
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photodissociation dynamics of N,N-dimethylformamide at 225 nm and 245 nm
Dennis
Milesevic
 ,
Divya
Popat
,
Patrick
Robertson
and
Claire
Vallance
*
,
Divya
Popat
,
Patrick
Robertson
and
Claire
Vallance
*
Department of Chemistry, University of Oxford, Chemistry Research Laboratory, 12 Mansfield Rd, Oxford, OX1 3TA, UK. E-mail: claire.vallance@chem.ox.ac.uk
First published on 11th November 2022
Abstract
N,N-Dimethylformamide, (CH3)2NCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, is the simplest tertiary amide and a model compound for investigating the photofragmentation of peptide bonds. We report the results of a velocity-map imaging study into the photodissociation dynamics of DMF following excitation at 225 nm and 245 nm. Excitation at either wavelength generates a variety of products, with the primary dissociation pathways involving cleavage of either the N–CO amide bond or an N–CH3 bond. Excitation at 225 nm is predominantly to the S2 21A′′ state via a parallel transition, with dissociation of the amide bond occurring either on this state or on a lower singlet surface following internal conversion. The topographies of all of the potential energy surfaces involved result in dissociation from a range of planar (apart from the methyl-group hydrogen atoms) and non-planar molecular geometries. Dissociation from planar geometries leads to little product internal excitation, correspondingly high photofragment velocities, and near-limiting values of the recoil-anisotropy parameter β. Dissociation from non-planar geometries leads to significant product internal excitation, with correspondingly lower photofragment velocities and breakdown of the axial recoil approximation to give reduced values of β. Excitation at 245 nm involves the same excited-state surfaces, but at the longer wavelength the S2 state can only be reached from non-equilibrium geometries of the ground state, leading to a reduction in the recoil anisotropy parameter relative to excitation at 225 nm. The potential energy curves associated with cleavage of the N–CH3 bond are less well characterised. However, the pathway is characterised by an isotropic angular distribution and a TKER distribution peaking at low energies, both of which can be rationalised in terms of the molecular geometry and the orientation of the transition dipole involved in the excitation step.
O, is the simplest tertiary amide and a model compound for investigating the photofragmentation of peptide bonds. We report the results of a velocity-map imaging study into the photodissociation dynamics of DMF following excitation at 225 nm and 245 nm. Excitation at either wavelength generates a variety of products, with the primary dissociation pathways involving cleavage of either the N–CO amide bond or an N–CH3 bond. Excitation at 225 nm is predominantly to the S2 21A′′ state via a parallel transition, with dissociation of the amide bond occurring either on this state or on a lower singlet surface following internal conversion. The topographies of all of the potential energy surfaces involved result in dissociation from a range of planar (apart from the methyl-group hydrogen atoms) and non-planar molecular geometries. Dissociation from planar geometries leads to little product internal excitation, correspondingly high photofragment velocities, and near-limiting values of the recoil-anisotropy parameter β. Dissociation from non-planar geometries leads to significant product internal excitation, with correspondingly lower photofragment velocities and breakdown of the axial recoil approximation to give reduced values of β. Excitation at 245 nm involves the same excited-state surfaces, but at the longer wavelength the S2 state can only be reached from non-equilibrium geometries of the ground state, leading to a reduction in the recoil anisotropy parameter relative to excitation at 225 nm. The potential energy curves associated with cleavage of the N–CH3 bond are less well characterised. However, the pathway is characterised by an isotropic angular distribution and a TKER distribution peaking at low energies, both of which can be rationalised in terms of the molecular geometry and the orientation of the transition dipole involved in the excitation step.
1 Introduction
Peptide bonds form the backbone of proteins and are of key interest for biochemical processes. Understanding the photostability and photofragmentation of such bonds provides insight into UV-radiation damage to biological systems. Monopeptides have been considered previously as useful model compounds for investigating the peptide bond fragmentation dynamics in a variety of experimental1–7 and theoretical8–11 studies. N,N-Dimethylformamide (DMF) is the simplest tertiary amide, a planar molecule12–15 (apart from the hydrogen atoms on the two methyl groups), and its amide bond offers an ideal candidate to model the photodissociation dynamics of peptide bonds. Additionally, DMF has been postulated to play an important role in the formation of adenine in astrochemical environments.16Experimental and theoretical studies of DMF have focused primarily on dynamics following excitation at wavelengths below 200 nm. Summarising first the theoretical studies, a number of authors have reported potential energy surfaces (PESs) for the ground and electronically excited states. Based on experimental gas-phase spectra of DMF,17,18 Serrano-Andrés and Fülscher19 used the complete active space with second order perturbation theory (CASPT2) to determine vertical excitation energies from the ground state. Liu et al.8 discussed the photolysis mechanism of DMF following excitation at 193 nm based on complete active space self consistent field (CASSCF) calculations of the relevant PESs. Eckert-Maksić and Antol9 also performed CASSCF calculations in the context of understanding DMF photolysis, and were able to characterize several significant points such as energy minima, saddle points, and conical intersections and to determine how the geometry of DMF changes to reach those points. Lipciuc et al.7 determined transition dipole moments for excitations to the first three valence singlet states via CASSCF calculations, and generated cuts through the PESs along N–CO and N–CH3 bonds based on CASPT2 calculations. The arguably most comprehensive theoretical study on DMF has been conducted by Shastri et al.11 These authors correlated the electronic absorption spectrum over the wavelength range 125–221 nm with results from density functional calculations, providing an in-depth discussion of relevant molecular orbitals, vibrational frequencies, and electronically excited singlet and triplet states. They reported potential energy curves for the first five excited states of DMF, including valence and Rydberg states, as a function of bond length. These potential energy curves guide the interpretation of the results reported in this paper, and are summarised in Fig. 1. Note that we include in this figure the 21A′′ Rydberg state, which was not found in the valence-state calculations carried out by Lipciuc et al.7 This now becomes the S2 state in place of the higher-lying 21A′ state that was identified as such by Lipciuc et al. Note that the first two excited singlet states exhibit energy barriers along the N–CO bond, impeding prompt dissociation along that coordinate.
