
Surface chemistry mediates the tumor entrance of nanoparticles probed using single-molecule dual-imaging nanodots†
Huiming
Ren
 a,
Qiuhui
Hu
ab,
Yuji
Sun
a,
Xiaoxuan
Zhou
b,
Yincong
Zhu
a,
Qiuyang
Dong
a,
Linying
Chen
a,
Jianbin
Tang
a,
Hongjie
Hu
b,
Youqing
Shen
a and
Zhuxian
Zhou
*a
a,
Qiuhui
Hu
ab,
Yuji
Sun
a,
Xiaoxuan
Zhou
b,
Yincong
Zhu
a,
Qiuyang
Dong
a,
Linying
Chen
a,
Jianbin
Tang
a,
Hongjie
Hu
b,
Youqing
Shen
a and
Zhuxian
Zhou
*a
aKey Laboratory of Biomass Chemical Engineering of Ministry of Education and Zhejiang Key Laboratory of Smart Biomaterials, College of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310027, China. E-mail: zhouzx@zju.edu.cn
bDepartment of Radiology, Sir Run Run Shaw Hospital (SRRSH) of School of Medicine, Zhejiang University, Hangzhou, Zhejiang 310027, China
First published on 22nd August 2023
Abstract
The active transport of nanoparticles into solid tumors through transcytosis has been recognized as a promising way to enhance tumor accumulation and penetration, but the effect of the physicochemical properties of nanoparticles remains unclear. Herein, we develop a type of single-molecule dual imaging nanodot by divergent growth of perylenediimide (PDI)-dye-cored polylysine dendrimers and internal orthogonal conjugation of Gd(III)-based macrocyclic probes for fluorescence imaging and magnetic resonance imaging (MRI) of surface chemistry-dependent tumor entrance. The MRI and fluorescence imaging show that sixth-generation nanodots with acetylated (G6-Ac) and oligo ethylene glycol (G6-OEG) surfaces exhibit similar high tumor accumulation but different intratumor distribution. Cellular uptake and transport experiments suggest that G6-Ac nanodots have lower lysosomal entrapment (61% vs. 83%) and a higher exocytotic rate (47% vs. 29%) than G6-OEG. Therefore, G6-Ac is more likely to undergo intercellular transport through cell transcytosis, and is able to reach a tumor area distant from blood vessels, while G6-OEG mainly enters the tumor through enhanced permeability and retention (EPR) effect-based passive transport, and is not able to deliver to distant tumor areas. This study suggests that it is possible to boost the tumor entrance of nanoparticles by engineering surface chemistry for active transport.
Introduction
The preferential accumulation of nanoparticles into solid tumors through leaky blood vessels has been the central paradigm in traditional cancer nanomedicine.1 However, the low frequency and high heterogeneity of vascular gaps lead to poor tumor accumulation.2 Moreover, the passive diffusion of nanoparticles from blood vessels to distinct tumors is limited due to a highly dense extracellular matrix and increased fluid pressure.3 Limited tumor accumulation and penetration have explained the predicament of nanomedicines for clinical translation in the past 30 years.4–6 According to a recent report, the active transport of nanoparticles through cellular transcytosis is an overarching way of tumor accumulation, even up to 97%.7 The active transport of nanoparticles into solid tumors has shown great promise in enhancing cancer therapeutic efficacy,8–11 which has been recognized as a new opportunity for cancer nanomedicine,6,12 but the mechanism and influencing factor behind the process remain unclear, and it is urgent and challenging to explore them.The surface chemistry of nanoparticles primarily governs nano–bio interactions and determines their pharmacokinetics and tumor entrance.13–15 Surface modification of polyethylene glycol (PEG) has been a standard way to develop clinical cancer nanomedicines. PEGylation can minimize protein absorption, prolong blood circulation time, and enhance tumor accumulation.16 However, PEGylation also minimizes the interaction with cancer cells, reduces cellular uptake, and generates potential immunogenicity after repeated administrations.17 Moreover, PEGylated nanomedicines, such as Doxil (PEGylated liposomal doxorubicin), cannot penetrate deep tumor tissue through passive diffusion. We previously found that Doxil primarily entered cells through clathrin-mediated endocytosis and was trapped in the lysosome, thus unable to generate efficient intercellular transport.18 Therefore, PEGylation is not a suitable surface modification for developing active transporting nanomedicines. The surface chemistry of nanoparticles can significantly affect their endocytic pathway and determine their capability of inducing cell transcytosis.19,20 It is essential to explore the role of surface chemistry in tumor entrance and discover an appropriate surface modification to boost active tumor transportation.
Many studies have been conducted to explore the effect of surface chemistry of nanoparticles on their in vitro and in vivo fate via nanodots. However, the precise exploration of the structure–activity relationship is limited due to the many drawbacks of the current nanodots. The random surface conjugation of imaging probes such as dye molecules will produce mixed dye-conjugated nanodots with a Poisson distribution of the dye ratio.21 The fluorophore ratio on the dendrimer surface significantly impacts cellular uptake and intracellular fluorescence lifetime.21 Moreover, the physicochemical properties of the surface dye molecules will also affect the cellular uptake and biodistribution. It is reported that the surface conjugation of a polymer with cyanine 5 (Cy5) will induce cell mitochondrion targeting, and labeling water-soluble sulfo-Cy5 will result in lysosomal entrapment.22 It is also reported that some dye molecules, such as heptamethine, have the intrinsic property of tumor-specific targeting.23,24 We have developed a type of single-molecule dye-cored nanodot with a precise structure and excellent fluorescence, which is suitable for precisely exploring nano–bio interactions.25,26
Fluorescence imaging has the advantages of high sensitivity and convenience, but has limitations in imaging deep tissue.27 Magnetic resonance imaging (MRI), as a routinely used diagnostic tool in clinical settings, benefits from non-invasiveness, unrestricted tissue penetration depth, and high spatial resolution but has low sensitivity.28,29 The fluorescence–MRI dual-modal imaging strategy is complementary and excellent for biomedical imaging.30–32 Herein, we develop a type of single-molecule nanodot with fluorescence and MRI dual imaging capability to precisely explore the surface-dependent tumor entrance. The nanodots were obtained by divergent growth of perylenediimide (PDI)-cored polylysine dendrimers with internal conjugation of multiple Gd(III)-based macrocyclic probes (Fig. 1a). We showed the size-dependent blood circulation and tumor accumulation of the nanodots. Using dual-modal single-molecule imaging, we found that the sixth-generation nanodots with acetylated and oligo ethylene glycol (OEG) surfaces exhibited similar tumor accumulation but different tumor penetration and intra-distribution (Fig. 1b). G6-Ac could penetrate tumor areas deeply through cellular transcytosis while G6-OEG is mainly accumulated in the peripheral tumor via the EPR effect (Fig. 1b). This study suggests that an acetylated surface may be suitable for developing active transporting nanomedicines, and surface chemistry engineering can enhance the transcytosis-inducing capability. Moreover, the single-molecule dual-modal imaging probe is appropriate to reveal precise nano–bio interactions.
