
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ruthenium-based antitumor drugs and delivery systems from monotherapy to combination therapy
Yu
Lu
abd,
Di
Zhu
ab,
Quynh
Le
c,
Yuji
Wang
 *ab and
Wei
Wang
*cd
*ab and
Wei
Wang
*cd
aDepartment of Medicinal Chemistry, College of Pharmaceutical Sciences of Capital Medical University, Beijing 100069, P. R. China. E-mail: wangyuji@ccmu.edu.cn
bBeijing Area Major Laboratory of Peptide and Small Molecular Drugs, Engineering Research Center of Endogenous Prophylactic of Ministry of Education of China, Beijing Laboratory of Biomedical Materials, Beijing Laboratory of Oral Health, Beijing 100069, P. R. China
cCenter for Pharmacy, University of Bergen, P. O. Box 7803, 5020 Bergen, Norway. E-mail: wei.wang@uib.no
dDepartment of Chemistry, University of Bergen, P. O. Box 7803, 5020 Bergen, Norway
First published on 11th October 2022
Abstract
Ruthenium complex is an important compound group for antitumor drug research and development. NAMI-A, KP1019, TLD1433 and other ruthenium complexes have entered clinical research. In recent years, the research on ruthenium antitumor drugs has not been limited to single chemotherapy drugs; other applications of ruthenium complexes have emerged such as in combination therapy. During the development of ruthenium complexes, drug delivery forms of ruthenium antitumor drugs have also evolved from single-molecule drugs to nanodrug delivery systems. The review summarizes the following aspects: (1) ruthenium complexes from monotherapy to combination therapy, including the development of single-molecule compounds, carrier nanomedicine, and self-assembly of carrier-free nanomedicine; (2) ruthenium complexes in the process of ADME in terms of absorption, distribution, metabolism and excretion; (3) the applications of ruthenium complexes in combination therapy, including photodynamic therapy (PDT), photothermal therapy (PTT), photoactivated chemotherapy (PACT), immunotherapy, and their combined application; (4) the future prospects of ruthenium-based antitumor drugs.
1. Introduction
According to the latest statistics from the World Health Organization's International Agency for Research on Cancer (IARC), cancer incidence and death rates continued to increase globally in 2020.1,2 There are currently three main methods of cancer treatment: surgery, chemotherapy, and radiotherapy.3,4 In chemotherapy, antitumor drugs are important for treatment due to high patient compliance and a high price–performance ratio. The research and development of antitumor drugs are central fields in cancer treatment. Although the sales volume of metal antitumor drugs such as cisplatin and carboplatin ranks top ten in the global antitumor drug market,5,6 this type of drug can result in nausea, vomiting, bone marrow transplantation, liver and kidney toxicity and other side effects.7–10 In the search for safe metal antitumor drugs, ruthenium (Ru) complexes have become a big breakthrough in the field.11–14In comparison with cisplatin and its derivatives, ruthenium complexes have the following advantages. First, platinum complexes form a four-coordination plane square, whereas ruthenium complexes form six-coordination octahedron mode, and the range of oxidation states is wider than that of platinum (including II, III, IV valence states), which leads to abundant complex structures.15,16 Second, the increased safety of ruthenium complexes and their selective killing effect on tumor cells may be due to the fact that ruthenium and iron are from the same main group, and ruthenium may be transported in the body in the same way as iron, through transferrin in the blood, thus having faster absorption and metabolism, higher bioavailability and lower systemic toxicity.17–20 Third, ruthenium complexes may supplement the treatment of tumors resistant to cisplatin.21–23 Fourth, ruthenium complexes have rich photophysical and photochemical properties, making photodynamic therapy (PDT)24,25 and photothermal therapy (PTT)17 potential methods for cancer treatments.
At present, a few ruthenium-based antitumor drugs are under clinical studies, including NAMI-A,26KP1019,27KP133928 and TLD143324 (see Fig. 1 for the chemical structures). The research and development of ruthenium complexes has been limited by obstacles such as poor antitumor effect (NAMI-A29) and poor solubility (KP101927). So far, no ruthenium complex is in clinical use. In 2017, TLD1433 was in a phase IIa clinical study and showed therapeutic effect on human non-musculoinvasive bladder cancer.24,30 It is the first ruthenium photosensitizer for PDT in a clinical trial. From NAMI-A to KP1019, KP1339, and TLD1433, the development of ruthenium antitumor drugs is moving from monotherapy to combination therapy.31
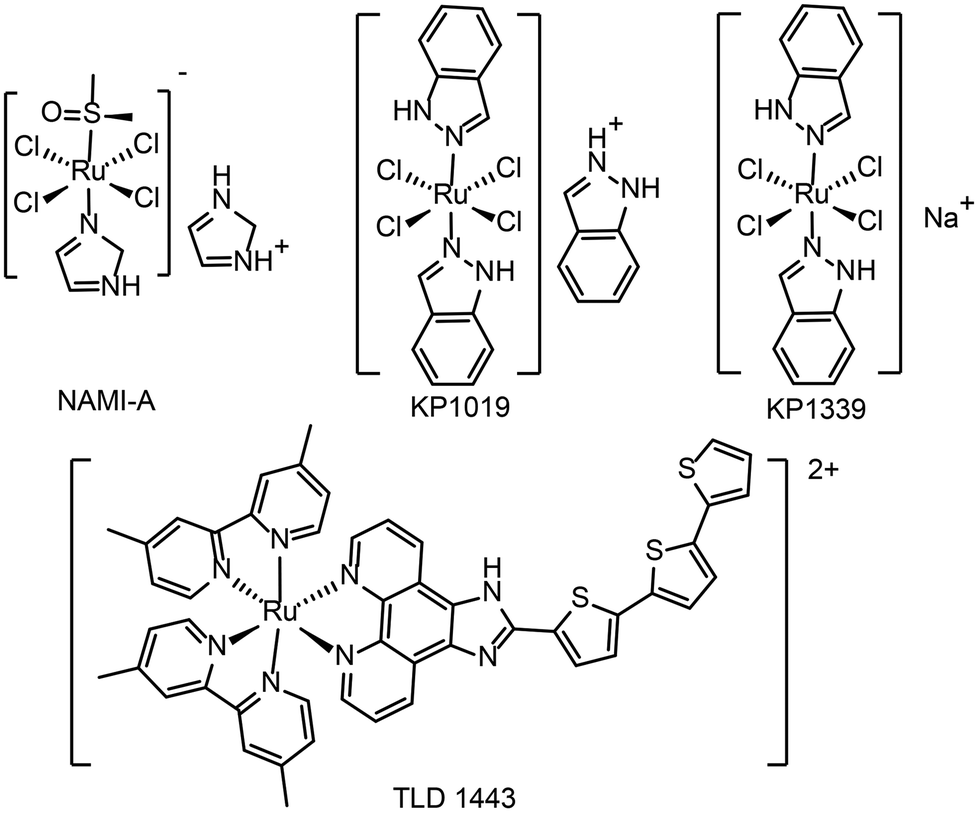 | ||
| Fig. 1 The chemical structures of NAMI-A, KP1019, KP1339 and TLD1433. | ||
Combination therapy for cancer includes the combination of multiple drugs and the combination of multiple modalities,32–34 including chemotherapy, radiotherapy, photodynamic therapy, photothermal therapy, immunotherapy and gene therapy, etc.3,35–37 The synergistic effect of multiple modalities not only enhances the therapeutic effect, but also solves many problems arising from the use of a single drug (including drug resistance, tumor recurrence, systemic toxicity).38–40
A few featured reviews have been published between 2017 to 2022 on ruthenium complexes. Zhe-Sheng Chen et al.41 summarized the mechanism and structure–activity relationship of ruthenium complexes, as well as their catalytic activity, photoinduced activation and their applications as anticancer drugs. The review focused on the structure of ruthenium complexes. In this review, ruthenium compounds were still viewed as a drug for use alone in anticancer therapy. A review42 published in 2021 has the same approach in structuring contents. New ruthenium compounds and delivery strategies were summarized for Ru-based anticancer therapy. In both reviews, nano-delivery systems for ruthenium drugs were reviewed. However, the nano-delivery systems reviewed were formulated in a traditional drug–excipient fashion. In recent years, more excipient-free ruthenium nanomaterials have been developed. Many studies show that ruthenium compounds can form nanostructures without adding other additives, which consequently reduces the limitation of excipients which are approved by the FDA or EMA. This aspect was not covered by the previous reviews.
Thorfinnur Gunnlaugsson et al.43 focused on the absorption and localization of Ru(II) polypyridine complex in cells and the application of two-photon activation. The review extends to the photoimaging and photoactivation activities of ruthenium complexes, especially in photodynamic therapy and photoactivated chemotherapy. Tianfeng Chen et al.31 also discussed the functionalization and cancer-targeting designs of different ruthenium complexes in conjunction with different therapeutical methods, such as photodynamic therapy, photothermal therapy, radiosensitization, targeted therapy and nanotechnology for precise cancer therapy. However, much recent progress in photoactivated chemotherapy and immunotherapy was not covered. In addition, the advantages of ruthenium in combination therapy due to its diverse optical characteristics have been recently discovered. Moreover, Celine J. Marmion et al.44 summarized the multi-targeted ruthenium complexes, including DNA and other targets (such as enzymes, peptides, and intracellular proteins). The molecular targets of ruthenium complexes in cells are well covered in this review, but the ADME process is lacking.
This review is organized as follows (Fig. 2). In Section 2, we summarize the development of ruthenium complexes from monotherapy to combination therapy, including the development of single-molecule compounds, nanomedicine with excipients, and self-assembled excipient-free nanomedicine. In Section 3, we discuss the recent studies on ADME (absorption, distribution, metabolism and excretion) for ruthenium complexes. Section 4 presents the applications of ruthenium complexes in combination therapy, such as PDT, PTT, PACT, immunotherapy, and their combinations. Finally, in Section 5, we give our prospects on ruthenium-based antitumor drugs.
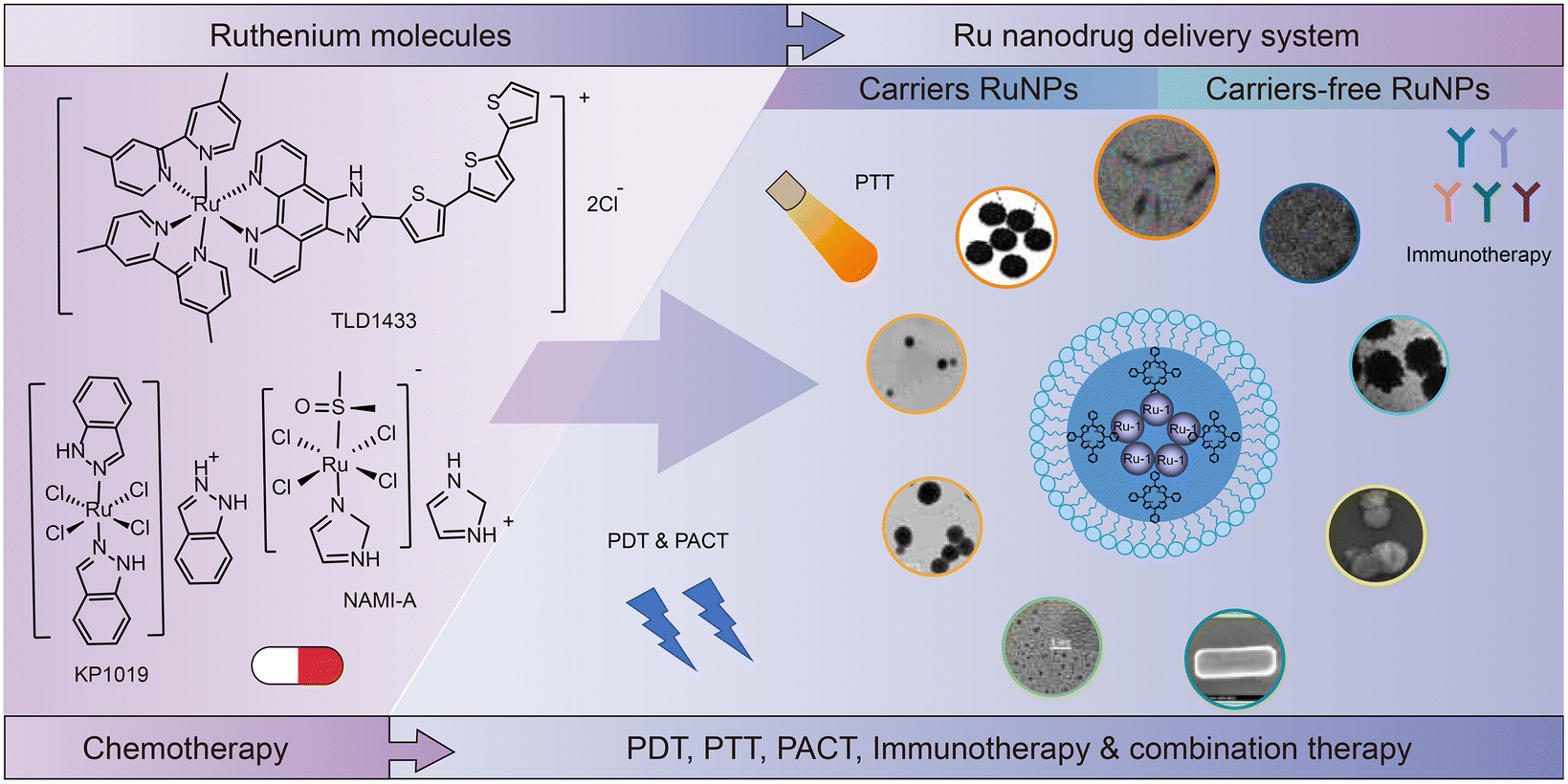 | ||
| Fig. 2 Ruthenium complexes from monotherapy to combination therapy. | ||
2. Development of ruthenium complex molecules
NAMI-A is the first ruthenium complex to enter clinical studies as a chemotherapeutic agent. Preclinical studies provided the evidence that the drug can inhibit secondary tumor metastasis in vitro and in vivo, instead of exerting a direct killing effect on tumor.45NAMI-A can inhibit the formation of new blood vessels.46 This neovascularization inhibition may be one of the mechanisms by which NAMI-A inhibits tumor metastasis. In addition, integrin α5β1 has also been proposed as the mechanism by which NAMI-A inhibits HCT116 cell metastasis.47KP1019 and KP1339 are different salt forms of the same ruthenium complex. In addition to anti-metastasis activity against secondary tumors, they also have a broad inhibitory effect on primary tumors.28KP1339 is a sodium salt form, which is more water-soluble than KP1019. In clinical studies, KP1339 has shown excellent therapeutic activity for solid tumors as a GRP78 protein inhibitor and is well tolerated with controllable side effects.28 The inhibitory mechanism of KP1339 is presented in Fig. 3.28KP1339 plays a role through down-regulation of GRP78, a key regulator of misfolding protein processing and tumor survival/anti-apoptosis. Down-regulation of GRP78 kills tumor cells through acute endoplasmic reticulum (ER) stress response.48 In addition, KP1339 disrupts intracellular ROS levels, induces apoptosis through mitochondria or the MAPK/P38 pathway, and blocks the cell cycle in the G2/M phase.
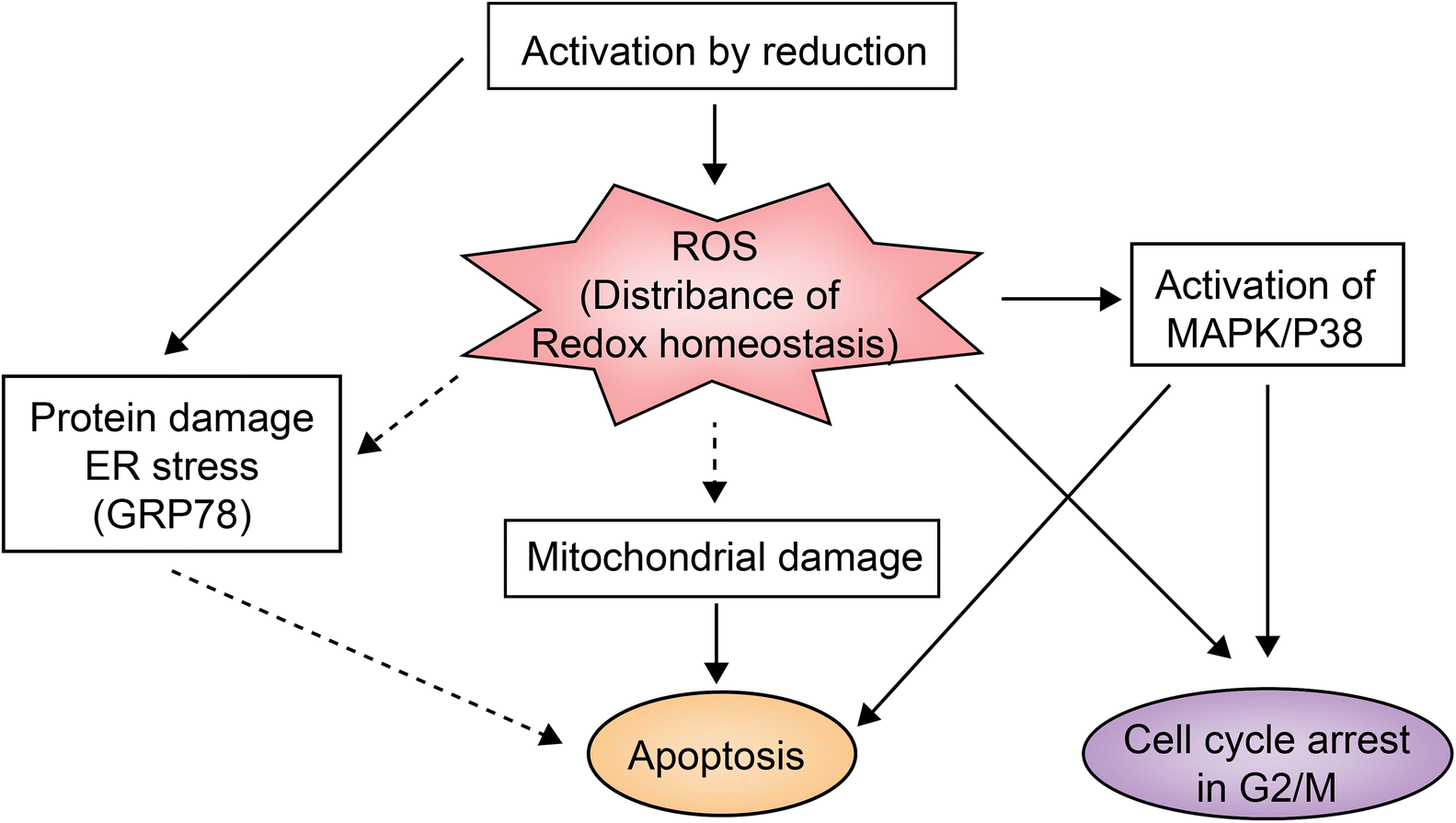 | ||
| Fig. 3 KP1339 clinical study mechanism.28 This figure has been reproduced from ref. 28 with permission from Wiley, copyright 2017. | ||
In addition to the ruthenium complexes in clinical studies, various ruthenium complexes were designed and synthesized as chemotherapy agents for the treatment of breast cancer,49 lung cancer50 and other common tumor types.51 These ruthenium complexes inhibit tumor growth and metastasis usually by inducing apoptosis, autophagy, necrotizing death, reactive oxygen species (ROS) generation, angiogenesis inhibition and cell cycle arrest.50 At present, these ruthenium complexes mainly include bipyridine52–55 and aromatic ruthenium complexes.56–60 There are excellent reviews41,42 about the structural–activity relationship and action mechanism of these bipyridine and aromatic ruthenium complexes. Therefore, they are not repeated here. Instead, we summarize these ruthenium complexes formed by natural ligands (Fig. 4, 5 and Table 1) due to the increasing number of studies on natural products and their derivatives as ligands for ruthenium complexes,61,62 such as β-carboline, isoquinoline, curcumin,63–68 amino acids,69 flavonoids,70,71 sugars,72,73etc.