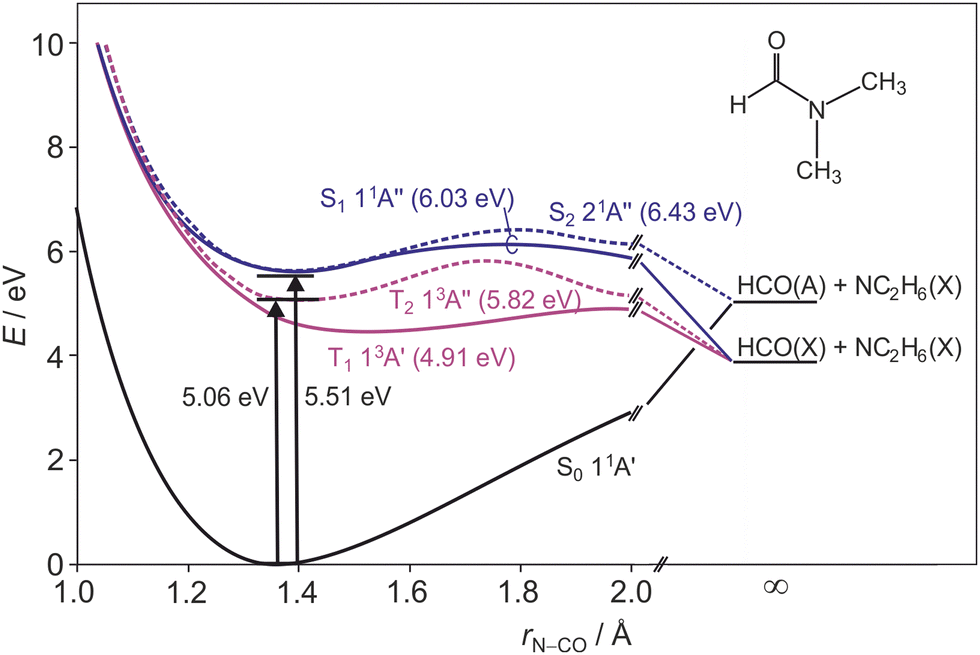 | ||
| Fig. 1 Schematic cuts through the ground state and first two excited singlet and triplet state potential energy surfaces of DMF along the N–CO stretch coordinate, adapted from ref. 11. Barrier heights for each surface are given in parentheses after the state labels, and the dissociation limit for each surface is indicated. | ||
There are comparatively fewer experimental studies on DMF. Forde et al.1,2 performed photodissociation studies at 193 nm and recorded recoil translational energy distributions and angular distributions for the dissociation products. The studies indicated two main dissociation channels: cleavage of the N–CO amide bond to form HCO and N(CH3)2; and fission of one of the two N–CH3 bonds, forming HCONCH3 and CH3. Cleavage of the N–CO bond was found to lead to formation of ground-state HCO(![[X with combining tilde]](https://www.rsc.org/images/entities/char_0058_0303.gif) ) with either ground state N(CH3)2() or electronically excited N(CH3)2(Ã). The products were measured to have a recoil anisotropy of β = 1.2 ± 0.2, implying a rapid bond dissociation to a state for which the transition dipole from the ground state lies predominantly parallel to the breaking bond.
) with either ground state N(CH3)2() or electronically excited N(CH3)2(Ã). The products were measured to have a recoil anisotropy of β = 1.2 ± 0.2, implying a rapid bond dissociation to a state for which the transition dipole from the ground state lies predominantly parallel to the breaking bond.
Lipciuc et al.7 investigated the 193 nm photolysis of DMF using velocity-map imaging. They observed the same N–CO and N–CH3 bond breaking channels as Forde et al., and measured complete scattering distributions for each observed photoproduct. For products associated with the N–CO bond breaking channels the β-parameter was found to show a strong dependence on the fragment velocity, with faster products exhibiting larger values of β. This was attributed to excitation of out-of-plane vibrations on the same timescale as dissociation. Excitation is primarily to the 31A′ state, via a transition with a transition dipole almost parallel to the N–CO bond. Dissociation occurs either on this state or following population transfer to the 11A′′ or 21A′ states. All three excited states have non-planar minima. For molecules dissociating rapidly from near-planar geometries with little out-of-plane nuclear motion, most of the available energy is released into product translation, leading to near-limiting values of β in good agreement with the axial recoil approximation. Conversely, molecules in which out-of-plane motions are excited will experience forces during dissociation that lead to rotational and vibrational excitation of the products, reducing their translational energy, and also to deviations from axial recoil, reducing β.
Previous experimental studies on the photoexcitation of DMF have neglected possible excitation to Rydberg states. However, DMF and related molecules exhibit sharp excitation bands corresponding to transitions to these states.20–23 The relevance of Rydberg and Rydberg/valence surfaces for amides has been discussed in detail elsewhere.24–26 The experimental studies described above have focused either on multi-photon absorption5 or on photolysis at wavelengths below 200 nm,1,2,7 perhaps because experiments at wavelengths longer than 200 nm are more challenging due to the lower UV absorption cross-section of the molecule.17,18 In the present study, we report data from a multimass velocity-map imaging study into the photofragmentation dynamics of DMF at excitation wavelengths of 225 nm and 245 nm. This work is complementary to our previous study of the dynamics at 193 nm.7
2 Methods
The experimental set-up is similar to that described by Hopkins et al.27 Briefly, helium (BOC, 99.9%) at a pressure of ∼1 bar was bubbled through liquid N,N-dimethylformamide (Sigma-Aldrich, 99.8%) to generate a ∼0.5% mixture of DMF in He. The gas mixture underwent a supersonic expansion through a pulsed solenoid valve (Parker Hannifin, Series 9, 10 Hz), which was collimated by a 1 mm skimmer before passing through the repeller plate of the ion optics assembly into the interaction region of a velocity-map imaging (VMI) time-of-flight (ToF) mass spectrometer. Here the molecular beam was intersected at right angles by the UV photolysis laser beam and VUV probe beam.The 225 nm or 245 nm photolysis beam used to initiate photolysis was generated using a tuneable, pulsed, frequency-doubled dye laser (Sirah Cobra Stretch), pumped by the third harmonic of a Nd:YAG laser (Continuum Surelite II). The beam was linearly polarised in the imaging plane (perpendicular to the time-of-flight axis). Following a short time delay of 15–18 ns, the neutral photofragments were ionised by the 118.2 nm VUV probe laser beam. The 118 nm radiation was produced by frequency tripling the third harmonic of a Nd:YAG laser (Continuum Surelite I) in a phase-matched mixture28 of 27 mbar Xenon (BOC, 99.9%) and 298 mbar Argon (BOC, 99.9%).29–31 The probe beam was linearly polarized along the time-of-flight axis (perpendicular to the imaging plane) to ensure that any anisotropy observed in the velocity-map images is caused by the photolysis laser polarization.
The velocity-map imaging ion lens used to map the ionised photofragments onto the detector consists of repeller, extractor, and ground electrodes. The repeller plate has a 4 mm diameter hole in the centre through which the molecular beam is admitted to the interaction region. The extractor and ground electrodes have 24 mm holes in the centre to transmit the expanding product scattering distributions. The ratio of potentials applied to the extractor and repeller plates is tuned to achieve velocity-mapping conditions,32,33 mapping the three-dimensional scattering distribution of the nascent photofragments via a 56 cm flight tube onto a two-dimensional position sensitive ion detector. The ion detector comprises a pair of chevron-mounted microchannel plates (MCPs) coupled to a P47 phosphor screen. Each incoming ion generates an optical signal on the phosphor, which is captured by a second-generation Pixel Imaging Mass Spectrometry (PImMS2) camera.34–36 The photofragment scattering distributions are read out from the camera in the form of an (x, y, t) data point for each detected ion. The data sets can be integrated over the (x, y) coordinates to obtain product time-of-flight spectra, or over the relevant arrival time ranges to generate velocity-map images for each photofragment.
Data sets were acquired under pump–probe conditions, and also, for the purposes of background subtraction, under pump-only and probe-only conditions. This allowed any one-laser signals to be subtracted from the overall signal in order to obtain the true pump–probe signal. Each ion signal generally excites multiple pixels and time bins within the PImMS sensor, which is corrected by running the data set through a centroiding algorithm. The velocity-map images are two-dimensional projections of the full three-dimensional scattering distribution for each photoproduct. The scattering distributions are cylindrically symmetrical about the polarisation vector of the photolysis laser, such that an inverse Abel transform can be used to obtain the central slice through each distribution, which contains all of the information required in order to extract product speed and angular distributions. The Abel inversion was performed using the Basex software package.37 From the inverted images we extracted radial and angular distributions, converting the radial distributions into kinetic energy distributions using a calibration determined in previous measurements on the 355 nm photolysis of Cl2.
