA using the same reaction conditions. Since LMA and GlyMA are both methacrylic monomers, they should exhibit similar comonomer reactivity ratios. Thus an approximately statistical distribution of GlyMA groups along the steric stabilizer chains is expected for this second precursor.For both syntheses, the polymerization was quenched after 15 h, with 1H NMR spectroscopy studies indicating 90% LMA conversion for PLMA63 and an overall comonomer conversion of 90% for P(LMA50-stat-GlyMA9). THF GPC analysis indicated an Mn of 12600 g mol−1 (Mw/Mn = 1.19) for PLMA63 and an Mn of 12300 g mol−1 (Mw/Mn = 1.19) for P(LMA50-stat-GlyMA9) (see Fig. S3†). In order to produce nanoparticles of comparable size, the PLMA63 precursor was chain-extended via RAFT dispersion polymerization of GlyMA in mineral oil targeting a degree of polymerization (DP) of 90 for the PGlyMA block while the P(LMA50-stat-GlyMA9) precursor was chain-extended using MMA targeting a mean PMMA DP of 70. 1H NMR analysis indicated a GlyMA conversion of 99% to afford PLMA63-PGlyMA89 nanoparticles while 95% MMA conversion produced P(LMA50-stat-GlyMA9)-PMMA67 nanoparticles. 1H NMR analysis also confirmed that essentially all the epoxy groups remained intact under these synthesis conditions (the ten aromatic protons assigned to the cumyl and dithiobenzoate end-groups at 7.10–8.00 ppm were compared to the epoxy methine proton at 3.12–3.33 ppm, see Fig. S2†). Similar observations were reported by Docherty et al. for the synthesis of PSMA-PGlyMA spherical nanoparticles in mineral oil.37 A relatively high blocking efficiency and narrow molecular weight distribution (Mw/Mn ≤ 1.18) was confirmed in each case by THF GPC analysis (see Fig. S3†). The PLMA63-PGlyMA89 nanoparticles had a z-average diameter of 28 nm (polydispersity index (PDI) = 0.03) while the P(LMA50-stat-GlyMA9)-PMMA67 nanoparticles exhibited a z-average diameter of 26 nm (PDI = 0.05), see Fig. 1. In summary, these nanoparticles were deemed suitable for studying the post-polymerization modification of their epoxy groups using various nucleophiles.
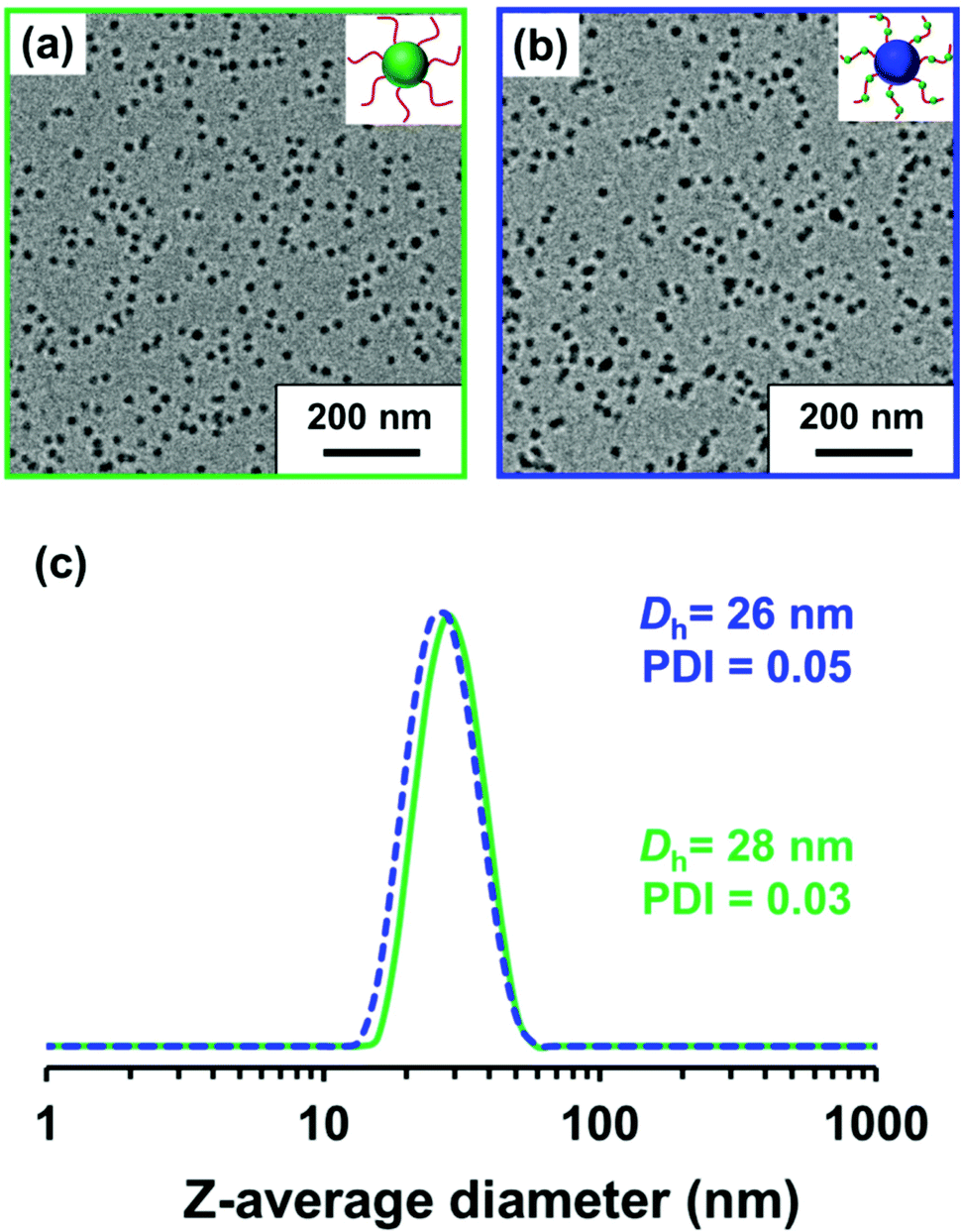 | ||
| Fig. 1 Representative TEM images obtained for (a) PLMA63-PGlyMA89 (green frame) and (b) P(LMA50-stat-GlyMA9)-PMMA67 (blue frame) nanoparticles prepared at 20% w/w solids in mineral oil. (c) DLS data recorded for 0.1% w/w dispersions of PLMA63-PGlyMA89 nanoparticles (green trace) and P(LMA50-stat-GlyMA9)-PMMA67 nanoparticles (blue trace) in mineral oil. A z-average hydrodynamic diameter (Dh) of 28 nm (DLS PDI = 0.03) was determined for the PLMA63-PGlyMA89 nanoparticles and 26 nm (DLS PDI = 0.05) for the P(LMA50-stat-GlyMA9)-PMMA67 nanoparticles. | ||
Post-polymerization modification of the epoxy groups within PLMA63-PGlyMA89 nanoparticles and P(LMA50-stat-GlyMA9)-PMMA67 nanoparticles using benzylamine
The post-polymerization modification of epoxy-functional diblock copolymer nanoparticles by amines has been previously studied for various aqueous PISA formulations.75,80,82,86 In such cases, epoxy-amine chemistry was used to introduce either disulfide82 or TEMPO75 functionalities or cationic character,75,80,82,86 with intra- or inter-nanoparticle crosslinking often being observed. Recently, the reaction of epoxy groups with N-methylaniline has been reported for PSMA-PGlyMA nanoparticles in mineral oil.37Herein we examine the derivatization of epoxy-functional PLMA63-PGlyMA89 and P(LMA50-stat-GlyMA9)-PMMA67 nanoparticles using benzylamine. This is a useful model compound because aromatic amines such as p-phenylenediamine can serve as anti-knock agents100 and/or anti-oxidants101,102 for automotive engine oil formulations. Initially, the reaction between the P(LMA50-stat-GlyMA9)-PMMA67 nanoparticles and benzylamine was examined using an [amine]/[epoxy] molar ratio of unity at 20% w/w solids in mineral oil at 70 °C. Subsequent studies involved using excess amine (ranging from two-fold to fifty-fold) under the same conditions. To identify the precise chemical shift for the new azamethylene signal produced by the epoxy ring-opening reaction, a model reaction between GlyMA monomer and benzylamine was examined in mineral oil (see Fig. S4†). Unfortunately, this new azamethylene signal overlaps with the original methylene signal arising from the epoxy ring at 2.60–2.90 ppm. Nevertheless, epoxy group consumption could be monitored by comparing the intensity of the (constant) satellite signal assigned to the PMMA backbone at 3.43–3.47 ppm to the methine signal of the epoxy ring at 3.12–3.30 ppm (see Fig. 2). Loss of 16% of