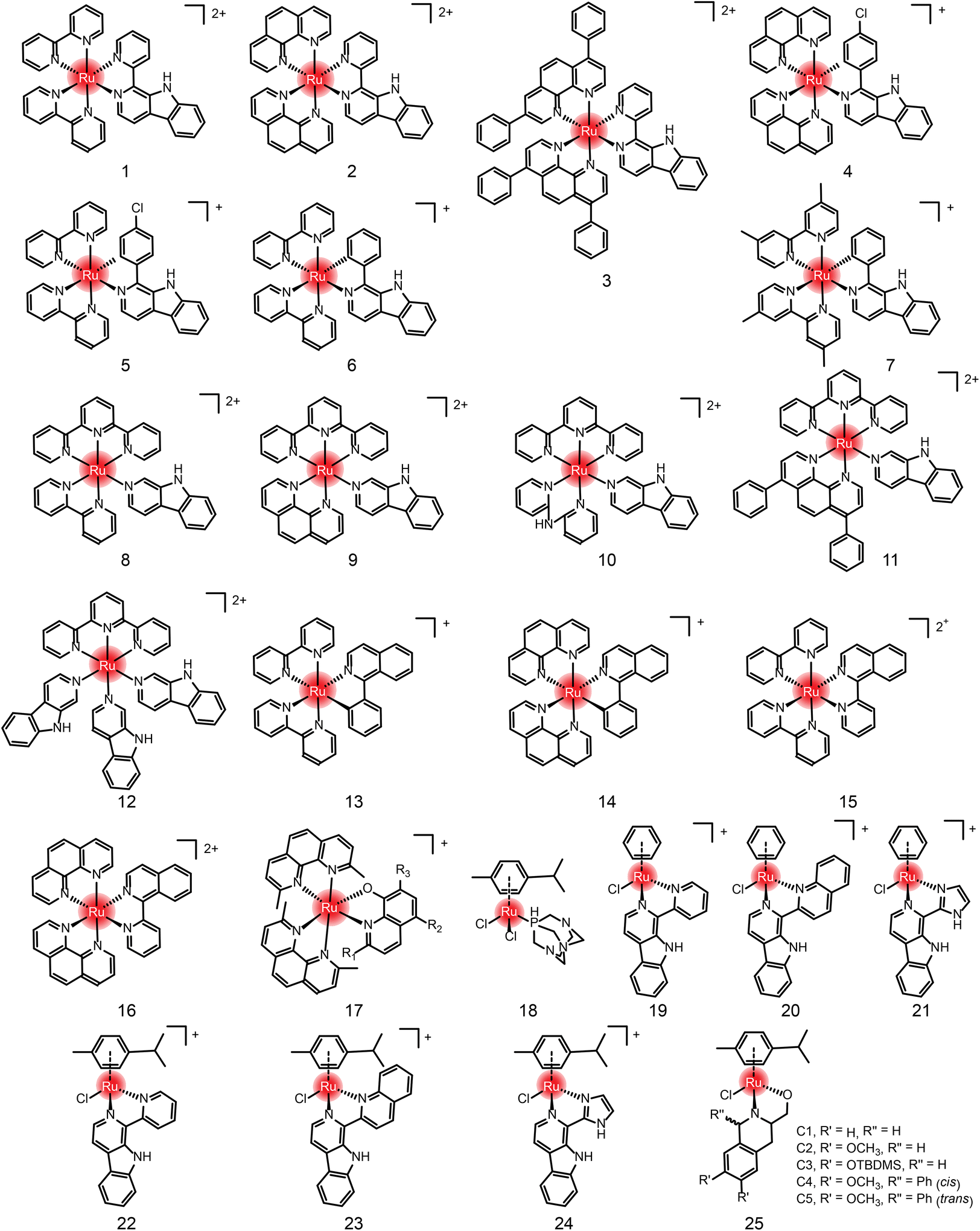 | ||
| Fig. 4 The chemical structures of compounds 1–25. | ||
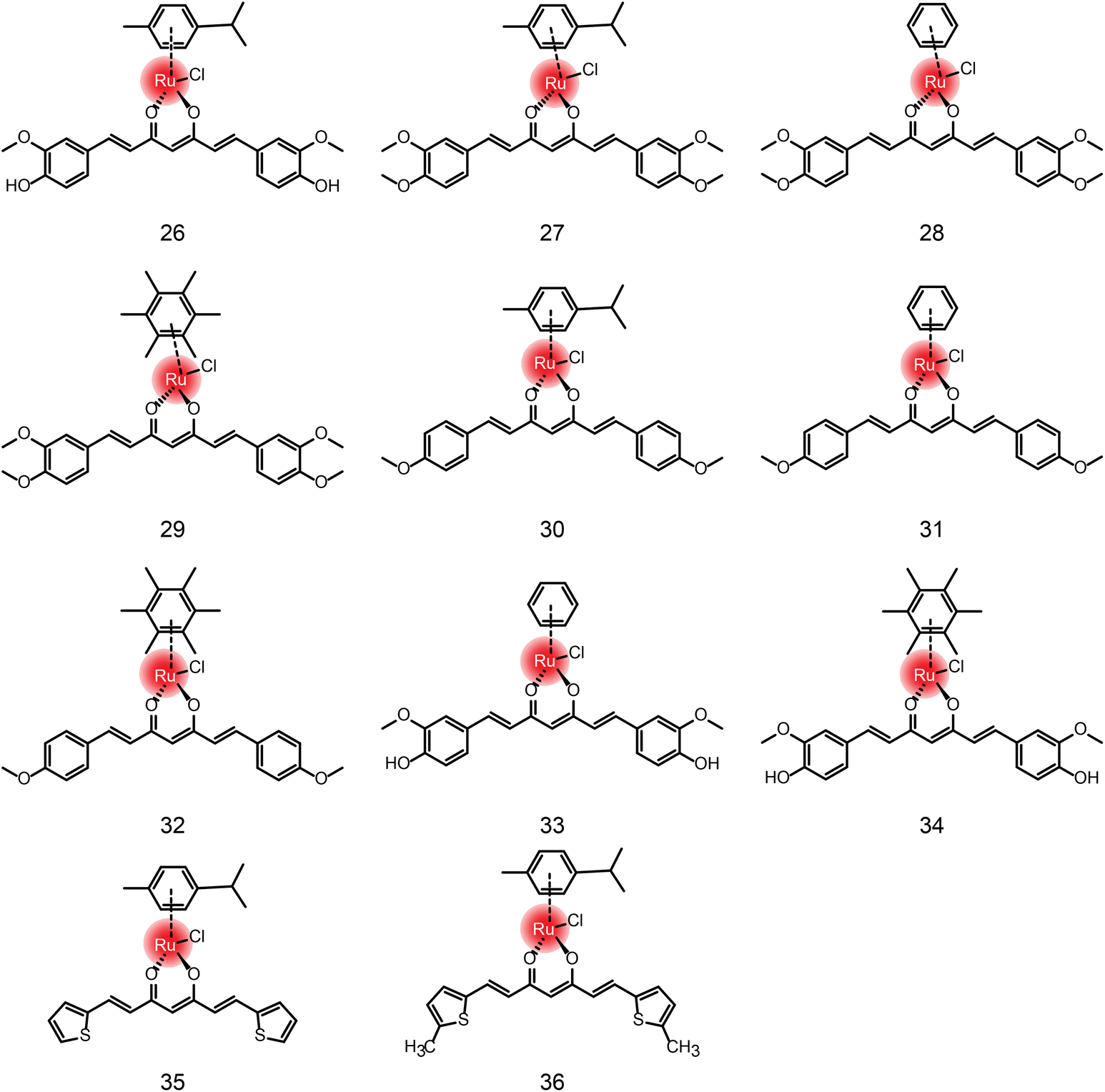 | ||
| Fig. 5 The chemical structures of compounds 26–36. | ||
| Compound | Activities in vivo and in vitro | Reference drug | Mechanism | Ref. |
|---|---|---|---|---|
| 1 | HeLa: 61.2 ± 3.9 μM | Cisplatin: 16.7 ± 2.5 μM (HeLa); NAMI-A: 625.8 ± 44.5 μM (HeLa) | DNA binding, ROS-dependent apoptosis and autophagy | 74 |
| 2 | HeLa: 20.2 ± 1.6 μM | |||
| 3 | HeLa: 1.9 ± 0.2 μM | |||
| 4 (RuβC-3) | HeLa: 3.2 ± 0.4 μM | Cisplatin: 18.1 ± 0.5 μM (HeLa) | G0/G1 cell cycle arrest and activation of mitochondria-dependent apoptosis via the ERK/Akt pathway | 75 |
| 5 (RuβC-4) | HeLa: 4.1 ± 0.6 μM | |||
| 6 | HeLa: 1.9 ± 0.4 μM | Cisplatin: 18.2 ± 1.2 μM (HeLa) | Regulating cell cycle-related proteins leads to cell cycle stagnation in G0/G1 phase. Mitochondrial dysfunction and ROS-mediated DNA damage induce apoptosis | 76 |
| 7 | HeLa: 3.4 ± 0.3 μM | |||
| 8 | HeLa: 48.3 ± 2.5 μM | Cisplatin: 20.2 ± 3.6 μM (HeLa) | Apoptosis was induced by mitochondrial pathway, loss of mitochondrial membrane potential and release of cytochrome C. The activation of p53 is caused by phosphorylation of SER-15 and SER-392. Accumulation of p53 is associated with increased p21 and caspase activation | 77 |
| 9 | HeLa: 41.8 ± 1.9 μM | |||
| 10 | HeLa: 51.2 ± 3.1 μM | |||
| 11 | HeLa: 35.8 ± 1.7 μM | |||
| 12 | Tumor cells: HeLa: 0.68 ± 0.3 μM, MCF-7: 1.86 ± 0.2; HepG2: 0.90 ± 0.2 μM, Bel-7402: 0.97 ± 0.1 μM, Normal cells: L02: 57.2 ± 4.1 μM, MCF-10A: 46.2 ± 3.6 μM, HEK-293: 52.3 ± 2.7 μM | Cisplatin: 20.2 ± 3.6 μM (HeLa); 22.8 ± 4.2 μM (MCF-7) | As compounds 8–11 | 78 |
| 13 (RuIQ-1) | NCI-H460: 2.1 ± 0.2 μM; HBE: 22 ± 0.8 μM; HeLa: 4.3 ± 0.3 μM; MCF: 7.2 ± 0.4 μM | Cisplatin: 15.0 ± 2.0 μM (HeLa); 21.8 ± 2.2 μM (MCF) | Regulation of cyclin induces S and G2/M bicycle arrest; reduce mitochondrial membrane potential (MMP), promote the production of intracellular ROS and induce DNA damage, leading to cell apoptosis | 79 |
| 14 (RuIQ-2) | NCI-H460: 1.8 μM; HBE: 20 ± 0.3 μM; HeLa: 4.7 ± 0.4 μM; MCF: 7.0 ± 0.3 μM | |||
| 15 (RuIQ-3) | NCI-H460: 69.1 ± 3.4 μM; HBE: 96.1 ± 3.4 μM; HeLa: 80.2 ± 4.1 μM; MCF: 72.0 ± 3.4 μM | |||
| 16 (RuIQ-4) | NCI-H460: 77.6 ± 4.1 μM; HBE: 97.6 ± 4.2 μM; HeLa: 82.2 ± 3.5; MCF: 88.7 ± 4.7 μM | |||
| 17 | See Table 2 | See Table 2 | Inhibition of protein synthesis | 80 |
| 18 (RAPTA-C) | EAC: 40 mg kg−1 decreased 50% EAC cells; human A2780 ovarian cells implanted in chicken chorioallantoic membrane : 0.2 mg kg−1 decreased about 75%; LS174T colorectal adenocarcinoma in nude mice: 100 mg kg−1 decreased about 50% | Not reported | Mitochondria and p53-JNK pathway induced apoptosis of EAC cells. Inhibition of angiogenesis | 81 and 82 |
| 19 | HeLa: 44.3 ± 3.7 μM | Cisplatin: 15.5 ± 1.7 μM (HeLa) | G2M phase was blocked by inhibiting CDK1; apoptosis is induced by mitochondrial-related pathways and intracellular reactive oxygen species (ROS) elevation | 83 |
| 20 | HeLa: 24.2 ± 2.4 μM | |||
| 21 | HeLa: 5.9 ± 0.9 μM | |||
| 22 | HeLa: 16.3 ± 1.5 μM | |||
| 23 | HeLa: 6.2 ± 0.7 μM | |||
| 24 | HeLa: 2.4 ± 0.2 μM | |||
| 25 (C1–C5) | MCF-7 cells: C1 – 54 ± 5.36 μM; C2 – 60 ± 4.56 μM; C3 – 68 ± 3.33 μM; C4 – 34 ± 0.29 μM; C5 – 218 ± 2.15 μM; normal cell MDBK with no activity | Cisplatin: 67 ± 5.0 μM (MCF-7) | Not reported | 84 |
| 26 (Ru-Cur) | MCF-7: 19.58 ± 2.367 μM; HCT116: 13.98 ± 1.503 μM; A2780: 23.38 ± 3.334 μM; CP8 platinum-resistant cells from ovarian cancer: 27.00 ± 2.332 μM; A549: 62.33 ± 8.934 μM | Cisplatin: 1.835 ± 0.237 μM (MCF-7)? | Interaction with DNA guanine Ru-N7 in vitro;64 dipeptidyl peptidase IV inhibitors (Ru-Cur = 97 ± 1.3 μM; curcumin ≥ 200 μM)67 | 64 and 67 |
| 27 | See Table 3 | See Table 3 | Caused apoptosis, but had no effect on cell cycle | 64 and 66 |
| 28 | ||||
| 29 | ||||
| 30 | ||||
| 31 | ||||
| 32 | ||||
| 33 | Not reported | Dipeptidyl peptidase IV inhibitors (DPPIV Ki: 20.2 μM) | 67 | |
| 34 | Dipeptidyl peptidase IV inhibitors (DPPIV Ki: 80.3 μM) | |||
| 35 | HeLa: 50 ± 1 μM; BEL-7404: 68 ± 1 μM; SMMC-7721: 89 ± 5 μM; normal cell HEK-293T: 53 ± 5 μM | Not reported | DNA binding | 68 |
| 36 | HeLa: 21 ± 1 μM; BEL-7404: 51 ± 5 μM; SMMC-7721: 50 ± 4 μM; normal cell HEK-293T: 29 ± 2 μM | |||
Anlong Xu et al.74 synthesized a group of Ru(II) complexes 1–3 containing β-carboline alkaloids. They can induce ROS-dependent autophagy and apoptosis in tumor cells (Fig. 6A). These Ru(II) complexes could penetrate into the nucleus and bind to DNA. The complexes had high inhibitory activity against tumor cells. Among these compounds, Compound 3 has higher antitumor activity than cisplatin. Based on this study, Lanmei Chen et al.75 synthesized two new Ru(II)-β-carboline complexes (RuβC-3 and RuβC-4) by introducing halogens into the carboline structure. The IC50 values of the compounds against HeLa cells were 3.2 ± 0.4 μM (compound 4, RuβC-3) and 4.1 ± 0.6 μM (compound 5, RuβC-4), respectively. An IC50 value of 4 μM was about 15 times smaller than that of compound 1, and 5 times smaller than that of compound 2. Mechanism studies showed (Fig. 6B) that two ruthenium complexes induced cell cycle arrest and apoptosis in HeLa cells by inhibiting ERK and Akt signaling. The research group also investigated the killing effect of N,C chelating complex on tumor cells,76 and designed N,C complexes Ru1 and Ru2 (Fig. 6C). The IC50 of A549 for HeLa and NCL-H460 cells ranged from 1.9 μM to 4.9 μM. The toxicity of Ru1 to HeLa cells was more than 30 times higher than that of compound 1 complexed with N,N, suggesting that the Ru-carboline derivatives complexed with N,C were more active.
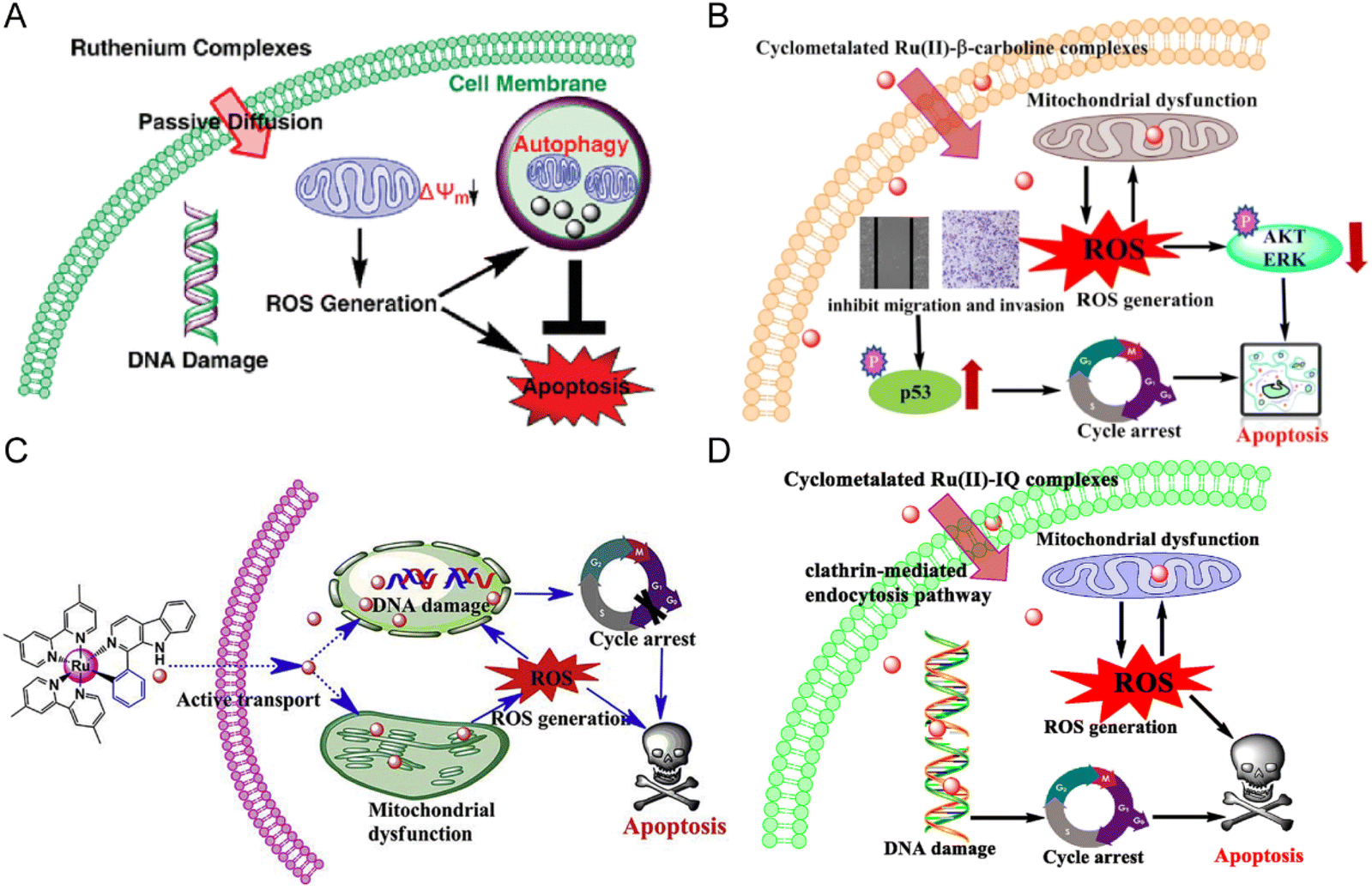 | ||
| Fig. 6 (A) The mechanism of compounds 1–3;74 this figure has been adapted from ref. 74 with permission from American Chemical Society, copyright 2010. (B) The mechanism of compounds 4–5;75 this figure has been adapted from ref. 75 with permission from Springer Berlin Heidelberg, copyright 2021. (C) The mechanism of compounds 6–7;76 this figure has been adapted from ref. 76 with permission from Elsevier, copyright 2017. (D) The mechanism of compounds 13–16;79 this figure has been adapted from ref. 79 with permission from Elsevier, copyright 2020. | ||
Lanmei Chen and Tao Wang et al.79 synthesized a group of novel Ru(II) complexes containing isoquinoline. RuIQ-1 and RuIQ-2 exhibited high cytotoxic activity against lung cancer cell lines (NCI-H460 and A549) and breast cancer cell lines (HeLa and MCF-7). Their IC50 values were one order of magnitude lower than cisplatin. These two complexes induced S and G2/M bicycle arrest by regulating cyclin-associated proteins (Fig. 6D). At the same time, they could reduce mitochondrial membrane potential (MMP). By promoting the production of intracellular reactive oxygen species (ROS), the complex caused DNA damage, leading to apoptosis-mediated cell death. More importantly, RuIQ-2 showed selective killing of tumor cells and low toxicity to both normal HBE cells in vitro and zebrafish embryos in vivo.
Edith C. Glazer et al.80 designed 22 complexes by chemically modifying the 2-, 5- and 7-sites of compound 17, and studied the structure–activity relationship for the antitumor effects. Shown in Table 2, the halogen modification at sites 2 and 7 can effectively improve the cytotoxicity of the complexes and reduce the IC50 of tumor cells to the level of nanomoles.
| Compounds | R1 | R2 | R3 | IC50 (μM) |
|---|---|---|---|---|
| 17-1 | H | H | H | 0.52 ± 0.06 |
| 17-2 | Me | H | H | 0.49 ± 0.07 |
| 17-3 | Br | H | H | 0.18 ± 0.015 |
| 17-4 | Cl | H | H | 0.11 ± 0.003 |
| 17-5 | o-Tolyl | H | H | 0.20 ± 0.069 |
| 17-6 | H | Br | H | 0.32 ± 0.013 |
| 17-7 | H | Cl | H | 0.43 ± 0.04 |
| 17-8 | H | o-Tolyl | H | 0.67 ± 0.19 |
| 17-9 | H | H | Br | 0.10 ± 0.018 |
| 17-10 | H | H | Cl | 0.09 ± 0.004 |
| 17-11 | H | H | o-Tolyl | 0.96 ± 0.11 |
| 17-12 | H | Me | Me | 0.18 ± 0.015 |
| 17-13 | H | Br | Br | 0.07 ± 0.008 |
| 17-14 | H | Cl | Cl | 0.11 ± 0.006 |
| 17-15 | H | Cl | Br | 0.09 ± 0.034 |
| 17-16 | H | Cl | I | 0.057 ± 0.002 |
| 17-17 | H | I | I | 0.12 ± 0.004 |
| 17-18 | Me | Me | Me | 0.25 ± 0.051 |
| 17-19 | Me | Br | Br | 0.08 ± 0.02 |
| 17-20 | Me | Cl | Cl | 0.12 ± 0.002 |
| 17-21 | Cl | Br | Br | 0.07 ± 0.009 |
| 17-22 Ru(II) dimer | H | — | Cl | 11.14 ± 0.658 |
In addition to the Ru-bipyridine antitumor chemotherapeutics, Ru-aromatic antitumor drugs are widely studied. Paul J. Dyson and Patrycja Nowak-Sliwinska et al.82 found that RAPTA-C reduced primary tumor growth in preclinical models of ovarian and colorectal cancer. When administered daily at a low dose (0.2 mg kg−1), RAPTA-C showed a significant reduction in the growth of A2780 ovarian cancer transplanted to the chicken chorioallantoic membrane model. Similar activity was observed at higher doses in a nude mouse model of LS174T colorectal cancer. In both models, significant inhibition of CD31-stained microvessel density was observed, confirming the previously found anti-angiogenic mechanism of RAPTA-C.
