
Green light LED activated ligation of a scalable, versatile chalcone chromophore†
Ishrath Mohamed
Irshadeen‡
 ab,
Kevin
De Bruycker‡
c,
Aaron S.
Micallef
b,
Sarah L.
Walden
ab,
Hendrik
Frisch
*ab and
Christopher
Barner-Kowollik
*ab
ab,
Kevin
De Bruycker‡
c,
Aaron S.
Micallef
b,
Sarah L.
Walden
ab,
Hendrik
Frisch
*ab and
Christopher
Barner-Kowollik
*ab
aSchool of Chemistry and Physics, Queensland University of Technology (QUT), 2 George Street, Brisbane, QLD 4000, Australia. E-mail: h.frisch@qut.edu.au; christopher.barnerkowollik@qut.edu.au
bCentre for Materials Science, Queensland University of Technology (QUT), 2 George Street, Brisbane, QLD 4000, Australia
cPolymer Chemistry Research Group, Centre of Macromolecular Chemistry (CMaC), Department of Organic and Macromolecular Chemistry, Faculty of Sciences, Ghent University, Krijgslaan 281-S4, Ghent, 9000, Belgium
First published on 21st June 2021
Abstract
Herein we introduce a photoreactive chalcone moiety that can be synthesized at a scale of several grams with ease, and efficiently undergoes a [2 + 2] photocycloaddition with light up to just below 500 nm as determined by an action plot. The peak chalcone reactivity is at 440 nm which is red-shifted by 25 nm compared to the absorption maximum at 415 nm. The chalcone was attached to a RAFT agent enabling reversible deactivation radical polymerization. The resulting polymer subsequently took part in a photoligation triggered by light from an LED centered at 505 nm. Thus, we introduce a chalcone that is capable of overcoming the synthetic disadvantages associated with styrylpyrenes and can readily undergo [2 + 2] photocycloadditon with visibile light.
Introduction
Tunable materials exhibit properties that can be adapted in response to external stimuli, and are highly desired in many applications, ranging from inks2 and coatings3 to biomedical materials.4 A variety of triggers can be used to alter materials such as temperature, pH, light and even magnetic fields.5Light is a powerful tool to modify material properties due to its precise spatio-temporal control, compared to thermally controlled processes. An additional degree of control arises from wavelength-specific activation of specific reactive moieties. Furthermore, light is a non-invasive trigger and, relative to a chemical trigger, does not leave residues or contaminants6 in the material. A variety of light sources are readily available, including conventional filaments or arc lamps, LEDs, lasers, and even chemiluminescence.7
[2 + 2] Photocycloadditions have been used in a range of applications due to their versatility and efficiency as well as their tolerance to a wide range of reaction conditions.8,9 [2 + 2] photocycloadditions are typically highly selective and most are catalyst-free, making them an attractive class of photoreactions to apply to the design of adaptable materials.10,11 However, a major drawback of the traditional library of [2 + 2] photocycloadditions is the requirement of UV light to initiate the reaction, which severely limits their applications due to potential photodamage to biological or chemical matrices and a very short penetration depth.12,13 Photoreactions that can be triggered in the visible spectrum are highly sought-after for biological applications, as these wavelengths can penetrate the cell matrix without lasting negative impacts on the cells. Consequently, in recent years, significant efforts have been directed towards identifying reactions triggered by visible light for drug delivery systems and as modulators to investigate the biochemical signaling cascade.14
The visible-light mediated [2 + 2] cycloaddition of styrylpyrene was initially reported in the 1980s,15 but was recently rediscovered and found a wide range of applications as a result of its benign activation wavelength (λ = 430 nm).1,7,10,16,17 However, large-scale applications of styrylpyrene are impeded by synthetic challenges. Indeed, functional styrylpyrenes are only obtained in relatively low yields and require expensive precursors preventing a straightforward upscaling (Scheme 1).1,7,10,16–19 To overcome the synthetic limitations of styrylpyrenes, a visible light reactive pyrene–chalcone (Py-chal) has recently been developed, which can be scaled to produce several grams of material with ease.20,21 While the Py-chal has proven its potential as a wavelength-gated binding site,20,21 an in-depth study of its photoreactivity has not been conducted thus far.