3 Results and discussion
3.1 Ultraviolet absorption spectrum of N,N-dimethylformamide
Several authors have reported UV/VIS spectra of DMF.11,17,18 However, these studies mainly reported results at shorter wavelengths, where the excitation cross-section is larger. A liquid-phase UV absorption spectrum of the DMF used in the present study, recorded over the wavelength range 190–350 nm on a Thermo Fisher Scientific Evolution 220 spectrometer, is shown in Fig. 2.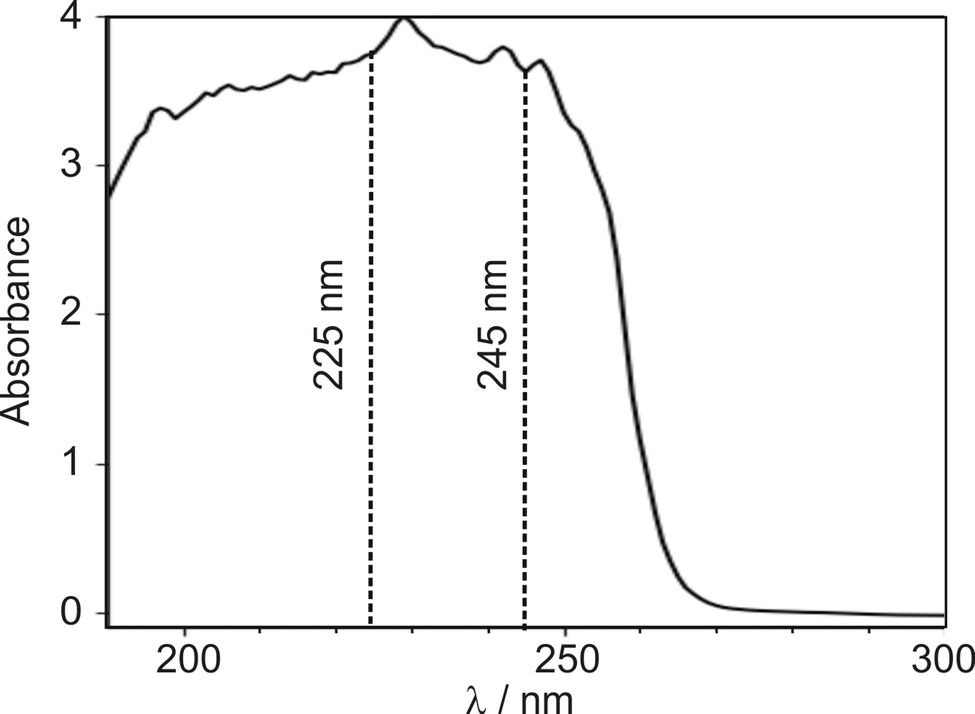 | ||
| Fig. 2 UV absorption spectrum of liquid DMF, recorded on a Thermo Fisher Scientific Evolution 220 spectrometer. Dashed vertical lines indicate the photolysis wavelengths of 225 and 245 nm chosen for the velocity-map imaging experiments. | ||
The absorption spectrum exhibits a peak between 220–230 nm with a wide shoulder on the red-edge extending to wavelengths of about 255 nm. We decided to investigate pump wavelengths of 225 nm, which is in the centre of the absorption peak, and 245 nm, marking the end of the shoulder, at which significant absorption is observed, while minimising potential contribution from the 225 nm feature.
3.2 Photolysis of DMF at 225 nm
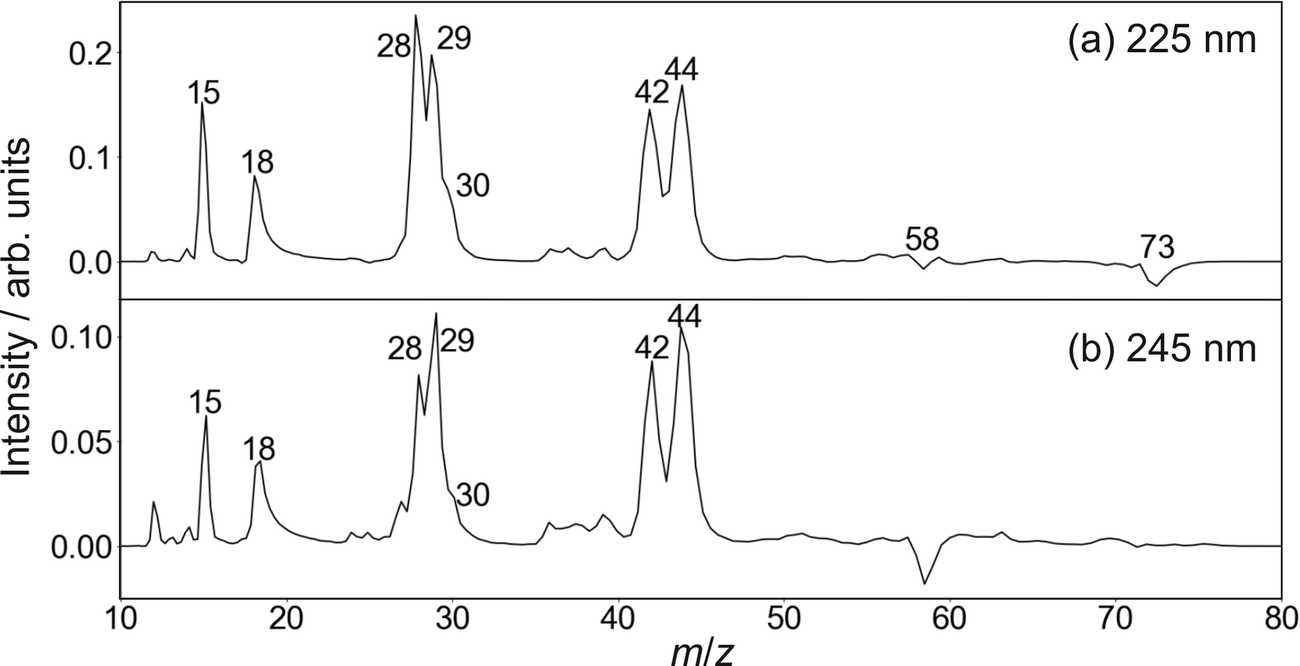 | ||
| Fig. 3 Time-of-flight mass spectra of the photofragments formed following photoexcitation of DMF at 225 nm (upper panel) and 245 nm (lower panel). | ||
Cleavage of the N–CO amide bond generates HCO and NC2H6 products, and we therefore expect to see signal arising from this pathway at m/z = 44 and 29. We do indeed see signal at these two m/z values. However, dissociative ionisation of the nascent fragments in the probe step, as well as neutral predissociation of some of the nascent HCO products,7,38 has the result that we see signal associated with the NC2H6 products at m/z = 44 (NC2H6+), 43 (NC2H5+), 42 (NC2H4+) and 18 (NH4+), and signal associated with the HCO fragment at m/z = 29 (HCO+) and 28 (CO+). We note that formation of NH4+ has been confirmed in isotope labelling experiments, and involves significant structural rearrangement of nascent internally excited N(CH3)2+ ions.7 Since there is little kinetic energy release during this secondary reaction, the NH4+ products essentially inherit the speed and angular distribution of the N(CH3)2+ ions from which they are formed, and the images resemble the central region of the images of these parent primary ions. The arrival time distribution for the NH4+ fragment has a significant tail extending to longer flight times, implying that the rearrangement and dissociation occur on a relatively long timescale, with the final products forming both within the interaction region of the experiment and along the flight tube between the interaction region and the detector.