Ru-aromatic carboline complexes 19–24 are another group of interesting compounds, among which carboline imidazole complexes had higher cytotoxicity and better activity than cisplatin.64 Inhibition of CDK1 led to G2/M phase blockade, and apoptosis was induced by mitochondrial-related pathways and intracellular ROS elevation. Ru-Tetrahydroquinoline complexes follow the structural general formula of compound 25 in Fig. 5.84 Five N, O coordination Ru-tetrahydroquinoline complexes C1–C5 showed moderate activity against MCF-7 cells, with IC50 values ranging from 34 μM to 218 μM, and no inhibition against normal cells. The selective killing effect of these compounds is superior in comparison with cisplatin. C5 had the lowest activity, while C4 had the highest activity, and they differed only by one diastereocenter.
In addition to the ruthenium complexes of carboline and quinoline, ruthenium also forms a complex with curcumin diketone. Francesco Caruso et al.64 synthesized Ru-curcumin aromatic complexes. The complexes exhibited strong anti-tumor activity against colorectal cancer cell line HCT116 (IC50 = 13.98 μM), and a mild effect on breast cancer cell line MCF-7 (19.58 μM) and ovarian cancer cell line A2780 (23.38 μM). Human glioblastoma cell line U87 and lung cancer cell line A549 were less sensitive to the complexes. Moreover, the activity of cisplatin-resistant ovarian cancer CP8 cells was similar to that of A2780, indicating that there was no cross-resistance between Ru-Cur and cisplatin. Meanwhile, in another study,66 compounds 27–32 showed that the antitumor activity of Ru-curcumin complex was increased in vitro when curcumin polarity was reduced (Table 3). The two OH groups in curcumin were replaced by methoxy groups or by H, resulting in higher antitumor activity than in the parent curcumin complex (compound 26). The complexation of ruthenium did not improve the activity of the ligand, but the modification of the aromatic hydrocarbon could change the activity of the complex to a substantial extent. Complexes 27 and 29 with cymene and hexamethylbenzene as the aromatic ligand showed better activity in tumor cells. We compared the DPPIV inhibitory activity of compounds 26, 33, 34in vitro, which are composed of the same curcumin ligand, except for the aromatics.67 Compound 33 showed the highest inhibitory activity of DPPIV using the phenyl as aromatic. Wei Su and Peiyuan Li et al.68 also designed complexation of ruthenium with curcumin analogues, such as compounds 35 and 36. The effect on tumor cells is twofold for compound 36 in comparison with compound 35, indicating that methyl substituents on ligands had positive anti-proliferation activity.
| A2780 | A549 | MCF-7 | HCT116 | U87 | |
|---|---|---|---|---|---|
| a p < 0.01 vs. 4. b p < 0.001 vs. 4. c p < 0.01 vs. 2. d p < 0.05 vs. 4. e p < 0.05 vs. 2. f p < 0.05 vs. HCurcI. g p < 0.01 vs. HCurcI. h p < 0.05 vs. HCurcII. i p < 0.01 vs. HCurcII. j p < 0.05 vs. curcumin. k p < 0.01 vs. curcumin. | |||||
| HCurcI | 1.5 ± 0.3i | 3.7 ± 0.5k,j | 1.76 ± 0.16i | 1.83 ± 0.20i | 9.3 ± 2.0i |
| HCurcII | 43.8 ± 1.9k | 53.2 ± 1.9k | 29.7 ± 6.8k | 24.2 ± 6.5j | 41.5 ± 9.7k |
| Curcumin | 4.3 ± 1.7 | 14.7 ± 1.8 | 10.5 ± 2.6 | 5.8 ± 0.8 | 12.7 ± 2.1 |
| Compound 27 | 9.4 ± 1.0c,d,f | 13.7 ± 0.6b,e,g,i | 10.6 ± 1.6a,i | 15.5 ± 2.7 | 9.4 ± 0.7i |
| Compound 28 | 28.3 ± 2.3g,i,k | 20.6 ± 0.7a,g,i | 11.6 ± 1.8a,i | 19.2 ± 0.5 | 21.4 ± 2.8h |
| Compound 29 | 11.4 ± 0.8c,d,g,i | 15.5 ± 1.7b,e,g,i | 9.7 ± 0.3b,i | 32.6 ± 5.7g,k | 10.9 ± 0.6i |
| Compound 30 | 21.2 ± 2.1g,i,k | 30.6 ± 1.1g,i | 25.2 ± 2.3g,k | 15.9 ± 3.1 | 21.2 ± 4.3h |
| Compound 26 | 23.4 ± 3.3 | 63.2 ± 8.9 | 19.6 ± 2.4 | 14.0 ± 1.5 | 62.3 ± 8.9 |
Collective analysis of these studies reveals the importance of aromatic hydrocarbons for the activity of ruthenium complexes. The ruthenium complexes with benzene as aromatic ligand had better enzyme activity in vitro because benzene had less spatial hindrance and entered the active pocket of enzymes more easily.67 In cells, however, the activity was reversed, possibly because cymene and hexamethylbenzene were more lipophilic and entered cells more easily. Therefore, a delicate balance between steric hindrance and lipophilicity needs to be considered when designing ruthenium complexes. In summarizing the structure–activity relationship, we found the following general principles for structure design: (1) most of the ruthenium complexes are more active than ligands; (2) the cytotoxic effect of ligands are often stronger after being modified by Cl, Br and I; (3) N,N-, N,C-, O,O-, N,O- and other coordination can form ruthenium complexes, among which the N,C-coordination ruthenium complexes are often more active; (4) the antitumor activities of ruthenium complex are mainly demonstrated by DNA binding, production of reactive oxygen species, the effect on cell cycle and apoptosis. However, we also found that the biological activities of ruthenium complexes evaluated by different research groups vary greatly, such as the IC50 of cisplatin for MCF-7 cells (Table 2), which may be related to cell status in different laboratories.
3. Research and development of ruthenium nanodrugs
Structural optimization of ruthenium complexes does not necessarily result in the best therapeutic effect in many cases. For example, it is difficult to satisfy the requirements of steric hindrance and lipophilicity in one compound.66 Therefore, combination with advanced drug delivery technology is necessary to further improve the antitumor activity of ruthenium complexes.85–87 The nano drug delivery system (NDDS) certainly offers many advantages over traditional methods.88 For example, firstly, NDDS can be used as a targeting delivery system for ruthenium complexes. Nanoparticles mediate the enhanced permeability and retention (EPR) effect,89–92 thus increase the passive targeting effect in tumor cells. NDDS can also be modified by receptors, proteins and enzymes for active targeting.93–97 Secondly, the increased selectivity of ruthenium compounds reduces systemic toxicity.98 Thirdly, the lipophilicity of ruthenium complexes can be increased by using NDDS rather than changing the structure of the compound, which promotes the delivery of ruthenium complexes to tumor cells, thus significantly improving the therapeutic effect.99 Fourthly, nanoparticles allow simultaneous delivery of multiple components, thus combining multiple therapeutic modalities.100Zhe-Sheng Chen et al.41 summarized studies on the encapsulation of Ru(II) complexes in NDDS, such as Ru(II)–selenium nanoparticles,101,102 Ru(II)–gold nanomaterials,103 Ru(II)–silicon dioxide composites104–106 and Ru(II)–carbon nanotubes,107etc. Encapsulation of Ru(II) complexes in these organic or inorganic vectors can improve their targeting and delivery to tumor cells. Christophe M. Thomas et al.108 also summarized nanomedicine for delivering Ru(II) polypyridine-based complexes of organic and inorganic materials. In addition to the encapsulation of ruthenium complex in nanocarriers, excipient-free nanodrugs3,109 have attracted the interest of researchers. Excipient-free nanodrugs refer to the fact that the matrix composed of nanomaterials is also a substance that exerts drug activity, and they are generally self-assembled by active pharmaceutical ingredients.110 Based on previous studies,109,111 the difference of excipient-free NDDS is based on whether the total amount of additives exceeds 80%.112,113 Therefore, in excipient-free NDDS, other additives may be present in addition to the active pharmaceutical ingredients. However, the additives in excipient-free NDDS may have a therapeutic effect rather than being as a carrier only, which is different from the traditional function of excipients in drug formulation. Here, we give an overview on Ru carrier nanoparticles and excipient-free nanodrugs.
3.1 Ru carrier nanoparticles
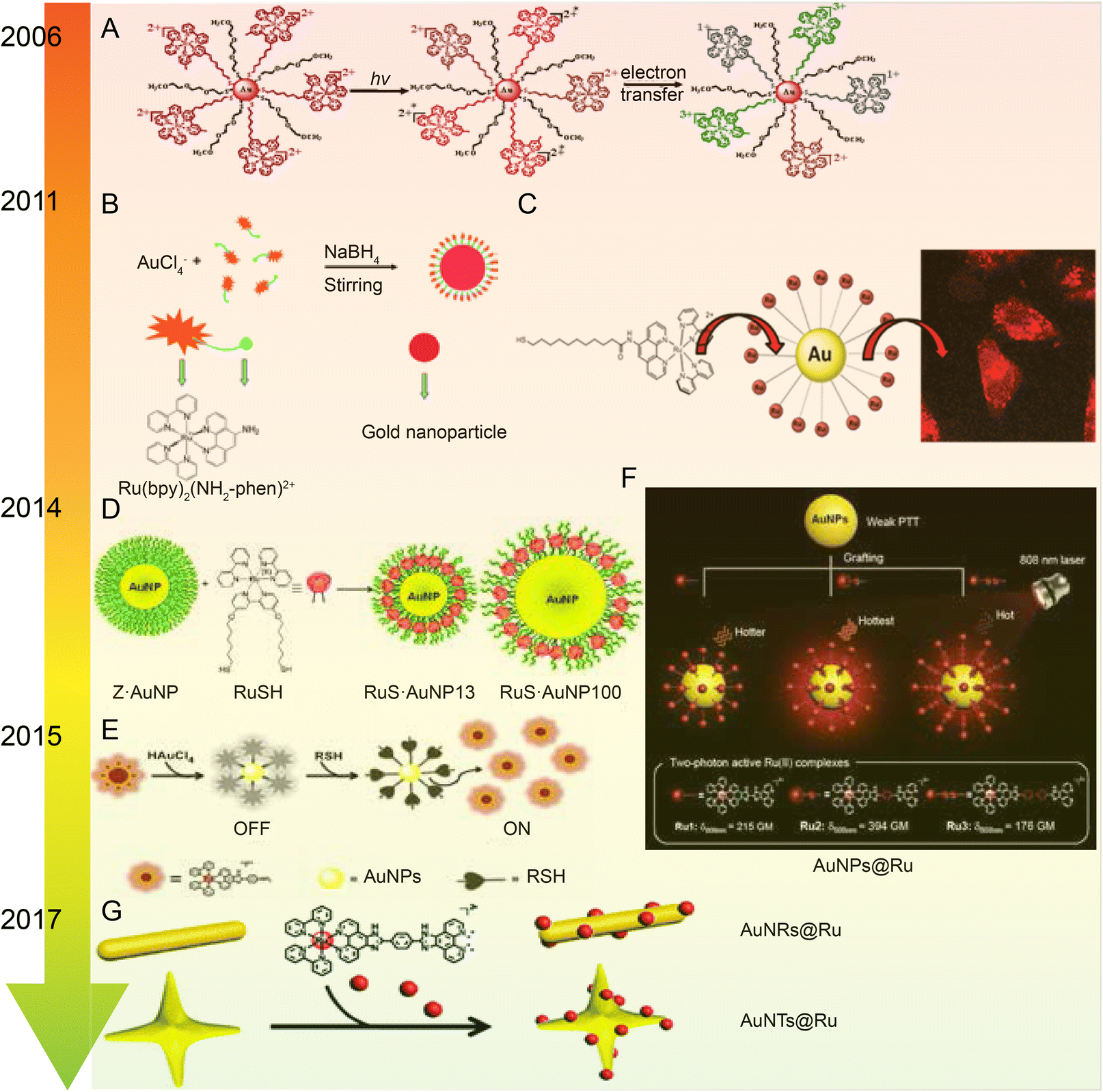 | ||
| Fig. 7 Development of Ru–Au nanoparticles. (A) Photochemistry of functionalized Ru tribipyridine on gold nanoparticles;125 this figure has been reproduced from ref. 125 with permission from the American Chemical Society, copyright 2006. (B) Schematic diagram of Ru(bpy)2(NH2-phen)2+ functionalized gold nanoparticles;126 this figure has been reproduced from ref. 126 with permission from the Royal Society of Chemistry, copyright 2011. (C) Luminescent Ru(II) polypyridine-functionalized gold nanoparticles as single-photon cell-imaging agents;128 this figure has been reproduced from ref. 128 with permission from the American Chemical Society, copyright 2011. (D) Gold nanoparticles coated with Ru(II) complexes are used for single-photon luminescence imaging in cells;129 this figure has been reproduced from ref. 129 with permission from Elsevier, copyright 2015. (E) Gold nanoparticles coated with Ru(II) complexes are used for single-photon luminescence imaging in cells;130 this figure has been reproduced from ref. 130 with permission from Elsevier, copyright 2014. (F) High photothermal conversion efficiency of gold nanospheres grafted by two-photon emitting Ru(II) complexes;131 this figure has been reproduced from ref. 131 with permission from the Royal Society of Chemistry, copyright 2012; (G) Schematic of Ru(II) complexes grafted to gold nanorods and nanostars;132 this figure has been reproduced from ref. 132 with permission from the Royal Society of Chemistry, copyright 2017. | ||
The same research group applied Ru–gold nanoparticles to tissues and animals. In 2014, they synthesized a RuNH2@AuNPs two-photon light-emitting probe130 (Fig. 7E), which formed mercaptan-responsive nanoparticles by using the principle that mercaptan can replace the Ru(II) complex on the AuNPs’ surface to release the luminescent RuNH2. The released Ru(II) complex showed strong two-photon luminescence and could penetrate living mouse tissues in depths of 80–170 μm using a two-photon microscope. In 2015, the research group grafted a Ru(II) poly-pyridine complex onto gold nanospheres (Ru@AuNPs)131 (Fig. 7F), which significantly improved the two-photon emission intensity and photothermal treatment efficiency of gold nanospheres. Under the irradiation of 808 nm laser with power density of 0.8 W cm−2 for 5 minutes, tumor size was significantly reduced and even ablated. In 2017, the research group designed functionalized gold nanorods (AuNRs@Ru) and gold nanorods (AuNTs@Ru) of Ru(II) complexes, showing higher photothermal stability and photothermal efficiency132 (Fig. 7G). AuNRs@Ru and AuNTs@Ru could retain the morphology and the properties of near infrared plasmon resonance absorption under 0.25 W cm−2 laser irradiation. Photothermal studies showed that AuNRs@Ru and AuNTs@Ru had more effective therapeutic effects in HeLa cells and in in vivo tumor models.
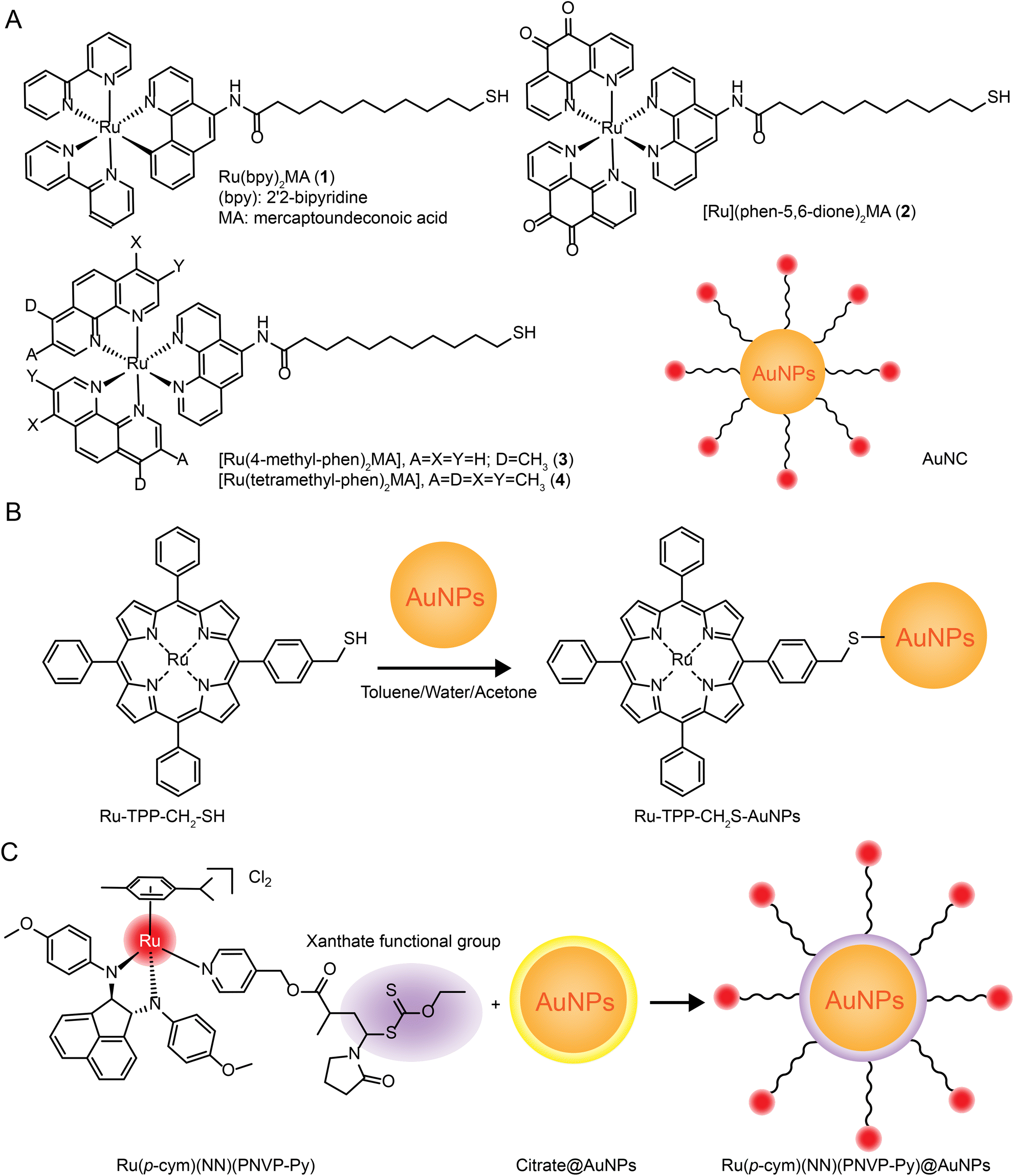
The number of studies on the modification of gold nanoparticles by ruthenium complexes has been increasing in recent years. Most of these studies involve the attachment of Ru polypyridine complexes on the surface of gold nanoparticles via sulfur atoms. For example, Balachandran Unni Nair et al.133 used functionalized gold nanoparticles of Ru(II) polypyridine complex (Fig. 8A) to stabilize the collagen of extracellular matrix under light irradiation. Francesca Limosani et al.120 explored the synthesis method of functionalized Ru porphyrin gold nanoparticles via sulfur/gold covalent bonds (Fig. 8B). Samarendra Maji and Mani Ganeshpandian et al.119 obtained Ru(p-cym)(NN)(PNVP-Py)@AuNPs by replacing some of citrate molecules on the surface of the gold nanoparticles with xanthate functional group Ru(η6-p-cym)(NN)(PNVP-Py)Cl2 (Fig. 8C). The surface-functionalized AuNPs of Ru(II)-arene complexes improved the solubility of the complexes, and prolonged blood circulation and the release of ruthenium complexes. Ru(p-cym)(NN)(PNVP-Py)@AuNPs showed low DNA-cutting ability but were more effective in colorectal adenocarcinoma cells than the complex alone, possibly because Ru(p-cym)(NN)(PNVP-Py)@AuNPs had better membrane permeability. In addition to the hydrophobicity of the aromatic ligands, long-chain polymers could also promote the nanoconjugates to cross the cell membrane and induce cell apoptosis.