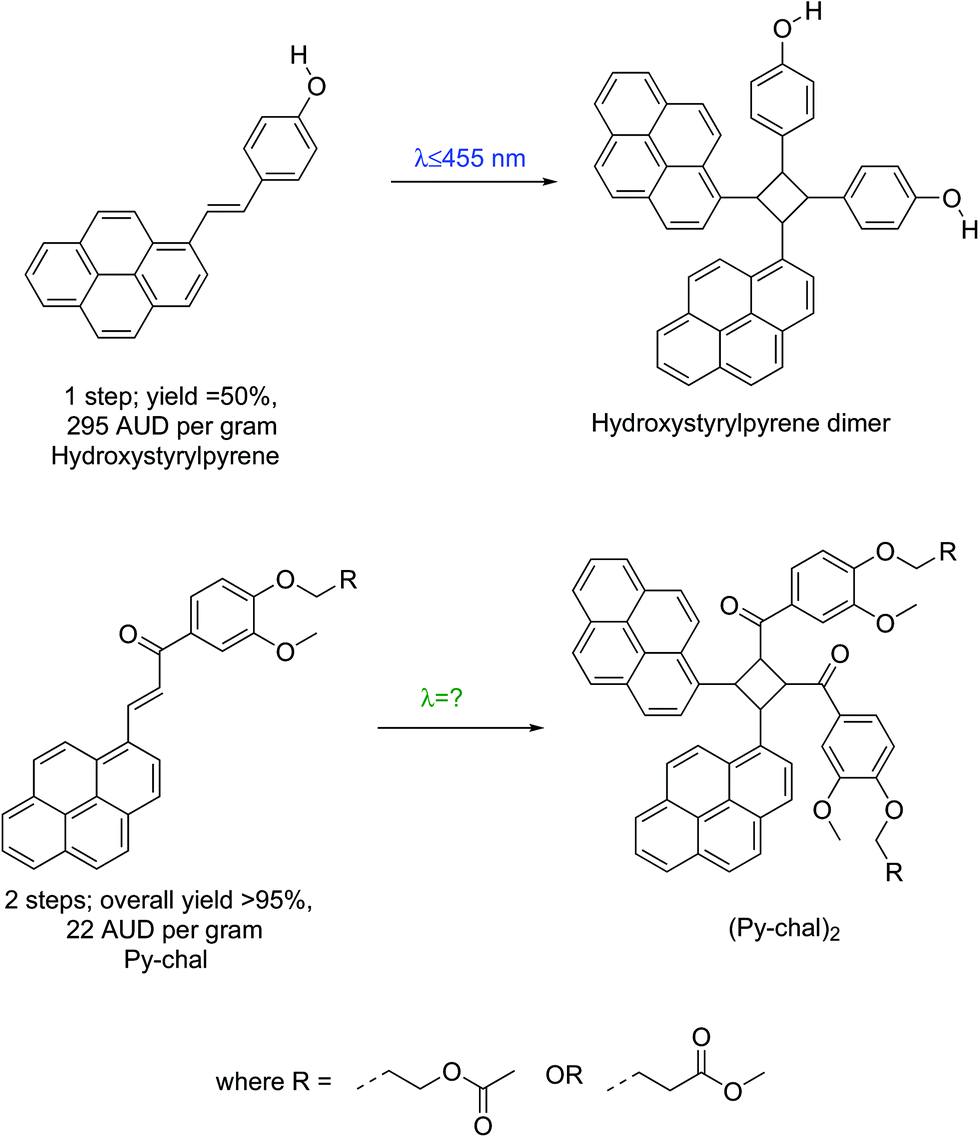 | ||
| Scheme 1 Schematic comparison between styrylpyrene1 and Py-chal, listing the overall required number of synthetic steps, yield and price per gram of each pyrene monomer, starting from commercial reagents. All prices were sourced from Sigma Aldrich©, quoted in Australian dollars, and were accurate at the time of publication. | ||
Over the past decade, our group has worked intensively to explore the wavelength-resolved covalent bond forming photochemistry of a wide array of reactions and discovered that the absorbance profile and reactivity of a photoreactive moiety do not always coincide. In other words, the wavelength at which maximum reactivity is observed is often offset by tens of nanometers relative to the absorption maximum.1,22,23 Using the concept of so-called ‘action plots’, the wavelength-dependent substrate conversion is experimentally mapped. This mapping of wavelength-dependent reactivity of the Py-chal chromophore is prerequisite knowledge for harnessing its photochemical reactions, allowing reactivity under the mildest possible conditions and the exploitation of wavelength-orthogonal chemistries.
