
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlternatives to fluorinated binders: recyclable copolyester/carbonate electrolytes for high-capacity solid composite cathodes†
Holly
Yeo
a,
Georgina L.
Gregory
 *a,
Hui
Gao
abc,
Kanyapat
Yiamsawat
a,
Gregory J.
Rees
bc,
Thomas
McGuire
a,
Mauro
Pasta
*bc,
Peter G.
Bruce
*bc and
Charlotte K.
Williams
*a
*a,
Hui
Gao
abc,
Kanyapat
Yiamsawat
a,
Gregory J.
Rees
bc,
Thomas
McGuire
a,
Mauro
Pasta
*bc,
Peter G.
Bruce
*bc and
Charlotte K.
Williams
*a
aDepartment of Chemistry, University of Oxford, Chemistry Research Laboratory, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: charlotte.williams@chem.ox.ac.uk
bDepartment of Materials, University of Oxford, Oxford, OX1 3PH, UK
cThe Faraday Institution, Quad One, Harwell Science and Innovation Campus, Didcot, OX11 0RA, UK
First published on 17th January 2024
Abstract
Optimising the composite cathode for next-generation, safe solid-state batteries with inorganic solid electrolytes remains a key challenge towards commercialisation and cell performance. Tackling this issue requires the design of suitable polymer binders for electrode processability and long-term solid–solid interfacial stability. Here, block-polyester/carbonates are systematically designed as Li-ion conducting, high-voltage stable binders for cathode composites comprising of single-crystal LiNi0.8Mn0.1Co0.1O2 cathodes, Li6PS5Cl solid electrolyte and carbon nanofibres. Compared to traditional fluorinated polymer binders, improved discharge capacities (186 mA h g−1) and capacity retention (96.7% over 200 cycles) are achieved. The nature of the new binder electrolytes also enables its separation and complete recycling after use. ABA- and AB-polymeric architectures are compared where the A-blocks are mechanical modifiers, and the B-block facilitates Li-ion transport. This reveals that the conductivity and mechanical properties of the ABA-type are more suited for binder application. Further, catalysed switching between CO2/epoxide A-polycarbonate (PC) synthesis and B-poly(carbonate-r-ester) formation employing caprolactone (CL) and trimethylene carbonate (TMC) identifies an optimal molar mass (50 kg mol−1) and composition (wPC 0.35). This polymer electrolyte binder shows impressive oxidative stability (5.2 V), suitable ionic conductivity (2.2 × 10−4 S cm−1 at 60 °C), and compliant viscoelastic properties for fabrication into high-performance solid composite cathodes. This work presents an attractive route to optimising polymer binder properties using controlled polymerisation strategies combining cyclic monomer (CL, TMC) ring-opening polymerisation and epoxide/CO2 ring-opening copolymerisation. It should also prompt further examination of polycarbonate/ester-based materials with today's most relevant yet demanding high-voltage cathodes and sensitive sulfide-based solid electrolytes.
Introduction
Solid-state batteries (SSBs) have recently emerged as a promising follow-up technology to commercial lithium-ion batteries.1 Replacing liquid electrolytes in the latter for less flammable solid alternatives offers conceivably safer next-generation devices with higher energy densities and improved long-term performance.2,3 To this end, a significant research achievement was the discovery of inorganic solid electrolytes (such as Li6PS5Cl) with competitive ionic conductivities to liquids.4,5 Unlike liquids, however, maintaining intimate contact between the electrolyte and electrode particles is difficult.1,6 Optimising these solid interfaces is particularly key in the cathode given it comprises multiple solid phases and its proper function dictates attainable capacity and capacity retention.7–10 While advancements have been made, for instance using high-capacity nickel-rich manganese–cobalt oxides (NMC), most recently in single-crystal (sc) form,11–13 a suitable polymer binder is needed for large-scale fabrication and commercial use of cells.1,14Typically, solid composite cathodes comprise a mixture of cathode particles, solid electrolyte, carbon and a polymer binder. The latter can hold solid phases together but also enables large-scale (i.e. roll-to-roll) electrode processability.15,16 Thus, they are an important consideration if solid-state battery technology is to be translated from the lab level to commercial development.17–20 In addition, elastomeric polymers can mitigate cathode volume changes during battery operation.21 The added complexity of even small volume changes (6 vol% for LiNi0.8Mn0.1Co0.1O2 cathodes, NMC811) requires pressure to be applied to the cell for long-term capacity retention.7,11 For many reports, this is unrealistically high (>50 MPa); acceptable cell pressures are 1–2 MPa or ideally lower.1
So far, traditional rubber binders (nitrile butadiene, NBR and styrene butadiene, SBR), require modification with polar functionalities to impart adhesiveness for appreciable cycling stability.22 For cathode composites integrating Li6PS5Cl, this prevents standard wet slurry-based cathode fabrication, owing to the poor compatibility of Li6PS5Cl with polar solvents.22–24 However, recent findings summarised by Lu et al. suggest dry-processed electrodes may be better for both performance and sustainability by reducing solvent use.25 Currently, polytetrafluoroethylene (PTFE) is the prevalent binder for producing these solvent-free cathode composites but alongside also commonly used polyvinylidene fluoride (PVDF) cathode binders, fluorinated polymers pose significant environmental concerns.26,27 Consequently, fluorinated substances are proposed to be banned by the EU following a recommendation by the European Chemicals Agency.28 Besides, given that providing effective pathways for ions is fundamental for maximising cathode capacity, the inherent non-conductive nature of these and most binders is also limiting.7,29 Indeed, recent work demonstrates that chemically modifying PTFE with ionomers to impart even modest ionic conductivities (1.6 × 10−5 S cm−1 at 25 °C) results in a 20% capacity gain over PTFE.30 Clearly, alternative polymer binders are needed to meet the demanding requirements for high-capacity cathode composites in solid-state batteries.31 Fluorinated binders offer high electrochemical stability and finding replacements will require identifying less environmentally persistent polymers with high-voltage stability.
Here, we propose all polycarbonate/ester-based polymers as high-voltage stable, ionically conducting binders. Poly(trimethylene carbonate), PTMC and poly(caprolactone), PCL are well-studied polycarbonate and polyester electrolytes, respectively.32–35 Of particular note are random copolymers of TMC and CL, P(CL-r-TMC) first reported by Mindemark et al. whereby a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 ratio of TMC:CL (20 mol% TMC) delivers optimal ionic conductivity (4.1 × 10−5 S cm−1 at 25 °C) due to disruption of PCL crystallinity.36–40 Specifically for their role as solid-state composite cathode binders, these Li-ion conductors are revered for their high oxidative stability (>4.5–5 V), placing them in the right range for use with NMC.41–43 Additionally, ester/carbonate linkages are known to enable recyclability to monomers which can be initiated by the addition of catalysts, heat and/or manipulation of reaction conditions.44–47
:4 ratio of TMC:CL (20 mol% TMC) delivers optimal ionic conductivity (4.1 × 10−5 S cm−1 at 25 °C) due to disruption of PCL crystallinity.36–40 Specifically for their role as solid-state composite cathode binders, these Li-ion conductors are revered for their high oxidative stability (>4.5–5 V), placing them in the right range for use with NMC.41–43 Additionally, ester/carbonate linkages are known to enable recyclability to monomers which can be initiated by the addition of catalysts, heat and/or manipulation of reaction conditions.44–47
On its own, P(CL-r-TMC) is a soft (low Tg) amorphous polymer. To modify the mechanical properties for optimal binder performance including accommodating volume changes caused by internal stresses at higher voltages, hard (high Tg) polymer blocks need to be introduced.21,48–51 Preparing phase-separated AB- or ABA-type block polymers (where A = hard and B = soft polymer) is a popular strategy where predictable microstructures correlate with particular mechanical behaviours.52 This study uses poly(4-vinyl cyclohexene carbonate) (PC) A-blocks as mechanical modifiers: whilst being a hard block, it is also oxygenated so may contribute towards Li-ion conductivity and its synthesis can sequester CO2.
