
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOperando PEPICO unveils the catalytic fast pyrolysis mechanism of the three methoxyphenol isomers†
Zeyou
Pan
 ab,
Andras
Bodi
a,
Jeroen A.
van Bokhoven
ab and
Patrick
Hemberger
*a
ab,
Andras
Bodi
a,
Jeroen A.
van Bokhoven
ab and
Patrick
Hemberger
*a
aPaul Scherrer Institute, 5232 Villigen, Switzerland. E-mail: patrick.hemberger@psi.ch
bInstitute for Chemical and Bioengineering, Department of Chemistry and Applied Biosciences, ETH Zurich, 8093 Zurich, Switzerland
First published on 9th September 2022
Abstract
The development of lignin valorization processes such as catalytic fast pyrolysis (CFP) to produce fine chemicals and fuels leads to a more sustainable future. The implementation of CFP is enabled by understanding the chemistry of lignin constituents, which, however, requires thorough mechanistic investigations by detecting reactive species. In this contribution, we investigate the CFP of the three methoxyphenol (MP) isomers over H-ZSM-5 utilizing vacuum ultraviolet synchrotron radiation and operando photoelectron photoion coincidence (PEPICO) spectroscopy. All isomers demethylate at first to yield benzenediols, from which dehydroxylation reactions proceed to produce phenol and benzene. Additional pathways to form benzene proceed over cyclopentadiene, methylcyclopentadiene, and fulvene intermediates. The detection of trace amounts of methanol in the product stream suggests a demethoxylation reaction to yield phenol. Guaiacol (2- or ortho-MP) exhibits slightly higher reactivity compared to 3-MP and 4-MP, due to the formation of the fulvenone ketene, which opens additional routes to benzene and phenol. When compared to benzenediol catalytic pyrolysis, the additional methyl group in MP leads to high conversion at lower reactor temperatures, which is mostly owed to the lower H3C–O vs. H–O bond energy and the possibility to demethoxylate to produce phenol.
Introduction
Lignin can be converted into fine chemicals and fuels via catalytic fast pyrolysis (CFP).1–3 Because lignin is a complex, three-dimensional, diversely functionalized, and amorphous macromolecule, its CFP is often poorly controllable and high selectivity towards targeted products is rarely achieved. To improve the selectivity targetedly and to reduce costs, it is critical to unveil the CFP reaction mechanism. In the bottom-up approach, common lignin model compounds, such as guaiacol, eugenol, and guaiacylglycerol-b-guaiacyl ether (GGGE), are investigated to understand the reaction mechanism in increasing order of complexity,5–9 which provides insights into the phenomena and trends reported in top-down experiments.10,11 Our group has studied the reaction mechanism of guaiacol over H-USY by photoelectron photoion coincidence (PEPICO) spectroscopy and observed the fulvenone ketene as a reactive intermediate, responsible for the branching of the reaction pathways into phenol and cyclopentadiene.5 Liu et al. compared GGGE pyrolysis with and without a catalyst by in situ atmospheric pressure photoionization high-resolution mass spectrometry. A phenolic pool on H-ZSM-5 catalyst was discovered.7 Recently, Liu et al. explored the pyrolysis chemistry of guaiacol and its derivatives 4-methylguaiacol, 4-vinylguaiacol, and vanillin using synchrotron photoionization mass spectrometry (PIMS) and proposed systematic reaction pathways.9An advanced understanding of the reaction mechanism of lignin model compounds can help us model and, eventually, control CFP. In general, the underlying chemistry of different isomers calls for advanced spectroscopic methods to disentangle the reaction mechanisms by isomer-selective detection of reactive species. For instance, in the catalytic pyrolysis of the three benzenediol isomers, catechol, resorcinol and hydroquinone,4 it was found that vicinal hydroxyl groups in the ortho isomer (catechol), can dehydrate at low temperature to form the fulvenone ketene, leading to a high conversion of o-benzenediol, while the more remote hydroxyl groups in m- and p-benzenediol will react individually and only at higher temperature, but still lead to a similar product distribution. This indicates, on the one hand, that the distance between the two hydroxyl groups has a strong effect on the reactivity and mechanisms without significantly affecting the product pool. On the other hand, Gerlach et al. pointed out that differences between the decomposition mechanism of the benzenediol isomers in non-catalytic pyrolysis also affect the product distribution.12 In m-xylene, it was found that the fuel radical, m-xylyl, cannot decompose by direct hydrogen loss, as this would result in the high-energy biradical m-xylylene species. Instead, the m-xylyl radical rearranges to the ortho and para isomers before a hydrogen is lost, as shown in pyrolytic microreactors13–15 and in flame sampling16 using vacuum ultraviolet (VUV) synchrotron radiation and photoelectron photoion coincidence detection (PEPICO). This technique permits us to measure photoion mass-selected threshold photoelectron spectra (ms-TPES), which are used in catalysis, combustion and atmospheric chemistry investigations to detect reactive species isomer-selectively and elucidate reaction mechanisms and kinetics.17–27 These examples emphasize the different reactivities and dynamics based on the substitution pattern. To get a comprehensive understanding of lignin CFP, it is important to refine the conversion mechanism of model compounds down to the individual isomers to align with top-down results,10,11 where lignin samples are directly investigated.
This study aims to understand the chemistry of catalytic pyrolysis of methoxyphenol (MP) isomers using H-ZSM-5 as catalyst. The pyrolysis-PEPICO technique allows us to detect elusive intermediates, such as radicals, and identify isomers, thereby revealing reaction pathways. In this contribution, we reveal how the position of the methoxy group influences the initial decomposition step, the conversion, and the product distribution of MP isomers. This work lays the groundwork for the study of more complicated hydroxyl/methoxy-containing derivatives and may guide the optimization of the process towards high selectivity to desired products.
Experimental
The PEPICO experiments have been performed utilizing the double imaging photoelectron photoion coincidence CRF-PEPICO endstation at the vacuum ultraviolet (VUV) beamline of the Swiss Light Source (SLS) at Paul Scherrer Institute, Switzerland.28–30 The three methoxyphenol MP isomers (Sigma-Aldrich, >97%) were placed in an in-vacuum sample container, packed within a quartz wool bed. The methoxyphenol sample temperature and thus the vapor pressure in the sample container was adjusted by a water thermostat (Huber Minichiller). The vaporized methoxyphenols were mixed with argon (PANGAS, 4.8) in the sample container and directed into a tubular quartz microreactor (2 mm inner diameter) with a 26 mm long catalyst bed (9–11 mg H-ZSM-5) embedded and fixed in position with quartz wool at a 20 sccm mass flow rate. The reactor assembly was resistively heated utilizing a thermocoax® wire wrapped around a cylindrical manifold over a length of 26 mm and an adjustable DC power supply (Voltcraft).4 The outer surface temperature was monitored by a type K thermocouple attached at the midpoint of the reactor and calibrated to obtain the catalyst temperature. The gas mixture containing the reactant, intermediates and products leaves the 1 mm microreactor outlet and forms an effusive molecular beam upon expansion into high vacuum (1–5 × 10−5 mbar). The beam is skimmed by a 2 mm skimmer, and the species along the centerline of the expansion enter the ionization chamber (10−6 mbar). There, the gas jet is intersected with monochromatic VUV synchrotron light. To obtain the ionizing radiation, bending magnet radiation was collimated, dispersed by a monochromator with a 150 mm−1 grating at an energy resolution of E/ΔE ≈ 1500, and focused onto the exit slit in the differentially pumped rare gas filter filled with 10 mbar of a mixture of Ar, Ne, and Kr to absorb higher order radiation.28 The VUV light then enters the ionization chamber, where it is absorbed by the gaseous sample to generate photoelectrons and photoions. These are accelerated by a constant, 218 V cm−1 electric field and detected in delayed coincidence by fast delay-line anode detectors in velocity map imaging (VMI) conditions.31 To identify the species, photoion mass-selected threshold photoelectron spectra (ms-TPES) were recorded by integrating only the electrons in the center of the detector, close to zero kinetic energy spot. The hot electron contribution was subtracted utilizing the method of Sztaray and Baer.32 The species were identified by comparing the ms-TPES with reference spectra and Franck–Condon (FC) simulations in conjunction with calculated ionization energies.Quantum chemical calculations were performed utilizing the Gaussian (16 rev. A.03) suite.33 Adiabatic ionization energies were calculated using the G4 composite method. Franck–Condon simulations of the photoelectron spectra were carried out using the optimized geometries and vibrational frequencies at the B3LYP/6-311++G(d,p) level. The stick spectra were convolved with a Gaussian function of 20–50 meV full width at half maximum.