the original epoxy groups was observed after 17 h at 70 °C when using a stoichiometric amount of benzylamine. In fact, complete loss of the epoxy groups (99%) was only achieved when using a 50-fold excess of benzylamine (see Fig. 2). In an attempt to enhance this sluggish epoxy-amine reaction, the reaction time, solids content and temperature were systematically varied while using an [amine]/[epoxy] molar ratio of unity. As shown in Table 1, an extended reaction time of 88 h resulted in a 30% reduction in the original epoxy group signal. In contrast, derivatizing the nanoparticles at 35% w/w solids only led to 23% of epoxy groups reacting with the benzylamine. Similar results (21%) were achieved when heating a 20% w/w nanoparticle dispersion to 90 °C for 17 h. Alternative aromatic amines such as N-methylaniline, 2-(dimethylamino)propylamine or triethylenetetramine were also examined but a discernible difference (i.e. 68% loss of the original epoxy groups) was only observed in the latter case (see Fig. S5†). Triethylenetetramine contains two primary and two secondary amine groups, which means that it can react with up to six epoxy groups. This leads to a high degree of inter-nanoparticle crosslinking: the apparent z-average diameter increased up to 59 nm, while the corresponding DLS PDI of 0.24 suggested significant broadening of the particle size distribution. Furthermore, using an alternative nucleophile such as 1-butanethiol only resulted in a 13% reduction of the epoxy groups even when using a 50-fold excess of this reagent, see Fig. S6.† These data suggest that a high degree of derivatization for the epoxy-functionalized P(LMA50-stat-GlyMA9)-PMMA67 nanoparticles can only be achieved when using a large excess of amine. This problem is most likely related to the relatively low epoxy group concentration (∼0.07 mol dm−3) under the reaction conditions. Indeed, similar observations were reported by Ratcliffe et al. for the derivatization of an aqueous dispersion of P(GMA65-stat-GlyMA1.7)-PHPMA140 worms via epoxy-amine chemistry using cystamine. In this prior study, a 20-fold excess of diamine was required to obtain primary amine-functionalized thermoresponsive worms.82
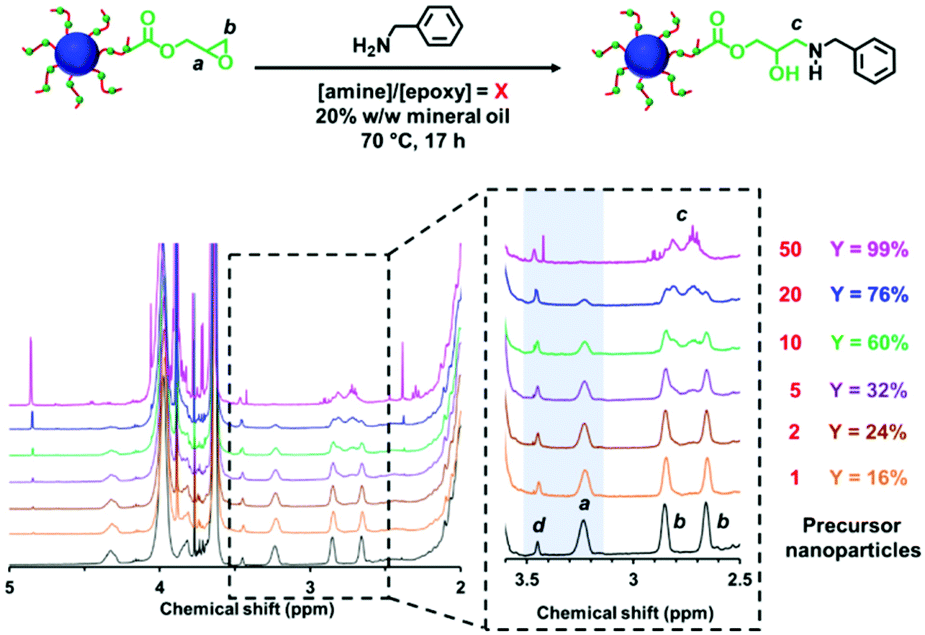 | ||
| Fig. 2 1H NMR spectra recorded in CD2Cl2 for P(LMA50-stat-GlyMA9)-PMMA67 before (black spectrum) and after reaction with benzylamine at 70 °C for 17 h at 20% w/w solids when using an [amine]/[epoxy] molar ratio (X) of 1 (orange spectrum), 2 (brown spectrum), 5 (purple spectrum), 10 (green spectrum), 20 (blue spectrum) and 50 (pink spectrum), respectively. Expansion of the 2.5–3.6 ppm region confirms the systematic loss of epoxy groups (note the gradual attenuation of methine signal a and the evolution of a new methylene signal c on increasing the [amine]/[epoxy] molar ratio. In each case, the epoxy loss (Y%) was determined by comparing the satellite signal d assigned to the PMMA backbone at 3.43–3.47 ppm to the methine signal a corresponding to the epoxide ring at 3.12–3.30 ppm. | ||
| Reaction code | Reaction time (h) | Solids content (%) | Reaction temperature (°C) | Y (%) |
|---|---|---|---|---|
| a | 17 | 20 | 70 | 16 |
| b | 40 | 20 | 70 | 23 |
| c | 64 | 20 | 70 | 28 |
| d | 88 | 20 | 70 | 30 |
| e | 17 | 25 | 70 | 19 |
| f | 17 | 30 | 70 | 21 |
| g | 17 | 35 | 70 | 23 |
| h | 17 | 20 | 25 | 9 |
| i | 17 | 20 | 50 | 13 |
| j | 17 | 20 | 90 | 21 |
Since benzylamine is a primary amine, there are various side-reactions that can occur during the attempted functionalization of P(LMA50-stat-GlyMA9)-PMMA67 nanoparticles (see Scheme 2).103 The initial epoxy ring-opening reaction produces a hydroxyl and a secondary amine. In principle, these nucleophilic groups can react further with the remaining epoxy groups, which inevitably results in intra-chain and inter-chain cross-linking. Indeed, THF GPC analysis indicates the appearance of a high molecular weight shoulder, which provides evidence for such crosslinking (see Fig. 3). Using an [amine]/[epoxy] molar ratio of unity resulted in an Mn of 24900 with an Mw/Mn of 1.79, and the latter value being a significant increase on that observed for the linear precursor nanoparticles (Mn = 21100; Mw/Mn = 1.17). However, systematically increasing the amount of excess benzylamine led to a gradual reduction in the high molecular weight shoulder (e.g., using a 50-fold excess of benzylamine afforded Mn = 23100; Mw/Mn = 1.31) (see Fig. 3). These data suggest that using a sufficiently large excess of amine can minimize crosslinking between the copolymer chains.
