
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSequence-defined antibody-recruiting macromolecules†
Resat
Aksakal
 a,
Corentin
Tonneaux
b,
Annemiek
Uvyn
c,
Mathieu
Fossépré
b,
Hatice
Turgut
a,
Nezha
Badi
*a,
Mathieu
Surin
*b,
Bruno G.
De Geest
*c and
Filip. E.
Du Prez
*a
a,
Corentin
Tonneaux
b,
Annemiek
Uvyn
c,
Mathieu
Fossépré
b,
Hatice
Turgut
a,
Nezha
Badi
*a,
Mathieu
Surin
*b,
Bruno G.
De Geest
*c and
Filip. E.
Du Prez
*a
aPolymer Chemistry Research Group, Centre of Macromolecular Chemistry (CMaC), Department of Organic and Macromolecular Chemistry, Faculty of Sciences, Ghent University, 9000 Ghent, Belgium. E-mail: nezha.badi@ugent.be; filip.duprez@ugent.be
bLaboratory for Chemistry of Novel Materials, Center of Innovation and Research in Materials and Polymers (CIRMAP), University of Mons-UMONS, 7000 Mons, Belgium. E-mail: mathieu.surin@umons.ac.be
cDepartment of Pharmaceutics, Ghent University, Ottergemsesteenweg 460, 9000 Ghent, Belgium. E-mail: br.degeest@ugent.be
First published on 22nd May 2023
Abstract
Antibody-recruiting molecules represent a novel class of therapeutic agents that mediate the recruitment of endogenous antibodies to target cells, leading to their elimination by the immune system. Compared to single-ligand copies, macromolecular scaffolds presenting multiple copies of an antibody-binding ligand offer advantages in terms of increased complex avidity. In this study, we describe the synthesis of sequence-defined macromolecules designed for antibody recruitment, utilising dinitrophenol (DNP) as a model antibody-recruiting motif. The use of discrete macromolecules gives access to varying the spacing between DNP motifs while maintaining the same chain length. This characteristic enables the investigation of structure-dependent binding interactions with anti-DNP antibodies. Through solid-phase thiolactone chemistry, we synthesised a series of oligomers with precisely localised DNP motifs along the backbone and a terminal biotin motif for surface immobilisation. Utilising biolayer interferometry analysis, we observed that oligomers with adjacent DNP motifs exhibited enhanced avidity for anti-DNP antibodies. Molecular modelling provided insights into the structures and dynamics of the various macromolecules, shedding light on the accessibility of the ligands to the antibodies. Overall, our findings highlight that the use of sequence-defined macromolecules can contribute to our understanding of structure–activity relationships and provide insights for the design of novel antibody-recruiting therapeutic agents.
Introduction
Nature makes use of amino acids as building blocks to synthesise discrete structures such as peptides and proteins.1 Their highly precise sequence allows to regulate their function as well as properties on a higher order. This motivated many polymer chemists recently to move away from disperse synthetic macromolecular structures and direct their research efforts to purely monodisperse structures in which the sequence is perfectly regulated.2–4 Since a long time, ionic, reversible deactivation radical or ROMP polymerisation techniques for example already allowed to synthesise low dispersity systems, tolerating a wide range of functional monomers.5,6 While such approaches permit to tailor polymer properties and to access multiple topologies,7 for certain application areas research in polymer synthesis is evolving towards obtaining strictly monodisperse macromolecules as these allow absolute control over the positioning of functional groups along the polymer backbone and to determine the exact structure–property relationships.8–12Multiple strategies have been developed in the last decade to achieve the synthesis of sequence defined macromolecules (SDMs).8,13–15 Thiolactone chemistry, for example, has been explored by our group and others16–18 for the synthesis of a wide range of discrete macromolecules including stereocontrolled oligomers, polyampholytes,11 and star-shaped macromolecules.19 This chemistry proved very efficient in advanced applications such as single-chain nanoparticles12 or data storage20–22 and it has been selected in this research for the design of antibody recruiting polymers.
Antibody-recruiting polymers contain multiple copies of a hapten ligand along their backbone and allow for binding of anti-hapten antibodies.23,24 The presence of a second ligand that binds to the surface of cells can mediate ternary complex formation between cell and anti-hapten antibodies, thereby recruiting the latter to the target cell surface. Clustering on the cell surface of endogenous anti-hapten antibodies, such as anti-galactose-α-1,3-galactose, anti-rhamnose and anti-dinitrophenol antibodies that are present in the blood of most human individuals, triggers destruction of the cell through complement activation and killing by macrophages and NK cells that recognise clustered antibody Fc fragments through their respective Fc-receptors.23 Several recent papers have demonstrated that compared to antibody-recruiting constructs that bear a single hapten ligand, polymers bearing multiple hapten ligands exhibit a vast increase in binding avidity (i.e. the strength) with the corresponding anti-hapten antibody, leading to an increase in antibody recruitment to cells in vitro and immune-mediated killing.25–27 However, polymers are typically disperse and vary in the degree of substitution without control over the distance between the hapten ligands.