Results and discussion
To prepare poly(carbonate-block-ester) binders featuring an ionically conducting P(CL-r-TMC) (B-block) combined with a PC mechanical modifier (A-block), a form of switchable polymerisation catalysis was selected (Fig. 1a).53–57 From a monomer mixture, this process applies a single catalyst (Scheme S1†) to direct polymerisations between lactone (CL)/cyclic carbonate (TMC) ring-opening polymerisation (ROP) and epoxide (vCHO)/CO2 ring-opening copolymerisation (ROCOP) by the presence/absence of CO2 (Scheme S2†).53,55,58 These switchable polymerisations are well-controlled, so the catalysis can be manipulated to systematically vary the polymer composition (A:B ratio), molar mass (Mn) and architecture.55 As such, this approach is attractive for designing new polymer binders as these parameters are expected to influence successful binder function.
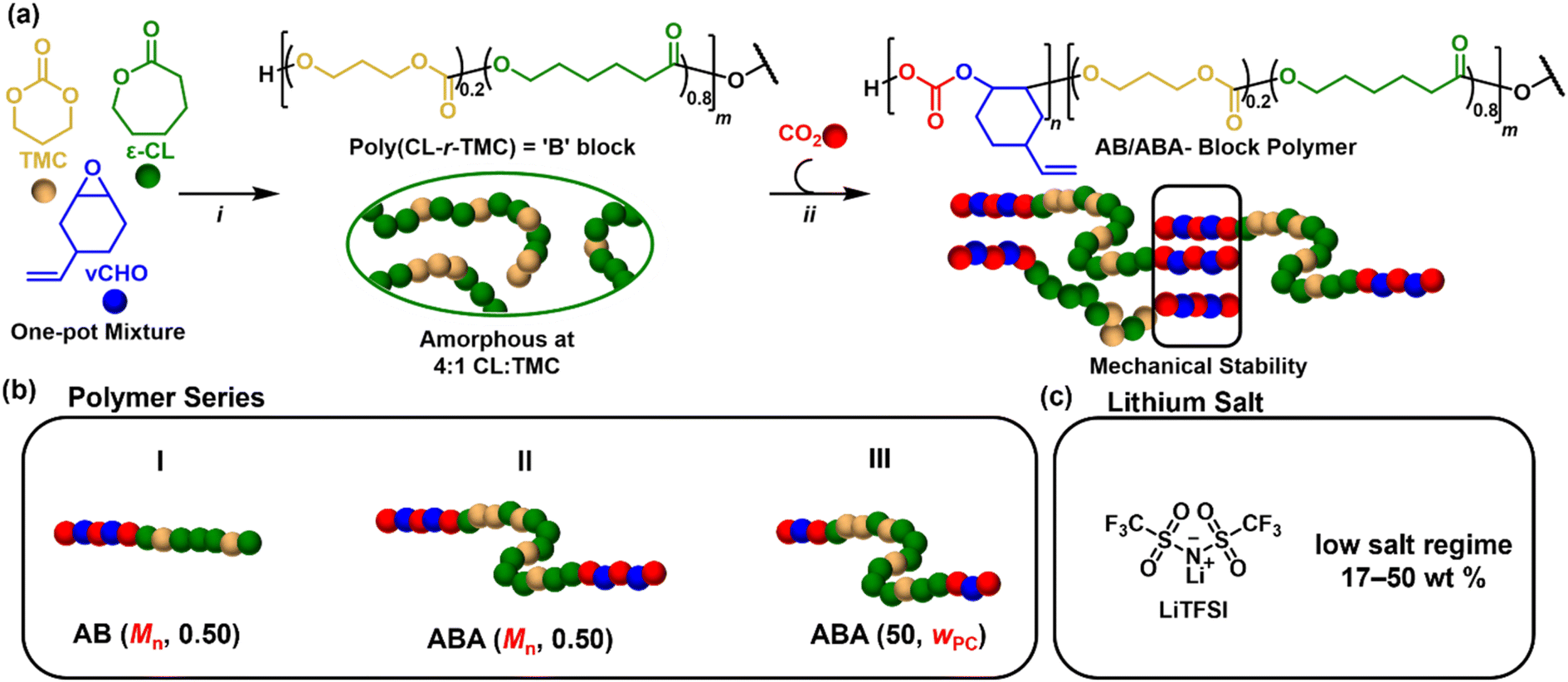 | ||
| Fig. 1 (a) Reaction scheme for switchable ROP/ROCOP polymerisation catalysis: (i) CL/TMC ROP at 100 °C for 1 h, [TMC + CL]0 = 2 M in toluene. For ABA polymers, initiator = BDM, for AB polymers, initiator = Me-BnOH. (ii) vCHO/CO2 ROCOP at 100 °C, 1 bar CO2, 16–48 h. See Table S1† for m and n values. (b) Synthesised polymer series, where Mn or the polycarbonate weight fraction (wPC) is systematically varied. (c) Electrolytes were prepared by mixing the polymer with LiTFSI salt. | ||
To control the architecture, two different alcohol initiators were used; these react rapidly in situ with the catalyst (Scheme S1†).
When applied with bifunctional alcohol, 1,4-benzenedimethanol (BDM), ABA triblock polymers are accessed. Whereas monofunctional alcohol, 4-methyl benzyl alcohol (Me-BnOH) yields AB polymers (Schemes S3 and S4†).54,59 To control the Mn and A:B ratio, the ratio of [CL + TMC]0 was adjusted with respect to the initiator, and the monomer conversions were varied (Table S1†). Accordingly, three systematic series of polymers were synthesised (Fig. 1b): Series I are AB (diblock) polymers with fixed A-block content expressed as a weight fraction, wPC = 0.5, but variable overall molar masses, Mn (35–66 kg mol−1); Series II are ABA (triblock) polymers with similarly fixed wPC = 0.5 and variable molar masses, Mn (26–69 kg mol−1); Series III feature ABA polymers with fixed molar mass (50 kg mol−1) but variable A-block content (0.26–0.62) (Table 1). Henceforth, the polymers are labelled as ABA/AB (Mn, wPC).
| Seriesa | Polymerb | M n (kg mol−1) | w PC | T g,B, Tg,Ad (°C) |
|---|---|---|---|---|
| a See Fig. 1 for Series I–III descriptors. b Polymers labelled as ABA/AB (Mn, wPC) where Mn = overall molar mass in kg mol−1 and wPC = weight fraction of PC. c Determined by 1H NMR integration of the purified polymer: initiator, PC (5.76 ppm) and P(CL-r-TMC) (2.00, 1.38 ppm) vs. 1,4-BDM (7.34 ppm) or Me-BnOH (7.17 ppm). d T g with 17 wt% LiTFSI from DSC, where Tg,B corresponds to P(TMC-r-CL) and Tg,A to PC. | ||||
| I | AB (26, 0.45) | 28 | 0.45 | −52, 96 |
| I | AB (32, 0.51) | 32 | 0.51 | −37, 84 |
| I | AB (45, 0.47) | 45 | 0.47 | −19, 102 |
| I | AB (69, 0.58) | 69 | 0.58 | 0, 94 |
| II | ABA (35, 0.52) | 35 | 0.52 | −49, 101 |
| II | ABA (44, 0.53) | 44 | 0.53 | −48, 102 |
| II | ABA (50, 0.47) | 50 | 0.47 | −48, 100 |
| II | ABA (66, 0.52) | 66 | 0.52 | −32, 100 |
| III | ABA (51, 0.26) | 52 | 0.26 | −38, 98 |
| III | ABA (47, 0.30) | 47 | 0.30 | −33, 66 |
| III | ABA (50, 0.35) | 50 | 0.35 | −40, 100 |
| III | ABA (50, 0.47) | 50 | 0.47 | −48, 100 |
| III | ABA (50, 0.62) | 50 | 0.62 | −32, 85 |
In all cases, the targeted 1:4 TMC:CL ratio of the B-block (set by the monomer feed) was verified by reaction aliquots taken just before CO2-triggered switching to vCHO/CO2 ROCOP (Fig. S1†). As mentioned earlier, this ratio is favourable for suppressing PCL crystallinity which is important to maximise conductivity. The polymers were readily isolated in good yield (>75%) by precipitation from methanol and both the purity and compositions were confirmed by NMR spectroscopy (Fig. S2–S5†); 13C{1H} NMR spectroscopy verified the random configuration of the TMC/CL mid-blocks for all samples. The overall block architecture was demonstrated by chain end-group titration and diffusion-ordered spectroscopy (DOSY) (Fig. S6 and S7†). Although polymer molar masses were calculated using 1H NMR spectroscopy (Mn,NMR) and size-exclusion chromatography, SEC (Mn,SEC), values discussed refer to Mn,NMR, which were comparable to those predicted (Mn,calc, Table S1†), indicating good polymerisation control (Fig. S8†).