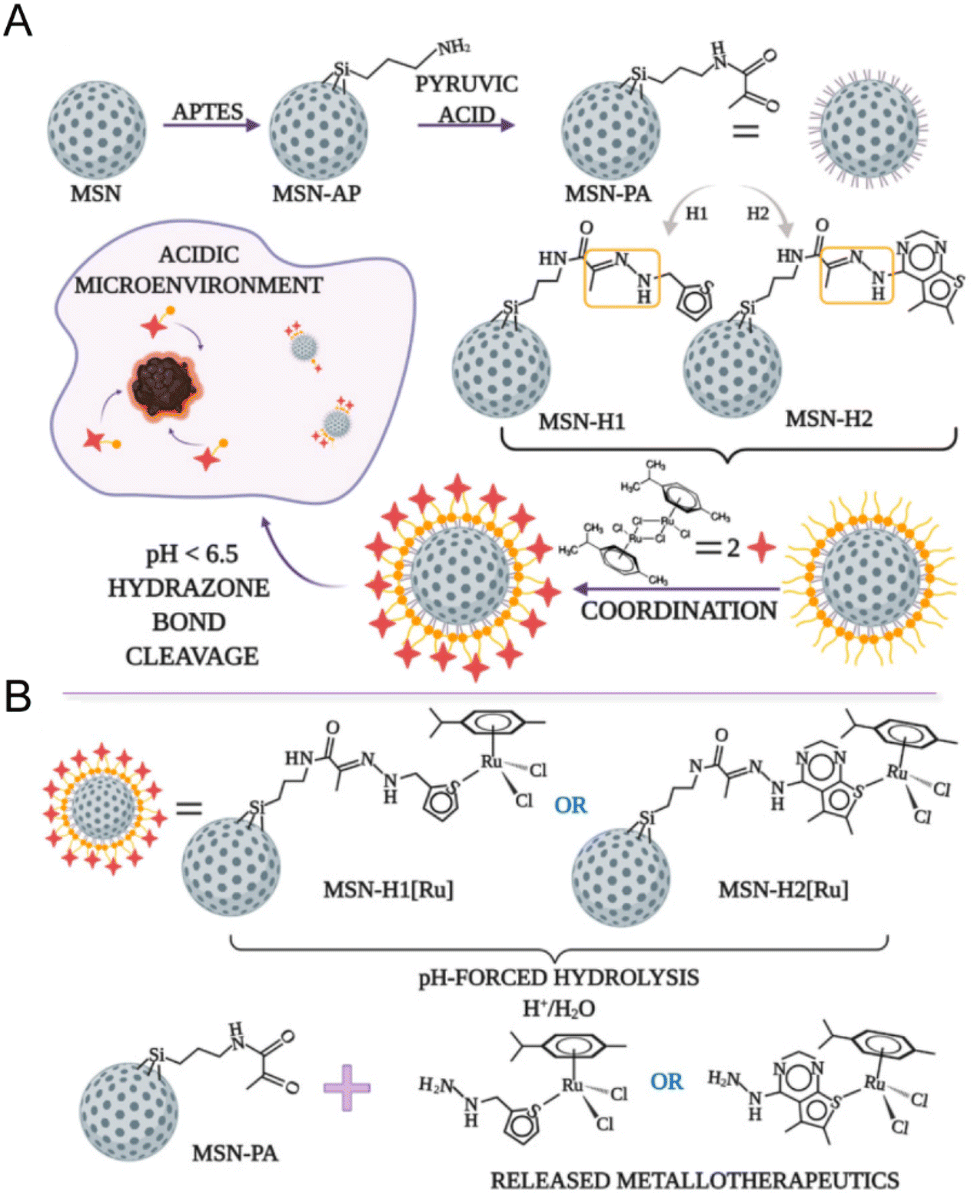
The pH-responsive delivery is also a common ruthenium complex type. The pH-responsive delivery depends on pH-sensitive chemical bonds or groups in the structure, such as hydrazone bonds and imidazole groups. Ling Qiu and Jianguo Lin et al.138 developed a pH-sensitive mesoporous silica nanocarrier RuNHC@MSNs–CTS–biotin (CTS = chitosan) for targeted delivery and controlled release of Ru(II) N-heterocyclic carbine (RuNHC) complex. CTS captures RuNHC complexes in the mesopores, and biotin enables the nanosystem to specifically target tumor cells through biotin receptor-mediated endocytosis. The release of RuNHC was pH-dependent, with a release rate of 59.71% at pH 5.0, but almost no release at pH = 7.4. In 2021, Goran N. Kaluđerović and Nikola Ž. Knežević et al.139 also constructed a pH-responsive mesoporous silica nanocarrier for the release of ruthenium metal therapeutics. In Fig. 9, the surface of mesoporous silica nanoparticles (MSN) is functionalized by two types of ligands, (2-thiophenyl methyl)hydrazine hydrochloride (H1) and (5,6-dimethylthiopheno[2,3-d]pyrimidine-4-)yl)hydrazine (H2). They are connected to the surface of MSN by pH-responsive hydrazone. Further coordination with the Ru(II) center resulted in two types of nanomaterials, MSN-H1[Ru] and MSN-H2[Ru]. The release of Ru(II) complexes is enhanced at low pH. The enhanced antitumor effect on metastatic B16F1 melanoma cells was confirmed under acidic conditions in vitro.
3.2 Ru excipient-free nanoparticles
Excipient-free nanoparticles are usually composed of pure drug molecules. Excipient-free nanoparticles often contain a small amount of surfactant, enabling self-assembly of nanomaterials, but the total drug load is higher than 80%.140 Two methods are commonly used to synthesize carrier-free nanoparticles.141 One method is the direct self-assembly of drug molecules through non-covalent interactions, such as hydrophobic interactions, π–π stacking and CH–π interactions. The self-assembly method can be a single drug or a combination of multiple drugs, such as chemotherapeutic drugs and photothermal conversion agents. The second method is to covalently link the drug with a helper molecule. Drug molecules in this category cannot directly self-assemble and form nanoparticles. Therefore, drug molecules are modified to form amphiphilic structures through connecting bridges with peptides, lipids, chemotherapy drugs, immunotherapy drugs and photothermal conversion agents through ester bonds, disulfide bonds and thioacetal bonds.Excipient-free nanoparticles containing metal compounds show excellent application potential in tumor imaging, drug delivery and tumor therapy. Metals such as iron, zinc, copper, gold and silver are the main metal components of biomedical nanoparticles,142 especially gold nanoparticles, which have been basically industrialized in biomedicine and can be controlled to form nanoparticles of various shapes and sizes. Different from the systematic study of gold nanoparticles, there is little research on excipient-free RuNPs, and the synthesis, morphology and size of excipient-free RuNPs have not been reviewed in detail. In addition to single ruthenium excipient-free nanoparticles, researchers also use covalent or non-covalent interactions to link ruthenium to large molecular proteins, small molecular drugs, peptides, etc., and self-assemble to form a variety of nanostructures. Therefore, we reviewed the research on single excipient-free RuNPs and the excipient-free nanoparticles by the coupling of Ru with small and large molecules in recent years.
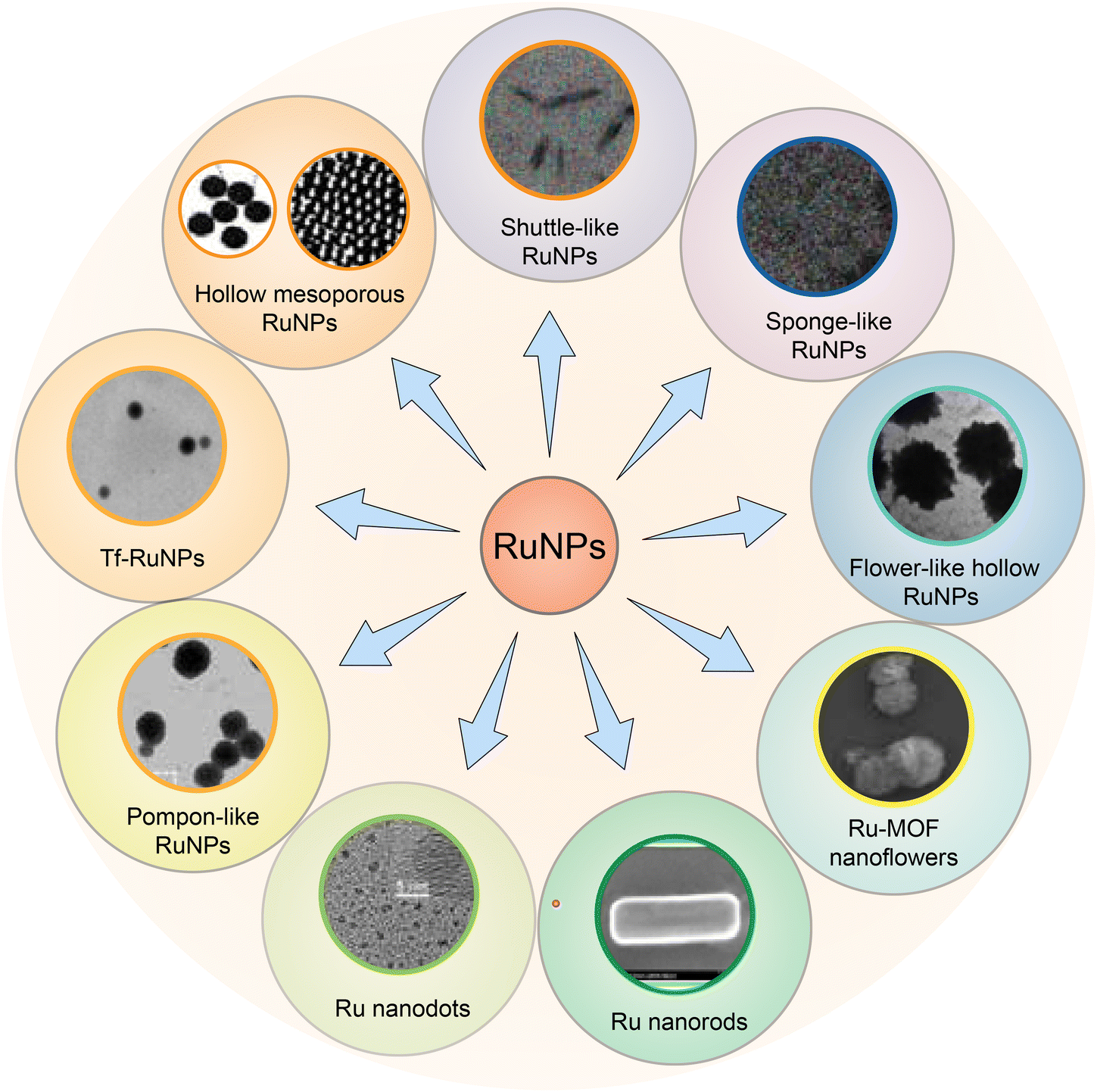 | ||
| Fig. 10 A summary of RuNPs with different morphology. | ||
3.2.1.1 Hollow mesoporous RuNPs. Sanghyo Kim et al.150 synthesized hollow mesoporous RuNPs with controllable sizes. The RuNPs were prepared by a double-template method. Poloxamer P407 and amino-functionalized silica particles were added into RuCl3 solution to form hollow mesoporous RuNPs, and the template material was dissolved by perchloric acid to obtain the RuNPs with controllable size. Furthermore, these authors also demonstrated that hollow mesoporous RuNPs exhibited cytotoxic effects above 100 μg mL−1, and that the synthetic RuNPs with negatively charged surfaces had high cellular absorption, high drug load, and pH-sensitive drug-release properties.
In 2018, the group of Jie Liu153 designed and synthesized a hollow mesoporous RuNPs with dual targeting function, RBT@MRN-SS-Tf/Apt (Fig. 11A and B). The anti-tumor drug [Ru(Bpy)2(TIP)]2+ (RBT) was loaded in the mesoporous ruthenium nanoparticles (MRN). The researchers used transferrin (Tf) and aptamer AS1411 (Apt) to modify the surface of MRN via disulfide bonds. RBT@MRN-SS-Tf/Apt could effectively penetrate the blood–brain barrier and target glioma, specifically killing glioma cells in vivo and in vitro. In addition, the antitumor drug [Ru(bpy)2(TIP)]2+ (RBT) could produce reactive oxygen species (ROS) and induce tumor cell apoptosis under laser irradiation.
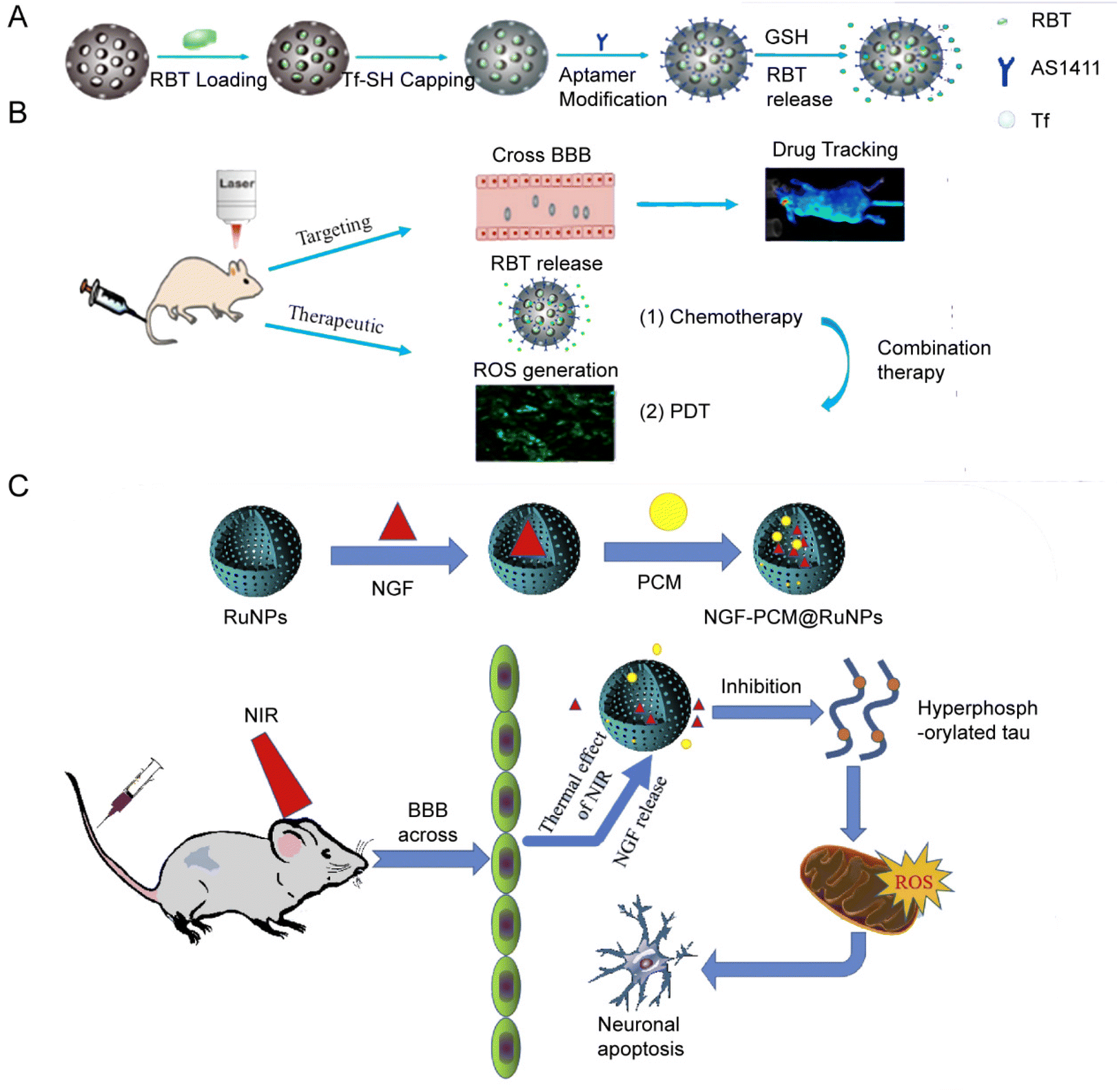 | ||
| Fig. 11 (A) Synthesis and (B) mechanism of RBT@MRN-SS-Tf/Apt. Adopted from ref. 153. This figure has been reproduced from ref. 153 with permission from Elsevier, copyright 2018; (C) NGF–PCM@RuNPs passed through the BBB under NIR irradiation, and PCM responds to thermal effects to trigger the release of NGF, thereby reducing ROS production and alleviating neuron damage by inhibiting tau hyperphosphorylation. Adopted from ref. 147. This figure has been reproduced from ref. 147 with permission from Elsevier, copyright 2020. | ||
3.2.1.2 Ruthenium nanospheres. The same research group17 also conducted a study on ruthenium nanospheres. Ruthenium nanospheres were synthesized by a classical double reduction method, in which Ru(III) was reduced by NaBH4 under the protection of polyethylene pyrrolidone (PVP). After purification, solid Ru nanospheres were yielded. Thioglycolic acid was used as a link to modify transferrin (Tf) onto the surface of Ru nanospheres to obtain Tf-RuNPs with high photothermal conversion properties and high targeting properties.
Tf modification was able to enhance the uptake of RuNPs by endocytosis. Meanwhile, Tf-RuNPs were able to inhibit or even ablate tumor tissues by PTT in vivo and in vitro. In addition, the photothermal conversion efficiency η of RuNPs was compared with that of AuNRs, a classical photothermal conversion agent. The temperature curve of RuNPs was similar to that of AuNRs under the irradiation of laser with the wavelength of 808 nm. The photothermal conversion efficiency η of RuNPs was 53.2%, lower than that of AuNRs (87.5%). However, when using a 660 nm laser, RuNPs had a slightly higher heating rate than AuNRs, and the η of RuNPs was 60.7%, slightly lower than that of AuNRs (67.4%). This was the first study to propose the use of Ru nanospheres in PTT and compare them with classical photothermal conversion agents.
3.2.1.3 Flower-like RuNPs. We found that there are two types of flower-like RuNPs reported at present. One is the hierarchical Ru-MOF nanoflower; the other is the flower-like mesoporous RuNPs. Yingzi Fu et al.146 reported a luminescent functionalized a metal–organic flower-like hierarchical nanostructure, which was prepared by one-step solvothermal method at low temperature154 for the detection of tryptophan with high sensitivity.
Jie Liu's group designed another flower-like RuNP.155 They synthesized flower-like RuNPs (FRu), spherical ruthenium (SRu) and spindle ruthenium (SPRu) nanoparticles. FRu had higher photothermal conversion efficiency (93.4%) than SRu (60.7%). FRu also mediated the synergistic effect between PTT and PDT, and presented no obvious toxicity in vivo and in vitro. In 2020, the research group147 used flower-shaped hollow nano-ruthenium as a carrier to load nerve growth factor (NGF) and sealed it with phase change material (PCM) (Fig. 11C). This NGF–PCM@Ru NPs could penetrate the blood–brain barrier (BBB) under near-infrared (NIR) irradiation and respond to the phase change of the lesion area. The release of NGF caused inhibition of tau hyperphosphorylation and reduction of oxidative stress. It further restored nerve damage and maintained neuronal morphology, and consequently improved learning and memorizing in a mouse model of Alzheimer's disease.
Cyclodextrin (CDs) is a cyclic oligosaccharide linked by 6–8 D-glucose units via α-1,4-glucoside bonds. Due to its good water solubility and low toxicity, CDs has been widely used as a component in supramolecular chemistry and can combine various guest molecules to form nanostructures.163,168,169 Zong-Wan Mao et al.162 designed a host–guest system by combining cyclodextrin-functionalized antitumor Ru(II) complex with adamantane attached tumor targeting peptide. The host–guest molecules self-assemble to form a stable phosphorescent nanostructure Ru–CD-RGD. Ru–CD forms C–N bond connection through the Mannich reaction,170 and the β-CD cavity could strongly bind to adamantine alkyl groups,171 thus Ru–CD-RGD was formed (Fig. 12A). Ru–CD-RGD selectively killed integrin αVβ3-enriched tumor cells U87MG. Further mechanism studies showed that Ru–CD-RGD was mainly distributed in lysosomes and induced apoptotic cell death through lysosome damage, ROS and caspase activation. Similarly, Yu Liu et al.163 designed self-assembly of adamantane-functionalized transferrin (Ad-TRF) and β-CD functionalized ruthenium complex (Ru-HOP-CD) (Fig. 12B). Ad-TRFs could be used as a target site for tumor cells, and the coordinated Ru(II) center could be used as a functional group for photodynamic therapy. Not only could Ad-TRF/Ru-HOP-CD target tumor cells and selectively kill tumor cells, but it also showed high PDT ability under visible light irradiation, combining chemotherapy with photodynamic therapy.