Discrete macromolecules provide precise control over the spacing between functional residues along their backbone, while maintaining the same chain length. This property enabled us, for the first time in the field of antibody-recruiting (macro)molecules, to explore the effect of hapten ligand positioning on the corresponding avidity of the antibody binding. Hereto, we synthesized a series of sequence-defined antibody-recruiting macromolecules via a solid-phase approach (Scheme 1), using a Rink-amide resin functionalised with a thiolactone (TLa) moiety. Upon aminolysis of the thiolactone group by ethanolamine, an amide is formed with concomitant generation of a free thiol moiety that is employed in a thiol–acrylate or thiol–halide reaction in a successive step. The thiol-X reaction allows the introduction and precise positioning of the functional moieties needed for the study, being dinitrophenyl (DNP) and biotin. DNP was selected as model hapten owing to the commercial availability of anti-DNP antibodies, which facilitate in vitro analysis of the ternary complex formation. At the oligomer chain end, biotin is introduced as a terminal functional moiety to couple the oligomers to streptavidin-coated biolayer interferometry (BLI) sensors, used to measure the avidity of the binding to anti-DNP antibodies (dashed box in Scheme 1).
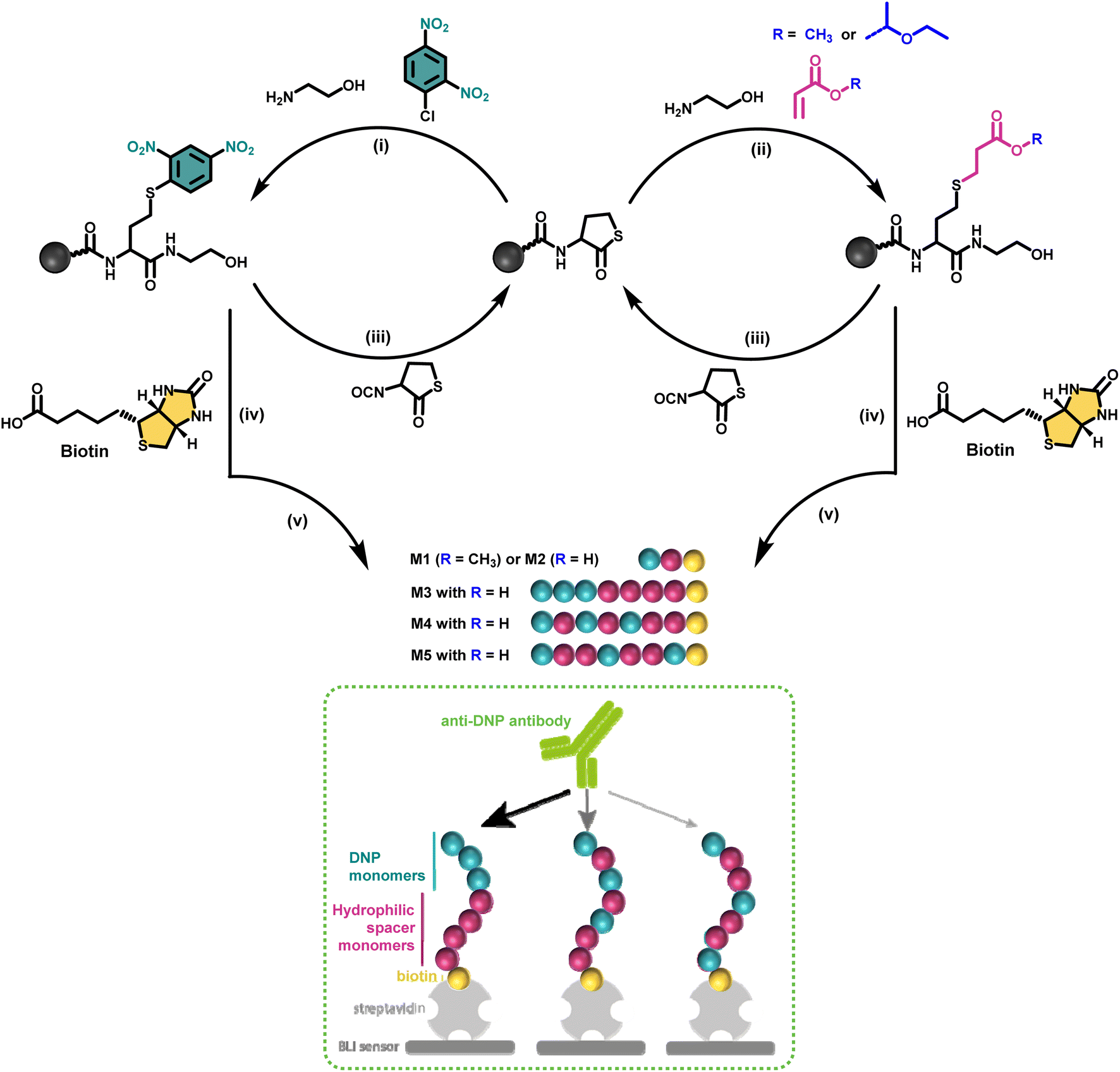 | ||
| Scheme 1 Iterative solid-phase synthesis of sequence-defined antibody recruiting macromolecules bearing DNP and non-functional monomers at different position along the backbone, as well as a biotin unit at one extremity (blue, pink and yellow balls, respectively): (i) aminolysis of the thiolactone ring with ethanolamine and nucleophilic aromatic substitution with DNP-Cl; (ii) aminolysis of the thiolactone ring with ethanolamine and thiol-Michael reaction using either methyl- or 1-ethoxyethyl- acrylate; (iii) chain extension with an isocyanate-functionalised thiolactone (TLa-NCO); (iv) addition of biotin as a terminal unit; (v) cleavage from the Rink amide support and simultaneous deprotection of the acetal unit when using the acetal-containing 1-ethoxyethyl acrylate (M2–M5). The dashed box schematizes the proposed concept of using sequence-defined macromolecules in the area of antibody recruiting. | ||
Additionally, molecular dynamics simulations were undertaken to understand the folding properties of these discrete macromolecules and thus assess the 3D positioning of the functional groups in relation to the BLI measurements.
