New trends in crystal engineering
Dario
Braga
a,
Lee
Brammer
b and
Neil R.
Champness
c
aDepartment of Chemistry G. Ciamician, University of Bologna, Via F. Selmi 2, 40126 Bologna, Italy. E-mail: dario.braga@unibo.it
bDepartment of Chemistry, University of Sheffield, Brook Hill, Sheffield, UK S3 7HF. E-mail: lee.brammer@sheffield.ac.uk
cSchool of Chemistry, University of Nottingham, University Park, Nottingham, UK NG7 2RD. E-mail: Neil.Champness@nottingham.ac.uk
First published on 4th January 2005
Abstract
The articles and highlights presented at the second CrystEngComm meeting, held in Nottingham in September 2004 are reviewed. The discussion has highlighted the current development of crystal engineering and allowed to emerge some of its future potentials. In particular, the papers described in this highlight focus on four fundamental aspects: (i) intermolecular interactions: evaluation and application to crystal design; (ii) networks: design and applications; (iii) approaches to crystal synthesis; (iv) polymorphism, solvates and chiral crystal resolution.
 Dario Braga | Dario Braga is Professor of Chemistry at the University of Bologna. He received the Raffaello Nasini Prize from the Italian Society of Chemistry in 1988 for his studies on solid state dynamic processes, and the FEDERCHIMICA Prize in 1995 for his research on the intermolecular interactions in organometallic systems. Presently, his main scientific interests are in the crystal engineering exploitation of hydrogen bonding interaction between ions, in the investigation of crystal polymorphism and in solvent-free gas–solid and solid–solid reactions. He has published more than 300 papers and reviews and organized several international conferences and schools on crystal engineering. Dario Braga is the Master of the Collegio Superiore of the University of Bologna. He is member of the international editorial board of Chemical Communications and the Scientific Editor of CrystEngComm. |
 Lee Brammer | Lee Brammer obtained his Ph.D. in Inorganic Chemistry from the University of Bristol. Following a NATO postdoctoral fellowship at the University of New Orleans and Brookhaven National Laboratory in New York, he embarked on his independent academic career in 1990 at the University of Missouri-St. Louis, returning to UK in 2001 to take up an appointment at the University of Sheffield. He is a recipient of the S. S. Sidhu Award from the Pittsburgh Diffraction Society for contributions to the field of crystallography and diffraction. He has been a co-editor for Acta Crystallographica, Section B since 1995 and currently serves as chair of the Editorial Board of CrystEngComm and of the IUCr Commission on Structural Chemistry. His research interests within the field of crystal engineering include the study of intermolecular interactions, particularly hydrogen bonding and halogen bonding. The design and synthesis of metal-containing network solids through the use of non-covalent and/or coordination bonds remains a prominent research focus. The design and application of functional porous and lamellar materials are the principal targets of these approaches. |
 Neil R. Champness | Neil Champness is the Professor of Chemical Nanoscience at the University of Nottingham. He began his academic career at the University of Southampton with a B.Sc. in Chemistry (1989) and Ph.D. (1993), under the supervision of Prof. W. Levason. Following postdoctoral studies in Southampton with Dr G. Reid, he moved to the University of Nottingham in 1995 as a Teaching Fellow in Inorganic Chemistry. He took up a Lecturership in Inorganic Chemistry in 1998 and was promoted to Reader in Chemistry in 2003. He was appointed to the Chair of Chemical Nanoscience in 2004. He has been a member of the Editorial Board of CrystEngComm since 2002. His research is concerned with all aspects of molecular organization, including the understanding of solution phase self-assembly processes, surface supramolecular assembly and organization in the solid-state via crystal engineering. Particular targets of his work are guest molecular entrapment and nanostructure formation in solid, solution or surface environments with an emphasis on applications in information storage and sustainable energy sources. |
1. Introduction
The first CrystEngComm Discussion meeting was held in Bristol in 2002. The meeting ended with the (semi-rhetorical) question: What is crystal engineering in 2002?1 The papers presented in Bristol covered mainly molecular materials and supramolecular chemistry, theoretical evaluation of crystal structures and solid-state methods showing that crystal engineering involves the study and understanding of intermolecular interactions and their application in the bottom-up construction of sophisticated solid state superstructures starting from suitably chosen ionic and/or molecular building blocks. The breadth and depth of the discussion, however, also demonstrated that some facets were taking shape, namely crystal engineering2 had evolved from its roots in organic solid-state chemistry2a,b to become “an all-purpose mature discipline, a science without borders, where the motivations can well be utilitarian and economical, but also aesthetical (when not artistic) and/or fuelled by pure, quintessential, scientific curiosity”.1 This open-minded attitude was evident in the lectures of Gavezzotti,3 Toda,4 Davey,5 and Coppens6 as well as in the many papers presented on that occasion.Where do we stand two years later, at the end of 2004?
The papers and highlights published on the occasion of CrystEngComm Discussion 2 (CECD2) held in Nottingham mark the rapid development of this discipline in such a short period of time as will be apparent from the following overview of both keynotes and papers presented at the meeting. In this review article it will be clear that modern crystal engineering has continued to develop in an interdisciplinary way, drawing ideas and including input from scientists in all areas of chemistry; and while relying on crystallography as a primary characterization method, increasingly invokes an array of physical methods needed to explore the many property-oriented studies that are being undertaken. Thus, crystal engineering is making important contributions in materials science and increasingly at the biological interface.
The aim of crystal engineering is that of controlling collective crystal properties by controlling the way molecular building blocks are assembled in the desired (designed) superstructure. Thus, crystal engineering has at its heart a synthetic endeavour that begins with the synthesis or identification of suitable molecular building blocks which can be combined in a directed manner by self-assembly methods to yield crystalline products. The Nottingham discussion has highlighted not only strategies for crystal synthesis, but practical aspects and theoretical underpinnings of the discipline allowing a snapshot of the current development of crystal engineering and of its future potential. In particular, the meeting has focused on four fundamental aspects of crystal engineering which were also expanded upon in the accompanying highlight articles by Price,7 Ward,8 Poeppelmeier,9 Erk10 and their respective collaborators (see below). The main topics discussed in Nottingham were: (i) intermolecular interactions: evaluation and application to crystal design; (ii) networks: design and applications; (iii) approaches to crystal synthesis; and (iv) polymorphism, solvates and chiral crystal resolution.
2. Intermolecular interactions: evaluation and application to crystal design
Understanding why molecules adopt the crystals structures they do and harnessing this understanding to be able to predict crystal structures to design new crystalline materials is of paramount importance in organic solid-state chemistry and crystal engineering. There are also widespread commercial implications, particularly in the pharmaceutical and pigment industries where understanding the relationship between different polymorphic forms of commercial products and their physical properties is crucial to their development.Often in crystal engineering, as seen throughout many of the papers associated with CECD2, interactions between molecules are analyzed in terms of hydrogen bonds and other directional non-covalent interactions and crystal structures are viewed in terms of building blocks (tectons11) that are linked via supramolecular synthons.12 While such an analysis can be highly effective, a quantitative theoretical model for intermolecular forces shows that the situation is often not so simple. Similarly, it is also often important to be able to evaluate the nature of extended structures and to appreciate structural features such as topology and interpenetration,13 and it is now becoming increasingly possible for this to be achieved using computational approaches.
In her highlight article Price7 provides a lucid introduction and overview of the different types of intermolecular forces that must be considered when developing an intermolecular potential model that can accurately explain or predict the packing of organic molecules.7 The limitations of widely used empirical isotropic atom–atom potentials are discussed and the development and application of non-empirical anisotropic potentials is illustrated with the case of chlorine-containing organic molecules (see Fig. 1).
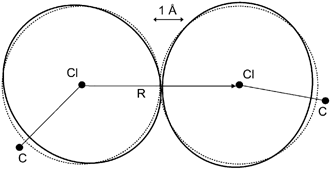 | ||
| Fig. 1 The anisotropy in the intermolecular overlap of the charge distribution of two organic Cl atoms. The contours represent an overlap of 0.002 au factorized between the atoms, as modeled by an anisotropic fit to the overlap (in black). The corresponding best isotropic fit is given in grey. This model was derived from monochlorobenzene,14 and shows the polar flattening and increased repulsion in the lone pair direction shown by all the Cl⋯Cl interactions studied.7 | ||
In particular, the structures of the chlorobenzenes and some chloroazoaromatics are examined with reference to the transferability of atom potentials from one molecule to another. Satisfactory anisotropic atom potentials were developed using a distributed multipole analysis of the charge density and overlap models. These were found to be transferable within classes of very similar molecules such as the chlorobenzenenes. Consideration of one member of this family, p-dichlorobenzene illustrates another major issue that has only just begun to be addressed regarding crystal structure prediction. Lattice energy minimization calculations using the high quality anisotropic atom–atom potential indicate that the three known polymorphs are among the four predicted lowest energy crystal structures.14 However, the global minimum in this search is unknown experimentally. This situation can be accounted for by modelling crystal growth rates using an attachment energy model. Such calculations suggest that the growth rate for the thermodynamically most crystal stable form is appreciably slower than for the three observed forms. Understanding the kinetics of crystal growth is in its infancy, but will clearly become important in developing a complete picture of why certain crystal forms are observed.
