
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Operando X-ray absorption spectroscopy of Fe–N–C catalysts based on carbon black and biomass-derived support materials for the ORR†
Garlef
Wartner
 *ab,
Julia
Müller-Hülstede
c,
Hanna
Trzesniowski
a,
Michael
Wark
d,
Peter
Wagner
c and
Robert
Seidel
*a
*ab,
Julia
Müller-Hülstede
c,
Hanna
Trzesniowski
a,
Michael
Wark
d,
Peter
Wagner
c and
Robert
Seidel
*a
aHelmholtz-Zentrum Berlin für Materialien und Energie, Department of Atomic-Scale Dynamics in Light-Energy Conversion, Albert-Einstein-Str. 15, 12489 Berlin, Germany. E-mail: robert.seidel@helmholtz-berlin.de; garlef.wartner@helmholtz-berlin.de
bMax Planck Institute for Chemical Energy Conversion, Department of Heterogeneous Reactions, Stiftstr. 34-36, 45470 Mülheim an der Ruhr, Germany
cGerman Aerospace Center (DLR), Institute of Engineering Thermodynamics, Carl-von-Ossietzky Str. 15, 26129 Oldenburg, Germany
dCarl von Ossietzky University, Institute of Chemistry, Chemical Technology 1, Carl-von-Ossietzky Str. 9-11, 26129 Oldenburg, Germany
First published on 15th April 2024
Abstract
Iron nitrogen carbon (Fe–N–C) catalysts are among the most promising non-platinum group metal catalysts for the oxygen reduction reaction (ORR). Their activity and stability are considerably influenced by the structure of the C-support. New biochar materials offer native heteroatom doping, making them a promising precursor for Fe–N–C catalysts. In this study, we apply operando X-ray absorption spectroscopy at the Fe K-edge to characterize the atomic Fe-based active sites of a commercial Fe–N–C catalyst, a carbon black-based catalyst as well as a novel biomass-based Fe–N–C catalyst. We compare the density and the potential-dependent nature of the FeNx-type active sites during operation. Our results demonstrate that the novel biomass-based catalyst exhibits a higher active-site density compared to commercial and carbon black-based Fe–N–C catalysts. Moreover, dynamic detection of the Fe K-edge intensity during potential cycling reveals that their reversible iron redox potential is lower compared to that of conventional catalysts. Evaluation of the Fe K-edge shift as well as of the extended X-ray absorption fine structure (EXAFS) suggests hetero-atom doping and iron under-coordination as potential causes for the observed differences. These insights open the pathway to develop new optimization strategies for Fe–N–C catalysts based on biomass support materials.
Introduction
Transforming the energy supply towards renewable power sources increases the demand for long-term and flexible energy storage in regenerative fuels such as hydrogen. The slow kinetics of the oxygen reduction reaction (ORR) limits the efficiency of reconverting hydrogen in fuel cells to provide electricity.1 To make such systems available for a broad community, cheap and abundant transition metal-based ORR-catalysts need to be developed. One of the most promising classes of non-platinum group metal catalysts are iron–nitrogen–carbon (Fe–N–C) materials, most of whose active sites are Fe–Nx moieties incorporated in a graphitic carbon support.2,3 In general, different coordination environments of iron species such as FeN4, FeN3 and FeN2 can be present. Specifically, FeN4-type sites, in which iron is coordinated to four nitrogen neighbors similar to the D4h-geometry of porphyrin, or FeN4+1-type sites, in which a fifth ligand is coordinated perpendicular to the square planar nitrogen ligands, are thought to be the most active sites.4 The different implementations of these sites within the graphene support and consequent coordination geometries determine their activity for the ORR.5–7 Material optimization is focused on the enhancement of the intrinsic activity and stability as well as the number of active sites within the catalyst material. These properties are mainly influenced by the number and type of functional groups implemented in the carbon support used for synthesis. Thus, the choice of the support material is crucial for the activity and durability of these catalysts.3,8–10 Recently, new biomass-derived support materials have been investigated.11–18 The high amount of functional groups and heteroatoms present in biomass-based materials as well as their high porosity promises to be beneficial for electrocatalytic applications.19 Moreover, in contrast to conventional carbon materials made from fossil resources, biomass-substrates are regrowing and thus more sustainable.16,19In our previous studies we showed that the use of phosphoric acid activated rye straw or coconut shells as a carbon support for Fe–Nx sites results in Fe–N–C catalysts with high nitrogen contents (∼10 at%) and the presence of P-species (∼2 at%).18 Thin-film analysis showed that the biomass-based Fe–N–C catalysts have comparable mass activities to a carbon black-based Fe–N–C and a significantly higher stability against carbon corrosion.18 The implementation of biomass-based Fe–N–C catalysts and a carbon black-based Fe–N–C in a high temperature proton exchange membrane fuel cell showed lower performance compared to that of a commercial Fe–N–C catalyst which was mainly attributed to non-optimized electrode preparation and high hydrophilicity of the catalyst.20 To date, the nature of the active centers in the novel biomass-based Fe–N–C catalysts has not yet been studied and it remains unclear whether the same active centers as on carbon black based and commercial Fe–N–C catalysts are formed. Therefore, a better understanding of the correlation between the properties of the support material and the characteristics of the active moieties is necessary for systematic improvement.
In this contribution, we use operando X-ray absorption fine structure (XAFS) to characterize three different Fe–N–C catalysts based on biomass and conventional supports. Fe K-edge X-ray absorption near-edge structure (XANES) spectra as well as the Fourier transform of the EXAFS-region are used for fingerprinting to identify different iron-containing phases present in the catalysts. The amount of iron based active sites is further elucidated by a linear combination analysis of the Fe K-edge XANES-spectra as well as the Fe K-edge change upon oxidation. Moreover, operando cyclic voltammetry X-ray absorption at fixed energies at the Fe K-edge is used to identify the iron-site redox processes. Fitting the Fourier transform of the Fe K-edge EXAFS-region enables determining more details about the geometry of the active sites.
Materials and methods
Catalyst preparation
Details of the catalyst synthesis of the carbon black (Fe–N-oxBP)- and phosphoric acid activated coconut shell-based (Fe–N-aCoCo) catalysts can be found in our previous publication.18 In brief, support-based catalyst synthesis was applied using oxidized commercial Black Pearls® 2000 (Cabot) and phosphoric acid activated coconut shells.18 First, 100 mg of the support were impregnated with 421 mg of cyanamide (Sigma Aldrich) and 16.25 mg iron(II) acetate (Sigma Aldrich) in 4 mL ethanol (Carl Roth) in a sonication bath. After drying the impregnated carbon in a vacuum oven (16 h, 30 °C), a first pyrolysis step at 900 °C (5 °C min−1) was applied for 1 h under nitrogen flow in a tube furnace. Next, an acid leaching step with 2 M H2SO4 (Carl Roth) for 16 h at 90 °C was applied followed by washing until a neutral pH was reached, drying and a second pyrolysis similar to the first pyrolysis. Additionally, in this study we included a commercial Fe–N–C catalyst (PMF-011904) from Pajarito Powder (Fe–N-Comm). The catalyst powders were doctor blade coated on a glassy carbon plate (Sigradur K, thickness 100 μm, HTW GmbH) cell. For this, an ink consisting of 50 mg catalyst powder, 166.6 mg of 2-propanol and 62.5 mg of Nafion solution (20 wt% in lower aliphatic alcohols and water, Sigma Aldrich) was used. Loadings in the range of 1.2–3.7 mg cm−2 were reached.Operando XAS setup