Results
(1) Mass spectra
The non-catalytic unimolecular thermal decomposition of methoxyphenols from Custodis et al.34 and Scheer et al.35 is summarized in R1 and is initiated by methyl radical loss at temperatures above 600 °C. The resulting hydroxy phenoxy radical is prone to decarbonylation to yield the hydroxycyclopentadienyl radical.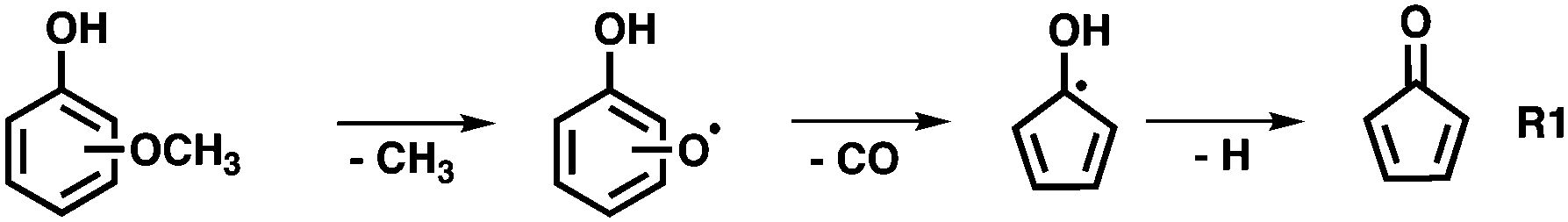
The latter dehydrogenates to produce cyclopentadienone and will finally yield C5H4 isomers. In the presence of a H-ZSM-5 catalyst, pyrolysis starts at the much lower temperature of ca. 440 °C for all three methoxyphenol isomers, as depicted in Fig. 1. Besides the methoxyphenol starting material (m/z 124), we observe mostly m/z 110, 108, 78 and also minor peaks at m/z 82 and 66 at lower temperatures. Upon increasing the temperature to 489 °C, guaiacol (2-MP) shows already high conversion to secondary and tertiary products. These are also observed in 3-MP and 4-MP but only at higher temperatures. Heavier species than MP are also found during CFP at, e.g., m/z 128, 138, and 142, while lighter compounds are observed as the reactor temperature is further increased. Blank experiments (Fig. S1, ESI†), where the catalyst was replaced by a quartz wool bed, show products at m/z 110, 80, 108 only above 580 °C, emphasizing the important role of the zeolite catalyst in this process.
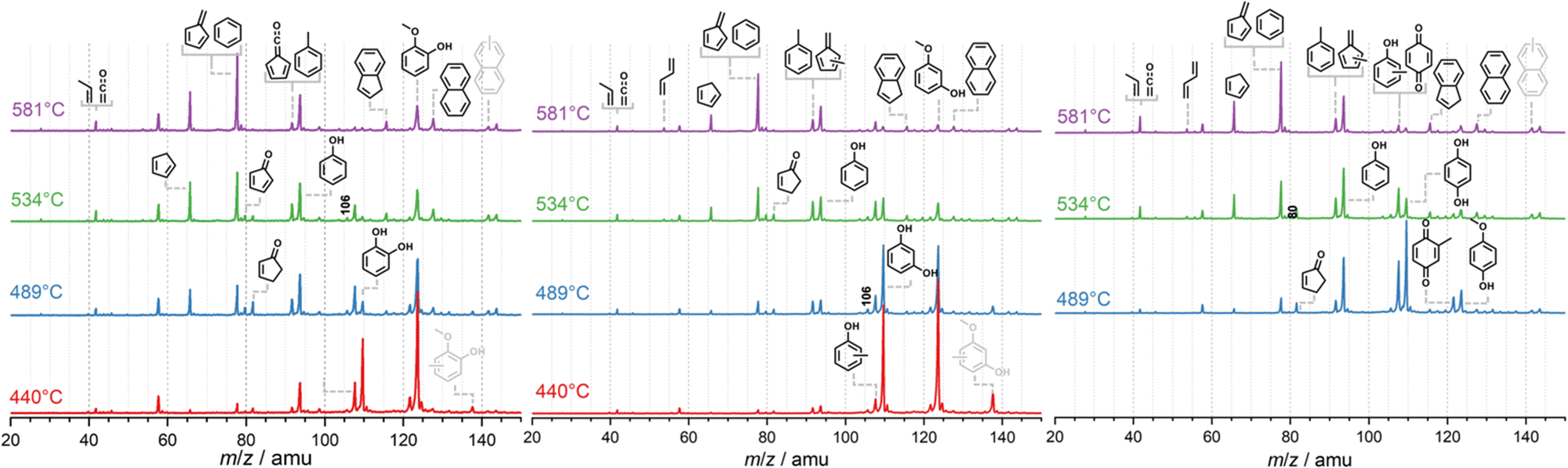 | ||
| Fig. 1 Mass spectra during the catalytic fast pyrolysis of 2-, 3- and 4-methoxyphenol as function of reactor temperature. Catalytic pyrolysis starts at around 440 °C. Conditions: hv = 10.5 eV; ∼0.01% sample in Ar; H-ZSM-5 (Si/Al = 25). The small peak at m/z 58 is assigned to acetone, an impurity in the detection chamber. | ||
(2) Species identification
In order to assign the mass speaks in Fig. 1, photoion mass-selected threshold photoelectron spectroscopy (ms-TPES) was utilized as an isomer-selective tool. Fig. 1 depicts ms-TPES of the stable reaction products appearing during the CFP of the three MP isomers. The spectra are colour coded for 2-MP (red), 3-MP (blue) and 4-MP (green). The ms-TPES of m/z 66 and 80 was assigned to cyclopentadiene and methyl-cyclopentadienes, based on the excellent agreement with the reference and FC simulated spectra.4,36 When reference spectra of reactive molecules are unavailable, the FC approach to model the vibrational transitions from the neutral into the ground electronic state of the cation has been proven to be an excellent alternative to assign the spectral carriers.24,27 The data for the m/z 78 mass channel shows rich features around 8.4 eV and 9.2 eV, which was identified as the two C6H6 isomers, fulvene (c-C5H4![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2) and benzene. Fulvene ionizes at 8.398 eV into the cation ground state (
CH2) and benzene. Fulvene ionizes at 8.398 eV into the cation ground state (![[X with combining tilde]](https://www.rsc.org/images/entities/b_char_0058_0303.gif) + 2A2 ← 1A1), while it is also responsible for the features at 9.55 eV, which results from transitions into the excited state (+ 2B1 ← 1A1).37 The sharp peak at 9.241 eV is assigned to the doubly degenerate + 2E1g cation ground electronic state of benzene.37,38 The experimental spectrum of m/z 78 agrees well with the reference spectra of fulvene and benzene and a very similar composition is found in all three methoxyphenol isomers.