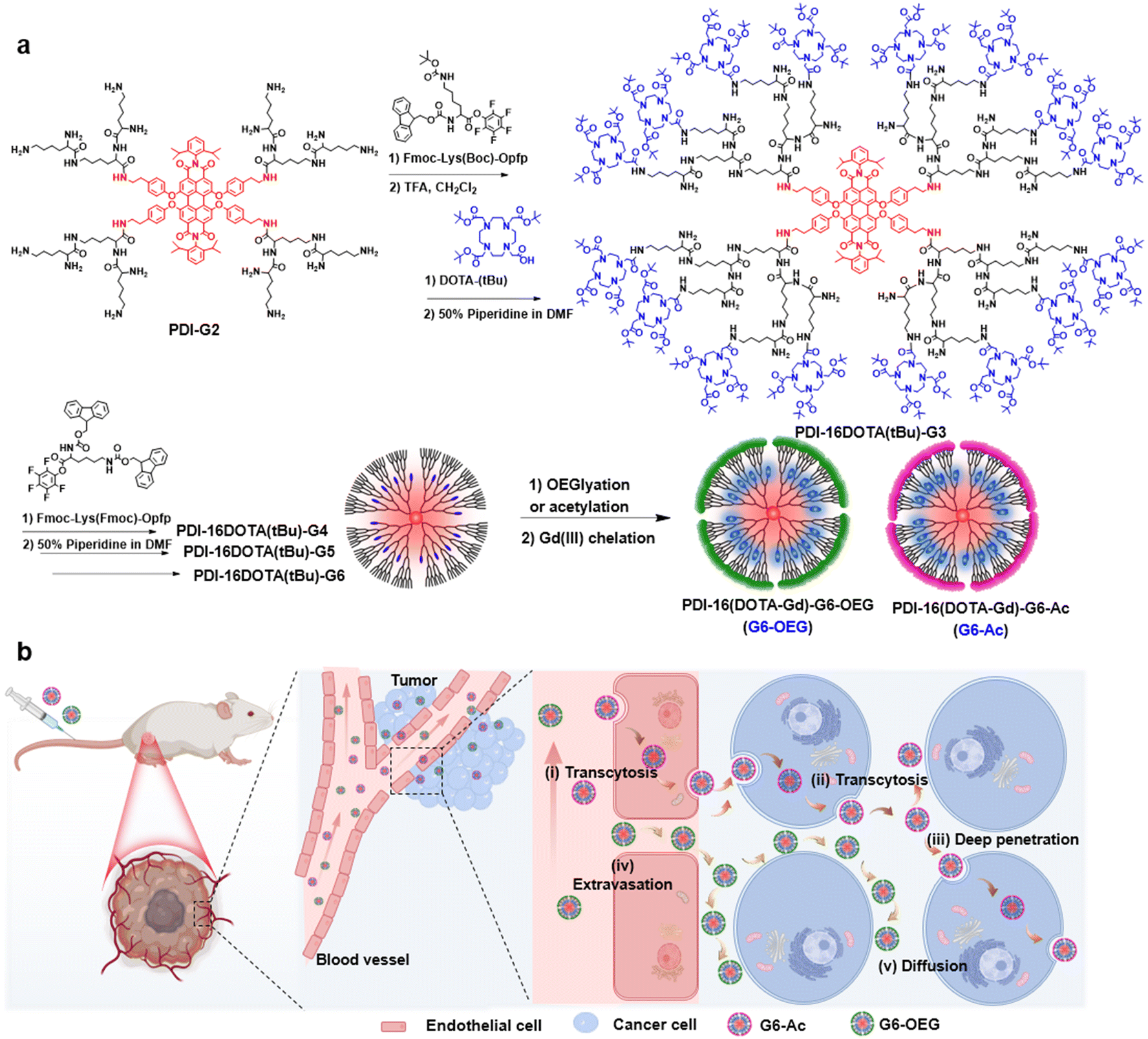 | ||
| Fig. 1 Synthesis and schematic illustration of the PDI-cored and (DOTA-Gd)-inlayed nanodots for dual-modal imaging of the tumor entrance. (a) Synthetic route to the fluorescence and MRI dual-imaging nanodots of the sixth-generation with a OEGylated or acetylated surface. (b) Illustration of the surface chemistry-dependent tumor entrance of nanodots primarily by active transcytosis (i–iii) or passive diffusion (iv–v). | ||
Results and discussion
Synthesis and characterization of the dual imaging nanodots
To obtain single-molecule multifunctional nanodots for dual-modal imaging, we designed PDI-cored and (DOTA-Gd)-inlayed PLL dendrimers. These dendrimeric nanodots were synthesized through orthogonal protection and selective conjugation using Fmoc- and Boc-protected lysine (Fig. 1a). The PDI-cored PLL dendrimer of the second generation (PDI-G2) was prepared according to our previous report25 and reacted with Fmoc–Lys(Boc)–OPFP (Fig. S1†), followed by the selective removal of the Boc-protection to obtain PDI-16Fmoc-G2.5 (Fig. S2 and Scheme S1†). The amine groups of PDI-16Fmoc-G2.5 were coupled with the carboxylate group of the macrocyclic chelator molecule, DOTA(tBu), followed by the removal of Fmoc to obtain PDI-16DOTA(tBu)-G3 (Scheme S2†). High generations of the nanodots were obtained by divergent synthesis, repeating the conjugation of Fmoc–Lys(Fmoc)–OPFP and Fmoc-deprotection (Fig. S3†). The dendrimers were surface modified with acetylation or OEG and the inner DOTA(tBu) groups were deprotected and chelated with Gd(III) to obtain PDI-cored and (DOTA-Gd)-embedded dual-modal imaging nanodots (Scheme S3†).The structures of the functional dendrimers and intermediate products were characterized by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectra (MS) (Fig. 2a, S5 and Table S1†), 1H NMR (Fig. S2 and S4†) and GPC (Fig. 2b). The MALDI-TOF MS showed that the targeted molecular weight values were consistent with their theoretical values, suggesting the successful preparation of the compounds. For example, PDI-16DOTA(tBu)-G3 showed a molecular weight peak at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 706.84 Da ([M]+), which is close to the theoretical value of 13707.13 Da. PDI-16Fmoc-G2.5 showed a molecular weight peak at 4841.97 Da, which is approximately equal to the theoretical value of 4840.44 ([M − 16Fmoc + H]+), with the Fmoc groups removed under intense laser pulses. In the gel permeation chromatography (GPC) traces (Fig. 2b), the molecular weight of the dendrimers increased with the dendrimer generations. In addition, all dendrimers showed a single peak with a polydispersity index (PDI, Mw/Mn) less than 1.15 (Table S2†), indicating their well-defined chemical structures. The high-performance liquid chromatography (HPLC) results showed that all the dendrimers exhibited a single peak (Fig. 2c). All the results proved the successful synthesis of the customized PDI-cored and DOTA-embedded nanodots with well-defined structures.
706.84 Da ([M]+), which is close to the theoretical value of 13707.13 Da. PDI-16Fmoc-G2.5 showed a molecular weight peak at 4841.97 Da, which is approximately equal to the theoretical value of 4840.44 ([M − 16Fmoc + H]+), with the Fmoc groups removed under intense laser pulses. In the gel permeation chromatography (GPC) traces (Fig. 2b), the molecular weight of the dendrimers increased with the dendrimer generations. In addition, all dendrimers showed a single peak with a polydispersity index (PDI, Mw/Mn) less than 1.15 (Table S2†), indicating their well-defined chemical structures. The high-performance liquid chromatography (HPLC) results showed that all the dendrimers exhibited a single peak (Fig. 2c). All the results proved the successful synthesis of the customized PDI-cored and DOTA-embedded nanodots with well-defined structures.