Fission of either N–CH3 bond in the parent DMF molecule generates the fragments CH3 and HCONCH3. Only the former is ionizable using one 118 nm photon,7 appearing at m/z = 15 in the mass spectrum. Low intensity signals, presumably associated with H-atom loss either from neutral or ionised CH3, can be observed at m/z = 12, 13, and 14.
Based on its mass-to-charge ratio, the small signal at m/z = 30, observed as a shoulder on the much larger signals at m/z = 28 and 29, may correspond either to C2H6+, HCOH+, or HNCH3+. While formation of C2H6+ would require significant molecular rearrangement, the latter two ions can be formed via quite plausible mechanisms. Whatever its identity, this minor fragment is not considered further in the following.
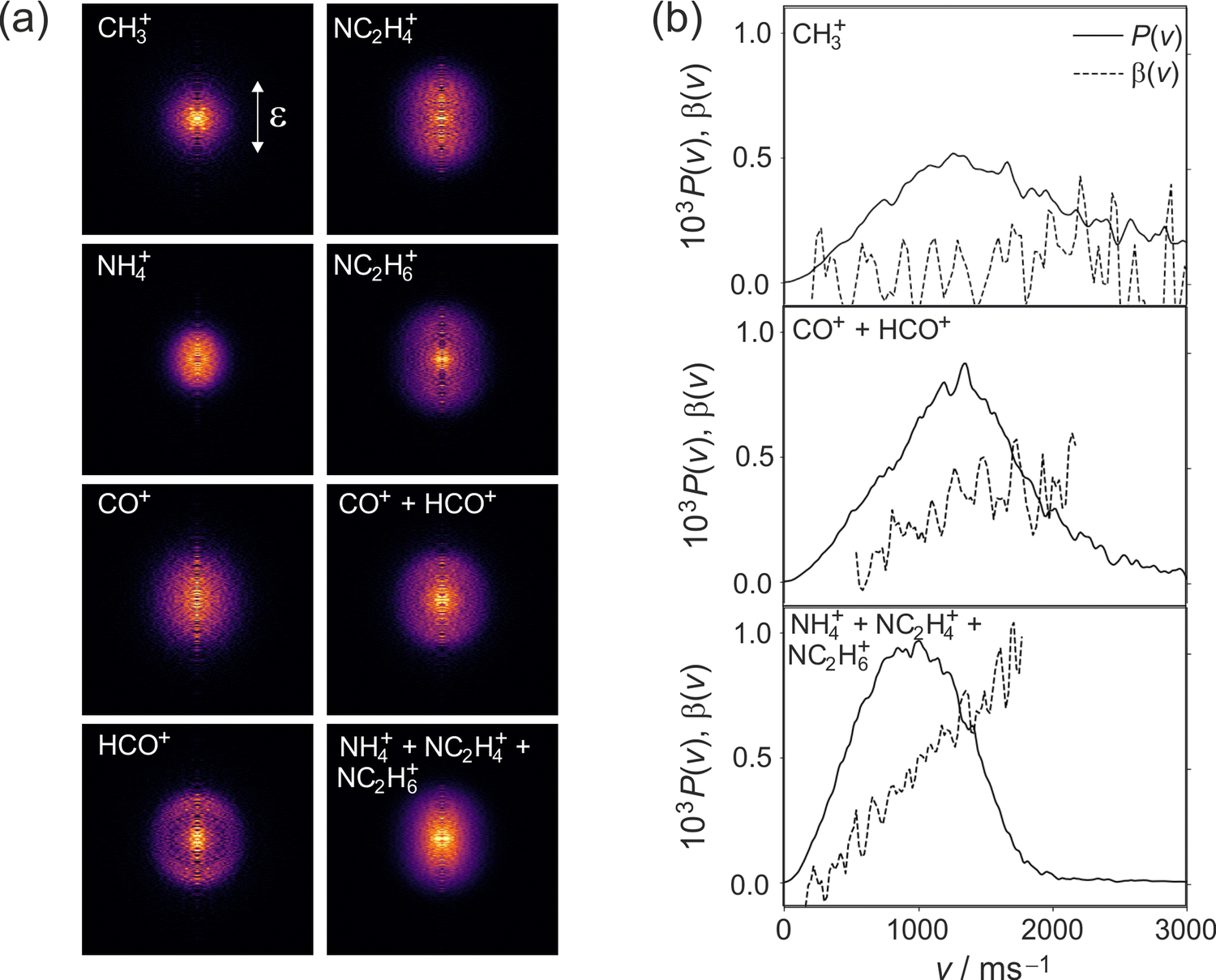 | ||
| Fig. 4 (a) Symmetrized and Abel-inverted velocity-map images for fragments observed following 225 nm photolysis of DMF; (b) speed and angular distributions for each product channel as explained in the text. | ||
| Species | m/z | β 225nm | β 245nm |
|---|---|---|---|
| CH3+ | 15 | 0.07 ± 0.17 | −0.05 ± 0.33 |
| NH4+ + NC2Hx | 18/42/43/44 | 0.52 ± 0.07 | 0.30 ± 0.09 |
| CO+ + HCO+ | 28/29 | 0.20 ± 0.09 | 0.17 ± 0.13 |
We note that the velocity distributions are all fairly broad, implying population of a wide range of rotational and vibrational states in the recoiling fragments. The β parameter for the CH3 fragments is close to zero, while the HCO and NC2H6 fragments exhibit non-zero β parameters that increase markedly with fragment velocity. These features of the speed and angular distributions will be discussed in more detail below.
Table 2 shows calculated vertical excitation energies to the three lowest lying electronic singlet states and two lowest lying triplet states of DMF, together with oscillator strengths for the associated transitions. Frequency calculations show the zero-point energies for the various states to be very similar, so we have not included these explicitly when considering the excitation energies. Two sets of values are given: the results of density functional theory calculations by Shastri et al.;11 and the results of CASPT2 calculations by Serrano-Andres et al.19 The recent DFT calculations by Shastri et al. indicate that the 11A′′ and 21A′′ states are likely to be energetically accessible at 225 nm, with the CASPT2 calculations of Serrano-Andres et al. predicting that the states lie slightly higher in energy. The (n → π*) transition to the first excited 11A′′ singlet state has a near-zero oscillator strength, making it unlikely to be populated directly by absorption of a 225 nm photon. In contrast, the (4a′′ → 3s) transition to the second excited 21A′′ singlet state has a much higher oscillator strength, implying that this state is most likely to be populated in the absorption step.
| Symmetry | Excitation energies/eV | Oscillator strength | |||
|---|---|---|---|---|---|
| DFTa | CASPT2b | DFTa | CASPT2b | ||
| a Shastri et al.11 b Serrano-Andres et al.19 | |||||
| 11A′ | (Ground state) | ||||
| 11A′′ | (n → π*) | 5.59 (222 nm) | 5.64 (220 nm) | <0.001 | 0.001 |
| 21A′′ | (4a′′ → 3s) | 5.62 (221 nm) | 5.92 (209 nm) | 0.021 | 0.005 |
| 21A′ | (16a′ → π*) | 5.99 (207 nm) | 6.48 (191 nm) | 0.009 | 0.002 |
| 13A′ | (n(σ) → π*) | 4.73 (262 nm) | 5.41 (229 nm) | — | — |
| 13A′′ | (n(π) → π*) | 5.08 (244 nm) | 5.06 (245 nm) | — | — |
The measured β parameters provide further insight into the excited states involved in the photodissociation. The transition dipole for the π ← n(σ) excitation to the first excited 11A′′ singlet state lies perpendicular to the breaking N–CO bond. This would give rise to a negative value for β, which approaches a value of −1 for a pure perpendicular transition. In contrast, excitation to the second excited 21A′′ singlet state involves movement of electron density from the n(π)-orbital located at the nitrogen atom to the Rydberg 3s-orbitals on the CH3-groups,11 resulting in a parallel transition and a correspondingly positive value of β between 0 and 2. As noted above, the measured values of β for both the HCO and NC2H6 products are positive. This is consistent with excitation to the second excited 21A′′ singlet state, in agreement with the predictions made above on the basis of the calculated oscillator strengths for the two transitions.