Herein, we probe the photoreactivity of Py-chalvia an action plot to determine the mildest possible conditions (i.e., longest wavelength irradiation) that can be applied for efficient polymer end group functionalization. The findings of the current study provide the required insights to exploit the Py-chal chromophore more widely.
Results and discussion
Styrylpyrene undergoes a [2 + 2] photocycloaddition over a limited visible light range,1,10,16–18 and has the synthetic disadvantage of limited yields. In contrast, synthesis of Py-chal is achieved on a multi-gram scale in 2 steps with relatively inexpensive and commercially available reagents in high yields (>95% across two steps). The first step involving a Williamson etherification readily produced over 20 g of product (ESI,†M1 and M4, yield = 98%), and the second step involving a Claisen–Schmidt condensation yielded close to 10 g of product (ESI,†M2 and M5, yield >95%). As such, we were able to synthesize several grams of product at once with relative ease, making Py-chal a very easily scalable chromophore.UV-vis absorbance and kinetic data
While our group has previously established that Py-chal forms photodimers at 415 nm, the detailed photo-characteristics of the chalcone and products are established herein.20,21 Firstly, the absorbance of Py-chal was recorded in a range of solvents to determine if there is a significant shift of the absorption characteristics in different reaction environments (ESI, Fig. 27†). The maximum molar extinction coefficient of the chalcone varies by 10–15% depending on the dielectric constant of the solvent. The Py-chal was also found to exhibit strong green fluorescence when excited with blue or UV light (φf = 0.11, λmax = 496 nm, ESI, Fig. 28†).The [2 + 2] photocycloaddition of Py-chal (415 nm, Fig. 1A) and its photocycloreversion (UV-B, Fig. 1C) were followed over time via UV-vis spectroscopy. The characteristic absorbances of Py-chal at 390 nm and 415 nm diminish when it is converted to the photodimer as the pyrene chromophore is deconjugated from the benzoyl system. In the photodimer, (Py-chal)2, the characteristic absorbances of pyrene at 335 nm and 353 nm are prominent. However, since the photodimerization proved to be a clean reaction according to 1H NMR spectroscopy (ESI, Fig. 14†) and the clean isosbestic point of the UV-vis profiles, the absorbance of the reaction mixture at any given wavelength is merely the sum of the absorbances of the monomer and dimer. Therefore, the dimer was isolated to determine its molar extinction coefficient, after which least a squares fit of the individual extinction coefficients to UV-vis spectra allowed for quantification of the concentration of each of the compounds as a function of time (ESI,† Extraction of kinetic data from UV-vis data).