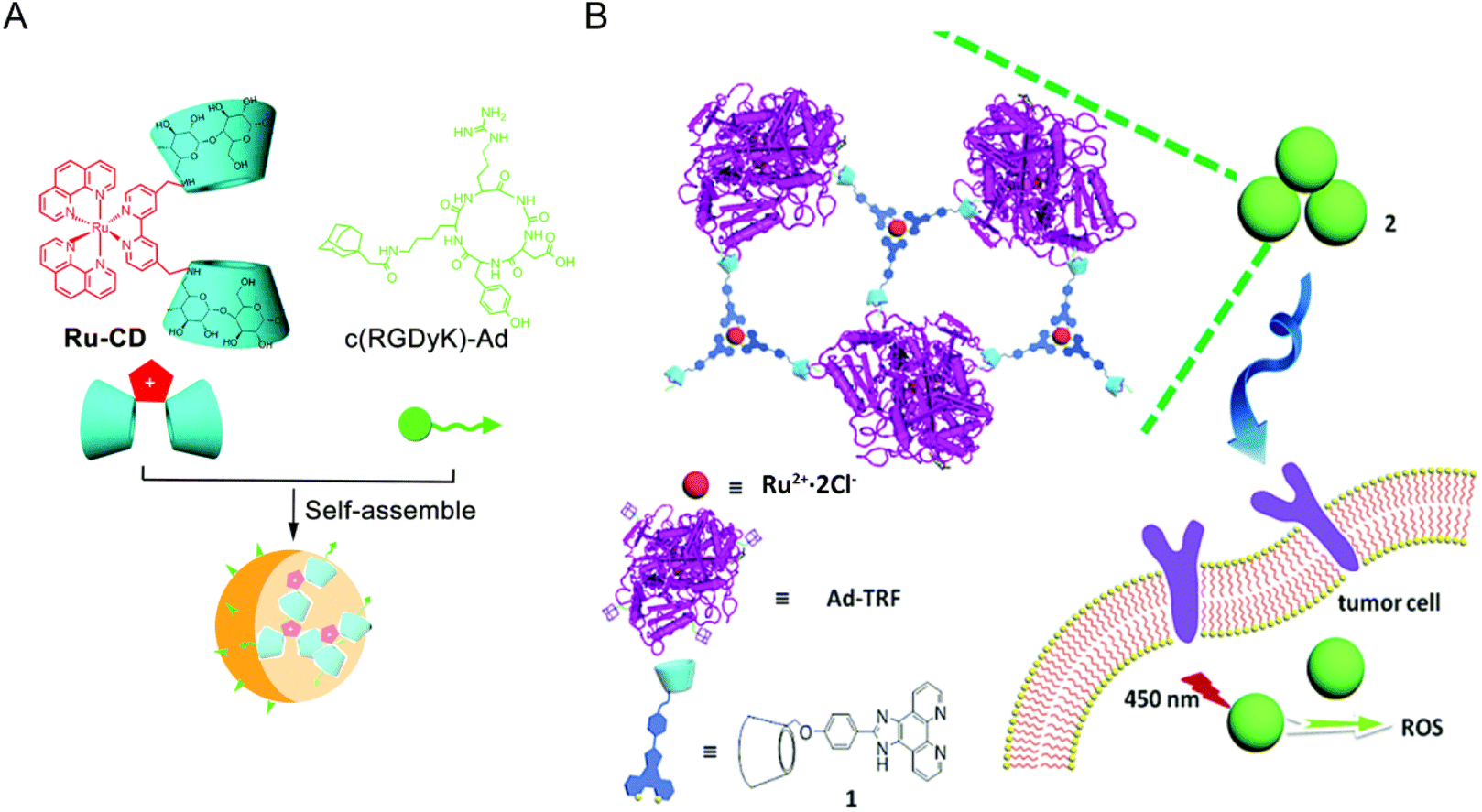 | ||
| Fig. 12 Schematic diagram of Ru and CD self-assembly. (A) Self-assembly process of Ru–CD-RGD.162 This figure has been adapted from ref. 162 with permission from the Royal Society of Chemistry, copyright 2017; (B) self-assembly process and mechanism of Ad-TRF/Ru-Hop-CD.163 This figure has been adapted from ref. 163 with permission from the Royal Society of Chemistry, copyright 2019. | ||
Jie Liu's group174 designed a fluorescent antitumor complex ([Ru(bpy)2(tip)]2+, RBT) and bispecific antibody conjugate (SS-Fc) using polyethylene glycol hollow mesoporous ruthenium nanoparticles (HMRu NPs) as carrier. HMRu NPs were synthesized using an improved dual-template method,175 loaded with RBT, which then linked to SS-Fc (anti-CD16, and anti-CEA) via polyethylene glycol. SS-Fc is a bispecific antibody with two different epitopes. On the one hand, its antitumor embryonic antigen (CEA) arm can recognize tumor-specific antigens, adding a targeting function to the delivery system;176 on the other hand, the anti-CD16 arm can bind NK cell receptors to activate immune responses.174 These functional designs enable HMRu@RBT-SS-Fc to be sensitive to near-infrared light and exhibit high tumor targeting and antitumor activity in vivo. HMRu@RBT-SS-Fc achieved the combined use of HMRu-based PTT, RBT-induced PDT and SS-Fc-mediated immunotherapy for colorectal cancer.
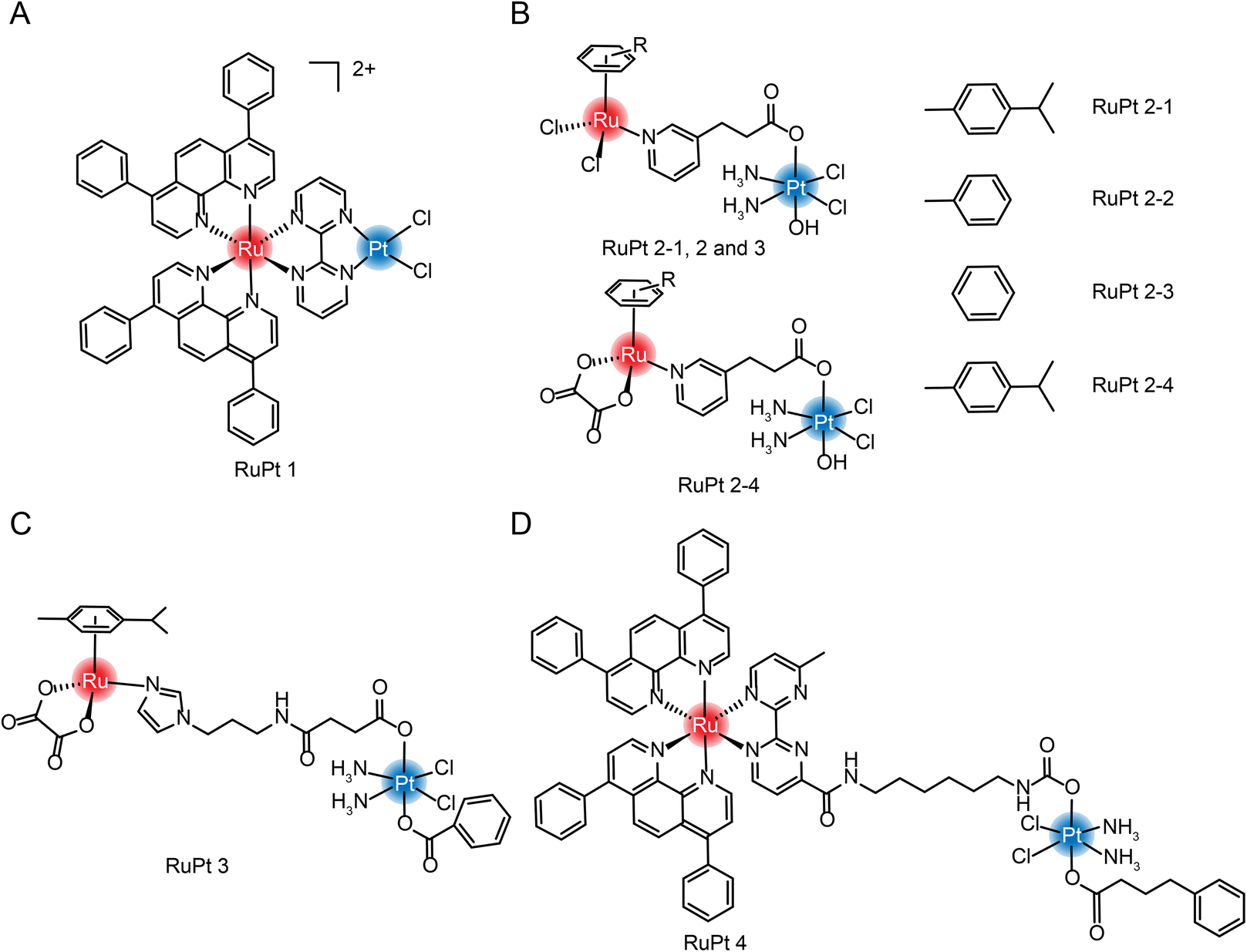 | ||
| Fig. 13 The structure of Ru/Pt complexes (A) RuPt 1; (B) RuPt 2-1, 2, 3 and 4; (C) RuPt 3 and (D) RuPt 4. | ||
Guangyu Zhu et al.182 were the first to design and synthesize four Ru(II)/Pt(IV) arene complexes (Fig. 13B). Although the self-assembly properties were not reported, complexation still resulted in enhanced cytotoxicity and increased anti-metastasis properties compared with aromatic Ru complexes. At the same time, the group also synthesized another dinuclear Ru(II)/Pt(IV) arene complex with connecting arms in the middle (Fig. 13C).183 In the tumor-normal cell co-culture system, the IC50 value of Pt(IV)–Ru(II) complex against normal cells is 7–10 times higher than that of cisplatin, which significantly improved the selectivity of tumor cells and reduced in vivo toxicity. The group also synthesized a multi-targeting Ru(II)–Pt(IV) polypyridyl complex combining cancer activation chemotherapy and photodynamic therapy to overcome drug-resistant cancers (Fig. 13D).184
Xing-Jie Liang and Si Wu et al.22 first proposed the self-assembly characteristics of Ru(II)/Pt(IV) and synthesized a dual-responsive Pt(IV)/Ru(II) bimetallic polymer (PolyPt/Ru). PolyPt/Ru was connected by three parts to form an amphiphilic structure (Fig. 14). The hydrophilic part was polyethylene glycol, and the hydrophobic part included reduction-responsive platinum precursor Pt(IV) and red-responsive Ru(II). PolyPt/Ru nanospheres could be absorbed by cisplatin-resistant cells. After irradiation with red light, the polymer degraded and released Ru(II) complex, cisplatin and 1O2, which could synergistically inhibit the growth of drug-resistant tumor cells in vivo and in vitro. In this study, the ruthenium complex was self-assembled with the prodrug of cisplatin conjugation to form a nanodrug delivery system, which can bind to the tumor site through EPR effect, achieving photoresponsiveness and glutathione reduction responsiveness. The synergic effect significantly improved the selective killing effect of tumors.
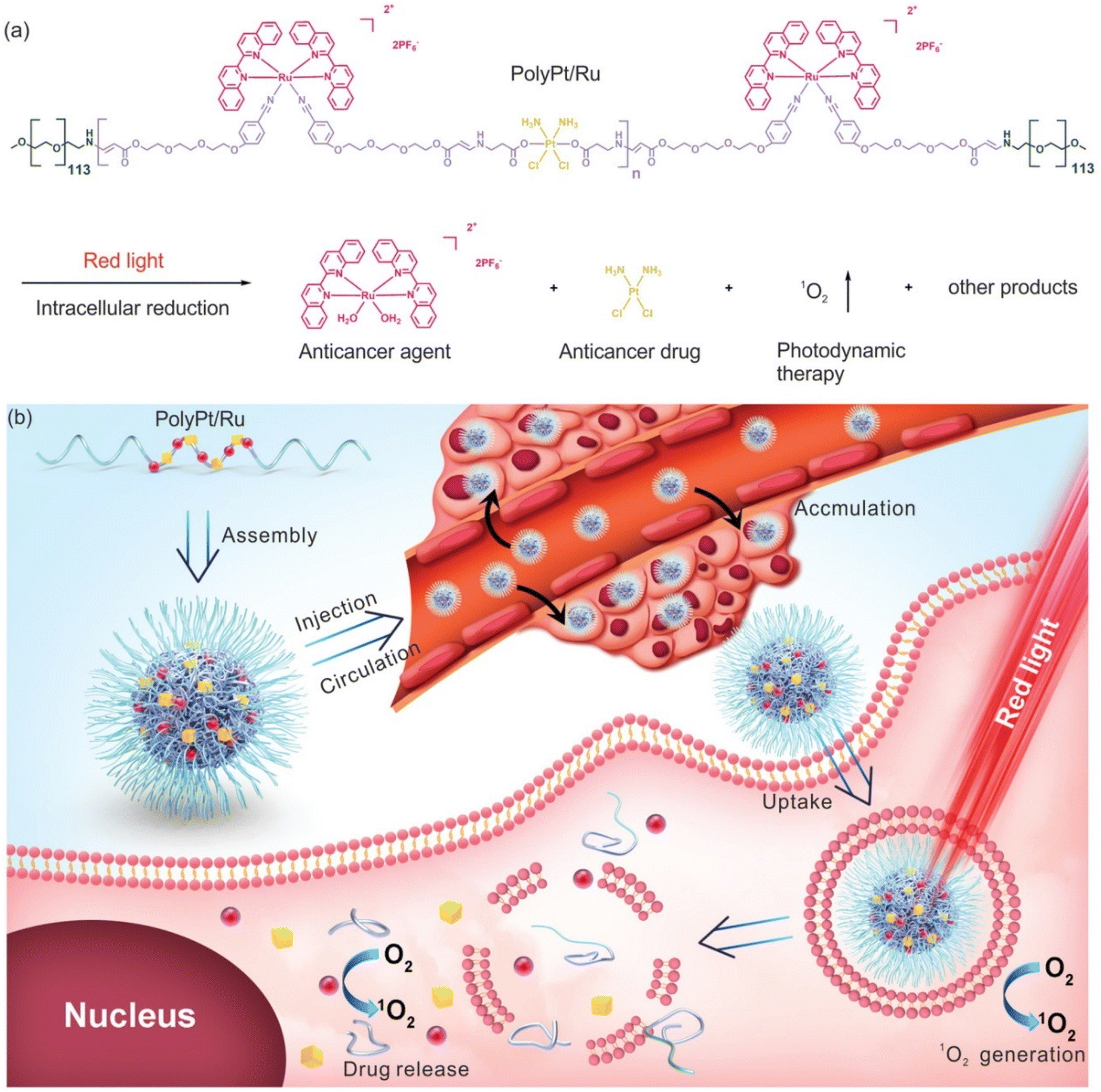 | ||
| Fig. 14 Structure of amphiphilic copolymer PolyPt/Ru. (a) Red light irradiation and intracellular reduction induced polymer degradation to produce 1O2, which released the antitumor drug cisplatin and antitumor agent [Ru(biq)2(H2O)2](PF6)2 (biq = 2,2′-biquinoline); (b) schematic diagram of self-assembly, extracellular and intracellular processes for antitumor therapy using PolyPt/Ru.22 This figure has been adapted from ref. 22 with permission from Wiley, copyright 2020. | ||
Both excipient-free nanodrugs and nanodrugs formed with helping agents are NDDS, that is, both involve carrier systems. The main basis for the division of conventional and excipient-free NDDS is whether the drug content is above 80%. The difference is whether the excipients play a therapeutic effect, rather than only play a role as a carrier, or even increase the excretory burden of the body. Besides, both conventional and excipient-free Ru nanodrugs are designed to achieve the combination of multiple therapeutic modalities, which is one of the potential directions for cancer treatment in the future. The development from conventional nanodrugs to excipient-free nanodrugs is an embodiment of the simplification of the combined treatment system for cancer. The applications of ruthenium nanomedicine in cancer combination therapy are discussed in detail in Section 5.
4. Absorption, distribution, metabolism and excretion (ADME) of ruthenium
The pharmacokinetics (PK) runs through the whole process of preclinical and clinical research for ruthenium complexes185 and its nanodrugs.186 There are a few studies on the complete ADME process of ruthenium complexes,187,188 but studies concerning one or two of the processes have become a standard practice in the research of ruthenium antitumor drugs. The ICP-MS and LC-MS/MS methods are the most common methods in PK studies.189 In this section, ADME studies of ruthenium drugs are reviewed (Fig. 15). | ||
| Fig. 15 ADME process of ruthenium complexes or nanoparticles. | ||
4.1 Absorption
The current method of administration of ruthenium complexes in living organisms is through intravenous injection.188 The drugs enter the internal circulation system directly and are transported to various parts of the body by the blood. Studies have found that ruthenium complexes can bind with albumin and transferrin in blood and be transported to tumor tissues. Once they reach the targeted site, they can be taken up by cells through different pathways.Drug uptake pathways by cells include simple diffusion, assisted diffusion, active transport, and endocytosis (Fig. 16).41,43,190 Both simple diffusion and assisted diffusion are passive, requiring no energy. Assisted diffusion requires the help of proteins, and simple diffusion does not. Active transport requires energy in the cell. When a cell needs to ingest large proteins, nanoparticles, or liquids, they are captured externally by the cell membrane and then separated from the cytoplasm by lipid membranes, an effect known as endocytosis.191 Endocytosis is divided into phagocytosis and pinocytosis. Pinocytosis is further divided into macropinocytosis, clathrin-mediated endocytosis, caveolin-mediated endocytosis, and clathrin- and caveolin-independent pathways based on the molecular mechanisms.192,193 Transferrin/transferrin receptor-mediated endocytosis is a typical clathrin-mediated endocytosis.
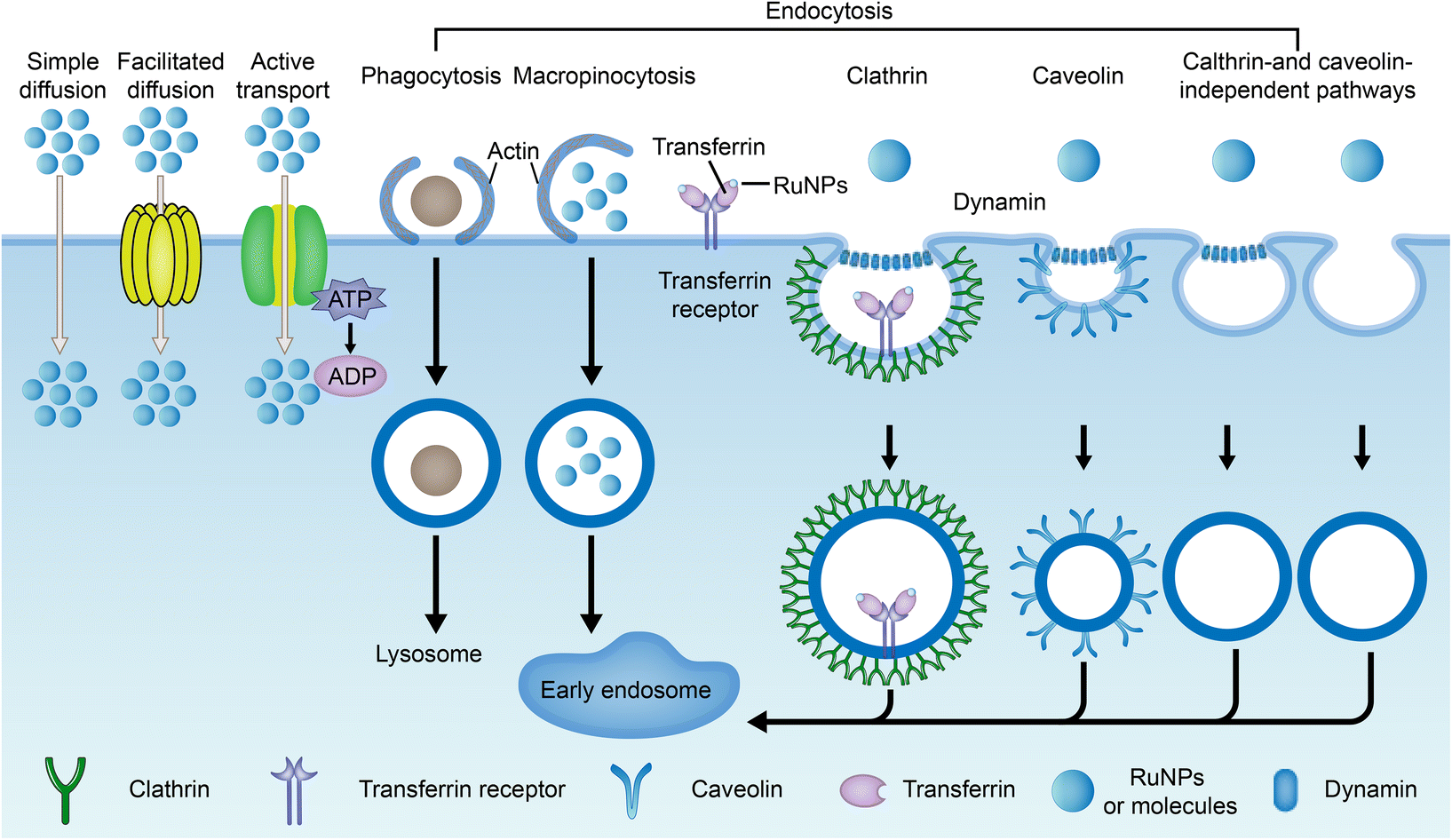 | ||
| Fig. 16 Cellular uptake of small molecule Ruthenium complexes and ruthenium nanoparticles. | ||
Small-molecule drugs (<1 kDa) can be taken up by cells in several ways, depending on molecular weight, lipophilicity, and charge carrying.192 The uptake of a ruthenium complex may depend on more than one pathway.194 Cellular uptake is determined primarily using metabolic inhibitors and transporter inhibitors. Jacqueline K. Barton et al.195 observed no reduction in the uptake of the ruthenium complex Ru(DIP)2DPPZ2+ (structure as Fig. 17A) in the case of metabolic inhibition with deoxyglucose and oligomycin, suggesting that the uptake of ruthenium complex into cells requires no energy input. The presence of organic cation transporter inhibitors also has a minor effect on absorption. However, cellular internalization of Ru(DIP)2DPPZ2+ was sensitive to membrane potential, and uptake decreased when cells were depolarized with a high-concentration potassium buffer and increased when cells were hyperpolarized with valamycin. These results suggest that Ru(DIP)2DPPZ2+ was taken up by cells by passive diffusion. Seungjo Park et al.196 synthesized three protonated ruthenium complexes (structure as Fig. 17B), and found that these ruthenium complexes entered cells through passive diffusion, but were actively expelled at the same time. When ATP production was inhibited, a significant increase in the average fluorescence intensity was noted in all cancer cell lines, suggesting that the compound may flow out of tumor cells in an energy-dependent manner under normal cell culture conditions.