+ 2A2 ← 1A1), while it is also responsible for the features at 9.55 eV, which results from transitions into the excited state (+ 2B1 ← 1A1).37 The sharp peak at 9.241 eV is assigned to the doubly degenerate + 2E1g cation ground electronic state of benzene.37,38 The experimental spectrum of m/z 78 agrees well with the reference spectra of fulvene and benzene and a very similar composition is found in all three methoxyphenol isomers.
The ms-TPE spectra of m/z 94 shows an excellent match with the phenol reference spectrum.4 The spectrum of m/z 108 can be associated with the three methyl phenol isomers (cresols). However, if 4-MP is used as precursor (Fig. 2, m/z 108, green trace), we find a sharp origin transition at ca. 10 eV and a small vibrational feature at ca. 10.2 eV, which agrees well with the reference spectrum of p-benzoquinone. At higher mass-to-charge ratios, we identify m/z 116 and 128 as indene and naphthalene, respectively, based on their reference spectra and well-defined ionization energies between 8.1 and 8.2 eV.39–41
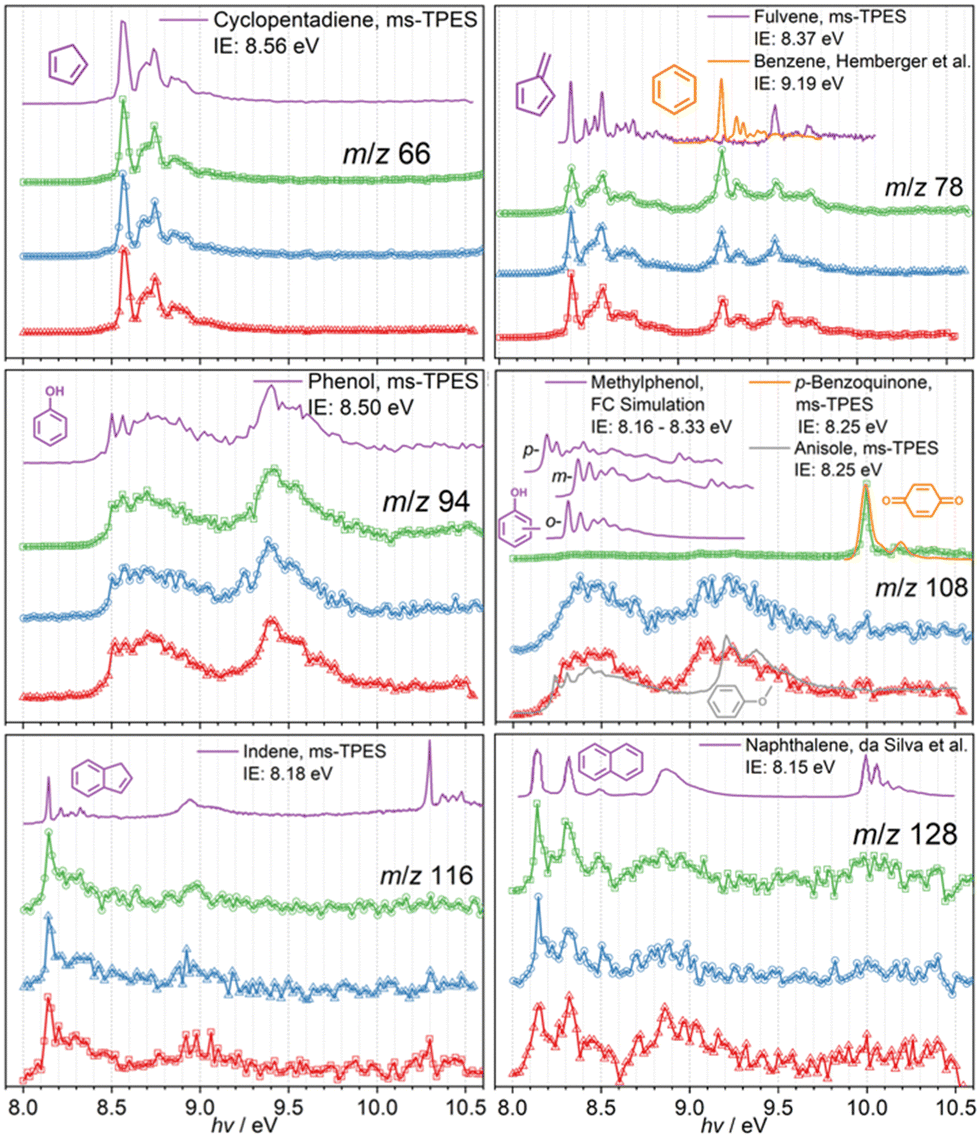 | ||
| Fig. 2 Photoion mass-selected threshold photoelectron spectra of the most abundant stable reaction products of the catalytic fast pyrolysis of the three methoxyphenol isomers (2-MP (red), 3-MP (blue), and 4-MP (green)). Reference spectra and FC simulations yield unambiguous assignments for cyclopentadiene (m/z 66), fulvene and benzene (m/z 78), phenol (m/z 94), ortho-, meta-, and para-methylphenols, as well as p-benzoquinone (m/z 108), indene (m/z 116), and, finally, for naphthalene (m/z 128). | ||
Reactive intermediates, also identified using ms-TPE spectra, are depicted in Fig. 3. The sharp origin transition of the methyl radical at a photon energy of 9.84 eV proves its presence in the CFP of 3- and 4-MP, while the signal is likely lost in the noise in 2-MP. The mass spectra in Fig. 1 do not display methyl radicals, likely because of the insufficient signal-to-noise ratio due to the false coincidence background, but the dynamic range of the experiment can be improved by plotting the ms-TPE close to the ionization onset, where the methyl radical has resonant transitions, to identify this reactive species in the complex reaction mixture.