Having established that excitation at 225 nm is most probably to the 21A′′ singlet state, we now explore the subsequent processes leading to cleavage of the N–CO and N–CH3 bonds and production of the observed photofragments.
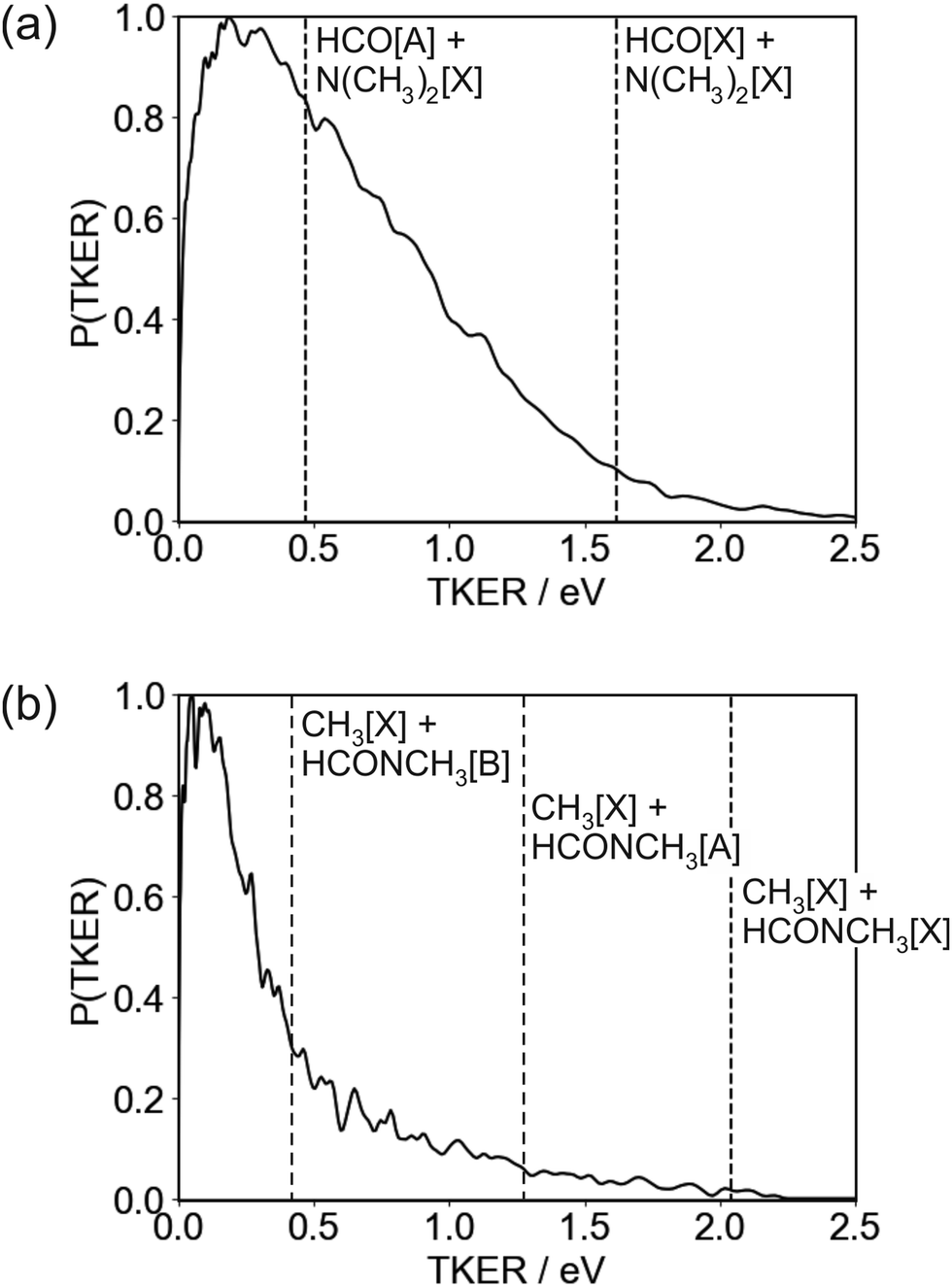 | ||
| Fig. 5 Total kinetic energy release (TKER) distribution for the (a) N–CO and (b) N–CH3 bond fission channels following 225 nm photolysis of DMF. The total available energies for the possible product channels are indicated by dashed vertical lines. | ||
The TKER distribution peaks at low energies, implying that the majority of photofragments are formed with significant amounts of internal energy. The lower energy part of the distribution, below 0.47 eV, is consistent with formation of HCO in either its electronic ground or first excited state, with the excited state not accessible for products with TKER above this value. While product internal excitation is clearly a key feature of the fragmentation dynamics, the long tail extending out to the maximum available energy indicates that for a significant proportion of products the majority of the available energy is released into relative translation of the recoiling fragments. A small proportion of fragments are formed with energies higher than the supposed maximum. A small amount of ‘extra’ energy can often be explained by the presence of a small fraction of vibrationally excited DMF in the molecular beam. However, in this case we believe the high energy tail extends too far above the nominal maximum available energy for this to be the explanation. Trajectory studies by Eckert-Makzic and coworkers9 indicate that following excitation to low-lying excited singlet states, a significant fraction of trajectories become trapped in minima on these states. We therefore postulate that in our experiment a small number of these ‘trapped’ excited-state molecules survive long enough to undergo dissociative ionisation by the probe laser, and that this is the source of the high energy tail on the TKER distribution for the N–CO bond cleavage channel. While ‘probe-only’ dissociative ionisation is removed from our signal during the background-subtraction process, this second dissociative ionisation signal requires both pump and probe lasers, so is not removed when the one-laser signals are subtracted.