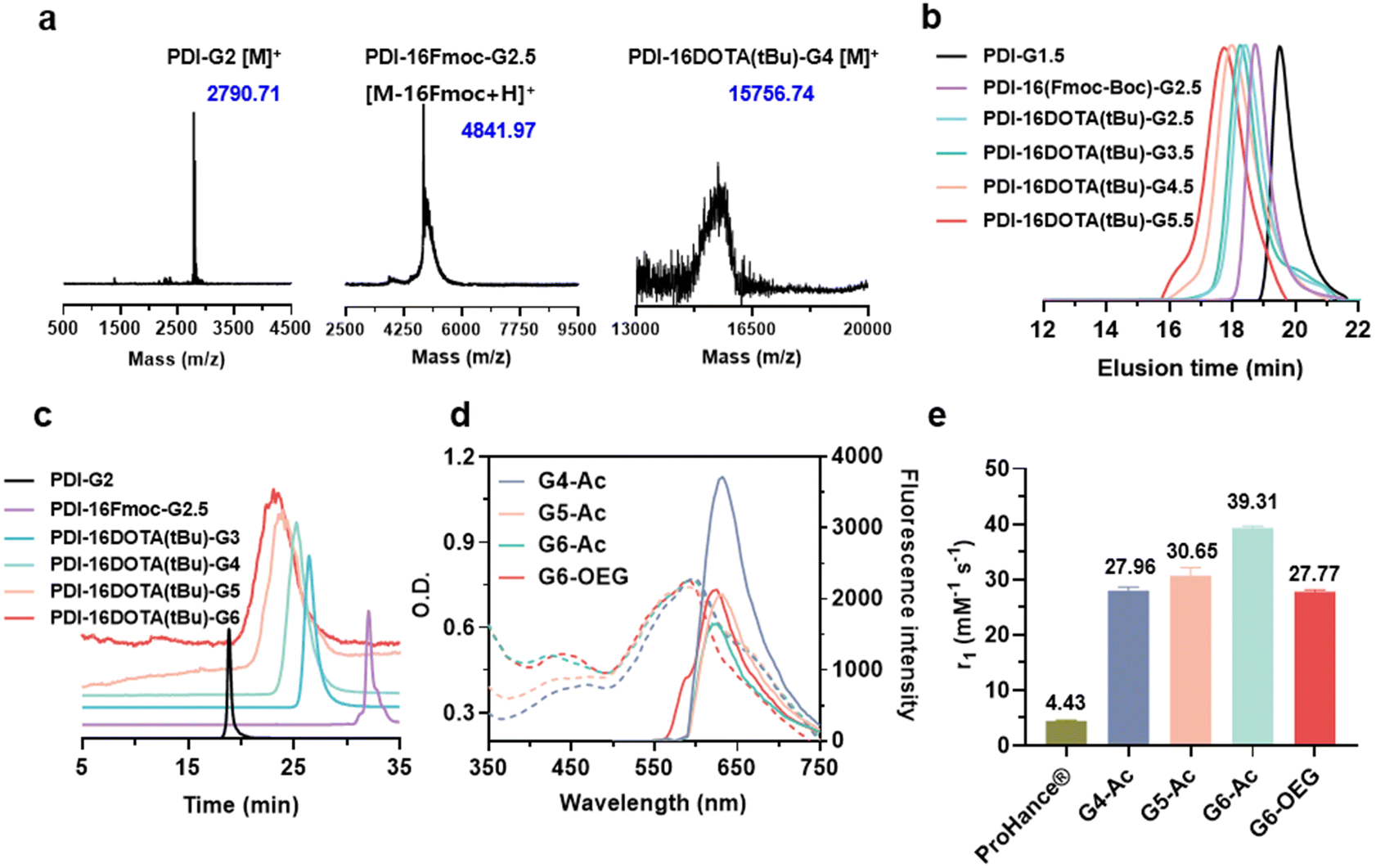 | ||
| Fig. 2 Characterization and physiochemical properties of the PDI-cored and (DOTA-Gd)-embedded dual-modal imaging nanodots. (a) MALDI-TOF mass spectra of the representative dendrimers. (b) Representative gel permeation chromatography (GPC) trace spectra (water, 50 °C, 0.8 mL min−1, Gx.5 stands for the Fmoc-protected dendrimers). (c) Representative high-performance liquid chromatography (HPLC) trace spectra. (d) Absorption and emission spectra of different nanodots at a concentration of 40 mM. (e) Longitudinal relaxivities (r1) values of ProHance® and nanodots. | ||
We obtained the dual-modal imaging nanodots with acetylated (Gn-Ac, n = 4–6) and OEGylated (G6-OEG) surfaces (Fig. 1a and Scheme S3†). The acetylated nanodots of different dendrimer generations were used for studying the size-dependent tumor entrance. The nanodots of the sixth generation with either an acetylated or OEGylated surface were used for exploring the surface chemistry-dependent tumor entrance. The average hydrodynamic diameters of G4-Ac, G5-Ac, and G6-Ac were 4.9 ± 0.6 nm, 6.4 ± 0.8 nm, and 8.3 ± 1.0 nm, respectively, as determined by dynamic light scattering (DLS) (Fig. S6†). The morphology and particle size of G6-Ac were further studied by transmission electron microscopy (TEM) (Fig. S7†). These nanodots were uniform and well-dispersed with a spherical morphology and an average diameter of 6.9 ± 0.8 nm, which was smaller than that measured by DLS due to the different forms of the nanodots in dryness and aqueous solution. All the nanodots were slightly negatively charged (−2 to −15 mV) after Gd(III) chelation (Fig. S8†), favorable for long blood circulation.33
We first studied the optical properties of the nanodots. All the nanodots showed maximum UV absorbance at about 595 nm and maximum emission at 625 nm (Fig. 2d). The fluorescence intensity of high-generation nanodots (G5 and G6) decreased compared to that of the lower generation nanodots (G4), probably due to the perturbation of inner DOTA moieties on the electronic network of the PDI molecule at high generations. We next studied the relaxivities of the nanodots, which are essential for MRI probes to provide high contrast enhancement.34
The longitudinal relaxivities (r1) of Gn-Ac (n = 4–6) ranged from 27.96 to 39.31 mM−1 s−1, exhibiting an increase with dendrimer size due to the enhancement of structural rigidity and the restriction of the rotational motion of gadolinium ions.35 The r1 of the nanodots was about 6.3–8.9 fold that of the commercialized small molecular contrast agent, ProHance® (4.43 mM−1 s−1). Interestingly, we found that surface chemistry also affected the relaxivity, so the r1 value of G6-OEG (27.77 mM−1 s−1) was about 70% that of G6-Ac (Fig. 2e, Fig. S9, and Table S3†). This may be related to the better hydrophilicity of the OEG surface, resulting in a more open and soft inner microenvironment in aqueous solution.
Fluorescence and MRI imaging of size and surface chemistry-dependent tumor accumulation
The nanodots’ strong and stable PDI fluorescence allows in vivo cancer fluorescence imaging. We studied the tumor entrance of the nanodots with different sizes (Gn-Ac, n = 4–6) and surface chemistry (G6-Ac and G6-OEG) in mice bearing orthotopic 4T1 breast tumors after intravenous injection (Fig. 3a, Fig. S10 and S11†). G4-Ac and G5-Ac with small sizes were quickly eliminated from the blood circulation, excluded through renal filtration, and accumulated in the bladder. In comparison, G6-Ac and G6-OEG showed long circulation in the blood, gradually accumulated at the tumor sites, reached the maximum fluorescence intensity at 48 h post-injection, and maintained high signals in tumors over a week (Fig. 3a). Quantitative fluorescence analysis revealed that tumor accumulation of G6-Ac and G6-OEG was similar, but the fluorescence intensity of G6-OEG in normal tissues was higher than that of G6-Ac (Fig. 3b). The tumor-to-normal ratio (TNR) analysis showed that G6-Ac had a much higher TNR than G6-OEG at all times. For instance, the TNR of G6-Ac was 3.32 at 48 h, which was 1.6-fold that of G6-OEG (TNR = 2.04) at the same time. The fluorescence imaging suggested that the OEGylated nanodots had better blood retention, resulting in high fluorescence in the background of normal tissue. These results indicated that although the OEGylated and acetylated nanodots showed similar tumor accumulation, their tumor entrance mechanisms might be different.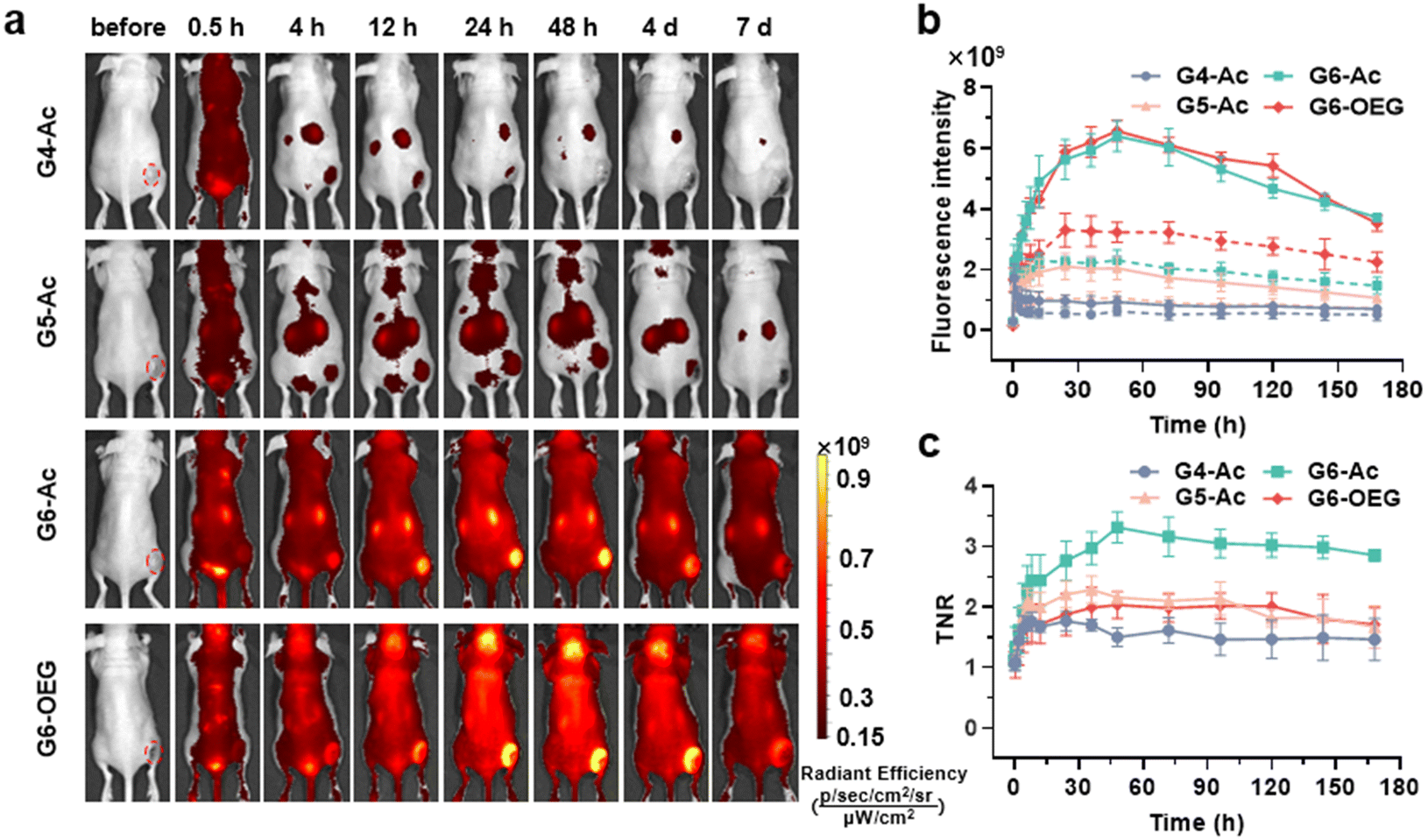 | ||