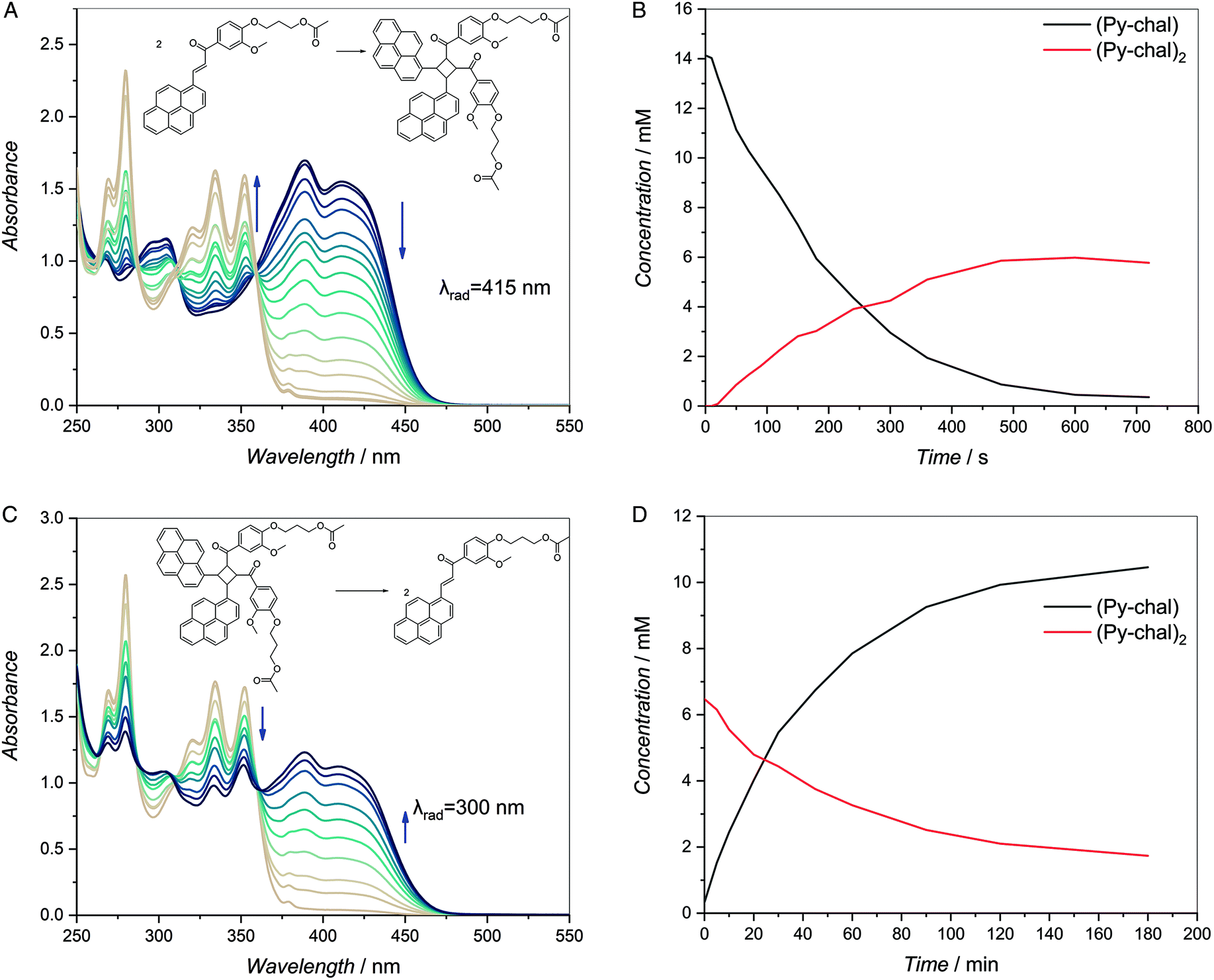 | ||
| Fig. 1 (A) Py-chal in DCM (14 mM) and degassed with argon for 10 min before being irradiated with a 10 W LED (λmax = 415 nm). The mixture was characterized via UV-vis spectroscopy measurements spanning across 12 min. The characteristic chalcone peaks at 390 and 415 nm decrease over time while the two pyrene peaks at 335 and 353 nm, increase. (B) The concentration of Py-chal and (Py-chal)2 over time during the forward reaction shows the gradual depletion of the monomer and concomitant increase in the dimer concentration over time. (C) (Py-chal)2 (DCM, 7 mM) was irradiated with UV-B light to revert it to the monomer and the sample was monitored with UV-vis spectroscopy over 180 min. The pyrene peaks at 335 and 353 nm decrease and the chalcone peaks at 390 and 415 nm simultaneously increase indicating conversion back to the monomer. Conversion was incomplete over the 180 min as indicated by residual pyrene peaks. (D) The concentration of the monomer and dimer over time during the reverse reaction shows the gradual depletion of the dimer and increase in the concentration of the monomer over time under UV-B light. | ||
Fig. 1(A and B) indicates that the forward reaction proceeds to near-completion within 12 min of irradiation with a 415 nm LED. The peaks at 390 nm and 415 nm decrease, and the characteristic pyrene absorbance of the cycloadduct appears prominently at 335 nm and 353 nm. In contrast—according to the UV-vis data—the cycloreversion of the dimer is much slower and even after 180 min of irradiation with UV-B light, only ∼75% of the dimer has been reverted to the monomer (Fig. 1C and D) (ESI,† UV-B reactions). We propose that this is due to the photostationary state as UV light can induce both the forward and reverse reactions of Py-chal, thus the rate and conversion of the reverse reaction are likely impeded by the competing forward reaction.24,25
Action plot and wavelength resolved reactivity
To fully exploit the reactivity of the chalcone, it is critical to establish the optimum dimerization wavelength and establish its wavelength dependent reactivity. In prior studies, we have quantitatively established that the maximum reactivity does not necessarily coincide with the maxima of the absorption band.1 Rather, peak reactivity is often red-shifted relative to the absorption maximum.A so called 'action plot' analysis employs a tunable laser to investigate the wavelength dependent reactivity of a photoactive moiety. It is imperative that the photoreactions conducted at different wavelengths maintain an identical photon count, while keeping other factors, such as concentration, solvent, and temperature constant. For these measurements, the chalcone was dissolved in deuterated acetonitrile (1 mmol L−1) and irradiated with monochromatic laser light at a range of wavelengths between 300 and 500 nm.
The resulting conversion from monomer to photodimer was determined via1H NMR spectroscopy. The integral of the resonance at δ = 8.92 ppm, which is assigned to one of the alkene protons of Py-chal (Fig. 4 (top) proton 8) relative to an internal standard (1,3,5-trimethoxybenzene), was used to determine the depletion of the monomer and quantify reactivity at each wavelength. The resonances at δ = 5.15 and δ = 5.35 ppm correspond to the protons of the cyclobutane ring in the primary cycloadduct (ESI, Fig. 14†). The depletion of the monomer relative to the wavelength of irradiation for Py-chal is presented in Fig. 3.