The high amount of internal excitation in the majority of products is explained by the topography of the excited state potential energy surface(s) over which the system evolves during dissociation. The 21A′′ surface to which DMF is initially excited correlates to NC2H6() + HCO(Ã) products, and exhibits a significant energy barrier of around 0.81 eV (see Fig. 1) along the N–CO bond stretching coordinate,11 with the energy maximum corresponding to a bond length of ∼1.8 Å. In contrast to the planar structure of ground-state DMF, the energy minimum of the 21A′′ surface is non-planar. The dissociating molecules are therefore likely to follow a variety of trajectories across the potential energy surface. Some of these trajectories will surmount the barrier with little out-of-plane motion, in which case there will be few torsional forces acting and correspondingly little internal excitation of the products, with the majority of the energy being released into product translation. For these products the lack of out-of-plane motion also maintains the spatial relationship between the polarisation vector of the excitation light and the axis of the dissociating bond (i.e. the axial recoil approximation holds), yielding relatively large positive values for the recoil anisotropy parameter β. Other trajectories will explore out-of-plane geometries that ‘skirt around’ the barrier. The torsional forces acting during dissociation in these cases lead to rotational and/or vibrational excitation of the photofragments, and therefore a correspondingly smaller amount of energy released into translation. The out-of-plane motion also leads to breakdown of the axial recoil approximation and a corresponding reduction in β. The correlation between product internal excitation and breakdown of the axial recoil approximation explains the observed increase in β with increasing photofragment velocity. Similar behaviour has been seen previously in the dissociation of DMF on the higher-lying 31A′ singlet surface following photoexcitation at 193 nm.7
As noted above, the TKER distribution for the HCO + NC2H6 products is not consistent with dissociation only on the 21A′′ surface, since many of the HCO products must be formed in their electronic ground state in order to account for the products formed with TKER greater than 0.47 eV. Coupling between the 21A′′ and 11A′′ surfaces can lead to transfer of population to the lower excited singlet surface, which correlates to ground state products. The topographies of the two singlet surfaces are qualitatively similar, with a barrier along the N–CO stretching coordinate and a non-planar minimum-energy geometry, so we see a similar increase in β with increasing product velocity and decreasing product internal excitation. Population transfer from the 11A′′ first excited singlet state to the 11A′ ground state is also possible via a conical intersection at highly non-planar geometries, as is intersystem crossing to the 13A′ first excited triplet state.8 In common with the second excited singlet state, the former of these pathways correlates to formation of ground state NC2H6 and electronically excited HCO, while the T1 state correlates to ground state products.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ distribution of the N–CO axis about the polarisation vector of the photolysis laser. In the case of the 193 nm photolysis studied by Lipciuc et al., the excitation was via a parallel transition to the 31A′ state rather than the 21A′′ state, but the same arguments apply. Ejection of CH3 at an angle of approximately 60 degrees to the N–CO axis, as determined by the molecular geometry, combined with this cos2θ distribution of molecular orientations, yields an almost isotropic distribution of CH3 velocity vectors relative to the photolysis polarisation axis, as observed.
θ distribution of the N–CO axis about the polarisation vector of the photolysis laser. In the case of the 193 nm photolysis studied by Lipciuc et al., the excitation was via a parallel transition to the 31A′ state rather than the 21A′′ state, but the same arguments apply. Ejection of CH3 at an angle of approximately 60 degrees to the N–CO axis, as determined by the molecular geometry, combined with this cos2θ distribution of molecular orientations, yields an almost isotropic distribution of CH3 velocity vectors relative to the photolysis polarisation axis, as observed.
The TKER distribution for the N–CH3 bond cleavage products is sharply peaked at low kinetic energy, indicating that the majority of the available energy is released into internal excitation. Following excitation to the 21A′′ state there are various possible pathways by which the molecule can evolve to the separated CH3 + NCOCH3 products. At 193 nm, the dissociation is thought to proceed via excitation to the 31A′ state and subsequent coupling to and dissociation on the 21A′ state. Couplings involving the 21A′′ state which is initially excited at 225 nm have not been comprehensively studied, but it is clear that several potential dissociation pathways are possible, involving either direct dissociation over a small barrier on the 21A′′ state to form electronically excited products, or coupling to a lower-lying state. Three product channels are energetically accessible at an excitation wavelength of 225 nm, corresponding to ground-state CH3 partnered by HCONCH3 in its electronic ground, first, or second excited state, respectively. None of these are ruled out by the form of the TKER distribution, though the sharp peak at low kinetic energies below the energy cutoff for production of CH3[] + HCONCH3[![[B with combining tilde]](https://www.rsc.org/images/entities/char_0042_0303.gif) ] does perhaps suggest a significant contribution from this product channel. If this is the case then it is certainly not the only product channel, as the TKER distribution extends out to the energy thresholds of both of the lower energy channels. Given the geometry of the DMF molecule (see inset to Fig. 1, it is also very likely that a considerable amount of the internal excitation is in rotational and vibrational modes of the products). Assuming the NCH3 bond cleavage is reasonably rapid, which it must be to compete with the N–CO bond dissociation pathway, the force exerted away from the centre of mass of the HCONCH3 fragment is likely to result in considerable rotational excitation of this fragment. Significant excitation of the umbrella vibrational mode in CH3 as well as other vibrational modes in both fragments is also highly likely.
] does perhaps suggest a significant contribution from this product channel. If this is the case then it is certainly not the only product channel, as the TKER distribution extends out to the energy thresholds of both of the lower energy channels. Given the geometry of the DMF molecule (see inset to Fig. 1, it is also very likely that a considerable amount of the internal excitation is in rotational and vibrational modes of the products). Assuming the NCH3 bond cleavage is reasonably rapid, which it must be to compete with the N–CO bond dissociation pathway, the force exerted away from the centre of mass of the HCONCH3 fragment is likely to result in considerable rotational excitation of this fragment. Significant excitation of the umbrella vibrational mode in CH3 as well as other vibrational modes in both fragments is also highly likely.