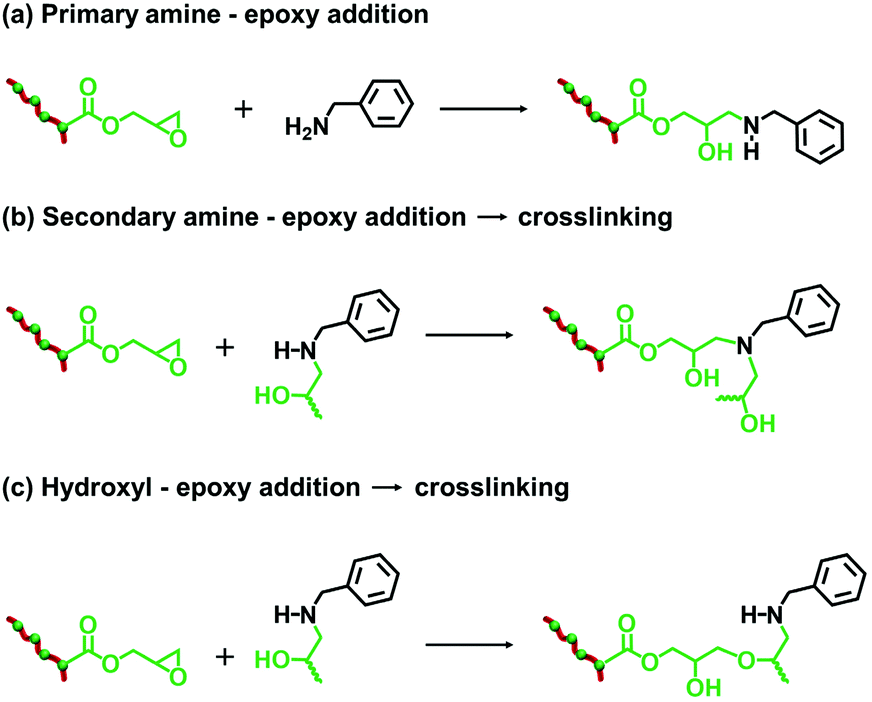 | ||
| Scheme 2 Summary of the three possible reactions that can occur when reacting P(LMA50-stat-GlyMA9)-PMMA67 nanoparticles with benzylamine. (a) Epoxy ring-opening results in the formation of a secondary amine and a hydroxyl group. (b) This secondary amine can then react with remaining epoxy groups. (c) Alternatively, the hydroxyl group can react with epoxy groups. Side-reactions (b) and (c) can lead cross-linking between and/or within stabilizer chains. In practice, the rate of reaction for (c) is expected to be slower than for (b). | ||
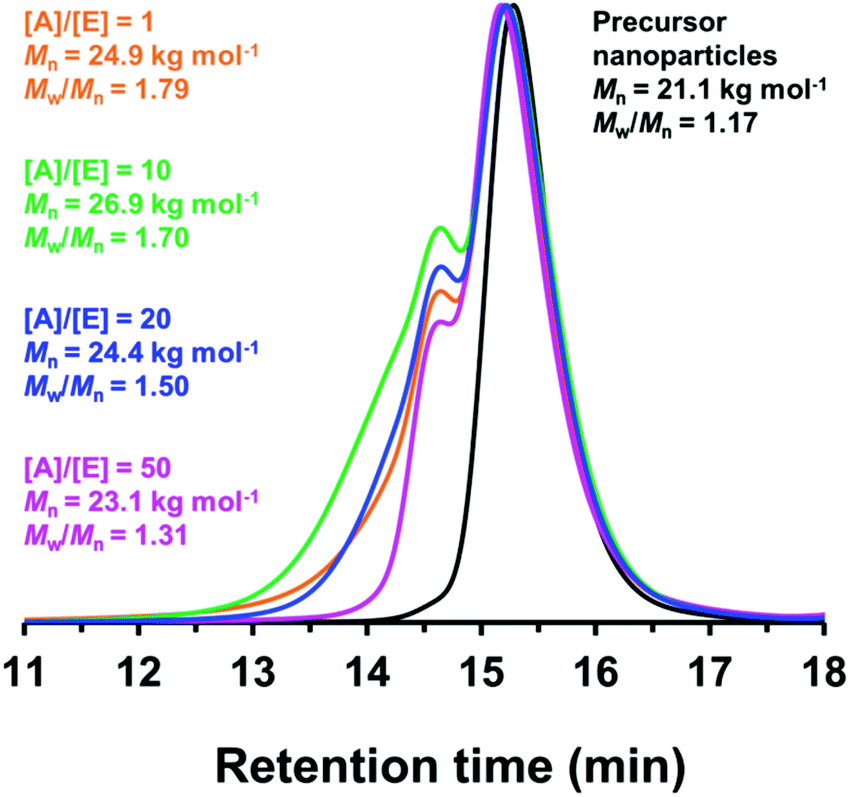 | ||
| Fig. 3 GPC curves (vs. a series of near-monodisperse poly(methyl methacrylate) calibration standards) recorded using a refractive index detector for P(LMA50-stat-GlyMA9)-PMMA67 (prepared in mineral oil at 20% w/w solids at 90 °C) (black curve) and the benzylamine-derivatized P(LMA50-stat-GlyMA9)-PMMA67 produced when using an [amine]/[epoxy] molar ratio of 1 (orange curve), 10 (green curve), 20 (blue curve) or 50 (pink curve). A systematic reduction in Mw/Mn was observed when increasing the [amine]/[epoxy] molar ratio. | ||
The derivatization of PLMA63-GlyMA89 nanoparticles with benzylamine could not be assessed by 1H NMR spectroscopy. In this case, the epoxy groups are much more closely located to each other within the nanoparticle cores. Thus using an [amine]/[epoxy] molar ratio of unity inevitably led to extensive core-crosslinking, which rendered the derivatized copolymer chains insoluble in deuterated solvents and hence precluded 1H NMR analysis. Instead, copolymer powders were analyzed by FT-IR spectroscopy (after extended purification to remove the unreacted amine and mineral oil) to determine the extent of reaction of the epoxy groups. When using a stoichiometric amount of benzylamine, the asymmetric/symmetric ring deformation bands assigned to the epoxy groups at 850 and 910 cm−1 are fully attenuated while a new strong band corresponding to the out-of-plane aromatic C–H bending of the conjugated benzylamine is evident at 699 cm−1 (see Fig. 4).
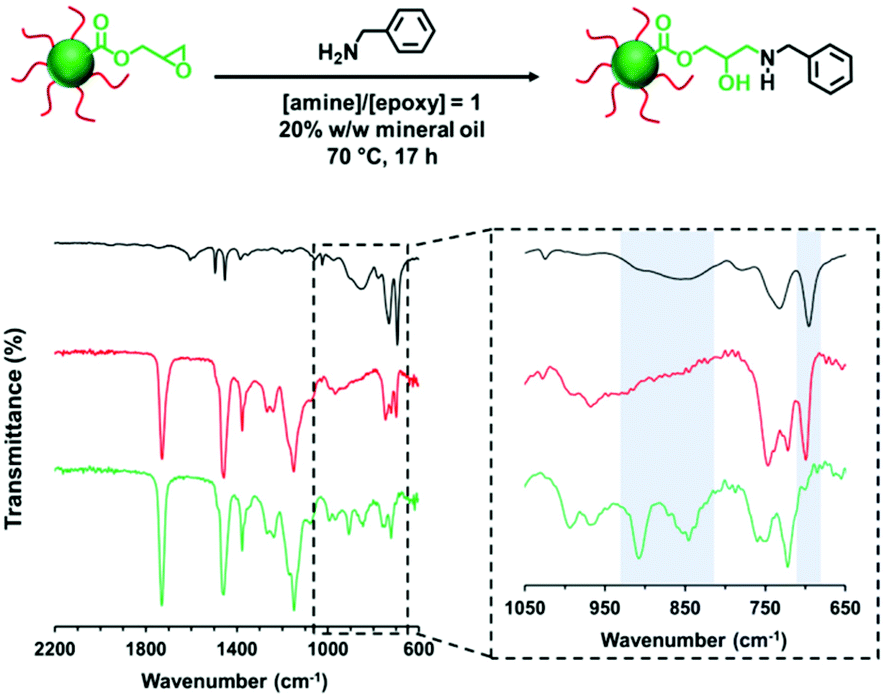 | ||
| Fig. 4 FT-IR spectra recorded for PLMA63-PGlyMA89 diblock copolymer prior to functionalization (green spectrum), PLMA63-PGlyMA89 diblock copolymer after functionalization with benzylamine (red spectrum) and benzylamine alone (black spectrum). The reaction conditions used are summarized in the corresponding chemical reaction. | ||
Ring-opening of epoxy groups in PLMA63-PGlyMA89 and P(LMA50-stat-GlyMA9)-PMMA67 nanoparticles by reaction with water
Zheng et al. reported that core-crosslinked all-acrylic diblock copolymer spherical nanoparticles significantly reduced the friction between metal surfaces within the boundary lubrication regime.104 Derry and co-workers reported similar performance for an analogous all-methacrylic formulation comprising poly(stearyl methacrylate)–poly-(benzyl methacrylate)–poly(ethylene glycol dimethacrylate) triblock copolymer spherical nanoparticles prepared directly in mineral oil.38 In principle, such nanoparticles could be potentially useful for the formulation of next-generation, ultralow-viscosity automotive engine oils.38Wan et al.105 and Yuan et al.106 demonstrated that ultrafine magnetite nanoparticles are produced by coprecipitation of ferric and ferrous salts in the presence of poly(glycerol monomethacrylate) (PGMA) homopolymer or PGMA-based diblock copolymers on addition of ammonium hydroxide. The PGMA chains adsorb strongly onto the magnetite nanoparticles because their pendent cis-diol groups can act as a bidentate chelating ligand for the surface Fe atoms.105,106 In the context of the present study, ring-opening of the epoxy groups with water should produce the same GMA repeat units.88 In principle, such GMA-functionalized nanoparticles may exhibit enhanced adsorption onto stainless steel, which might be expected to reduce friction between moving parts within an automotive engine.38
Ratcliffe et al. reported that GlyMA monomer can be readily converted into GMA by reaction with water at 80 °C.88 Accordingly, the ring-opening of epoxy groups within the steric stabilizer chains of P(LMA50-stat-GlyMA9)-PMMA67 nanoparticles was examined by simply heating a 20% w/w dispersion of such nanoparticles in mineral oil up to 110 °C. 1H NMR analysis enabled the loss of epoxy groups over time to be monitored as they reacted with trace amounts of water. This approach indicated an extent of reaction of 72% within 17 h and 100% after 40 h (see Fig. 5a). However, the initial free-flowing 20% w/w dispersion was transformed into a highly viscous gel (see Fig. S7†). Moreover, the z-average diameter of the nanoparticles increased from 26 nm (PDI = 0.05) to 117 nm (PDI = 0.43), which indicated extensive crosslinking between neighboring nanoparticles (see Fig. 6a). TEM analysis also provided direct evidence for such nanoparticle aggregation (see arrows in Fig. 6c). Moreover, THF GPC analysis indicated the appearance of a high molecular weight shoulder after 17 h (see Fig. 5b).