| Fig. 3 Fluorescence imaging of the size and surface chemistry-dependent tumor entrance via the nanodots in mice bearing 4T1 orthotopic breast tumors. (a) Whole-body fluorescence imaging of mice bearing 4T1 breast tumors after intravenous injection of the nanodots (100 nmol). (b) Quantitative fluorescence intensity signals in the tumor (red circles indicate tumor ROI) at different imaging times. The solid line denotes the tumor tissue and the dotted line means the normal tissue at a symmetrical position in relation to the tumor. (c) Tumor-to-normal ratio (TNR) at different imaging times. Data are represented as mean ± SD (n = 3). | ||
We further studied the effect of surface chemistry on the tumor entrance of the nanodots by MRI, which allowed us to image the spatial distribution of nanodots. T1-weighted MRI images were taken at designated times after intravenous injection of G6-Ac, G6-OEG, or ProHance® at 0.1 mM Gd kg−1 (Fig. 4a). Both the nanodots provided prolonged and enhanced contrast in the tumor tissue. In contrast, the small molecular contrast agent only showed contrast enhancement at 15 min, and the signal quickly decreased to a background level. The contrast-to-noise ratio (CNR) was employed to quantitatively assess the contrast enhancement in the tumor tissue (Fig. 4b). The CNRs of the G6-Ac and G6-OEG groups gradually and continuously increased within 48 h, reaching a maximum value of 58.4 and 41.2, respectively. In comparison, the small molecular contrast could quickly extravasate from blood vessels to accumulate in the tumor but was also quickly eliminated from the tumor, with the CNR decreasing from 15.6 to 5.3 in 2 h. The time-dependent tumor accumulation behavior of the nanodots was consistent with that observed in the fluorescence imaging. We further analyzed the intratumor distribution of the nanodots. Both the nanodots accumulated in the tumor periphery at the beginning and gradually penetrated the deep tumor. G6-Ac showed better tumor penetration and provided more robust contrast enhancement in the tumor interior than G6-OEG (Fig. 4c). We also tracked the normal tissue distribution of nanodots using whole-body MRI imaging and quantitatively analyzed the images considering the signal enhancement ratio (ER) (Fig. S12†). The ER in the artery was 3.5 and 2.6 for the G6-OEG and G6-Ac groups at 15 min, respectively, gradually decreasing with time. The superior ER of G6-OEG compared to that of G6-Ac is due to its better blood retention (Fig. S12a†). Both nanodots showed a decreased ER in the kidneys from 2.0–2.4 to 1.3–1.4 in 48 h, indicating their gradual clearance from the body via kidney filtration (Fig. S12b†). Both nanodots had an ER of less than 1.1 in the liver during imaging, suggesting their low liver accumulation (Fig. S12c†).
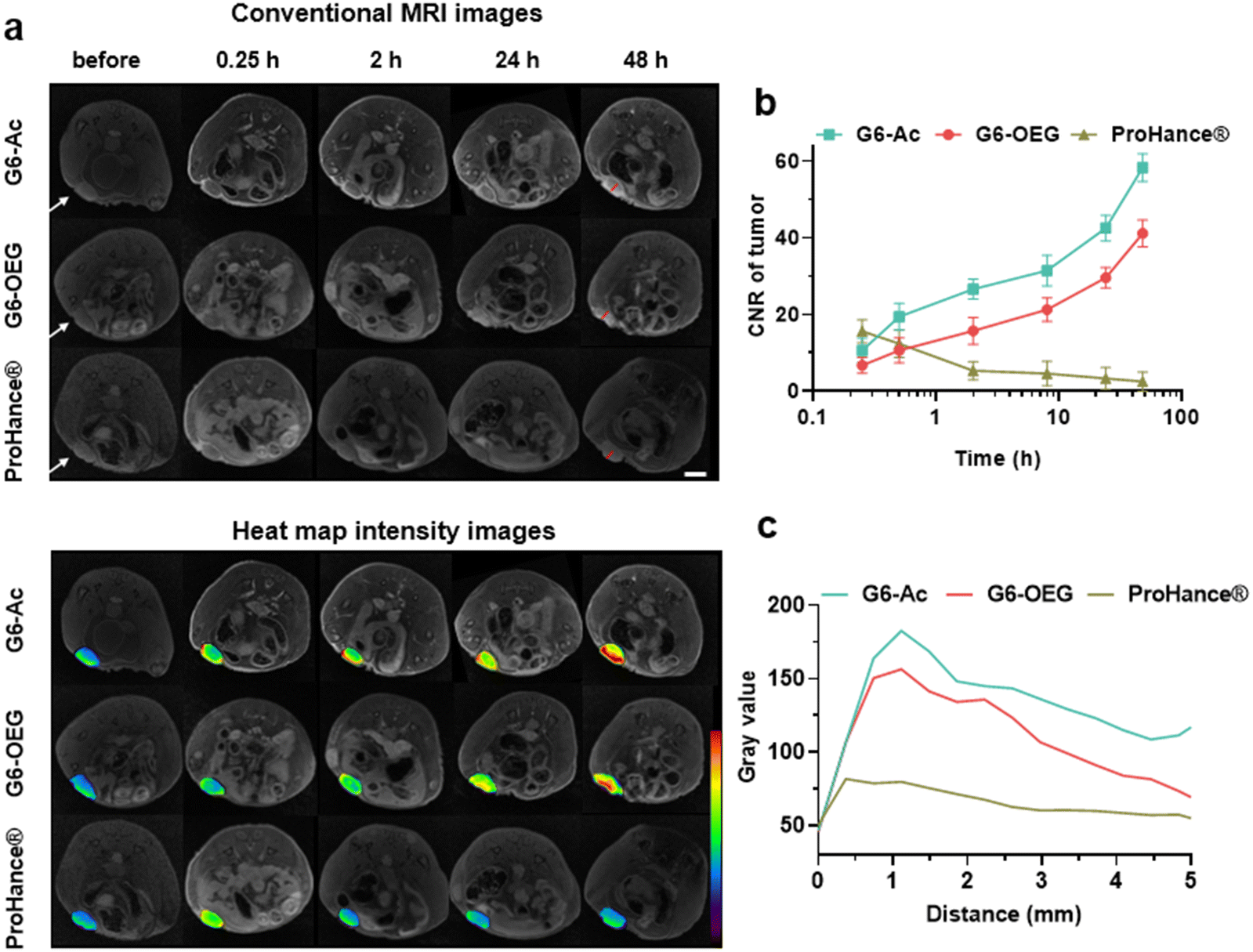 | ||
| Fig. 4 MR imaging of the surface chemistry-dependent tumor entrance via the nanodots in mice bearing 4T1 orthotopic breast tumors. (a) Representative T1-weighted MR images and their pseudo-color map images before (pre) and after injection of ProHance® and nanodots, at 0.1 mmol Gd kg−1 (scale bar = 10 mm, the tumor is indicated with the white arrow). (b) Contrast-to-noise ratio (CNR) in the tumor at different times. (c) Gray value from the tumor rim to the tumor interior, as indicated by red lines in (a). | ||
The tumors from mice 48 h post-injection of the nanodots were sectioned and subjected to immunofluorescence staining with CD31 to visualize the vasculature. The distribution and co-localization of the nanodots and tumor vasculature were imaged by confocal fluorescence imaging (Fig. 5). G4-Ac and G5-Ac showed low fluorescence in tumor tissue due to fast clearance (Fig. 5a–c), consistent with the live fluorescence imaging (Fig. 3a). G6-Ac and G6-OEG had strong and similar fluorescence intensities in all the tumor slides. We delineated the vascular-rich and avascular areas according to the vascular fluorescence immunostaining (Fig. 5a) and analyzed the distribution of the nanodots (Fig. 5c). Interestingly, we found that the fluorescence of G6-Ac was much more localized in the avascular region than that in the vasculature-rich region (78% vs. 22%), whereas the fluorescence of G6-OEG was mainly distributed in the vasculature-rich area (70%). The results showed that G6-OEG was primarily localized around blood vessels, but G6-Ac could travel to areas far from blood vessels and penetrate the tumor deeply, consistent with the MR images. The results also suggested that the tumor entrance mechanisms of G6-OEG and G6-Ac might be different.