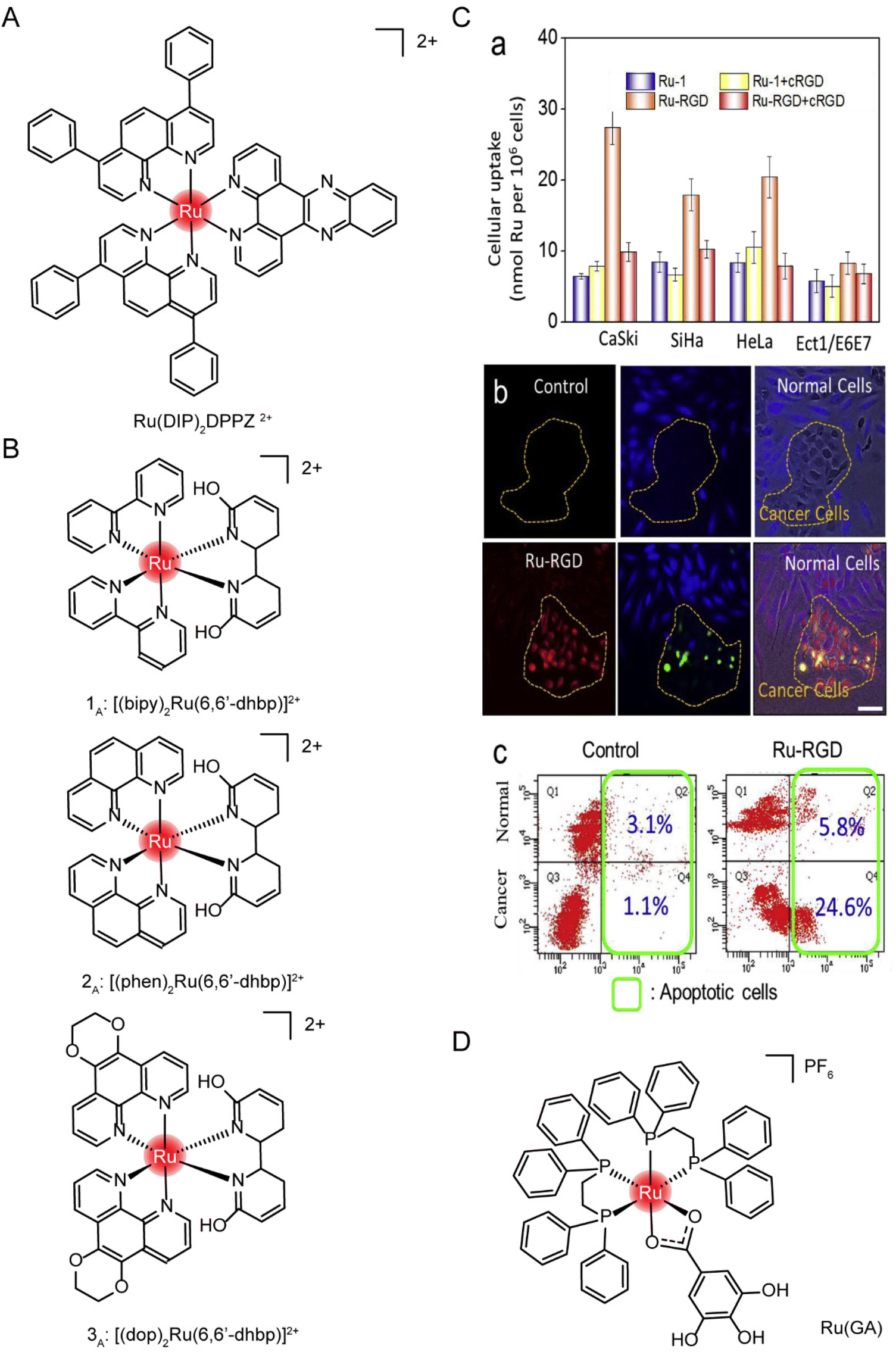 | ||
| Fig. 17 The structure of Ru complexes in absorption studies. The structure of (A) Ru(DIP)2DPPZ2+ and (B) three protonated ruthenium complexes; (C) selective accumulation of Ru-RGD in cervical cancer cells. (a) A series of cell lines was incubated with Ru complex (20 μM) for 4 h with or without cRGD (10 μg mL−1) pretreatment. (b and c) Ru-RGD (20 μM, 16 h) detected by TUNEL/CellTracker Blue staining selectively induced apoptosis in CaSki cancer cell and Ect1/E6E7 normal cell co-culture models.164 This figure has been adapted from ref. 164 with permission from Elsevier, copyright 2019; (D) the structure of [Ru(GA)(DPPE)2]PF6 (Ru(GA)). | ||
Endocytosis of nanoparticles is usually determined by endocytosis inhibitors, such as chlorpromazine (CPZ), which is often used for clathrin-mediated endocytosis. Methyl-β-cyclodextrin (M-β-CD)/filipin/mycorycin are inhibitors of caveolin-mediated endocytosis, while amiloride inhibits macropinocytosis.197,198 Many RuNPs are commonly ingested simultaneously through multiple pathways.194 Lanmei Chen and Tao Wang et al.79 designed isoquinoline Ru-bipyridine complexes (RuIQ-1 and RuIQ-2, structure as Fig. 5 compound 13 and 14). In order to further study the endocytosis pathways of RuIQ-1 and RuIQ-2, two endocytosis inhibitors were used, namely nystatin (caveolin-mediated endocytosis) and sucrose (clathrin-mediated endocytosis). The cell uptake of RuIQ-1 and RuIQ-2 was significantly decreased in the sucrose pretreatment group and only moderately decreased in the nystatin group, suggesting that the transmembrane transport of RuIQ-1 and RuIQ-2 was through clathrin-mediated endocytosis.
Many ruthenium nanoparticles enter the cell via receptor-mediated endocytosis, including cell adhesion molecule receptors (including integrin and calcemin), G-protein-coupled receptors, epidermal growth factor receptors, and other emerging molecular receptors (including progesterone receptor and glucose transporter).199 In addition, transferrin/transferrin receptor-mediated endocytosis is one of the important endocytosis modes of ruthenium complexes.20,200,201 Methods such as ICP-MS, confocal laser microscopy and flow cytometry can be used to quantify the uptake of nanoparticles by cells. These receptor-mediated endocytosis routes are mediated by tumor-specific or highly expressed receptor types, which also play a role in targeted transport.202
Tianfeng Chen et al.164 designed a RGD-functionalized and bio-responsive ruthenium prodrug (Ru-RGD, structure as Fig. 21C), in which the cyclic Arg–Gly–Asp (cRGD) peptide has high affinity and specificity for integrin αVβ3, enabling ruthenium to enter cells through integrin protein-mediated endocytosis. To verify this design, cell uptake was first measured after incubation with Ru-RGD (20 μM) for 4 h, with or without pretreatment with cRGD (10 μg mL−1). The results (Fig. 17C) showed that Ru-RGD accumulated preferentially in tumor cells compared with normal cells, and the preincubation with excessive cRGD blocked the uptake of Ru-RGD. In the CaSki tumor cells and Ect1/E6E7 normal cells co-culture model, Ru-RGD (20 μM, 16 h) was detected by TUNEL/CellTracker Blue staining to selectively induce tumor cell apoptosis without affecting normal cell growth. These results indicated that Ru-RGD can selectively kill tumor cells through integrin-mediated endocytosis.
Marcia R. Cominetti et al.201 synthesized a ruthenium complex containing galic acid (GA) as ligand [Ru(GA)(DPPE)2]PF6 (Ru(GA), structure as Fig. 17D). In order to study whether Ru(GA) was absorbed by cells through transferrin/transferrin receptor-mediated endocytosis, the spontaneous and strong interaction between transferrin or albumin with Ru(GA) was determined by the fluorescence quenching method in vitro. To further evaluate the interference of albumin and transferrin on Ru(GA) transport and cell interaction, tumor cells MDA-MB-231 and normal mammary epithelial cells MCF-10A were co-incubated with Ru(GA), and albumin and human lipid-carrying transferrin were added simultaneously. The activity of MDA-MB-231 was not significantly affected with the increase of BSA concentration. However, with the increase of lipid-carrying transferrin, cell viability was significantly decreased compared with Ru(GA)-treated cells, suggesting that Ru(GA) may be absorbed by tumor cells through transferrin-mediated endocytosis. Although albumin binds Ru(GA), ruthenium uptake was not affected. Finally, validation was performed on TfR-silenced MDA-MB-231 cells. TfR silencing in MDA-MB-231 cells reduced Ru(GA) cytotoxicity. These studies suggested that Ru(GA) entered tumor cells through transferrin/transferrin receptor endocytosis.
4.2 Distribution and targets
The distribution of ruthenium complexes in vivo can be quantified by ICP-MS or LC-MS/MS.203 Specific elements graphite furnace atomic absorption spectrometry and capillary electrophoresis have also been used for quantitative determination of ruthenium content.204 Ulrich Jaehde et al.187 determined the PK characteristics of KP1019 in human subjects in phase I clinical trials for the first time. The plasma concentration–time curve of ruthenium under multiple dosing conditions (Fig. 18A) showed that KP1019 was characterized by a small distribution volume, low clearance rate, and a long half-life (the average final half-life was about 100 hours). The area under the curve (AUC) increased with dose after a single dose (Fig. 18B), indicating that the PK profile of KP1019 is linear. In addition, KP1019 did not reach the steady-state condition after the fifth dose (Fig. 18C), so steady-state pharmacokinetics could not be measured.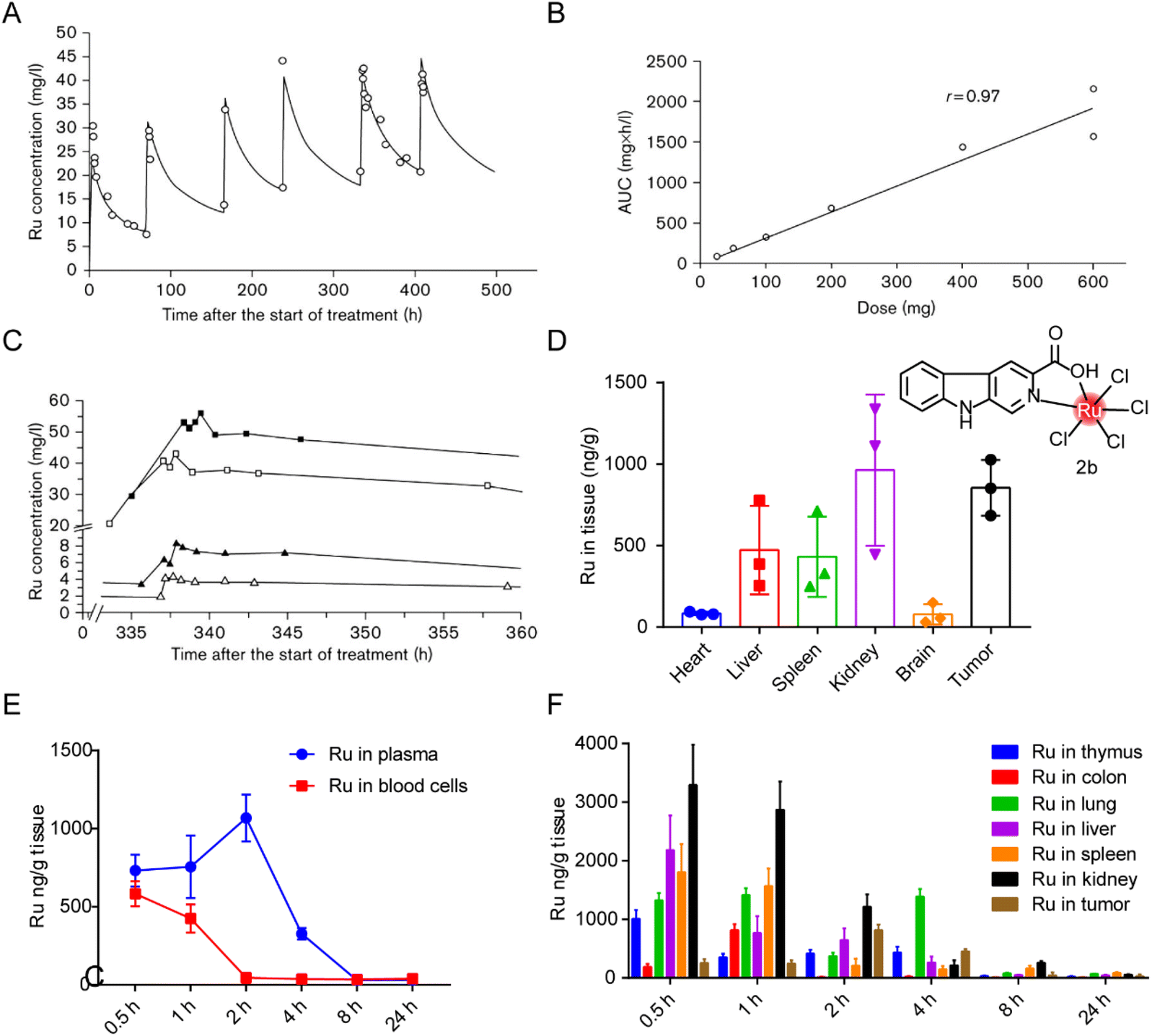 | ||
| Fig. 18 Distribution of ruthenium complex in vivo. (A) Ruthenium concentrations in plasma of patient 5 after six administrations of 400 mg KP1019.187 This figure has been adapted from ref. 187 with permission from the Royal Society of Chemistry, copyright 2009; (B) correlation between ruthenium area under the curve (AUC) and dose after the first administration of KP1019;187 (C) ruthenium plasma concentrations versus time curve after the fifth administration of 50 mg KP1019 (patient 2, △), 100 mg KP1019 (patient 3, ▲), 400 mg KP1019 (patient 5, □) and 600 mg KP1019 (patient 7, ■).187 (D) Body distribution of 2b (5.0 mg kg−1), represented by the mean ± SD (ng) of ruthenium per g of organ (n = 6).20 (E and F) Degradation of ruthenium over time in plasma, blood cells and organs after administration of a single 2b dose of 5 mg kg−1.20 This figure has been adapted from ref. 20 with permission from BioMed Central, copyright 2021. | ||
In our previous studies,20 distribution of a tumor-targeted ruthenium complex 2b (structure as Fig. 18D) in tissues was reported after continuous administration (Fig. 18D) and in blood or tissues after a single administration (Fig. 18E and F). After nine days of continuous administration in tumor-bearing mice, ruthenium levels in the kidneys and tumors were about two to three times higher than those in the liver and spleen. After single administration, ruthenium was evenly distributed in plasma and blood cells 30 min after administration, and reached the maximum plasma concentration around 2 hours after administration, while the concentration in blood cells decreased to almost zero. Ruthenium levels declined gradually over time, suggesting that the liver and kidneys were constantly metabolizing ruthenium complex. The maximum ruthenium concentration in the tumor was reached in 2 hours after administration. In 4 hours, ruthenium was detected in tumors and lungs, and most of the ruthenium in normal organs had been expelled. After 24 hours, all ruthenium was eliminated from the body. Bang-Ping Jiang et al.166 designed a hybrid ruthenium nanoaggregate (HA-Ru NAs) of hyaluronic acid (HA). These ruthenium nanoparticles had good biocompatibility and degradability, and had a biological half-life of 3 hours in blood. Biological distribution results showed high concentrations of ruthenium in liver and spleen, suggesting that HA-Ru NAs accumulated in the reticuloendothelial system. Meanwhile, HA-Ru NAs also showed progressively effective tumor enrichment, with the ruthenium content reaching 15.0% ID g−1 within 24 hours.
The targets of ruthenium complexes are closely related to the distribution of ruthenium.44 Confocal laser microscopy, inductively coupled plasma mass spectrometry (ICP-MS), flow cytometry, and transmission electron microscopy are commonly used to determine the intracellular distribution of ruthenium complexes.194 Ruthenium complexes and their nanoparticles are found in the nucleus, mitochondria, lysosome, endoplasmic reticulum, Golgi apparatus, etc. (Fig. 19), and exert anti-tumor effects through different targets.205
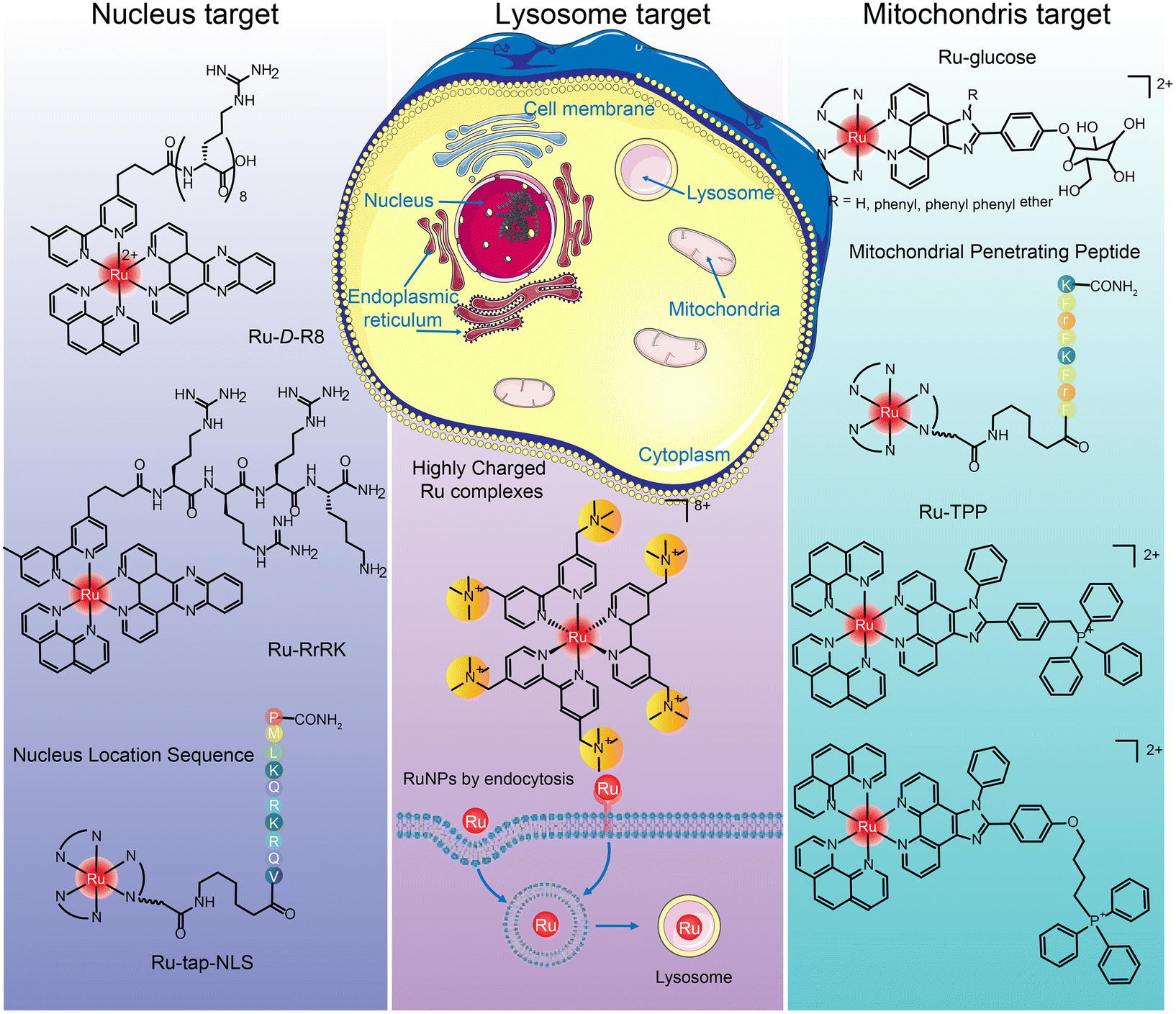 | ||
| Fig. 19 Intracellular distribution of ruthenium complexes. | ||
Many positively charged Ru complexes could be localized in mitochondria due to the negative potential of the mitochondrial inner membrane attracting lipophilic cations.216,217 Hui Chao et al.216 designed and synthesized a ruthenium–glucose complex, which can preferentially target tumor cells and mitochondria. Under near-infrared light irradiation at 810 nm, a large number of ROS can be produced to kill tumor cells. In addition, ruthenium complex can target mitochondria after binding with TPP218,219 and MPPs.220–222 Tia E. Keyes et al.222 designed binuclear Ru complexes [(Ru(bpy)2phen-Ar)2-MPP]7+·4(ClO4)− by linking [Ru(bpy)2phen-Ar-COOH]2+ with mitochondrial penetrating peptide FrFKFrFK–CONH2 through caproic acid. The co-localization study confirmed the accurate targeting in mitochondria, and the oxygen concentration in mitochondria was determined semi-quantitatively. Hui Chao et al.219 designed a series of Ru(II) polypyridinyl complexes containing 4,7-diphenyl-1,10-phenanthroline (DIP) and TPP. These Ru(II) complexes were almost non-toxic to cells and 3D cell spheres in the dark, but singlet oxygen could be produced under single and two-photon irradiation and trigger cell death.