The operando X-ray absorption spectroscopy measurements were carried out at the KMC-3 beamline at the BESSY II synchrotron radiation facility in Berlin, Germany, using the CryoEXAFS experimental station.21 The samples were measured in a home-designed cell, which implements the catalyst-coated glassy carbon membranes as the working electrode, an Ag/AgCl reference electrode (ItalSens) and a Pt-wire (thermocouple quality, Sigma Aldrich) as the counter electrode. The cell was filled with 0.1 M HClO4 as the electrolyte. The electrolyte was stirred and purged with oxygen gas during the measurements in order to maintain oxygen saturation. The electrode potential was controlled using a BioLogic SP-300 potentiostat.Operando XAS characterization
X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine-structure (EXAFS) were measured at the Fe K-edge of the samples in fluorescence mode with a constant electrode potential applied. Static experiments were carried out at electrode potentials of 1.0, 0.8, 0.6, 0.4 and 0.2 V vs. RHE, respectively, starting from the highest electrode potential. At least 5 individual spectra were recorded for each potential and averaged in order to optimize counting statistics. During operando cyclic voltammetry (OPCV) measurements the incident photon energy was fixed at 7124 eV and the fluorescence yield (FY) was recorded while sweeping the potential between 1.2 V vs. RHE and 0.1 V vs. RHE with 10 mV s−1. The recorded FY-data were smoothed based on binomial filtering and the derivative was computed using the Igor Pro software.22XANES and EXAFS data evaluation
The photon energy was calibrated based on the Fe K-edge position of metallic Fe foil (7112 eV). The FY signal of the XA spectra was divided by the intensity I0 of the incoming X-ray beam and normalized to an edge-height of 1. The Athena software23 was used for calculating the Fourier-transform of the EXAFS-region (FT-EXAFS, FT(χ(k))) of the XA spectra. Local structure models were fitted to the EXAFS regions using the Artemis software with FEFF 6 implemented.23 These models are based on FeN4-type functional groups, which become reversibly oxidized by chemisorption of oxygen at the Fe center. The Fe–O bond length was assumed to be equal to the Fe–N bond length and the coordination number of nitrogen in the reduced and oxidized state was also assumed to be equal. The amplitude reduction factor S02 was assumed to be 1. The model thus comprises seven free parameters: the average bond lengths of the first shell in the oxidized and reduced state ROx and RRed, the mean square radial displacements SOx and SRed of the oxidized and reduced state, the average coordination number of oxygen N(Fe–O) as well as nitrogen N(Fe–N) within the first shell and the photon energy shift E0. In the structure model, the nitrogen ligands were optionally substituted by phosphorous, which has been demonstrated to be present in Fe–N-aCoCo.18Results and discussion
Three Fe–N–C catalysts were compared including Fe–N–C catalysts based on oxidized black pearls and phosphoric acid activated coconut shells as well as commercial Fe–N–C. All Fe–N–C have similar bulk iron contents of around 1.3 wt%.20 An electrochemical characterization of these catalysts can be found in our previous publication.18Iron-phase composition
As a first step the different iron-containing phases within the samples were identified. Fig. 1A shows the near-edge region of the Fe K-edge spectra of the catalysts in the oxidized state (solid lines), i.e., with an electrode potential of +1.0 V vs. RHE applied, in comparison with different reference spectra taken from the literature (dashed lines). One observation is that the shape of the XANES-region of Fe–N-aCoCo is distinctly different from that of Fe–N-oxBP and Fe–N-Comm, whose spectra are quite similar. The spectrum of Fe–N-aCoCo exhibits a pre-edge feature at about 7112 eV, which can be attributed to the Fe 1s → 3d transition. As this excitation is formally dipole-forbidden, the intensity results from the weaker quadrupole transition, or from hybridization of 3d and 4p states facilitating the partially allowed Fe 1s → 3d4p transition. The latter can be caused by deviation from a centrosymmetric coordination geometry like D4h.24,25 The feature at about 7112 eV was previously observed for FeIII-porphyrin complexes26,27 and is also present in the two reference spectra of Fe–N–C catalysts in Fig. 1A. The spectrum reported by Jia et al.27 characterizes an Fe–N–C catalyst prepared by pyrolyzing Fe-porphyrins mixed with black pearls, and the spectrum reported by Fei et al.28 characterizes an Fe–N–C material prepared by implementing iron–nitrogen functional groups into graphene frameworks. Both spectra can be modeled by only FeNx species incorporated in a carbon matrix, and there are no other iron phases present in the sample, as demonstrated by the authors.27,28 The spectral shape measured on Fe–N-aCoCo is similar to both references, although some distinct differences between the spectral shapes in the white-line region are present in all three spectra. This suggests that the Fe–N-aCoCo catalyst may also contain mainly FeNx species. The different shapes of the spectra probably reflect the considerable influence various types of distributions of FeNx-sites has on the Fe K-edge XANES-region as demonstrated by Jia et al.27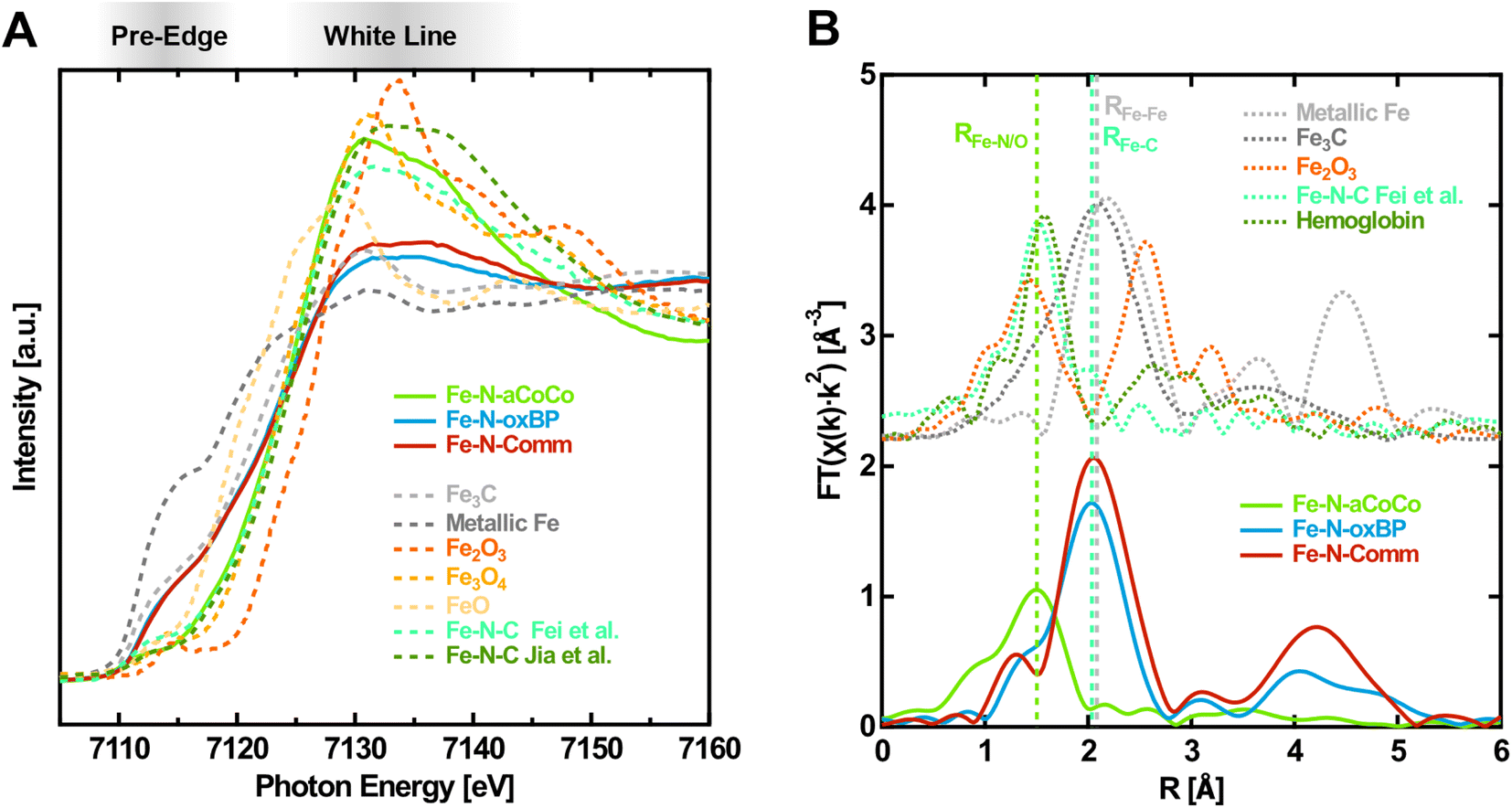 | ||
| Fig. 1 Fingerprinting at the Fe K-edge of the Fe–N–C catalysts in the oxidized state (1.0 V vs. RHE applied). (A) XANES-region of the Fe K-edge with references of iron carbide29 (Fe3C), metallic iron, iron oxides30(Fe2O3, Fe3O4, and FeO) and different Fe–N–C materials (from Fei et al.28 and Jia et al.27). (B) Fourier transform of the EXAFS-region with references of iron carbide29 (Fe3C), metallic iron, iron oxide23 (Fe2O3) and an exemplary Fe–N–C material from Fei et al.28 and glycated hemoglobin.31 | ||
The spectra of Fe–N-oxBP and Fe–N-Comm are quite distinct from the Fe–N–C reference spectra by Jia et al. and Fei et al. (Fig. 1A) and exhibit, in contrast to these spectra, a shoulder in the rising edge at around 7115 eV. Such a shoulder is not present in the spectra of iron oxides, but is characteristic for metallic iron or iron carbide, as can be seen in Fig. 1A. A shoulder at 7117 eV was previously found to be a fingerprint for FeIIN4 complexes in D4h coordination.26,27 As the spectra were measured with oxidizing electrode potentials applied, the active centers are in their oxidized FeIIINx state. Even active centers, which are not in electrical or ionic contact, are expected to be oxidized due to the presence of oxygen.32 Consequently, FeIIN4-species cannot account for the shoulder in the spectra of Fe–N-oxBP and Fe–N-Comm. This shoulder can thus rather be explained by a considerable amount of iron carbide or iron being present in the catalysts, which is also in agreement with the iron-rich, nitrogen-free domains found previously in the TEM-EDX mappings of both materials.18,33 The composition of the catalysts can further be elucidated from the Fourier transform of the EXAFS-region of the Fe K-edge spectra shown in Fig. 1B. As demonstrated by the reference spectra of Fe–N–C by Fei et al.28 and hemoglobin, the first shell of Fe–N-aCoCo appears at the same distance as the Fe–N or Fe–O bond (RFe–N/O) in oxidized Fe–N–C catalysts or iron porphyrins. This further proves the main iron species in Fe–N-aCoCo to be FeNx-type complexes. The smaller scattering contribution of the Fe–C bond-distance (RFe–C) identified by Fei et al.28 to the Fourier transformed spectrum of Fe–N-aCoCo again indicates different implementation types of the FeNx-sites within the carbon material dominating the site-distributions of both catalysts. In contrast, Fe–N-oxBP and Fe–N-Comm exhibit only a small shoulder at RFe–N/O, suggesting that bond-distances corresponding to the Fe–N bond of FeNx-type species are not the dominating contribution to the first coordination-shell of iron. The maximum of the first coordination shell of Fe–N-oxBP and Fe–N-Comm appears at about the same scattering distance as the Fe–Fe bond (RFe–Fe) in iron carbide or iron, which in turn is an indication of the presence of such phases in these catalysts. Furthermore, comparison with the reference spectrum of hematite indicates that large contributions of iron oxide phases can be excluded for all three catalysts since there are no significant oxidic Fe–Fe scattering contributions in their Fourier transformed EXAFS region.