The action plot indicates that the highest reactivity was obtained at 440 nm (61% conversion for 18 μmol of photons). The full conversion of Py-chal when irradiated with monochromatic light at 440 nm gives a single isomer as the major photoproduct with some evidence of a very minor product (Fig. 2). The longest wavelength at which any reactivity is still observed is 490 nm. Towards the shorter wavelength regime, the reactivity does not decrease below 15% (at 360 nm). As both the forward and reverse reactions are initiated by UV light, these are expected to compete at these shorter wavelengths, ultimately leading to a photostationary state.24,25
 | ||
| Fig. 2 1H NMR spectrum of the photodimer (Py-Chal)2 in deuterated acetonitrile after irradiation with monochromatic light at 440 nm. The minor 1H resonances highlighted in orange boxes suggest the formation of a potential minor photodimer. 1,3,5-Trimethoxybenzene was employed as an internal standard. | ||
The action plot for Py-chal confirms that the photochemical reactivity and absorbance are not necessarily congruent. The peak reactivity occurs at 440 nm, which is red shifted by 25 nm compared to the absorption (λmax = 415 nm). We suggest that the bathochromic shift in reactivity is, in part, due to the lower molar absorptivities and therefore increased penetration depths at long wavelengths.26 For example, >99% of the incident light is absorbed within the first millimeter of the sample for wavelengths ≤440 nm (ESI, Fig. 3†). With no stirring mechanisms, the only mixing of the sample solution during the reaction will be due to diffusion, likely leading to an under-reporting of the conversions in this wavelength region. In addition, in earlier studies we have hypothesized that conical intersections might allow transitions to occur more efficiently in regions of low absorptivity.26 This red-shifted reactivity can be advantageous in biological applications, for example, as it is less likely to produce unspecified photodamage to the biological matrix.22,27–30
Reactivity and end group modification at 505 nm
According to the action plot for the [2 + 2] photocycloaddition, the longest wavelength at which there is still significant reactivity is 490 nm. The dimerization of the Py-chal monomer was tested by irradiating a solution of deuterated acetonitrile (1.42 mM) with a green LED centered (λmax = 505 nm, Fig. 3) for three days. 1H NMR spectroscopic analysis of the product mixture showed near-quantitative conversion of the monomer to the dimer (Fig. 4). These results confirm that the Py-chal photodimerization is one of the few current examples of catalyst-free photocycloadditions that can be induced by green LED irradiation. | ||
| Fig. 3 Action plot of Py-chal, where conversion was determined by the depletion of monomer (black squares) and plotted against the absorbance spectrum of Py-chal (red line, inset at top-left shows zoomed in molar extinction at 490 nm). The chalcone monomer was irradiated with 18 μmol of photons from a tuneable laser at each wavelength in deuterated acetonitrile (1 mmol L−1). The conversion was determined via1H NMR spectroscopy using the integral of the Py-chal double bond resonance at δ = 8.93–8.95 ppm in deuterated acetonitrile (600 MHz). The normalized emission spectrum of the 505 nm green LED (green curve) shows that it emits light in the 490 nm region, where the most red-shifted reactivity of Py-chal is observed. | ||