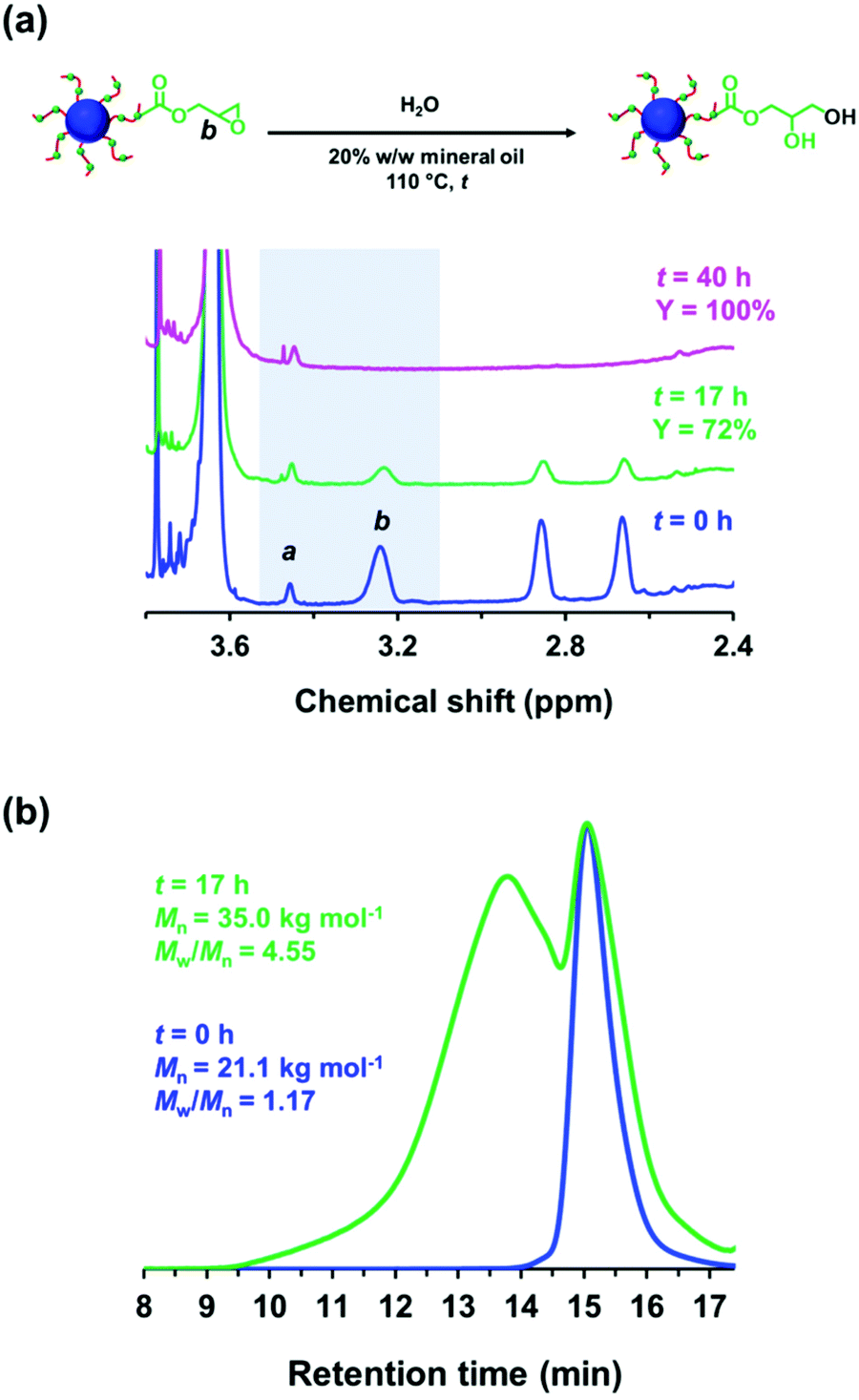 | ||
| Fig. 5 (a) 1H NMR spectra recorded in CD2Cl2 for P(LMA50-stat-GlyMA9)-PMMA67 chains before (t = 0 h; blue spectrum) and after heating the corresponding 20% w/w nanoparticle dispersion at 110 °C for either 17 h (green spectrum) or 40 h (pink spectrum). The epoxy loss (Y%) was determined by comparing the satellite signal a assigned to the PMMA backbone at 3.43–3.47 ppm to the methine signal b corresponding to the epoxide ring at 3.12–3.30 ppm. (b) GPC curves (refractive index detector vs. a series of near-monodisperse poly(methyl methacrylate) calibration standards) recorded for the P(LMA50-stat-GlyMA9)-PMMA67 chains prior to heating (blue trace) and after heating to 110 °C for 17 h (green trace). The prominent high molecular weight shoulder indicates that extensive inter-chain crosslinking occurs under such conditions. | ||
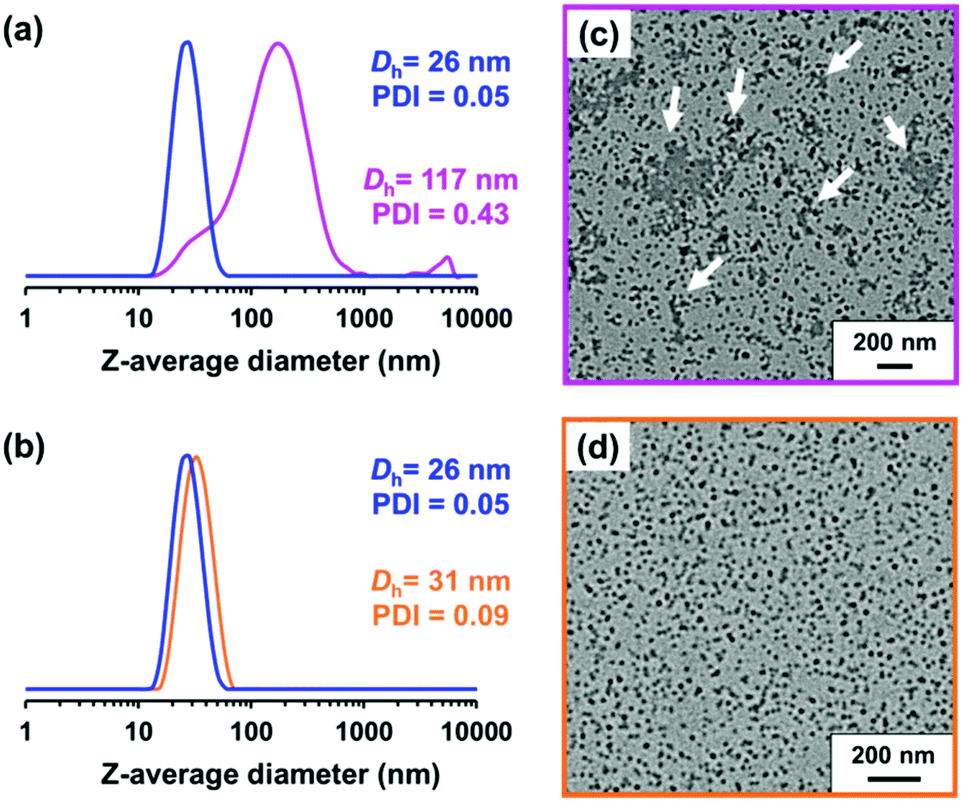 | ||
| Fig. 6 DLS particle size distributions recorded for P(LMA50-stat-GlyMA9)-PMMA67 nanoparticles both prior to heating (blue trace) and after heating to 110 °C for 40 h at (a) 20% w/w solids (pink trace) or (b) 1.0% w/w solids (orange trace). Corresponding TEM images obtained for (c) the 20% w/w dispersion and (d) the 1.0% w/w dispersion after heating to 110 °C for 40 h. | ||
In principle, water can either originate from the mineral oil itself (which contains ≤ 0.004% water) or from the atmosphere (≤ 4%, depending on the relative humidity). To identify the source, the heating experiment was repeated under a N2 atmosphere after deoxygenating the dispersion for 30 min using a stream of dry N2 gas prior to heating. In this case, only ∼21% of the original epoxy groups were lost within 88 h at 110 °C, which suggests that the major source of water is from the atmosphere (see Fig. S8†). Moreover, using a relatively high reaction temperature appears to be essential because similar experiments conducted at 40 °C, 70 °C or 90 °C did not result in any discernible loss of the epoxy groups. In such cases, the GPC curves recorded for the copolymer chains before and after heating overlapped, which suggests that no ring-opening occurred under these milder conditions (see Fig. S9†). As