A crystal structure prediction study of chlorothalonil (2,3,5,6-tetrachloro-1,3-benzenedicarbonitrile) combined with an experimental search for new polymorphs highlights two further limitations of current models used in crystal structure prediction.15 Chlorothalonil can crystallize in both a disordered form (form 2, see Fig. 2) and adopt a crystal structure with Z′
= 3 (form 3, see Fig. 3). Both situations lie outside the capabilities of current computational approaches. Finally, the danger of relying on anticipated hydrogen bond formation in designing new molecular crystalline materials is illustrated by the case of alloxan, whose crystal structure contains no traditional-length hydrogen bonds despite the number of strong hydrogen bond donor (N–H) and acceptor groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O).16 However, its packing arrangement can be rationalized by full consideration of intermolecular forces. Thus, it is clear that more accurate potentials improve the possibility of discriminating between sometimes many predicted low energy structures for a particular compound. The recently developed SCDS-Pixel method of Gavezzotti has shown considerable promise in this area.17
O).16 However, its packing arrangement can be rationalized by full consideration of intermolecular forces. Thus, it is clear that more accurate potentials improve the possibility of discriminating between sometimes many predicted low energy structures for a particular compound. The recently developed SCDS-Pixel method of Gavezzotti has shown considerable promise in this area.17
 | ||
Fig. 2 The layer structure of the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m disordered structure of chlorothalonil form 2.7 m disordered structure of chlorothalonil form 2.7 | ||
 | ||
| Fig. 3 The crystal structure of form 3 of chlorothalonil showing the symmetry independent molecules.7 | ||
Another substantial theoretical challenge is represented by the hydrogen bond. There is no doubt that the hydrogen bond plays a fundamental role in crystal engineering.18 The characteristic qualities of transferability (from crystal to crystal), reproducibility (in terms of cohesive contribution to crystal packing) and ease of chemical manipulation (molecular functionalization with hydrogen bonding donor/acceptor systems) make it the interaction par excellence in crystal engineering. These features coincide with the crucial importance of hydrogen bonds in biology and physics. It comes as no surprise, hence, that hydrogen bonds have attracted, and continue to attract, the interest of experimentalists as well as of theoreticians in the effort to rationalize the many intriguing aspects of this supramolecular interactions.19
One of the controversial aspects of the hydrogen bond is the role of this interaction when charged species (i.e. ions, and molecular ions) are involved. In their paper,20 Novoa and D'Oria expand on previous studies of the so-called “strength–length” relationship in O–H⋯O hydrogen bonds in the light of ab initio computations (MP2, 6-31+G(d), BSSE, and IMPT). The analysis is based on the calculation of interaction energies between pairs of molecules or ions as a function of the O–H⋯O interaction. Crystal structures determined by neutron diffraction containing hydrogen-bonded pairs of molecules or ions with short O–H⋯O contacts where metrics were consistent with hydrogen bonds were selected from the Cambridge Crystallographic Database (CSD).21a
The main outcome of this study is the absence of correlation between the H⋯O distance and the MP2 interaction energy calculated for the isolated pair of molecules or ions at the geometries found in the crystal structures. While the intermolecular interaction energy will not be the same in the gas phase as in the solid state due to the modifying effect of neighbouring molecules or ions in the latter case, the difference is most marked in the case of interactions between ions where the mediating effect of counterions is removed in gas phase dimer calculations. Thus, in the dimer calculations interactions between charged molecules obey a q1q2/r law, while interactions between uncharged molecules have a different dependence upon the distance between the centres of mass of the fragments. Consequently, no correlation between the intermolecular separation determined in the solid state and interaction energy calculated for the isolated dimer is found when hydrogen bonds, albeit with the same metrics, belong to different classes, particularly if charged⋯charged (i.e. interionic) and neutral⋯neutral (i.e. intermolecular) complexes are mixed. These findings support the conclusion that looking at the shortest contacts between molecules in a crystal does not always provide a clear indication of the energetics of the crystal. It has also been pointed out that the repulsive nature of the interaction energy of isolated cation⋯cation or anion⋯anion pairs does not rule out the manifestation of properties that normally would have been associated with the presence of bonds when such pairings are found in the solid state.22 The reason is that these bond properties are due to overlap between the orbitals of the two moieties, and this overlap is also seen in dimolecular systems that present repulsive interaction energies, whose existence is only due to the presence of cation⋯anion interactions that outweigh that repulsive interaction. In other words, this overlap is induced by the cation⋯anion interactions instead of originating from the intrinsic stability of the dimolecular system. Bonds of this type probably exist in anionic dimolecular systems having short X–H⋯O interactions contacts,23 but are also common in Cu⋯Cu short contacts,24 or in contacts between ions where lone pairs are overlapped (e.g., Pt⋯Pt).25
In another study of hydrogen bonding Czugler and Báthori have investigated two salts of maleic acid, namely N,N-dimethyl-benzylammonium maleate and N,N-dimethyl-benzylammonium oxalate.26 Crystals of the former salt are chiral (space group P212121) and contain the characteristic helices of salt pairs linked through the mediating hydrogen bonding of the anion. Both prochiral sites in the N,N-dimethyl-benzylammonium cation behave in the crystal as if they were differently substituted. Such contacts are indications of non-uniformity in the crystal space of the prochiral groups, whether as a consequence or a cause of chiral crystallization, spontaneous or directed resolution. One may thus conclude that building a crystal space is like a chemical substitution, a behaviour leading to conglomerate crystallization. Following the fundamental logic of supramolecular chemistry such a crystallization may be described as yielding enantiomeric crystal forms, i.e. as an expression of stereoisomerism at the supramolecular level. This is a close analogy to that of the definitions of molecular chemistry where a stereomer has the same meaning as a supramer may have in the world of supramolecular chemistry. As the interactions portrayed here largely depend on the interplay of spatial and electronic requirements, it is not surprising that the smaller and more symmetric oxalic acid in N,N-dimethyl-benzylammonium oxalate does not induce chiral crystallization. The resulting crystal seems to show more flat building preferences, thus providing convenient interfaces for centrosymmetric induction.
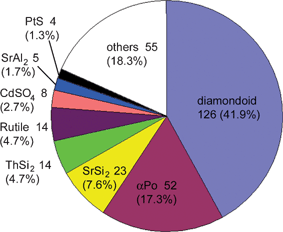
Turning now to the application of computational approaches to network descriptions, Proserpio and colleagues report a comprehensive analysis of the CSD21a and ICSD21b structural databases for the occurrence of interpenetrating metal-organic and inorganic networks.27 A novel version of the program package TOPOS28 was employed, where interpenetrating net recognition is based on the representation of a crystal structure as a finite reduced graph. A comprehensive list of 301 interpenetrating metal-organic 3D structures from the CSD were analyzed on the basis of their topologies and distinct classes of interpenetrating nets have been envisaged, depending on the relationships of the individual motifs (see Fig. 4). The topology of interpenetration observed in the nets is presented in terms of the degree of interpenetration Z and of the modes of interpenetration. The authors are able to recognize distinct classes of interpenetration corresponding to the different modes in which individual identical motifs can interpenetrate, namely (i) translational, (ii) non-translational and (iii) both translational and non-translational. It was found that the large majority of inorganic and metal-organic networks are found to exhibit a limited number of ‘common’ topologies. The distribution of the 47 different topologies (see Fig. 5) shows that the most common interpenetrating nets are by far the diamondoid nets (42%, including supertetrahedral nets), followed by the α-polonium nets (17%), but also many uncommon and new topologies are observed within interpenetrating networks.
 | ||
| Fig. 4 Distribution of the topologies within the 301 interpenetrated structures observed in the CSD and ICSD.27 | ||
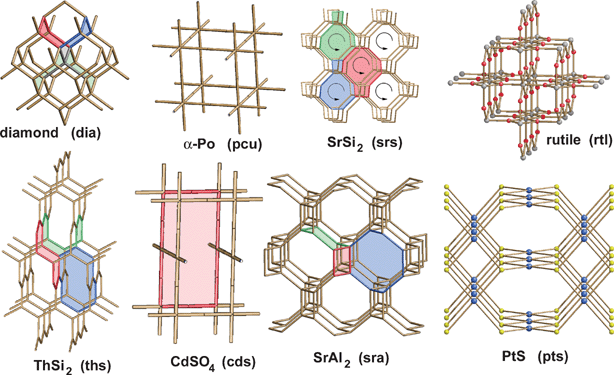 | ||
| Fig. 5 The most frequent topologies observed within the 301 interpenetrated structures; some of the rings characterizing each topology are indicated in colour.27 | ||
3. Networks: design and application
The design of crystal structures can often require the planned synthesis not only of localized interactions, or supramolecular synthons, but a greater appreciation of the whole three-dimensional structure of a crystal. Whereas intermolecular interactions are used to arrange the constituent building-blocks of a material the extended structure of those building-blocks is commonly responsible for the material properties of the designed crystalline array. The synergy between these two themes is key to the successful design of molecular networks.The relationship between extended structure and properties does not apply only to crystalline materials but also to other materials such as surfactants or block-copolymers and the relationship between the structural features of these compounds is elegantly discussed by Ward.8 The themes of synthon and building block design is further expanded in contributions by Orpen,29 Burrows,30 Valdés-Martínez,31 Bourne,32 Ruiz-Perez33 and James.34 Although the nature of the synthon applied to network construction varies from weak hydrogen bonds to strong coordination bonds, common themes are developed illustrating the importance of an appreciation of network structure.
The Highlight article by Ward discusses the nature of the structural relationships that connect molecular crystals, surfactants and block-copolymers.8 The authors observe that although these materials are properly classified as “soft matter”, they are usually regarded as distinct owing to their different properties and applications. They also note that the length scales of translational order in typical molecular crystals are typically at least one order of magnitude smaller than in often highly symmetrical organized microstructures of surfactant assemblies and block copolymers (see Fig. 6). Though the crystal structures of most molecular systems have been typically characterized for the purpose of understanding molecular structure, the recent emergence of crystal engineering as a distinct discipline1,2,35 has targeted the characterization of the factors that determine crystal packing and crystal design strategies. Interestingly, the authors note that the development of crystal engineering has coincided with a growing interest in surfactants and block copolymers with properties that can be tuned at the molecular level. Thus the authors point out that “it seems opportune to begin examining the structural relationships that connect these classes of materials.” The intent of this highlight is to provoke interest in such comparisons through examples of molecular crystals that exhibit features which mimic microstructures observed in surfactant assemblies and block copolymers. The authors identify a suitable range of compounds for analysis that are characterized by molecular components with some degree of amphiphilic character and supramolecular structural elements that (i) enforce topologies that are predestined to form certain high symmetry structures, (ii) introduce conformational softness that permits curvature at small length scales, (iii) form aggregates (i.e., through hydrogen bonding) which effectively increase the length scale so that curved surfaces required for high symmetry lattices can be formed with minimal energetic penalties (see Fig. 7). Through this class of compounds the authors find evidence that the relationship between length scale, energy, curvature, and crystal symmetry requires further examination.
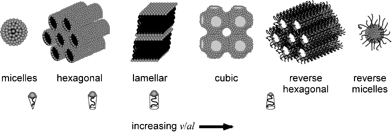 | ||
| Fig. 6 Schematic representation of surfactant microstructures.8 | ||
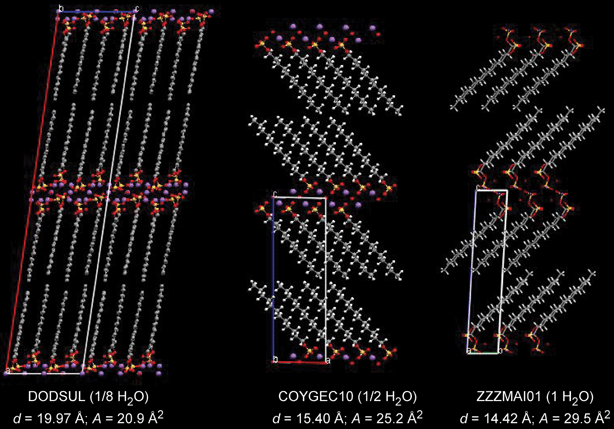 | ||
| Fig. 7 The crystal structures observed for the three hydrates of sodium dodecylsulfate. The addition of H2O beyond the monohydrate results in the formation surfactant phases, initially with lamellar architectures. The CSD reference codes, the degree of hydration, the interlamellar d-spacing (d), and the area (A) of the polar head group are provided beneath each structure.8 | ||
The systematic design of hydrogen-bonded crystal structures has been approached by Orpen and coworkers through the combination of metallate anions containing specific hydrogen bond acceptor groups with rigid organic cations containing spatially well-defined NH+ (4,4′-bipyridinium) or NH2+ (4,4′-bipiperidinium) hydrogen bond donor groups.29,36 Systematic elaboration of these ionic building blocks has been undertaken, in this case focusing on the anions, through use of a sequence of square planar metallate complexes with peripheral CN groups. In order of increasing size these are [M(CN)4]2−, [M(SCN)4]2− and [M(mnt)4]2− (M = Pt, Pd; mnt = 1,2-dithiomaleonitrile). The resulting structures are free of solvent inclusion and comprise hydrogen-bonded networks that result from N–H⋯NC hydrogen bonds that exhibit approximately linear H⋯NC geometries due to the directional properties of the cyano group lone pair (see Fig. 8).37
![The 2-D sheet motif found in [4,4′-H2bipip][PtCl4] showing the pores created in the network.29](/image/article/2005/CE/b417413e/b417413e-f8.gif) | ||
| Fig. 8 The 2-D sheet motif found in [4,4′-H2bipip][PtCl4] showing the pores created in the network.29 | ||
The results of this approach to crystal design and synthesis, termed synthetic crystallography by the authors, has been analyzed in terms of four hierarchically arranged goals, namely control over (i) chemical composition, (ii) hydrogen bond and synthon formation, (iii) periodic motif formation and (iv) the entire crystal structure. A successful synthetic strategy using salt metathesis in non-aqueous solvents has been highly successful in controlling composition. The combination of cyano acceptor groups and N–H donor groups reinforced by the electrostatic association of the ions has led to consistency in hydrogen bond geometries though some flexibility in synthon formation was still present. Complete control over periodic motif formation was shown to be still elusive as a number of different 4-connected nets were formed by using different combinations of tectons. Such control will be necessary in order to achieve the ultimate objective of complete control of the crystal structure formed.