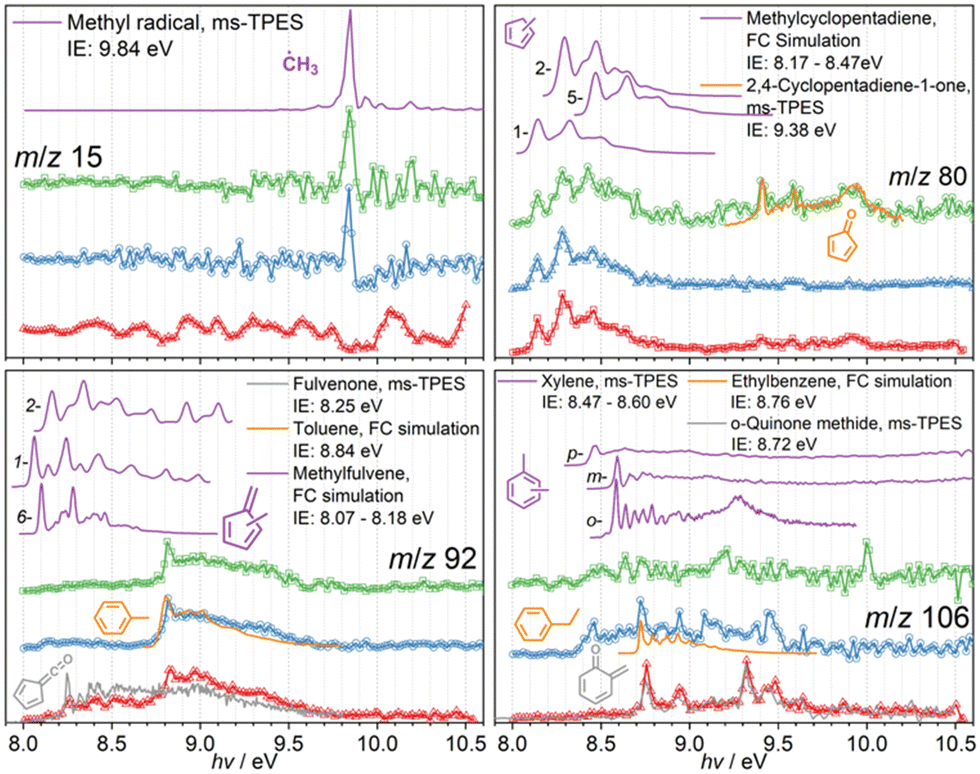 | ||
| Fig. 3 Photoion mass-selected threshold photoelectron spectra (ms-TPES) of reactive species in the catalytic pyrolysis of three methoxyphenol isomers 2-MP (ortho, red), 3-MP (meta, blue), and 4-MP (para, green). Methyl radicals (m/z 15) are evidently found in the meta and para isomer, while methylcyclopentadienes and traces of cyclopentadienone are assigned to m/z 80. While in the 3-MP (blue) and 4-MP (green) isomer, we find toluene and xylenes and traces of ethylbenzene in the ms-TPES of m/z 92 and 106, respectively, the 2-MP isomer shows strong contributions of fulvenone and ortho-quinone methide. | ||
The ms-TPES of m/z 80, as shown in Fig. 3, can be reproduced well using 1-, 2-, and 5-methyl cyclopentadiene Franck–Condon simulations, responsible for the features in the 8.2–8.7 eV range, while minor contributions of cyclopentadienone (c-C5H4O) are also identified based on the ionization energy at 9.41 eV and its reference spectrum.42 While the ms-TPE spectra of m/z 92 in the 3-MP and 4-MP CFP are identical and can be assigned to toluene based on the FC-simulation, the 2-MP (red trace) isomer sticks out and shows signal already at 8.25 eV. These features are associated with the fulvenone ketene (c-C5H4CO) and compare well with the literature spectrum.43 The m/z 106 signal is very low in 3- and 4-MP CFP and can tentatively be assigned to ortho-, meta- and para-xylene. Here again, 2-MP shows a different behavior and strong features at 8.75, 8.95, 9.3 and 9.5 eV appear in the ms-TPE spectrum. It was found that the o-quinone methide, also a highly reactive species, is responsible for these features.44 The ms-TPES of the remaining species at m/z 40, 42, 52, 54, 82, 110, 122, and 124 are shown in the ESI† (Fig. S2) and are associated with propyne, ketene & propene, vinylacetylene, 1,3-butadiene, and cyclopentenone, respectively.
(3) Temperature dependence & comparison to benzenediol reactivity
Hydrocarbons, such as cyclopentadiene (m/z 66), benzene (m/z 78), indene (m/z 116), and naphthalene (m/z 128) are produced and desorbed from the catalyst surface in larger quantities at high reactor temperatures (see Fig. 4). Phenols (m/z 94) and methylphenols (m/z 108, cresols or anisole) are probably consumed in secondary reactions as indicated by their decreasing signal as the temperature increases. A similar trend is also observed for methylcyclopentadienes at m/z 80, cyclopentenone (m/z 82) and species at m/z 92, such as fulvenone (in the case of 2-MP) and toluene. Small amounts of methanol (m/z 32) are detected in all three isomers at lower reactor temperatures, but its signal vanishes at higher temperatures (Fig. 4).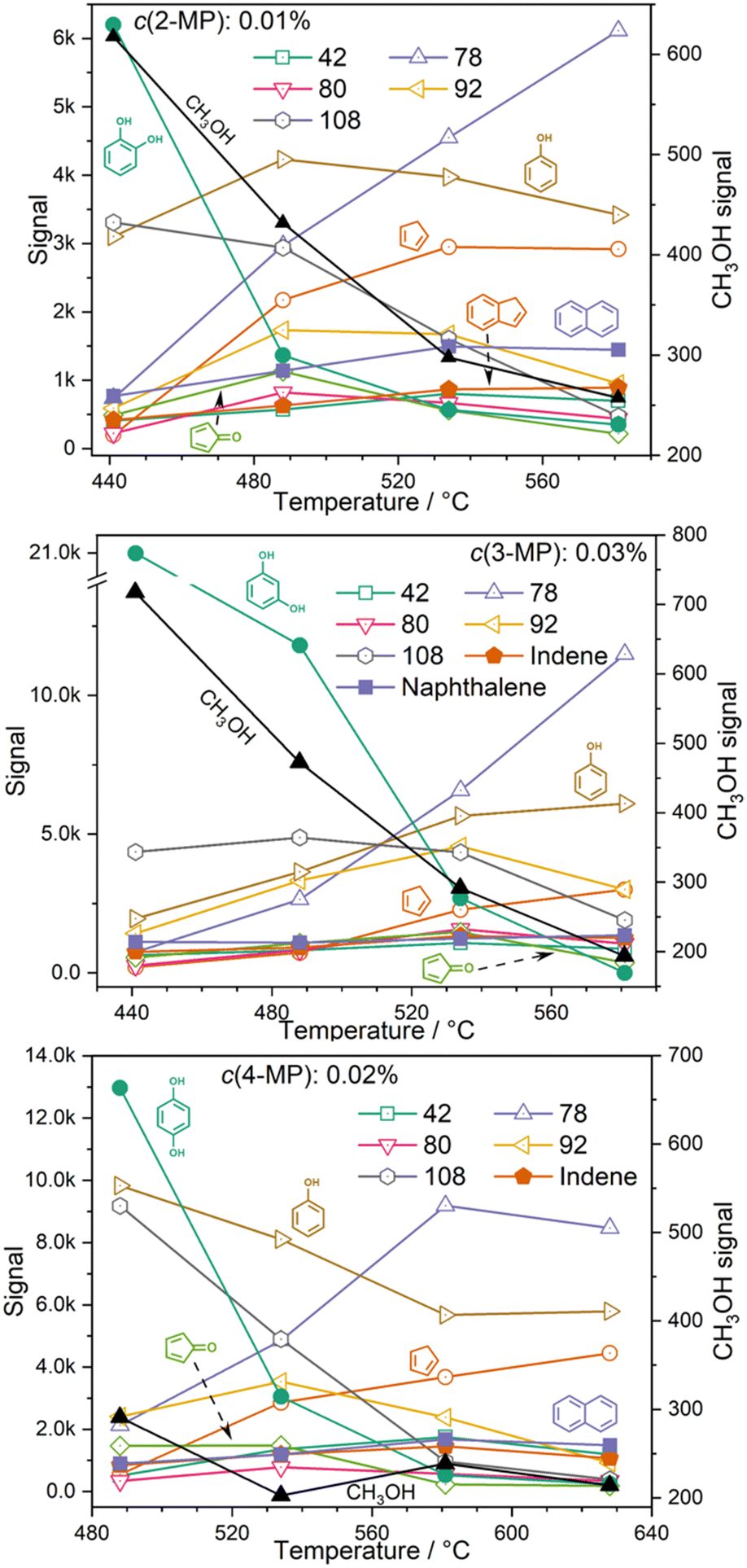 | ||
| Fig. 4 Temperature dependence of MP CFP. A similar pattern is observed across the isomers, with a decreasing signal intensity at m/z 110 (benzenediols), and m/z 94 (phenol) while the C6H6 (m/z 78, benzene and fulvene), indene (m/z 116) and naphthalene (m/z 128) signals steadily increase with increasing temperature. | ||
In general, the O–H bond energy is around 100 kJ mol−1 higher than that of the O–CH3 bond, which should lead to an increase in the MP reactivity with respect to benzenediols. Indeed, by comparing the resorcinol reactivity4 with 3-MP at ca. 500 °C reactor temperature using H-ZSM-5, it was found that the latter shows nearly 50% selectivity to resorcinol and a much higher conversion (Fig. 5). Thus, methylation leads to an increase in reactivity. Considering the temperature dependent behavior as well as the comparison of the 3-MP CFP results with those on resorcinol, we can elucidate the pyrolysis mechanism below.