Results and discussion
Macromolecular design
In a first synthetic strategy, an amine functionalised DNP (DNP-NH2) was employed, which would have allowed to install the DNP functionality when ring opening the TLa moiety. To design the backbone, 2-hydroxyethyl acrylate (2HEA) was introduced to react the acrylate with the liberated thiol. Unfortunately, the obtained structures suffered from impurities and poor water solubility. To mitigate the latter, 2-hydroxyethyl acrylamide was used instead of 2HEA, to replace the ester groups with amides. However, this second approach increased the presence of impurities, which is attributed to the lower efficiency of the thia-Michael addition using acrylamides.28 Furthermore, the overall chain length was found to be limited to a trimer using either protocol, despite the use of fresh reactants and extensive efforts to improve reaction conditions (e.g. various solvents including DMF, THF and chloroform have been tested). Finally, the fact that DNP-NH2 synthesis required already three steps persuaded us to re-design the approach.Alternatively, using ethanolamine for the TLa ring opening, the DNP units could be introduced via a nucleophilic aromatic substitution,29 by employing DNP-Cl, which resulted in quantitative conversion already within 15 minutes of reaction (data not shown). In this approach, methyl acrylate (MA) was employed as a non-functional comonomer. MA was chosen for two reasons. First, it is not expected to contribute to the binding affinity of the macromolecule, which is desired; secondly, MA resulted in quantitative conversion and has no functionality that can interfere with the further elongation of the backbone (i.e. addition of TLa-NCO to elongate the backbone). Hence, for initial experiments to optimise reaction conditions, the obtained structure consisted of only two monomers, i.e. DNP and MA. Next, biotin was conjugated to the –OH end of the DNP-MA dimer via a previously reported procedure involving a DIC/DMAP coupling to form M1 (see Fig. 1 and S1 for complete structure and ESI† for experimental procedures).19
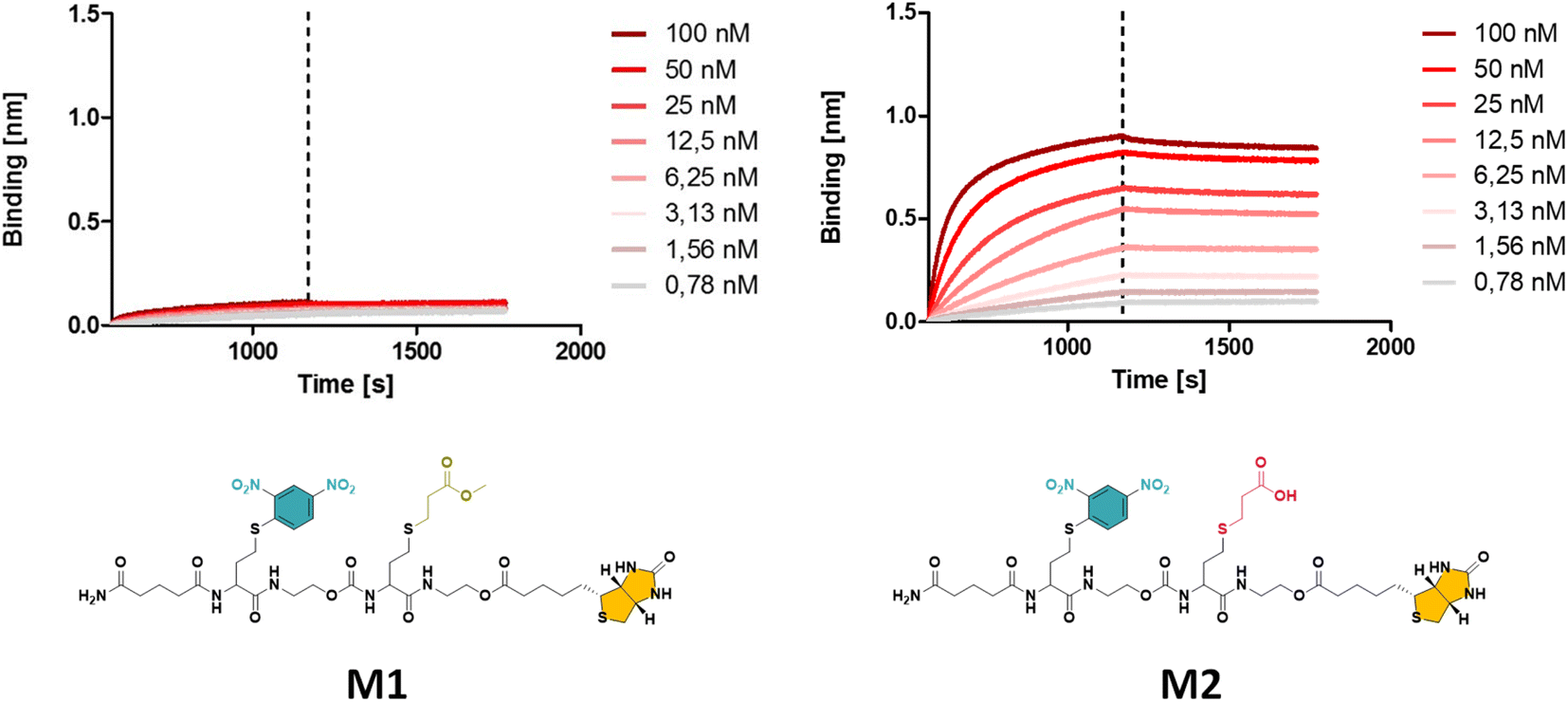 | ||
| Fig. 1 The obtained BLI results for oligomer M1 with a methyl ester (left), displaying a significantly lower binding affinity compared to M2 with a carboxylic acid (right) side group (see ESI† for the synthesis procedure). | ||
While the use of MA is straightforward in terms of the thia-Michael chemistry, we speculated that the presence of a methyl ester in the side chain could decrease the overall hydrophilicity of the sequence-defined macromolecules and therefore negatively impact the binding of anti-DNP antibody. To increase water solubility, attempts to hydrolyse the methyl ester into a carboxylic acid would have been ineffective, as the biotin is conjugated through an identical