discussed above, hydroxyl groups produced via epoxy ring-opening can react further with the remaining epoxy groups. Such reactions can either occur between chains within an individual nanoparticle to produce intra-chain and/or inter-chain crosslinking or between two or more nanoparticles to produce incipient aggregation (see Scheme 3). In order to minimize the latter problem, the heating protocol was also conducted at a much lower nanoparticle concentration. As expected, heating a 1.0% w/w dispersion of P(LMA50-stat-GlyMA9)-PMMA67 nanoparticles in mineral oil led to ring-opening of all the epoxy groups within 17 h at 110 °C (see Fig. S10a†). THF GPC analysis of the resulting copolymer chains produced a similar chromatogram to that recorded for the 20% w/w dispersion after heating (see Fig. S10b†). However, in this case DLS studies indicated only relatively minor changes in the nanoparticle size distribution (Dh = 31 nm; PDI = 0.09), while TEM analysis confirmed a well-defined spherical morphology for the final nanoparticles (see Fig. 6b and d).
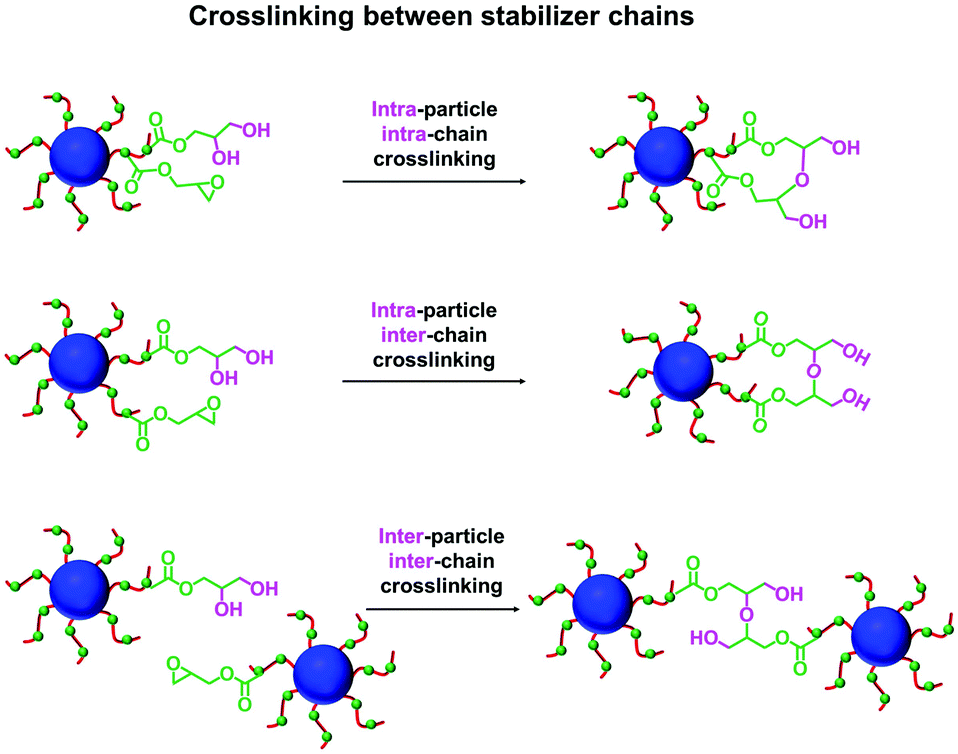 | ||
| Scheme 3 Schematic representation of the various reactions that can occur when reacting P(LMA50-stat-GlyMA9)-PMMA67 nanoparticles with trace water by heating a 20% w/w copolymer dispersion in mineral oil to 110 °C. Following the initial epoxy ring-opening reaction, the resulting hydroxyl groups can react with the remaining epoxy groups. This leads to both intra-particle and/or inter-particle crosslinking. | ||
Heating a 20% w/w dispersion of PLMA63-PGlyMA89 nanoparticles up to 70 °C for 17 h led to no discernible change in the epoxy NMR signals, see Fig. S11.† Significant loss of epoxy groups was observed at 90 °C but the copolymer remained soluble in CD2Cl2 for 1H NMR analysis. Raising the reaction temperature to 110 °C resulted in core-crosslinking and hence insoluble copolymer chains, which precluded 1H NMR analysis. However, FT-IR spectroscopy studies of the purified copolymer isolated after heating to 110 °C for 17 h indicated a relatively small amount of residual epoxy groups, as judged by the weak asymmetric/symmetric ring deformation bands at 850 and 910 cm−1 (see Fig. S12†).