 | ||
| Fig. 4 1H NMR of Py-chal [top] and (Py-chal)2 [bottom] in deuterated acetonitrile illustrating the near-quantitative conversion of Py-chal into (Py-chal)2 after irradiation with a green LED (λmax = 505 nm) for three days. | ||
To translate the green light activated [2 + 2] photocycloaddition of Py-chal into the realm of polymer chemistry, Py-chal-terminated poly(methyl methacrylate) (PMMA, P1′, ESI† Synthesis of P1′) was synthesized via reversible addition–fragmentation chain transfer (RAFT) polymerization with a chain transfer agent containing the chalcone unit (M7, ESI† Chalcone RAFT CTA).31 The successful synthesis of P1 also demonstrates the ability of Py-chal to survive radical polymerizations at elevated temperatures. The benzodithioate moiety of P1′ (Mn = 3.2 kg mol−1, Mw = 4.1 kg mol−1, Đ = 1.3) was subsequently oxidized (P1) to prevent any further reaction between the sulfur groups in the RAFT agents, such as the formation of disulfide bonds, which could alter the molar mass distribution.32 The polymer was finally irradiated with a green LED (λmax = 505 nm) in a solution of deuterated acetonitrile containing a five-fold excess of free Py-chal before being characterized via Size Exclusion Chromatography coupled with Electron Spray Ionisation Mass Spectrometry (SEC-ESI-MS) (Fig. 5).
 | ||
| Fig. 5 (A) Extract of mass spectrum showing the peaks associated with the starting polymer (P1) in the mixture before ligation [top], which disappear completely after ligation and [Bottom] the simulation for the polymer with 17 MMA repeat units. (B) Extract of mass spectrum showing the peaks associated with the end-group modified product polymer (17 MMA repeat units) (P2) in the mixture after the photoligation [Top] as well as the identical mass range of the starting material for comparison, showing the appearance of product distinctly after the ligation reaction. [Bottom] Simulation of the polymer mass spectrum for the photoligated product. | ||
The end group fidelity of the polymer was characterized before and after irradiation via SEC-ESI-MS to detect the dimerization of the end group chalcone with the free chalcone. The isotopic pattern of the macromolecule containing 17 MMA units was simulated before and after the end group ligation and compared to the measured mass spectra to determine whether the ligation was successful. The isotopic pattern of P1, evident in the pre-irradiated solution, is absent in the post-irradiation polymer (Fig. 5). Extracted ion chromatograms (XIC) of the reaction mixture before and after irradiation (Fig. 6) show a significant shift toward earlier elution times as a result of green light irradiation. It is well known that chromatographic fractions corresponding to higher molar mass elute faster than that with a lower molar mass, providing strong evidence that the product P2 is formed.33 Therefore, the comparison of the SEC-ESI-MS polymer profiles and XIC traces before and after the photochemical reaction shows that the polymer end group modification via [2 + 2] photocycloaddition with green light was successful.