The contribution of the prominent shoulder within the spectra of iron carbide and iron, which is not present in the spectra of the oxide phases or FeN4-type species, can be exploited to quantitatively estimate the contributions of these phases to the Fe K-edge spectra of the catalysts. Thus, to estimate the different iron-phase contributions (Table 1), a linear combination fitting analysis of the spectra within the near-edge region between 7107 eV and 7133 eV was conducted.
| FeNxC | Fe3C | Metallic Fe | Reduced χ2 | |
|---|---|---|---|---|
| Fe–N-aCoCo | 100 | 0 | 0 | 0.0011869 |
| Fe–N-oxBP | 20.3 ± 0.9 | 74.6 ± 2.7 | 5.1 ± 2.4 | 0.0002187 |
| Fe–N-Comm | 30.5 ± 1.0 | 43.8 ± 4.2 | 25.7 ± 2.8 | 0.0002577 |
Fe–N-aCoCo was only fitted with the Fe–N–C reference by Fei et al.,28 because adding oxide-, nitride-, carbide- or iron reference spectra to the fit did not improve the less-ideal fitting-result around the pre-edge and the white-line region (Fig. S1A†). The apparent deviations between the spectrum of Fe–N-aCoCo and the spectrum of the catalysts synthesized by Fei et al. might be explained by a different distribution of different FeNx-type sites being present in the two materials. Adding hematite or magnetite reference spectra does not improve the fitting result of all three catalysts. Thus, significant contribution of the thermodynamically most stable iron oxides can be excluded, which agrees with the absence of significant iron oxide Fe–Fe scattering-contributions to the Fourier transformed EXAFS region. Likewise including the FeN spectrum reported by Tayal et al.34 as a reference component does not improve the fitting results of all three materials. Consequently, no significant amounts of electrochemically inactive35 iron nitride species are present in the samples. Both the spectra of Fe–N-oxBP and Fe–N-Comm can best be fitted with the Fe–N–C reference spectra by Fei et al.,28 the iron carbide reference spectra as well as the metallic iron reference spectra. The resulting amounts of FeNx-type sites are about 20% of the iron species in Fe–N-oxBP and about 30% of the iron species in Fe–N-Comm. However, these quantitative results are influenced by qualitative errors arising from the choice of reference spectra, which is reflected in the fitting-deviations documented in Table 1. The weighted sum of squared deviations (reduced χ2) is higher by a factor of 5 for the fit of Fe–N-aCoCo compared to the fit of Fe–N-oxBP and Fe–N-Comm. As already alluded to, the variety of distributions of FeNx-type sites within differently prepared Fe–N–C catalysts makes it difficult to find an ideal fitting-reference for FeNx-species limiting the accuracy of the quantitative result. As the discerning feature between the FeNx-species and iron or iron carbides is the shoulder at 7115 eV, the resulting amount of these species should be not too far off from the actual value. Moreover, the structural boundaries between iron and iron carbide are blurred, as carbon can get dissolved in the iron phase. Iron carbides are thus polymorphous and the cementite (Fe3C) reference spectrum used in the fits is only one possible carbide phase.36,37 As the XANES regions of the different iron carbides differ significantly depending on their carbon content,38 the differentiation between iron and cementite is only a very rough estimate. In fact, iron carbide may be present in different compositions. Iron carbides as well as metallic α- or γ-iron have been previously identified in Fe–N–C catalysts as a byproduct created during synthesis, which can be present as particles covered by a graphitic shell.39 While such particles were reported to boost the ORR activity of Fe–Nx sites in alkaline media by decreasing the charge density on the Fe site and increasing the binding energy to O2,40 they are unstable and inactive towards the ORR in acidic media.4,17,35,39,41,42 The overall iron content of all three catalysts was previously determined to be 1.3 wt% based on ICPMS measurements.20 Due to the equal iron content, the relative contributions of the different iron species resulting from the linear combination fitting are a direct measure of the respective site densities of these species. Although the Fe–N-aCoCo catalyst shows the highest amount of Fe–Nx sites, the mass activity determined in our previous study (1.64 A g−1 at 0.8 V vs. RHE in 0.1 M HClO4)18 is only slightly higher compared to that of Fe–N-oxBP (1.34 A g−1)18 and lower compared to that of Fe–N-Comm (2.12 A g−1).33 This indicates a different nature in terms of accessibility and activity (turnover frequency, TOF) in the catalyst with Fe–N-Comm having a smaller amount of more active FeNx sites compared to Fe–N-aCoCo. This is in agreement with the findings of Primbs et al. who reported highly active sites with low site density for a similar type of Pajarito powder-based catalyst.43
Static potential dependent XANES
As a second step, to gain more evidence about the amount and nature of the redox-active FeNx-type sites, the edge shift observed during the operando-measurements was analyzed. Fig. 2A shows the Fe K-edge XANES spectra of the Fe–N–C catalysts measured in the oxidized and reduced state with a potential of 1.0 V vs. RHE and 0.2 V vs. RHE applied, respectively. From high to low applied potential, the Fe K-edges are shifted to lower photon energies reflecting the decrease in the Fe oxidation state. Fe–N-aCoCo exhibits the most distinct potential dependent shift while the shift of the other two catalysts is less pronounced. This is also visible in the difference spectra (dashed lines in Fig. 2A), which reflect the spectral change upon reduction. The latter is similar for Fe–N-oxBP and Fe–N-Comm but larger and qualitatively different for Fe–N-aCoCo. The qualitative difference is caused by the intensity of the white-line, which grows upon reduction for Fe–N-oxBP and Fe–N-Comm and decreases for Fe–N-aCoCo.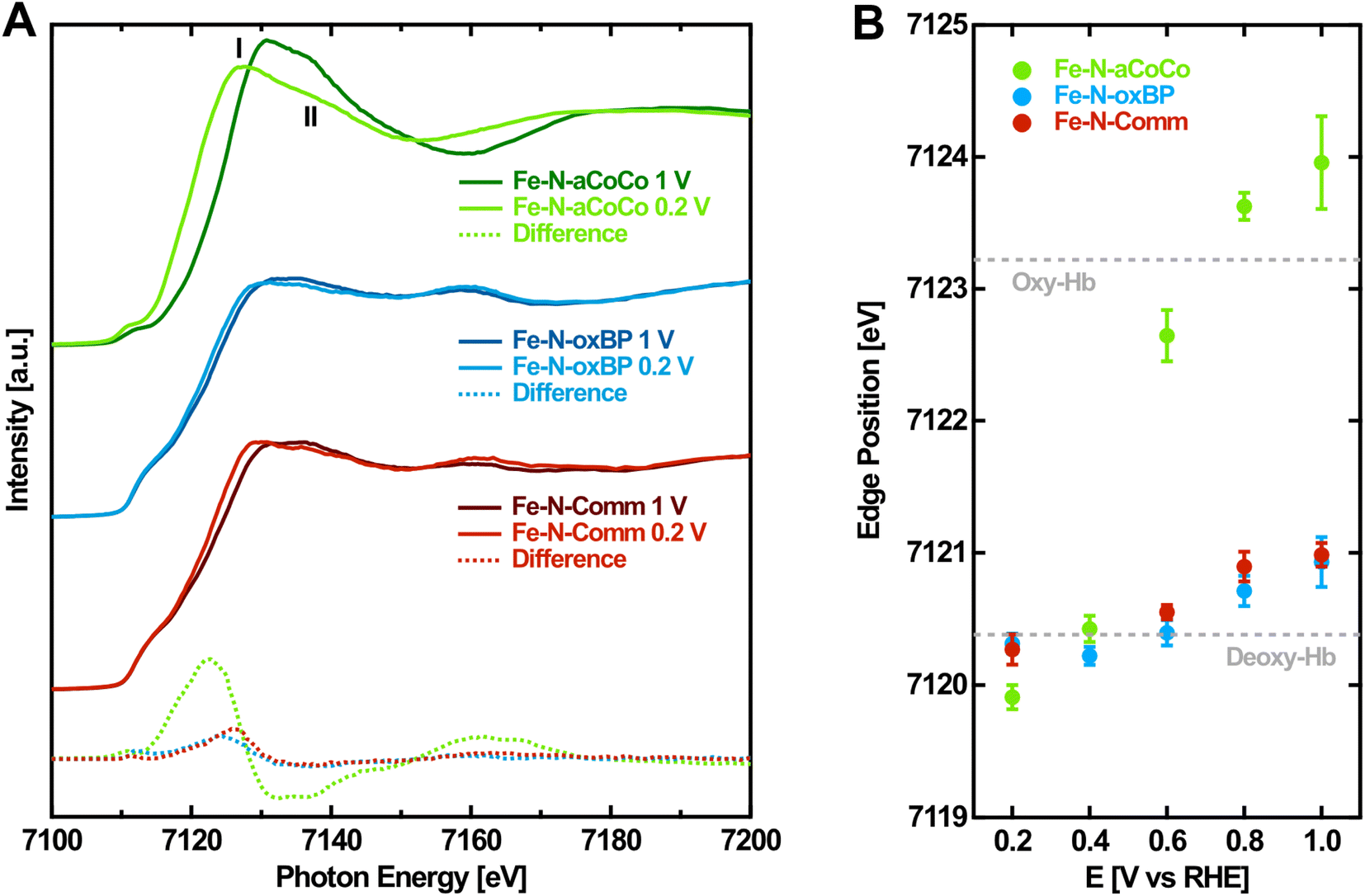 | ||
| Fig. 2 Potential-dependence of the Fe K-edge of Fe–N–C catalysts. (A) Fe K-edge of Fe–N–C catalysts measured at oxidizing and reducing electrode potentials. (B) Fe K-edge position of Fe–N–C catalysts determined by the half-height of the edge jump depending on the applied electrode potential. The dashed lines indicate the Fe K-edge position of oxidized (oxy-Hb) and reduced (deoxy-Hb) hemoglobin according to Wilson et al.44 | ||
To quantitatively evaluate the potential-dependent edge change, the edge position at the half-height of the edge jump from the spectra measured on different electrode potentials was determined. Fig. 2B shows the evaluated Fe K-edge positions depending on the applied electrode potential. For all three catalysts, there is no certain threshold redox potential. Instead, the redox-process proceeds over almost the full potential range probed, reflecting the variety of different redox-active species present in the samples. As noticed before, the overall edge shift of Fe–N-aCoCo between +1.0 V vs. RHE and +0.2 V vs. RHE is by a factor of about 6 larger compared to the almost identical edge shift of Fe–N-oxBP and Fe–N-Comm (see Table 2). In order to better interpret the different edge shifts, it is useful to compare them to values observed for similar, well-known compounds. One example is the protein complex hemoglobin which contains redox-active porphyrin-type FeN4 centers. As indicated by the dashed lines in Fig. 2B, the redox-induced edge shift in Fe–N-aCoCo is even larger compared to the Fe-edge shift between deoxy- and oxy-hemoglobin reported by Wilson et al.44 The shift is determined by two influences: Firstly, by the amount of redox-active iron species present in the sample (compare with Table 1), and secondly, by the covalency of the iron–oxygen bond formed upon oxidation.44,45 The larger edge shift of Fe–N-aCoCo compared to hemoglobin, which comprises 100% of redox-active species, might thus be explained by more ionic bonds being formed between the active centers and oxygen adsorbates.44,46Table 2 compares the edge shifts reported in the literature for different Fe–N–C catalysts. All of the Fe–N–C catalysts exhibit edge shifts, which are similar or smaller compared to the edge-shift reported for hemoglobin (ΔEph = 2.8 eV). In contrast, the edge shift of iron oxides with the oxidation state (ΔEph = 5.5 eV) is larger compared to that of hemoglobin, and larger compared to that of Fe–N-aCoCo (ΔEph = 4.05 ± 0.36). This suggests that the metal to ligand charge-transfer effect and thus the covalency of the iron–oxygen bond in Fe–N-aCoCo is smaller compared to those of iron oxides, but larger compared to those of hemoglobin and other Fe–N–C catalysts. As reported in our previous publications, Fe–N-aCoCo contains about 10.6 at% of oxygen and 1.9 at% of phosphorous.18 A smaller ligand-to-metal charge transfer effect compared to that of other Fe–N–C catalysts or hemoglobin could thus be explained by an electron withdrawing effect of the large amount of oxygen species within the carbon matrix4 of Fe–N-aCoCo or by the incorporation of oxygen within FeNxOy-type or phosphorous within FeNxPy-type redox-active species. Another explanation for the larger edge shift could be a lower average oxidation state of iron caused by a part of the iron species getting reduced to Fe0, as previously observed in Fe–O–C catalysts for CO2 reduction.45 However, this would be accompanied by a shape change of the rising edge according to the shoulder at around 7115 eV being typical for metallic iron, which is not observed (compare with Fig. 1 and 2).