Ring-opening of epoxy groups on PLMA63-PGlyMA89 by reaction with 50% v/v aqueous acetic acid
It is well-known that carboxylic acids can act as nucleophiles in epoxy ring-opening reactions to produce a β-hydroxypropyl ester.94,107–109 Such reactions are often catalyzed by base. However, carboxylic acids can also be employed as acid catalysts to promote epoxy ring-opening by water.110 Herein, the ring-opening of the epoxy groups in PLMA63-PGlyMA89 nanoparticles was examined by heating a 50% v/v aqueous acetic acid solution at 110 °C and passing the resulting hot vapor through a 20% w/w copolymer dispersion (also heated to 110 °C) using the experimental setup shown in Fig. S1† under a N2 atmosphere. Disappearance of the characteristic epoxy bands at 850 and 910 cm−1 shown in Fig. 7 suggests that complete loss of the epoxy groups can be achieved within 17 h. Moreover, the appearance of a broad band corresponding to a hydroxyl stretch at 3000–3650 cm−1 plus a primary alcohol C–O stretch at 1041cm−1 confirmed the presence of hydroxyl groups within the final copolymer. A z-average diameter of 38 nm (DLS PDI = 0.31) was determined for the derivatized PLMA63-PGlyMA89 nanoparticles after dilution with n-dodecane. This increase in nanoparticle size and polydispersity indicated some degree of inter-particle crosslinking. Furthermore, DLS analysis after dilution with THF (z-average diameter = 43 nm; PDI = 0.28) confirmed core-crosslinking of these nanoparticles because THF is a good solvent for the linear diblock copolymer precursor. Apart from the addition of water to the epoxy ring and the subsequent hydroxyl-epoxy reaction, there are two further side-reactions that can occur: (i) epoxy ring-opening by acetic acid and esterification between acetic acid and the pendent hydroxyl groups (see Scheme S1†).94,109 However, the presence of 50% v/v water appears to minimize such side-reactions.110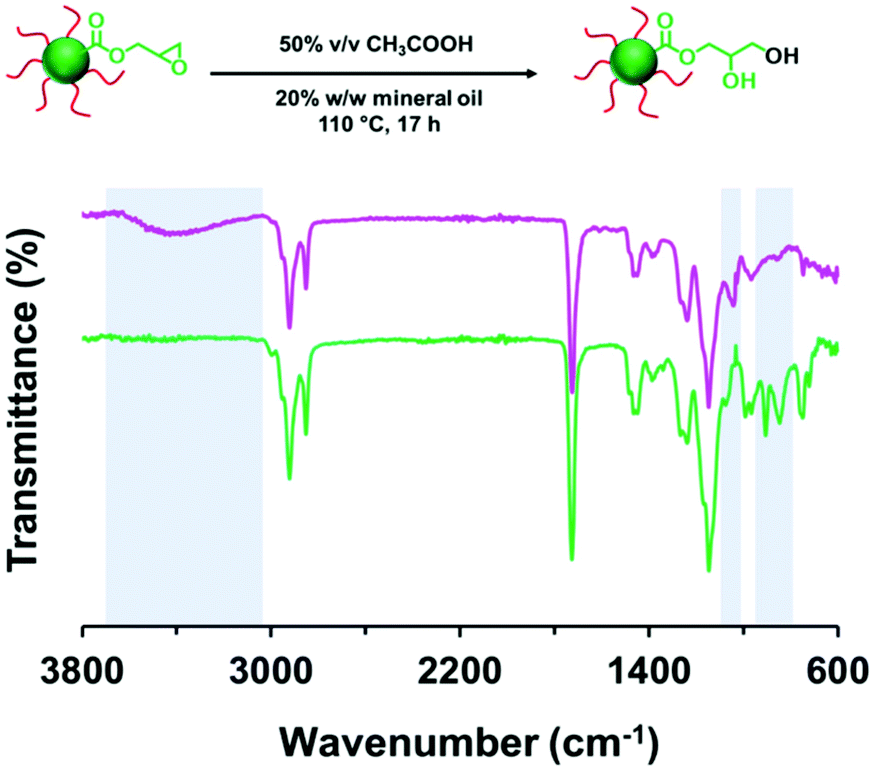 | ||
| Fig. 7 FT-IR spectra recorded for PLMA63-PGlyMA89 prior to (green spectrum) and after reacting with 50% v/v aqueous acetic acid at 110 °C for 17 h (pink spectrum). In principle, this simply involves acid-catalyzed epoxy ring-opening with water but in practice side-reactions also occur, which leads to extensive core-crosslinking of the nanoparticles. | ||
Conclusions
Epoxy-functional P(LMA50-stat-GlyMA9)-PMMA67 and PLMA63-PGlyMA89 spherical nanoparticles of comparable size were prepared at 20% w/w solids in mineral oil via RAFT-mediated PISA. Both types of nanoparticles were characterized using 1H NMR spectroscopy, THF GPC, DLS and TEM. Post-polymerization modification of these nanoparticles was studied using benzylamine as a model compound. Using a stoichiometric amount of amine relative to the epoxy groups was sufficient for complete derivatization of the PLMA63-PGlyMA89 nanoparticles. However, achieving a similar extent of reaction for the P(LMA50-stat-GlyMA9)-PMMA67 nanoparticles required a 50-fold excess of benzylamine. This is attributed to the relatively low molar concentration of the epoxy groups (0.07 mol dm−3vs. 0.52 mol dm−3) in the latter case. Furthermore, the epoxy ring-opening reaction of these spheres using a trace amount of water was also studied. Complete loss of epoxy groups was observed for the P(LMA50-stat-GlyMA9)-PMMA67 nanoparticles on heating a 20% w/w dispersion at 110 °C for 17 h. This protocol led to crosslinking between neighboring nanoparticles but this problem could be minimized by diluting the nanoparticles to 1.0% w/w solids prior to heating. On simply heating a 20% w/w dispersion of PLMA63-PGlyMA89 nanoparticles to 110 °C for 17 h, a minor fraction of unreacted epoxy groups was observed by FT-IR spectroscopy. However, when using 50% v/v aqueous acetic acid instead of water, complete loss of epoxy groups was achieved under the same conditions. In the case of the PLMA63-PGlyMA89 spheres, derivatization always led to core-crosslinking owing to the ring-opening of epoxy groups by neighboring hydroxyl groups. In principle, such epoxy-functional nanoparticles may be useful additives for designing next-generation ultralow-viscosity engine oils.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC for a CDT PhD studentship for C. G. (EP/L016281) and Lubrizol Ltd. (Hazelwood, Derbyshire, UK) for financial support of this project and for permission to publish these results. S. P. A. also acknowledges an EPSRC Established Career Particle Technology Fellowship (EP/R003009). The authors thank Christopher Hill and Dr Svetomir Tzokov at the University of Sheffield Biomedical Science Electron Microscopy suite for their technical assistance.References
- J. Chiefari, Y. K. B. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS
.
- G. Moad, E. Rizzardo and S. H. Thang, Acc. Chem. Res., 2008, 41, 1133–1142 CrossRef CAS PubMed
- D. J. Keddie, Chem. Soc. Rev., 2014, 43, 496–505 RSC
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS PubMed
- M. J. Derry, L. A. Fielding and S. P. Armes, Prog. Polym. Sci., 2016, 52, 1–18 CrossRef CAS
- A. B. Lowe, Polymer, 2016, 106, 161–181 CrossRef CAS
- J. Yeow and C. Boyer, Adv. Sci., 2017, 4, 1700137 CrossRef
- F. D'Agosto, J. Rieger and M. Lansalot, Angew. Chem., Int. Ed., 2020, 59, 8368–8392 CrossRef
- J. Cao, Y. Tan, Y. Chen, L. Zhang and J. Tan, Macromol. Rapid Commun., 2021, 42, 2100498 CrossRef CAS PubMed
- C. J. Ferguson, R. J. Hughes, B. T. T. Pham, B. S. Hawkett, R. G. Gilbert, A. K. Serelis and C. H. Such, Macromolecules, 2002, 35, 9243–9245 CrossRef CAS
- M. Manguian, M. Save and B. Charleux, Macromol. Rapid Commun., 2006, 27, 399–404 CrossRef CAS
- J. Rieger, F. Stoffelbach, C. Bui, D. Alaimo, C. Jérôme and B. Charleux, Macromolecules, 2008, 41, 4065–4068 CrossRef CAS
- Y. Li and S. P. Armes, Angew. Chem., Int. Ed., 2010, 49, 4042–4046 CrossRef CAS PubMed