Burrows and coworkers have also pursued the construction of ionic hydrogen-bonded materials. They report the synthesis and structures of a new family of guanidinium sulfonates (GS).30 GS materials have been widely studied by Ward and shown to adopt structures with highly predictable metrics based upon hydrogen-bonded layers that arise from the complementary hydrogen bond donor and acceptor capabilities of the guanidnium and sulfonate ions, respectively.38 While GS have already been used to design of polar crystals for NLO and related applications39 and as recyclable hosts for separations of isomers of chemical feedstocks,40 the new systems reported by Burrows are aimed at studying the potential of designing colourimetric solid-state sensors. A series of four hydrogen-bonded salts are reported using the anion of indicator methyl orange in combination with guanidinium or alkylguanidinium cations. The structure of [C(NH2)3]
[O3SC6H4NNC6H4NMe2] adopts a bilayer arrangement (see Fig. 9) based on hydrogen-bonded sheets analogous to related GS structures with monosulfonate anions.
![The interactions between the cations and anions in [C(NH2)2(NH2)][O3SC6H4NNC6H4NMe2] to form GS hydrogen-bonded sheets. Only part of the anions are shown for clarity.30](/image/article/2005/CE/b417413e/b417413e-f9.gif) | ||
| Fig. 9 The interactions between the cations and anions in [C(NH2)2(NH2)][O3SC6H4NNC6H4NMe2] to form GS hydrogen-bonded sheets. Only part of the anions are shown for clarity.30 | ||
Notably, despite the restriction on hydrogen bond formation that results from mono- or dialkylation of the guanidinium cation, use of [C(NH2)2(NHMe)]+, [C(NH2)2(NHEt)]+ or [C(NH2)2(NMe2)]+ cations leads to similar pseudo-bilayer structures, though N–H⋯O hydrogen bonding now propagates tapes rather than sheets. Treatment of crystals of [C(NH2)3][O3SC6H4NNC6H4NMe2] with gaseous HCl results in a marked colour change from orange to red that is reversible upon treatment with NH3 gas. This process has been monitored by diffuse reflectance UV-visible spectroscopy and by IR spectroscopy wherein protonation/deprotonation of the central diazo unit causes changes in the NN stretch. The reactions were also monitored by powder diffraction demonstrating that the product of reaction of [C(NH2)3][O3SC6H4NNC6H4NMe2] with HCl is crystalline, but not simply a mixture of [C(NH2)3]Cl and O3SC6H4NHNC6H4NMe2. Thus, a first step towards colourimetric sensing has been demonstrated.
Valdés-Martínez, Aakeröy and coworkers have also employed neutral coordination complexes as hydrogen-bonding building blocks.31 Their strategy allows reliable construction of 1D assemblies through mutual recognition of amide groups of trans disposed isonicotinamide or 4-pyridine. This approach resembles earlier studies in which pyridine-based ligands (L) with amide, carboxyl or oxime hydrogen bonding substituents have been employed.41 However, while earlier reports have typically involved cationic building blocks based on either homoleptic complexes ([ML2]n+, [ML3]n+ or [ML4]n+)42 or [M(PR3)2L2]n+ species43 the deliberate strategy employed here of using neutral complexes resulting from the coupling of M(II) ions with two monoanionic chelating ligands avoids any potential competition with counteranions for hydrogen bond sites. The 1,3-diphenylpropanedionato ligands force the amide-bearing ligands to adopt the desired mutually trans geometry and also provide solvent cavities in a manner resembling a polymeric version of the wheel-and-axle approach44 to designing solvated crystals.
Öhrström and co-workers report45 an interesting hydrogen-bonded network structure, [Co(Hbiim)2(H2biim)]2(p-OOCC6H4COOH)2·H2O, constructed via the bridging of the Co(II) 2,2′-biimidazole cation, [Co(Hbiim)2(H2biim)]+, by the mono-deprotonated anion of terephthalic acid and water molecules (see Fig. 10). The nature of the deprotonated dicarboxylic acid bridge was assigned on the basis of the C–O bond lengths determined via single crystal X-ray diffraction studies. The resultant 3-connected net exhibits five-fold interpenetration and was determined to have a vertex symbol of 4 124 122 122, 4 124 124, 124 126 126. However it was noted that depending on the interpretation of the hydrogen bond pattern, this structure may also be interpreted as a single self-interpenetrating network if the bridges formed by the water molecules are included in the topological evaluation.
![An idealized representation of the hydrogen-bonded bridge reinforced by charge interactions between the [Co(H2biim)3]3+ cations and dicarboxylate anions.45](/image/article/2005/CE/b417413e/b417413e-f10.gif) | ||
| Fig. 10 An idealized representation of the hydrogen-bonded bridge reinforced by charge interactions between the [Co(H2biim)3]3+ cations and dicarboxylate anions.45 | ||
Interestingly, the authors discuss the meaning of “a net” in crystal structures and suggest that a possible definition could be that a path along the net should follow the strongest intermolecular bonds and be significantly stronger than all other intermolecular interactions. The authors comment that when ions are involved this definition fails as the Coulombic forces are much stronger than all other intermolecular interactions and are also long-range. However, as in the case reported in the paper, it is clear that nets exist also in these structures, due to the short-range energy minimization by the formation of the most favourable hydrogen or coordinative bond patterns, i.e. directional energies. The role of the charge-charge interaction is to provide overall stability in the crystal, and not only through the interaction with nearest neighbours. The authors use quantum chemical calculations (DFT and semi-empirical methods) to investigate which is the more appropriate description of a net by estimation of the directional forces in their structure and it was found that this approach favours the interpretation of the structure of [Co(Hbiim)2(H2biim)]2(p-OOCC6H4COOH)2·H2O as five interpenetrating nets rather than a single self-penetrating net (see Fig. 11).
![The five interpenetrating nets in [Co(Hbiim)2(H2biim)]2(p-OOCC6H4COOH)2·H2O.45](/image/article/2005/CE/b417413e/b417413e-f11.gif) | ||
| Fig. 11 The five interpenetrating nets in [Co(Hbiim)2(H2biim)]2(p-OOCC6H4COOH)2·H2O.45 | ||
Mukherjee and coworkers report46 the analysis of hydrogen bonding, in particular C–H⋯Cl–M hydrogen bonds, in the crystal structures of two neutral coordination compounds. This work builds upon earlier studies by Brammer, Orpen and colleagues in which the efficacy of metal halides as hydrogen bond acceptors with either strong (N–H, O–H) and weak (C–H) donors was established and typical metrics of such hydrogen bonds were reported.47 Perhalometallates have been used in the systematic construction of hydrogen bonded ionic crystals using either cationic N–H hydrogen bond donors48,49 or C–H donors.50 Here Mukherjee and coworkers report the crystal structures of two (Lx)MCln (Lx = tridentate ligand) complexes in which C–H⋯Cl–M hydrogen bonding is examined. In the crystal structure of [MnCl2(L3)(EtOH)] (L3 = 2-[3-{2′-pyridyl}pyrazol-1-ylmethyl]pyridine the coordinated ethanol molecule serves as a multiple hydrogen bond donor forming one O–H⋯Cl and three C–H⋯Cl hydrogen bonds. Molecules are linked to neighbours via an O–H⋯Cl and a C–H⋯Cl hydrogen bond to give tapes that the authors describe as a ‘staircase’ arrangement (see Fig. 12). These tapes form in parallel pairs that adopt a bilayer arrangement allowing peripheral aromatic groups of the L3 ligands to form offset π-stacks with adjacent bilayers. However, in the structure of a related trichloroiron(III) compound only one rather long intermolecular C–H⋯Cl hydrogen bond is observed and provides a cross-link between helically-stacked molecules.
![View of the staircase of [(2-[3-(2-pyridyl)pyrazol-1-ylmethyl]pyridine)MnCl2(EtOH)] molecules linked via C–H⋯Cl and O–H⋯Cl hydrogen bonding interactions.46](/image/article/2005/CE/b417413e/b417413e-f12.gif) | ||
| Fig. 12 View of the staircase of [(2-[3-(2-pyridyl)pyrazol-1-ylmethyl]pyridine)MnCl2(EtOH)] molecules linked via C–H⋯Cl and O–H⋯Cl hydrogen bonding interactions.46 | ||
Bourne et al. and Ruiz-Perez et al. have reported network materials constructed from a combination of coordination bond linkages and hydrogen bonds. Bourne and coworkers report the synthesis of five inorganic–organic layered composites based on compounds of copper(II) chloride and lead(II) chloride with 1,4-phenylenediamine and benzidine that have a 1 : 1 inorganic : organic layer structure (see Fig. 13).32 These systems resemble the organic-inorganic perovskite layer compounds reported by Mitzi that have enabled the exploration of a number of important physical properties (conducting, magnetic, optical).51
![{CuCl42−}n sheets in CuCl4·(H2(1,4-phenylenediamine)]. Above and below the plane are two short Cu–Cl bonds. (a) Organic cations are visible through the holes in the inorganic sheet. (b) View down the b-axis.32](/image/article/2005/CE/b417413e/b417413e-f13.gif) | ||
| Fig. 13 {CuCl42−}n sheets in CuCl4·(H2(1,4-phenylenediamine)]. Above and below the plane are two short Cu–Cl bonds. (a) Organic cations are visible through the holes in the inorganic sheet. (b) View down the b-axis.32 | ||
The copper(II) chloride structure with p-biphenylamine is a 1 : 2 inorganic : organic layer structure which undergoes an order–disorder transition at ca. 200 K. Crystallization of benzidine in the presence of lead(II) chloride or cobalt(II) chloride at elevated temperature produced only the chloride salt of benzidine. For the hybrid inorganic–organic compounds reported in the study the size of the organic cation is limited by the size of the hole into which it must fit, thus the authors state that “while the extension of the organic layer may vary, the molecule must fit within the “footprint” set by the 2-D inorganic framework (i.e. within the area defined by the terminal halides from four adjacent octahedra). Thus structures reported which contain corner or face-sharing polyhedra all have similar unit cell parameters (ca. 7 Å) in the 2 dimensions defined by the inorganic sheets, the length of the organic cation defining the third dimension. The M⋯M second-neighbour distance within the metal halide sheets varies from ca. 5 Å to 7.7 Å. M⋯M distances between adjacent layers correspond to the relevant unit cell length (either a or c in four of the compounds) except for one compound in which the structure adopts a double layer of mono-ammonium cations where it corresponds to c/2. This observation suggests the possibility of tuning the M⋯M separation by adjusting the length of the organic cation, possibly even the incorporation of a mixed cationic layer.
Ruiz-Pérez et al. report33 three new compounds of formula one of formula [Cu(Hmal)2] and two of formula [Cu(H2O)(H2mal)(mal)] in which the three species of the malonate (malonic acid, H2mal, hydrogen malonate anion, Hmal−, and malonate dianion, mal2−) are present. All the compounds have corrugated malonate layers of [Cu(Hmal)2] or [Cu(H2O)(H2mal)(mal)] units bridged by carboxylate–malonate groups which are linked by hydrogen bonding to form 3-D networks (see Fig. 14 for compound [Cu(Hmal)2]). The magnetic properties of all three compounds have been investigated (see Fig. 15) and it is found that the effectiveness of the exchange magnetic coupling is related to the degree of protonation of the malonate, being negligible for H2mal, very weak for Hmal− and more effective for mal2−. Meanwhile the Cu–O distances also depend on the degree of protonation, being shorter for the Cu–O(mal) and longer for the Cu–O(H2mal), as expected. The authors show using the three new compounds that the versatility of the malonate ligand is enhanced not only by the coordination and bridging modes it can display but also because its protonated species can also afford complexes and mediate magnetic coupling between the magnetic centres.