 | ||
| Fig. 6 XIC traces of the polymer with 17 MMA repeat units in the starting material and product shows that the product elutes before the starting material. The earlier elution time indicates that the product is larger than the starting material, which signifies the successful end group ligation of the terminal chalcone after irradiation with a green LED (λmax = 505 nm) for 3 days. | ||
Conclusions
Based on an in-depth study of the photochemical reactivity of a pyrene chalone moiety Py-chal, we demonstrate that its photodimerization is activated with wavelengths up to 490 nm. The most efficient wavelength for Py-chal photodimerization, was 440 nm, which is red-shifted by 25 nm relative to its absorption maximum. As a result of the photoreactivity, a quantitative photodimerization of Py-chal yielding (Py-chal)2 was achieved using commercially available green LEDs centered around 505 nm. The photoreactive chalcone moiety was subsequently attached to a RAFT agent (M7), which was employed in a reversible deactivation radical polymerization of PMMA to afford chalcone-terminated polymer P1. Under green LED irradiation, the chalcone moieties of P1 were readily available for post-polymerization end group functionalization with Py-chal yielding P2. In summary, we demonstrate Py-chal to be efficient under mild photochemical conditions and accessible in high yields with relatively inexpensive starting materials, making it a viable candidate for biological applications.Author contributions
I. M. I. undertook the writing of the manuscript as well as the synthesis and characterization of all components involving polymers and the action plot. K. D. B. synthesized all the small molecules, measured all the UV-vis data, and performed the related analyses. S. L. W. assisted with the setup of the tunable laser as well as the analysis of the 1H NMR spectra related to the action plot. A. S. M. assisted with the analysis of NMR spectra to establish a primary photoproduct. H. F. and C. B.-K. co-conceptualized the study and experimental design. All authors contributed to the manuscript editing and review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. B.-K. acknowledges funding from the Australian Research Council (ARC) in the form of a Laureate Fellowship (FL170100014) enabling his photochemical research program as well as continued key support from the Queensland University of Technology (QUT). C. B.-K. acknowledges additional support via an ARC Discovery grant targeted at red-shifting photoligation chemistry. I. M. I. gratefully acknowledges QUT for a PhD Research Scholarship. H. F. acknowledges support by the ARC in the form of a DECRA Fellowship.The Central Analytical Research Facility (CARF) at QUT is gratefully acknowledged for access to analytical instrumentation.Notes and references
- D. E. Marschner, H. Frisch, J. T. Offenloch, B. T. Tuten, C. R. Becer, A. Walther, A. S. Goldmann, P. Tzvetkova and C. Barner-Kowollik, Macromolecules, 2018, 51(10), 3802–3807 CrossRef CAS.
- C. Deng, Z. Yang, Z. Zheng, N. Liu and J. Ling, J. Mater. Chem. C, 2015, 3(15), 3666–3675 RSC.
- M. Yamamura, K. Orihashi, Y. Mawatari and H. Kage, Drying Technol., 2007, 26(1), 97–100 CrossRef.
- A. Banstola, T. T. Pham, J.-H. Jeong and S. Yook, Drug Delivery, 2019, 26(1), 629–640 CrossRef CAS PubMed.
- A. Chan, R. P. Orme, R. A. Fricker and P. Roach, Adv. Drug Delivery Rev., 2013, 65(4), 497–514 CrossRef CAS PubMed.
- K. Jung, N. Corrigan, M. Ciftci, J. Xu, S. E. Seo, C. J. Hawker and C. Boyer, Adv. Mater., 2020, 32(18), 1903850 CrossRef CAS PubMed.
- K. B. Kockler, H. Frisch and C. Barner-Kowollik, Macromol. Rapid Commun., 2018, 1800516 CrossRef PubMed.
- D. Sarkar, N. Bera and S. Ghosh, Eur. J. Org. Chem., 2020, 2020(10), 1310–1326 CrossRef CAS.
- S. Poplata, A. Tröster, Y.-Q. Zou and T. Bach, Chem. Rev., 2016, 116(17), 9748–9815 CrossRef CAS PubMed.
- H. Frisch, F. R. Bloesser and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2019, 58(11), 3604–3609 CrossRef CAS PubMed.