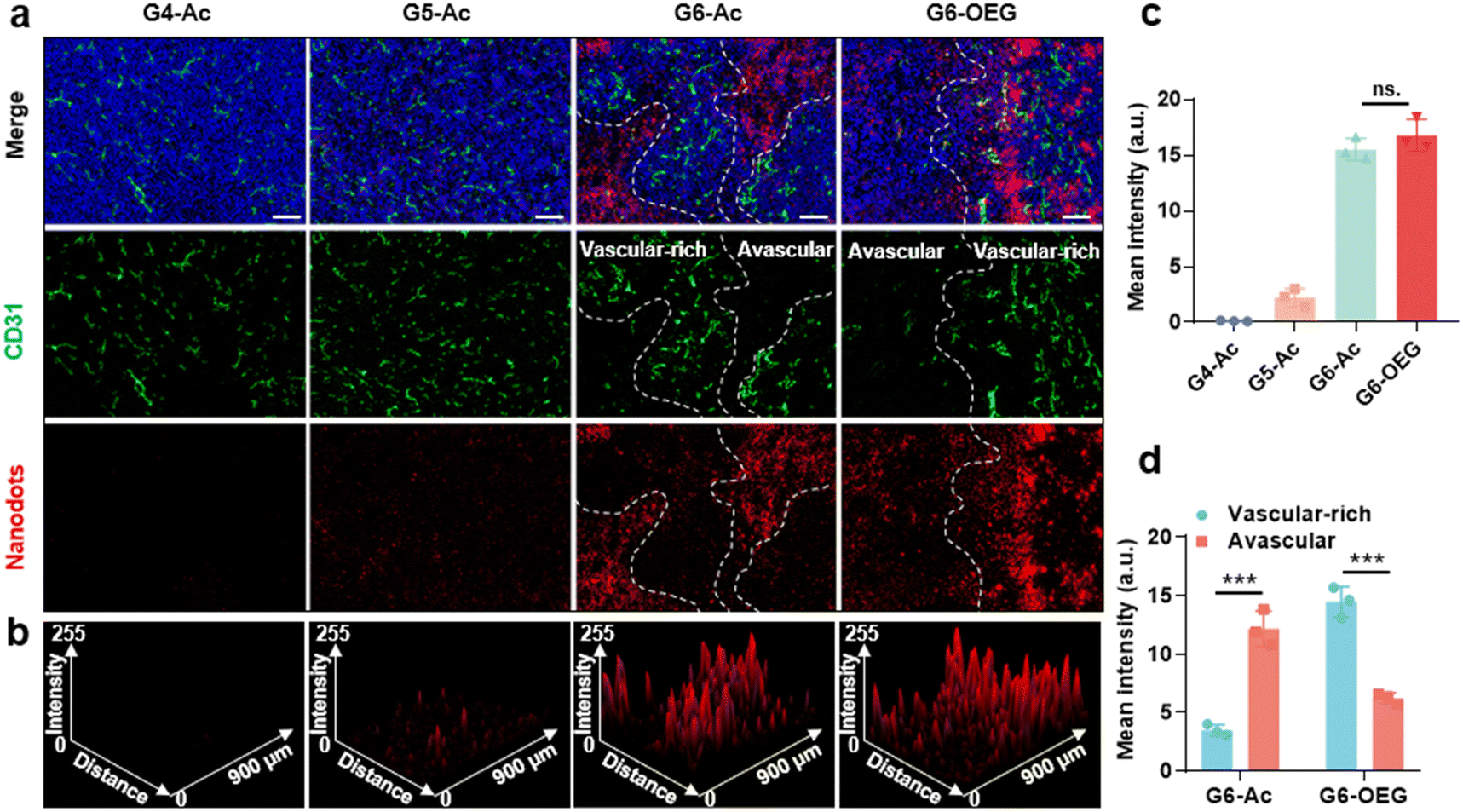 | ||
| Fig. 5 Intratumor distribution of the nanodots. (a) Representative confocal fluorescence imaging of the nanodots and blood vessels in the tumor tissues. The tumor was collected at 48 h after intravenous injection of the nanodots. Vasculature was subjected to immunofluorescence staining with an antibody against CD31 (green), the cell nuclei were labeled with DAPI (blue), and nanodots are shown as red (scale bar = 100 μm). (b) The ImageJ 3D plot of fluorescence intensity of the nanodots. (c) The fluorescence intensity in the confocal fluorescence images of (a). (d) The fluorescence intensity of the vascular-rich and avascular areas. Data are represented as mean ± SD, n = 3, and significances are determined by two-tailed Student's t-test, ns means no significant difference, *** P < 0.001. | ||
Transcytosis-inducing capability of the nanodots with different surfaces
We further investigated the cellular uptake and transcytosis-inducing capability of the nanodots in 4T1 cells by flow cytometry and confocal laser scanning microscopy (CLSM). The endocytic fluorescence intensity was determined by flow cytometry after culturing cells for 24 h. The exocytotic fluorescence was calculated by comparing the fluorescence of the treated groups before and after exocytosis in a fresh medium for another 24 h. Cells internalized with nanodots were also incubated with a fresh medium for 24 h, and the remaining uptake was determined by flow cytometry (Fig. S13†). The fluorescence of each group was normalized to the relative value of the endocytotic fluorescence of G4-Ac (Fig. 6a). The results showed that the endocytosis of different Gn-Ac (n = 4–6) was close (92%–100%), about 1.6–1.7 fold that of G6-OEG (58%). The exocytosis of different Gn-Ac was also close (43%–50%), about 2.4–2.8 fold that of G6-OEG (18%). These results showed that the particle size had a small effect on the cellular internalization and exocytosis of these nanodots, but the surface chemistry greatly affected these values. The exocytosis/endocytosis ratio of G6-Ac (47%) was 1.6 times that of G6-OEG (29%) (Fig. 6b). The cellular uptake of different nanodots was further imaged by CLSM after culturing cells for 12 h. More intracellular fluorescence was observed for the cells treated with G6-Ac than those treated with G6-OEG (Fig. S14†), consistent with the flow cytometry results. We further used the “cell infection” method to study the transcytosis of nanodots (Fig. 6c).9 G6-Ac could efficiently transfer from the old cells to the newly added cells, whereas G6-OEG could barely transfer to the newly added cells in 24 h.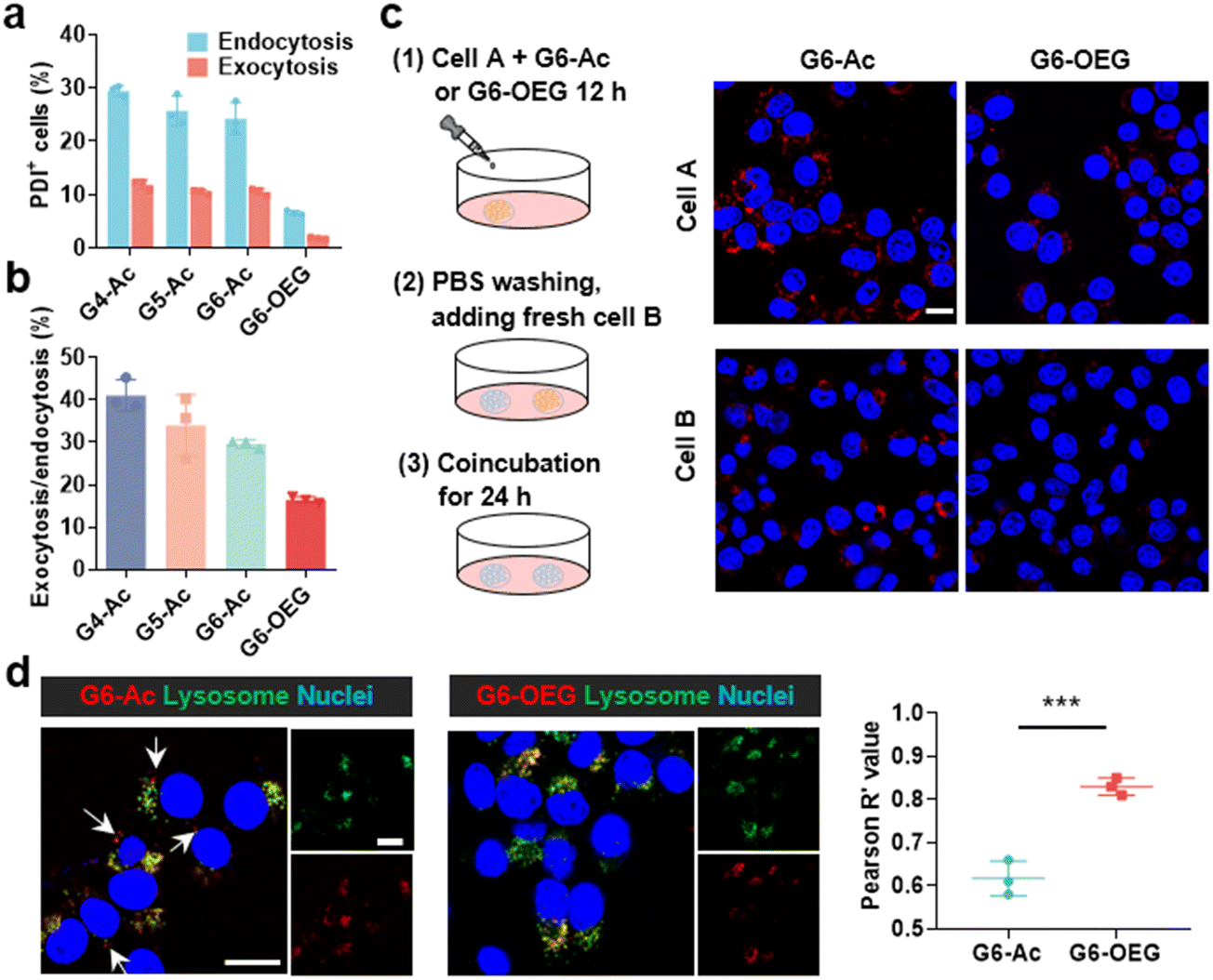 | ||
| Fig. 6 Cellular uptake and transcytosis-inducing capability of the nanodots. (a) Quantification analysis of cellular uptake and exocytosis of nanodots by flow cytometry. (b) The exocytosis in relation to the endocytosis rates of the nanodots. (c) Transcellular transport of nanodots observed by confocal laser scanning microscope (CLSM). The cell nuclei were stained with Hoechst 33342 (scale bar = 15 μm). (d) Colocalization and Pearson correlation coefficients of nanodots in the lysosome after culturing cells for 12 h (scale bar = 15 μm). Data are represented as mean ± SD, n = 3, and significances are determined by two-tailed Student's t-test, *** P < 0.001. | ||
It is reported that nanocarriers should avoid lysosomal entrapment to realize efficient transcytosis.8 Thus, we further studied the subcellular distribution of G6-Ac and G6-OEG, and quantified their colocalization with lysosomes using Pearson correlation coefficients (Fig. 6d). The results showed that G6-Ac nanodots had a lower Pearson correlation coefficient (61%) than G6-OEG (83%), indicating that G6-Ac nanodots were more likely to escape from lysosomes. The result could explain the superior transcytosis-inducing capability of G6-Ac compared to G6-OEG. All the results suggested that the surface chemistry was dominant in affecting the cellular uptake and transcytosis of the nanodots, thus greatly influencing their way of entering the tumor. Nanoparticles can enter tumors either by passive accumulation through leaky blood vessels or by an active process through cell transcytosis. We concluded that G6-Ac had more opportunities to enter tumors by the active process, thus showing better tumor penetration. In contrast, G6-OEG could accumulate in the tumor mainly through the EPR effect benefiting from their prolonged blood circulation, but could not penetrate deep tumor36 (Fig. 1b).