Hui Chao and Gilles Gasser et al.229 designed a structure by adding tertiary ammonium groups in the core of Ru bipyridine to improve water solubility and increase binding affinity between complex and negatively charged cell membrane. Three highly positively charged Ru(II) polypyridine complexes were synthesized that can specifically accumulate in lysosomes by endocytosis. This is the first report of Ru(II) complexes as lysosomal-targeted two-photon PDT reagents. They have high 1O2 quantum yield, excellent photostability, remarkable two-photon properties and low dark cytotoxicity. The lysosomal-targeting cation Ru(II)-conjugated oligomer Ru-OTE designed by Yanli Tang et al.228 also introduced tertiary ammonium groups to increase the lysosomal targeting of ruthenium complexes.
Joon Myong Song et al.224 designed a self-assembled nanoparticle Ru-1@TPP–PEG–biotin that encapsulated Ru-1 with multifunctional tetraphenylporphyrin (TPP) conjugated PEG and biotin (TPP–PEG–biotin) (see structure in Fig. 20A). TPP–PEG–biotin inhibited glucose-regulatory protein 78 (GRP78) and induced ER stress, leading to lysosomal membrane instability by reducing lysosomal ceramide expression. In addition, lysosome was decomposed by the photosensitizer located in the membrane of lysosome during PDT and thus participated in the autophagy of tumor cells.
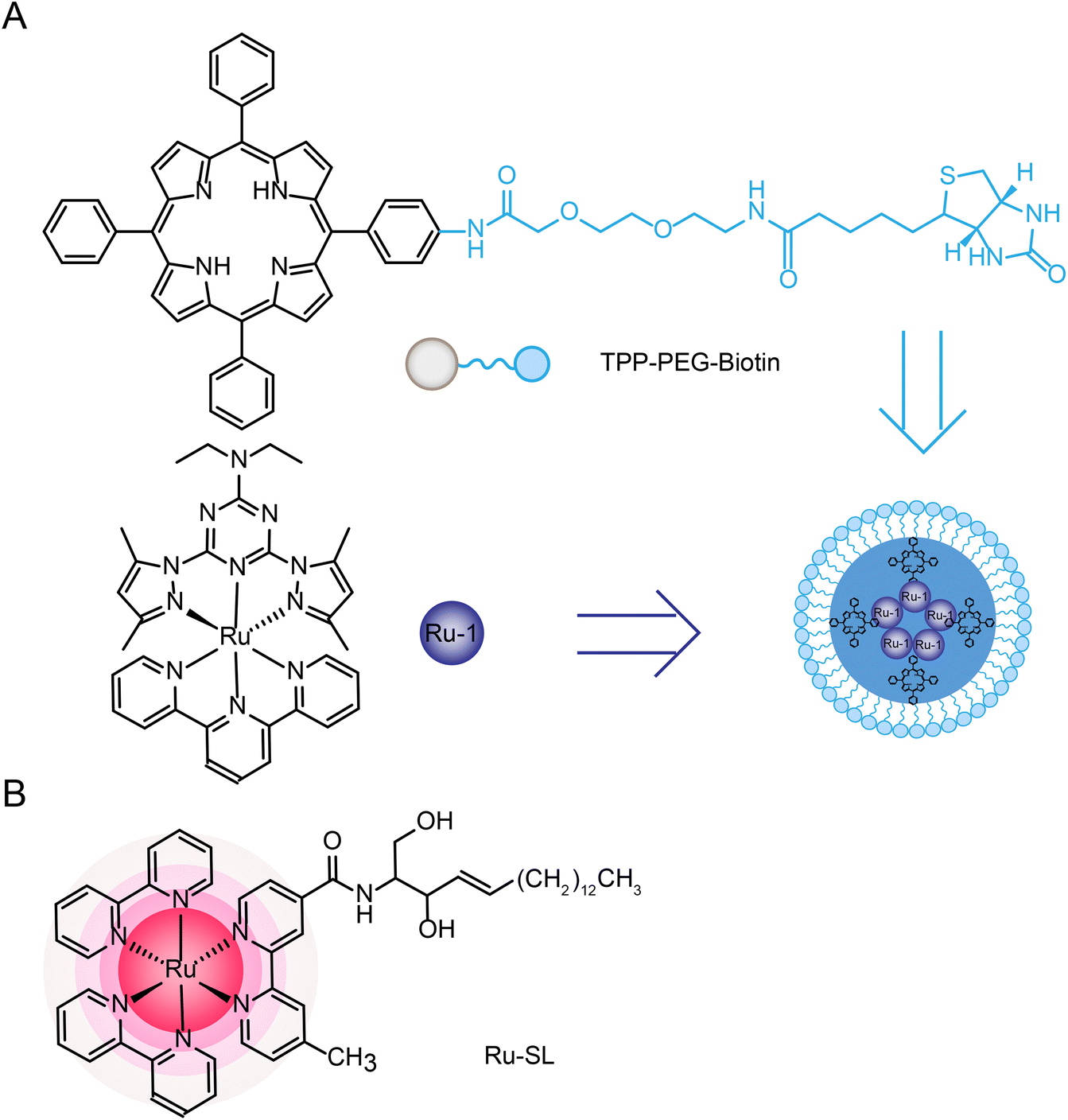
In addition, multi-targeted Ru complexes have also attracted the attention of researchers.44,238 These complexes are generally linked to active ligands, which can be released under physiological conditions and can target multiple intracellular targets, including DNA and other targets (such as enzymes, peptides, and intracellular proteins). In the process of moving from a single approach to a multi-targeted approach, multi-targeted Ru complexes showed more efficient effects than single ligands or complexes. And, the released ligands can also bring the effect of combination therapy, which provides a theoretical basis for the design and application of combination therapy ruthenium complex. Celine J. Marmion44 and Zdeněk Trávníček235 have fully discussed multi-targeted ruthenium complexes, and we will add some more recent cases in Section 5.
4.3 Metabolism
Most cisplatin remains stable due to relatively high chloride concentrations in the blood.239,240 But when it enters cells with a fairly low chloride concentration,241,242 cisplatin goes though hydrolyzation and loses chloride ions. However, the diaqua and dihydroxo hydrolysates are almost non-existent. The hydrolysates are an active substance that forms bonds with DNA bases. Because of their cis conformation, these bonds readily cross-link with bases on guanine and adenine (Fig. 21A), and these cross-links block cellular DNA replication.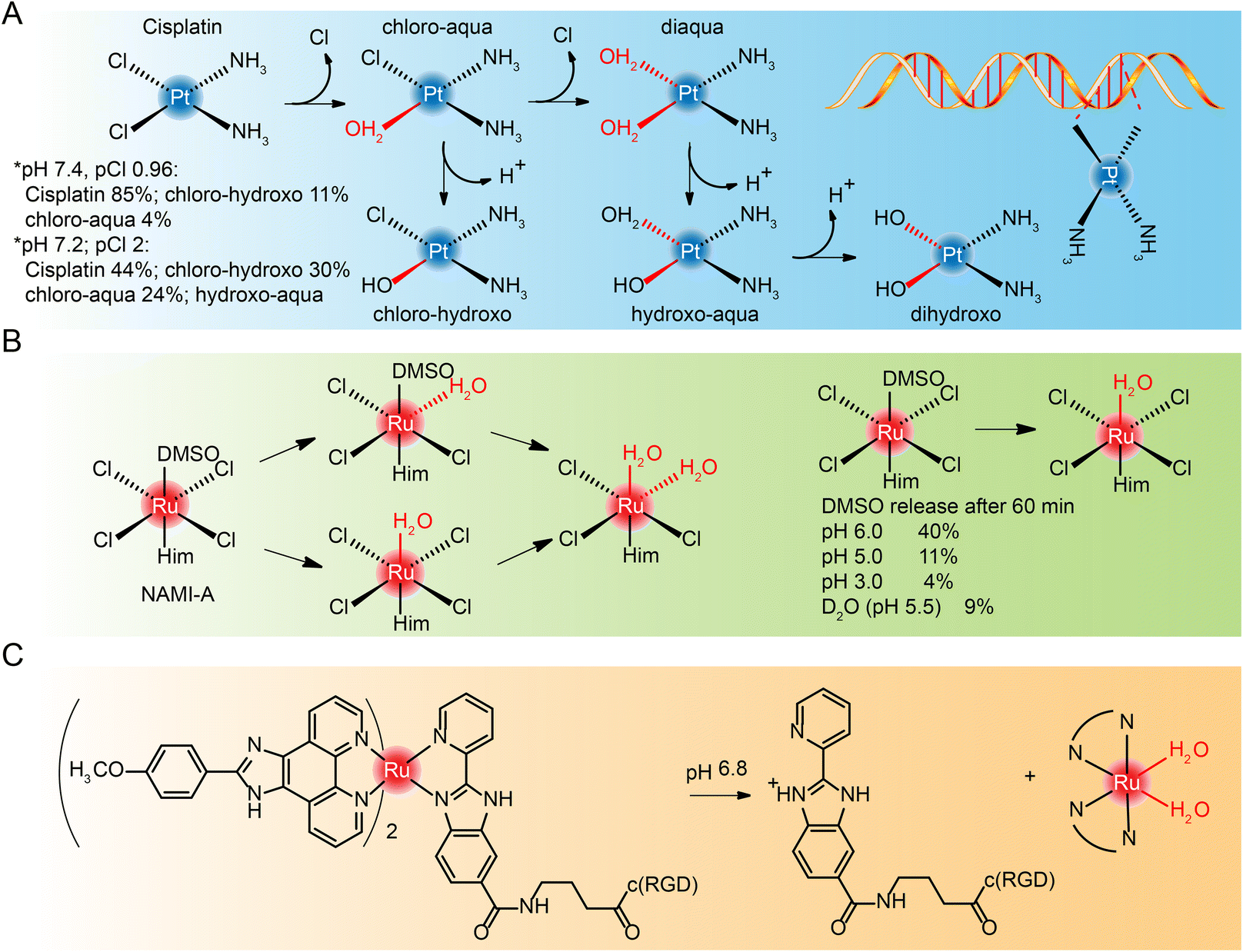 | ||
| Fig. 21 Hydrolysis process of (A) cisplatin, (B) NAMI-A and (C) Ru-RGD. | ||
Hydrolysis is a key activation step in cells before drugs reach the intracellular DNA target.243 Studies of NAMI-A244–246 and KP1019,247,248 which entered clinical studies earlier, also found that they interact with DNA, although DNA may not be the only target of their anti-metastasis activity. The hydrolysis process of NAMI-A was studied by NMR (Fig. 21B),244 and it was found that the percentage of DMSO released at different pH values decreased with the decrease of pH, and the hydrolysate could coordinate with 9-methyl adenine of DNA.
The hydrolysis of other novel ruthenium complexes was also studied.64,249 Tianfeng Chen et al.164 reported an RGD peptide-functionalized and bioresponsive ruthenium prodrug (Ru-RGD) (see structure in Fig. 21C). The benzimidazolidyl ligand of the complex is sensitive to acidic conditions, so that after the complex reaches the tumor microenvironment, protonation within the imidazole ring results in dissociation of the ruthenium complex and the release of the therapeutic drug.
4.4 Excretion
The accumulation of metal drugs in vivo is a major cause of their toxicity, so the excretion of ruthenium complexes has always been a concern.250,251 Ruthenium complexes can be excreted through the kidneys.252 Methods such as ICP-MS,152 magnetic resonance imaging/photoacoustic imaging MRI/PAI,152,253,254 fluorescence imaging255,256 and SPECT/CT257 can be used to determine the excretion of ruthenium complex from a body. Liang Cheng et al.152 designed an ultra-small metal–organic coordination polymer nanodot (Ru-Phen CPNs) based on Ru3+/Phen (Fig. 22A). PA imaging and ICP-MS were used to detect the scavenging behavior of Ru-Phen CPNs. After intravenous administration of Ru-Phen CPNs, PA signals appeared rapidly in the kidneys (Fig. 22B and C), and the kidneys were collected 2 hours after injection for in vitro PA imaging. Strong PA signals from Ru-Phen were also observed in these kidneys (Fig. 22D). This result suggested that the ultra-small nanoparticles may be excreted through the kidneys. In addition, the content of Ru in the main organs at different time points after injection was determined by ICP-MS. The highest concentration of Ru was found in the kidneys, while low levels of ruthenium were also observed in all the organs examined (Fig. 22E), indicating that most Ru-Phen CPNs were excreted from the body. The excreta of mice after intravenous administration of Ru-Phen CPNs were collected, and the content of ruthenium was analyzed by ICP-MS. Ru-Phen CPNs were found to be excreted by the kidneys for a short period of time, but biological distribution results showed relatively high concentrations of Ru in liver and spleen, suggesting that Ha-Ru NAs accumulate in the reticuloendothelial system. These accumulated Ru residues further clear out through feces. Within two weeks, nearly 86.95 ± 6.72% of Ru-Phen CPNs were eliminated from the body. Finally, hematology and H&E staining demonstrated that Ru-Phen CPNs had no significant in vivo toxicity in mice with the tested dose.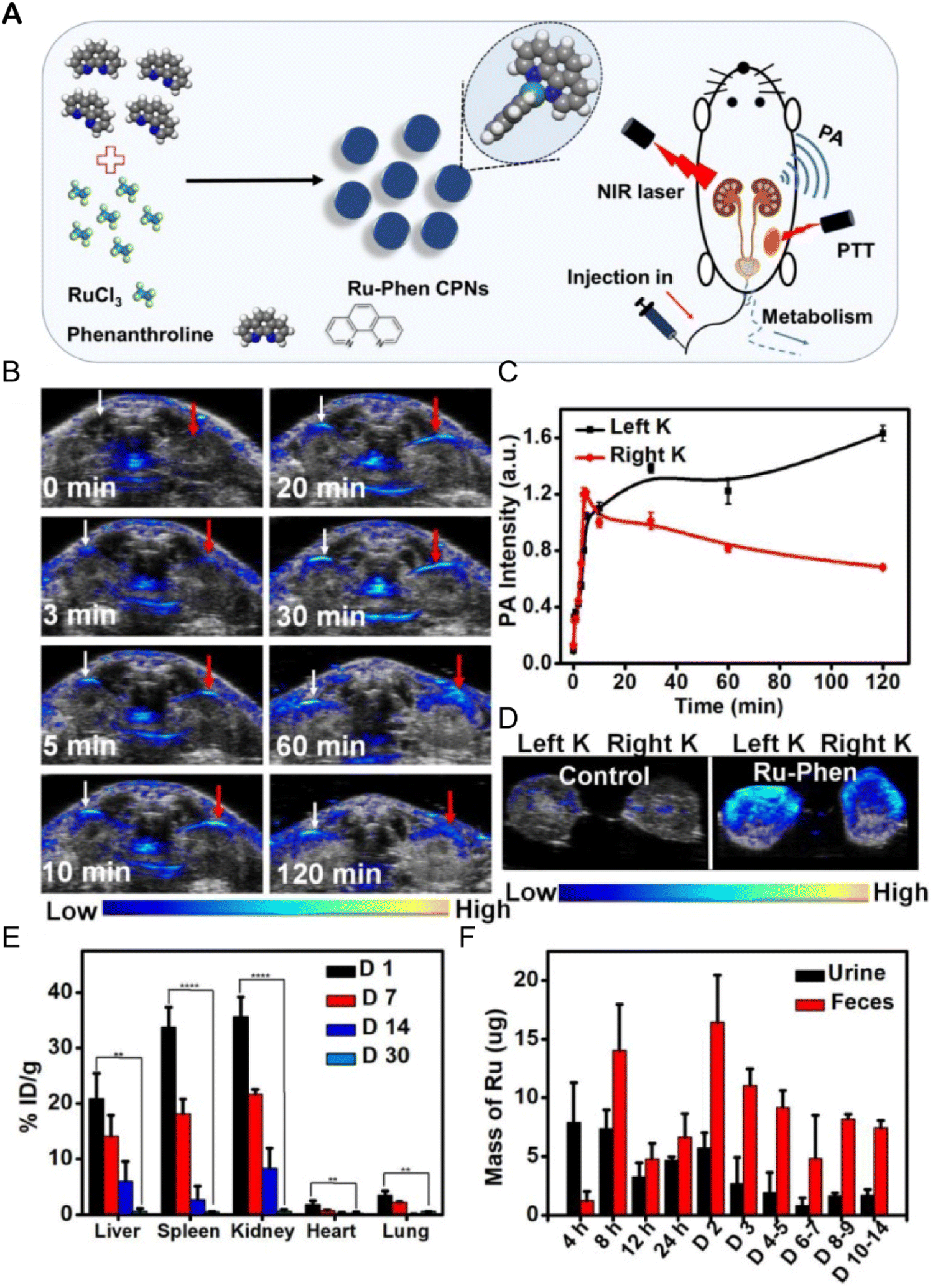 | ||
| Fig. 22 Synthesis and renal clearance of Ru-Phen CPNs. (A) Ru-Phen CPNs synthesis pathway, photothermal therapy and renal clearance behavior; (B) photoacoustic (PA) images of mouse kidneys at different time points after i.v. of Ru-Phen CPNs; (C) relative PA signal of kidney at different time points after Ru-Phen CPNs injection; (D) in vitro PA image of mouse kidney 2 hours after i.v. of Ru-Phen CPNs; (E) biological distribution of Ru-Phen CPNs in mice determined by ICP-MS at 1, 7, 14, and 30 days after i.v.; (F) Ru levels in feces and urine at different time points after i.v. of Ru-Phen CPNs (injection dose: Ru = 7 mg kg−1).152 This figure has been reproduced from ref. 152 with permission from Ivyspring International Publisher, copyright 2019. | ||
The study of the ADME process of ruthenium is an essential step for each ruthenium compound before it becomes a drug. Based on the above-reported studies on ADME, we find that the following points should be paid attention to in future studies: (1) the absorption of ruthenium is closely related to its structural design. In addition to the molecular weight, lipophilicity and carrying charge, the influence of receptor–ligand interaction should also be considered. The introduction of specific ligands into the structural design of ruthenium complex may not only improve the targeting ability, but also increase the absorption of ruthenium. However, it is worth noting that some studies have also shown that the introduction of some ligands (such as RGD) can reduce the absorption of drugs while increasing the targeting ability; (2) the distribution of ruthenium in vivo and in cells is quite different. At present, multi-target complexes have gradually become a research hotspot, but we should consider the side effects caused by multi-target complexes; (3) metabolic activation may be a necessary step for ruthenium complexes to function. However, few studies have been conducted on ruthenium complex, which may be related to the experimental cost and difficulty; (4) the ruthenium complexes require a suitable excretion rate. The half-life may be too short for the drug to be effective, or the half-life may be too long and cause toxicity. Special attention should be paid to the long half-life of metal drugs.
5. Combination therapies
A growing number of studies have shown that combination therapy is an effective way to eliminate tumors.258,259 Common treatments include chemotherapy, radiation therapy, photodynamic therapy, photothermal therapy, chemodynamic therapy, immunotherapy, gene therapy, etc., and in a combination therapy, two or more of these methods are used. Combination therapy has been widely used in clinical practice; for example, chemotherapy combined with radiotherapy is used in cancer treatment,260 and their combination with hyperthermia has been carried out for many years.261 New combination therapies are being developed to reduce their systemic toxicity and unsatisfactory therapeutic effect.In order to achieve a certain purpose of combination therapy, a variety of drugs or means are required in the process, including chemotherapy drugs,262 photosensitizers,263 lasers,264 monoclonal antibody or antibody-coupled drugs,265,266 genes,267etc. The emergence of nanodrugs provides a new idea for the realization of combination therapy and provides a platform for the combination of two or more drugs.3,268 However, these complicated nanocomponents may cause systemic toxicity, and the load capacity also limits the type and quantity of drugs, thus limiting the use of combination therapy. The emergence of ruthenium complexes, especially multi-targeted ruthenium complexes, has improved the situation.42 In addition to their property as a chemotherapeutic drug, ruthenium complexes offer various other properties, such as photokinetic properties, photothermal properties, and chemical kinetic properties (Fig. 23). Moreover, ruthenium complexes can be self-assembled into nanoparticles; thus, the usage of ruthenium complexes can reduce the composition of nanoparticles in combination therapy, offering the best formulation for combination therapy.