| Material | Preparation | Secondary phases | Source | ΔEph | Oxidizable Fe percentage | |
|---|---|---|---|---|---|---|
| Fe–N-aCoCo [%] | Hemoglobin [%] | |||||
| Hemoglobin | — | No | Wilson et al.44 | 2.8 | — | — |
| TPP-800 | Porphyrin pyrolysis | No | Jia et al.27 | 2.8 | — | — |
| Fe–N–C | MOF-based | (No) | Osmieri et al.7 | 2.7 | — | — |
| Fe–N–C | MOF-based | (No) | Li et al.10 | 2.7 | — | — |
| Fe–N–C | MOF-based | (No) | Zitolo et al.32 | 1.8 | — | — |
| Fe–N–C | MOF-based | (No) | Santori et al.47 | 1.9 | — | — |
| Fe–oxides | — | No | Görlin et al.48 | 5.5 | — | — |
| Fe–N-aCoCo | Biomass-based | (No) | This work | 4.05 ± 0.36 | 100 | 143 ± 13 |
| Fe–N-oxBP | Conv. support | Yes | This work | 0.62 ± 0.20 | 15 ± 5 | 21 ± 7 |
| Fe–N-Comm | Commercial | Yes | This work | 0.72 ± 0.15 | 18 ± 4 | 25 ± 5 |
The edge shift of Fe–N-oxBP and Fe–N-Comm is limited by the amount of redox-active species, which is only about 20% and 30%, respectively (see Table 1). In addition to the linear combination analysis, the amount of redox-active species can also be estimated from the edge shift upon reduction. For this, the full edge shift of 100% of redox-active species present in the sample was assumed to be either equal to the value of hemoglobin (ΔEph = 2.8 eV) or equal to the average value of Fe–N-aCoCo (ΔEph = 4.05 eV). The resulting amounts of redox-active species being present in Fe–N-oxBP and Fe–N-Comm are summarized in Table 2. For a full edge shift of ΔEph = 2.8 eV, the determined values agree with the amount of FeNx-sites obtained from linear combination analysis, while the resulting amounts of redox-active species are lower compared to the values obtained by linear combination fitting, if an edge shift equal to Fe–N-aCoCo is assumed. Consequently, it is consistent with the data that the edge-shifts of the Fe–N-oxBP and Fe–N-Comm active centers are more similar to the edge shift reported for hemoglobin or other Fe–N–C than the edge shift observed for Fe–N-aCoCo. However, it cannot be fully ruled out that a portion of the FeNx is not redox-active if some of these sites are located within pores or cavities that are not exposed to the electrolyte.49 Thus, it is possible that the amount of redox-active species is not equal to the overall amount of FeNx-type species. As this method determines only the redox-active amount, it represents a lower limit for the total amount of FeNx-type species. This may explain the tendentially lower amount of FeNx estimated from the edge shift compared to the value obtained by linear combination analysis.
More information on the nature of the active centers in Fe–N-aCoCo can be gained from the shape of the reduced XANES spectra (Fig. 2A). Jia et al. demonstrated that in the reduced state feature I is expected to be larger compared to feature II marked in Fig. 2A, if the probed FeN4-type species exhibit D4h symmetry. With increasing distortion of the centrosymmetric square planar geometry, the intensity of feature I decreases relative to that of feature II.27 Accordingly, the geometry of the FeNx-type species present in Fe–N-aCoCo clearly deviates (on average) from the square planar symmetry, as feature I is distinctly larger compared to feature II in the XANES spectrum.
Static potential dependent EXAFS
In order to further characterize the average local geometry of the active centers, a local-structure model was fitted to the EXAFS regions. This analysis was restrained to Fe–N-aCoCo, as in the case of the other catalysts, the large contributions of iron or iron carbide species superimpose with the contributions of the FeNx-type species making a proper analysis impossible. Based on a simple model, the first shell of the Fe FT-EXAFS of Fe–N-aCoCo was fitted in the oxidized and reduced state simultaneously. As can be seen in Fig. S2,† the intensity of the first shell increases upon oxidation, which likely reflects additional oxygen species being present in the first shell in the oxidized state compared to the reduced state. Thus, the first shell in the reduced state was assumed to consist solely of equal nitrogen scattering paths, while in the oxidized state a scattering path for the adsorbed oxygen species was added to the first shell. Further details on the fitting model used for EXAFS data evaluation can be found in the Materials section.Fig. 3 shows the fitting-results for this model and results for the fitted model parameters are summarized in Table S1.† The good agreement between data and the model suggests that the first shell may indeed be governed by nitrogen and oxygen paths. The average bond distance is not significantly different in the oxidized and reduced states (ROx = 2.02 ± 0.01 Å and RRed = 2.04 ± 0.01 Å). A very similar oxidation state dependent average first-shell bond distance was also observed by Jia et al. for Fe-porphyrin complexes mixed with black pearls and pyrolyzed at 800 °C.27 The resulting oxygen coordination number N(Fe–O) is 0.9 ± 0.6 and thus close to one, which is in agreement with one oxygen species adsorbing on each active center on average in the oxidized state. The resulting nitrogen coordination number N(Fe–N) is 3.1 ± 0.5. Hence, the iron centers might be slightly undercoordinated compared to ideal FeN4-type sites indicating some FeN3- or FeN2-type moieties to be incorporated in the catalyst, as previously suggested by Asset et al.4 The applied model does not account for Fe–C scattering paths, which may overlap with the first N/O-shell, as shown for the Fe–N–C material characterized by Fei et al. in Fig. 1B. This may cause the observed deviations between the model and the measured spectrum on the far side of the first shell. However, adding different specific carbon paths deteriorates the fitting results. This suggests that a variety of different implementations of the FeNx-type moieties contributes to the first shell, which makes it difficult to consider these contributions in the fit. Despite the small deviations, the result is in good agreement with the presence of FeNx-type active sites (with x ≈ 3.1 ± 0.5), on which oxygen species adsorbs upon oxidation. Moreover, due to the similar shape of the oxygen and nitrogen next-nearest neighbor scattering paths, the result is also consistent with the presence of oxygen heteroatoms at FeNxOy-type sites. This may explain the smaller average metal to ligand charge transfer effect concluded from the larger edge shift compared to that of hemoglobin. Furthermore, as the Fe–N-aCoCo catalyst contains about 1.9 at% of phosphorus, the EXAFS-fitting model was extended to implement phosphorous next-nearest neighbors within FeNxPy-type moieties. For this, the phosphorous coordination number and the Fe–P bond distance were added to the fitting model. As shown in Fig. S3,† this model also leads to a satisfying fitting result. The resulting average Fe–N bond distance is only slightly decreased (see Table S1†), and the average Fe–P bond distance of 2.34 ± 0.02 Å is in good agreement with the value observed by Yuan et al. (2.35 ± 0.02 Å).50 However, the small average coordination numbers of N(Fe–N) = 2.5 ± 0.3 and N(Fe–P) = 0.3 ± 0.1 suggest that the iron centers within FeNxPy-type moieties are, on average, highly undercoordinated compared to ideal FeN4-type moieties.