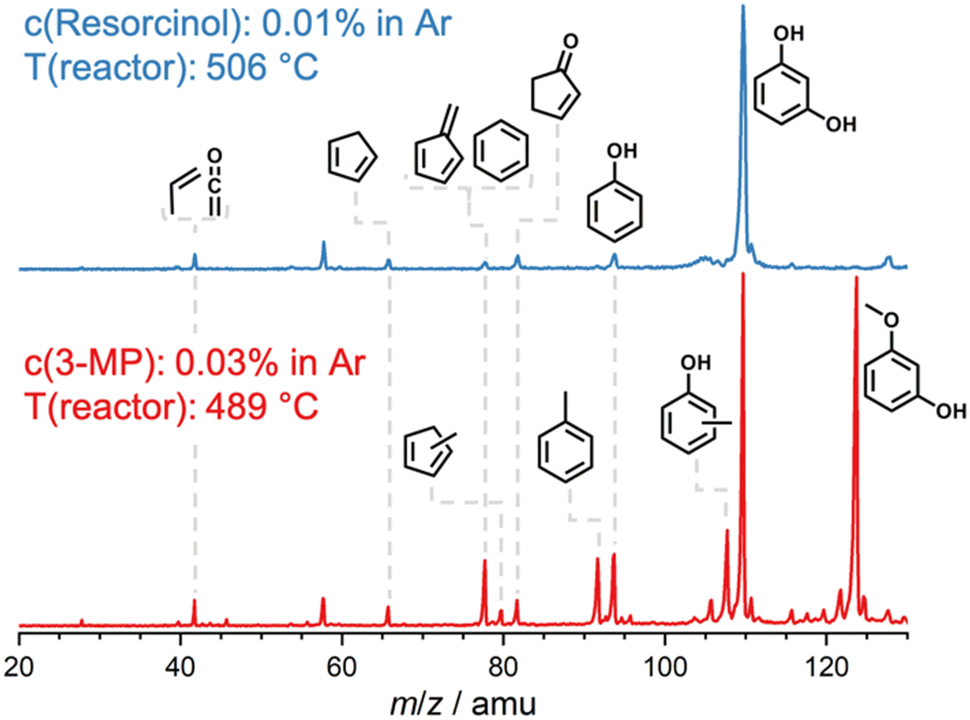 | ||
| Fig. 5 Catalytic pyrolysis mass spectra of resorcinol4 and 3-MP over H-ZSM-5 at ca. 500 °C. The higher 3-MP conversion as compared to resorcinol is associated with the lower H3C–O bond energy (see text). | ||
Discussion
The catalytic fast pyrolysis of the 2-, 3- and 4-MP isomers over H-ZSM-5 is initiated by a demethylation reaction, yielding surface bound hydroxy-phenoxy and methyl species. This initial reaction step is similar to the flash vacuum pyrolysis as described in R1, except that demethylation of the methoxyphenols, followed by hydrogen addition, takes place on the zeolite surface according to reaction R2. This pathway is proven by the unequivocal detection of the desorbed methyl radicals at high reactor temperatures (see m/z 15 ms-TPES in Fig. 2).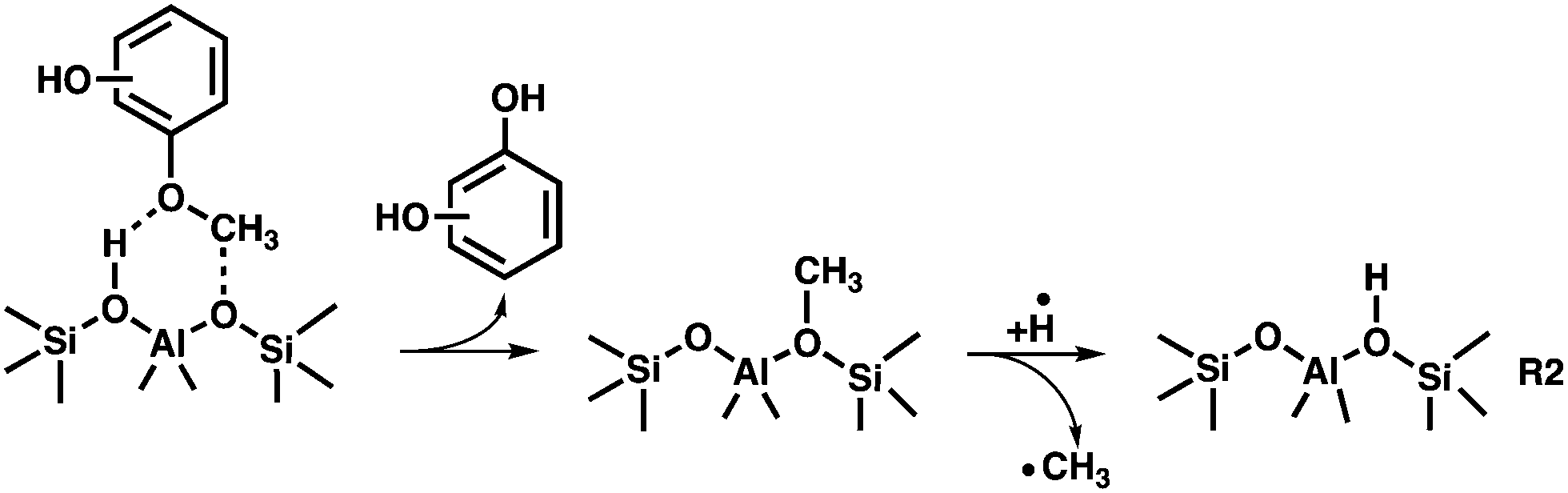
Consequently, the three benzenediols (catechol, resorcinol, and hydroquinone) are formed on the zeolite surface followed by desorption into the gas phase. Alternatively, the benzenediols may further decompose following diffusion to another active site (see below). If we compare the temperature dependence of the CFP of the benzenediols4 with that of the MPs, a higher conversion can be achieved at lower reactor temperatures in the latter case (see Fig. 5). Due to the lower bond energy of O–CH3vs. O–H, the methyl group increases the reactivity, which significantly lowers the activation energy to initiate the first decomposition reaction on the zeolite surface. Despite the early-onset formation of the three benzenediols, high phenol signals are found, which suggest that it, too, belongs to the primary reaction products. Starting from the activated methoxyphenol complex on the surface, a demethoxylation reaction to produce phenol is likely. This is rationalized by the detection of traces of methanol (m/z 32) formed at intermediate reactor temperatures (R3). Due to the high reactivity of methanol as methylation agent, it vanishes at higher reactor temperatures. Furthermore, methanol may also participate in the classical methanol-to-olefins (MTO) cycle,45–48 which explains the formation of ethylene, propylene and the ethenone ketene (see Fig. S2, ESI†).