Conclusions
In summary, we developed PDI-cored and (DOTA-Gd)-inlayed single-molecule dendrimeric nanodots for dual-modal imaging to investigate the surface chemistry dependent tumor entrance. These nanodots showed bright fluorescence and high r1 relaxivity, excellent for fluorescence and MR imaging. Using these nanodots, we found that the acetylated and OEGylated G6 nanodots showed similar tumor accumulation but different intratumor distribution. G6-Ac showed deeper tumor penetration and was mainly distributed in the avascular areas more likely through cell transcytosis-based active transport. In contrast, G6-OEG mostly accumulated in vascular-rich areas of the tumor via the EPR effect-based passive transport. These results suggested that the surface chemistry of nanoparticles could dominate the ways of tumor entrance. This work can also guide the design of efficient cancer drug delivery systems, especially for active transporting nanocarriers.Experimental section
Materials
Nα-Fmoc-Nε-Boc-L-lysine (Fmoc–Lys(Boc)–OH), N-α,ε-di-Fmoc-L-lysine (Fmoc–Lys(Fmoc)–OH), trifluoroacetic acid (TFA), 2,3,4,5,6-pentafluorophenol (PFP-OH), dicyclohexylcarbodiimide (DCC), N,N-diisopropylethylamine (DIPEA), O-benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluorophosphate (HBTU) and N,N-diethylethanamine (TEA) were purchased from Energy Chemical Reagent Co., Ltd (Shanghai, China). 1,4-Dioxane and isopropyl ether were purchased from Macklin Biochemical Co., Ltd (Shanghai, China). tert-Butyloxycarbonyl DOTA (DOTA(tBu)) was purchased from Mimotopes Peptides Company (Wuxi, China). Acetic anhydride, ethyl acetate, sodium chloride, anhydrous sodium sulfate, dichloromethane (CH2Cl2, DCM), N,N-dimethylformamide (DMF), methyl alcohol, diethyl ether and piperidine were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China).Instruments
1H NMR spectra were acquired on a 400 MHz Varian Gemini NMR spectrometer in a deuterated solvent as noted. Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectra were obtained on a Bruker UltrafleXtreme MALDI-TOF MS spectrometer with 2,5-dihydroxybenzoic acid (2,5-DHB) as the matrix. Gel permeation chromatography (GPC) was performed on a Wyatt GPC/SEC-MALS (Wyatt Technology Corporation, Santa Barbara, CA, USA) system fitted with a PLgel 5 μm 500 Å column from Agilent Technology at 25 °C. The eluent used was DMF containing 50 mM LiBr with a flow rate of 0.8 mL min−1. High-performance liquid chromatography (HPLC) was performed on an Agilent 1260 Infinity II HPLC system fitted with a 1525 binary pump, 2475 multi-λ-fluorescence detector, 2998 photodiode array detector, and SunFireTM C18 (4.6 mm × 250 mm, 5 μm) column. Eluant A was water containing 0.1% TFA and eluant B was acetonitrile. The mobile phase was run from 0% B to 100% B within 50 min at a flow rate of 1.0 mL min−1 and detected at 595 nm. The UV–vis absorption and fluorescence spectra of nanodots in aqueous solutions were recorded on a microplate reader (Molecular Devices, SpectraMax). The Gd(III) contents were determined by inductively coupled plasma mass spectrometry (ICP-MS, PerkinElmer NexION 300X). The particle sizes of nanodots in aqueous solutions were measured by dynamic light scattering (DLS) on Malvern Zen3600 instrument at 25 °C with a scattering angle of 173° and a laser of 633 nm and the measurement procedure was performed in triplicate. The nanodots were imaged using a transmission electron microscope (TEM, JEOL JEM-1010) at a voltage of 80 kV and the average particle size was analyzed using ImageJ software from 100 individual particles. The cell endocytosis, exocytosis, subcellular distribution and transcellular endocytosis were analyzed using a flow cytometer (Becton Dickinson FACSCalibur) and a confocal laser scanning microscope (CLSM, Nikon-A1).Synthesis and characterization
The synthesis of the fluorescence and MRI dual-imaging nanodots is shown in Fig. 1a and Schemes S1–S3.†:1, v/v, 15 mL) with stirring for 4 hours at RT. After concentration by vacuum rotary evaporation, the residue was precipitated in cold diethyl ether (50 mL), and the solid was collected by filtration and washed with cold diethyl ether (10 mL) three times. A red powder of PDI-16Fmoc-G2.5·TFA (0.92 g, 0.09 mmol) was obtained after vacuum drying with a yield of 90% (Scheme S1†).