ester bond, which would very likely also hydrolyse during the reaction. On the other hand, acrylic acid could not be employed as the reactivity of its double bond towards a thiol is much lower, which potentially could lead to incomplete reactions. In addition, the presence of a carboxylic acid on the side chain during the conjugation of biotin (using DIC/DMAP) could yield impurities as the hydroxyl end group of one structure could react with the carboxylic acid containing side chain of another one, thus resulting in the coupling of multiple chains. Alternatively, the use of an acetal-protected monomer, i.e. 1-ethoxyethyl acrylate (1-EA), proved to be ideal, as the hemiacetal ester protecting group can easily be removed without changing the overall structure of the macromolecule. Treatment with trifluoroacetic acid (TFA) is sufficient to deprotect the carboxylic acid and release volatile ethyl vinyl ether. Moreover, the deprotection step can be carried out simultaneously during the cleavage of the macromolecule from the resin in the presence of 10% TFA (in dichloromethane). M2 has been prepared following this final strategy (see Fig. 1 and S2† for complete structure of M2). In the last step of the synthesis, the cleaved macromolecules were lyophilised twice overnight to remove residual TFA.
Next, the short DNP/Biotin containing structures with respectively a methyl ester (M1) and a carboxylic acid (M2) residue in the side chain have been compared in terms of binding affinity using BLI. Initial results showed that binding to anti-DNP antibodies was very low for M1 (Fig. 1, left). While MA was an appropriate choice as a non-functional comonomer in terms of the synthetic approach, the insolubility of the dimer prevented proper interaction with anti-DNP antibody. On the other hand, the BLI analysis of the water soluble carboxylic acid-containing dimer M2 revealed a 5-fold increase in affinity to anti-DNP antibody, compared to the MA-containing counterpart. This observation confirmed our hypothesis that the choice of the comonomer has a strong impact on the binding behaviour and that a more hydrophilic backbone allows for improved antibody binding. After this preliminary study on dimers, a library of longer oligomers (heptamers) with varying positioning of the DNP unit has been synthesised using DNP-Cl and the earlier described 1-EA strategy.
Introducing structural variations to regulate binding properties