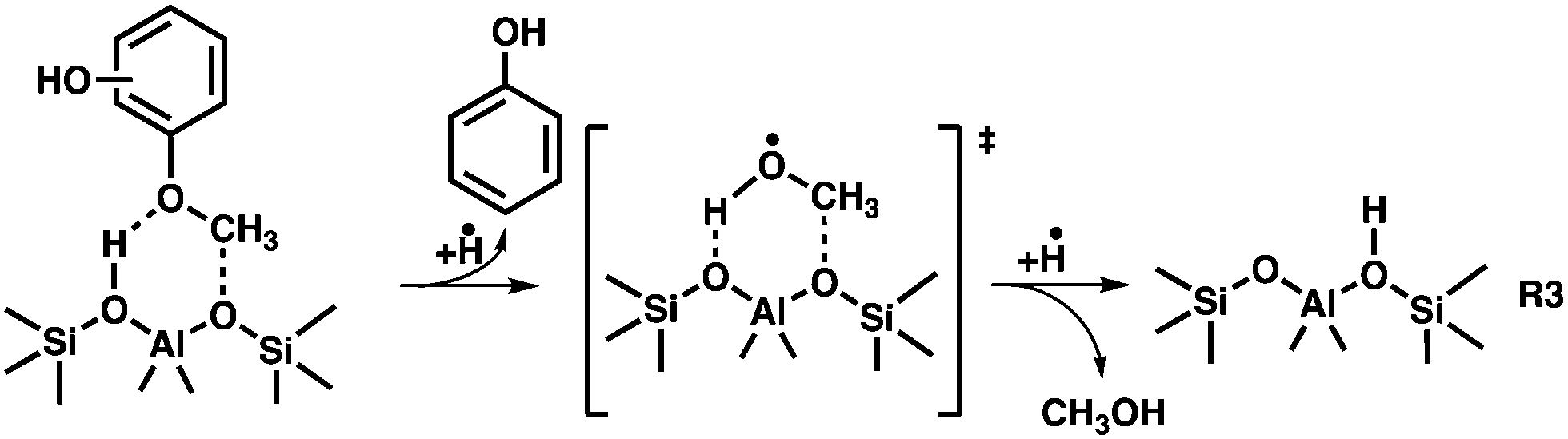
The dominance of the benzenediol primary products means that we can employ and extend the catechol, resorcinol and hydroquinone CFP surface catalysed reaction mechanisms4 to account for the methoxyphenol processes as shown in Fig. 6. Isomer 1 is in equilibrium with tautomer 2, which readily decarbonylates to hydroxycyclopentadiene 3. Hydroxycyclopentadiene is desorbed from the catalyst surface and observed as cyclopentenone 4. In our previous studies, we found 4 to be an excellent precursor of cyclopentadiene 6 following dehydroxylation over H-ZSM-5.4
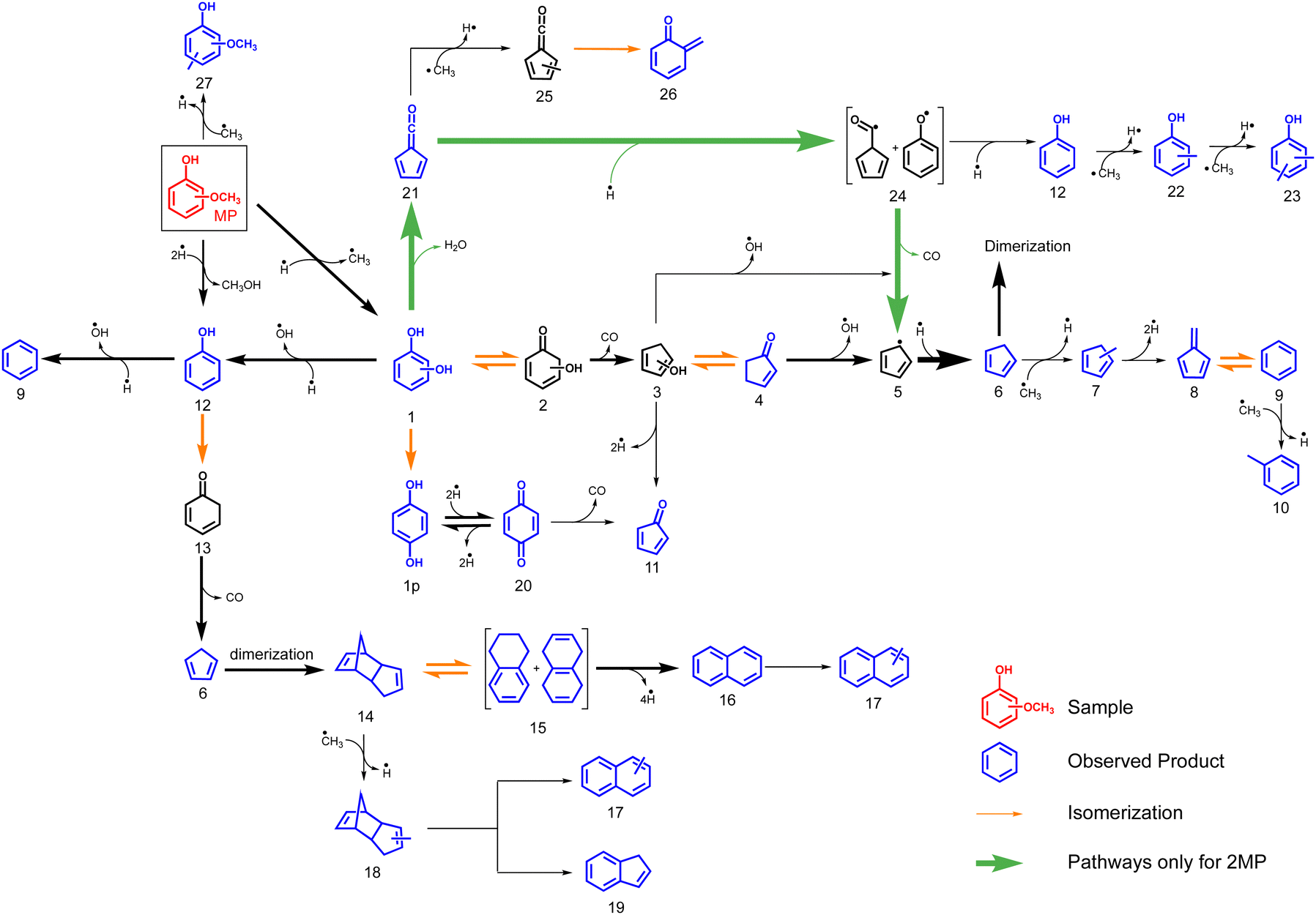 | ||
| Fig. 6 Overview of the reaction mechanism of the catalytic fast pyrolysis of 2–4-methoxyphenol (n-MP, n = 2–4) over H-ZSM-5 catalyst, based on the benzenediol CFP mechanism.4 | ||
Due to the large abundance of methyl species on the H-ZSM-5 surface, 6 is preferentially methylated to 7 and dehydrogenates to yield fulvene 8. This rationalizes the temperature-dependent behaviour seen in Fig. 4. At these conditions, fulvene 8 also rearranges to benzene 9,37 which is methylated to yield toluene 10 (Fig. 6). The minor formation of cyclopentadienone 11 is in contrast to the non-catalytic pyrolysis of the methoxyphenols (R1), which yields this reactive species in large quantities.34,35
Based on the benzenediol 1 catalytic fast pyrolysis mechanism, a second deoxygenation channel is identified.4 Catechol, resorcinol and hydroquinone 1 are dehydroxylated over H-ZSM-5 to form phenol 12. Phenol tautomerizes to intermediately form 13, which decarbonylates to cyclopentadiene 6, a reaction also observed in non-catalytic thermal decomposition (Fig. 6).49 Alternatively, phenol 12 dehydroxylates to form benzene 9, which marks a second pathway besides the 7 → 8 → 9 methylation and rearrangement reaction, proven by 13C-labeling of the methoxy group in the catalytic pyrolysis of guaiacol.5
We also investigated the mechanism of the cyclopentadiene dimerization 6 → 14 over H-ZSM-5 recently, which explains the initiation of polycyclic aromatic hydrocarbon (PAH) formation in the methoxyphenol catalytic pyrolysis.4 Here, dicyclopentadiene 14 rearranges to yield dihydronaphthalenes 15 at m/z 132, which subsequently lose hydrogens to finally yield naphthalene 16. Indeed, species at m/z 130 and 132 are observed at lower temperatures, which decrease with increasing temperature, evidencing this pathway (see Fig. 1). Since surface methyl species are readily available, methylation of naphthalene can be trivially explained to form 17. Indene 19, on the other hand, was only observed in smaller amounts during the CFP of the three benzenediols, while appreciable concentrations can be seen from the mass spectra in Fig. 1. This suggests that the abundant methyl species on the H-ZSM-5 surface may be responsible for the formation of indene. One pathway may proceed via a subsequent grow of the C3 chain at the benzene and ring closure. However, the small xylene signal at m/z 106, in comparison to benzene (m/z 78), does not justify this stepwise CH3 addition pathway. We speculate that a feasible alternative is the methylation of the dicyclopentadiene 14, to yield 18, which subsequently loses hydrogen and ethylene to produce 19. However, the missing intermediate signals mean that this pathway remains elusive.