Next, the copolymers were rendered Li-ion conductive by mixing (in the first instance) with LiTFSI salt at 17 wt% vs. overall polymer mass (Fig. 1c and S9†).49 Transparent films (∼250 μm thick) were prepared through a solution casting technique using THF (see ESI for details†). Prior to conductivity measurements, differential scanning calorimetry (DSC) of the dry solid polymer electrolytes confirmed, in all cases, that they were fully amorphous and A:B phase-separated. A Tg well below room temperature was consistent with a soft random PTMC/CL B-block phase and a second Tg close to 100 °C was associated with the hard PC A-block phase (Table 1). Phase separation of the A and B blocks is essential to deliver the targeted elastomeric mechanical properties. A low Tg for the primary conducting phase improves ionic conductivity, which increases with greater segmental motion.60 Thermal stability is also notable for processability and battery safety. Thermogravimetric analysis (TGA) reassured that Td,5% was sufficiently above 200 °C for all polymer compositions, leaving a wide window for processing before degradation (Table S2 and Fig. S10†).
Ionic conductivities were measured by electrochemical impedance spectroscopy (EIS) (Table S3, Fig. S11 and S12†). Comparing polymers in Series I (AB) and II (ABA) with wPC ∼ 0.5 allowed an understanding of the optimum structure and Mn (Fig. 2a). The block structure had a marked impact, with the overall trends being quite different for the AB vs. ABA type polymers. For AB polymers in Series I, the ionic conductivity decreased with increasing Mn (Fig. 2a), whereas the opposite trend was observed for the ABA Series II. For the AB polymers, the decreasing conductivity as molar mass increased is linked to soft B-block Tg,B which increases from −52 °C for AB (26, 0.45) to 0 °C for AB (69, 0.58). A good fit of the temperature dependence of the ionic conductivity to the Vogel–Fulcher–Tammann (VFT) model supports ionic conductivity being assisted by polymer chain segmental motion, which is of course, related to Tg (Fig. S13†). For the ABA polymers in Series II, the reverse relationship was observed: conductivity increased with Mn despite constant or increased Tg of the soft segment. This difference is attributed to slower chain mobility due to the pinning at both ends by the rigid PC blocks.61 This restricted segmental motion at the PC/P(TMC-r-CL) interface is expected to be less influential at higher Mn where the fraction of these interfaces is lower.62
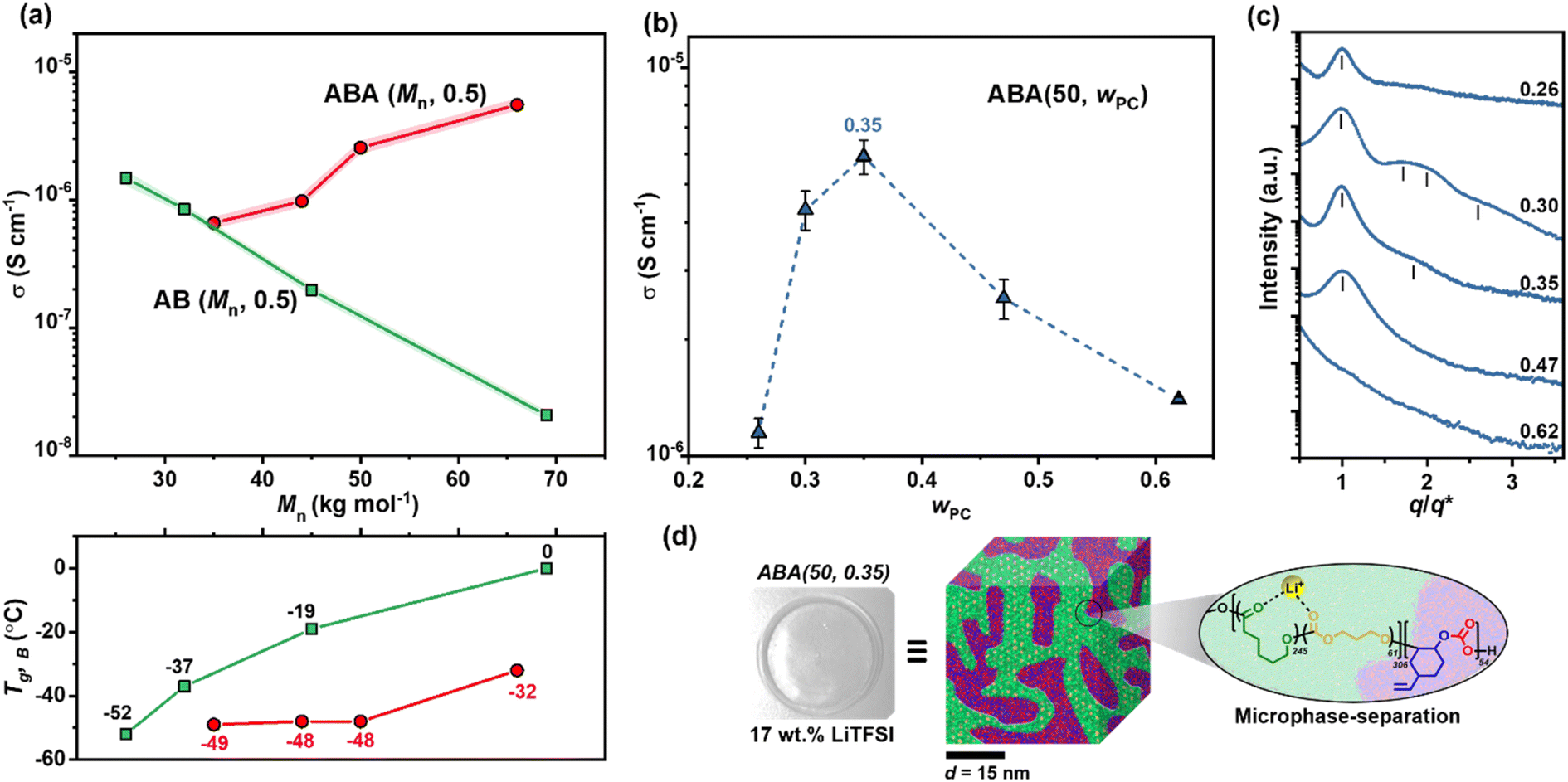 | ||
| Fig. 2 (a) (Top) Mn-dependence of ionic conductivity (σ) at T = 30 °C for AB vs. ABA polymers/17 wt% LiTFSI at fixed wPC ∼ 0.5. (Bottom) Corresponding DSC Tg values for P(TMC-r-CL) B-block. (b) Ionic conductivity with variable A-content (wPC) for ABA polymers with constant Mn = 50 kg mol−1. (c) SAXS data for as-prepared ABA (50, wPC)/17 wt% LiTFSI polymer systems. (d) ABA (50, 0.35) electrolyte film and schematic illustration of phase-separation behaviour. Nanostructure domain spacing, d is estimated from the principal scattering peak (q*) in the SAXS pattern (d = 2π/q*). For all conductivity measurements, shading or error bars represent standard error for N = 3 repeats. | ||
Since the ABA polymers show the most promising conductivity values, the influence of A-block content for these polymers was then investigated (Series III). Keeping the overall molar mass constant at 50 kg mol−1 and changing only the A-block content, higher conductivity was achieved for wPC 0.3–0.35 (Fig. 2b). It is proposed that at these compositions the block polymers form a common microstructure which maximises ion transport. Small-angle X-ray scattering (SAXS) measurements confirmed this hypothesis since ABA (50, 0.30) and ABA (50, 0.35) formed weakly-ordered microstructures; either body-centred cubic or hexagonal (Fig. 2c and S14†). It is feasible that these microstructures provide channels that accelerate ion transport (Fig. 2d). This is consistent with work conducted by Balsara and co-workers which showed that for PS-b-PEO electrolytes, weakly ordered morphologies improved conductivity.61
From this systematic investigation of block polymer parameters, the optimal ionic conductivity was identified for ABA (50, 0.35) of 2.2 × 10−4 S cm−1 at 60 °C (Fig. 3a). Subsequently, this polymer formed the focus of the forthcoming investigations. From VFT analysis, the barrier for ion transport was favourably low, with an activation energy of 17.4 (±0.7) kJ mol−1 (Fig. S13†). We attribute this low barrier to both blocks being able to coordinate Li-ions, the occurrence of which is consistent with the presence of two resonances in the solid-state 7Li NMR spectroscopy (deconvolution given in Fig. 3b). By comparing against spectra collected for the constituent homopolymers, these resonances are assigned to the PC and P(CL-r-TMC) domains (with shifts of 2.7 and 0.9 ppm respectively) (Fig. S15†).
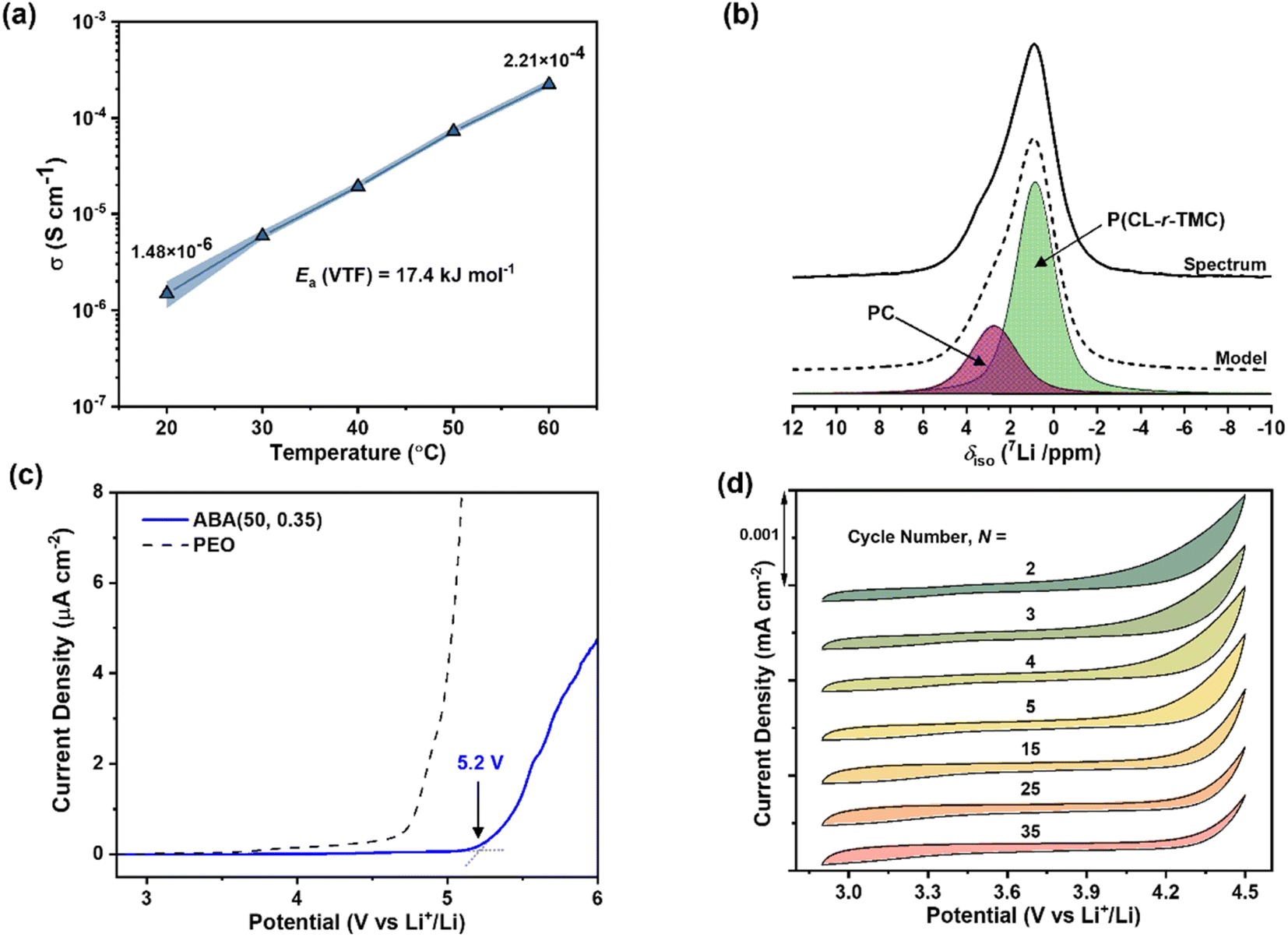 | ||
| Fig. 3 (a) Temperature-dependence of ionic conductivity for ABA (50, 0.35)/17 wt% LiTFSI. (b) Solid-state 7Li NMR spectrum with peak deconvolution to PC and P(CL-r-TMC) phases. (c) Oxidative stability vs. Li metal and stainless-steel counter electrode measured by LSV at 60 °C, 0.1 mV s−1 scan rate. (d) CV at 0.5 mV s−1 scan rate recorded over 35 cycles. | ||
To assess the Li-ion coordination strength of the domains and infer the fraction of Li-ions contributing to the total ionic conductivity (the lithium transference number, tLi+) pulsed-field gradient (PFG) 7Li and 19F NMR spectroscopy was used (see ESI for details†). Assuming fully dissociated LiTFSI salt (likely at the low salt content), the measured self-diffusion coefficients for Li- and F-containing species correlate to Li+ and TFSI−. For species in the P(CL-r-TMC)-rich domain, tLi+ = 0.44 while for the PC-rich domain, tLi+ = 0.52 (Fig. S16 and Table S4†). An overall tLi+ of 0.47 accounts for the weighted average of the two constituent tLi+. This is 1.5 times greater than the equivalent ABA polymer with PC A-blocks and PEO B-blocks measured using the same technique.21 The increase is consistent with carbonyl Li+ coordination being weaker than the complexation by the ether oxygen in PEO.40,63,64
Moreover, the polyester/carbonate-based electrolytes given here have superior oxidative stability. This is crucial for enabling the use of the highest-performance cathodes (such as Ni- and Mn-rich oxides) resulting in higher-capacity batteries.65 Linear sweep voltammetry (LSV) revealed that ABA (50, 0.35) was stable up to 5.2 V (vs. Li metal), even at 60 °C and a slow scan rate of 0.1 mV s−1 (Fig. 3c). To test the performance further, cyclic voltammetry (CV) experiments conducted from 3 to 4.5 V, at a scan rate of 0.5 mV s−1, revealed that ABA (50, 0.35) showed consistently stable behaviour over at least 35 cycles (Fig. 3d). Good chemical stability of the polyester/carbonate against Li6PS5Cl was also confirmed by EIS measurements over multiple periods (Fig. S17†).
Though encouraging, this oxidative and chemical stability of ABA (50, 0.35) is not in itself a guarantee of beneficial binder function when integrated into the more complex multicomponent cathode composite. As the cathode particles also experience mechanical stress and strain during charge/discharge, the mechanical properties of polymer binders are also crucial. In particular, an adaptive and elastic nature should aid any void filling between components and sustain contact during cycling. Based on the A:B block ratio and Mn of ABA (50, 0.35) it would be expected to behave as a thermoplastic elastomer. Indeed, the stress–strain behaviour of ABA (50, 0.35)/LiTFSI supported the formation of a stretchable material that can repeatably recover after stress is applied and removed (Fig. S18†). In comparison, PTFE samples, prepared equivalently, showed very low elastic recovery and were not elastomeric (Fig. S19†). Peel force testing, on alumina, using ABA (50, 0.35)/LiTFSI also showed promising adhesive properties with a peel force of 0.45 N mm−1 (Fig. S20†). The adhesive properties are comparable to a previously reported series of polycarbonate-b-ether-b-carbonate binders for solid state batteries.56
Before cell fabrication, the viscoelastic properties of ABA (50, 0.35)/LiTFSI were also probed by temperature and frequency sweep rheological experiments. The elastic properties, expressed as the shear storage modulus (G′), and the viscosity, related to the loss modulus (G′′), were observed to crossover (G′ = G′′) at 73 °C (Fig. 4a). Below this temperature, the Li-ion conducting binder behaves as an elastic solid with a low value of G′ (2–0.2 MPa), resulting in a soft, more resilient polymer filler. Above this temperature and at sufficiently slow shear rates (<0.2 Hz (Fig. 4b and S21†)), the binder becomes processable. This is a very accessible processing window compared to for example PTFE binders, which have very high melting temperatures.66
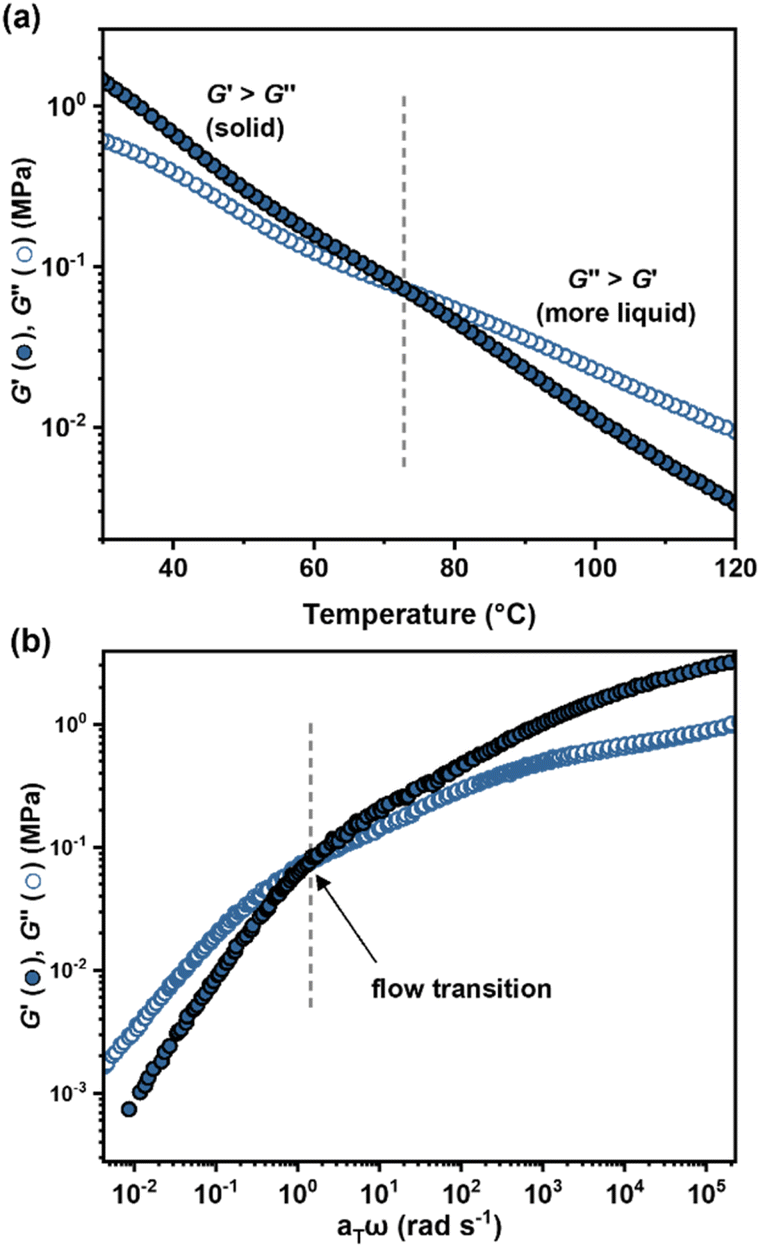 | ||
| Fig. 4 Viscoelastic mechanical properties for ABA (50, 0.35)/LiTFSI. (a) Temperature-dependence of G′ and G′′. (b) Master curve of the frequency dependence of G′ and G′′. | ||
Subsequently, the ABA (50, 0.35)/LiTFSI polymer electrolyte film could be cryo ball milled to produce a fine powder and dry-mixed into a free-standing composite cathode. The polymer was integrated in 5 wt% with high-performance cathode material sc-NMC811, ceramic electrolyte Li6PS5Cl and carbon nanofibre, CNF, in a 70:23:2 wt% ratio (Fig. 5a). Evidence of sufficient mixing of the cathode components was evaluated by SEM/XPS (Fig. S22 and S23†). The resulting cathode composite was then densified at 250 MPa, before being assembled into a full solid-state cell with a Li4Ti5O12 (LTO) anode and Li6PS5Cl solid electrolyte. LTO was chosen due to its superior power density to graphite and its chemical and electrochemical stability with Li6PS5Cl.67
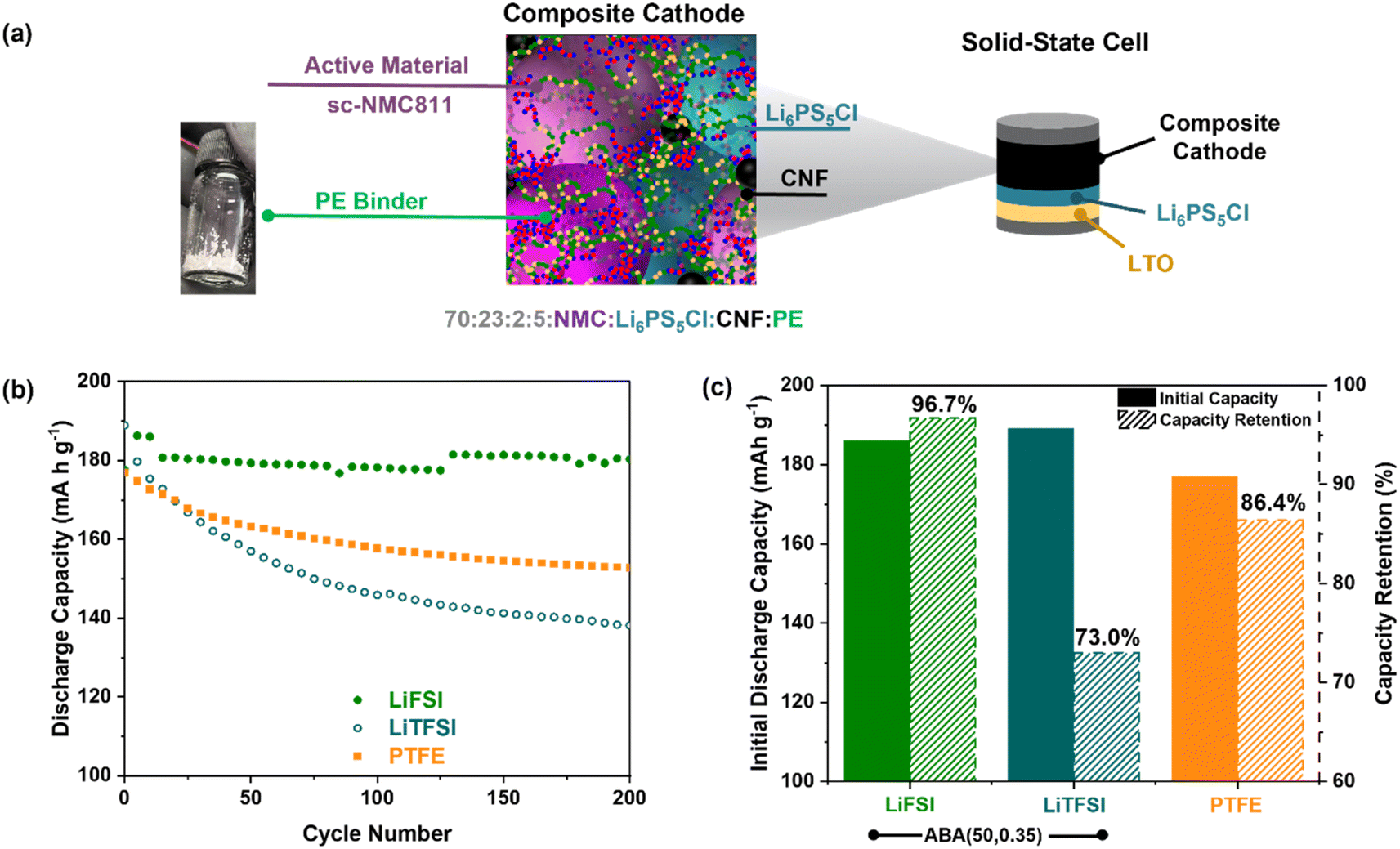 | ||
| Fig. 5 (a) Fabrication of composite cathode and solid-state cell configuration, LTO|Li6PS5Cl|NMC–PE composite. (b) Specific discharge capacity vs. cycle number at T = 60 °C, 1 MPa stack pressure, 1 mA cm−2. (c) Initial discharge capacity, and capacity retention after 200 cycles (labelled). | ||
To evaluate the binder performance, the cell was then cycled at 60 °C using a low 1 MPa applied pressure (Fig. 5b, c and S24,† areal capacity = 3 mA h cm−2). A high initial discharge capacity of 186 mA h g−1 was observed compared to 177 mA h g−1 with analogous cells prepared with PTFE binder. As in previous studies, we investigated the new binder with both FSI and TFSI anions. Whereas the capacity with the TFSI anion declined with the number of charge/discharge cycles (73% retention after 200 cycles), this was not observed with FSI which resulted in 96.7% after 200 cycles (Fig. 5b, c and S25†). This could be attributed to the TFSI anion forming unstable interphases with the cathode.68,69 Further detailed investigations of this inorganic chemistry will form the focus of subsequent work. Regardless, this high cycling stability with LiFSI whilst still delivering a good initial capacity (183 mA h g−1) is promising. In particular, it outperforms those obtained with PTFE binder, which retains 86.4% capacity over 200 cycles starting at 177 mA h g−1 (Fig. 5b and c). This can be attributed to the ionic conductivity and mechanical properties of the ABA (50, 0.35) electrolyte; PTFE is neither conductive nor elastomeric.17 Furthermore, the composite cathode also showed a superior coulombic efficiency of >99.7% (Fig. S26†). When cycled at 30 °C, the cell had a lower initial discharge capacity (111 mA h g−1); this could be optimised as part of further work (Fig. S27†).
Lastly, given the promising cell performances, proof-of-principle chemical recycling of the polymer electrolyte binder to component monomers was explored.45,47 The polymer was extracted from the composite with toluene (Fig. 6a). Although this is also achievable with fluorinated binders, it typically requires harsher solvents, namely NMP. Furthermore, these polar solvents will likely result in breakdown of the Li6PS5Cl ceramic electrolyte. Next, catalysed depolymerisation of a pure sample of ABA (50, 0.35) was demonstrated and at 200 °C allowed for 90% recovery of monomers after 16 h (Fig. 6b, S26, Scheme S5, see ESI for optimised depolymerisation conditions†); such depolymerisation is not possible with PTFE. The depolymerisation resulted in 90% conversion to small molecules, which were identified using spectroscopy. These molecules were the recovered monomers vCHO, TMC and CL, and a cyclic carbonate which is also a potential monomer for polycarbonates. These monomers were formed in the expected ratios based on the polymer composition (Fig. 6c and Table S5†). The 10% residual mass is attributed to a small amount of crosslinked polymer, likely formed at higher temperatures via reactions between the vinyl groups (N.B. experiments were conducted using neat polymer). Also observed by FTIR and NMR analyses was trans-cyclic carbonate which can be polymerised to the analogous polycarbonate as that produced from vCHO/CO2 ROCOP (Scheme S5†).70 Further separation of the monomers is unnecessary as the switch catalysis used to make these polymers works from a mixture of monomers, with the introduction of CO2 triggering the formation of the A-block polymers.
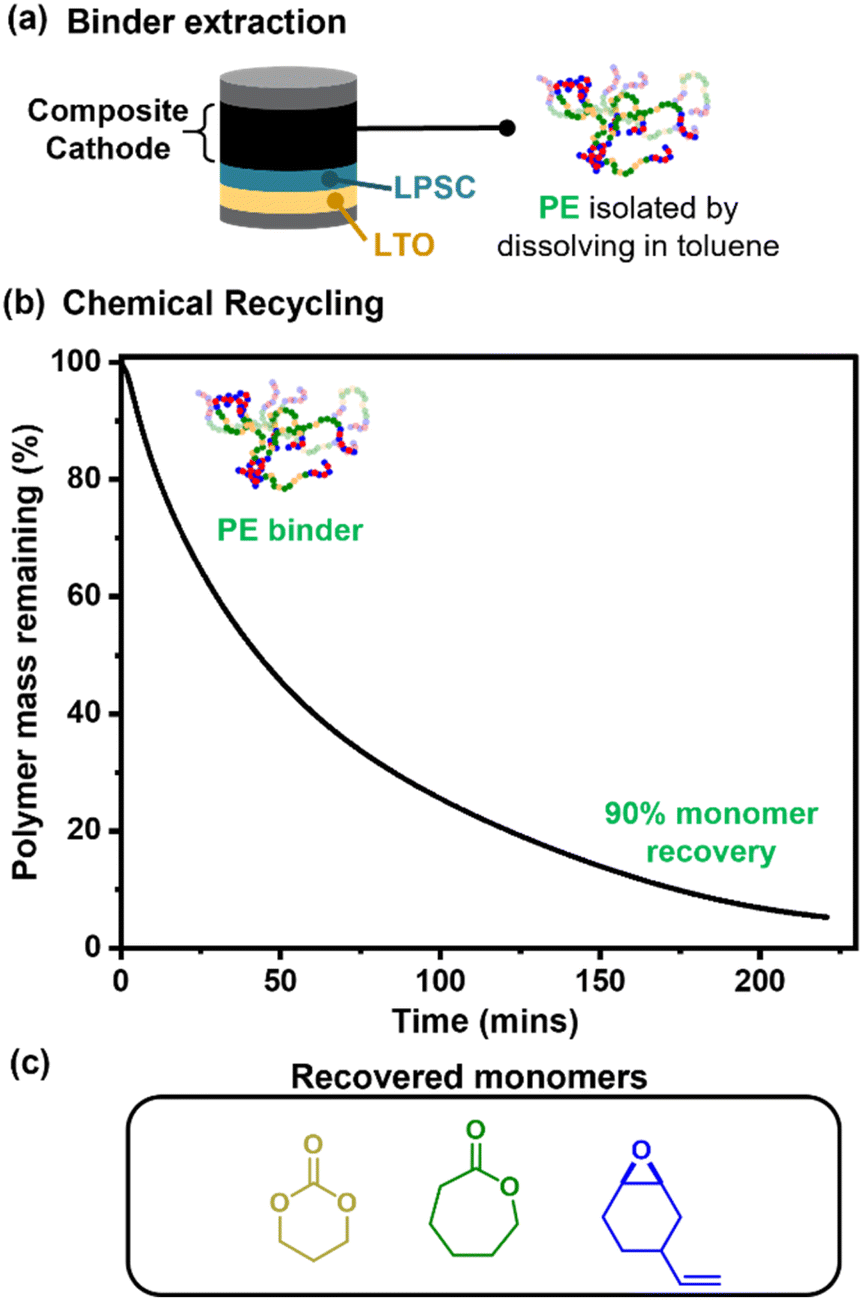 | ||
| Fig. 6 Chemical recycling of the polymer electrolyte binder, catalysed by Zn(Oct)2/GEO ([Zn(Oct)2]0:[GEO]0:[polymer]0 = 1:1.13:1000) at 200 °C. Monomers were recovered by distillation under 1 mbar pressure, with the mixture heated to 200 °C. (a) Schematic depicting how the polymer binder can be recovered from the cell. (b) Polymer degradation vs. time, throughout the chemical recycling process. (c) The monomers recovered in the chemical recycling process. | ||
Conclusions
A series of ABA- and AB-block polymers, where A = poly(vinyl cyclohexene carbonate) (PC), and B = poly(ε-caprolactone-r-trimethylene carbonate) (P(CL-r-TMC)) were tested as polymer electrolyte binders for solid composite cathodes. The polymers were prepared using a one-pot, one-catalyst switchable polymerisation allowing for control over the molar mass, composition and end-group chemistry. This allowed systematic investigations of the polymer structure (AB vs. ABA), molar mass (26 < Mn < 69 kg mol−1) and PC-block content (0.26 < A-block content < 0.62). The best electrolyte, comprising ABA-polymer and lithium salt, showed high oxidative stability allowing for use with high-capacity cathodes (5.2 V), good Li-ion conductivity (2.2 × 10−4 S cm−1 at 60 °C) and viscoelastic behaviour. As such, the polymer was a compatible binder with a leading cathode (sc-NMC811) and an inorganic solid electrolyte (Li6PS5Cl). The resulting full solid-state cell capacities were high, measuring 186 mA h g−1, and showed impressive capacity retention of 96.7% over 200 cycles. These results outperformed the use of PTFE, which is the leading binder for dry-processed solid-state composites, resulting in capacities of 177 mA h g−1 and 86.4% capacity retention over 200 cycles. Moreover, chemical recycling of the polyester/carbonate binder allowed for 90% monomer recovery, establishing a route to cell component recycling. Overall, these polymers are an alternative to currently relied-upon fluorinated polymers, which have growing associated environmental concerns.Finally, establishing commercial solid-state batteries demands improved performance of the cathode. Switchable polymerisation is an excellent tool for the ongoing optimisation of composite electrodes, via polymeric binder design. There is a wealth of commercial monomers available which apply to this approach and thus are capable of leading to diverse, heteroatom-rich, therefore Li-ion conductive, polymeric binders.
Author contributions
HY, HG, KY, TM and GJR: investigation, methodology, validation, formal analysis, data curation. GLG, MP, PGB and CKW: conceptualization, supervision, funding acquisition, writing.Conflicts of interest
CKW is a director of Econic Technologies.Acknowledgements
The Engineering and Physical Sciences Research Council (CW: EP/S018603/1; EP/R027129/1; EP/V003321/1; PB: EP/P003532/1), The Faraday Institute (FIRG056, SOLBAT as well as FIRG007, FIRG008, LiSTAR FIRG058) and The Oxford Martin School (Future of Plastics) are acknowledged for funding. The Diamond Light Source for Rapid Access DL-SAXS (P38) measurements through proposal SM29810-1. The Henry Royce Institute for Advanced Materials [EP/R00661X/1, EP/S019367/1, EP/R010145/1]. The Royal Thai Government DPST scholarship (K. Y.).References
- J. Janek and W. G. Zeier, Nat. Energy, 2023, 8, 230–240 CrossRef.
- S. Randau, D. A. Weber, O. Kötz, R. Koerver, P. Braun, A. Weber, E. Ivers-Tiffée, T. Adermann, J. Kulisch, W. G. Zeier, F. H. Richter and J. Janek, Nat. Energy, 2020, 5, 259–270 CrossRef CAS.
- Q. Zhao, S. Stalin, C.-Z. Zhao and L. A. Archer, Nat. Rev. Mater., 2020, 5, 229–252 CrossRef CAS.
- T. Famprikis, P. Canepa, J. A. Dawson, M. S. Islam and C. Masquelier, Nat. Mater., 2019, 18, 1278–1291 CrossRef CAS PubMed.
- K. J. Kim, M. Balaish, M. Wadaguchi, L. Kong and J. L. M. Rupp, Adv. Energy Mater., 2021, 11, 2002689 CrossRef CAS.
- D. H. S. Tan, A. Banerjee, Z. Chen and Y. S. Meng, Nat. Nanotechnol., 2020, 15, 170–180 CrossRef CAS PubMed.
- X. Gao, B. Liu, B. Hu, Z. Ning, D. S. Jolly, S. Zhang, J. Perera, J. Bu, J. Liu, C. Doerrer, E. Darnbrough, D. Armstrong, P. S. Grant and P. G. Bruce, Joule, 2022, 6, 636–646 CrossRef CAS.
- J. Zhang, Z. Chen, Q. Ai, T. Terlier, F. Hao, Y. Liang, H. Guo, J. Lou and Y. Yao, Joule, 2021, 5, 1845–1859 CrossRef CAS.
- Z. Zeng, J. Cheng, Y. Li, H. Zhang, D. Li, H. Liu, F. Ji, Q. Sun and L. Ci, Mater. Today Phys., 2023, 32, 101009 CrossRef CAS.
- S. Y. Kim, H. Cha, R. Kostecki and G. Chen, ACS Energy Lett., 2023, 8, 521–528 CrossRef CAS.
- C. Doerrer, I. Capone, S. Narayanan, J. Liu, C. R. M. Grovenor, M. Pasta and P. S. Grant, ACS Appl. Mater. Interfaces, 2021, 13, 37809–37815 CrossRef CAS PubMed.
- X. Fan, G. Hu, B. Zhang, X. Ou, J. Zhang, W. Zhao, H. Jia, L. Zou, P. Li and Y. Yang, Nano Energy, 2020, 70, 104450 CrossRef CAS.
- G. Qian, Y. Zhang, L. Li, R. Zhang, J. Xu, Z. Cheng, S. Xie, H. Wang, Q. Rao, Y. He, Y. Shen, L. Chen, M. Tang and Z.-F. Ma, Energy Storage Mater., 2020, 27, 140–149 CrossRef.
- S. Sen, E. Trevisanello, E. Niemöller, B.-X. Shi, F. J. Simon and F. H. Richter, J. Mater. Chem. A, 2021, 9, 18701–18732 RSC.
- H. Chen, M. Ling, L. Hencz, H. Y. Ling, G. Li, Z. Lin, G. Liu and S. Zhang, Chem. Rev., 2018, 118, 8936–8982 CrossRef CAS PubMed.
- Y. Shi, X. Zhou and G. Yu, Acc. Chem. Res., 2017, 50, 2642–2652 CrossRef CAS PubMed.
- F. Zou and A. Manthiram, Adv. Energy Mater., 2020, 10, 2002508 CrossRef CAS.
- D. Mecerreyes, L. Porcarelli and N. Casado, Macromol. Chem. Phys., 2020, 221, 1900490 CrossRef CAS.
- Y. Ma, J. Ma and G. Cui, Energy Storage Mater., 2019, 20, 146–175 CrossRef.
- R. Schlem, C. F. Burmeister, P. Michalowski, S. Ohno, G. F. Dewald, A. Kwade and W. G. Zeier, Adv. Energy Mater., 2021, 11, 2101022 CrossRef CAS.
- G. L. Gregory, H. Gao, B. Liu, X. Gao, G. J. Rees, M. Pasta, P. G. Bruce and C. K. Williams, J. Am. Chem. Soc., 2022, 144, 17477–17486 CrossRef CAS PubMed.
- L. Rao, X. Jiao, C.-Y. Yu, A. Schmidt, C. O'Meara, J. Seidt, J. R. Sayre, Y. M. Khalifa and J.-H. Kim, ACS Appl. Mater. Interfaces, 2022, 14, 861–872 CrossRef CAS PubMed.
- K. Lee, J. Lee, S. Choi, K. Char and J. W. Choi, ACS Energy Lett., 2019, 4, 94–101 CrossRef CAS.
- J. Lee, K. Lee, T. Lee, H. Kim, K. Kim, W. Cho, A. Coskun, K. Char and J. W. Choi, Adv. Mater., 2020, 32, 2001702 CrossRef CAS PubMed.
- Y. Lu, C.-Z. Zhao, H. Yuan, J.-K. Hu, J.-Q. Huang and Q. Zhang, Matter, 2022, 5, 876–898 CrossRef CAS.
- W. Dou, M. Zheng, W. Zhang, T. Liu, F. Wang, G. Wan, Y. Liu and X. Tao, Adv. Funct. Mater., 2023, 2305161 CrossRef CAS.
- A. Rensmo, E. K. Savvidou, I. T. Cousins, X. Hu, S. Schellenberger and J. P. Benskin, Environ. Sci.: Processes Impacts, 2023, 25, 1015–1030 RSC.
- X. Lim, Nature, 2023, 620, 24–27 CrossRef CAS PubMed.
- A. Bielefeld, D. A. Weber and J. Janek, ACS Appl. Mater. Interfaces, 2020, 12, 12821–12833 CrossRef CAS PubMed.