While multivalent polymers can contain multiple binding units, their positioning along the backbone is random, regardless of the (post-)polymerisation technique used.30 In a random copolymer, depending on their copolymerisation behaviour, the binding monomers (i.e. DNP units here) can cluster in certain regions, as well as be distantly separated in other regions along the backbone. At the same time, the measured binding affinity of a polymer is an average value for all chains. Therefore, the direct effect of a small variation in the structure (e.g. the effect of the DNP unit positioning) cannot be assessed with disperse (co)polymers.To investigate the effect of the sequence of DNP units along a macromolecule on its antibody binding properties, we designed a small library of 3 heptamers with identical mass, which only varied in the spacing between the DNP units (Fig. 2). Three DNP motifs were introduced along every heptamer, while 1-EA-containing monomers were used to fill the other positions with carboxylic acid functions after deprotection. Initially, we expected that the avidity of these oligomers would increase with increasing spacing between the DNP units, since a larger interspacing would favour the accessibility of DNP motifs to antibodies and favour multivalent interactions.31,32 On the other hand, the highest affinity was observed for M3 for which no spacing monomer was introduced between the three DNP units, with an equilibrium dissociation constant (KD) of 2.6 × 10−10 M, indicating that adjacent positioning of DNP motifs confers a stronger anti-DNP antibody binding (Table 1 and Fig. 3).32
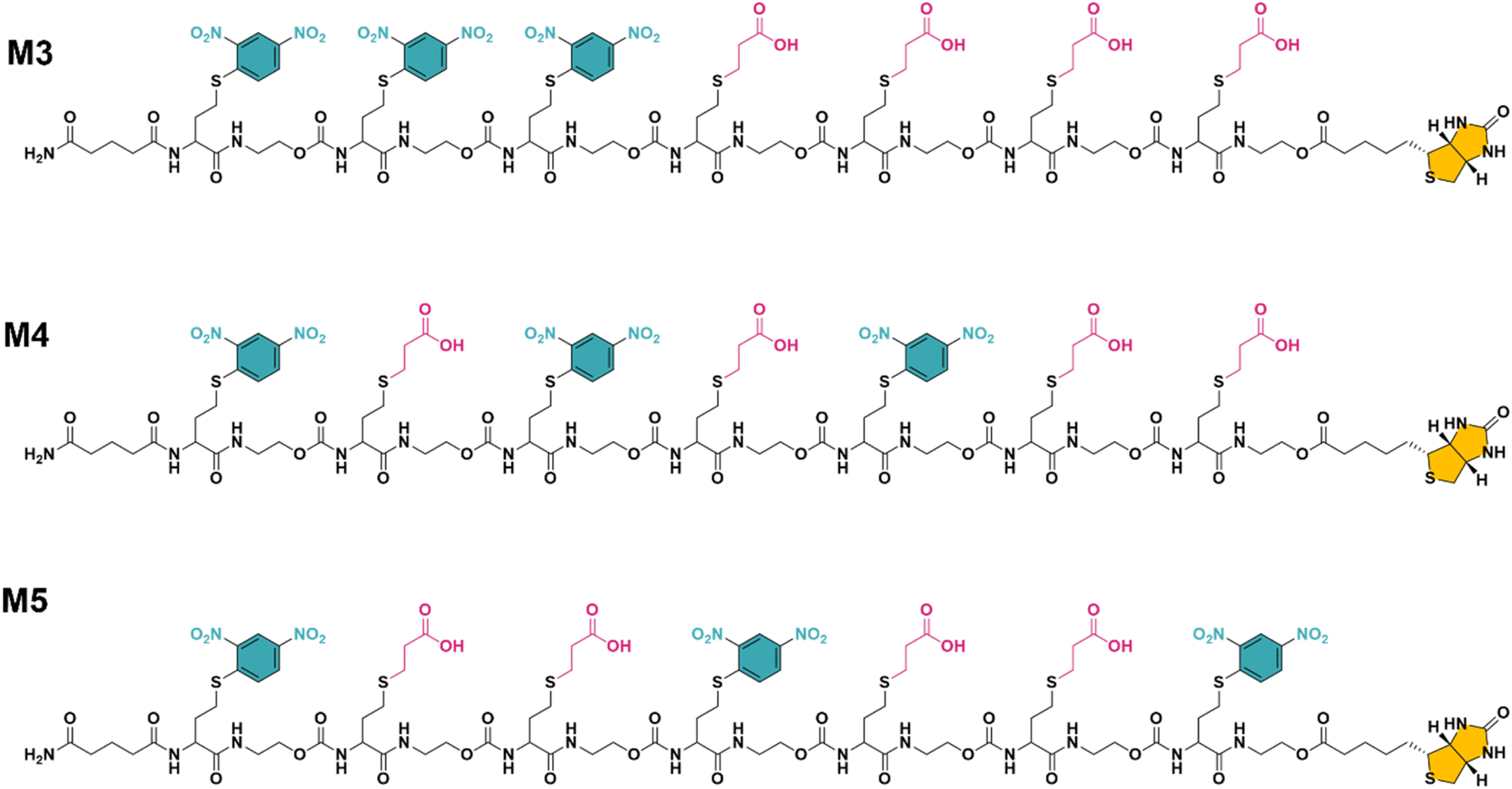 | ||
| Fig. 2 The final structure of three unique heptamers (same exact mass = 2528 Da) synthesised using an optimised procedure with DNP-Cl and 1-EA. Note the increase in spacing between the DNP units from M3 to M5 (see Fig. S3–S5 and ESI† for their synthesis). | ||
| Ligand | K D [M] | R 2 |
|---|---|---|
| M3 | 2.60 × 10−10 | 0.9978 |
| M4 | 8.29 × 10−10 | 0.9990 |
| M5 | 1.05 × 10−9 | 0.9992 |
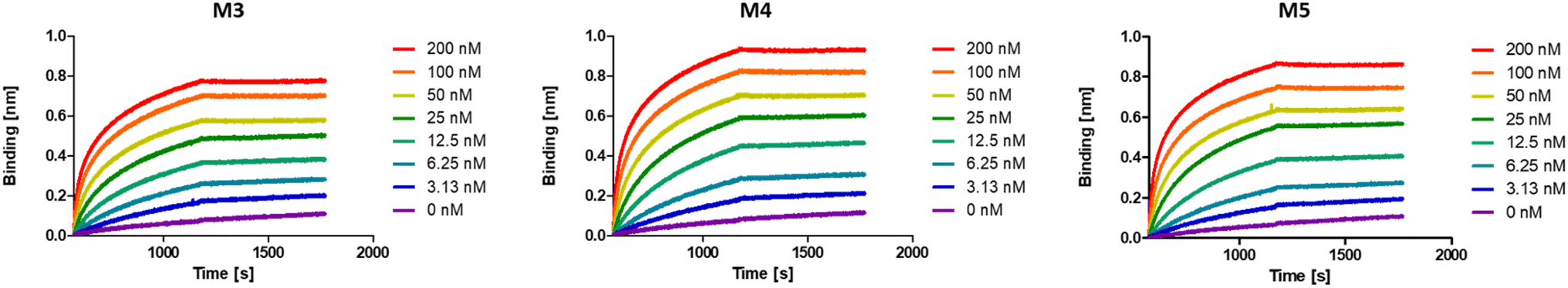 | ||
| Fig. 3 BLI sensorgrams of anti-DNP antibody binding to oligomers M3–M5, displaying the highest binding affinity for M3. | ||
Interestingly, the lowest binding affinity was measured for M5 (KD = 1.05 × 10−9 M), in which two non-binding units were introduced between the DNP units. Moreover, the size of this non-binding spacer does not seem to have a significant influence on the binding avidity since only a small difference could be observed when measuring the binding affinity of M4, where the DNP unit was separated by only one non-binding unit (KD = 8.29 × 10−10 M). Although the KD values do not differ dramatically between the 3 heptamers, they do indicate that the positioning and therefore the interplay between the aromatic groups on these structures are likely of great importance and thus that the sequence effect should be taken into consideration in the design of antibody-recruiting macromolecules. Potentially, stacking between the DNP groups could occur, which would sterically hamper the access to the DNP units. This would in turn influence the binding behaviour of these macromolecules (Fig. 3).