![View of three adjacent layers of [Cu(Hmal)2]
(hydrogen malonate anion, Hmal−) down the b-axis.33 Hydrogen bonds are drawn as broken lines.](/image/article/2005/CE/b417413e/b417413e-f14.gif) | ||
| Fig. 14 View of three adjacent layers of [Cu(Hmal)2] (hydrogen malonate anion, Hmal−) down the b-axis.33 Hydrogen bonds are drawn as broken lines. | ||
![Thermal dependence of the χMT product for the two compounds (red and blue circles) of formula [Cu(H2O)(H2mal)(mal)]
(malonic acid, H2mal, malonate dianion, mal), the solid lines are the best-fit. The inset shows the magnetization versus H plot at 2.0 K for 2: (•) experimental; (—) Brillouin function for a spin doublet.33](/image/article/2005/CE/b417413e/b417413e-f15.gif) | ||
| Fig. 15 Thermal dependence of the χMT product for the two compounds (red and blue circles) of formula [Cu(H2O)(H2mal)(mal)] (malonic acid, H2mal, malonate dianion, mal), the solid lines are the best-fit. The inset shows the magnetization versus H plot at 2.0 K for 2: (•) experimental; (—) Brillouin function for a spin doublet.33 | ||
Turning now to coordination frameworks, James et al. report34 an interesting study of the nature of coordination polymer formation itself. Building on previous work the authors elaborate on the relationship between the formation of discrete multi-metallic architectures and their ring-opening polymerization to form coordination polymer structures. In the reported study unusual discrete bowl-shaped {Ag4L3} complexes have a formal ring-opening polymerization relationship with a previously reported {Ag4L3}n porous 2-D polymer, where L is a triphosphine ligand (see Fig. 16). The discrete complexes were isolated for triphosphines based on heterocyclic cores whereas the previously-reported polymer was isolated for the corresponding benzene-cored ligand 1,3,5-tris(diphenylphosphino)benzene.
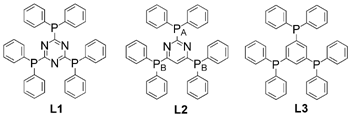 | ||
| Fig. 16 The rigid triphosphines based on triazine (L1), pyrimidine (L2) and triphosphine based on a benzene core (L3).34 | ||
This polymer was crystallized from solutions of the benzene-cored triphosphine 1,3,5-tris(diphenylphosphino)benzene, L3, and silver triflate.52 The structure of the polymer and its formal relationship to the bowl complexes are shown schematically in Fig. 17. The polymer consists of ‘chickenwire’ layers with very large hexagonal rings. AgP3 centres act as the trigonal nodes, and Ag2(L3)2 units form the connecting units. Overall, the polymer stoichiometry is {Ag4L3}. Formal disconnection of the Ag–P bonds at the AgP2 centres around the rim of the bowl, followed by closure of 12-membered rings between the complexes, as indicated in the figure, would give the connecting units in the polymer. If this were to occur on all three sides of the bowl, then the hexagonal polymer structure would result, in which the AgP3 centres of the bowls would become the AgP3 nodes of the polymer. This suggests that the bowl complexes isolated for L1 and L2 may represent intermediates in the formation of the polymer based on L3.
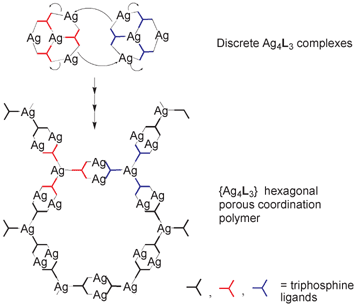 | ||
| Fig. 17 Schematic representation of the relationship between the discrete bowl-shaped {Ag4L3} complexes and chickenwire {Ag4L3}n polymers.34 | ||
4. Approaches to crystal synthesis
Beyond the design of building blocks and the consideration of intermolecular forces and supramolecular synthons, making new materials through crystal engineering necessarily requires a choice of method for crystal synthesis itself. Simple solution phase methods such as solvent evaporation or diffusion and slow cooling are widely used and can include a variety of sophisticated variations in such methods. However, methods of crystal synthesis such as solvothermal synthesis, solvent-free grinding of components sometimes known as mechanochemical synthesis, crystal growth at elevated pressures and at very low temperatures are less well known to most who are involved in crystal synthesis. These methods are discussed and their applications to specific problems illustrated in the series of articles by Poeppelmeier,9 Braga,53 Kuroda,54 Pulham,55 Sermon56 and Boese57 and respective collaborators.The highlight by Poeppelmeier and coworkers9 addresses an important question in the design of functional materials, namely how to engineer noncentrosymmetric or specifically polar and/or chiral crystal structures. Such materials are required for applications such as piezo-, pyro- or ferroelectricity, and for second harmonic generation (SHG).
A commercially important acentric crystalline material used in SHG applications is LiNbO3, in which acentric oxoniobate (distorted) octahedra give rise to the asymmetric crystal structure that permits the non-linear optical (NLO) properties. The approach of Poeppelmeier involves using the strictly acentric octahedral oxofluorometallate anions MOF52− (M = V, Nb, Ta) and cis-MO2F42− (M = Mo, W) or MF62− analogues (M = Ti, Zr, Hf) and developing strategies to incorporate these anions into acentric crystal structures.58 Specifically in this highlight the design of coordination chains of vertex-linked octahedra is explored and a strategy for making these chains helical and thus polar is discussed. In this context the relationship between linear chains, zigzags and helices has been examined (see Fig. 18). The coordination chains involve bridging fluoride ligands that link the early transition metals of the octahedral anions with late transition metals such as Cu, Zn, or Cd, which are themselves crosslinked through pyrazine ligands. The two remaining coordination sites at the late transition metal centre are occupied by water ligands, which form O–H⋯F hydrogen bonds to neighbouring oxofluorometallate moieties in four neighbouring chains. All of these components are important in arriving at the final crystal structure.
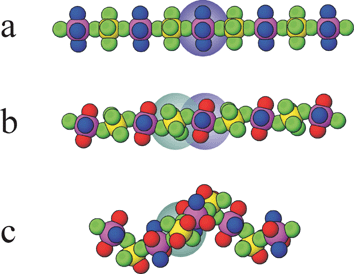 | ||
| Fig. 18 The progression of a linear chain as in (a) Cu(py)4MX6 (M = TiF62−, ZrF62−, HfF62−, NbOF52−, TaOF52−, WO2F42−; py = pyridine) to a zigzag chain in (b) Cu(py)2(H2O)2WO2F4, and Cu(pyz)(H2O)2TiF6 (pyz = pyrazine) to a helical chain in (c) M(pyz)(H2O)2MoO2F4 (M = Zn, Cd).9 The cation and anion octahedra are highlighted with purple and blue shadings, respectively. | ||
A key observation has been that coordination of the metallate anion involves two trans sites at the late transition metal, but coordination at the metallate node in the chain can be trans or cis (see Fig. 19). Careful examination of the three classes of metallate anion reveals that the MF62− and MOF52− ions are trans directing consistent specifically in the latter with the greatest accumulation of negative electrostatic potential being at the oxide site and its trans fluoride. However, cis-MO2F42− anions are found to make cis linkages within the chains with bridging to the neighbouring late transition metal centre occurring through the fluoride ligands that lie trans to the oxides. This cis connection in the chain promotes zigzag or, with the right choice of ligands at the late transition metal site, helical chains.
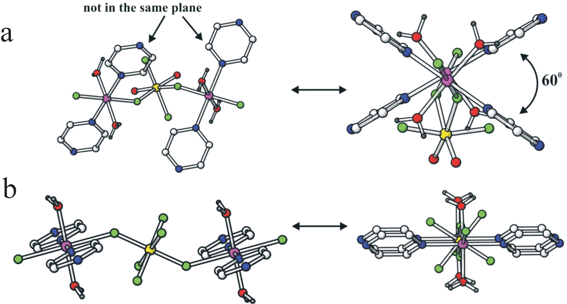 | ||
| Fig. 19 Comparison of (a) the cis-trans chains in Zn(pyz)(H2O)2MoO2F4 and (b) the trans-trans chains in Cu(pyz)(H2O)2TiF6, aligned horizontally (left) and perpendicular to the page (right).9 | ||
The crystals containing these coordination chains are synthesized in a one-pot solvothermal approach in which oxides of the two metals are solubilized in aqueous HF, which simultaneously supplies the required fluoride ions. Multiple trials are typically required for such a synthetic method to establish not only the correct composition space to yield the desired product, but also to identify the necessary heating and cooling cycle (and implicitly the pressure conditions). This necessity has been overcome by an ingenious approach in which multiple syntheses with different concentrations/stoichiometries can be pursued simultaneously under essentially identical conditions. Thus rather than single vessel syntheses, reactions are performed within sealed Teflon pouches within a single pressure vessel.59
Turning now to solventless reactions, it has been pointed out that direct reactions between two crystals or between crystals and molecules in the vapour phase are viable routes to the preparation of molecular crystals,60 and hence of interest in crystal engineering. Braga et al. report an application of this strategy to the preparation of coordination polymers.53 The coordination polymer Ag[N(CH2CH2)3N]2[CH3COO]·5H2O has been obtained by co-grinding in the air crystalline powders of silver acetate and of N(CH2CH2)3N in a 1 : 2 ratio. The exact nature of the product has been established by comparing its experimental powder diffractogram with that calculated on the basis of the crystal structure from single crystal obtained by a different crystallization method. Fig. 20 shows the Ag–[N(CH2CH2)3N]–Ag–[N(CH2CH2)3N]–Ag chain present in Ag[N(CH2CH2)3N]2 [CH3COO]·5H2O. Each silver atom carries an extra pendant N(CH2CH2)3N ligand and a coordinated water molecule resulting in tetrahedral coordination geometry.
![The coordination network in Ag[N(CH2CH2)3N]2[CH3COO]·5H2O. Note the chain of Ag–[N(CH2CH2)3N]–Ag–[N(CH2CH2)3N]–Ag with each silver atom carrying an extra pendant N(CH2CH2)3N ligand and a coordinated water molecule giving a tetrahedral coordination geometry.53](/image/article/2005/CE/b417413e/b417413e-f20.gif) | ||
| Fig. 20 The coordination network in Ag[N(CH2CH2)3N]2[CH3COO]·5H2O. Note the chain of Ag–[N(CH2CH2)3N]–Ag–[N(CH2CH2)3N]–Ag with each silver atom carrying an extra pendant N(CH2CH2)3N ligand and a coordinated water molecule giving a tetrahedral coordination geometry.53 | ||
The coordination polymer Zn[N(CH2CH2)3N]Cl2 on the other hand has been obtained by reacting N(CH2CH2)3N with ZnCl2. Single crystals of Zn[N(CH2CH2)3N]Cl2 could be obtained by layering a solution of ZnCl2 in methanol over a solution of N(CH2CH2)3N in CH2Cl2. Fig. 21 shows that the structure of Zn[N(CH2CH2)3N]Cl2 is based on a 1-D coordination network constituted of alternating N(CH2CH2)3N and ZnCl2 units, joined by Zn–N bonds. Contrary to the silver complex, however, co-grinding leads initially to formation of a new compound of unknown stoichiometry, which can be transformed into the known anhydrous phase Zn[N(CH2CH2)3N]Cl2 by prolonged grinding. It seems reasonable to infer that the intermediate phase is a hydrated system, wherefrom water is eliminated by further mechanical stress.