- X. Zhang, S. Boissé, W. Zhang, P. Beaunier, F. D'Agosto, J. Rieger and B. Charleux, Macromolecules, 2011, 44, 4149–4158 CrossRef CAS
- I. Chaduc, W. Zhang, J. Rieger, M. Lansalot, F. D'Agosto and B. Charleux, Macromol. Rapid Commun., 2011, 32, 1270–1276 CrossRef CAS
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS PubMed
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS
- J. Tan, H. Sun, M. Yu, B. S. Sumerlin and L. Zhang, ACS Macro Lett., 2015, 4, 1249–1253 CrossRef CAS PubMed
- W. M. Wan, X. L. Sun and C. Y. Pan, Macromolecules, 2009, 42, 4950–4952 CrossRef CAS
- N. Zaquen, W. A. A. W. Azizi, J. Yeow, R. P. Kuchel, T. Junkers, P. B. Zetterlund and C. Boyer, Polym. Chem., 2019, 10, 2406–2414 RSC
- G. Desnos, A. Rubio, C. Gomri, M. Gravelle, V. Ladmiral and M. Semsarilar, Polymers, 2021, 13, 2502 CrossRef CAS PubMed
- W. M. Wan and C. Y. Pan, Polym. Chem., 2010, 1, 1475–1484 RSC
- W. M. Wan, X. L. Sun and C. Y. Pan, Macromol. Rapid Commun., 2010, 31, 399–404 CrossRef CAS PubMed
- M. Semsarilar, E. R. Jones, A. Blanazs and S. P. Armes, Adv. Mater., 2012, 24, 3378–3382 CrossRef CAS PubMed
- E. R. Jones, M. Semsarilar, A. Blanazs and S. P. Armes, Macromolecules, 2012, 45, 5091–5098 CrossRef CAS
- D. Zehm, L. P. D. Ratcliffe and S. P. Armes, Macromolecules, 2013, 46, 128–139 CrossRef CAS
- Y. Pei and A. B. Lowe, Polym. Chem., 2014, 5, 2342–2351 RSC
- W. Zhao, G. Gody, S. Dong, P. B. Zetterlund and S. Perrier, Polym. Chem., 2014, 5, 6990–7003 RSC
- Y. Pei, N. C. Dharsana and A. B. Lowe, Aust. J. Chem., 2015, 68, 939–945 CrossRef CAS
- L. Houillot, C. Bui, M. Save, B. Charleux, C. Farcet, C. Moire, J. A. Raust and I. Rodriguez, Macromolecules, 2007, 40, 6500–6509 CrossRef CAS
- L. Houillot, C. Bui, C. Farcet, C. Moire, J. A. Raust, H. Pasch, M. Save and B. Charleux, ACS Appl. Mater. Interfaces, 2010, 2, 434–442 CrossRef CAS
- M. J. Derry, O. O. Mykhaylyk and S. P. Armes, Angew. Chem., Int. Ed., 2017, 56, 1746–1750 CrossRef CAS
- E. J. Cornel, S. Van Meurs, T. Smith, P. S. O. Hora and S. P. Armes, J. Am. Chem. Soc., 2018, 140, 12980–12988 CrossRef CAS PubMed
- M. J. Rymaruk, S. J. Hunter, C. T. O'Brien, S. L. Brown, C. N. Williams and S. P. Armes, Macromolecules, 2019, 52, 2822–2832 CrossRef CAS
- M. J. Rymaruk, C. T. O'Brien, S. L. Brown, C. N. Williams and S. P. Armes, Macromolecules, 2019, 52, 6849–6860 CrossRef CAS
- P. J. Docherty, M. J. Derry and S. P. Armes, Polym. Chem., 2019, 10, 603–611 RSC
- M. J. Derry, T. Smith, P. S. O'Hora and S. P. Armes, ACS Appl. Mater. Interfaces, 2019, 11, 33364–33369 CrossRef CAS
- M. J. Rymaruk, C. T. O'Brien, S. L. Brown, C. N. Williams and S. P. Armes, Macromolecules, 2020, 53, 1785–1794 CrossRef CAS
- E. J. Cornel, P. S. O'Hora, T. Smith, D. J. Growney, O. O. Mykhaylyk and S. P. Armes, Chem. Sci., 2020, 11, 4312–4321 RSC
- E. J. Cornel, G. N. Smith, S. E. Rogers, J. E. Hallett, D. J. Growney, T. Smith, P. S. O'Hora, S. Van Meurs, O. O. Mykhaylyk and S. P. Armes, Soft Matter, 2020, 16, 3657–3668 RSC
- P. J. Docherty, C. Girou, M. J. Derry and S. P. Armes, Polym. Chem., 2020, 11, 3332–3339 RSC
- L. A. Fielding, M. J. Derry, V. Ladmiral, J. Rosselgong, A. M. Rodrigues, L. P. D. Ratcliffe, S. Sugihara and S. P. Armes, Chem. Sci., 2013, 4, 2081–2087 RSC
- B. R. Parker, M. J. Derry, Y. Ning and S. P. Armes, Langmuir, 2020, 36, 3730–3736 CrossRef CAS
- B. Darmau, M. J. Rymaruk, N. J. Warren, R. Bening and S. P. Armes, Polym. Chem., 2020, 11, 7533–7541 RSC
- C. György, S. J. Hunter, C. Girou, M. J. Derry and S. P. Armes, Polym. Chem., 2020, 11, 4579–4590 RSC
- C. György, M. J. Derry, E. J. Cornel and S. P. Armes, Macromolecules, 2021, 54, 1159–1169 CrossRef PubMed
- C. György, C. Verity, T. J. Neal, M. J. Rymaruk, E. J. Cornel, T. Smith, D. J. Growney and S. P. Armes, Macromolecules, 2021, 54, 9496–9509 CrossRef
- I. R. Dorsman, M. J. Derry, V. J. Cunningham, S. L. Brown, C. N. Williams and S. P. Armes, Polym. Chem., 2021, 12, 1224–1235 RSC
- E. Raphael, M. J. Derry, M. Hippler and S. P. Armes, Chem. Sci., 2021, 12, 12082–12091 RSC
- M. J. Derry, O. O. Mykhaylyk and S. P. Armes, Soft Matter, 2021, 17, 8867–8876 RSC
- C. György, T. J. Neal, T. Smith, D. J. Growney and S. P. Armes, Macromolecules, 2022, 55, 4091–4101 CrossRef PubMed
- Y. Pei, O. R. Sugita, L. Thurairajah and A. B. Lowe, RSC Adv., 2015, 5, 17636–17646 RSC
- Y. Pei, L. Thurairajah, O. R. Sugita and A. B. Lowe, Macromolecules, 2015, 48, 236–244 CrossRef CAS
- Y. Pei, J. M. Noy, P. J. Roth and A. B. Lowe, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2326–2335 CrossRef CAS
- M. J. Derry, L. A. Fielding and S. P. Armes, Polym. Chem., 2015, 6, 3054–3062 RSC
- L. P. D. Ratcliffe, B. E. McKenzie, G. M. D. Le Bouëdec, C. N. Williams, S. L. Brown and S. P. Armes, Macromolecules, 2015, 48, 8594–8607 CrossRef CAS
- V. J. Cunningham, S. P. Armes and O. M. Musa, Polym. Chem., 2016, 7, 1882–1891 RSC
- M. J. Derry, L. A. Fielding, N. J. Warren, C. J. Mable, A. J. Smith, O. O. Mykhaylyk and S. P. Armes, Chem. Sci., 2016, 7, 5078–5090 RSC
- A. Blanazs, A. J. Ryan and S. P. Armes, Macromolecules, 2012, 45, 5099–5107 CrossRef CAS
- J. Rosselgong, A. Blanazs, P. Chambon, M. Williams, M. Semsarilar, J. Madsen, G. Battaglia and S. P. Armes, ACS Macro Lett., 2012, 1, 1041–1045 CrossRef CAS PubMed
- M. Semsarilar, E. R. Jones and S. P. Armes, Polym. Chem., 2014, 5, 195–203 RSC
- B. Karagoz, L. Esser, H. T. Duong, J. S. Basuki, C. Boyer and T. P. Davis, Polym. Chem., 2014, 5, 350–355 RSC
- W. Zhou, Q. Qu, W. Yu and Z. An, ACS Macro Lett., 2014, 3, 1220–1224 CrossRef CAS PubMed
- Y. Pei, J. M. Noy, P. J. Roth and A. B. Lowe, Polym. Chem., 2015, 6, 1928–1931 RSC
- C. A. Figg, A. Simula, K. A. Gebre, B. S. Tucker, D. M. Haddleton and B. S. Sumerlin, Chem. Sci., 2015, 6, 1230–1236 RSC
- W. Zhou, Q. Qu, Y. Xu and Z. An, ACS Macro Lett., 2015, 4, 495–499 CrossRef CAS PubMed
- W. Zhao, H. T. Ta, C. Zhang and A. K. Whittaker, Biomacromolecules, 2017, 18, 1145–1156 CrossRef CAS PubMed