Molecular modelling of the heptamers
For a more in-depth understanding of the folding properties of these macromolecules and the positioning of the functional groups, molecular dynamics (MD) simulations were performed with positional restraints on the biotin moiety for approaching the conditions of BLI measurements. Single oligomers were considered in a water solvent model (see ESI† for computational details), considering the excellent solubility in water and the attachment of isolated molecules to the surface in the conditions of BLI experiments. It was observed that the three different sequences M3, M4, and M5 adopt a globular shape in solution, as a result of an important folding in water (Fig. 4a). The size and the global shape of the folded structure do not significantly vary by modifying the sequence, with rather similar radii of gyration (see Fig. S6†). The intramolecular interactions driving the folding are the hydrogen bonding interactions (occurring between carbamates, amides and carboxylate groups) and the aromatic interactions between DNP units. The final MD snapshots, depicted in Fig. 4a, show that the DNPs for oligomers M4 and M5 are located within the core of folded structure, whereas the DNPs of oligomer M3 are located at its periphery (see positions of DNPs indicated by blue balls).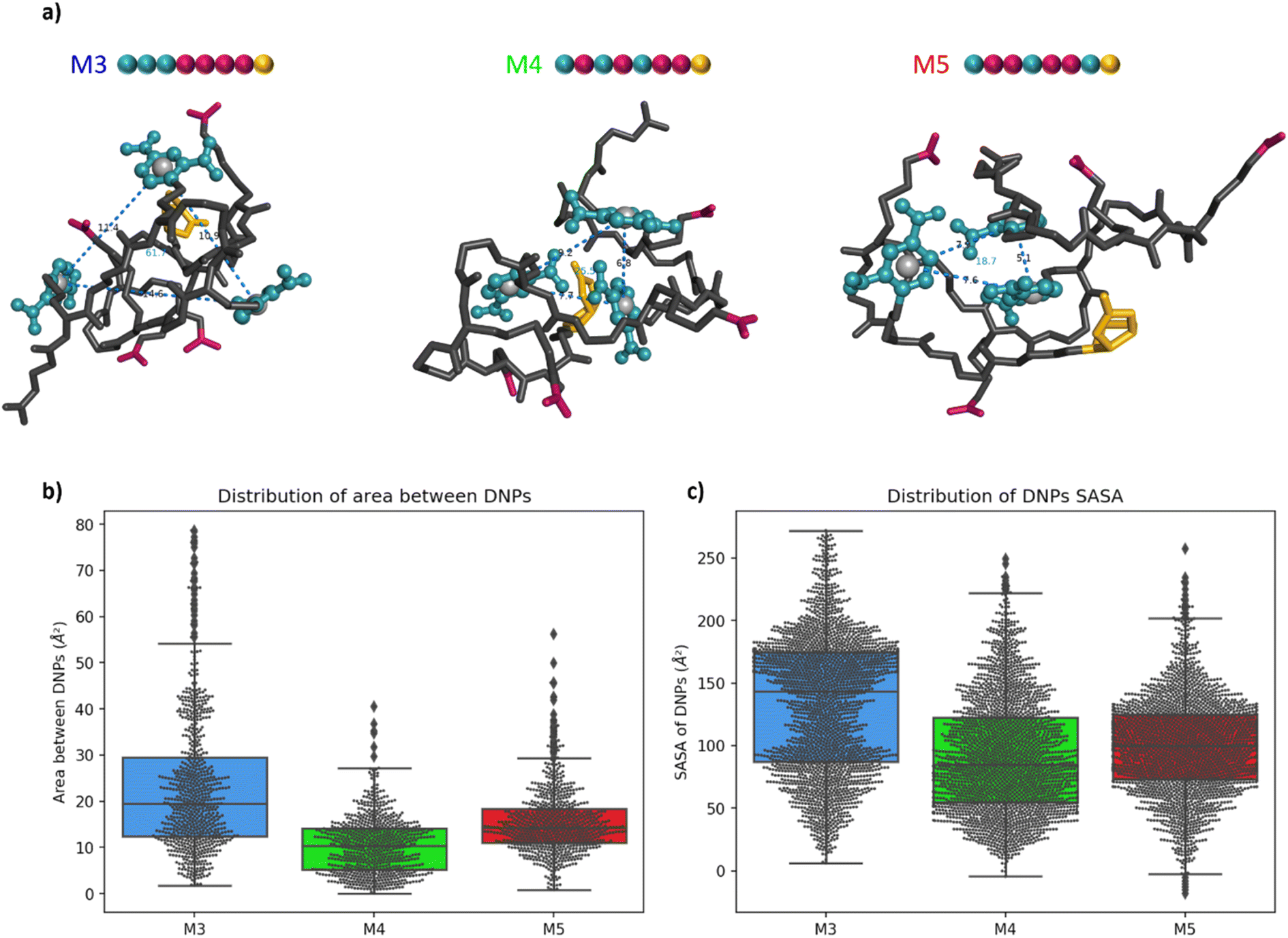 | ||
| Fig. 4 (a) Snapshot of the final MD frame (water molecules not shown for clarity). The DNP ligands are coloured in blue, the carboxylate residues in dark pink and the biotin moieties in gold. The grey spheres are located at the geometric centre of the aromatic cycle of the DNPs. The blue dashed lines measure the distances between the DNP with the labelled value in Å. The blue value inside the triangle represents the area of the triangle in Å2. (b) Box-and-whisker plot of the area between DNPs for the 3 oligomers. The first line of the box represents the first quartile (Q1), the second represents the median (Q2) and the third represents the third quartile (Q3). The small diamonds indicate the outliers, i.e. data values below Q1 − 1.5 × IQR or above Q3 + 1.5 × IQR (c) box-and-whisker plot of the SASA of each DNP for the 3 oligomers. | ||
The accessibility of DNPs is a key parameter to understand the binding of the oligomers to the antibody.33,34 In this context, we focused our analysis on two parameters: the spacing between DNPs and the accessibility. The spacing between the different DNP units is quantified by the area of a triangle that is formed by the geometric centres of the three DNP aromatic cycles. The plots of these areas for M3, M4, and M5 are reported in Fig. 4b. Molecule M3 shows a wide distribution with an interquartile range (IQR) of 17 Å2, which is a significantly larger value compared to molecules M4 and M5, respectively 9 and 7 Å2. The related histograms are given in Fig. S7.† The median of the data is 19.5 Å2 for M3, 10.3 Å2 for M4 and 14.3 Å2 for M5. MD snapshots of heptamer M3 are 68% and 55% beyond the upper quartile of heptamers M4 and M5, respectively. Therefore, oligomer M3 shows in average a larger spacing between the three DNPs compared to the two other structures. Those results suggest that, despite the proximity of the DNP residues in the primary structure, oligomer M3 tends to fold in such a way that the DNPs are far from each other in their 3D conformations compared to M4 and M5.