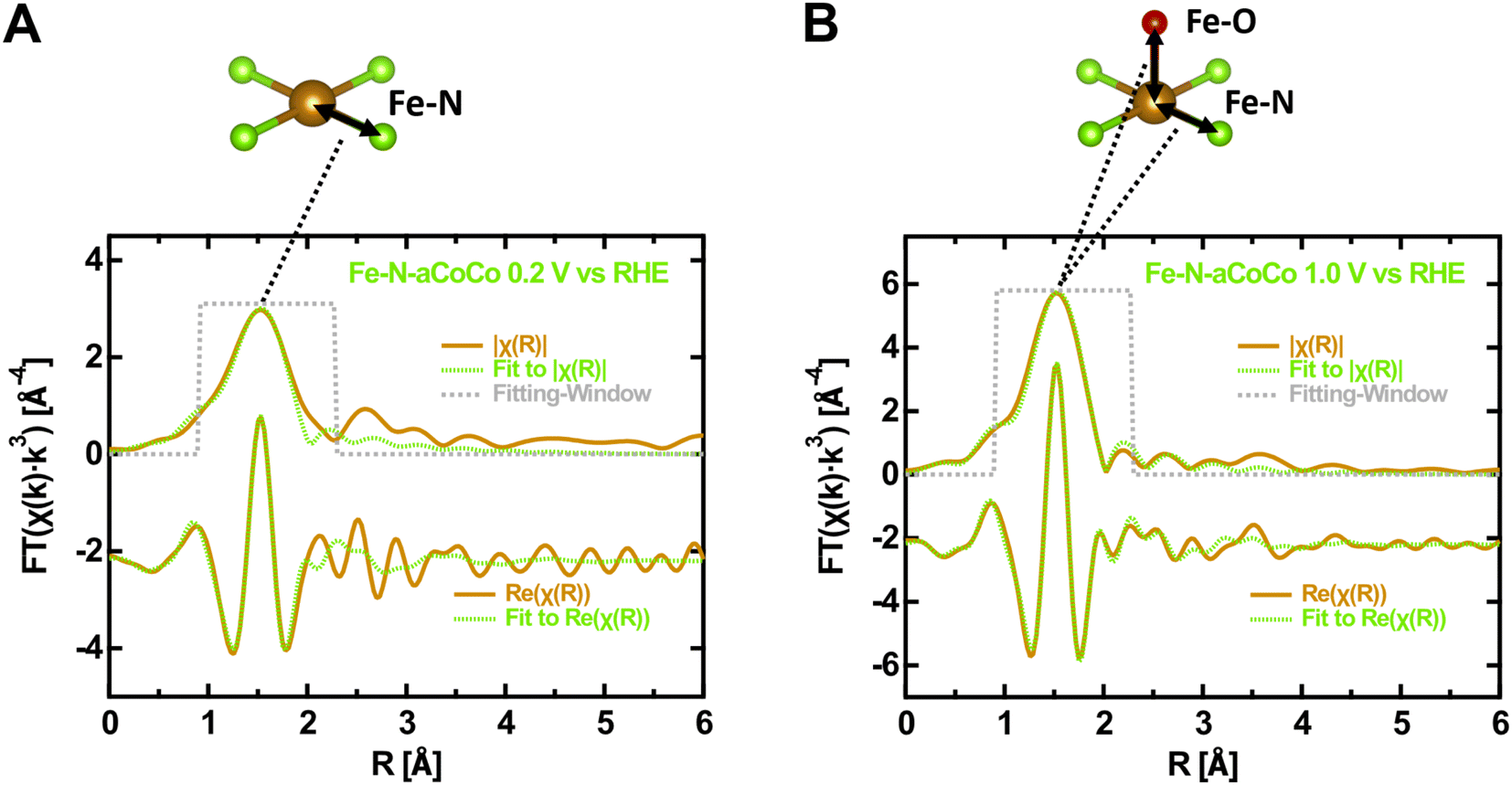 | ||
| Fig. 3 Modeling of the EXAFS-region of the Fe K-edge of Fe–N-aCoCo. (A) 0.2 V vs. RHE. (B) 1.0 V vs. RHE. To illustrate the fitting-model, an exemplary D4h nearest neighbor nitrogen (green) coordination of iron (brown) is shown above, which is supplemented by an oxygen or intermediate species (red) binding out of plane to the iron center. | ||
Potentiodynamic XANES
As a fourth step, OPCV measurements were performed to further evaluate the Fe redox transition with a higher resolution in electrode potential. Hereby, the fluorescence-yield (FY) intensity at a fixed photon energy of 7124 eV was recorded while cycling the electrode potential between 1.2 V vs. RHE and 0.1 V vs. RHE. The intensity obtained at this particular photon energy increases upon reduction, as can be seen from the difference spectra between oxidizing and reducing electrode potentials shown in Fig. 2A. It is therefore a good measure for the overall number of reduced species being present in the sample.The observed FY-intensity changes from its minimum to its maximum value within the wide potential range of +1.1 V vs. RHE to +0.2 V vs. RHE (upper panel of Fig. 4). This agrees with the results of the edge shift observed in the static potential dependent XANES-measurements in the same potential range. Hysteresis occurs between the FY-signal of the anodic and the cathodic scan, which is influenced by the rate of the redox transition relative to the chosen scan rate. Along with the FY-intensity, the electrochemical current was measured during potential cycling (lower panel of Fig. 4). The CV is dominated by capacitive currents, faradaic currents resulting from the ORR as well as carbon related redox currents such as from the hydroquinone/quinone redox transition.51 The rather small iron-related redox currents, which are coupled to catalytic transitions through the ORR mechanism, are obscured by these dominating contributions.52
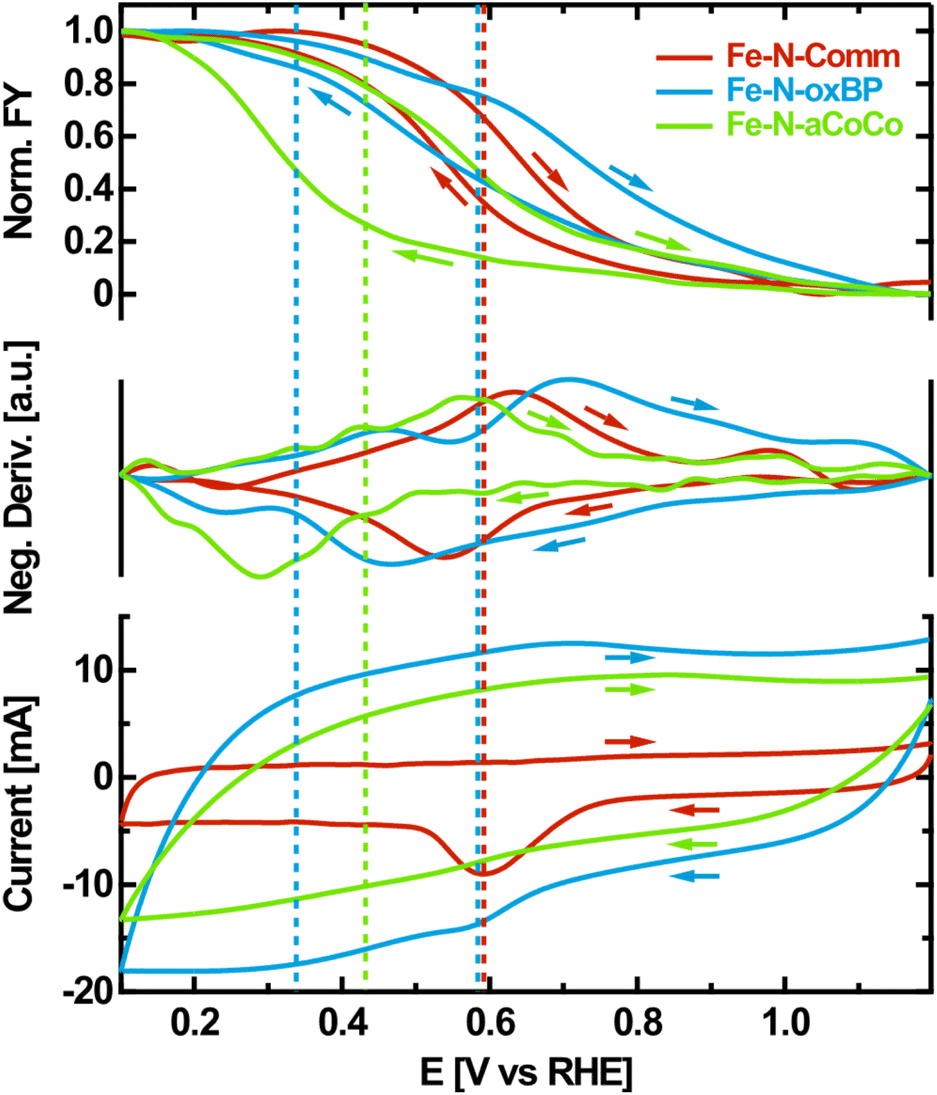 | ||
| Fig. 4 OPCV of Fe–N–C catalysts. Normalized, smoothed FY at 7124 eV photon energy depending on the applied electrode potential as well as the negative potential-derivative of the FY compared to the electrode-current measured during OPCV. The electrolyte was purged with oxygen during the measurement and a scan rate of 10 mV s−1 was used. The arrows indicate the scanning direction. | ||
To correlate the iron oxidation-state change with electrochemical currents, the negative potential-derivative of the FY-intensity was determined (middle-panel of Fig. 4). As the FY-intensity at 7124 eV is proportional to the amount of reduced iron species present in the sample, the negative potential-derivative of the FY-intensity is a selective measure of the iron-induced redox current.