![The zig-zag coordination network present in crystals of Zn[N(CH2CH2)3N]Cl2 as obtained from solution crystallization.53](/image/article/2005/CE/b417413e/b417413e-f21.gif) | ||
| Fig. 21 The zig-zag coordination network present in crystals of Zn[N(CH2CH2)3N]Cl2 as obtained from solution crystallization.53 | ||
A related set of experiments has been described by Kuroda et al.54 The study is an extension of the investigation of solid-state co-grinding of two or three different aromatic compounds to produce coloured adduct crystals.61 The inter-solid reactions between benzoquinone (BQ) and the diol crystals racemic bis-β-naphthol (rac-BN), 2,2′-biphenol (2BP), and 4,4′-biphenol (4BP) have been thoroughly investigated by quantitative mixing/co-milling and monitored over the course of the reaction by X-ray powder diffraction measurements. The adduct crystals rac-BN-BQ, 2BP-BQ and 4BP-BQ have all been obtained by slow and gradual molecular diffusion in the solid-state. The crystal-to-crystal transformation occurs without going through an amorphous intermediate indicating that molecules diffuse and rearrange via hydrogen bond breaking and forming, yielding crystalline order. Interestingly, the processes are independent of the melting point of the reactants, since the mp of 4BP is higher than and 2BP is lower than rac-BN. In the cases of 2BP-BQ and 4BP-BQ adduct formation simply requires contact between the microcrystals of the two compounds, while in the case of rac-BN-BQ a sheering power above a particular threshold was required to initiate the process (see Fig. 22). In all cases, the reaction proceeded slowly and continued for a surprisingly long period of time. The different speed and initial power required seem to reflect the different lattice energy of the diol crystals and the formation energy of the adduct crystals.
 | ||
| Fig. 22 (a) A cross section of a disk formed by applying pressure to layered powders of rac-BN (top) and BQ (bottom). (b) A cross section of a disk formed by applying pressure to layered powders of 2BP (top) and BQ (bottom). (c) Softly layered 2BP (top) and BQ (bottom) powders as observed initially, after 5 min and overnight following the layering. (d) Softly layered 4BP (top) and BQ (bottom) powders as observed overnight, after 3 days and after 1 week following the layering.54 | ||
Co-grinding and milling are mechanochemical methods to obtain new crystalline materials. These methods exploit the effect of stress, pressure and attrition to activate reaction. Pulham and collaborators55 investigate the effect of pressure directly on crystal formation by cooling a liquid or a solution inside a diamond-anvil cell under high pressure. In earlier applications the technique has been successfully used to prepare new polymorphs of simple molecular organic and inorganic compounds such as ketones, alcohols, and mineral acids are readily obtained.62 In the present study, the technique is applied to the quest of a new polymorph of phenanthrene (obtained from dichloromethane at a pressure of 0.7 GPa), and of novel dihydrate of paracetamol (obtained from water at a pressure of 1.1 GPa).
As the authors point out, pressure is very effective at generating new polymorphs and solvates because it favours formation of denser structures in which molecules must pack together more efficiently. Fig. 23 shows how the crystal structure of phenanthrene is dominated by the classical herring-bone packing motif. The high-pressure phase is 12% denser. Indeed, this paper demonstrates that high-pressure recrystallisation can be used to prepare polymorphs that are metastable under ambient conditions. It is important to appreciate, however, that high-pressure recrystallisation is fundamentally different from a pressure-induced solid–solid transformation.
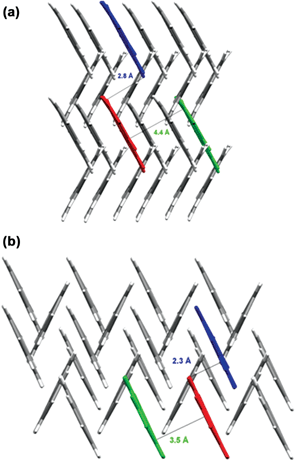 | ||
| Fig. 23 Herring-bone type structure of phenanthrene (a) at ambient pressure and (b) at high pressure.54 | ||
While the two papers above discuss methods that involve treatment of molecular crystals (whether organic or metalloorganic) in their paper, Sermon et al. discuss56 how to prepare nanocrystals of number of inorganic solids, which are usually produced from vapours by high-energy methods or from solutions by low energy methods. It is argued that “…colloid science, in the context of crystal engineering, may no longer be the world of neglected dimensions but could be the basis of a new business area of nanotechnology.” In particular, the authors present a method to obtain nanoribbons of BaSO4. The method is based on a molecular mangle that delivers continuous ribbons of selected crystals with nm thickness and features a number of other applications.
Fig. 24 shows an example of commercial BaSO4 while Fig. 25 shows how the nanoribbons are formed (for details of the reactor see the original paper in ref. 56). The nanoribbons have a width of 50–60 nm, but are formed continuously over significant lengths. The interfacial crystallization reactor used in this experiment may also be utilized in the design of metastable inorganic nanoribbons other than BaSO4.
 | ||
| Fig. 24 Commercial AW2 barite powder from Viaton Industries. Scale = 1 µm.56 | ||
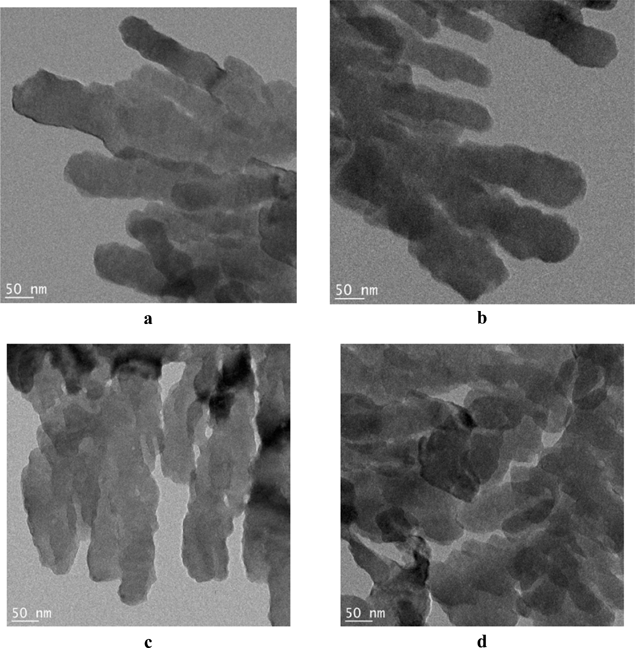 | ||
| Fig. 25 TEM images of BaSO4 nanoribbons grown in the surface film (nanomangle) in the reactor.56 | ||
Boese et al. report57 an interesting series of compounds that exploit C–H⋯N interactions to make co-crystals of acetylene with a range of azacyclic N-acceptors. The acetylene can be introduced under increased pressure, forming molecular complexes with the N-acceptors which were crystallized and investigated by X-ray methods. This in situ method of crystallization has been successfully applied by Boese in a number of elegant studies.63 Five co-crystals of acetylene with pyridine and derivatives of pyridine and pyrazine were crystallized in situ to study the competition of the different C–H donors for the N-atom acceptor (as an example, the co-crystal with pyridine is shown in Fig. 26). In some of the examples studied water molecules are also included in the crystal structures providing competition for the hydrogen-bonding interactions.
It was found that often the chosen stoichiometry of the co-crystals cannot be rationalized, as they are a consequence of the versatility and size of acetylene to fit the available donors and of the optimization of the overall packing. Even if acetylene only forms one interaction with each acidic hydrogen atom, the dimensionality of the resulting network is difficult to predict. The structures include trimeric adducts, linear chains, zigzag chains and, with consideration of aromatic C–H⋯N interactions, higher dimensional networks are observed. It was concluded that co-crystallization as a representative of supramolecular synthesis has lots of experimental difficulties, especially if small and gaseous components are involved. However when successful, much information is provided about the stability of networks, preferred motifs and preferences of donor–acceptor functionalities.
5. Polymorphism, solvates and chiral crystal resolution
Crystal polymorphism64 has emerged as an area of enthusiastic research effort associated with modern crystal engineering. On the other hand, as pointed out during the discussion, it can be argued that crystal polymorphism may also be regarded as the nemesis of this field of research.2b,65 Indeed, the existence of crystal polymorphs ultimately demonstrates that the design of a desired crystal architecture is often a multi-solution problem because the minimization of the global free energy of a crystal (the free energy of the molecule convoluted with the free energy of the supramolecular crystal structure) permits different compromises between the various components.It is also true, however, that the investigation of crystal polymorphism affords invaluable insight into the factors responsible for crystal cohesion and for molecular recognition.66 Moreover, this problem has an extremely relevant commercial impact as it carries implications for marketing and patenting of drugs formulated as crystalline solids.67
Polymorphism and other utilitarian materials chemistry aspects of crystal engineering, in particular those in the area of industrial pigments, are outlined in the highlight by Erk.10 Erk and collaborators discuss examples of the application of crystal engineering strategies to the development of various industrial products, e.g. pigments, dyes, agrochemicals and drugs. As they point out, crystal engineering protocols, based on the possibility of directing synthetic chemistry towards the design of molecules with required and useful solid state properties, have long been used by companies. The interest of industries in collective molecular crystal properties begun in the mid-seventies mainly in the area of pigments or related functional materials, e.g. DuPont, ICI, Eastman Kodak, Ciba-Geigy, BASF and Hoechst. However, intuitive and experience-based crystal engineering has been employed since the beginning of the 20th century. Among the first documented examples are the development of azo pigments and the early investigations into polymorphism of copper phthalocyanine.
There are a number of areas in which design and evaluation of molecular crystals is at the core of industrial applications. Erk lists a few. For example, high conductivity could be obtained by crystallizing molecular materials in segregated stacks of donors and acceptors as in the classical TTF-TCNQ. However, chemically sensible variations of TCNQ-like acceptors were limited because of the bulky C(CN)2 group which causes bending of substituted and annealed quinoid TCNQ-like acceptor molecules such as TCNAQ (see Fig. 27). Quinoid acceptors bearing the smaller N–CN group can tolerate vicinal substituents as well as condensed benzene rings and stay planar as in the case of DCNAQI. This approach to design a new class of organic acceptors, DCNQIs, has been successful in the synthesis of a number of highly conducting charge transfer complexes of substituted benzo- and even naphthoquinones, which do not have counterparts in the TCNQ family for the reasons given above.
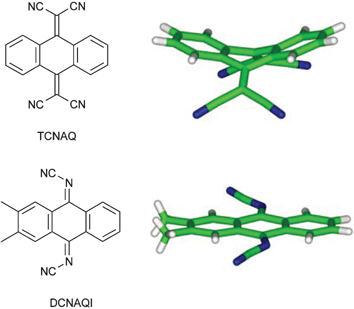 | ||
| Fig. 27 Molecular geometries of TCNAQ and DCNAQI building blocks.10 | ||
DCNQI acceptors also gave access to some of the highest normal-conducting organic materials known to date, radical anion salts of the type [2,5-R,R′-DCNQI]2M (M = Cu, Ag, alkaline metal, ammonium, R, R′ = CH3, Cl, Br, I, OCH3). An important crystal engineering result is the discovery that, while most salts undergo a phase transition which breaks down the conductivity by several orders of magnitude, the copper salts bearing very large substituents do not distort and remain metallic down to the lowest temperatures.
In the field of pigments, Erk et al. mentions Pigment Yellow 139, synthesized in the 1960s and first marketed by BASF in 1973 as a high performance colourant for paints and plastics, as a good example of the use of hydrogen bonds in pigment design (see Fig. 28). This isoindoline pigment crystallizes in a layered structure forming a complete 2D hydrogen-bonded network, which makes the pigment completely insoluble.
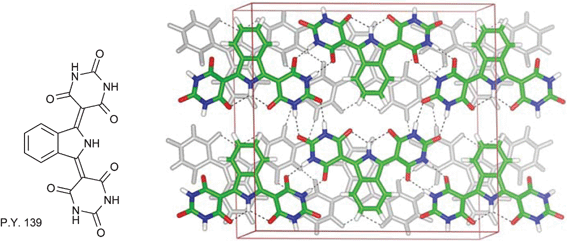 | ||
| Fig. 28 Hydrogen bonding network in Pigment Yellow 139.10 | ||
Monoazo pigments like Pigment Yellow 74, which also packs in a layered structure, were among the first organic pigments and are still of considerable commercial importance today. The structures of Pigment Yellow 74 and of Pigment Red 208 are shown in Fig. 29.