3.3 Photolysis of DMF at 245 nm
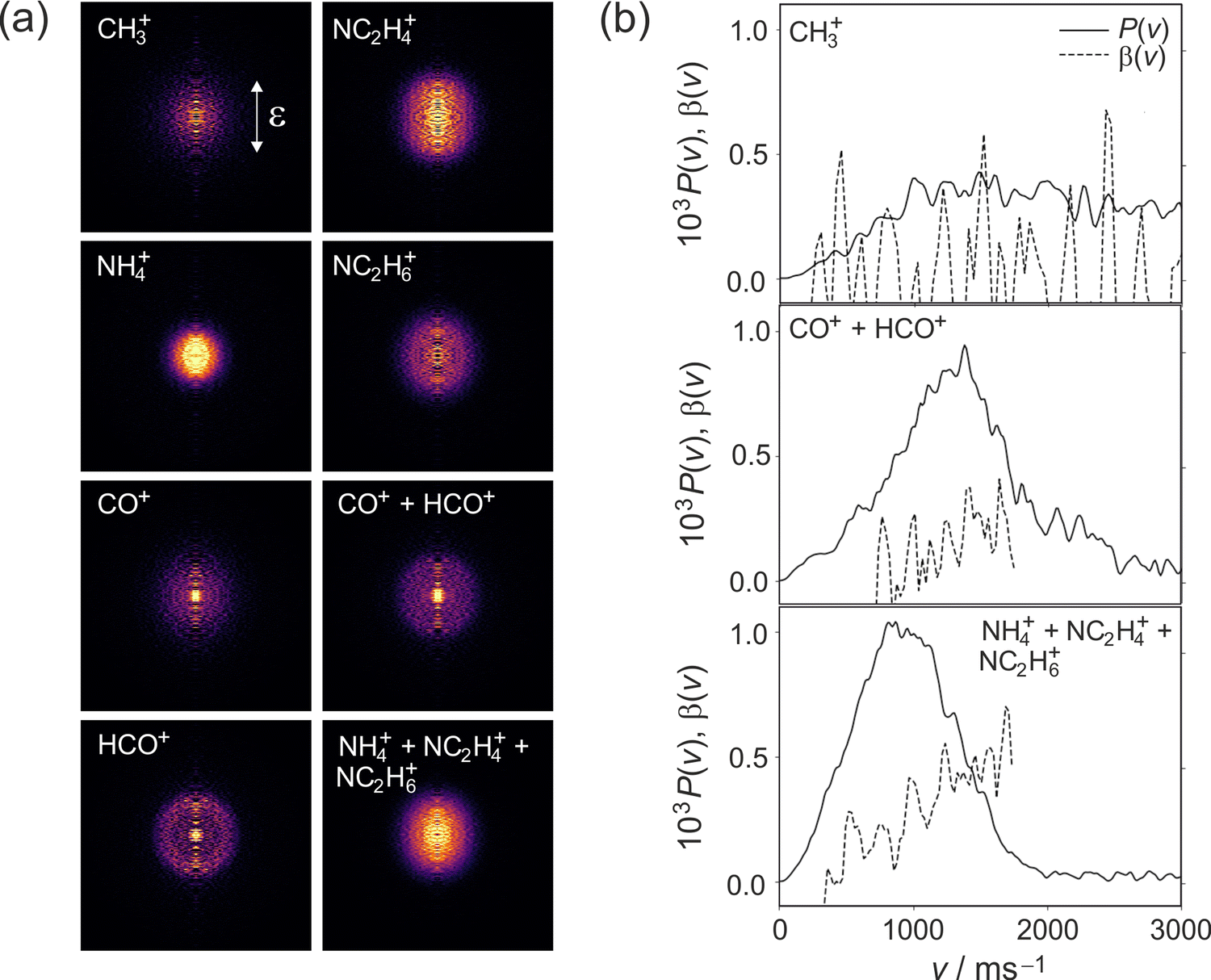 | ||
| Fig. 6 (a) Symmetrized and Abel-inverted velocity-map images for fragments observed following 245 nm photolysis of DMF; (b) speed and angular distributions for each product channel as explained in the text. | ||
Referring to the excitation energies in Table 2 we see that for ground-state molecules at their equilibrium geometry all of the singlet states lie significantly higher in energy than the 5.06 eV energy of a 245 nm photon. However, the strong similarities between the scattering distributions observed at 225 and 245 nm, coupled with the very similar absorption intensities at the two wavelengths, lead us to believe that excitation is to the S2 state in both cases. The apparent energy shortfall is explained by the fact that the 2.749 eV zero-point energy of ground-state DMF (calculated at UB3LYP/6-311++g(d,p) level) means that the vibrational ground state of the molecule spans a significant range of molecular geometries. Distortion of the molecular geometry towards the equilibrium geometry of the S2 state, which involves significant out of plane motion of the molecular backbone, reduces the excitation energy to the S2 state considerably.9 Based on the smaller β parameters observed at the longer wavelength, the geometrical distortion results in a rotation of the S0 → S2 transition dipole away from the C–N bond axis. As noted above, though the maximum β parameters are roughly halved at 245 nm compared with photolysis at 225 nm, we see a similar increase in β with photofragment velocity. As before, we believe this arises as a result of dissociation from a range of molecular geometries. Dissociation from more planar geometries yields higher photofragment velocities and larger values of β, and dissociation from significantly non-planar geometries results in a higher degree of internal excitation and therefore lower photofragment velocities and β parameters.
It is also possible that excitation to the S1 state plays a small role at molecular geometries away from the equilibrium structure. This state also becomes energetically accessible at such geometries, and the S0 → S1 transition dipole lies perpendicular to the amide bond even at the equilibrium molecular geometry, which would result in a reduction in the observed value of β. However, the oscillator strength for the transition to S1 is at least an order of magnitude lower than that for excitation to the S2 state, so any involvement of this state is unlikely to account for the observed two-fold reduction in β. Shastri et al.11 suggested based on the calculated vertical excitation energies that the two low-lying triplet states may also make a contribution to the absorption band under study. The excitations to the T1 and T2 states are respectively perpendicular and parallel in character relative to the amide bond, but on the basis of oscillator strength arguments any such contributions are likely to be minor. As noted previously for dissociation at 245 nm, involvement of the S1, T1, and S0 states in the exit channel, accessed via internal conversion or intersystem crossing, is certainly likely and has been proposed previously.8
The TKER distribution for the HCO + NC2H6 products is shown in Fig. 7(a). The calculated maximum kinetic energy is shown as a vertical dashed line on the plot. The TKER distribution measured at 245 nm is very similar to that observed following photolysis at 225 nm, peaking at low kinetic energies but with a long tail extending out beyond the nominal maximum available energy. As for the 225 nm data, we believe the high-energy tail to be caused by dissociative ionisation of a small number of molecules that become trapped within minima on the low-lying singlet surfaces. As already noted, dissociation occurs from a range of molecular geometries, with dissociation from planar geometries yielding little internal excitation and relatively fast products, and dissociation from non-planar geometries leading to a higher degree of photofragment internal excitation and correspondingly lower photofragment velocities.
 | ||
| Fig. 7 Total kinetic energy release (TKER) distribution for the (a) N–CO and (b) N–CH3 bond fission channels following 245 nm photolysis of DMF. The total available energies for the possible product channels are indicated by dashed vertical lines. | ||
4 Conclusions
We have reported the results of a comprehensive experimental study on the photofragmentation dynamics of N,N-dimethylformamide following excitation at 225 nm and 245 nm, respectively. To our knowledge, this is the first study into the dissociation dynamics of DMF in this wavelength region. At both excitation wavelengths, we observe products corresponding to cleavage of both the N–CO amide bond and one of the N–CH3 bonds, as well as products arising from secondary fragmentation or dissociative ionisation of the primary products.The velocity-map images allow us to determine speed and angular distributions for each fragment, as well as total kinetic energy releases associated with each fragmentation channel. We have interpreted these data in light of complementary theoretical studies by Shastri et al.11 and Serrano-Andres et al.,19 and by comparison with results from a previous study performed in our own group into the photolysis of DMF at 193 nm,7 in order to determine the electronic states involved in the photofragmentation at each wavelength. We conclude that excitation at both 225 nm and 245 nm is predominantly to the S2 21A′′ state via a parallel transition, with dissociation along the N–CO bond occurring either on this state or on a lower singlet surface following internal conversion. At the shorter wavelength, the S2 state is energetically accessible via a vertical transition from the equilibrium geometry of the molecule, while geometrical distortion of the molecular structure is required in order to access this state at the longer wavelength. Dissociation occurs either on the S2 surface or following population transfer to S1. Both singlet surfaces, as well as the low lying T1 and T2 triplet surfaces, share similar topographical features, exhibiting a barrier along the N–CO stretching coordinate, and possessing non-planar minimum-energy geometries. As a result, dissociation occurs from a variety of molecular geometries. Dissociation from planar geometries leads to little product internal excitation and correspondingly high photofragment velocities, with near-limiting values of the recoil-anisotropy parameter β. In contrast, dissociation from non-planar geometries leads to significant product internal excitation, with correspondingly lower photofragment velocities and breakdown of the axial recoil approximation to give reduced values of β.
Less detailed information is available on the potential energy curves along the N–CH3 bond coordinate than for the N–CO coordinate. However, at both wavelengths we see an isotropic angular distribution for the CH3 product, and a TKER distribution peaking at low kinetic energies, indicating significant internal excitation in the fragments. This is similar to the behaviour observed previously at 193 nm,7 and can be rationalised in terms of the geometry of the molecule and the orientation of the breaking bond relative to the transition dipole for the initial excitation.