The resulting curves exhibit distinct maxima in the anodic and the cathodic potential scan, representing the iron redox waves which are not detectable in the electrochemical measurement. If the iron redox transition proceeding at the FeNx-type sites is assumed to be reversible, its average equilibrium potential can approximately be located at the center between the peak potentials of the corresponding anodic and the cathodic redox waves identified in the negative potential-derivative of the FY-intensity. This is indicated by the dashed lines in Fig. 4. According to this evaluation, Fe–N-Comm exhibits one well-defined but broad redox wave with an average redox potential at about +0.59 V vs. RHE. In contrast, the redox-processes occurring on the surface of Fe–N-oxBP are more heterogeneous. At least two different average redox potentials are observed at about +0.58 V vs. RHE and +0.34 V vs. RHE. For Fe–N-aCoCo the average of the majority of redox potentials can be identified at around +0.42 V vs. RHE, although apparent undulating variations in the derivative probably indicate small amounts of redox transitions occurring at distinctly different redox potentials.
The redox potential of an active site is thought to be directly related to the catalytic ORR activity scaling with the binding strength of intermediates according to Sabatier's principle.7,27,53 The binding energy of intermediates to the active sites decreases as the redox potential increases. As redox potentials below the onset of the ORR are assumed to be on the high-energy side of the volcano plot, the observed ORR activity of the FeNx-type active sites is expected to increase with the redox potential within this regime.27 In other words, the ORR activity increases as the redox transition involved in the ORR appears at a lower overpotential. Current studies suggest that reduced FeIINx sites are the starting point of the 4-step ORR-mechanism. This active center becomes oxidized as it binds to molecular oxygen. In the subsequent proton-coupled electron transfer steps the oxygen decomposes, while the iron center passes different oxidation states. In the last step, the FeIINx is regenerated, which is necessary for ongoing turnover.54 This only occurs at a high probability if the Fe-redox potential of the active site is undercut. Therefore, based on the results obtained from OPCV, the reported trends in the electrochemical activity can be understood as follows: about 25% to 30% of highly ORR-active iron sites in Fe–N-Comm cause this catalyst to exhibit the highest catalytic activity. About 100% of the less ORR-active iron sites in Fe–N-aCoCo lead to a medium catalytic activity. About 15% to 20% of heterogeneous iron sites in Fe–N-oxBP are partially highly ORR-active and partially exhibit very sluggish ORR kinetics, resulting in the lowest catalytic activity of the investigated materials.
To explain the different properties of the active sites in Fe–N-aCoCo, three different scenarios can be suggested based on our findings: firstly, FeN4-type moieties exhibit a distorted D4h symmetry and undercoordinated FeNx-type moieties are responsible for ORR-activity. The binding strength to oxygen is influenced by the undercoordination, a possible electron withdrawing effect of the carbon basal plane being modified by oxygen and phosphorus bound to the carbon species4,55 and the magnitude of the distortion of the active sites modified by the details of their implementation geometry.27 Secondly, the oxygen binding properties of the FeN4-type and undercoordinated FeNx-type active sites are modified by heteroatoms being coordinated to the iron species themselves, i.e. the formation of FeNxOy-type or FeNxPy-type sites. Specifically the involvement of oxygen seems to be likely, as it could well explain the modified ligand-to-metal charge transfer effect. Alternatively, it may also be possible that the heteroatoms are bound to the ligands to form e.g. NO-type ligands. Thirdly, different types of complexes, like agglomerated FexNy-type sites might be present, which exhibit modified adsorption properties and decreased Fe–N coordination numbers compared to FeNx-type sites.
Conclusion
Three different Fe–N–C catalysts were analyzed for their iron sites and coordination environment including a commercial Fe–N–C and two support-based Fe–N–C catalysts consisting of black pearls and phosphoric acid activated coconut shells. The biomass-based catalyst (Fe–N-aCoCo) is the only one in which solely redox-active iron species were identified. Both other catalysts contain around 70% to 80% of inactive iron- or iron carbide phases. This underpins the potential of coconut-shell based, nitrogen and heteroatom-rich biochar to reach higher active-site densities compared to conventional substrates, making it a promising precursor for future Fe–N–C catalysts. However, the active sites of Fe–N-aCoCo exhibit a lower redox potential and thus a higher binding energy to oxygen species compared to the commercial (Fe–N-Comm) and the black pearl based (Fe–N-oxBP) catalysts, which decreases the turnover frequency of each site. The identified differences in the amount and binding strength of the active sites lead to the following trend in activity: Fe–N-Comm > Fe–N-aCoCo > Fe–N-oxBP. Furthermore, our data are consistent with the oxidation-induced edge shift of the iron active centers being similar to edge shifts reported previously for other Fe–N–C materials or hemoglobin. In contrast, the edge shift of the iron active centers in Fe–N-aCoCo is unconventionally large and lies between that of iron–nitrogen complexes like hemoglobin and iron oxides. This indicates a lower ligand-to-metal charge transfer effect, which is probably caused by heteroatom doping. Modeling the EXAFS-region of the Fe K-edge suggests that undercoordinated FeNx-type moieties (x < 4) are present in Fe–N-aCoCo, which are potentially modified by oxygen or phosphorous heteroatom substitution.Based on our exemplary study, it is possible to develop specific strategies to improve Fe–Nx incorporation in Fe–N–C catalysts. In particular, further Fe–N–C development strategies should consider the influence of heteroatom doping, e.g., with P-species.
Author contributions
The manuscript was written through contributions of all the authors. All authors have given approval to the final version of the manuscript. J. M., P. W. and M. W. conceptualized and optimized the sample preparation procedures. J. M. prepared the samples. P. W. and M. W. supervised the sample preparation. R. S. and G. W. conceptualized the operando XAS experiments. G. W. and H. T. conducted the operando XAS experiments. G. W., J. M. and R. S. interpreted the results. G. W. wrote the original manuscript. R. S., M. W., P. W., J. M., H. T. and G. W. reviewed and edited the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank the staff of Helmholtz-Zentrum Berlin for their support during the beamtimes at BESSY II. G. Wartner, and R. Seidel gratefully acknowledge financial support from the German Research Foundation (DFG) through an Emmy-Noether grant (project SE 2253/3-1). H. Trzesniowski acknowledges support from the German Federal Ministry of Education and Research in the framework of the project Catlab (grant 03 EW0015A/B). J. Müller-Hülstede and P. Wagner would like to thank the Federal Ministry for Economic Affairs and Climate Action for funding (grant number 03ETB016A).References
- W. T. Hong, M. Risch, K. A. Stoerzinger, A. Grimaud, J. Suntivich and Y. Shao-Horn, Toward the rational design of non-precious transition metal oxides for oxygen electrocatalysis, Energy Environ. Sci., 2015, 8(5), 1404–1427, 10.1039/c4ee03869j
.