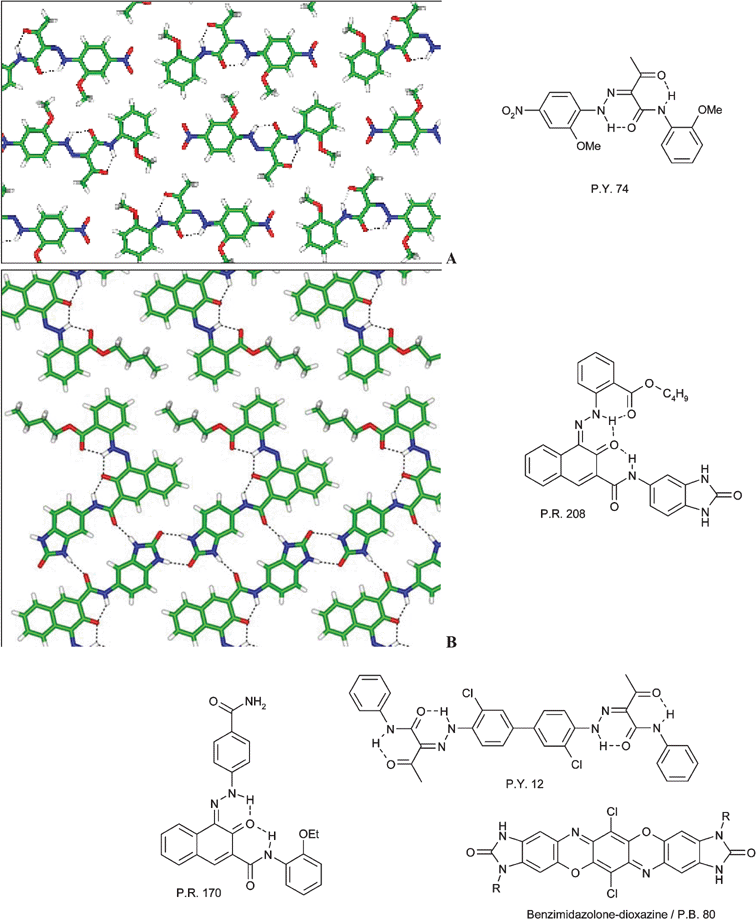 | ||
| Fig. 29 Examples for the advancement of organic pigments by intuitive and intentional crystal engineering. (A) Layered structure of Pigment Yellow 74 (P.Y. 74); (B) layered structure of Pigment Red 208 (P.R. 208).10 | ||
Another key area of industrial interest is that of crystal polymorphism. Erk et al point out that research in this area has afforded industrial innovation. Indeed, beside the importance of polymorphism problems in the preparation and marketing of drugs, polymorphism also is extremely important for pigments. Linear trans-quinacridone (Pigment Violet 19) was first synthesized by Liebermann in 1935. The potential of quinacridone as a high performance pigment was recognised 20 years later by Struve and his team at DuPont, who discovered two new polymorphs with remarkably different colour properties. The synthesis of quinacridone yields the reddish α-form which was not commercialised because of its lack of fastness properties. Treatment in high boiling organic solvents at elevated temperature converts the α-quinacridone to the β- or γ-form, depending on the nature of the solvent. Non-polar solvents promote the formation of the violet β-phase, while polar solvents direct to pink γ-quinacridone. Fig. 30 shows the different electronic spectra of quinacridone polymorphs. Black: α-; blue: β-; red: γ-form.
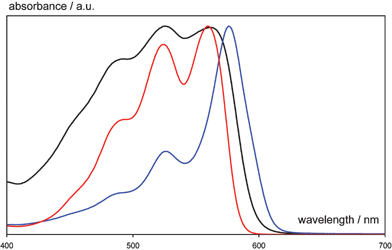 | ||
| Fig. 30 The different electronic spectra of quinacridone polymorphs. Black: α-; blue: β-; red: γ-form.10 | ||
Another important class of pigments where polymorphism is a major player is that of phthalocyanines. After a serendipitous discovery at Scottish Dyes in 1928, the ability to control polymorphism of copper phthalocyanine (CuPc, Pigment Blue 15) paved its way into the market. Erk et al. point out that copper phthalocyanine pigments have revolutionised colour printing. The β-form CuPc pigments correspond to the CIE 12–66 standard cyan shade of the European Colour Scale for offset and letterpress printing. No other blue pigment offers a better economy and such high tinctorial strength. This finally enabled the widespread use of four-colour printing.
On the technical side, Erk alerts readers to the strong need to develop methods for ab-initio structure determination from powder diffraction, which is very important for insoluble pigments, because it is often impossible to grow crystals suitable for single crystal X-ray experiments. As an example, the authors cite P.Y. 110, which was obtained from Ciba SC (Cromophtal® Yellow 2RLP). Finally, Erk et al. mention the importance of studying solvation and solvate crystals. While moving from one component in a crystal to two or more components increases the complexity of the system under investigation, binary crystals like solvates or hydrates often form because of better packing options by involving solvent molecules and by making use of specific interactions between the solvent and the solute. The control of solvate formation in formulations is often a challenge, either because of the formulation stability or because of the formulation's function. Plastic solar cells are prepared by spin-coating from a liquid formulation of a conjugated polymer and a fullerene derivative (PCBM). The change from toluene to chlorobenzene as the solvent used for casting the photoactive blend has lead to an increase of the photovoltaic efficiency from 1 to 2.5%. The increase in efficiency originates from different morphologies of the final film in which both components are phase segregated.
As well illustrated by Erk's highlight, prediction and understanding of the factors affecting polymorph formation is one of the more important areas in crystal engineering.10 Nucleation is one of the key steps in crystal formation,5 but it is very poorly understood as is the connection between small solution phase aggregates (nuclei) and the final crystal form. Hunter, Spitaleri and colleagues have developed a method for structure determination of such aggregates based upon 1H NMR complexation-induced changes in chemical shift.68 The structure of these aggregates (here dimers) has then been compared with those observed in the known crystal forms of molecules studied. The structure determination method uses a genetic algorithm to search conformational space and thus optimize a calculated (solution phase) structure giving a suitable fit to the measured changes in 1H NMR chemical shift. Two sterically similar molecules have been studied. The first has limited hydrogen-bonding options while the second although having only one hydrogen bond donor has multiple hydrogen bond acceptor sites. In both cases an excellent agreement was found between the solution phase dimer structure determined by NMR methods and the molecular aggregation observed in the crystal structure (one polymorph in the case of the second compound, the drug sulfamerazine; see Fig. 31). Although the authors point to shortcomings of the method that needed to be overcome through the use of distance constraints in the study of the polymorphic sulfamerazine, this approach represents a significant step forward in bridging our gulf in understanding between nucleation and crystal structure.
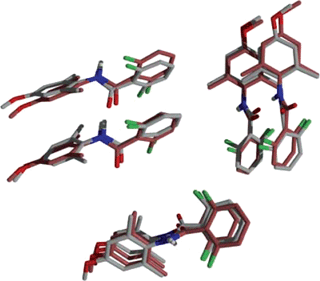 | ||
| Fig. 31 Three orthogonal views of the sulfamerazine dimer. The solution NMR structure is shown in grey and the X-ray crystal structure is shown in brown.68 | ||
In their paper Scott and collaborators expand on their work on the phenol/ether “Horning-crowns” macrocycles,69 which are characterized by the presence of at least one 2,6-bis(benzyl)phenol unit, reminiscent of calixarenes and at least one polyether moiety, typical of crown ethers. These supermolecules are obtained by convergent building block approach via a number simple synthetic steps and transformations. In this study they show that a macrocycle containing phenol and ether functionality is able to twist via intramolecular hydrogen bonding to provide a “wrung out” conformation with a large cleft (see Fig. 32), which self-associates to form dimers under a range of different crystallization conditions. The interactions of the macrocycles with a variety of guest molecules (such as pyridine, toluene and ethanol but also anthracene-9-carboxaldehyde, heptane, dichloromethane, etc.) does not disrupt the highly complementary dimers, but leads to subtle changes in hydrogen bond geometry and to twisting about the axis approximately perpendicular to the internal π–π stacked aromatic rings, yielding dimeric pairs with differing π–π interactions and external shapes.
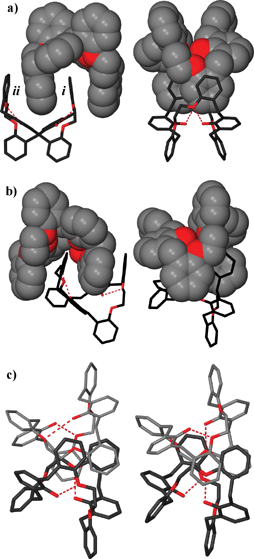 | ||
| Fig. 32 A pair of intramolecular hydrogen bonds between phenol donor and ether acceptor twists the macrocycle, closing the cavity and forming a structure with a distinct cleft. These associate as clasped dimers composed of pairs of twisted, intramolecularly hydrogen-bonded macrocycles. Four salicylyl moieties participate in π–π interactions. The two macrocycles forming the dimer are independent molecules and the dimer is not symmetrical but twisted about the axis of the π-stacked central rings.69 | ||
Herradón and collaborators70 have reported the results of a crystallization-induced dynamic atroposelective resolution of the peptide-biphenyl hybrids: methyl(R,R,R,R)-2-[2-({2′-[1-(1-methoxycarbonylethylcarbamoyl)-3-methyl-butylcarbamoyl]biphenyl-2-carbonyl}amino)-4-methylpentanoylamino]propanoate, methyl (S,S)-2-{[2′-(1-methoxycarbonyl-2-phenylethylcarbamoyl)biphenyl-2-carbonyl]amino}-3-phenylpropanoate, methyl(R,R)-2-{[2′-(1-methoxycarbonyl-2-phenylethylcarbamoyl)biphenyl-2-carbonyl]amino}-3-phenylpropanoate. The crystal engineering idea is based on the simultaneous exploitation in packing design of arene-arene interactions and of N–H⋯OC hydrogen bonds between the peptide CO and N–H groups
Resolution has been achieved by crystallization from methanol solution. These hybrids crystallize in highly ordered structures kept by an extensive array of intramolecular and intermolecular hydrogen bonds. The solid-state molecular structures are shown to depend on the substituents on the α-carbon of the amino acid, which also control the helical superstructures whose orientation depends on the senses of the chirality of the α-carbon of the amino acid residues and the aryl-aryl interaction. Due to the presence of the stereogenic centres in the substituents the aryl-aryl interaction is a diastereogenic element, leading to formation of atropisomers. In solution, the conformational equilibria in the peptide-biphenyl hybrids are much slower than in other 2,2′-disubstituted biphenyls, with one atropisomer predominating over the other as detected by 1H NMR. The shift of the dynamic equilibrium between the atropisomers is controlled by the efficient packing of the molecules of one of the isomers that, in turn, is determined by favourable intermolecular interactions. In the crystalline state, these molecules possess three stereochemical levels: the chiral carbons at the amino acid residues, the stereogenic aryl-aryl interaction, and the supramolecular helix. Examples of the supramolecular arrangements are provided in Figs. 33 and 34.
![Crystal packing of methyl(R,R,R,R)-2-[2-({2′-[1-(1-methoxycarbonylethylcarbamoyl)-3-methyl-butylcarbamoyl]biphenyl-2-carbonyl}amino)-4-methylpentanoylamino]propanoate showing the supramolecular left-handed helix (M-configuration).70](/image/article/2005/CE/b417413e/b417413e-f33.gif) | ||
| Fig. 33 Crystal packing of methyl(R,R,R,R)-2-[2-({2′-[1-(1-methoxycarbonylethylcarbamoyl)-3-methyl-butylcarbamoyl]biphenyl-2-carbonyl}amino)-4-methylpentanoylamino]propanoate showing the supramolecular left-handed helix (M-configuration).70 | ||
![View along the a-axis of the crystal packing of methyl (S,S)-2-{[2′-(1-methoxycarbonyl-2-phenylethylcarbamoyl)biphenyl-2-carbonyl]amino}-3-phenylpropanoate showing the array of H-bonds.70](/image/article/2005/CE/b417413e/b417413e-f34.gif) | ||
| Fig. 34 View along the a-axis of the crystal packing of methyl (S,S)-2-{[2′-(1-methoxycarbonyl-2-phenylethylcarbamoyl)biphenyl-2-carbonyl]amino}-3-phenylpropanoate showing the array of H-bonds.70 | ||
6. Conclusions
Crystal engineering can trace its roots back in the '60s if not earlier. Schmidt's studies of photocyclization reactions in the solid state are recognized as the first attempts to rationally design crystals with a purpose,2a the purpose being that of organizing molecules at a distance and with the appropriate orientation for chemical reaction.71 It is however under the stimulus and pressure of supramolecular chemistry72 that crystal engineering has become a booming field of research that spans across all traditional sub-disciplinary barriers of chemistry (organic, inorganic, coordination etc.). The papers reviewed in this highlight clearly show how cross-fertilization is taking place: building blocks (molecules, ligands, means and geometry of coordination, ions etc.) from these sub-areas are used simultaneously and cooperatively to produce new hybrid aggregates with novel structural features. This also applies to methods. Molecular chemists are learning to approach the products of their crystal engineering efforts with less traditional tools, such as thermogravimetric analysis, calorimetry, methods for gas absorption and release, solid state NMR spectroscopy, etc as well as to exploit non-solution methods (grinding, milling, etc.) as a means to prepare new crystalline materials. And yet the transformation of the field is not over yet. If ten years ago the main concern was with the role and relevance of non-covalent interactions fuelled by the success of supramolecular chemistry today's crystal engineering has ambitions that look well beyond the use and exploitation of non-covalent bonds to make crystals. “Modern” crystal engineering is not only making crystals with a purpose but is now also defining new purposes to make crystals. Here is where we have found new barriers, perhaps expected perhaps not. We know quite well how to make crystals (whether by solution or non-solution methods), though complete control over the final structure remains elusive in many cases. We know how to characterize even the most complicated structures, and the range of accessible techniques has increased and methodology has advanced (e.g. structure solution from powder diffraction). We know how to work with complex multicomponent systems, but we have only in a limited manner and in a limited number of cases, yet been able to design collective properties of crystalline materials from the molecular properties of their components.Erk's words10 make the point: “Controlling the formation of crystalline materials is a lively science, bearing a huge potential as an originator for innovations within its classical subjects but also in new highly interdisciplinary fields of research and technology, where the solid state may be a common ground… Many challenges in crystal engineering are still on the Ångstrom scale, but it may be extended to a controlled formation of nanostructured systems and even into the mesoscopic range, where optoelectronics and photonics open up new horizons for organic solids.”