The accessibility of each DNP was further assessed by estimating its so-called Solvent-Accessible Surface Area (SASA).35 A larger SASA represents a more accessible DNP, which could result in better binding to the antibody.33 The distributions of SASA for all DNPs in Fig. 4c shows that DNPs of oligomer M3 have a larger SASA compared to the DNPs of oligomers M4 and M5 (see Fig. S8† for the distribution plots). The distribution between upper and lower quartile for oligomer M3 spreads on 88 Å2 while it is only 68 and 51 Å2 for oligomers M4 and M5, respectively. The DNPs are more accessible to the solvent in M3, with a median SASA at 143 Å2, which is significantly larger compared to oligomers M4 (85 Å2) and M5 (100 Å2). Moreover, the average SASA of individual DNPs is always larger for M3 than for M4 and M5 (see Table S1†). In summary, DNPs of oligomer M3 are more accessible to the water solvent, and therefore could be more prone to the antibody binding than for the two other cases.
The root-mean-square deviation (RMSD), showing the dynamics of the structures, also reflects this behaviour (see Fig. S9†). For oligomer M3, RMSD profiles of DNPs are larger than that for the whole molecule, showcasing an important flexibility of those residues as a result of their localisation at the periphery of the structure. On the other hand, for oligomers M4 and M5, the DNPs are in the core of the globular structures, resulting in RMSD of DNPs globally equal (for M4) or inferior (for M5) to the global RMSD of the whole oligomers. For the 3 oligomers, it should be noted that a multivalent binding of antibodies is in principle possible given that the spacing between 2 DNPs in extended conformation exceeds the minimum distance between two antibodies (i.e. 1.6 nm, as estimated from analysis of the 1BAF structure in the Protein Data Bank).34,36 However, given the experimental conditions in BLI experiments, where the oligomers are immobilized on a surface at one extremity, we believe that the differences of KD reported in Table 1 are related to accessibility changes of DNP units to the antibody.
Altogether, these results suggest that, counter-intuitively with respect to the sketched 2D structures (Fig. 2), the 3D structures of oligomer M3 show a much better accessibility of DNPs to the antibody binding compared to oligomers M4 and M5, for which DNPs are located at the core of the folded structure. This fully rationalises the KD measurements, which showed a higher affinity (lower KD) for oligomer M3 compared to both M4 and M5, as a result of a higher accessibility of the DNP ligand to the antibodies. In other words, we could demonstrate for the first time through a combined experimental and molecular modelling study that the exact position of the hapten ligand on a macromolecular backbone can allow tuning the binding avidity.
Conclusion
In this study, we report the first sequence-defined antibody recruiting macromolecules that show a structure-dependent binding affinity related to the precise positioning of the binding DNP units. The highest affinity was observed when the binding units were positioned close to each other along the oligomer sequence, whereas the placing of hydrophilic spacer units between the binding units results in a significant drop in binding avidity. Those counter-intuitive results show that 2D structures might be misleading to understand the properties of SDMs, and that the 3D structures and dynamic aspects obtained from MD simulations can help in rationalising the binding affinity results through a molecular view of the ligand accessibility to the antibodies. This knowledge could be used in forthcoming studies to maximise the activity of antibody-recruiting precision macromolecules by a thorough control over their sequence of ligand units.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
This manuscript has been prepared through the contribution of all authors.Conflicts of interest
There is no conflict to declare.Acknowledgements
The authors would like to acknowledge financial support from the Research Foundation Flanders (FWO) and Fonds de la Recherche Scientifique (FNRS) under EOS-project 30650939. B. G. D. G. acknowledges funding from FWO (research grant no. G022521N). N. B., B. G. D. G. and F. E. D. P. thank BOF-UGent for GOA funding. Computational resources have been provided by the Consortium des Équipements de Calcul Intensif (CÉCI), funded by FNRS under grant no 2.5020.11 and by the Wallonia Region. The authors would like to thank Irene de Franceschi and Matthieu Soete for fruitful discussions.Notes and references
- E. Maron, J. H. Swisher, J. J. Haven, T. Y. Meyer, T. Junkers and H. G. Börner, Angew. Chem., Int. Ed., 2019, 58, 10747–10751 CrossRef CAS PubMed.
- R. Aksakal, C. Mertens, M. Soete, N. Badi and F. Du Prez, Adv. Sci., 2021, 8, 2004038 CrossRef CAS PubMed.
- M. A. R. Meier and C. Barner-Kowollik, Adv. Mater., 2019, 31, 1806027 CrossRef PubMed.