- U. I. Kramm, A. Shahraei and I. Martinaiou, Metall-N-C-Katalysatoren in der Elektrokatalyse, Nachr. Chem., 2017, 65(11), 1096–1099, DOI:10.1002/nadc.20174055316
- U. Martinez, S. Komini Babu, E. F. Holby, H. T. Chung, X. Yin and P. Zelenay, Progress in the Development of Fe-Based PGM-Free Electrocatalysts for the Oxygen Reduction Reaction, Adv. Mater., 2019, 31(31), 1–20, DOI:10.1002/adma.201806545
- T. Asset and P. Atanassov, Iron-Nitrogen-Carbon Catalysts for Proton Exchange Membrane Fuel Cells, Joule, 2020, 4(1), 33–44, DOI:10.1016/j.joule.2019.12.002
- U. I. Kramm, J. Herranz, N. Larouche, T. M. Arruda, M. Lefèvre, F. Jaouen, P. Bogdanoff, S. Fiechter, I. Abs-Wurmbach, S. Mukerjee and J. P. Dodelet, Structure of the catalytic sites in Fe/N/C-catalysts for O2-reduction in PEM fuel cells, Phys. Chem. Chem. Phys., 2012, 14(33), 11673–11688, 10.1039/c2cp41957b
- A. Zitolo, V. Goellner, V. Armel, M. T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Identification of catalytic sites for oxygen reduction in iron- and nitrogen-doped graphene materials, Nat. Mater., 2015, 14(9), 937–942, DOI:10.1038/nmat4367
- L. Osmieri, R. K. Ahluwalia, X. Wang, H. T. Chung, Xi Yin, A. J. Kropf, J. Park, D. A. Cullen, K. L. More, P. Zelenay, D. J. Myers and K. C. Neyerlin, Elucidation of Fe-N-C electrocatalyst active Site functionality via in-situ X-ray absorption and operando determination of oxygen reduction reaction kinetics in a PEFC, Appl. Catal., B, 2019, 257, 117929, DOI:10.1016/j.apcatb.2019.117929
- C. H. Choi, H. K. Lim, M. W. Chung, G. Chon, S. Ranjbar, A. Nastaran, S. Abdulrahman, M. Tahar, L. Stievano, H. S. Oh, E. S. Park, F. Luo, P. Strasser, G. Dražić, K. J. J. Mayrhofer and F. Kim, The Achilles’ heel of iron-based catalysts during oxygen reduction in an acidic medium, Energy Environ. Sci., 2018, 11(11), 3176–3182, 10.1039/c8ee01855c
- H. Singh, S. Zhuang, B. Ingis, B. B. Nunna and E. S. Lee, Carbon-based catalysts for oxygen reduction reaction: a review on degradation mechanisms, Carbon, 2019, 151, 160–174, DOI:10.1016/j.carbon.2019.05.075
- J. Li, M. T. Sougrati, A. Zitolo, J. M. Ablett, I. C. Oguz, T. Mineva, I. Matanovic, P. Atanassov, Y. Huang, I. Zenyuk, A. Di Cicco, K. Kumar, L. Dubau, F. Maillard, G. Dražić and F. Jaouen, Identification of durable and non-durable FeNx sites in Fe-N-C materials for proton exchange membrane fuel cells, Nat. Catal., 2021, 4(1), 10–19, DOI:10.1038/s41929-020-00545-2
- K. Wang, H. Wang, S. Ji, H. Feng, V. Linkov and R. Wang, Biomass-derived activated carbon as high-performance non-precious electrocatalyst for oxygen reduction, RSC Adv., 2013, 3(30), 12039–12042, 10.1039/c3ra41978a
- C. Z. Guo, W. L. Liao and C. G. Chen, Design of a non-precious metal electrocatalyst for alkaline electrolyte oxygen reduction by using soybean biomass
as the nitrogen source of electrocatalytically active center structures, J. Power Sources, 2014, 269, 841–847, DOI:10.1016/j.jpowsour.2014.07.024
- J. Zhang, S. Wu, X. Chen, K. Cheng, M. Pan and S. Mu, An animal liver derived non-precious metal catalyst for oxygen reduction with high activity and stability, RSC Adv., 2014, 4(62), 32811–32816, 10.1039/c4ra06495j
- X. Zhang, R. Liu, Y. Zang, G. Liu, S. Liu, G. Wang, Y. Zhang, H. Zhang and H. Zhao, Shrimp-shell derived carbon nanodots as precursors to fabricate Fe,N-doped porous graphitic carbon electrocatalysts for efficient oxygen reduction in zinc-air batteries, Inorg. Chem. Front., 2016, 3(7), 910–918, 10.1039/c6qi00059b
- Z. Xu, J. Ma, M. Shi, Y. Xie and C. Feng, Biomass based iron and nitrogen co-doped 3D porous carbon as an efficient oxygen reduction catalyst, J. Colloid Interface Sci., 2018, 523, 144–150, DOI:10.1016/j.jcis.2018.03.092
- Y. Liu, M. Su, D. Li, S. Li, X. Li, J. Zhao and F. Liu, Soybean straw biomass-derived Fe-N co-doped porous carbon as an efficient electrocatalyst for oxygen reduction in both alkaline and acidic media, RSC Adv., 2020, 10(12), 6763–6771, 10.1039/c9ra07539a
- J. Hülstede, D. Schonvogel, H. Schmies, P. Wagner, F. Schröter, A. Dyck and M. Wark, Relevant Properties of Carbon Support Materials in Successful Fe-N-C Synthesis for the Oxygen Reduction Reaction: Study of Carbon Blacks and Biomass-Based Carbons, Materials, 2021, 14(1), 1–19, DOI:10.3390/ma14010045
- J. Müller-Hülstede, D. Schonvogel, H. Schmies, P. Wagner, A. Dyck and M. Wark, Incorporation of Activated Biomasses in Fe-N-C Catalysts for Oxygen Reduction Reaction with Enhanced Stability in Acidic Media, ACS Appl. Energy Mater., 2021, 4(7), 6912–6922, DOI:10.1021/acsaem.1c01018
- Q. Chen, X. Tan, Y. Liu, S. Liu, M. Li, Y. Gu, P. Zhang, S. Ye, Z. Yang and Y. Yang, Biomass-derived porous graphitic carbon materials for energy and environmental applications, J. Mater. Chem. A, 2020, 8(12), 5773–5811, 10.1039/c9ta11618d
- J. Müller-Hülstede, T. Zierdt, H. Schmies, D. Schonvogel, Q. Meyer, C. Zhao, P. Wagner and M. Wark, Implementation of different Fe-N-C catalysts in high temperature proton exchange membrane fuel cells - effect of catalyst and catalyst layer on performance, J. Power Sources, 2022, 537, 231529, DOI:10.1016/j.jpowsour.2022.231529
- G. Schuck and I. Zisak, CryoEXAFS: X-ray absorption spectroscopy station with cryogenic or in-beam operando electrochemistry sample conditions at BESSY II, J. Large-scale Res. Facil., JLSRF, 2020, 6, 139, DOI:10.17815/jlsrf-6-176
-
WaveMetrics Inc., Igor Pro 9.0, Portland, USA, 2022 Search PubMed
- B. Ravel and M. Newville,
ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT, J. Synchrotron Radiat., 2005, 12(4), 537–541, DOI:10.1107/S0909049505012719
- W. M. Heijboer, P. Glatzel, K. R. Sawant, R. F. Lobo, U. Bergmann, R. A. Barrea, D. C. Koningsberger, B. M. Weckhuysen and F. M. F. de Groot, Kβ-Detected XANES of Framework-Substituted FeZSM-5 Zeolites, J. Phys. Chem. B, 2004, 108(28), 10002–10011, DOI:10.1021/jp048368w
- T. E. Westre, P. Kennepohl, J. G. DeWitt, B. Hedman, K. O. Hodgson and E. I. Solomon, A Multiplet Analysis of Fe K-Edge 1s → 3d Pre-Edge Features of Iron Complexes, J. Am. Chem. Soc., 1997, 119(27), 6297–6314, DOI:10.1021/ja964352a
- I. T. Bae, D. A. Tryk and D. A. Scherson, Effect
of Heat Treatment on the Redox Properties of Iron Porphyrins Adsorbed on High Area Carbon in Acid Electrolytes: An in Situ Fe K-Edge X-ray Absorption Near-Edge Structure Study, J. Phys. Chem. B, 1998, 102(21), 4114–4117, DOI:10.1021/jp972605w
- Q. Jia, N. Ramaswamy, H. Hafiz, U. Tylus, K. Strickland, G. Wu, B. Barbiellini, A. Bansil, E. F. Holby, P. Zelenay and S. Mukerjee, Experimental Observation of Redox-Induced Fe-N Switching Behavior as a Determinant Role for Oxygen Reduction Activity, ACS Nano, 2015, 9(12), 12496–12505, DOI:10.1021/acsnano.5b05984
- H. Fei, J. Dong, Y. Feng, C. S. Allen, C. Wan, B. Volosskiy, M. Li, Z. Zhao, Y. Wang, H. Sun, P. An, W. Chen, Z. Guo, C. Lee, D. Chen, I. Shakir, M. Liu and Y. Hu, General synthesis and definitive structural identification of MN4C4 single-atom catalysts with tunable electrocatalytic activities, Nat. Catal., 2018, 1(1), 63–72, DOI:10.1038/s41929-017-0008-y
- L. Avakyan, A. Manukyan, A. Bogdan, H. Gyulasaryan, J. Coutinho, E. Paramonova, G. Sukharina, V. Srabionyan, E. Sharoyan and L. Bugaev, Synthesis and structural characterization of iron-cementite nanoparticles encapsulated in carbon matrix, J. Nanopart. Res., 2020, 22, 30, DOI:10.1007/s11051-019-4698-8