So, what will crystal engineering be in the near future? Developments can be expected in diverse and complementary areas: design of nano- and meso-cavities (for reactions and catalysis); molecular storage (e.g. fuel storage and transportation); perhaps nano-computing; biomimetic and biocompatible materials; molecular sieves, sensors and traps (with an impact on environmental chemistry); polymorphism (which impacts on drug production, delivery and uptake), just to mention a few possibilities. The steps forward, however, can be made only if the crystal engineer seeks a synergistic interaction across disciplinary with physicists, biologists and engineers. This open-minded approach will allow a focus upon problems that are “non-chemical” in nature, but that can be successfully tackled only by those who know how do deal with bonds between atoms and between molecules, with molecular ions as well as with metal–ligand interactions and molecular aggregates, i.e. the chemists.
Acknowledgements
The authors wish to thank the local organizing committee of the CrystEngComm Discussion on New Trends in Crystal Engineering. It was a pleasure to collaborate with Dr Jamie Humphrey, managing editor of CrystEngComm, and Dr Claire Darby in the preparation of this meeting. We are grateful to Diane Hort for her efforts towards a smooth organization of the meeting. We thank the University of Nottingham and its staff (notably Dr Peter Hubberstey) for support in the local organization of the meeting and for providing an excellent site for the conference. Finally, we acknowledge the Royal Society of Chemistry for having recognized crystal engineering as an interdisciplinary field of research that deserves investment and support. This foresight is witnessed by the success of CrystEngComm (http://www.rsc.org/is/journals/current/crystengcomm/cecpub.htm) and by the recent launch of CrystEngCommunity, the web portal for crystal engineering (http://www.rsc.org/is/journals/current/crystengcomm/crystengcommunity)/.References
- D. Braga, J. Miller, G. R. Desiraju, A. G. Orpen and S. L. Price, CrystEngComm, 2002, 4, 500 RSC.
- (a) G. M. J. Schmidt, Pure Appl. Chem., 1971, 27, 647 CAS; (b) G. R. Desiraju, Crystal Engineering: The Design of Organic Solids, Elsevier, Amsterdam, 1989 Search PubMed; (c) C. B. Aakeröy, Acta Crystallogr., 1997, B53, 569 CAS; (d) Proceedings of the Dalton Discussion on Inorganic Crystal Engineering, J. Chem. Soc., Dalton Trans., 2000, 3705–3998 (the whole issue, no. 21) Search PubMed; (e) Crystal Engineering: from Molecules and Crystals to Materials, ed. D. Braga, F. Grepioni and A. G. Orpen, Kluwer Academic Publishers, Dordrecht, 1999 Search PubMed; (f) B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629 CrossRef CAS; (g) D. Braga, F. Grepioni and G. R. Desiraju, Chem. Rev., 1998, 98, 1375 CrossRef CAS; (h) A. J. Blake, N. R. Champness, P. Hubberstey, W. S. Li, M. A. Withersby and M. Schröder, Coord. Chem. Rev., 1999, 183, 117 CrossRef CAS; (i) L. Brammer, Chem. Soc. Rev., 2004, 33, 476 RSC; (j) C. Janiak, Dalton Trans., 2003, 2781 RSC; (k) M. D. Hollingsworth, Science, 2002, 295, 2410 CAS.
- A. Gavezzotti, CrystEngComm, 2002, 4, 343 RSC.
- F. Toda, CrystEngComm, 2002, 4, 215 RSC.
- R. J. Davey, K. Allen, N. Blagden, W. I. Cross, H. F. Lieberman, M. J. Quayle, S. Righini, L. Seton and G. J. T. Tiddy, CrystEngComm, 2002, 4, 257 RSC.
- P. Coppens, B. Ma, O. Gerlits, Y. Zhang and P. Kulshrestha, CrystEngComm, 2002, 4, 302 RSC.
- S. L. Price, CrystEngComm, 2004, 6, 344 RSC.
- M. D. Ward and M. J. Horner, CrystEngComm, 2004, 6, 401 RSC.
- P. A. Maggard, A. L. Kopf, C. L. Stern and K. R. Poeppelmeier, CrystEngComm, 2004, 6, 451 Search PubMed.
- P. Erk, H. Hengelsberg, M. F. Haddow and R. van Gelder, CrystEngComm, 2004, 6, 474 Search PubMed.
- (a) M. W. Hosseini, CrystEngComm, 2004, 6, 318 RSC; (b) T. Maris and J. D. Wuest, J. Org. Chem., 2004, 69, 1776 CrossRef CAS.
- G. R. Desiraju, Angew. Chem. Int. Ed., 1995, 34, 2311 CrossRef CAS.
- (a) R. Robson, J. Chem. Soc. Dalton Trans., 2000, 3735 RSC; (b) S. R. Batten and R. Robson, Angew. Chem. Int. Ed., 1998, 37, 1461 CrossRef; (c) S. R. Batten, CrystEngComm, 2001, 3, 67 RSC; (d) L. Carlucci, G. Ciani, D. M. Proserpio and S. Rizzato, CrystEngComm, 2002, 4, 121 RSC; (e) S. A. Barnett and N. R. Champness, Coord. Chem. Rev., 2003, 246, 145 CrossRef CAS.
- G. M. Day and S. L. Price, J. Am. Chem. Soc., 2003, 125, 16434 CrossRef CAS.
- M. Tremayne, L. Grice, J. C. Pyatt, C. C. Seaton, B. M. Kariuki, H. H. Y. Tsui, S. L. Price and J. C. Cherryman, J. Am. Chem. Soc., 2004, 126, 7071 CrossRef CAS.
- (a) W. Bolton, Acta Crystallogr., 1964, 17, 147 CrossRef CAS; (b) G. R. Desiraju, CrystEngComm, 2002, 4, 499 RSC.
- (a) A. Gavezzotti, CrystEngComm, 2003, 4, 429 RSC; (b) A. Gavezzotti, CrystEngComm, 2003, 4, 439 RSC.
- (a) G. A. Jeffrey and W. Saenger, Hydrogen Bonding in Biological Structures, Springer-Verlag, Berlin, 1991 Search PubMed; (b) L. J. Prins, D. N. Reinhoudt and P. Timmerman, Angew. Chem. Int. Ed., 2001, 40, 2382 CrossRef CAS; (c) G. Gilli and P. Gilli, J. Mol. Struct., 2000, 552, 1 CrossRef CAS; (d) T. Steiner, Angew. Chem. Int. Ed., 2002, 41, 48 CrossRef CAS; (e) G. R. Desiraju, Acc. Chem. Res., 2002, 35, 565 CrossRef CAS; (f) G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond in Structural Chemistry and Biology, Oxford University Press, Oxford, 1999 Search PubMed; (g) D. Braga, F. Grepioni, K. Biradha, V. R. Pedireddi and G. R. Desiraju, J. Am. Chem. Soc., 1995, 117, 3156 CrossRef CAS; (h) M. Nishio, M. Hirota and Y. Umezawa, The CH/π interaction: Evidence, Nature, and Consequences, Wiley-VCH, New York, 1998 Search PubMed.
- (a) H. Umeyama and K. Morokuma, J. Am. Chem. Soc., 1977, 99, 1316 CrossRef CAS; (b) J. Emsley, O. P. A. Hoyte and R. E. Overill, J. Am. Chem. Soc., 1978, 100, 3303 CrossRef CAS; (c) V. Bertolasi, P. Gilli, V. Ferretti and G. Gilli, Chem. Eur. J., 1996, 8, 925; (d) L. Turi and J. J. Dannenberg, J. Am. Chem. Soc., 1993, 97, 12197 CAS; (e) L. Turi and J. J. Dannenberg, J. Am. Chem. Soc., 1994, 116, 8714 CrossRef CAS; (f) M. S. Gordon and J. H. Jensen, Acc. Chem. Res., 1996, 29, 536 CrossRef.
- E. D'Oria and J. J. Novoa, CrystEngComm, 2004, 6, 367 Search PubMed.
- (a) F. H. Allen, Acta Crystallogr., 2002, B58, 380 CAS; (b) Inorganic Crystal Structure Database (ICSD), Fachinformationszentrum (FIZ) Karlsruhe and Gmelin Institut, Release 2000/2.
- (a) J. J. Novoa, P. Lafuente, R. E. Del Sesto and J. S. Miller, Angew. Chem. Int. Ed., 2001, 40, 2540 CrossRef CAS; (b) J. J. Novoa, P. Lafuente, R. E. Del Sesto and J. S. Miller, CrystEngComm, 2002, 4, 373 RSC.
- (a) D. Braga, F. Grepioni and J. J. Novoa, Chem. Commun., 1998, 1959 RSC; (b) J. J. Novoa, I. Nobeli, F. Grepioni and D. Braga, New J. Chem., 2000, 24, 5 RSC; (c) D. Braga, L. Maini, F. Grepioni, F. Mota, C. Rovira and J. J. Novoa, Chem. Eur. J., 2000, 6, 4536 CrossRef CAS; (d) D. Braga, J. J. Novoa and F. Grepioni, New J. Chem., 2000, 25, 226 Search PubMed; (e) D. Braga, E. D'Oria, F. Grepioni, F. Mota, J. J. Novoa and C. Rovira, Chem. Eur. J., 2002, 8, 1163 CrossRef.
- M. A. Carvajal, S. Alvarez and J. J. Novoa, Chem. Eur. J., 2004, 10, 2117 CrossRef CAS.
- J. J. Novoa, G. Aullón, P. Alemany and S. Alvarez, J. Am. Chem. Soc., 1995, 117, 7169 CrossRef CAS.
- M. Czugler and N. Báthori, CrystEngComm, 2004, 6, 494 Search PubMed.
- V. A. Blatov, L. Carlucci, G. Ciani and D. M. Proserpio, CrystEngComm, 2004, 6, 377 RSC.
- (a) TOPOS 3.2/4.0: a program package for multipurpose geometrical and topological analysis of crystal structures. See http://www.topos.ssu.samara.ru/; (b) V. A. Blatov, A. P. Shevchenko and V. N. Serezhkin, J. Appl. Crystallogr., 2000, 33, 1193 CrossRef CAS.
- P. C. Crawford, A. L. Gillon, J. Green, A. G. Orpen, T. J. Podesta and S. V. Pritchard, CrystEngComm, 2004, 6, 419 RSC.