- W. J. Zhang, C. Y. Hong and C. Y. Pan, Biomacromolecules, 2017, 18, 1210–1217 CrossRef CAS PubMed
- N. J. W. Penfold, A. J. Parnell, M. Molina, P. Verstraete, J. Smets and S. P. Armes, Langmuir, 2017, 33, 14425–14436 CrossRef CAS PubMed
- Q. Xu, Y. Zhang, X. Li, J. He, J. Tan and L. Zhang, Polym. Chem., 2018, 9, 4908–4916 RSC
- C. György, J. R. Lovett, N. J. W. Penfold and S. P. Armes, Macromol. Rapid Commun., 2018, 40, 1800289 CrossRef PubMed
- X. Dai, L. Yu, Y. Zhang, L. Zhang and J. Tan, Macromolecules, 2019, 52, 7468–7476 CrossRef CAS
- S. J. Hunter, K. L. Thompson, J. R. Lovett, F. L. Hatton, M. J. Derry, C. Lindsay, P. Taylor and S. P. Armes, Langmuir, 2019, 35, 254–265 CrossRef CAS PubMed
- F. L. Hatton, A. M. Park, Y. Zhang, G. D. Fuchs, C. K. Ober and S. P. Armes, Polym. Chem., 2019, 10, 194–200 RSC
- X. Dai, Y. Zhang, L. Yu, X. Li, L. Zhang and J. Tan, ACS Macro Lett., 2019, 8, 955–961 CrossRef CAS PubMed
- F. L. Hatton, M. J. Derry and S. P. Armes, Polym. Chem., 2020, 11, 6343–6355 RSC
- J. Cao, Y. Tan, X. Dai, Y. Chen, L. Zhang and J. Tan, Polymer, 2021, 230, 124095 CrossRef CAS
- P. Chambon, A. Blanazs, G. Battaglia and S. P. Armes, Langmuir, 2012, 28, 1196–1205 CrossRef CAS PubMed
- J. R. Lovett, L. P. D. Ratcliffe, N. J. Warren, S. P. Armes, M. J. Smallridge, R. B. Cracknell and B. R. Saunders, Macromolecules, 2016, 49, 2928–2941 CrossRef CAS PubMed
- N. J. W. Penfold, Y. Ning, P. Verstraete, J. Smets and S. P. Armes, Chem. Sci., 2016, 7, 6894–6904 RSC
- L. P. D. Ratcliffe, K. J. Bentley, R. Wehr, N. J. Warren, B. R. Saunders and S. P. Armes, Polym. Chem., 2017, 8, 5962–5971 RSC
- H. Yao, Y. Ning, C. P. Jesson, J. He, R. Deng, W. Tian and S. P. Armes, ACS Macro Lett., 2017, 6, 1379–1385 CrossRef CAS PubMed
- Z. Sadrearhami, J. Yeow, T. K. Nguyen, K. K. K. Ho, N. Kumar and C. Boyer, Chem. Commun., 2017, 53, 12894–12897 RSC
- J. Tan, D. Liu, C. Huang, X. Li, J. He, Q. Xu and L. Zhang, Macromol. Rapid Commun., 2017, 38, 1–7 Search PubMed
- F. L. Hatton, J. R. Lovett and S. P. Armes, Polym. Chem., 2017, 8, 4856–4868 RSC
- M. C. Jones, P. Tewari, C. Blei, K. Hales, D. J. Pochan and J. C. Leroux, J. Am. Chem. Soc., 2006, 128, 14599–14605 CrossRef CAS PubMed
- L. P. D. Ratcliffe, A. J. Ryan and S. P. Armes, Macromolecules, 2013, 46, 769–777 CrossRef CAS
- E. M. Muzammil, A. Khan and M. C. Stuparu, RSC Adv., 2017, 7, 55874–55884 RSC
- H. Gao, M. Elsabahy, E. V. Giger, D. Li, R. E. Prud'Homme and J. C. Leroux, Biomacromolecules, 2010, 11, 889–895 CrossRef CAS PubMed
- F. J. Xu, M. Y. Chai, W. B. Li, Y. Ping, G. P. Tang, W. T. Yang, J. Ma and F. S. Liu, Biomacromolecules, 2010, 11, 1437–1442 CrossRef CAS PubMed
- M. Benaglia, A. Alberti, L. Giorgini, F. Magnoni and S. Tozzi, Polym. Chem., 2013, 4, 124–132 RSC
- S. De and A. Khan, Chem. Commun., 2012, 48, 3130–3132 RSC
- W. J. Blank, Z. A. He and M. Picci, J. Coat. Technol., 2002, 74, 33–41 CrossRef CAS
- V. Tsyalkovsky, V. Klep, K. Ramaratnam, R. Lupitskyy, S. Minko and I. Luzinov, Chem. Mater., 2008, 20, 317–325 CrossRef CAS
- N. V. Tsarevsky, S. A. Bencherif and K. Matyjaszewski, Macromolecules, 2007, 40, 4439–4445 CrossRef CAS
- K. D. Safa and M. H. Nasirtabrizi, Polym. Bull., 2006, 57, 293–304 CrossRef CAS
- J. Cao, Y. Tan, Y. Chen, L. Zhang and J. Tan, Macromol. Rapid Commun., 2021, 42, 2100333 CrossRef CAS PubMed
- J. S. Trent, Macromolecules, 1984, 17, 2930–2931 CrossRef CAS
- J. E. Brown, F. X. Markley and H. Shapiro, Ind. Eng. Chem., 1955, 47, 2141–2146 CrossRef CAS
- K. Varatharajan, M. Cheralathan and R. Velraj, Fuel, 2011, 90, 2721–2725 CrossRef CAS
- G. Balaji and M. Cheralathan, J. Sci. Ind. Res., 2014, 73, 177–180 CAS
- M. R. Acocella, C. E. Corcione, A. Giuri, M. Maggio, A. Maffezzoli and G. Guerra, RSC Adv., 2016, 6, 23858–23865 RSC
- R. Zheng, G. Liu, M. Devlin, K. Hux and T. C. Jao, Tribol. Trans., 2010, 53, 97–107 CrossRef CAS
- S. Wan, Y. Zheng, Y. Liu, H. Yan and K. Liu, J. Mater. Chem., 2005, 15, 3424–3430 RSC
- J. J. Yuan, S. P. Armes, Y. Takabayashi, K. Prassides, C. A. P. Leite, F. Galembeck and A. L. Lewis, Langmuir, 2006, 22, 10989–10993 CrossRef CAS PubMed
- L. Shechter, J. Wynstra and R. P. Kurkjy, Ind. Eng. Chem., 1957, 49, 1107–1109 CrossRef CAS
- S. C. Chang, S. J. Chiu, C. Y. Hsu, Y. Chang and Y. L. Liu, Polymer, 2012, 53, 4399–4406 CrossRef CAS
- K. S. Jang, Y. S. Eom, K. S. Choi and H. C. Bae, Polym. Int., 2018, 67, 1241–1247 CrossRef CAS
- J. Kalal, F. Svec and V. Marousek, J. Polym. Sci., Part C: Polym. Symp., 1974, 47, 155–166 CAS
Footnote |
| † Electronic supplementary information (ESI) available: Schematic illustration of the experimental setup used for nanoparticle derivatization using 50% v/v aqueous acetic acid, assigned 1H NMR spectra recorded for P(LMA50-stat-GlyMA9)-PMMA67 and PLMA63-PGlyMA89; GPC curves recorded for PLMA63, P(LMA50-stat-GlyMA9), P(LMA50-stat-GlyMA9)-PMMA67 and PLMA63-PGlyMA89; 1H NMR spectra recorded for the model reaction of GlyMA with benzylamine and the reaction of P(LMA50-stat-GlyMA9)-PMMA67 with various amines or 1-butanethiol; digital photograph recorded for a 20% w/w dispersion of P(LMA50-stat-GlyMA9)-PMMA67 nanoparticles prior to and after heating to 110 °C for 17 h; 1H NMR spectra for P(LMA50-stat-GlyMA9)-PMMA67 after heating to 110 °C for 88 h under N2; GPC curves for P(LMA50-stat-GlyMA9)-PMMA67 after heating to 40, 70 or 90 °C for 17 h; 1H NMR and GPC data obtained after heating a 1% w/w dispersion of P(LMA50-stat-GlyMA9)-PMMA67 nanoparticles to 110 °C for 17 h; 1H NMR spectra for PLMA63-PGlyMA89 after heating to 40, 70 or 90 °C for 17 h; FT-IR spectra recorded for PLMA63-PGlyMA89 before and after heating to 110 °C for 17 h; summary of possible side-reactions for the derivatization of PLMA63-PGlyMA89 with 50% v/v aqueous acetic acid. See DOI: https://doi.org/10.1039/d2py00559j |
| This journal is © The Royal Society of Chemistry 2022 |