:1) with stirring at RT for 5 hours, and the solvent was concentrated by rotary evaporation. The residue was dropped in cold diethyl ether (50 mL) to precipitate the product, and the solid was collected by filtration and washed with cold diethyl ether (10 mL) three times. A purple powder of PDI-16DOTA(tBu)-G3 (0.51 g, 0.037 mmol) was obtained after vacuum drying with a yield of 90% (Scheme S2†). MALDI-TOF (Fig. S5†) Calc. [M]+: 13707.13, found [M]+: 13706.84.
PDI-16DOTA(tBu)-Gn (n = 4–6) were synthesized by repeating the reaction with Fmoc–Lys(Fmoc)–OPFP and deprotection in piperidine/DMF (v/v, 1:1). All the dendrimers had a yield of over 85%.
Cell culture and animals
The mouse 4T1 breast cancer cell lines were purchased from the American Type Culture Collection (ATCC) and were cultured in RPMI-1640 medium with 10% fetal bovine serum (FBS) and 1% antibiotic/antimycotic solution (Sigma A9909) at 37 °C in a 5% CO2 environment. BALB/c mice were obtained from the Zhejiang University Animal Center and all the animal experiments were in agreement with the approval of the Animal Care and Use Committee of Zhejiang University (license number: 17611).In vivo fluorescence imaging
Female BALB/c nude mice (6 to 8 weeks old, 20 g) were subcutaneously inoculated with 1 × 107 4T1 breast cancer cells (0.1 mL of cell suspension) on their right leg to develop an orthotopic breast tumor. Within 1 week, the mice bearing a tumor with a volume of about ∼100 mm3 were chosen for further experiments. The mice were anesthetized with 2% isoflurane and intravenously injected with nanodots at a dose of 5 mM kg−1 (200 μL, with equal fluorescence intensity). The whole-body fluorescence images were taken at 0.5 h, 2 h, 4 h, 12 h, 24 h, 48 h, 72 h, 96 h, 5 days, 6 days and 7 days after administration on a Maestro FLEX In Vivo Imaging System (Cambridge Research & Instrumentation, Inc. Woburn, MA, USA) with an excitation filter of 640 nm, an emission filter of 720 nm and an exposure time of 5 s. The average fluorescence intensity data of images were analyzed by ROI measurement of Living Image software 4.5.2 using the TNR formula: mean signal in tumor/mean signal in normal tissue (left flank muscle). All data are expressed as mean ± SD.In vivo MRI imaging
| CNR = (Si − Spre)/σn |
Signal enhancement ratios (ERs) were calculated in the artery, kidneys, and liver using a different equation:
| ER = Si/Spre |
Tumor tissue immunofluorescence
The tumor tissue was resected from the BALB/c mice after the tail-vein injection of nanodots for 48 hours and frozen with liquid nitrogen for 10 seconds before storing at −80 °C. The frozen tissue was embedded in an optimum cutting temperature (OCT) block for cryostat sections. Frozen tissue slices (6 μm thick) were restored to RT, treated with 0.1% cell magic solution for 15 minutes and blocked for 30 minutes with 10% donkey serum in TBST before staining with the primary antibodies against CD31 (1:2000) in the antibody reaction buffer at 4 °C overnight. Then, the sections were washed with TBST three times and incubated with the Alexa Fluor 488 secondary antibody (1:400) at RT for 1 hour. The cell nuclei were stained with DAPI. The fluorescence of nanodots was observed with an excitation laser at 593 nm, and the emission laser was read from 603 to 618 nm. CD31 for the blood vessels was excited with a 488 nm laser and signals were collected from 495 to 545 nm. DAPI for the nucleus was read using a 405 nm laser, and the emission was read from 410 to 440 nm. The fluorescence intensity data were analyzed with ImageJ software.
Cell endocytosis and exocytosis of nanodots
For flow cytometry analysis, 4T1 cells were cultured in two 24-well plates (plate I and plate II) with 5 × 104 cells per well overnight. Gn-Ac (n = 4–6) or G6-OEG was added into the wells at a concentration of 10 μM and cultured for 24 hours. The cells on plate I were washed with PBS and collected in 300 μL of PBS after trypsinizing. Cells on plate II were washed and cultured with a fresh medium for another 24 hours before trypsinizing, washing and resuspending in PBS. All the cells were immediately determined by flow cytometry (Becton Dickinson, FACSCalibur, San Jose, CA, USA), and at least 10000 events were collected for every sample. The data were analyzed using FlowJo software. Cell endocytosis was achieved by the fluorescence intensity of plate I and cell exocytosis was obtained by subtracting the fluorescence intensity of plate I from the fluorescence intensity of plate II.
For confocal imaging, 4T1 cells were seeded in glass-bottom Petri dishes at a density of 8 × 104 cells per dish and cultured overnight. G6-Ac or G6-OEG was added into the dish at a dose of 20 μM for 12 hours. The cells were washed with PBS, added with a fresh medium, and stained with Hoechst 33342 for 15 minutes at 37 °C for observation by CLSM. Nanodots were observed using a 561 nm laser, and Hoechst 33342-labeled nuclei were observed with a 405 nm laser. The fluorescence intensity data were analyzed using ImageJ software.
Subcellular distributions of nanodots
4T1 cells were seeded in glass-bottom Petri dishes at a density of 8 × 104 cells per dish and cultured overnight. G6-Ac or G6-OEG was added to the dish at a dose of 20 μM for 12 hours. Then, the cells were washed with PBS and added with a fresh medium. Lysosomes were stained with LysoTracker Green for 30 minutes and the cell nuclei were stained with Hoechst 33342 for 15 minutes before confocal imaging. The Pearson correlation coefficients were analyzed using ImageJ software. Nanodots were observed using a 561 nm laser, LysoTracker Green-stained lysosomes were observed with a 488 nm laser and Hoechst 33342-labelled nuclei were observed with a 405 nm laser.Transcytosis-inducing behaviors of nanodots
4T1 cells were seeded on the coverslips and cultured overnight. The cells A on the coverslip were first incubated with G6-Ac or G6-OEG (20 μM) for 12 hours. Then, the cells A on the coverslip were washed with PBS and co-incubated with fresh cells B on another coverslip in a fresh medium for 24 hours. Afterwards, the cells were washed with PBS and the cell nuclei were stained with Hoechst 33342 for 15 minutes before confocal imaging. Hoechst 33342-labeled nuclei were observed with a 405 nm laser.Author contributions
Huiming Ren: investigation (lead), methodology (lead), data curation, and writing – original draft. Qiuhui Hu, Yuji Sun, Xiaoxuan Zhou, Yincong Zhu, Qiuyang Dong, and Linying Chen: methodology (supporting) and investigation (supporting). Jianbin Tang, Hongjie Hu, and Youqing Shen: conceptualization (supporting), funding acquisition (supporting), supervision (supporting), and writing – review & editing (supporting). Zhuxian Zhou: conceptualization (lead), funding acquisition (lead), supervision (lead), and writing – review & editing (lead).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Key Research and Development Program of China (2021YFA1201200), the National Natural Science Foundation of China (21875211 and 52073249), and the Zhejiang Provincial Key Research and Development Program (2020C01123). We also thank Zhaoxiaonan Lin from the Core Facilities, Zhejiang University School of Medicine, and Chao Sun from the Analysis Center of Agrobiology and Environmental Sciences, Zhejiang University, for their technical support.References
- R. Sun, J. Xiang, Q. Zhou, Y. Piao, J. Tang, S. Shao, Z. Zhou, Y. H. Bae and Y. Shen, Adv. Drug Delivery Rev., 2022, 191, 114614 CrossRef CAS PubMed
.