- N. J. Burke, A. D. Burrows, M. F. Mahon and S. J. Teat, CrystEngComm, 2004, 6, 429 RSC.
- C. B. Aakeröy, J. Desper and J. Valdés-Martínez, CrystEngComm, 2004, 6, 413 RSC.
- S. A. Bourne and Z. Mangombo, CrystEngComm, 2004, 6, 437 Search PubMed.
- F. S. Delgado, J. Sanchiz, C. Ruiz-Pérez, F. Lloret and M. Julve, CrystEngComm, 2004, 6, 443 Search PubMed.
- P. Miller, M. Nieuwenhuyzen, J. P. H. Charmant and S. L. James, CrystEngComm, 2004, 6, 408 RSC.
- D. Braga, Chem. Commun., 2003, 2751 RSC.
- A. L. Gillon, G. R. Lewis, A. G. Orpen, S. Rotter, J. Starbuck, X.-M. Wang, Y. Rodríguez-Martín and C. Ruiz-Pérez, J. Chem. Soc., Dalton Trans., 2000, 3897 RSC.
- (a) S. Ferlay, V. Bulach, O. Felix, M. W. Hosseini, J.-M. Planeix and N. Krytsakas, CrystEngComm, 2002, 4, 447 RSC; (b) S. Ferlay, O. Felix, M. W. Hosseini, J.-M. Planeix and N. Krytsakas, Chem. Commun., 2002, 702 RSC; (c) S. Ferlay and M. W. Hosseini, Chem. Commun., 2004, 788 RSC.
- See, for instance, (a) V. A. Russell, C. C. Evans, W. J. Li and M. D. Ward, Science, 1997, 276, 575 CrossRef CAS; (b) K. T. Holman, A. M. Pivovar, J. A. Swift and M. D. Ward, Acc. Chem. Res., 2001, 34, 107 CrossRef CAS.
- (a) K. T. Holman, A. M. Pivovar and M. D. Ward, Science, 2001, 294, 1907 CrossRef CAS; (b) J. A. Swift and M. D. Ward, Chem. Mater., 2000, 12, 1501 CrossRef CAS.
- A. M. Pivovar, K. T. Holman and M. D. Ward, Chem. Mater., 2001, 13, 3018 CrossRef.
- See, for instance, (a) A. M. Beatty, CrystEngComm, 2001, 3, 243 RSC; (b) L. Brammer, in Perspectives in Supramolecular Chemistry, Vol. 7 – Crystal Design: Structure and Function, ed. G. R. Desiraju, Wiley, Chichester, 2003, pp. 1–75 Search PubMed.
- (a) C. B. Aakeröy and A. M. Beatty, Chem. Commun., 1998, 1067 RSC; (b) C. B. Aakeröy, A. M. Beatty and D. S. Leinen, J. Am. Chem. Soc., 1998, 120, 7383 CrossRef; (c) C. B. Aakeröy, A. M. Beatty and B. A. Helfrich, J. Chem. Soc., Dalton Trans., 1998, 1943 RSC; (d) C. B. Aakeröy, A. M. Beatty and D. S. Leinen, Angew. Chem. Int. Ed., 1999, 38, 1815 CrossRef CAS; (e) J. C. Mareque Rivas and L. Brammer, New J. Chem., 1998, 22, 1315 RSC.
- (a) Z. Qin, H. A. Jenkins, S. J. Coles, K. W. Muir and R. J. Puddephatt, Can. J. Chem., 1999, 77, 155 CrossRef CAS; (b) Z. Qin, M. C. Jennings and R. J. Puddephatt, Inorg. Chem., 2001, 40, 6220 CrossRef CAS; (c) C. J. Kuehl, F. M. Tabellion, A. M. Arif and P. J. Stang, Organometallics, 2001, 20, 1956 CrossRef CAS.
- (a) Comprehensive Supramolecular Chemistry, ed. D. D. MacNicol, F. Toda and R. Bishop, Pergamon, Oxford, 1996, vol. 6, pp. 14–15, 310–313, 467–470, 602–605 Search PubMed; (b) R. K. R. Jetti, F. Xue, T. C. W. Mak and A. Nangia, J. Chem. Soc., Perkin Trans. 2, 2000, 1223 RSC and refs. therein.
- K. Larsson and L. Öhrström, CrystEngComm, 2004, 6, 354 Search PubMed.
- V. Balamurugan, W. Jacob, J. Mukherjee and R. Mukherjee, CrystEngComm, 2004, 6, 396 RSC.
- (a) G. Aullón, D. Bellamy, L. Brammer, E. A. Bruton and A. G. Orpen, Chem. Commun., 1998, 653 RSC; (b) L. Brammer, E. A. Bruton and P. Sherwood, Cryst. Growth Des., 2001, 1, 277 CrossRef CAS.
- (a) G. R. Lewis and A. G. Orpen, Chem. Commun., 1998, 1873 RSC; (b) A. Angeloni and A. G. Orpen, Chem. Commun., 2001, 343 RSC; (c) B. Dolling, A. L. Gillon, A. G. Orpen, J. Starbuck and X.-M. Wang, Chem. Commun., 2001, 567 RSC.
- (a) J. C. Mareque Rivas and L. Brammer, Inorg. Chem., 1998, 37, 4756 CrossRef; (b) L. Brammer, J. K. Swearingen, E. A. Bruton and P. Sherwood, Proc. Nat. Acad. Sci. U. S. A., 2002, 99, 4956 CrossRef CAS.
- (a) F. Neve and A. Crispini, CrystEngComm, 2003, 5, 265 RSC; (b) M. Felloni, P. Hubberstey, C. Wilson and M. Schröder, CrystEngComm, 2004, 6, 87 RSC.
- (a) D. B. Mitzi, J. Chem. Soc., Dalton Trans., 2001, 1 RSC; (b) D. B. Mitzi, Prog. Inorg. Chem., 1999, 48, 1 CAS.
- (a) S. L. James, Macromol. Symp., 2004, 209, 119 CrossRef CAS; (b) S. L. James, X. Xu and R. V. Law, Macromol. Symp., 2003, 196, 187 CrossRef CAS.
- D. Braga, S. L. Giaffreda, F. Grepioni and M. Polito, CrystEngComm, 2004, 6, 458 Search PubMed.
- R. Kuroda, K. Higashiguchi, S. Hasebe and Y. Imai, CrystEngComm, 2004, 6, 463 Search PubMed.
- F. P. A. Fabbiani, D. R. Allan, W. I. F. David, S. A. Moggach, S. Parsons and C. R. Pulham, CrystEngComm, 2004, 6, 504 Search PubMed.
- P. A. Sermon, N. Mason McLellan and I. R. Collins, CrystEngComm, 2004, 6, 469 Search PubMed.
- M. T. Kirchner, R. Boese, A. Gehrke and D. Bläser, CrystEngComm, 2004, 6, 360 Search PubMed.
- See, for example, (a) P. A. Maggard, C. L. Stern and K. R. Poeppelmeier, J. Am. Chem. Soc., 2001, 123, 7742 CrossRef CAS; (b) M. E. Welk, A. J. Norquist, C. L. Stern and K. R. Poeppelmeier, Inorg. Chem., 2001, 40, 5479 CrossRef CAS; (c) P. A. Maggard, A. L. Kopf, C. L. Stern and K. R. Poeppelmeier, Inorg. Chem., 2002, 41, 4852 CrossRef CAS.
- W. T. A. Harrison, T. M. Nenoff, T. E. Gier and G. D. Stucky, Inorg. Chem., 1993, 32, 2437 CrossRef CAS.
- (a) K. Tanaka and F. Toda, Chem. Rev., 2000, 100, 1025 CrossRef CAS; (b) K. Tanaka, Solvent-free Organic Synthesis, Wiley-VCH, 2003 Search PubMed; (c) D. Bradley, Chem. Brit., September, 2002, 42 Search PubMed; (d) G. W. V. Cave, C. L. Raston and J. L. Scott, Chem. Commun., 2001, 2159 RSC; (e) G. Rothenberg, A. P. Downie, C. L. Raston and J. L. Scott, J. Am. Chem. Soc., 2001, 123, 8701 CrossRef CAS; (f) L. R. Nassimbeni, Acc. Chem. Res., 2003, 36, 631 CrossRef CAS; (g) G. Kaupp, CrystEngComm, 2003, 5, 117 RSC; (h) D. Braga and F. Grepioni, Angew. Chem. Int. Ed., 2004, 43, 4002 CrossRef CAS.
- (a) R. Kuroda, Y. Imai and N. Tajima, Chem. Commun., 2002, 2848 RSC; (b) Y. Imai, N. Tajima, T. Sato and R. Kuroda, Chirality, 2002, 14, 604 CrossRef CAS; (c) E. Y. Cheung, S. J. Kitchin, K. D. M. Harris, Y. Imai, N. Tajima and R. Kuroda, J. Am. Chem. Soc., 2003, 125, 14658 CrossRef CAS; (d) K. D. M. Harris, M. Tremayne and B. M. Kariuki, Angew. Chem. Int. Ed., 2001, 40, 1626 CrossRef CAS; (e) K. D. M. Harris, M. Tremayne, P. Lightfoot and P. G. Bruce, J. Am. Chem. Soc., 1994, 116, 3543 CrossRef CAS.
- (a) D. R. Allan, S. J. Clark, R. M. Ibberson, S. Parsons, C. R. Pulham and L. Sawyer, Chem. Commun., 1999, 751 RSC; (b) D. R. Allan, S. J. Clark, S. Parsons and M. Ruf, J. Phys. Condens. Matter, 2002, 12, L613 CrossRef.
- See for instance, R. Boese, M. T. Kirchner, W. E. Billups and L. R. Norman, Angew. Chem. Int. Ed., 2003, 42, 1961 Search PubMed; M. T. Kirchner, R. Boese, W. E. Billups and L. R. Norman, J. Am. Chem. Soc., 2004, 126, 9407 CrossRef CAS; R. Boese and M. Nussbaumer, in Organic Crystal Chemistry, ed. J. B. Garbarczyk and D. W. Jones, Oxford University Press, Oxford, 1994, pp. 20–37 CrossRef CAS.
- J. Bernstein, Polymorphism in Molecular Crystals, Oxford University Press, Oxford, 2002 Search PubMed.
- See also, A. Gavezzotti, Acc. Chem. Res., 1994, 27, 309 Search PubMed.
- (a) J. Bernstein, R. J. Davey and J.-O. Henck, Angew. Chem. Int. Ed. Eng., 1999, 38, 3440 CrossRef; (b) See also: N. Bladgen and R. J. Davey, Chem. Brit., 1999, 35, 44 Search PubMed.
- (a) A. Burger, Topics in Pharmaceutical Sciences, ed. D. D. Breimer and P. Speiser, Elsevier: Amsterdam, 1983, p. 347 Search PubMed; (b) T. L. Threlfall, Analyst, 1995, 120, 2435 RSC; (c) S. R. Byrn, Solid State Chemistry of Drugs, Academic Press, New York, 1982, p. 79 Search PubMed.
- A. Spitaleri, C. A. Hunter, J. F. McCabe, M. J. Packer and S. L. Cockroft, CrystEngComm, 2004, 6, 489 Search PubMed.
- L. T. Higham, U. P. Kreher, J. L. Scott and C. R. Strauss, CrystEngComm, 2004, 6, 484 Search PubMed.
- B. Herradón, A. Montero, E. Mann and M. A. Maestro, CrystEngComm, 2004, 6, 513 RSC.
- See also L. R. McGillivray, CrystEngComm, 2002, 4, 37 Search PubMed for a recent survey of topochemical reactions.
- (a) J. M. Lehn, Angew. Chem., Int. Ed. Engl., 1990, 29, 1304 CrossRef; (b) J. M. Lehn, Supramolecular Chemistry: Concepts and Perspectives, VCH, Weinheim, 1995 Search PubMed; (c) J. W. Steed and J. L. Atwood, Supramolecular Chemistry, Wiley & Sons, 2000 Search PubMed.
| This journal is © The Royal Society of Chemistry 2005 |