Additionally, we find species particular to the catalytic pyrolysis of 2-MP and 4-MP, namely p-benzoquinone 20 and fulvenone 21. These are formed during dehydrogenation from hydroquinone 1p and dehydration of catechol 1o, respectively.420 is a known precursor of cyclopentadienone 11, which was observed in trace amounts herein.50 As known from the benzenediol CFP mechanism, fulvenone 21 is only observed in the ortho isomer, which correlates with the higher reactivity at lower reactor temperatures in the 2-MP case (see Fig. 1). This channel is responsible for the phenol 12 and cyclopentadiene 6 formation over the intermediates 24.4,5 Methylation of phenol 12 explains the appearance of cresols 22 and xylenols 23, while methylation of fulvenone 21 to methyl fulvenone 25, may be responsible for the observation of the o-quinone methide 26.
At lower reactor temperatures, secondary and tertiary reactions are only observed in the 2-MP case (Fig. 1), an effect readily attributed to the unique formation of fulvenone 21 in the ortho isomer, which boosts the reactivity by opening up third reaction channel (green pathway Fig. 6). However, this effect is less pronounced in MPs than in benzenediols, which is owed to the methyl group at the oxygen, giving rise to demethoxylation reactions to produce phenol (MP → 12, Fig. 6). This proves the higher reactivity of methoxy groups as compared to the bare alcohols in lignin model compounds.
Summary & conclusion
We have investigated the catalytic fast pyrolysis of the ortho-, meta- and para-methoxyphenol (2–4-MP) isomers over H-ZSM-5 in the 400 to 600 °C temperature range. At lower reactor temperatures, benzenediols, phenol, cresols, and anisole are detected by operando photoelectron photoion coincidence spectroscopy. Mechanistically, the reaction is initiated by demethylation to yield benzenediols and further proceeds via stepwise dehydroxylation to phenol and benzene. The highest reactivity of the three MP was observed in 2-MP (guaiacol), which we attribute to the formation of fulvenone (c-C5H4CO), a reactive intermediate that can only be accessed in the ortho isomer. Fulvenone ketene is either hydrogenated to form the phenoxy radical and phenol, or it can decarbonylate to yield cyclopentadiene. The primary reaction products will decompose at higher reactor temperatures to yield aromatics (benzene, toluene) and polycyclic aromatic hydrocarbons (PAHs), such as naphthalene, indene, and their methylated analogues. Furthermore, methylation of all intermediates and products gives rise to alternative reaction pathways to benzene formation, following the cyclopentadiene → methyl-cyclopentadiene → fulvene → benzene sequence. If we compare the CFP of isomeric benzenediols to the analogous MPs, we find higher conversion already at lower reaction temperatures in the latter, which indicates that the additional methyl group increases the reactivity, due to the lower H3C–O bond energy, giving rise to activated surface species. Surface-bound methyl desorbs at high temperatures and is detected and is also responsible for abundant surface catalysed methylation reactions, leading to, e.g., methylcyclopentadiene, methylfulvene and toluene. The weak but unambiguous methanol signal in the gas phase gives insights into demethoxylation pathways, which can additionally boost the reactivity towards phenols, and are also found together with methanol already at lower reactor temperatures. This rationalizes the increased reactivity of the methoxy phenols, as compared to the benzenediols.
In this study, we expanded on previous insights into the methoxyphenols CFP mechanism by investigating the 3- and 4-MP isomers in addition to guaiacol, 2-MP, and connecting the CFP mechanism to that of the well-characterized benzenediol chemistry to understanding the role of isomerism. This is particularly important in the bottom-up approach to obtain a detailed mechanistic knowledge of the chemistry and reactivity of model compounds to identify the driving forces and elementary reactions in lignin CFP. In the case of MP isomers, the observed product pool was not isomer-dependent. However, the methylation increases the reactivity compared to the benzenediols and helps to reduce the reaction temperatures. The formation of the fulvenone ketene in the 2-MP case is still beneficial for the overall reactivity, but demethoxylation in 3- and 4-MP can similarly increase the reactivity. Thus, tailoring lignin towards methoxy functionalities may help to reduce reaction temperatures to make the process more controllable and avoid high temperature products such as PAHs, which are the precursors of carbonaceous depots, reducing the activity of the zeolite.
Author contributions
ZP: investigation, visualization, validation, formal analysis, writing manuscript. AB: discussion of the data, corrections to the manuscript. JAvB: discussion of the data, corrections to the manuscript, thesis supervisor. PH: conceptualization, data analysis, writing manuscript, resources, supervision, project administration, funding acquisition.Data availability
Data presented in the main figures of the manuscript are publicly available through the repository: https://doi.psi.ch/detail/10.16907%2F1fd4c4aa-21fb-4d51-8bfb-9c606aae830d.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The measurements were performed at the VUV beamline of the Swiss Light Source located at Paul Scherrer Institute, Villigen, Switzerland. P. H. and Z. P. are grateful for the funding by Swiss National Science Foundation (SNSF, 200021_178952). We thank Patrick Ascher for technical assistance.Notes and references
- W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- A. I. Osman, N. Mehta, A. M. Elgarahy, A. Al-Hinai, A. A. H. Al-Muhtaseb and D. W. Rooney, Environ. Chem. Lett., 2021, 19, 4075–4118 CrossRef CAS.
- J. A. Poveda-Giraldo, J. C. Solarte-Toro and C. A. Cardona Alzate, Renewable Sustainable Energy Rev., 2021, 138, 110688 CrossRef CAS.
- Z. Pan, A. Puente-Urbina, A. Bodi, J. A. van Bokhoven and P. Hemberger, Chem. Sci., 2021, 12, 3161–3169 RSC.
- P. Hemberger, V. B. F. Custodis, A. Bodi, T. Gerber and J. A. van Bokhoven, Nat. Commun., 2017, 8, 15946 CrossRef PubMed.
- J.-Y. Kim and J. W. Choi, Fuel, 2019, 240, 92–100 CrossRef CAS.
- C. Liu, X. Chen, X. Liu, C. Cui, Z. Zhou, L. Jia and F. Qi, Angew. Chem., Int. Ed., 2021, 60, 2643–2647 CrossRef CAS PubMed.
- N. Nastasiienko, T. Kulik, B. Palianytsia, J. Laskin, T. Cherniavska, M. Kartel and M. Larsson, Appl. Sci., 2021, 11, 7205 CrossRef CAS.
- P. Liu, J. Huang, K. Yang, H. Zhuang, L. Chen, Y. Pan, J. Yang and L. Jia, Fuel, 2022, 312, 122874 CrossRef CAS.
- A. Dufour, J. Weng, L. Jia, X. Tang, B. Sirjean, R. Fournet, H. L. Gall, N. Brosse, F. Billaud, G. Mauviel and F. Qi, RSC Adv., 2013, 3, 4786–4792 RSC.
- L. Jia, Y. Le Brech, G. Mauviel, F. Qi, M. Bente-von Frowein, S. Ehlert, R. Zimmermann and A. Dufour, Energy Fuels, 2016, 30, 1555–1563 CrossRef CAS.
- M. Gerlach, A. Bodi and P. Hemberger, Phys. Chem. Chem. Phys., 2019, 21, 19480–19487 RSC.