- H. Frisch, D. E. Marschner, A. S. Goldmann and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2018, 57(8), 2036–2045 CrossRef CAS PubMed.
- F. R. de Gruijl, H. J. van Kranen and L. H. F. Mullenders, J. Photochem. Photobiol., B, 2001, 63(1), 19–27 CrossRef CAS.
- R. P. Sinha and D.-P. Häder, Photochem. Photobiol. Sci., 2002, 1(4), 225–236 CrossRef CAS PubMed.
- J. P. Olson, M. R. Banghart, B. L. Sabatini and G. C. R. Ellis-Davies, J. Am. Chem. Soc., 2013, 135(42), 15948–15954 CrossRef CAS PubMed.
- N. P. Kovalenko, A. Abdukadirov, V. I. Gerko and M. V. Alfimov, J. Appl. Spectrosc., 1980, 32(6), 607–612 CrossRef.
- S. Bialas, L. Michalek, D. E. Marschner, T. Krappitz, M. Wegener, J. Blinco, E. Blasco, H. Frisch and C. Barner-Kowollik, Adv. Mater., 2019, 31(8), 1807288 CrossRef PubMed.
- H. Frisch, J. P. Menzel, F. R. Bloesser, D. E. Marschner, K. Mundsinger and C. Barner-Kowollik, J. Am. Chem. Soc., 2018, 140(30), 9551–9557 CrossRef CAS PubMed.
- H. Frisch, D. Kodura, F. R. Bloesser, L. Michalek and C. Barner-Kowollik, Macromol. Rapid Commun., 2020, 41(1), 1900414 CrossRef CAS.
- L. Michalek, S. Bialas, S. L. Walden, F. R. Bloesser, H. Frisch and C. Barner-Kowollik, Adv. Funct. Mater., 2020, 30(48), 2005328 CrossRef CAS.
- M. Van De Walle, K. De Bruycker, J. P. Blinco and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2020, 59(33), 14143–14147 CrossRef CAS PubMed.
- M. Van De Walle, C. Petit, J. P. Blinco and C. Barner-Kowollik, Polym. Chem., 2020, 11(40), 6435–6440 RSC.
- K. Kalayci, H. Frisch, V. X. Truong and C. Barner-Kowollik, Nat. Commun., 2020, 11(1), 4193 CrossRef.
- K. Kalayci, H. Frisch, C. Barner-Kowollik and V. X. Truong, Adv. Funct. Mater., 2020, 30(15), 1908171 CrossRef CAS.
- M. Liu, W. Wenzel and H. Frisch, Polym. Chem., 2020, 11(41), 6616–6623 RSC.
- D. Kehrloesser, R.-P. Baumann, H.-C. Kim and N. Hampp, Langmuir, 2011, 27(7), 4149–4155 CrossRef CAS PubMed.
- J. P. Menzel, B. B. Noble, A. Lauer, M. L. Coote, J. P. Blinco and C. Barner-Kowollik, J. Am. Chem. Soc., 2017, 139(44), 15812–15820 CrossRef CAS PubMed.
- A. Ghasemi-Kahrizsangi, J. Neshati, H. Shariatpanahi and E. Akbarinezhad, Prog. Org. Coat., 2015, 85, 199–207 CrossRef CAS.
- L. D. Suits and Y. G. Hsuan, Geotext. Geomembr., 2003, 21(2), 111–122 CrossRef.
- Z. Zhang, S. Wang and J. Zhang, Polym. Compos., 2014, 35(12), 2365–2375 CrossRef CAS.
- N. Grassie, M. I. Guy and N. H. Tennent, Polym. Degrad. Stab., 1986, 14(3), 209–216 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31(16), 5559–5562 CrossRef CAS.
- M. Dietrich, M. Glassner, T. Gruendling, C. Schmid, J. Falkenhagen and C. Barner-Kowollik, Polym. Chem., 2010, 1(5), 634–644 RSC.
- T. Gruendling, M. Guilhaus and C. Barner-Kowollik, Anal. Chem., 2008, 80(18), 6915–6927 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py00533b |
| ‡ These authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |