
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A systematic study on the processes of lignin extraction and nanodispersion to control properties and functionality†
Ekaterina
Sheridan
a,
Svitlana
Filonenko
 *a,
Alexander
Volikov
a,
Juho Antti
Sirviö
b and
Markus
Antonietti
*a
*a,
Alexander
Volikov
a,
Juho Antti
Sirviö
b and
Markus
Antonietti
*a
aMax Planck Institut für Kolloid- und Grenzflächenforschung Am Mühlenberg 1, 14476 Potsdam, Germany. E-mail: svitlana.filonenko@mpikg.mpg.de; office.cc@mpikg.mpg.de
bFibre and Particle Engineering Research Unit, University of Oulu, P.O. Box 4300, 90014 Oulu, Finland
First published on 17th January 2024
Abstract
This article reviews a variety of literature-reported lignin isolation procedures that bring partly minor but important changes in the produced extracts. Five lignins were produced or acquired from various sources and characterized based on their thermal properties, molecular weights, chemical structures, as well as elemental compositions, which were all found to depend on their extraction methods. These extracts were then used to produce 5 different nanolignins using 2 similar facile solvent shifting methods with the difference being an alteration in pH. The nanolignins were characterized after the solvent shifting accounting for differences in pH. Nanolignins generated via only solvent exchange exhibited diameters ranging from 58 nm to 95 nm, while nanolignins precipitated using a solvent shifting method along with a shift in pH exhibited diameters ranging from 10 nm to 34 nm and dissimilar morphology, size, and surface charge. Those differences highlight the control options for nanolignin preparetion on the colloidal length scale. Understanding how different lignins can be used to generate nanolignins is mandatory for unlocking the possibilities of nanolignin for functional material applications. These nanolignins can be applied in construction, medical, and/or agricultural industries as renewable, biodegradable alternatives to existing polymer dispersions.
1. Introduction
Wood is a polymer composite material made up of hemicellulose, lignin, and cellulose microfibrils interconnected via covalent and hydrogen bonding.1,3 Lignin is the second most abundant hydrocarbon biopolymer in nature, the second most readily available biomass on our planet, and the most abundant source of phenolic and aromatic compounds.4,5 It is used by plants to provide plasticity to their cell wall structures and to regulate water and solute transport through their vasculature system.5 Lignin functions in plants also include anti-bacterial and anti-phytopathogenic activities along with inhibition of enzymatic degradation.6 Because lignin makes up a large percentage of lignocellulosic biomass (10–30%), a sizeable amount of it is harvested via agricultural processes and the practice of foresting, and it is even separated in the paper manufacturing process.4 It is estimated that annually, approximately 60–120 million tons of lignin is isolated worldwide.7 The vast majority of it is treated as a low-value product and burned for energy, while only about 2% of lignin is used for high-value applications such as chemical precursors, fillers, antioxidants, adsorbents, emulsifiers, adhesives, and dispersants.4,8–11Depending on the species of lignocellulose, lignin is a complex 3D-branched phenolic polymer made up of p-hydroxyphenyl (H), guaiacyl (G), and syringyl (S) moieties linked together via aromatic and aliphatic ether bonds as well as non-aromatic C–C linkages.4,12 Lignin contains a variety of functional groups, including methoxyl, carboxyl, aliphatic hydroxyl, and phenolic hydroxyl groups.3 Partially owing to its vast array of functional groups accessible for surface modification, lignin has found use as an alternative to fossil-fuel-based chemicals, as a composite dispersant, as a drug carrier, as a UV-blocker, as an antibacterial coating, and so on.5,9 Technical lignin is classified based on the modification-extraction process and has at least 5 major types: lignosulfonate, organosolv, Kraft, soda, and enzymatic hydrolysis lignin.9,13 Lignin can possess molecular masses in the range from 1000 to 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, and it is a highly branched heteropolymer with no clearly defined chemical structure.14 In lignin, chains composed of phenyl propane units are cross-linked to form an amorphous 3D sub-phase glued to cellulose fibrils via hemicellulose. The problem with lignin, which is also a chemical advantage, is its diversity; lignin exhibits variations in its composition and properties, greatly depending on the type of plant from which it was attained, its age, conditions that the plant experienced during growth, and the manner in which it was extracted from the plant.15 While these natural variations in lignin are more difficult to control, the chemistry of the isolation process provides a tool for the modification of the properties of the isolated polymer.
000 g mol−1, and it is a highly branched heteropolymer with no clearly defined chemical structure.14 In lignin, chains composed of phenyl propane units are cross-linked to form an amorphous 3D sub-phase glued to cellulose fibrils via hemicellulose. The problem with lignin, which is also a chemical advantage, is its diversity; lignin exhibits variations in its composition and properties, greatly depending on the type of plant from which it was attained, its age, conditions that the plant experienced during growth, and the manner in which it was extracted from the plant.15 While these natural variations in lignin are more difficult to control, the chemistry of the isolation process provides a tool for the modification of the properties of the isolated polymer.
The large majority of commercially produced lignin is Kraft lignin, generated through a cooking process of lignocellulosic biomass at approximately 165–175 °C for 1–2 hours in the presence of sodium sulphite and sodium hydroxide.11,13 Due to the relatively harsh nature of this procedure in terms of temperature, pH alteration, and pressure, the involved changes in the natural structure during extraction are substantial.11 It is not the most attractive choice for a circular bio-economy because it contains sulfur and tends to be insoluble in water and most organic solvents, making it difficult to work with or to combine with other materials.11 On the other side of the spectrum, the extraction of organosolv lignin only involves solubilization in mixtures of organic solvents and water at comparably mild temperatures (80 °C, and up to 190 °C), and is thereby assumed to be close to its native structure, while it does not contain sulfur or other unwanted reaction aids.6,11 In comparison to Kraft lignin, it exhibits a lower ash-content, lower molecular weight, and higher purity.14 While Kraft lignin has molecular weights of 1000–15000 Da, a raised glass transition temperature, and low malleability and solubility, organosolv lignin is soluble in a wide range of organic solvents, and typically yields lower molecular weights of 500–5000 Da.14 Organosolv lignin is quite attractive for the production of biomaterials due to favorable properties such as its purity, solubility, chemical functionality, and biodegradability.4,11 It contains a relatively high content of reactive functional groups, like hydroxyl and carboxyl groups, which are favorable for the alteration and production of new biomaterials with specific properties and functionalities.4,10 The addition of an acid catalyst to this process would cleave more ether linkages but produce lignin with a more complex structure due to some intramolecular condensation reactions.1
Lignin's availability, biodegradability and biocompatibility have made it a material of high interest in the realm of biomaterials, cosmetics, and nanomaterials.2,10 The complicated structure of lignin, poor processability, limited solubility, and brittleness have all been issues when industrializing or homogenizing lignin.5 The issue of limited solubility and, thereby, the reactivity of lignin is addressed in a variety of fashions. According to Österberg et al. and Chen et al., there have been attempts at fine-tuning lignin's solubility and reactivity including sulfonation, carboxymethylation, demethylation, phenolation, and hydroxymethylation.10,16 Another method of overcoming these technical barriers without changing solubility is moving to two-phase systems or polymer dispersions by generating lignin nanoparticles, also called nano-lignin or LNPs. LNPs have emerged as a simple way to apply lignin from an aqueous dispersion onto a material matrix while using the expanded surface-to-volume ratio to tune the properties and take advantage of the quantum size effect of nanomaterials.8,17 According to Raman et al., several methods have been researched to produce lignin nanoparticles such as water-in-oil micro-emulsion methods, sonication, CO2 saturation, solvent exchange, flash-precipitation, as well as non-solvent precipitation methods.9 LNPs tend to have a significant specific surface area, high dispersibility, and negative surface charge. Since lignin nanoparticles are readily dispersed in water, attractive blending properties within a host matrix are opened, as well as structural and morphological control of the lignin.14 Lignin, lignin nanoparticles, and their nanocomposites have, therefore, gained massive scientific and economic interest because they are a model case for the change in polymer chemistry towards sustainability and a circular economy, and could replace some synthetic nanoparticles used in the polymer industry.10 Raman et al. stated that LNPs have found use as adsorbents for the removal of heavy metal ions and dyes, anticorrosive nanofillers, drug delivery agents, and UV blocking agents.9
Nanolignin is, however, not to be misunderstood as a specific compound; its properties vary greatly depending on the lignin source, processing, and even the person producing it. It is, therefore, the purpose of this article to discern some of the “hidden parameters” involved in the isolation and generation of nanolignin, the control of which would allow reproducible manufacturing. Understanding how the differences in extracted lignins may be used to reproducibly generate nanolignin dispersion is one key to more advanced nanolignin applications in functional materials.
The method of solvent shifting is simple and effective for the production of nanolignin since the liquid intermediate states drive the formation of spherical particles when introduced into a non-solvent phase.10 This method takes advantage of the hydrophobic and hydrophilic moieties of the lignin, as well as the lignin's reduced solubility in water or an anti-solvent, to self-assemble the spheres.10 The aggregation and dispersion behaviour of the LNPs should be considered and tuned by the use of pH, additives, concentrations, and solvents.10
Several reactions may occur during lignin isolation that can lead to modifications that produce functional groups such as carbonyl groups, catechols, epoxides, acidic phenolic hydroxyl groups, quinones, as well as carboxylic acid groups, giving the extracted lignin higher reactivity as well as higher availability to chemical modifications and reactions. Such reactions may be oxidative cleavage, demethoxylation, hydrolysis, oxidative degradation, and/or alkaline treatment. These functional groups also make the pH an essential aspect of nanolignin production. The effect of the pH of the non-solvent was tested by Leskinen et al. and shown to have a profound effect on nanolignin size, with the size of the particles having a nearly linear relationship with pH, decreasing with increasing pH.18 Deprotonation of the carboxylic acid groups is likely responsible for these phenomena, increasing the surface charge and electrostatic stabilization between particles.18 Zeta potential and the dispersity of the particle population were also greatly affected by the pH of the (aqueous) antisolvent being used.19
At low pH, the lignin's functional groups, such as carboxylic acids (although not a part of the literature structure), quinone and hydroxyl groups are protonated, leading to less electrostatic repulsion and final colloidal stability. As the pH is increased, deprotonation of the functional groups occurs, changing the molecular conformations, weakening aggregation, and increasing solubility. Lignin, however, is likely to undergo chemical modifications throughout its extraction under either very acidic or alkaline conditions.12,20 In hot alkaline conditions, lignin may undergo alkaline degradation of the chain, leading to the cleavage of chemical bonds, resulting in changes in chemical structure and molecular weight.12 Highly acidic conditions, on the other hand, can promote ether hydrolysis as well as phenol dealkylation.12
Another aspect that must be considered when producing nanolignin is the concentration of the solvent solution before solvent shifting/precipitation in anti-solvent is performed. It has been shown that if the concentration is too high, aggregation of particles or interlinked particle networks may occur.18 Molecular weight has been found to influence the formation of nanolignin as well as the glass transition temperature of the lignin itself. It has been proposed by Sipponen et al. that the molecules with the larger molecular weight act as nuclei for the growing nanoparticles, while the smallest lignin molecules finish the precipitation process, leaving the particle surfaces enriched with hydrophilic moieties such as carboxylic acid groups.21 Some small, water-soluble components even stay dissolved in the water after particle formation.18 There have been efforts to increase the molecular weight of lignin by using grafting or cross-linking.10
Despite some research being done on the influence of the lignin solvent used and conditions of the solvent shift, the preparation methods of nanolignin still lack full linkage between the lignin chemistry and the possibility of controlling the particle size, especially to achieve small-sized nanoparticles that offer homogeneity, morphology, and surface functionalities that would make them viable for reproducible industrial use as fillers, adhesives, or additives. In this work, we investigate several types of lignin with tuned chemistry and molecular weight, using different isolation methods, and follow the influence of the molecular weight and functional groups on the size and charge of the LNP and the possibility of tuning the NP size by changing the ionization state of the lignin functionality via pH adjustment.
2. Experimental section
2.1 Materials
The lignins acquired from outside sources, which were used in the experiments, were BioPiva 100 Kraft lignin (UPM, Finland) and Organosolv lignin (Fraunhofer Gmbh, Germany). Beechwood sawdust was acquired from Goldspan GmbH (Goldenstedt, Germany). Spruce sawdust was obtained in the never-dried form (around 50% dry matter content) from Keitele Forests Oy. All chemicals were bought from Sigma-Aldrich and were used as received.2.2 Lignin isolation procedures
:4 v/v ethanol and DI water (Evoqua GmbH, Barsbuttel, Germany) mixture, respectively. The autoclave was heated at a rate of 7 °C min−1, stirred at a rate of 200 rpm at 190 °C for 1 hour, and then shut off and cooled by quenching with room temperature water. The resulting mix of sawdust and solvent was vacuum filtered through a paper filter and the solids were washed with 200 mL DI water. A further 200 mL of DI water was added to the product, and the mixture was allowed to precipitate in an ice bath for 1 hour. The resulting mix was centrifuged at 12298 G for 20 minutes using a Universal 320 centrifuge (Hettlich, Tuttlingen, Germany). The resulting solid was dried overnight in a vacuum oven (VTR 5036, Heraeus Instruments, Hanau, Germany) until all solvent was removed. The resulting powder was ground with mortar and pestle, resulting in a homogenous dark brown powder. This sample is referred to as Lab-Org.
:2 ethanol:water mixture, 4 mL of 37% HCl solution along with a magnetic stir bar. A reflux condenser was placed onto the flask and the mixture was rapidly stirred in an oil bath at 90 °C for 5 hours. The mixture was allowed to cool to room temperature and filtered. The residue was washed 4 times with 25 mL ethanol. The liquor was concentrated using rotary evaporation. The solid was dissolved in acetone and an ultrasonic bath was used to ensure solvation. The lignin was precipitated by the addition of 600 mL water. The lignin was filtered, washed with 100 mL, and air-dried overnight.
:10) were weighed into a 250 mL beaker and placed in an oil bath at temperature 80 °C. When clear liquid was formed, 10 g of never-dried sawdust was added. The mixture was stirred for 60 min followed by the addition of around 100 ml of ethanol to quench the reaction. The ethanolic mixture was filtered and washed with 50 ml ethanol. The resulting mixture was then dispersed in a beaker in 200 ml ethanol for 15 min, followed by filtration and washing with an additional 50 ml of ethanol. MSA in the ethanolic mixture was neutralized by adding 3.86 g of NaOH dissolved in 20 ml of water. Then, 1000 ml of water was added to the ethanolic mixture to precipitate lignin. The resulting solid was then filtered on the polyvinylidene fluoride (PVDF) membrane (pore size 0.65 μm) and washed with 500 ml of water. During the first filtration, some portion of lignin appeared as a dark liquid due to the formation of a deep eutectic supramolecular polymer with phenol. If some of the liquid was passed through the membrane, it was collected after discarding the washing liquid. After first washing with water, the lignin was purified by dissolution and precipitation: lignin on the top of the membrane was dissolved in ethanol and allowed to drop into a large excess of water (1000 ml), followed by filtration and washing with 500 ml of water. Dissolution and precipitation were conducted three times. In the end, a pinkish lignin powder was collected and dried under vacuum at room temperature.
2.3 Preparation of lignin nanoparticles
Adapted from Henn et al.7 In a closed flask, a mixture of 30.7 wt% ethanol, 34.6 wt% analytical-grade acetone, and 30.0 wt% deionized water was prepared, and 4.7 wt% lignin was added. The pH of the starting solution was between 3 and 4. pH adjustment was done by adding the appropriate amount of 0.01 M NaOH dropwise while stirring. The mixture was swirled for three hours before being vacuum-filtered using 589/3-grade ashless filter papers. The precipitation of water-dispersible nanoparticles was then started by quickly adding the solution to 1.72 times its mass of rapidly stirred deionized water by using a glass pipette. The acetone and ethanol were extracted from the dispersion by rotary evaporation at 40 °C and 30 mbar after 15 minutes of stirring. Centrifugation was used to concentrate the solvent-free colloidal nanolignin dispersion. Then, 45–50 mL of the dispersion was centrifuged in 50 mL Eppendorf tubes for 30 min at 10500 rpm using a Universal 320 tabletop centrifuge (Hettlich, Tuttlingen, Germany) and the supernatant was removed. The particles were redispersed by vortexing using a VWR digital vortex mixer. The nanolignins produced in this manner were labelled “NAME.pH”, where NAME is a sample code described above. The preliminary procedure of solvent shifting is shown in Fig. 1.
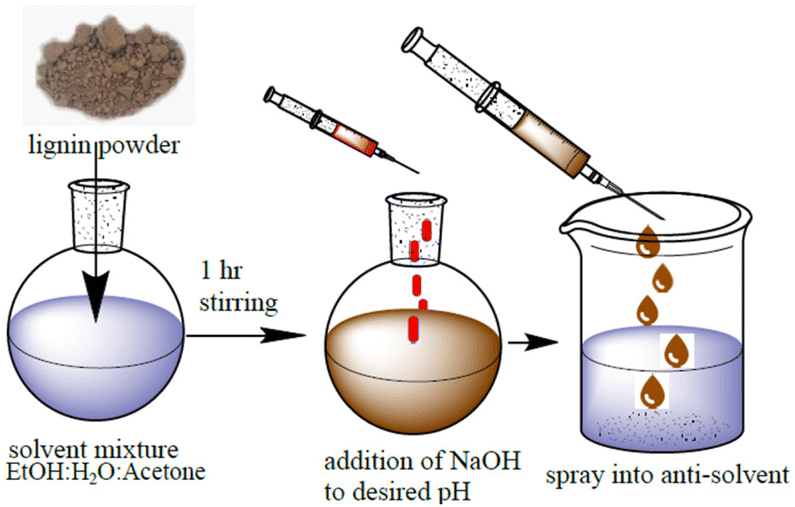 | ||
| Fig. 1 Production of nanolignin through solvent shifting. | ||
2.4 Characterizations
SEM images were recorded on the Gemini 1550, Zeiss AG, (Oberkochen, Germany), at an accelerating voltage of 3.00 kV. The samples were placed onto an aluminum stage covered by a layer of carbon, and no additional alteration.
| % KL = ((Dried solid)/(Original Lignin weight)) × 100. |
:1.6 deuterated chloroform and pyridine mixture was used to produce an internal standard solution using 2-chloro-4,4,5,5-tetramethyl-1,3-2-dioxaphospholane (TMDP) at a concentration of 5 mg mL−1 and N-hydroxy-5-norbornene-2,3-dicarboximide (NHND) at a concentration of 18 mg mL−1. TMDP was used for the phosphitylation of hydroxyl groups and NHND was used as an internal standard for the evaluation of the spectra. Here, 0.1 mL of the internal standard solution was added to 30 mg lignin and 0.5 mL of the chloroform/pyridine solution. This solution was stirred until the lignin was dissolved completely, and then NMR spectra were obtained by using a 31P decoupling pulse process with a spectral width of 100 ppm, acquisition time of 0.8 s, relaxation delay of 10 s, 120 scans, and the center of the spectrum, at 140 ppm.
3. Results and discussion
For the present comparative study, we selected 5 different lignin extracts to precipitate nanolignin in an already optimized solvent-shifting process, and the influence of lignin properties on particle nucleation, growth, and stability is analysed. Some lignins were freshly isolated from raw biomass in our laboratory, while others were obtained from trusted external sources (UPM and Fraunhofer). For simplicity, they will be referred to as Lab-Org, Fraun-Org, UPM-Kraft, Acid-Org and Phenol-AH. The method of converting these lignins into nanolignins using non-toxic, recyclable solvents was adapted from Henn et al.7For the experiments in this report, various lignins were picked and produced to show an array of lignins that may be used for the production of nanolignin. Organosolv lignin, one produced by adapting the procedure of Margellou et al.,4 and one obtained from a technical batch provided by the Fraunhofer Society, were used for the experiments. In the present context we, however, also underline that the pH of the material is near neutral, it contains metal counterions, and is negatively charged, which again limit solubility in pure organic media.
Additionally, a flash-organosolv process was used to produce lignins in which the natural buffer equilibria were kept as stable as possible, while oxidative post-stresses were minimized. Adding acids throughout the extraction process gives the so-called Phenol-AH lignin. Here, most phenols are in their acidic form, and the typical brownish color of phenolates is missing, leaving the lignin with a light, whitish color. A variation to stronger, oxidizing HCl-treatment results in the so-called Acid-Org lignin, with a light pink color, which was adapted from the procedure of Zijlstra et al.20 Acid-catalyzed depolymerization is known to be relatively mild and spares the natural ether linkages from cleavage, allowing for the extraction of more native lignin.20 In this method, the monolignols are prone to partially demethylating to the corresponding biphenols, as discussed below in the results section.
UPM-Kraft lignin was used to represent the vast majority (90%) of technical lignin isolated in industry and could give a good idea of how lignin could realistically be employed for industrial purposes.10,25
3.1 Analysis of the structure and composition of lignin
The 2D HSQC NMR spectra were separated into two spectral regions according to Mattsson et al.,27 namely the aromatic region (δC/δH 100.0–130/6.0–7.4 ppm), and the inter-unit aliphatic lignin region/oxygenated aliphatic region (δC/δH 50–90.0/3–5.7 ppm). The resulting spectra for all lignins are shown in Fig. 2.
 | ||
| Fig. 2 2D NMR spectra of the various lignins; inter-unit aliphatic lignin region/oxygenated aliphatic region (left) and aromatic region (right). | ||
The inter-unit aliphatic lignin/oxygenated aliphatic region is where the carbohydrate sugars, as well as carbons in proximity to ether, alcohol, and aryl methoxy groups, resonate.27
For a more quantitative calculation, 31P NMR was also performed to back up these findings. The NMR spectra nicely reflect the structural changes lignin may experience depending on its extraction process. The most abundant lignin bonds are ether-type linkages (β-O-4), making up approximately 50% of all linkages, while C–C (β–β, β-5) bonds appear much less.4 Through harsh processing, natural lignin will lose many of its ether-type linkages due to a variety of degradation reactions.11,20,25
Fig. 3 shows the main linkages and components that were identified using 2D NMR. Table 1 summarizes the calculated integrated intensities of the major lignin components according to Zijlstra et al.20 Not surprisingly, the data reflect our assumption that the UPM-Kraft lignin underwent an intense extraction method, experienced partial cleavage of β-O-4 bonds, and therefore contains the fewest β-O-4 units.20,25 It also appears to have a high G unit content, which could indicate a lower reactivity than lignins with a high percentage of S units, which contain more methoxy functionalities.12,28 Acid hydrolysis was performed in order to extract Phenol-AH lignin, and it seems from the high purity content that the process allowed for the efficient separation of lignin from cellulose without extensive degradation of the lignin.22 The methanesulfuric acid, or MSA, disrupts the aromatic unit network of lignin and cleaves ether linkages, resulting in an altered lignin structure. The exact chemical structure of Phenol-AH lignin is not yet known and is currently under more thorough investigation. Nevertheless, in nature, the softwood lignin has been identified to contain mainly coniferyl (G) units, and as was shown in HSQC and 13P analysis, Phenol-AH lignin contains a large quantity of p-hydroxyphenyl (H) units. Therefore, it can be assumed that phenolation takes place during the fractionation process (in a similar manner to that reported for the phenolation of isolated lignin described in the literature, as seen in the work of Jiang et al.).29 Since Phenol-AH and Fraun-Org have a relatively small amount of β-O-4 units, it may be postulated that the Phenol-AH process, as well as the Fraunhofer Organosolv process, were somewhat harsher or more degrading than expected.20
 | ||
| Fig. 3 Common linkages and structures in lignin as identified by 2D NMR. Adapted from Zijlstra.20 | ||
| Lignin samples | S (%) | G (%) | H (%) | β-O-4 | β–β | β-5 |
|---|---|---|---|---|---|---|
| /100 Ar | /100 Ar | /100 Ar | ||||
| Lab-Org | 76.1 | 23.2 | 0.7 | 34.0 | 6.8 | 3.0 |
| Fraun-Org | 75.9 | 24.1 | 0.1 | 11.3 | 1.7 | 0.2 |
| UPM-Kraft | 36.7 | 58.6 | 4.8 | 7.2 | 3.4 | 0.7 |
| Acid-Org | 69.2 | 29.8 | 1.0 | 57.0 | 15.3 | 3.1 |
| Phenol-AH | 7.4 | 39.3 | 53.3 | 12.0 | 0.2 | 0.2 |
β–β and β-5 linkages seen in Fig. 3 are also known to decrease in number as the extraction method becomes harsher, which agrees well with the results, supporting that Lab-Org and Acid-Org had the milder extractions, while UPM-Kraft underwent the most notable degradation during its extraction.20 The Scondensed signal also indicates in these samples a significant decrease in β-O-4 linkages and degradation of the native structure.20 The lignins that exhibited a noticeable signal in this area were Fraun-Org and UPM-Kraft, coinciding perfectly with the significant lack of β-O-4 linkages in these lignins. Acid-Org was a relatively mild method of lignin extraction, which can be seen by the retention of the mentioned linkages. The Gcondensed signal overlaps with G5, meaning that full condensation of G units will have likely occurred if very little or no G2 or G5 signals are present.20 Phenol-AH has a noticeably high amount of H units within its structure, supported by further testing via31P NMR, as shown in Table 2.
| Aliphatic OH (mmol g−1) | C5 substituted OH (mmol g−1) | Syringyl (S) (mmol g−1) | Guaiacyl OH (G) (mmol g−1) | p-Hydroxyphenyl OH (H) (mmol g−1) | Carboxylic acid OH (mmol g−1) | |
|---|---|---|---|---|---|---|
| Lab-Org | 4.6 | 1.5 | 1.1 | 0.9 | 0.1 | 0.3 |
| Fraun-Org | 1.4 | 2.6 | 1.6 | 0.7 | 0.2 | 0.1 |
| UPM-Kraft | 2.1 | 1.1 | 0.5 | 2.4 | 0.4 | 0.6 |
| Acid-Org | 3.9 | 0.9 | 0.6 | 0.8 | 0.2 | 0.1 |
| Phenol-AH | 1.8 | 0.4 | 0.1 | 1.2 | 3.3 | 0.1 |
These ratios may influence the types of application for each lignin. The noticeable presence of G-type units within UPM-Kraft lignin, as well as its high phenolic content, may indicate that it would contain many reactive potential active sites for further functionalization, cross-linking, or polymerization.12,30 Lignins with a higher S-type unit content, such as Lab-Org, Fraun-Org, and Acid-Org may exhibit better reducing power, resulting in greater antioxidant properties.12
The primary mechanism of lignin extraction, even a milder extraction process such as the organosolv process, is the cleavage of Cβ–O bonds from β-O-4′ structures and the cleavage of Cα–O bonds from α-O-4′ structures.26 The aryl–aryl–ether bonds of lignin are shown to be labile under the given extraction conditions, and hydrolysis creates two open phenols from one C–O–C. Electron-rich phenols such as those from monolignoles, are strong antioxidants and care should be taken in the lignin isolation protocol to avoid the presence of dioxygen and other oxidants. In the other case, the hydrolysate can transfer one electron to form a semichinone, which then forms a new linkage with a second semichinone radical.20 The C–C formation of two phenolic carbon radicals is unwanted because it does not occur in natural lignin formation and is hardly reversible. This side reaction is also expected to increase glass transition and limit the repeated recycling of lignin products. The spectra reflect the dominance of the syringyl (S) units within the lignins extracted from beechwood, a hardwood.4 It is not exactly known to us from which biomasses Fraun-Org and UPM-Kraft are extracted, but the ratios would suggest that Fraunhofer is also extracted from hardwood, while it is known whether UPM Kraft is obtained from softwood, also confirmed by the fact that the HSQC spectra are richer in signals of guaiacyl (G) units and p-hydroxyphenyl (H).31 These particular units contain free C3 and/or C5 positions, allowing for rapid conversion, cross-linking, and functionalization.31 Lignins obtained from softwood have often been the focus of lignin studies because these more reactive sites for functionalization or synthesis are available.28 The large amount of phenolic hydroxyl groups within lignin makes it a possible replacement for phenol-formaldehyde resins, a toxic compound often employed in construction and wood-based materials.30,32
Quantitative 31P NMR spectroscopy was performed for the analysis of hydroxyl groups by using the procedure of Meng et al.24 This method is based on the interaction of the 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane with the lignin as well as the internal standard (N-hydroxy-5-norbornene-2,3-dicarboximide). The spectra are shown in Fig. 4, and the calculated values of OH group contents are given in Table 2.
 | ||
| Fig. 4 31P NMR spectra with outlined regions; signal interpretation is adapted from Meng et al.24 | ||
It was found that the results aligned quite well with the results of 2D-NMR, reflecting the high ratio of S units in Lab-Org, S units in Fraun-Org, a high proportion of G units within UPM-Kraft, a high amount of S and G units in Acid-Org, and a high proportion of H units in Phenol-AH. This information obtained through 31P NMR can also give a hint about the reactivity of the lignins. p-Hydroxyphenyl, guaiacyl, and syringyl phenolic –OH groups have 0, 1 and 2 adjacent methoxy groups, respectively.3,30 Their reactivity follows in the same order, with p-hydroxyphenyl –OH groups being the most reactive, and syringyl –OH groups being the least, due to steric hindrance.3 If only considering these S/G/H contributions, this would indicate that Phenol-AH lignin should have the highest reactivity, while Fraun-Org lignin should have the least; however, the other OH-groups will greatly affect reactivity, as well as pH. It is considered that the next most reactive OH groups to reactions such as oxidation, condensation, and esterification would be aliphatic, followed by carboxylic acid OH groups.33 If considering the number of aliphatic OH groups, Lab-Org and Acid-Org have the most notable amounts, and if considering carboxylic OH groups, UPM-Kraft outnumbers the rest. It is good to understand the contributions of various OH groups, however, it is difficult to draw immediate conclusions about their reactivity.
12,34,35
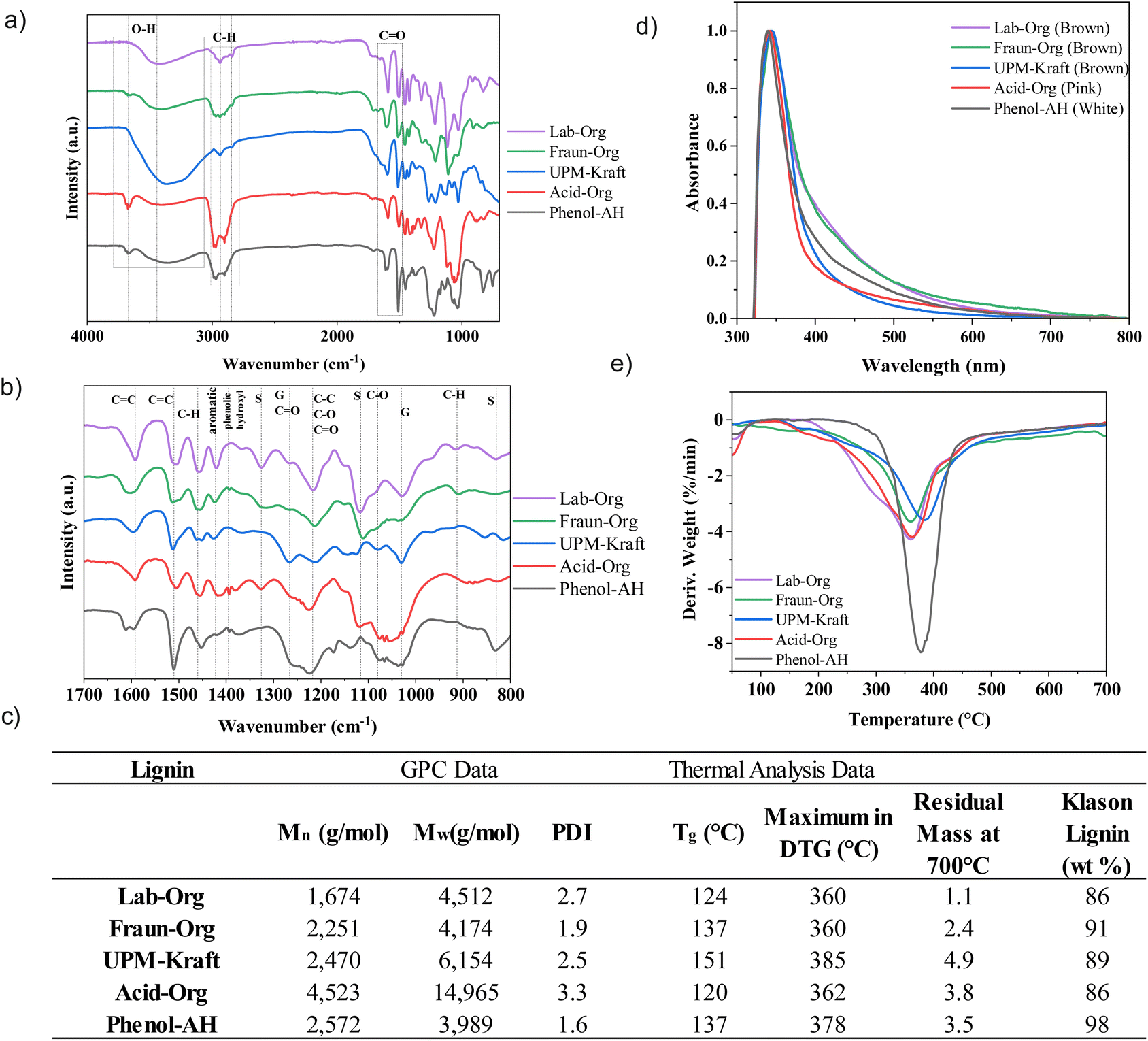 | ||
| Fig. 5 (a) FTIR spectra, (b) FTIR fingerprint region, (c) GPC data, glass transition temperature, degradation temperature, residual mass at 700 °C (ash content), Klason Lignin content, (d) UV-vis data, (e) DTG curves derived from TGA data. | ||
One of the main appeals of extracted lignin is its abundance of alcoholic and phenolic hydroxyl groups, which allow for functionalization, polymerization, as well as electrostatic stabilization with the use of pH changes.12,30 Lignin extraction procedures may lead to oxidation through elevated temperature, oxygen exposure, metal ions, or acidic conditions.25 The harsher the procedure, the more oxidation is expected to occur, leading to oxidized functional groups such as aldehydes, carboxylic acids, and quinones. While the lignins differ from one another in certain aspects, these functional groups may be observed in all 5 lignins.10 One of the most significant peaks exhibited by all lignins is at approximately 3450 cm−1, which is attributed to the stretching vibrations of the –OH in phenolic and alcoholic hydroxyl groups.4,35 It can be seen that the Kraft process led to the most intense peak at this wavenumber. The cleavage of lignin–carbohydrate bonds during the harsh alkaline pulping process leads to lignin with a relatively high hydroxyl group content.12 During the alkaline pulping process, with the assistance of NaOH and Na2S at high temperatures, cellulose, lignin and hemicellulose are broken apart, and hydrosulfide and hydroxide anions cleave α- and β-aryl ether linkages between phenylpropane units, breaking the lignin into smaller fragments.12,25 These saponification and cleavage reactions result in the exposure of hidden hydroxyl groups, as well as the formation of new ones. This high –OH content may be reflected in a markedly high glass transition temperature, as described below.25 This high –OH content may make UPM-Kraft lignin more reactive and applicable.11,12 It can be seen from the FTIR spectra that both Acid-Org as well as Phenol-AH exhibit a considerable amount of characteristic peaks in the range of 3200–3600 cm−1 (OH stretching vibrations), which correspond to the OH-stretching vibrations of alcohols, phenols, as well as other hydroxyl groups in phenolic and aliphatic structures.35,36 Other lignin samples are characterized by a lower signal of hydroxyl groups.
The small peak at 3670 cm−1 experienced by Fraun-Org, Acid-Org, and Phenol-AH is attributed to free hydroxyl groups that are not involved in hydrogen bonding and are quite common in lignin.37 These free hydroxyl groups could be a side product from the cleavage of ether linkages during the extraction process. Organosolv lignin, however, is extracted through the dissolution of lignin under relatively milder conditions in organic solvents, sometimes with the addition of a base or an acid.11,25 This results in a lignin with a structure closer to its native structure, and a lower free hydroxyl content, also reflected in FTIR spectra.
The peaks around 2935 and 2845 cm−1 are attributed to the C–H asymmetric stretching vibrations in aromatic methoxyl groups and methyl and methylene groups of side chains.4,36 These peaks are particularly pronounced for Fraun-Org, Acid-Org, and Phenol-AH. The stretching vibrations of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds of conjugated aldehydes, non-conjugated ketones and carboxylic esters in the aromatic ring appear at around 1700 cm−1.4,36 This peak is particularly intense for Lab-Org, Fraun-Org, and UPM-Kraft, and includes a shoulder at approximately 1670 cm−1 from conjugated carbonyl–carbonyl stretching, indicating that the (presumably thermal) cleavage of β-O-4′ linkages led to the formation of carbonyl groups.4,36
O bonds of conjugated aldehydes, non-conjugated ketones and carboxylic esters in the aromatic ring appear at around 1700 cm−1.4,36 This peak is particularly intense for Lab-Org, Fraun-Org, and UPM-Kraft, and includes a shoulder at approximately 1670 cm−1 from conjugated carbonyl–carbonyl stretching, indicating that the (presumably thermal) cleavage of β-O-4′ linkages led to the formation of carbonyl groups.4,36
Interpreting spectra below 1400 cm−1 may be quite difficult as peaks become more complex and begin to contribute to each other's intensity.36 A characteristic peak for hardwood and non-wood lignins at 1326 cm−1 is due to the C–O stretching in carboxylic esters, ethers, or alcohols related to syringyl units,4,36 while the aromatic C–H in-plane deformation at 1116 cm−1 is also attributed to syringyl units.4,30 Both of these peaks are particularly apparent for those lignins with high syringyl content: Lab-Org, Fraun-Org, and Acid-Org, as corroborated by 2D NMR spectra. The peak at approximately 1214 cm−1 is attributed to the C–C, C–O, and CO stretching, and in general, carbohydrate vibrations can often be seen in association with other vibrations in the range of 1000–1300 cm−1.30,36 The aromatic C–H in-plane deformation mainly from G aromatic units is seen at 1031 cm−1, while the C–H out of plane vibration in aromatic units is at 911 cm−1, and the peak at 831 cm−1 shows the out-of-plane vibrations of C–H on the syringyl aromatic units of lignin.4,30 Kraft lignin, which has a high proportion of G-type units, exhibits peaks at 854 cm−1 and 817 cm−1, showing C–H out of plane vibrations in positions 2, 5, and 6 of guaiacyl units, which agree well with the NMR analysis.36
FTIR spectra of organosolv lignin and nanolignin (ESI Fig. S1†) showed that the chemical structure of the lignin remains mostly unchanged before and after the production of nanolignin, indicating that the structure and functional groups and chemical structure are unaltered during the processing of nanolignin.
According to GPC analysis, Lab-Org has the smallest molecular weight, indicating a milder reaction procedure, while Acid-Org exhibits the largest molecular weight, while also appearing to have conserved much of the original lignin structure as seen by FTIR and later by 2D-HSQC NMR.12 This indicates that this procedure can extract large molecules from the wood without incurring too much oxidative degradation during the procedure.20 Phenol-AH lignin has the lowest PDI, whilst exhibiting a similar molecular weight to Fraun-Org and UPM-Kraft lignins. The harsher the method of extraction, the larger the molecular weight is expected to be, as cleavage of β-O-4 and β–β linkages first break down the primary chains, but rapid recondensation (cross-linking and repolymerization reactions between molecules of lignin) then leads to a more condensed lignin structure with an increased molecular weight.10–12,25 Another possible explanation for this increase in molecular weight could be that harsher extraction methods can remove larger molecules from the wood, while also breaking some of them apart, raising the PDI along the way.20 The lignins used for these experiments mostly follow this trend, with PDI going up along with the molecular weight.
In general, lignin with high molecular weight and low water solubility produces smaller nanolignin particles, as these large molecules act as the nuclei for the nanoparticles, while the smaller molecules endowed with hydrophilic functions such as carboxylic acids coat the outside of the LNPs.10 Faster, more intense nucleation then gives more and smaller particles. The lignins that we tested did not precisely follow this trend, and although they did not have the lowest molecular weight, UPM-Kraft lignin produced the nanolignin with the smallest size, as well as the smallest size dispersity. Chemical factors overlap with the molecular weight dependence of dissolution, and the resulting behavior is more complex.
The harsher extraction procedures of lignin should not only result in a larger molecular weight but a higher percentage of carbon should also be found in elemental analysis.20,25 To our surprise, the relative carbon and oxygen content of the lignins are rather constant throughout the samples, with only Phenol-AH showing a somewhat higher carbon content. This is because harsher methods remove more non-lignin materials from the biomass, such as proteins and carbohydrates, chemical modifications lead to the formation of new carbon–oxygen bonds and C–C bonds, and lastly, condensation reactions occur during harsh extraction methods resulting in a more condensed lignin structure.20,25
The degradation of lignin must be considered for its potential applications in building and packaging materials. Thermogravimetric analysis was performed on the various lignins to understand the thermal stability of the corresponding nanolignins. TGA was performed both under nitrogen and synthetic air atmospheres to understand degradation temperature and ash content. TG curves of lignin and most woody biomasses typically undergo three regions of thermal degradation, starting with the evaporation of low molecular weight components and water around 50–200 °C.12,17 In the second stage between 250–400 °C, due to the formation of char, monomeric phenols would be liberated into the vapour phase.38 The last stage of deep pyrolysis is associated with the decomposition of carbohydrates as well as the condensation of the earlier formed carbonaceous material after 400.4,38 Methoxy groups fragmented between 400 °C and 600 °C, while CO and C–O–C bonds likely fragmented after 600 °C.38 The TGA results indicate that the lignins had quite varying onset temperatures, but quickly degraded and evaporated in the temperature range of approximately 355 °C to 385 °C, after which the lignols decomposed and carbonization occurred. Thermogravimetric analysis data (table in Fig. 5c) showed that UPM-Kraft has the highest thermal stability, while Lab-Org and Fraun-Org have the lowest. Some thermally stable char was left after the TGA analysis up to 700 °C, likely due to the formation of highly branched and condensed aromatic structures within the materials.12
The amount of ash should be typical for an isolation pathway. Ash may represent extracted inorganic compounds present within the lignin, such as silica or iron oxides, as well as the alkaline and alkaline earth counter-ions of the carboxylic acids, extracted under neutral or alkaline conditions.39 The ash value was the lowest for the lab-extracted Organosolv lignin, at only 1.1% ash content, and was the highest for UPM-Kraft lignin at nearly 5 wt%. The organosolv process typically leads to high-purity lignin with less than 5 wt% residual content, which is seen for Lab-Org and Fraun-Org.11,26 The Kraft process is known to have a somewhat greater presence of inorganics, particularly sodium content.1,11 The values also go quite nicely with the mineral content overall, indicating that Kraft-lignin is a substantial counter ion carrier.39 These ash content values align closely with the literature values of ash content for Kraft and Organosolv lignins.5,11 For simplicity, assuming K2CO3 (pot-ash) as a typical mineral combustion leftover, calculations revealed 1 carboxylate unit per 10 monolignol units, i.e., the isolated lignin is unexpectedly highly charged. This also explains the stability of nanolignin particles without further added stabilizers.
DSC. The glass transition temperature (Tg) is an important factor for pre-evaluating the possible applications of lignin and lignin nanoparticles as fillers or precursors in biocomposite materials. Tg is typically an indicator of how mobile macromolecular chains are, and this depends on several factors, including the molecular weight of the polymer, its degree of chemical and physical crosslinking, as well as the strength of the intermolecular interactions, and the cohesion energy density. Below the glass transition temperature, the polymer chains cannot move and a glassy solid is obtained.40 At the glass transition temperature, the glassy immobilization breaks down, and the lignin becomes softer, allowing for a plastic material deformation.
Differential scanning calorimetry was used to determine the glass transition temperature (Tg) of the 5 lignins, shown in Fig. 5c, indicating the temperature at which the amorphous polymers undergo a transition from glassy to the viscoelastic state.12,40 The values range between approximately 126 °C and 154 °C. The Tg values of technical lignin are typically found between 90 °C and 180 °C, so the values agree nicely with previous reports.11,38 The higher the molecular weight of the lignin, the fewer chain ends will be present, leading to lower mobility and a higher glass transition temperature (Tg).12,25
From the Tg data acquired, UPM-Kraft lignin appears to have the highest glass transition temperature, with Phenol-AH/Fraun-Org, Lab-Org and Acid-Org following in order. From FTIR data, it was seen that UPM-Kraft lignin contains a relatively large amount of the hydroxyl group within its structure, indicating the presence of alcohols and phenols and thereby potential hydrogen bridges and increased intermolecular cohesion.11,25 2D HSQC data also indicate that UPM lignin contains a larger amount of aliphatic entities, providing a second reason for a lowered Tg. Kraft lignin has been found to generally have a higher Tg than the organosolv lignins from literature.12 Although Acid-Org had the highest molecular weight, it did not exhibit the highest glass transition temperature.
Besides UPM-Kraft, the higher the molecular weight of the lignin, the higher the glass transition temperature appears to be, indicating the expected correlation.25,40
A proposed method for lowering the glass transition temperature of lignin in order to expand applications, e.g., for melt gluing, would be to add low molecular weight diluents, potentially even water.40 It should be noted that as the moisture content increases within the wood, the Tg goes down in lignin until it reaches its water-saturated point.41 This highly depends on the number of hydroxyl groups available within lignin to bind water, and it is based on structure; therefore, it can be assumed that UPM-Kraft lignin would be the most likely to swell due to water adsorption.11
The presence of certain functional groups also has an effect on the Tg. Functional groups with high polarity such as hydroxyl and carbonyl groups tend to increase the overall polymer Tg due to the promotion of intermolecular interactions between the chains, increasing the energy barrier for segmental motion, thereby raising the Tg. This is likely the reason why UPM-Kraft has such an increased Tg, detected in the dry state. Lignins with many rigid phenolic side groups on the main chain tend to have a higher Tg, while those with higher methoxy content tends to have a lower Tg.41 Bulky, non-flexible side groups are also considered to be a barrier to the free motion of polymer chains, increasing the Tg. Any crosslinking may also hinder the softening of a polymer because a tough 3D network may be formed within the polymer, restricting mobility. More flexible groups such as alkyl chains, on the other hand, lower Tg. Therefore, cross-linking, hydrogen bonds, the isolation method, species, thermal prehistory, degree of branching, and the presence of other functional groups affect the glass transition temperature.41
It is important to consider the carbohydrate content of lignins when manufacturing nanolignin because of glycosylation or other binding effects every step along the way. Solubility and precipitation will be altered by the carbohydrate content because they tend to be more soluble in polar solvents that can form hydrogen bonds with the hydroxyl groups in carbohydrates, such as water and ethanol.43 The solubility of the lignin is altered by the carbohydrate content, leading to a different size or shape of the LNPs. If carbohydrate-rich regions of lignin exhibit hydrogen bonding with the water in the solvent mixture, the interactions may influence aggregation and/or self-assembly behaviour. These regions may also lead to a more heterogeneous set of particles due to incomplete lignin purification and/or extraction. Carbohydrates may also modify the surface properties of the nanoparticles, affecting their interaction with other substances, dispersibility, and/or reactivity.35,43 If one considers the hydrophobic nature of lignin, and the hydrophilic nature of carbohydrates, the amphiphilic properties of a lignin with a substantial amount of carbohydrates could be taken advantage of for the assembly of a variety of structures.35
Nanocomposites containing nanolignin have been shown to have enhanced UV resistance due to these transitions within lignin, which can convert photon energy into heat and release it.8 During the oxidative polymerization of monolignols to generate lignin, the electronic conjugation of the double bond in the propanoid-chain para to the phenolic OH will vanish, resulting in the generation of UV chromophores at coupling sites.6 These chromophoric groups give lignin the ability to absorb in the UV/Visible region, as well as its color.44 Due to the slightly skin-like color, nanolignin has found application in UV-blocking fabrics, as well as in cosmetics and sunscreen.8,9,44
Dispersion into nanolignin. All 5 nanolignins could be prepared successfully from their respective starting material, with and without a change in pH as seen in Fig. 6. They are displayed along with their original lignin powder counterparts in Fig. 6 (lower left). Observation with the naked eye supports a residue-free dispersion for all nanolignins.
 | ||
| Fig. 6 (Top left): From left to right: Lab-Org.NL, Fraun-Org.NL, UPM-Kraft, Acid-Org.NL and Phenol-AH.NL (middle left): Lab-Org.pH.NL, Fraun-Org.pH.NL, UPM-Kraft.pH.NL, Acid-Org.pH.NL, and Phenol-AH.pH.NL. (Lower left): Corresponding lignins: Lab-Org, Fraun-Org, UPM-Kraft, Acid-Org, and Phenol-AH. (Upper right) Particle sizes of nanolignins produced from the various lignins, with and without pH alterations. | ||
We chose pH 7, at which the stabilization of lignin nanoparticles was observed. The neutral pH facilitates the application of the resulting nanomaterials. Without pH changes, the pH was approximately 3.5.
These experiments made it clear that a change in solvent pH affects the size and appearance of the nanolignin dispersions. Visually, the nanolignin formed without pH modification was much brighter in colour than their counterparts, having a milky appearance, and settling much more quickly to the bottom of the vial due to their lower zeta potential and large size. The sizes were determined using SEM analysis and will be further discussed. This difference in appearance can be attributed to the Tyndall effect and the settling can be attributed to a lower surface charge, and therefore stability. The nanolignin formed with a change in pH had a darker, much more see-through appearance, as well as higher stability, not easily settling to the bottom of the vial and forming aggregates. The chromophores present in lignin, as well as the varying phenolate content, could explain the wide range of appearances.44
To understand more clearly how the pH affects the nanolignin produced, Fraunhofer lignin (the least glycosylated) was used to precipitate 6 different nanolignins, only altering the pH of the solvent before precipitation into the anti-solvent. The results in Fig. 7 show that the colour of the nanolignin dispersion becomes darker as the pH is raised, and that the size of the nanoparticles decreases as the pH increases. The sizes and surface charges of the resulting nanolignins are shown in Fig. 7. After one week of production, the nanolignins with lower pH aggregated more readily on the bottom of the vials, indicating and reflecting the lower zeta potential of the colloidal suspension, and in turn, a lower stability. It seemed that using pH 7 was, in fact, a good choice for the production of nanolignins, showing a decently high colloidal stability.
 | ||
| Fig. 7 (Left) 3 wt% Fraun-Org.NL. pH 4–9 before (above) and after (below) 1 week of undisturbed rest; (right) particle size and zeta potential for Fraun.Org. pH 4–9. | ||
In general, a zeta potential more negative than −30 mV indicates sufficient repulsion energy and therefore colloidal stability.38 Large negative charges indicate a strong electrostatic repulsion between the particles and a high stability of the dispersion. Nanolignins contain acidic phenolic groups, as well as carboxylic acid functional groups, on their surfaces, which give the nanolignin a negative surface charge and enable the formation of an electric double layer stabilizing the LNPs. In weakly acidic conditions, phenol groups remain in their protonated state, which is less water-soluble. Under basic conditions, phenols experience deprotonation, letting a proton go from the hydroxyl group, producing negatively charged phenolate ions. The darkening effect with increasing pH is explained by the phenols of lignin. In its pure form, phenol is a white solid, while phenolates are brownish.
 | ||
| Fig. 8 SEM images of the LNPs. From left to right: Lab-Org.NL, Fraun-Org.NL, UPM-Kraft.NL, Acid-Org.NL, and Phenol-AH.NL. | ||
 | ||
| Fig. 9 SEM images of the LNPs. From left to right: Lab-Org.NL.pH, Fraun-Org.NL.pH, UPM-Kraft.NL.pH, Acid-Org.NL.pH, and Phenol-AH.NL.pH. | ||
 | ||
| Fig. 10 SEM images of nanolignins Fraun-Org.pH 4–9 after being air-dried on a carbon slide. | ||
 | ||
| Fig. 11 (upper left) 5 wt% nanolignins after 1 week of sitting at rest. From left to right: Lab-Org.NL, Fraun-Org.NL, UPM-Kraft.NL, Acid-Org.NL, and Phenol-AH.NL. (lower left) 5 wt% nanolignins after 1 week of sitting at rest. From left to right: Lab-Org.NL.pH, Fraun-Org.NL.pH, UPM-Kraft.NL.pH, Acid-Org.NL.pH, and Phenol-AH.NL.pH. (right) zeta potentials of nanolignins after production. | ||
The nanolignins were produced in the method acquired directly from the work of Henn et al., as well as with a change in solvent pH in order to discern the difference that the pH alteration makes on the size and charge of the acquired nanolignin.7 From the results in Fig. 7, it can be seen that pH adjustment significantly reduces the size of the nanolignin particles; on average approximately 4.2 times smaller nanolignins were achieved by a simple change in pH. The most significant changes in size were achieved by UPM-Kraft.NL and Acid-Org.NL, with a size difference of more than 5 times. It was observed in our case that the molecular weight does not have a significant effect on the size of the resulting nanolignin.
The extremely small diameter of UPM-Kraft LNPs is to be discussed as it can be as small as 10 nm and deviates from the behavior of all other samples. There are some possible explanations for this extreme result, including some extra impurities and/or extractives left within the Kraft lignin during the extraction procedure. For example, tall oil is a by-product of the pulp and paper production process, especially in the Kraft pulping process of softwood. It is composed of resins, resin acids, fatty acid derivatives, as well as other organic compounds and is certainly able to assist lignin in the formation of very small LNPs by acting as an ultra-hydrophobe and surfactant.27 Another possible explanation for the small size of UPM-Kraft nanolignin could be its high carboxylic acid content, which quite effectively allows changes to be made through the change in pH.
Fig. 8 and 9 depict the various nanolignins dried and self-assembled into films with different surface textures. This is likely due to differences in solubility within the original solvent mixture (acetone, ethanol, water). Rao et al. reported observable differences in organosolv lignin nanoparticles through the variation of ethanol within the starting solvent throughout drying.35 In conjunction with a homogenization treatment, they found that the ideal ethanol ratio created round spheres, while other ratios with lower or higher ethanol content form globular clusters.35
When the lignin solvent mixture is briskly injected into the water, the hydrophobic polymers form a densely packed core. The formation of these droplets is kinetically controlled, rather than being thermodynamically driven.18 UPM-Kraft Lignin is quite hydrophilic and water-swollen even throughout film formation, thus creating near-homogeneous, structured films, the observation of which tells us little about the original dispersion. Fraun-Org Lignin gives a tight packing of well-separated particles, ca. 20 nm in size. The particles are not roundish, but rather irregularly packed onto each other; i.e., we can assume a certain particle swelling with ethanol, notwithstanding the high capillary drying pressures in the final phase of film formation.
For the other lignins, the specific solvent/non-solvent system and procedure for the production of nanolignin gives clear spheres, however, with a discernible size dispersity between 10 and 50 nm. The affinity of hydrophobic and hydrophilic moieties in the solvent is likely to influence which shape/size the nanolignin will end with.35 The carbohydrate content of the lignins is thought to influence the formation of nanolignin through the influence of solvent interactions.10,43
Fig. 10 shows that the change in pH had a great effect on the size of the resulting LNP. Below a pH of 4.5, lignin nanoparticles often experience aggregation and instability, clumping together to form large aggregates.38 This again underlines the role of included standard carboxylic acids as the active stabilizer, which are effectively neutralized below their pKa value.
If a strongly amphiphilic molecule like Kraft lignin is unable to self-assemble into spheres due to inner polarity “frustrations”, it may form irregular and non-spherical shapes. The organization of hydrophobic and hydrophilic moieties is quite important when minimizing the cohesion energy of the overall system.35,43 As mentioned above, the chemical and molecular heterogeneity of lignin is important, i.e., high molecular weights and/or more hydrophobic fractions precipitate first and constitute the nucleus or the inner part of the particle, while a further decline of solvent quality also precipitated the more polar compounds, which then also stabilized the particles.35 More homogeneous size distributions in general rely on controlled, fast nucleation and then extended growth phases.35 Our current protocol is not optimized for minimizing particle dispersity. Centrifugation techniques are, however, useful for the narrowing of size within a nanolignin dispersion, or at least cutting the very large and fine particles of the dispersion.45
It should be noted that the sizes of the nanolignins produced differ quite notably from the current literature on nanolignins.1 This could be due to the measuring technique used in this report, which varies from other common uses of DLS.10,35 The nanolignins were dropped on a carbon slide, allowed to dry, and imaged using SEM. After drying, the nanolignins surely lost much of their weight and shrank, which should be considered; however, as all samples were prepared in the same way, the results should be consistent in relation to one another.
For the reproducible production of nanolignin, it would be important to know how extraction protocols change the chemical nature of the extracted fraction of lignocellulosic biomass. The properties of the lignins extracted depend on the plant species, the part of the plant it is extracted from, as well as the extraction protocol.28 It was shown that the different lignins analyzed were indeed chemically varied and consecutively led to a variety of nanolignins.
4. Conclusions
We analyzed 5 different lignins produced via various extraction protocols by employing a combination of analytical techniques. It was observed in all cases that pH- and temperature-induced degradation, depolymerization, and repolymerization events had occurred, the extent of which depends on the extremity of the processing conditions. We identified several partly known, but usually neglected, “hidden parameters” in the real materials, which can seriously change the properties of the lignin. One of them is the natural conjugation/glycosylation of lignin with sugars and glycans, which is more pronounced in the isolated product the more gentle the extraction protocol is. Another is the rather high carboxylate content of all lignins, which also is responsible for the otherwise surfactant-free formulation of the nanolignins. The carboxylates usually come with alkaline and alkaline earth counter ions, which can make up as high as 6 wt% of the isolated lignin in the gentler extraction protocols, in excellent correspondence with the ash content of full wood. In this work, it remains an open question whether these carboxylic groups are a part of the sugar grafts, e.g. as glucuronic acid (as known from hemicelluloses), or if they are due to the oxidation of the propionic fragments of lignin.A third potential source of complexity, especially for softwood, is the contained tall oil, which would be co-extracted but contains resins, abietic acids, and fatty acid derivatives.27
The typical oversimplified textbook schemes of all-phenolic lignin are misleading because they give a wrong impression of the polymer structure involved and they suggest overly complicated processes to transfer them into an applicable form.
The characterized five different lignins were used for the facile production of nanolignin. A general method for generating nanolignin dispersions using non-toxic and recyclable solvents was fine-tuned in order to produce nanolignins of smaller sizes and size distributions. All five lignins reproducibly generated stable and sediment-free nanodispersions with a diameter of about 80–100 nm using solvent shifting methods, while additional pH adjustment (considering the phenolic residues and the usually non-listed carboxyl acid content) enabled us to go down to a diameter 10–34 nm. This is about four times smaller than usual, or a seventy times higher particle number. Although the lignins used were of different sources and compositions, the nanolignins produced were quite consistent in size and colloidal stability. Zeta potential measurements showed highly negative surface changes, which can be related to carboxylic acids, while phenolic groups act as co-surfactants. When the pH of the lignins was raised, the resulting nanolignin also exhibited a higher zeta potential, and in turn, higher stability.
For materials applications of such nanolignins, e.g., for gluing or in hot molds, polymer properties are important. The lignin that exhibited the lowest glass transition temperature, as well as the lowest molecular weight, was Lab-Org, the lignin freshly extracted in a relatively mild manner in our lab. The nanolignin that is likely to show the most industrial potential is the UPM-Kraft.NL, as the vast majority of produced lignin is Kraft lignin, and through the facile production process, a very small nanolignin was achieved. This small nanolignin, along with its small size dispersibility could make it viable for materials in which small defects must be avoided. The glass transition temperature of Kraft lignin is -without water- the highest of the ones tested. It also had the highest degradation temperature and high thermal stability, which could make it a practical filler for composite materials. Its small and consistent size makes its excellent in terms of dispersibility as well as consistency, and as we speculated above that the differences from all other lignins could be potentially due to tall-oil-based surfactants that theoretically can assist UPM-Kraft in the stabilization, size control, solubilization and phase transfer of the nanolignins.27
Such nanolignin particles have high potential in the world of green polymers and materials as they can be made using a facile and green production method and come from renewable and abundant sources. In our work, we currently focus on the combination or incorporation of LNPs with other biomaterials and utilize them for their UV-blocking, antibacterial, and/or adhesive properties.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Group leader for Sustainable Solvents: Dr Svitlana Filonenko. Director of the Max Planck Institute for Colloids and Interfaces, Dr Professor Markus Antonietti. Open Access funding provided by the Max Planck Society.References
- S. Iravani and R. S. Varma, Greener synthesis of lignin nanoparticles and their applications, Green Chem., 2020, 22(3), 612–636, 10.1039/C9GC02835H
.
- O. u. Rahman, S. Shi, J. Ding, D. Wang, S. Ahmad and H. Yu, Lignin nanoparticles: synthesis, characterization and corrosion protection performance, New J. Chem., 2018, 42(5), 3415–3425, 10.1039/C7NJ04103A
- L. D. Antonino, J. R. Gouveia, R. R. de Sousa Júnior, G. E. S. Garcia, L. C. Gobbo, L. B. Tavares and D. J. dos Santos, Reactivity of Aliphatic and Phenolic Hydroxyl Groups in Kraft Lignin towards 4,4′ MDI, Molecules, 2021, 26(8), 2131 CrossRef CAS PubMed
- A. G. Margellou, P. A. Lazaridis, I. D. Charisteidis, C. K. Nitsos, C. P. Pappa, A. P. Fotopoulos, V. d. Bosch, S. Sels, B. F. Triantafyllidis and K. S, Catalytic fast pyrolysis of beech wood lignin isolated by different biomass (pre)treatment processes: Organosolv, hydrothermal and enzymatic hydrolysis, Appl. Catal., A, 2021, 623, 118298, DOI:10.1016/j.apcata.2021.118298
- M. S. Ganewatta, H. N. Lokupitiya and C. Tang, Lignin Biopolymers in the Age of Controlled Polymerization, Polymer, 2019, 11(7), 1176 Search PubMed
- D. Kai, M. J. Tan, P. L. Chee, Y. K. Chua, Y. L. Yap and X. J. Loh, Towards lignin-based functional materials in a sustainable world, Green Chem., 2016, 18(5), 1175–1200, 10.1039/C5GC02616D
- K. A. Henn, N. Forsman, T. Zou and M. Österberg, Colloidal Lignin Particles and Epoxies for Bio-Based, Durable, and Multiresistant Nanostructured Coatings, ACS Appl. Mater. Interfaces, 2021, 13(29), 34793–34806, DOI:10.1021/acsami.1c06087
- Z. Zhang, V. Terrasson and E. Guénin, Lignin Nanoparticles and Their Nanocomposites, Nanomaterials, 2021, 11(5), 1336 CrossRef CAS PubMed
- A. Raman, A. Sankar, S. D. A, A. Anilkumar and A. Saritha, Insights into the Sustainable Development of Lignin-Based Textiles for Functional Applications, Macromol. Mater. Eng., 2022, 307(8), 2200114, DOI:10.1002/mame.202200114
- M. Österberg, M. H. Sipponen, B. D. Mattos and O. J. Rojas, Spherical lignin particles: a review on their sustainability and applications, Green Chem., 2020, 22(9), 2712–2733, 10.1039/D0GC00096E
-
S. Bhattacharyya, L. Matsakas, U. Rova and P. Christakopoulos, Melt Stable Functionalized Organosolv and Kraft Lignin Thermoplastic, in Processes, 2020, vol. 8 Search PubMed
- N. Latif, N. Brosse, I. Ziegler-Devin, L. Chrusciel, R. Hashim and M. H. Hussin, A Comparison of Alkaline and Organosolv Lignin Extraction Methods from Coconut Husks as an Alternative Material for Green Applications, BioResources, 2021, 17, 469–491, DOI:10.15376/biores.17.1.469-491
- J. Karthäuser, V. Biziks, C. Mai and H. Militz, Lignin and Lignin-Derived Compounds for Wood Applications—A Review, Molecules, 2021, 26(9), 2533 CrossRef PubMed
- P. Figueiredo, K. Lintinen, J. T. Hirvonen, M. A. Kostiainen and H. A. Santos, Properties and chemical modifications of lignin: Towards lignin-based nanomaterials for biomedical applications, Prog. Mater. Sci., 2018, 93, 233–269, DOI:10.1016/j.pmatsci.2017.12.001
- J. Rencoret, A. Gutiérrez, E. Castro and J. C. d. Río, Structural characteristics of lignin in pruning residues of olive tree (Olea europaea L.), Holzforschung, 2019, 73(1), 25–34, DOI:10.1515/hf-2018-0077
- Y. Chen, X. Gong, G. Yang, Q. Li and N. Zhou, Preparation and characterization of a nanolignin phenol formaldehyde resin by replacing phenol partially with lignin nanoparticles, RSC Adv., 2019, 9(50), 29255–29262, 10.1039/C9RA04827H
- X. Wang, X. Yang, Z. Wu, X. Liu, Q. Li, W. Zhu, Y. Jiang and L. Hu, Enhanced Mechanical Stability and Hydrophobicity of Cellulose Aerogels via Quantitative Doping of Nano-Lignin, Polymer, 2023, 15(5), 1316 Search PubMed
- T. Leskinen, M. Smyth, Y. Xiao, K. Lintinen, M.-L. Mattinen, M. A. Kostiainen, P. Oinas and M. Osterberg, Scaling up production of colloidal lignin particles, Nord. Pulp Pap. Res. J., 2017, 32, 586–596, DOI:10.3183/NPPRJ-2017-32-04-p586-596
- T. Leskinen, M. Smyth, Y. Xiao, K. Lintinen, M.-L. Mattinen, M. A. Kostiainen, P. Oinas and M. Österberg, Scaling Up Production of Colloidal Lignin Particles, Nord. Pulp Pap. Res. J., 2017, 32(4), 586–596, DOI:10.3183/npprj-2017-32-04_p586-596_leskinen
- D. S. Zijlstra, A. de Santi, B. Oldenburger, J. de Vries, K. Barta and P. J. Deuss, Extraction of Lignin with High β-O-4 Content by Mild Ethanol Extraction and Its Effect on the Depolymerization Yield, JoVE, 2019,(143), e58575, DOI:10.3791/58575
- M. H. Sipponen, H. Lange, M. Ago and C. Crestini, Understanding Lignin Aggregation Processes. A Case Study: Budesonide Entrapment and Stimuli Controlled Release from Lignin Nanoparticles, ACS Sustainable Chem. Eng., 2018, 6(7), 9342–9351, DOI:10.1021/acssuschemeng.8b01652
- J. A. Sirviö, I. Romakkaniemi, J. Ahola, S. Filonenko, J. P. Heiskanen and A. Ämmälä, Supramolecular interaction-driven delignification of lignocellulose, Green Chem., 2024, 26(1), 287–294, 10.1039/D3GC03857B
-
A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton and D. Crocker, Determination of Structural Carbohydrates and Lignin in Biomass, DE-AC36-08GO28308, Colorodo, United States, 2012 Search PubMed
- X. Meng, C. Crestini, H. Ben, N. Hao, Y. Pu, A. J. Ragauskas and D. S. Argyropoulos, Determination of hydroxyl groups in biorefinery resources via quantitative 31P NMR spectroscopy, Nat. Protoc., 2019, 14(9), 2627–2647, DOI:10.1038/s41596-019-0191-1
- C. Wang, S. S. Kelley and R. A. Venditti, Lignin-Based Thermoplastic Materials, ChemSusChem, 2016, 9(8), 770–783, DOI:10.1002/cssc.201501531
- J. Bergrath, J. Rumpf, R. Burger, X. T. Do, M. Wirtz and M. Schulze, Beyond Yield Optimization: The Impact of Organosolv Process Parameters on Lignin Structure, Macromol. Mater. Eng., 2023, 308(10), 2300093, DOI:10.1002/mame.202300093
- C. Mattsson, S.-I. Andersson, T. Belkheiri, L.-E. Åmand, L. Olausson, L. Vamling and H. Theliander, Using 2D NMR to characterize the structure of the low and high molecular weight fractions of bio-oil obtained from LignoBoost™ kraft lignin depolymerized in subcritical water, Biomass Bioenergy, 2016, 95, 364–377, DOI:10.1016/j.biombioe.2016.09.004
-
L. Ana and P. Helena, Compositional Variability of Lignin in Biomass, in Lignin, ed. P. Matheus, IntechOpen, 2017, ch. 3 Search PubMed
- X. Jiang, J. Liu, X. Du, Z. Hu, H.-m. Chang and H. Jameel, Phenolation to Improve Lignin Reactivity toward Thermosets Application, ACS Sustainable Chem. Eng., 2018, 6(4), 5504–5512, DOI:10.1021/acssuschemeng.8b00369
- N. A. Aziz, A. F. A. Latip, L. C. Peng, N. H. A. Latif, N. Brosse, R. Hashim and M. H. Hussin, Reinforced lignin-phenol-glyoxal (LPG) wood adhesives from coconut husk, Int. J. Biol. Macromol., 2019, 141, 185–196, DOI:10.1016/j.ijbiomac.2019.08.255
- T. V. Lourençon, S. Alakurtti, T. Virtanen, A.-S. Jääskeläinen, T. Liitiä, M. Hughes, W. L. E. Magalhães, G. I. B. Muniz and T. Tamminen, Phenol-formaldehyde resins with suitable bonding strength synthesized from “less-reactive” hardwood lignin fractions, Holzforschung, 2020, 74(2), 175–183, DOI:10.1515/hf-2018-0203
- P. R. Sarika, P. Nancarrow, A. Khansaheb and T. Ibrahim, Bio-Based Alternatives to Phenol and Formaldehyde for the Production of Resins, Polymer, 2020, 12(10), 2237 CAS
- A. Lisý, A. Ház, R. Nadányi, M. Jablonský and I. Šurina, About Hydrophobicity of Lignin: A Review of Selected Chemical Methods for Lignin Valorisation in Biopolymer Production, Energies, 2022, 15(17), 6213 CrossRef
- T. Rashid, C. F. Kait and T. Murugesan, A “Fourier Transformed Infrared” Compound Study of Lignin Recovered from a Formic Acid Process, Procedia Eng., 2016, 148, 1312–1319, DOI:10.1016/j.proeng.2016.06.547
- X. Rao, Y. Liu, Q. Zhang, W. Chen, Y. Liu and H. Yu, Assembly of Organosolv Lignin Residues into Submicron Spheres: The Effects of Granulating in Ethanol/Water Mixtures and Homogenization, ACS Omega, 2017, 2(6), 2858–2865, DOI:10.1021/acsomega.7b00285
- C. Boeriu, D. Bravo, R. Gosselink and J. Dam, Characterisation of structure-dependent functional properties of lignin with infrared spectroscopy, Ind. Crops Prod., 2004, 20, 205–218, DOI:10.1016/j.indcrop.2004.04.022
- E. S. Esakkimuthu, D. DeVallance, I. Pylypchuk, A. Moreno and M. H. Sipponen, Multifunctional lignin-poly (lactic acid) biocomposites for packaging applications, Front. Bioeng. Biotechnol., 2022, 10, 1025076, DOI:10.3389/fbioe.2022.1025076
- U. P. Perera, M. L. Foo and I. M. L. Chew, Synthesis and characterization of lignin nanoparticles isolated from oil palm empty fruit bunch and application in biocomposites, Sustainable Chem. Clim. Action, 2023, 2, 100011, DOI:10.1016/j.scca.2022.100011
- D. Smołka-Danielowska and M. Jabłońska, Chemical and mineral composition of ashes from wood biomass combustion in domestic wood-fired furnaces, Int. J. Environ. Sci. Technol., 2022, 19(6), 5359–5372, DOI:10.1007/s13762-021-03506-9
-
D. A. I. Goring, Thermal softening, adhesive properties and glass transitions in lignin, hemicellulose and cellulose, in Consolidation of the Paper Web, Trans. of the IIIrd Fund. Res. Symp. Cambridge, 1965, ed. F. Bolam, 1965, pp. 555–568. DOI:10.15376/frc.1965.1.555
- Z. Börcsök and Z. Pásztory, The role of lignin in wood working processes using elevated temperatures: an abbreviated literature survey, Eur. J. Wood Wood Prod., 2021, 79(3), 511–526, DOI:10.1007/s00107-020-01637-3
- H. G. Jung, D. R. Mertens and A. J. Payne, Correlation of acid detergent lignin and Klason lignin with digestibility of forage dry matter and neutral detergent fiber, J. Dairy Sci., 1997, 80(8), 1622–1628, DOI:10.3168/jds.S0022-0302(97)76093-4
- H. V. Sadeghifar, R. A. Venditti, J. L. Pawlak and J. Jur, Bi-component carbohydrate and lignin nanoparticle production from bio-refinery lignin: A rapid and green method, BioResources, 2019, 14(3), 6179–6185, DOI:10.15376/biores.14.3.6179-6185
- Y. Zhang and M. Naebe, Lignin: A Review on Structure, Properties, and Applications as a Light-Colored UV Absorber, ACS Sustainable Chem. Eng., 2021, 9(4), 1427–1442, DOI:10.1021/acssuschemeng.0c06998
- J. Wang, W. Chen, D. Yang, Z. Fang, W. Liu, T. Xiang and X. Qiu, Photonic Lignin with Tunable and Stimuli-Responsive Structural Color, ACS Nano, 2022, 16(12), 20705–20713, DOI:10.1021/acsnano.2c07756
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc04299e |
| This journal is © The Royal Society of Chemistry 2024 |