- P. Hemberger, A. J. Trevitt, E. Ross and G. da Silva, J. Phys. Chem. Lett., 2013, 4, 2546–2550 CrossRef CAS.
- P. Hemberger, A. J. Trevitt, T. Gerber, E. Ross and G. da Silva, J. Phys. Chem. A, 2014, 118, 3593–3604 CrossRef CAS PubMed.
- M. Steglich, G. Knopp and P. Hemberger, Phys. Chem. Chem. Phys., 2019, 21, 581–588 RSC.
- T. Bierkandt, P. Hemberger, P. Oßwald, M. Köhler and T. Kasper, Proc. Combust. Inst., 2017, 36, 1223–1232 CrossRef CAS.
- J. H. D. Eland, Photoelectron Spectroscopy, Butterworths, London, 2nd edn, 1984 Search PubMed.
- J. Kruger, G. A. Garcia, D. Felsmann, K. Moshammer, A. Lackner, A. Brockhinke, L. Nahon and K. Kohse-Hoinghaus, Phys. Chem. Chem. Phys., 2014, 16, 22791–22804 RSC.
- P. Oßwald, P. Hemberger, T. Bierkandt, E. Akyildiz, M. Köhler, A. Bodi, T. Gerber and T. Kasper, Rev. Sci. Instrum., 2014, 85, 025101 CrossRef PubMed.
- D. Felsmann, K. Moshammer, J. Krüger, A. Lackner, A. Brockhinke, T. Kasper, T. Bierkandt, E. Akyildiz, N. Hansen, A. Lucassen, P. Oßwald, M. Köhler, G. A. Garcia, L. Nahon, P. Hemberger, A. Bodi, T. Gerber and K. Kohse-Höinghaus, Proc. Combust. Inst., 2015, 35, 779–786 CrossRef CAS.
- D. Schleier, P. Constantinidis, N. Faßheber, I. Fischer, G. Friedrichs, P. Hemberger, E. Reusch, B. Sztáray and K. Voronova, Phys. Chem. Chem. Phys., 2018, 20, 10721–10731 RSC.
- G. Zichittella, M. Scharfe, B. Puértolas, V. Paunović, P. Hemberger, A. Bodi, L. Szentmiklósi, N. López and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2019, 58, 5877–5881 CrossRef CAS PubMed.
- J. Bourgalais, Z. Gouid, O. Herbinet, G. A. Garcia, P. Arnoux, Z. Wang, L. S. Tran, G. Vanhove, M. Hochlaf, L. Nahon and F. Battin-Leclerc, Phys. Chem. Chem. Phys., 2020, 22, 1222–1241 RSC.
- P. Hemberger, J. A. van Bokhoven, J. Pérez-Ramírez and A. Bodi, Catal. Sci. Technol., 2020, 10, 1975–1990 RSC.
- D. P. Mukhopadhyay, D. Schleier, I. Fischer, J. C. Loison, C. Alcaraz and G. A. Garcia, Phys. Chem. Chem. Phys., 2020, 22, 1027–1034 RSC.
- G. Zichittella, P. Hemberger, F. Holzmeier, A. Bodi and J. Pérez-Ramírez, J. Phys. Chem. Lett., 2020, 11, 856–863 CrossRef CAS.
- P. Hemberger, A. Bodi, T. Bierkandt, M. Köhler, D. Kaczmarek and T. Kasper, Energy Fuels, 2021, 35, 16265–16302 CrossRef CAS.
- M. Johnson, A. Bodi, L. Schulz and T. Gerber, Nucl. Instrum. Methods Phys. Res., Sect. A, 2009, 610, 597–603 CrossRef CAS.
- X. Tang, G. A. Garcia, J.-F. Gil and L. Nahon, Rev. Sci. Instrum., 2015, 86, 123108 CrossRef PubMed.
- B. Sztáray, K. Voronova, K. G. Torma, K. J. Covert, A. Bodi, P. Hemberger, T. Gerber and D. L. Osborn, J. Chem. Phys., 2017, 147, 013944 CrossRef PubMed.
- A. Bodi, B. Sztáray, T. Baer, M. Johnson and T. Gerber, Rev. Sci. Instrum., 2007, 78, 084102 CrossRef PubMed.
- B. Sztáray and T. Baer, Rev. Sci. Instrum., 2003, 74, 3763–3768 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- V. B. F. Custodis, P. Hemberger, Z. Ma and J. A. van Bokhoven, J. Phys. Chem. B, 2014, 118, 8524–8531 CrossRef CAS.
- A. M. Scheer, C. Mukarakate, D. J. Robichaud, M. R. Nimlos and G. B. Ellison, J. Chem. Phys. A, 2011, 115, 13381–13389 CrossRef CAS.
- P. J. Derrick, L. Åsbrink, O. Edqvist, B. Ö. Jonsson and E. Lindholm, Int. J. Mass Spectrom. Ion Phys., 1971, 6, 161–175 CrossRef CAS.
- J. Savee, B. Sztáray, P. Hemberger, J. Zádor, A. Bodi and D. L. Osborn, Faraday Discuss., 2022 10.1039/D2FD00028H.
- P. Hemberger, X. Wu, Z. Pan and A. Bodi, J. Chem. Phys. A, 2022, 126, 2196–2210 CrossRef CAS PubMed.
- D. A. da Silva Filho, R. Friedlein, V. Coropceanu, G. Öhrwall, W. Osikowicz, C. Suess, S. L. Sorensen, S. Svensson, W. R. Salaneck and J.-L. Brédas, Chem. Commun., 2004, 1702–1703, 10.1039/B403828B,.
- P. M. Mayer, V. Blanchet and C. Joblin, J. Chem. Phys., 2011, 134, 244312 CrossRef.
- B. West, A. Sit, A. Bodi, P. Hemberger and P. M. Mayer, J. Chem. Phys. A, 2014, 118, 11226–11234 CrossRef CAS PubMed.
- T. K. Ormond, P. Hemberger, T. P. Troy, M. Ahmed, J. F. Stanton and G. B. Ellison, Mol. Phys., 2015, 113, 2350–2358 CrossRef CAS.
- P. Hemberger, Z. Pan, A. Bodi, J. A. van Bokhoven, T. K. Ormond, G. B. Ellison, N. Genossar and J. H. Baraban, ChemPhysChem, 2020, 21, 2217–2222 CrossRef CAS.
- M. B. Prendergast, B. B. Kirk, J. D. Savee, D. L. Osborn, C. A. Taatjes, P. Hemberger, S. J. Blanksby, G. da Silva and A. J. Trevitt, Phys. Chem. Chem. Phys., 2019, 21, 17939–17949 RSC.
- W. Song, J. F. Haw, J. B. Nicholas and C. S. Heneghan, J. Am. Chem. Soc., 2000, 122, 10726–10727 CrossRef CAS.
- P. Tian, Y. Wei, M. Ye and Z. Liu, ACS Catal., 2015, 5, 1922–1938 CrossRef CAS.
- Z. Shi, M. Neurock and A. Bhan, ACS Catal., 2021, 11, 1222–1232 CrossRef CAS.
- A. Cesarini, S. Mitchell, G. Zichittella, M. Agrachev, S. P. Schmid, G. Jeschke, Z. Pan, A. Bodi, P. Hemberger and J. Pérez-Ramírez, Nat. Catal., 2022, 5, 605–614 CrossRef CAS.
- A. M. Scheer, C. Mukarakate, D. J. Robichaud, M. R. Nimlos, H.-H. Carstensen and G. B. Ellison, J. Chem. Phys., 2012, 136, 044309 CrossRef PubMed.
- D. J. Robichaud, A. M. Scheer, C. Mukarakate, T. K. Ormond, G. T. Buckingham, G. B. Ellison and M. R. Nimlos, J. Chem. Phys., 2014, 140, 234302 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp02741k |
| This journal is © the Owner Societies 2022 |