- J.-F. Lutz, Macromol. Rapid Commun., 2017, 38, 1700582 CrossRef PubMed.
- S. Aksakal, R. Liu, R. Aksakal and C. R. Becer, Polym. Chem., 2020, 11, 982–989 RSC.
- R. B. Grubbs and R. H. Grubbs, Macromolecules, 2017, 50, 6979–6997 CrossRef CAS.
- K. Matyjaszewski and J. Spanswick, Mater. Today, 2005, 8, 26–33 CrossRef CAS.
- P. Nanjan and M. Porel, Polym. Chem., 2019, 10, 5406–5424 RSC.
- A. J. DeStefano, R. A. Segalman and E. C. Davidson, JACS Au, 2021, 1, 1556–1571 CrossRef CAS PubMed.
- S. Kardas, M. Fossépré, V. Lemaur, A. E. Fernandes, K. Glinel, A. M. Jonas and M. Surin, J. Chem. Inf. Model., 2022, 62, 2761–2770 CrossRef CAS PubMed.
- C. Mertens, R. Aksakal, N. Badi and F. E. Du Prez, Polym. Chem., 2021, 12, 4193–4204 RSC.
- M. A. Reith, S. Kardas, C. Mertens, M. Fossépré, M. Surin, J. Steinkoenig and F. E. Du Prez, Polym. Chem., 2021, 12, 4924–4933 RSC.
- S. C. Solleder, R. V. Schneider, K. S. Wetzel, A. C. Boukis and M. A. R. Meier, Macromol. Rapid Commun., 2017, 38, 1600711 CrossRef.
- X. Wang, X. Zhang and S. Ding, Polym. Chem., 2021, 12, 2668–2688 RSC.
- S. A. Hill, C. Gerke and L. Hartmann, Chem. Asian J., 2018, 13, 3611–3622 CrossRef CAS.
- N. Illy and E. Mongkhoun, Polym. Chem., 2022, 13, 4592–4614 RSC.
- S. Martens, J. Van den Begin, A. Madder, F. E. Du Prez and P. Espeel, J. Am. Chem. Soc., 2016, 138, 14182–14185 CrossRef CAS PubMed.
- I. De Franceschi, C. Mertens, N. Badi and F. Du Prez, Polym. Chem., 2022, 13, 5616–5624 RSC.
- M. A. Reith, I. De Franceschi, M. Soete, N. Badi, R. Aksakal and F. E. Du Prez, J. Am. Chem. Soc., 2022, 144, 7236–7244 CrossRef CAS PubMed.
- M. Soete, C. Mertens, R. Aksakal, N. Badi and F. Du Prez, ACS Macro Lett., 2021, 10, 616–622 CrossRef CAS PubMed.
- M. Soete, K. De Bruycker and F. Du Prez, Angew. Chem., Int. Ed., 2022, 61, e202116718 CAS.
- M. Soete, C. Mertens, N. Badi and F. E. Du Prez, J. Am. Chem. Soc., 2022, 144, 22378–22390 CrossRef CAS PubMed.
- P. J. McEnaney, C. G. Parker, A. X. Zhang and D. A. Spiegel, ACS Chem. Biol., 2012, 7, 1139–1151 CrossRef CAS PubMed.
- B. P. M. Lake, R. G. Wylie, C. Bařinka and A. F. Rullo, Angew. Chem., Int. Ed., 2023, 62, e202214659 CrossRef CAS PubMed.
- A. Uvyn, R. De Coen, M. Gruijs, C. W. Tuk, J. De Vrieze, M. van Egmond and B. G. De Geest, Angew. Chem., Int. Ed., 2019, 58, 12988–12993 CrossRef CAS PubMed.
- R. De Coen, L. Nuhn, C. Perera, M. Arista-Romero, M. D. P. Risseeuw, A. Freyn, R. Nachbagauer, L. Albertazzi, S. Van Calenbergh, D. A. Spiegel, B. R. Peterson and B. G. De Geest, Biomacromolecules, 2020, 21, 793–802 CrossRef CAS PubMed.
- S. Achilli, N. Berthet and O. Renaudet, RSC Chem. Biol., 2021, 2, 713–724 RSC.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- M. Soete and F. E. Du Prez, Angew. Chem., Int. Ed., 2022, 61, e202202819 CAS.
- E. Zumbro and A. Alexander-Katz, ACS Omega, 2020, 5, 10774–10781 CrossRef CAS PubMed.
- P. Hoyos, A. Perona, O. Juanes, Á. Rumbero and M. J. Hernáiz, Chem.–Eur. J., 2021, 27, 7593–7624 CrossRef CAS PubMed.
- H. Jung, T. Yang, M. D. Lasagna, J. Shi, G. D. Reinhart and P. S. Cremer, Biophys. J., 2008, 94, 3094–3103 CrossRef CAS PubMed.
- K. N. Houk, A. G. Leach, S. P. Kim and X. Zhang, Angew. Chem., Int. Ed., 2003, 42, 4872–4897 CrossRef CAS.
- A. Uvyn, C. Tonneaux, M. Fossépré, A. Ouvrier-Buffet, B. de Geest and M. Surin, Chem.–Eur. J., 2023, e202300474 CrossRef.
- J. Weiser, P. S. Shenkin and W. C. Still, J. Comput. Chem., 1999, 20, 217–230 CrossRef CAS.
- A. T. Brünger, D. J. Leahy, T. R. Hynes and R. O. Fox, J. Mol. Biol., 1991, 221, 239–256 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc01507f |
| This journal is © The Royal Society of Chemistry 2023 |