- F. Bardelli, G. Barone, V. Crupi, F. Longo, G. Maisano, D. Majolino, P. Mazzoleni and V. Venuti, Iron speciation in ancient attic pottery pigments: a non-destructive SR-XAS investigation, J. Synchrotron Radiat., 2012, 19(5), 782–788, DOI:10.1107/S0909049512023990
- Z. Wu, R. E. Benfield, Y. Wang, L. Guo, M. Tan, H. Zhang, Y. Ge and D. Grandjean, EXAFS study on the local atomic structures around iron in glycosylated haemoglobin, Phys. Med. Biol., 2001, 46(3), N71–N77, DOI:10.1088/0031-9155/46/3/403
- A. Zitolo, N. Ranjbar-Sahraie, T. Mineva, J. Li, Q. Jia, S. Stamatin, G. F. Harrington, S. M. Lyth, P. Krtil, S. Mukerjee, E. Fonda and F. Jaouen, Identification of catalytic sites in cobalt-nitrogen-carbon materials for the oxygen reduction reaction, Nat. Commun., 2017, 8, 957, DOI:10.1038/s41467-017-01100-7
-
J. Müller-Hülstede, Non-Precious Metal Catalysts Based on Activated Biochar for the Oxygen Reduction Reaction in High Temperature Proton Exchange Membrane Fuel Cell, Carl von Ossietzky Universität Oldenburg, 2022 Search PubMed
- A. Tayal, M. Gupta, A. Gupta, P. Rajput and J. Stahn, Origin of anomalous diffusion in iron mononitride thin films, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92(5), 1–7, DOI:10.1103/PhysRevB.92.054109
- T. Sun, Y. Jiang, Q. Wu, L. Du, Z. Zhang, L. Yang, X. Wang and Z. Hu, Is iron nitride or carbide highly active for oxygen reduction reaction in acidic medium?, Catal. Sci. Technol., 2017, 7(1), 51–55, 10.1039/c6cy01921h
- X. W. Liu, S. Zhao, Y. Meng, Q. Peng, A. K. Dearden, C. F. Huo, Y. Yang, Y. W. Li and X. D. Wen, Mössbauer Spectroscopy of Iron Carbides: From Prediction to Experimental Confirmation, Sci. Rep., 2016, 6, 26184, DOI:10.1038/srep26184
- S. Boccato, R. Torchio, S. Anzellini, E. Boulard, F. Guyot, T. Irifune, M. Harmand, I. Kantor, F. Miozzi, P. Parisiades, A. D. Rosa, D. Antonangeli and G. Morard, Melting properties by X-ray absorption spectroscopy: common signatures in binary Fe-C, Fe-O, Fe-S and Fe-Si systems, Sci. Rep., 2020, 10, 11663, DOI:10.1038/s41598-020-68244-3
- H. Zhao, J.-X. Liu, C. Yang, S. Yao, H.-Y. Su, Z. Gao, M. Dong, J. Wang, A. I. Rykov, J. Wang, Y. Hou, W.-X. Li and D. Ma, Synthesis of Iron-Carbide Nanoparticles: Identification of the Active Phase and Mechanism of Fe-Based Fischer-Tropsch Synthesis, CCS Chem., 2020, 2712–2724, DOI:10.31635/ccschem.020.202000555
- C. H. Choi, C. Baldizzone, G. Polymeros, E. Pizzutilo, O. Kasian, A. K. Schuppert, N. Ranjbar Sahraie, M. T. Sougrati, K. J. J. Mayrhofer and F. Jaouen, Minimizing Operando Demetallation of Fe-N-C Electrocatalysts in Acidic Medium, ACS Catal., 2016, 6(5), 3136–3146, DOI:10.1021/acscatal.6b00643
- W. J. Jiang, L. Gu, L. Li, Y. Zhang, X. Zhang, L. J. Zhang, J. Q. Wang, J. S. Hu, Z. Wei and L. J. Wan, Understanding the High Activity of Fe-N-C Electrocatalysts in Oxygen Reduction: Fe/Fe3C Nanoparticles Boost the Activity of Fe-Nx, J. Am. Chem. Soc., 2016, 138(10), 3570–3578, DOI:10.1021/jacs.6b00757
- R. Sgarbi, K. Kumar, F. Jaouen, A. Zitolo, E. A. Ticianelli and F. Maillard, Oxygen Reduction Reaction Mechanism and Kinetics on M-NxCy and M@N-C Active Sites Present in Model M-N-C Catalysts under Alkaline and Acidic Conditions, J. Solid State Electrochem., 2021, 25(1), 45–56, DOI:10.1007/s10008-019-04436-w
- K. Kumar, P. Gairola, M. Lions, N. Ranjbar-Sahraie, M. Mermoux, L. Dubau, A. Zitolo, F. Jaouen and F. Maillard, Physical and Chemical Considerations for Improving Catalytic Activity and Stability of Non-Precious-Metal Oxygen Reduction Reaction Catalysts, ACS Catal., 2018, 8(12), 11264–11276, DOI:10.1021/acscatal.8b02934
- M. Primbs, Y. Sun, A. Roy, D. Malko, A. Mehmood, M. T. Sougrati, P. Y. Blanchard, G. Granozzi, T. Kosmala, G. Daniel, P. Atanassov, J. Sharman, C. Durante, A. Kucernak, D. Jones, F. Jaouen and P. Strasser, Establishing Reactivity Descriptors for Platinum Group Metal (PGM)-Free Fe-N-C Catalysts for PEM Fuel Cells, Energy Environ. Sci., 2020, 13(8), 2480–2500, 10.1039/d0ee01013h
- S. A. Wilson, E. Green, I. I. Mathews, M. Benfatto, K. O. Hodgson, B. Hedman and R. Sarangi, X-Ray Absorption Spectroscopic Investigation of the Electronic Structure Differences in Solution and Crystalline Oxyhemoglobin, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(41), 16333–16338, DOI:10.1073/pnas.1315734110
- C. Genovese, M. E. Schuster, E. K. Gibson, D. Gianolio, V. Posligua, R. Grau-Crespo, G. Cibin, P. P. Wells, D. Garai, V. Solokha, S. Krick Calderon, J. J. Velasco-Velez, C. Ampelli and R. Pera,
Operando Spectroscopy Study of the Carbon Dioxide Electro-Reduction by Iron Species on Nitrogen-Doped Carbon, Nat. Commun., 2018, 9, 935, DOI:10.1038/s41467-018-03138-7
- S. A. Wilson, T. Kroll, R. A. Decreau, R. K. Hocking, M. Lundberg, B. Hedman, K. O. Hodgson and E. I. Solomon, Iron L-Edge X-Ray Absorption Spectroscopy of Oxy-Picket Fence Porphyrin: Experimental Insight into Fe-O2 Bonding, J. Am. Chem. Soc., 2013, 135(3), 1124–1136, DOI:10.1021/ja3103583
- P. G. Santori, F. D. Speck, J. Li, A. Zitolo, Q. Jia, S. Mukerjee, S. Cherevko and F. Jaouen, Effect of Pyrolysis Atmosphere and Electrolyte PH on the Oxygen Reduction Activity, Stability and Spectroscopic Signature of FeNx Moieties in Fe-N-C Catalysts, J. Electrochem. Soc., 2019, 166(7), F3311–F3320, DOI:10.1149/2.0371907jes
- M. Görlin, J. Halldin Stenlid, S. Koroidov, H. Y. Wang, M. Börner, M. Shipilin, A. Kalinko, V. Murzin, O. V. Safonova, M. Nachtegaal, A. Uheida, J. Dutta, M. Bauer, A. Nilsson and O. Diaz, Key Activity Descriptors of Nickel-Iron Oxygen Evolution Electrocatalysts in the Presence of Alkali Metal Cations, Nat. Commun., 2020, 11(1), 1–11, DOI:10.1038/s41467-020-19729-2
- F. Jaouen, M. Lefèvre, J. P. Dodelet and M. Cai, Heat-Treated Fe/N/C Catalysts for O2 Electroreduction: Are Active Sites Hosted in Micropores?, J. Phys. Chem. B, 2006, 110(11), 5553–5558, DOI:10.1021/jp057135h
- K. Yuan, D. Lützenkirchen-Hecht, L. Li, L. Shuai, Y. Li, R. Cao, M. Qiu, X. Zhuang, M. K. H. Leung, Y. Chen and U. Scherf, Boosting Oxygen Reduction of Single Iron Active Sites via Geometric and Electronic Engineering: Nitrogen and Phosphorus Dual Coordination, J. Am. Chem. Soc., 2020, 142(5), 2404–2412, DOI:10.1021/jacs.9b11852
- K. Mamtani and U. S. Ozkan, Heteroatom-Doped Carbon Nanostructures as Oxygen Reduction Reaction Catalysts in Acidic Media: An Overview, Catal. Lett., 2015, 145(1), 436–450, DOI:10.1007/s10562-014-1434-y
- U. I. Kramm, A. Zana, T. Vosch, S. Fiechter, M. Arenz and D. Schmeißer, On the Structural Composition and Stability of Fe-N-C Catalysts Prepared by an Intermediate Acid Leaching, J. Solid State Electrochem., 2016, 20(4), 969–981, DOI:10.1007/s10008-015-3060-z
- Q. Jia, E. Liu, L. Jiao, S. Pann and S. Mukerjee, X-Ray Absorption Spectroscopy Characterizations on PGM-Free Electrocatalysts: Justification, Advantages, and Limitations, Adv. Mater., 2019, 31(31), 2–9, DOI:10.1002/adma.201805157
- W. Zhong, Z. Wang, S. Han, L. Deng, J. Yu, Y. Lin, X. Long, M. Gu and S. Yang, Identifying the Active Sites of a Single Atom Catalyst with pH-Universal Oxygen Reduction Reaction Activity, Cell Rep. Phys. Sci., 2020, 1(7), 100115, DOI:10.1016/j.xcrp.2020.100115
- M. Kübler, S. Wagner, T. Jurzinsky, S. Paul, N. Weidler, E. D. Gomez Villa, C. Cremers and U. I. Kramm, Impact of Surface Functionalization on the Intrinsic Properties of the Resulting Fe-N-C Catalysts for Fuel Cell Applications, Energy Technol., 2020, 8, 2000433, DOI:10.1002/ente.202000433
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4se00342j |
| This journal is © The Royal Society of Chemistry 2024 |