- S.-B. Hong, Y.-J. Lee, U.-H. Kim, C. Bak, Y. M. Lee, W. Cho, H. J. Hah, Y.-K. Sun and D.-W. Kim, ACS Energy Lett., 2022, 7, 1092–1100 CrossRef CAS.
- N.-Y. Kim, J. Moon, M.-H. Ryou, S.-H. Kim, J.-H. Kim, J.-M. Kim, J. Bang and S.-Y. Lee, Adv. Energy Mater., 2022, 12, 2102109 CrossRef CAS.
- B. Sun, J. Mindemark, K. Edström and D. Brandell, Solid State Ionics, 2014, 262, 738–742 CrossRef CAS.
- D. Zhang, L. Zhang, K. Yang, H. Wang, C. Yu, D. Xu, B. Xu and L.-M. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 36886–36896 CrossRef CAS PubMed.
- C. P. Fonseca, D. S. Rosa, F. Gaboardi and S. Neves, J. Power Sources, 2006, 155, 381–384 CrossRef CAS.
- C. Polo Fonseca and S. Neves, J. Power Sources, 2006, 159, 712–716 CrossRef.
- J. Mindemark, B. Sun, E. Törmä and D. Brandell, J. Power Sources, 2015, 298, 166–170 CrossRef CAS.
- C. Sångeland, R. Younesi, J. Mindemark and D. Brandell, Energy Storage Mater., 2019, 19, 31–38 CrossRef.
- J. Mindemark, E. Törmä, B. Sun and D. Brandell, Polymer, 2015, 63, 91–98 CrossRef CAS.
- F. P. Nkosi, M. Valvo, J. Mindemark, N. A. Dzulkurnain, G. Hernández, A. Mahun, S. Abbrent, J. Brus, L. Kobera and K. Edström, ACS Appl. Energy Mater., 2021, 4, 2531–2542 CrossRef CAS.
- T. Eriksson, A. Mace, J. Mindemark and D. Brandell, Phys. Chem. Chem. Phys., 2021, 23, 25550–25557 RSC.
- H. Xu, J. Xie, Z. Liu, J. Wang and Y. Deng, MRS Energy Sustain., 2020, 7, E2 CrossRef.
- J. Zhang, J. Yang, T. Dong, M. Zhang, J. Chai, S. Dong, T. Wu, X. Zhou and G. Cui, Small, 2018, 14, 1800821 CrossRef PubMed.
- J. Mindemark, M. J. Lacey, T. Bowden and D. Brandell, Prog. Polym. Sci., 2018, 81, 114–143 CrossRef CAS.
- C. Li, L. Wang, Q. Yan, F. Liu, Y. Shen and Z. Li, Angew. Chem., Int. Ed., 2022, 61, e202201407 CrossRef CAS PubMed.
- T. M. McGuire, A. C. Deacy, A. Buchard and C. K. Williams, J. Am. Chem. Soc., 2022, 144, 18444–18449 CrossRef CAS PubMed.
- C. F. Gallin, W.-W. Lee and J. A. Byers, Angew. Chem., Int. Ed., 2023, 62, e202303762 CrossRef CAS PubMed.
- T. M. McGuire, A. Buchard and C. Williams, J. Am. Chem. Soc., 2023, 145, 19840–19848 CrossRef CAS PubMed.
- M. J. Lee, J. Han, K. Lee, Y. J. Lee, B. G. Kim, K.-N. Jung, B. J. Kim and S. W. Lee, Nature, 2022, 601, 217–222 CrossRef CAS PubMed.
- A. Bergfelt, G. Hernández, R. Mogensen, M. J. Lacey, J. Mindemark, D. Brandell and T. M. Bowden, ACS Appl. Polym. Mater., 2020, 2, 939–948 CrossRef CAS.
- A. Bergfelt, M. J. Lacey, J. Hedman, C. Sångeland, D. Brandell and T. Bowden, RSC Adv., 2018, 8, 16716–16725 RSC.
- R. Andersson, G. Hernández, J. See, T. D. Flaim, D. Brandell and J. Mindemark, ACS Appl. Energy Mater., 2022, 5, 407–418 CrossRef CAS.
- F. S. Bates, M. A. Hillmyer, T. P. Lodge, C. M. Bates, K. T. Delaney and G. H. Fredrickson, Science, 2012, 336, 434 CrossRef CAS PubMed.
- C. Romain and C. K. Williams, Angew. Chem., Int. Ed., 2014, 53, 1607–1610 CrossRef CAS PubMed.
- G. S. Sulley, G. L. Gregory, T. T. D. Chen, L. Peña Carrodeguas, G. Trott, A. Santmarti, K.-Y. Lee, N. J. Terrill and C. K. Williams, J. Am. Chem. Soc., 2020, 142, 4367–4378 CrossRef CAS PubMed.
- A. C. Deacy, G. L. Gregory, G. S. Sulley, T. T. D. Chen and C. K. Williams, J. Am. Chem. Soc., 2021, 143, 10021–10040 CrossRef CAS PubMed.
- G. L. Gregory, G. S. Sulley, J. Kimpel, M. Łagodzińska, L. Häfele, L. P. Carrodeguas and C. K. Williams, Angew. Chem., Int. Ed., 2022, 61, e202210748 CrossRef CAS PubMed.
- K. C. Poon, G. L. Gregory, G. S. Sulley, F. Vidal and C. K. Williams, Adv. Mater., 2023, 35, 2302825 CrossRef CAS.
- C. Romain, Y. Zhu, P. Dingwall, S. Paul, H. S. Rzepa, A. Buchard and C. K. Williams, J. Am. Chem. Soc., 2016, 138, 4120–4131 CrossRef CAS PubMed.
- G. L. Gregory, G. S. Sulley, L. P. Carrodeguas, T. T. D. Chen, A. Santmarti, N. J. Terrill, K.-Y. Lee and C. K. Williams, Chem. Sci., 2020, 11, 4367–4378 RSC.
- J. Lopez, D. G. Mackanic, Y. Cui and Z. Bao, Nat. Rev. Mater., 2019, 4, 312–330 CrossRef CAS.
- K. Timachova, H. Watanabe and N. P. Balsara, Macromolecules, 2015, 48, 7882–7888 CrossRef CAS.
- R. Bouchet, T. N. T. Phan, E. Beaudoin, D. Devaux, P. Davidson, D. Bertin and R. Denoyel, Macromolecules, 2014, 47, 2659–2665 CrossRef CAS.
- T. Eriksson, H. Gudla, Y. Manabe, T. Yoneda, D. Friesen, C. Zhang, Y. Inokuma, D. Brandell and J. Mindemark, Macromolecules, 2022, 55, 10940–10949 CrossRef CAS PubMed.
- M. P. Rosenwinkel, R. Andersson, J. Mindemark and M. Schönhoff, J. Phys. Chem. C, 2020, 124, 23588–23596 CrossRef CAS.
- P. K. Nayak, E. M. Erickson, F. Schipper, T. R. Penki, N. Munichandraiah, P. Adelhelm, H. Sclar, F. Amalraj, B. Markovsky and D. Aurbach, Adv. Energy Mater., 2018, 8, 1702397 CrossRef.
- T. Tomkovic and S. G. Hatzikiriakos, Can. J. Chem. Eng., 2020, 98, 1852–1865 CrossRef CAS.
- H. Zhang, Y. Yang, H. Xu, L. Wang, X. Lu and X. He, InfoMat, 2022, 4, e12228 CrossRef CAS.
- E. R. Logan, A. Eldesoky, E. Eastwood, H. Hebecker, C. P. Aiken, M. Metzger and J. R. Dahn, J. Electrochem. Soc., 2022, 169, 040560 CrossRef CAS.
- I. L. Johansson, C. Sångeland, T. Uemiya, F. Iwasaki, M. Yoshizawa-Fujita, D. Brandell and J. Mindemark, ACS Appl. Energy Mater., 2022, 5, 10002–10012 CrossRef CAS PubMed.
- A. K. Diallo, E. Kirillov, M. Slawinski, J.-M. Brusson, S. M. Guillaume and J.-F. Carpentier, Polym. Chem., 2015, 6, 1961–1971 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05105f |
| This journal is © The Royal Society of Chemistry 2024 |