- W. Stefan, J. T. Anthony, D. Qin, O. Seiichi, A. Julie, F. D. Harold and W. C. W. Chan, Nat. Rev. Mater., 2016, 1, 16014 CrossRef
- Z. Zhou and Z. R. Lu, Adv. Drug Delivery Rev., 2017, 113, 24–48 CrossRef CAS PubMed
- D. Rosenblum, N. Joshi, W. Tao, J. M. Karp and D. Peer, Nat. Commun., 2018, 9, 1410 CrossRef PubMed
- P. Wei and J. S. Moodera, Nat. Mater., 2020, 19, 481–482 CrossRef CAS PubMed
- S. Pandit, D. Dutta and S. M. Nie, Nat. Mater., 2020, 19, 478–480 CrossRef CAS PubMed
- S. Sindhwani, A. M. Syed, J. Ngai, B. R. Kingston, L. Maiorino, J. Rothschild, P. MacMillan, Y. Zhang, N. U. Rajesh, T. Hoang, J. L. Y. Wu, S. Wilhelm, A. Zilman, S. Gadde, A. Sulaiman, B. Ouyang, Z. Lin, L. Wang, M. Egeblad and W. C. W. Chan, Nat. Mater., 2020, 19, 566–575 CrossRef CAS PubMed
- Q. Zhou, S. Shao, J. Wang, C. H. Xu, J. Xiang, Y. Piao, Z. Zhou, Q. S. Yu, J. Tang, X. Liu, Z. Gan, R. Mo, Z. Gu and Y. Shen, Nat. Nanotechnol., 2019, 14, 799–811 CrossRef CAS PubMed
- R. Sun, Y. F. Zhang, X. W. Lin, Y. Piao, T. Xie, Y. He, J. J. Xiang, S. Q. Shao, Q. Zhou, Z. X. Zhou, J. B. Tang and Y. Q. Shen, Angew. Chem., Int. Ed., 2023, 62, e202217408 CrossRef CAS PubMed
- G. Wang, Y. Jiang, J. Xu, J. Shen, T. Lin, J. Chen, W. Fei, Y. Qin, Z. Zhou, Y. Shen and P. Huang, Adv. Mater., 2023, 35, 2207271 CrossRef CAS PubMed
- S. Q. Chen, Y. Zhong, W. F. Fan, J. J. Xiang, G. W. Wang, Q. Zhou, J. Q. Wang, Y. Geng, R. Sun, Z. Zhang, Y. Piao, J. G. Wang, J. Y. Zhuo, H. L. Cong, H. P. Jiang, J. Ling, Z. C. Li, D. D. Yang, X. Yao, X. Xu, Z. X. Zhou, J. B. Tang and Y. Q. Shen, Nat. Biomed. Eng., 2021, 5, 1019–1037 CrossRef CAS PubMed
- S. Mao, Science, 2020, 367, 638–639 CrossRef
- A. E. Nel, L. Mädler, D. Velegol, T. Xia, E. M. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed
- M. Sousa de Almeida, E. Susnik, B. Drasler, P. Taladriz-Blanco, A. Petri-Fink and B. Rothen-Rutishauser, Chem. Soc. Rev., 2021, 50, 5397–5434 RSC
- E. Song, A. Gaudin, A. R. King, Y. E. Seo, H. W. Suh, Y. Deng, J. J. Cui, G. T. Tietjen, A. Huttner and W. M. Saltzman, Nat. Commun., 2017, 8, 15322 CrossRef CAS PubMed
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Delivery Rev., 2016, 99, 28–51 CrossRef CAS PubMed
- B. M. Chen, T. L. Cheng and S. R. Roffler, ACS Nano, 2021, 15, 14022–14048 CrossRef CAS PubMed
- G. Wang, B. Wu, Q. Li, S. Chen, X. Jin, Y. Liu, Z. Zhou, Y. Shen and P. Huang, Small, 2020, 16, e2004172 CrossRef PubMed
- Q. Zhou, J. X. Huang, L. Q. Hao, Y. Geng, C. H. Xu, Z. X. Zhou, J. B. Tang, R. H. Zhou and Y. Q. Shen, Bioconjugate Chem., 2022, 33, 2132–2142 CrossRef CAS PubMed
- S. Chen, Q. Zhou, G. Wang, Z. Zhou, J. Tang, T. Xie and Y. Shen, Biomacromolecules, 2021, 22, 5139–5150 CrossRef CAS PubMed
- C. A. Dougherty, S. Vaidyanathan, B. G. Orr and M. M. Banaszak Holl, Bioconjugate Chem., 2015, 26, 304–315 CrossRef CAS PubMed
- A. M. Mahmoud, J. P. Morrow, D. Pizzi, A. M. Azizah, T. P. Davis, R. F. Tabor and K. Kempe, Biomacromolecules, 2020, 21, 3007–3016 CrossRef CAS PubMed
- X. Yang, C. Shi, R. Tong, W. Qian, H. E. Zhau, R. Wang, G. Zhu, J. Cheng, V. W. Yang, T. Cheng, M. Henary, L. Strekowski and L. W. Chung, Clin. Cancer Res., 2010, 16, 2833–2844 CrossRef CAS PubMed
- C. Zhang, T. Liu, Y. Su, S. Luo, Y. Zhu, X. Tan, S. Fan, L. Zhang, Y. Zhou, T. Cheng and C. Shi, Biomaterials, 2010, 31, 6612–6617 CrossRef CAS PubMed
- H. Cong, K. Wang, Z. Zhou, J. Yang, Y. Piao, B. Yu, Y. Shen and Z. Zhou, ACS Nano, 2020, 14, 5887–5900 CrossRef CAS PubMed
- J. Yang, K. Wang, Y. Zheng, Y. Piao, J. Wang, J. Tang, Y. Shen and Z. Zhou, Angew. Chem., Int. Ed., 2022, 61, e202202128 CrossRef CAS PubMed
- G. M. van Dam, G. Themelis, L. M. Crane, N. J. Harlaar, R. G. Pleijhuis, W. Kelder, A. Sarantopoulos, J. S. de Jong, H. J. Arts, A. G. van der Zee, J. Bart, P. S. Low and V. Ntziachristos, Nat. Med., 2011, 17, 1315–1319 CrossRef CAS PubMed
- H. Li, D. Luo, C. Yuan, X. Wang, J. Wang, J. P. Basilion and T. J. Meade, J. Am. Chem. Soc., 2021, 143, 17097–17108 CrossRef CAS PubMed
- P. Caravan, Chem. Soc. Rev., 2006, 35, 512–523 RSC
- H. Cai, X. Dai, X. Wang, P. Tan, L. Gu, Q. Luo, X. Zheng, Z. Li, H. Zhu, H. Zhang, Z. Gu, Q. Gong and K. Luo, Adv. Sci., 2020, 7, 1903243 CrossRef CAS PubMed
- C. Yao, Y. Li, Z. Wang, C. Song, X. Hu and S. Liu, ACS Nano, 2020, 14, 1919–1935 CrossRef CAS PubMed
- Y. Fan, W. Tu, M. Shen, X. Chen, Y. Ning, J. Li, T. Chen, H. Wang, F. Yin, Y. Liu and X. Shi, Adv. Funct. Mater., 2020, 30, 1909285 CrossRef CAS
- Z. Zhou, X. Liu, D. Zhu, Y. Wang, Z. Zhang, X. Zhou, N. Qiu, X. Chen and Y. Shen, Adv. Drug Delivery Rev., 2017, 115, 115–154 CrossRef CAS PubMed
- Z. Zhou and Z. R. Lu, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2013, 5, 1–18 CrossRef CAS PubMed
- J. E. Lemaster, Z. Wang, A. Hariri, F. Chen, Z. Hu, Y. Huang, C. V. Barback, R. Cochran, N. C. Gianneschi and J. V. Jokerst, Chem. Mater., 2019, 31, 251–259 CrossRef CAS PubMed
- H. Kang, S. Rho, W. R. Stiles, S. Hu, Y. Baek, D. W. Hwang, S. Kashiwagi, M. S. Kim and H. S. Choi, Adv. Healthcare Mater., 2020, 9, e1901223 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3bm01171b |
| This journal is © The Royal Society of Chemistry 2023 |