Having now established the photochemical pathways involved in the cleavage of the model peptide bond in DMF at a number of different photolysis wavelengths, future work is likely to focus on investigating the effects of the various different amino acid side chains in the photofragmentation of a series of simple di- and tri-peptides.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the EPSRC for funding through Programme Grants EP/L005913/1, EP/T021675/1, and EP/V026690/1.References
- N. R. Forde, T. L. Myers and L. J. Butler, Faraday Discuss., 1997, 108, 221–242 RSC.
- N. R. Forde and L. J. Butler, J. Chem. Phys., 1999, 110, 8954–8968 CrossRef CAS.
- S. Shin, A. Kurawaki, Y. Hamada, K. Shinya, K. Ohno, A. Tohara and M. Sato, J. Mol. Struct., 2006, 791, 30–40 CrossRef CAS.
- M. Ruzi and D. T. Anderson, J. Chem. Phys., 2012, 137, 194313 CrossRef PubMed.
- X. Qiu, Z. Ding, Y. Xu, Y. Wang and B. Zhang, Phys. Rev. A: At., Mol., Opt. Phys., 2014, 89, 033405 CrossRef.
- P. Salén, V. Yatsyna, L. Schio, R. Feifel, R. Richter, M. Alagia, S. Stranges and V. Zhaunerchyk, J. Chem. Phys., 2016, 144, 244310 CrossRef PubMed.
- M. L. Lipciuc, S. H. Gardiner, T. N. Karsili, J. W. Lee, D. Heathcote, M. N. Ashfold and C. Vallance, J. Chem. Phys., 2017, 147, 013941 CrossRef PubMed.
- D. Liu, W. Fang, Z. Lin and X. Fu, J. Chem. Phys., 2002, 117, 9241–9247 CrossRef CAS.
- M. Eckert-Maksić and I. Antol, J. Phys. Chem. A, 2009, 113, 12582–12590 CrossRef PubMed.
- M. Eckert-Maksić, M. Vazdar, M. Ruckenbauer, M. Barbatti, T. Müller and H. Lischka, Phys. Chem. Chem. Phys., 2010, 12, 12719–12726 RSC.
- A. Shastri, A. K. Das, S. Krishnakumar, P. J. Singh and B. N. Raja Sekhar, J. Chem. Phys., 2017, 147, 224305 CrossRef PubMed.
- M. B. Shundalau, P. S. Chybirai, A. I. Komyak, A. P. Zazhogin, M. A. Ksenofontov and D. S. Umreiko, J. Appl. Spectrosc., 2011, 78, 326–336 CrossRef CAS.
- X. Zhou, J. A. Krauser, D. R. Tate, A. S. VanBuren, J. A. Clark, P. R. Moody and R. Liu, J. Chem. Phys., 1996, 100, 16822–16827 CrossRef CAS.
- T. C. Jao, I. Scott and D. Steele, J. Mol. Spectrosc., 1982, 92, 1–17 CrossRef CAS.
- V. Renugopalakrishnan, G. Madrid, G. Cuevas and A. T. Hagler, Proc. - Indian Acad. Sci., Chem. Sci., 2000, 112, 35–42 CrossRef CAS.
- R. Saladino, C. Crestini, G. Costanzo, R. Negri and E. Di Mauro, Bioorg. Med. Chem., 2001, 9, 1249–1253 CrossRef CAS PubMed.
- H. D. Hunt and W. T. Simpson, J. Am. Chem. Soc., 1953, 75, 4540–4543 CrossRef CAS.
- K. Kaya and S. Nagakura, Theor. Chim. Acta, 1967, 7, 117–123 CrossRef CAS.
- L. Serrano-Andrés and M. P. Fülscher, J. Am. Chem. Soc., 1996, 118, 12190–12199 CrossRef.
- H. Basch, M. B. Robin and N. A. Kuebler, J. Chem. Phys., 1968, 49, 5007–5018 CrossRef CAS.
- J. M. Gingell, N. J. Mason, H. Zhao, I. C. Walker and M. R. Siggel, Chem. Phys., 1997, 220, 191–205 CrossRef CAS.
- D. H. Ter Steege, C. Lagrost, W. J. Buma, D. A. Leigh and F. Zerbetto, J. Chem. Phys., 2002, 117, 8270–8280 CrossRef CAS.
- M. A. Larsen, T. I. Sølling, R. Forbes, A. E. Boguslavskiy, V. Makhija, K. Veyrinas, R. Lausten, A. Stolow, M. M. Zawadzki, L. Saalbach, N. Kotsina, M. J. Paterson and D. Townsend, J. Chem. Phys., 2019, 150, 054301 CrossRef PubMed.
- M. N. Ashfold, G. A. King, D. Murdock, M. G. Nix, T. A. Oliver and A. G. Sage, Phys. Chem. Chem. Phys., 2010, 12, 1218–1238 RSC.
- G. M. Roberts and V. G. Stavros, Chem. Sci., 2014, 5, 1698–1722 RSC.
- M. J. Paterson and D. Townsend, Int. Rev. Phys. Chem., 2020, 39, 517–567 Search PubMed.
- W. S. Hopkins, M. L. Lipciuc, S. H. Gardiner and C. Vallance, J. Chem. Phys., 2011, 135, 034308 CrossRef PubMed.
- J. M. Gray, J. Bossert, Y. Shyur, B. Saarel, T. C. Briles and H. J. Lewandowski, J. Chem. Phys., 2021, 154, 024201 CrossRef CAS PubMed.
- A. H. Kung, J. F. Young and S. E. Harris, Appl. Phys. Lett., 1973, 22, 301–302 CrossRef CAS.
- A. H. Kung, J. F. Young and S. E. Harris, Appl. Phys. Lett., 1976, 28, 294 CrossRef.
- N. P. Lockyer and J. C. Vickerman, Laser Chem., 1997, 17, 139–159 CrossRef CAS.
- D. W. Chandler and P. L. Houston, J. Chem. Phys., 1987, 87, 1445–1447 CrossRef CAS.
- A. T. Eppink and D. H. Parker, Rev. Sci. Instrum., 1997, 68, 3477–3484 CrossRef CAS.
- A. Nomerotski, M. Brouard, E. Campbell, A. Clark, J. Crooks, J. Fopma, J. J. John, A. J. Johnsen, C. Slater, R. Turchetta, C. Vallance, E. Wilman and W. H. Yuen, J. Instrum., 2010, 5, C07007 Search PubMed.
- E. S. Wilman, S. H. Gardiner, A. Nomerotski, R. Turchetta, M. Brouard and C. Vallance, Rev. Sci. Instrum., 2012, 83, 013304 CrossRef PubMed.
- A. T. Clark, J. P. Crooks, I. Sedgwick, R. Turchetta, J. W. Lee, J. J. John, E. S. Wilman, L. Hill, E. Halford, C. S. Slater, B. Winter, W. H. Yuen, S. H. Gardiner, M. L. Lipciuc, M. Brouard, A. Nomerotski and C. Vallance, J. Phys. Chem. A, 2012, 116, 10897–10903 CrossRef CAS PubMed.
- V. Dribinski, A. Ossadtchi, V. A. Mandelshtam and H. Reisler, Rev. Sci. Instrum., 2002, 73, 2634–2642 CrossRef CAS.
- N. R. Forde, L. J. Butler, B. Ruscic, O. Sorkhabi, F. Qi and A. Suits, J. Chem. Phys., 2000, 113, 3088–3097 CrossRef CAS.
- M. E. Jacox, NIST Chemistry Webbook, NIST Standard Reference Database No. 69, National Institute of Standards and Technology, Gaithersburg, MD, 2022 Search PubMed.
| This journal is © the Owner Societies 2022 |