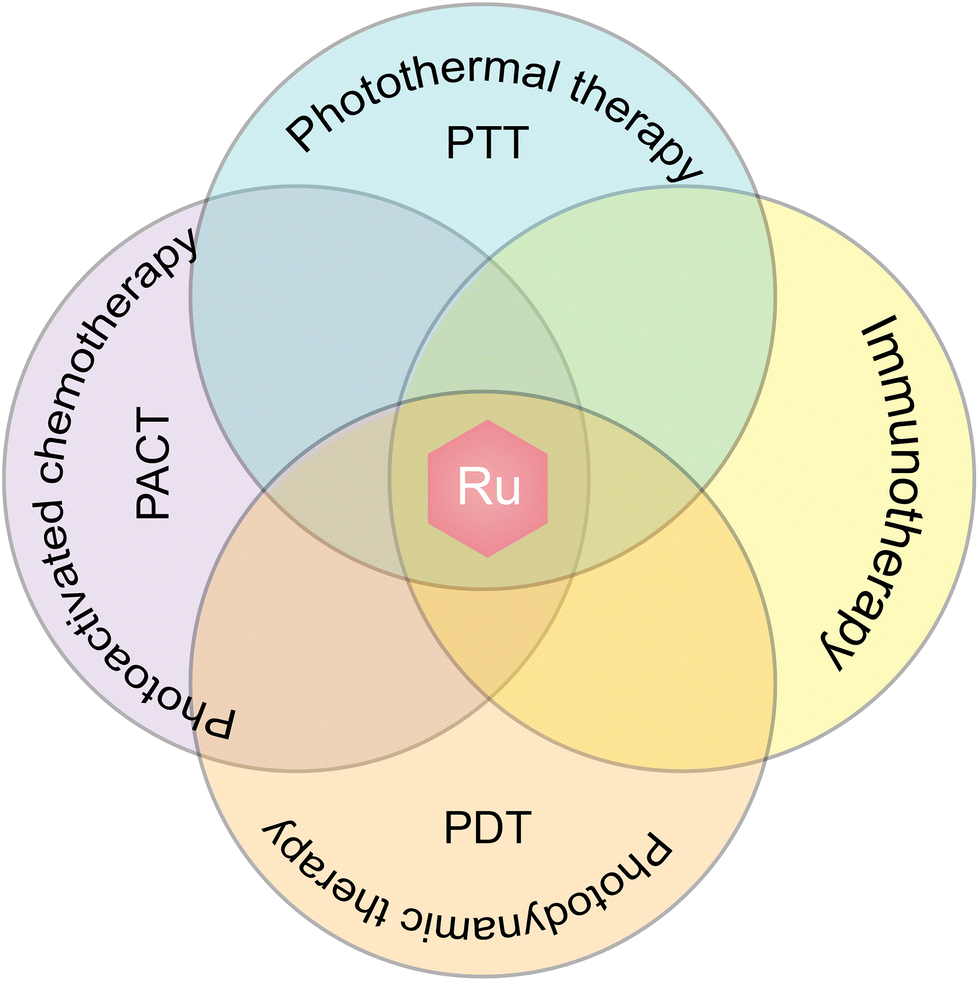 | ||
| Fig. 23 Combination therapy strategies for ruthenium complexes. | ||
5.1 Photodynamic therapy (PDT)
PDT is a treatment that kills tumor tissue by irradiating the tumor site with visible light at a specific range of wavelength (650–850 nm269) in the presence of oxygen and photosensitizers.270 The photosensitizer forms the excited triplet T1 under laser irradiation, which can further react with oxygen to form ROS. It is also possible to transfer its energy directly to molecular oxygen to form excited singlet oxygen. Both ROS and singlet oxygen species can damage tumor tissue in the region where they are produced, thus achieving a selective killing effect (Fig. 24).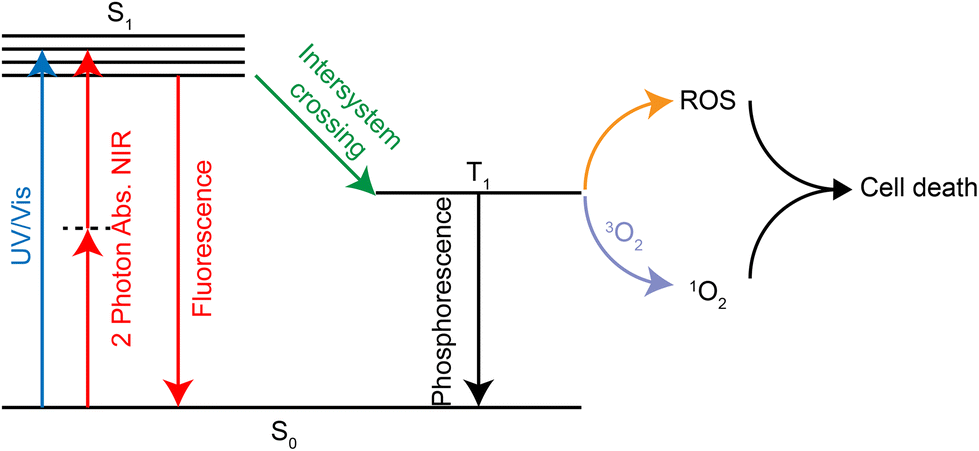 | ||
| Fig. 24 Mechanisms of photodynamic therapy with ruthenium complexes. | ||
The lowest energy transition in the UV/Vis spectrum of Ru(II) complexes is usually the metal-to-ligand charge transfer band of 1MLCT with a wavelength of about 450 nm, which shows poor photopenetration in tissues. However, the ruthenium complex can produce two-photon absorption (2PA) under confocal laser irradiation, which is located within the biooptical window (600–900 nm) of PDT. In addition, traditional organic photosensitizers, such as porphyrin derivatives, dihydrogen porphyrin, phthalocyanine and porphyrin, have poor solubility. Ruthenium complexes can significantly improve the solubility and increase cell absorption. Currently, a ruthenium polypyridine complex, TLD1433, has entered clinical trials due to good PDT effects.
Many new ruthenium complexes have been synthesized for photodynamic therapy.232,235,271–276 K. Muniyappa et al.277 synthesized four new ruthenium(II) azo-8-hydroxyquinoline complexes (Fig. 25 and Table 4 compounds 37–40), which showed enhanced cytotoxic activity under light irradiation. Moreover, ruthenium complexes can interact with DNA through groove-binding modes and show significant photonuclease activity when exposed to light. Tumor cells are killed by inducing apoptosis. Hui Chao and Gilles Gasser et al.278 synthesized a series of new Ru(II) polypyridine complexes as single-photon (595 nm) and two-photon (800 nm) photosensitizers (Fig. 25 and Table 4 compounds 41–44). These metal complexes can penetrate 3D cell structures under 595 nm and 800 nm laser irradiation and produce singlet oxygen in hypoxic centers, showing excellent phototoxicity.
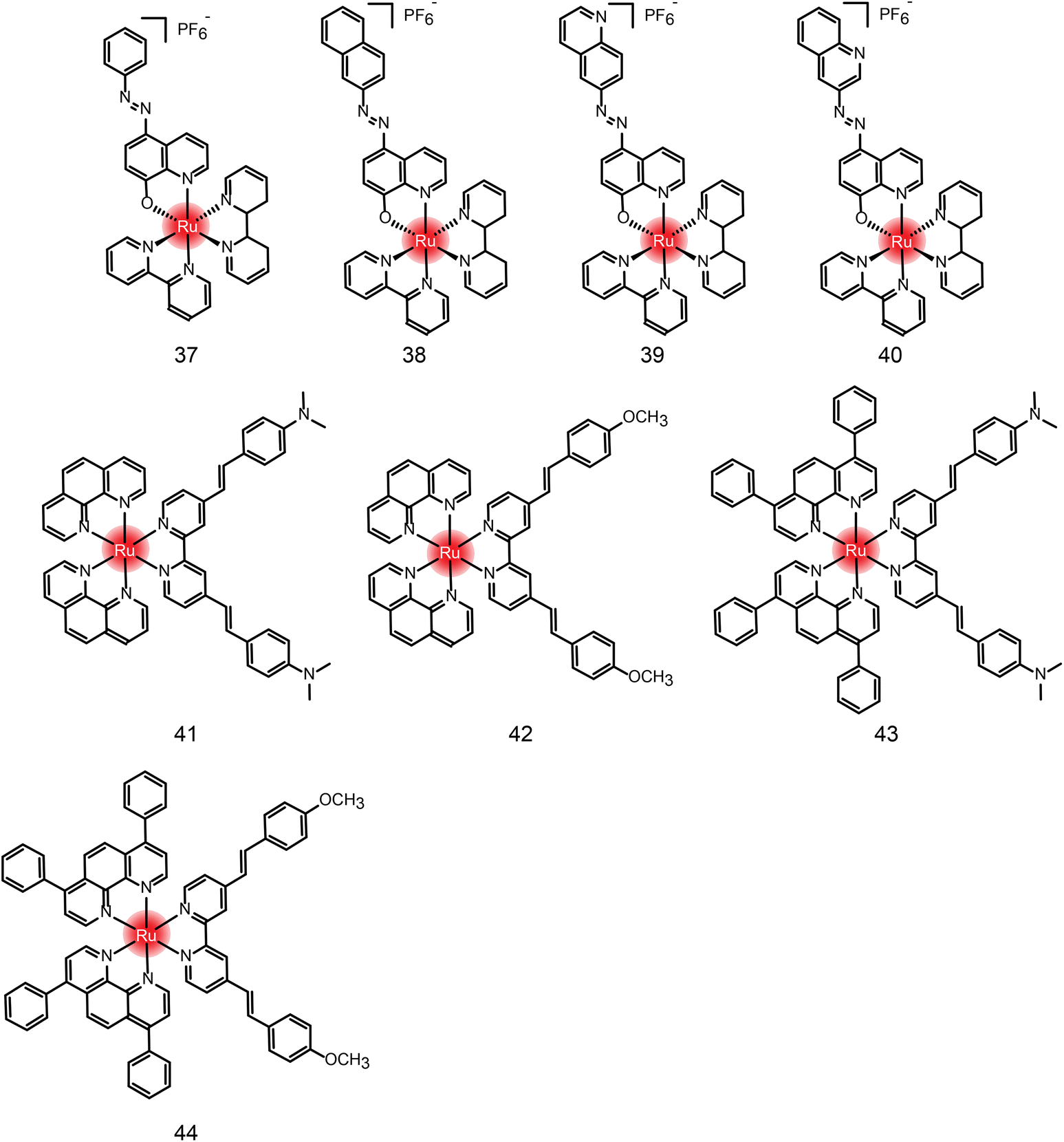 | ||
| Fig. 25 The chemical structures of compounds 37–44. | ||
| Compound | Activities | Mechanism | Ref. |
|---|---|---|---|
| 37 | MCF-7, U2OS HeLa (dark: 15.59 μM; light: 7.22 μM) | Interacting with DNA through embedding and groove-binding mode; photonuclease activity; inducing apoptosis | 277 |
| 38 | MCF-7, U2OS HeLa (dark: 22.25 μM; light: 11.88 μM) | ||
| 39 | MCF-7, U2OS HeLa (dark: 20.83 μM; light: 8.91 μM) | ||
| 40 | N.D. | ||
| 41 | HeLa 3D cells (dark: >300 μM; light 595 nm: 32.6 μM; light 800 nm: 27.8 nM) | N.D. | 278 |
| 42 | HeLa 3D cells (dark: >300 μM; light 595 nm: 7.5 μM; light 800 nm: 1.2 nM) | ||
| 43 | HeLa 3D cells (dark: 27.8 μM; light 595 nm: 8.9 μM; light 800 nm: 3.1 nM) | ||
| 44 | HeLa 3D cells (dark: 29.8 μM; light 595 nm: 3.8 μM; light 800 nm: 7.7 nM) |
5.2 Photothermal therapy (PTT)
PTT refers to a treatment method that converts light energy into heat energy by using a photothermal conversion agent (PTA) to raise the temperature of the surrounding environment and cause tumor cell death.279 PTA is critical in photothermal therapy, and the light absorption profile typically occurs between 750 and 1350 nm, including the first (750–1000 nm, NIR-I) and second (1000–1350 nm, NIR-II) near-infrared windows (Fig. 26). Noble metals are one of the most studied PTA materials, including Au, Ag, Pt and Pd.280,281 Ruthenium nanomaterials have emerged as a new PTA recently.282 | ||
| Fig. 26 Mechanism of photothermal therapy by ruthenium complexes. | ||
Jie Liu et al.17 synthesized ruthenium nanoparticles with simple spherical shape, which had an excellent photothermal effect. Transferrin-modified ruthenium nanoparticles (Tf-RuNPs) enhanced the absorption capacity of Ru nanoparticles absorbed by cells through endocytosis. They also showed low toxicity and high cell destruction ability in in vitro and in vivo ablative tumor therapy. In addition, Hui Chao et al.283 synthesized ultra-small chitosan-coating nanoparticles CS–RuO2NPs based on Ru(IV)O2. These showed strong absorption in NIR-II window and had high photothermal conversion efficiency. These nanoparticles can be localized in the nucleus. Under 1064 nm laser irradiation, CS–RuO2NPs could kill tumor cells at low temperature (41.9 °C), and showed significant DNA damage. CS–RuO2NPs also showed excellent tumor growth inhibition in vivo without damage to major organs.
5.3 Photoactivated chemotherapy (PACT)
PACT refers to the use of a certain wavelength of light to induce the dissociation or decomposition of photoactivators, and the resulting products have anti-tumor activity.284,285 In contrast to photodynamic therapy, this treatment does not require oxygen.286 Many Ru(II) complexes can undergo photoinduced ligand dissociation, demonstrating their potential as photoactivators.287–291 The mechanism of action292 (Fig. 27) is that the ruthenium complexes reach 1MLCT state and then 3MLCT state from the ground state under light irradiation, and the 3MLCT excited state can enter 3MC state (state of mind or ligand field in metal) by thermal activation. 3MC state can lead to ligand dissociation and produce Ru(II) hydrolysate which can bind DNA, exerting a cytotoxic effect. Currently, two-photon absorption is also used for photoactivation of ruthenium complexes.292,293 As for the application of Ru complexes in photoactivated chemotherapy, Qianxiong Zhou et al.292 discussed the development of Ru(II)-based photoactivated chemotherapy drugs, which is not discussed in detail here.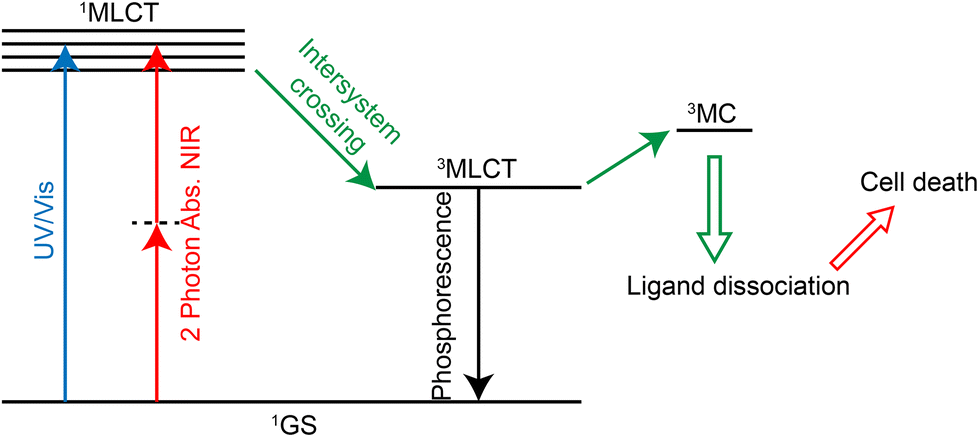 | ||
| Fig. 27 Mechanism of photoactivated chemotherapy. | ||
5.4 Immunotherapy
Immunotherapy for cancer refers to the treatment of cancer by activating the body's immune system with drugs to attack tumor cells through the body's natural immunity.294,295 Current approaches to tumor immunotherapy can be divided into checkpoint inhibitors, lymphocyte-promoting cytokines, engineered T cells (such as CAR T and T-cell receptor T cells), excitatory antibody receptors for costimulation, cancer vaccines, oncolytic viruses, and bispecific antibodies.296,297 Nanoparticles are often used to specifically deliver antigens, adjuvants, and therapeutic agents.298,299 Nanoparticles themselves can also regulate the immune microenvironment of tumors.300–303Many inorganic nanoparticles, such as gold nanoparticles304,305 and iron oxide nanoparticles,302 have been used in immunotherapy. With the development of ruthenium antitumor drugs, some ruthenium molecules and nanoparticles are considered new candidates for immunotherapy. Currently, the application of Ru in immunotherapy is still rare. Here, we summarize the modes of action and the potential action modes of Ru in immunotherapy (Fig. 28).
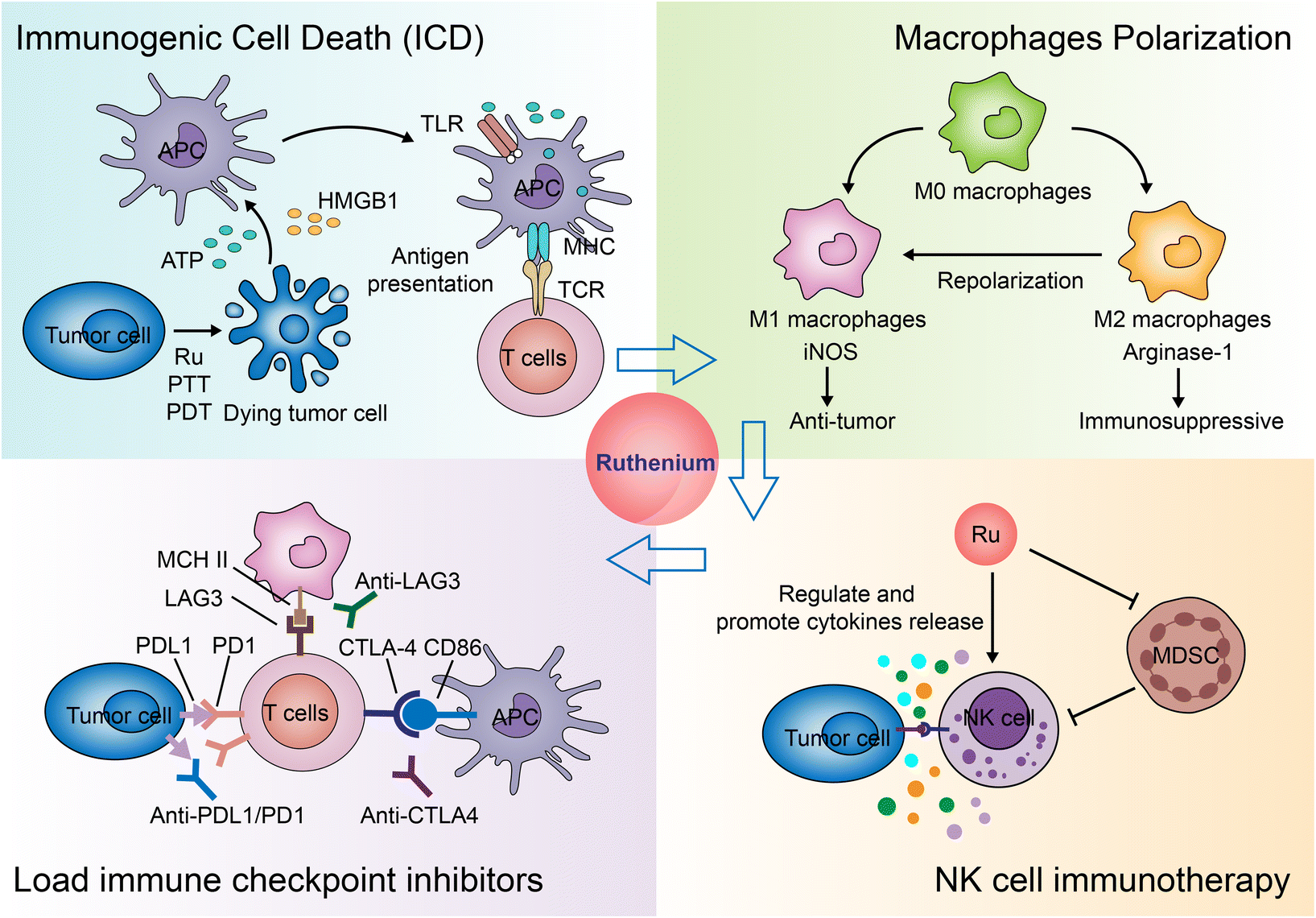 | ||
| Fig. 28 The role of ruthenium in immunotherapy. | ||
Another study screened a mononuclear ruthenium complex312 which selectively killed immunosuppressive M2 macrophages, sensitized macrophage/cancer cell 3D co-cultured spheres to chemotherapeutic drugs, and increased cell surface exposure to calreticulin.
5.5 The combination of PDT and PTT
The combination of PDT and PTT is a common strategy for Ru-mediated combination therapy. PDT&PTT is more effective in killing tumor cells than either PTT or PDT alone.107 Currently, Ru complexes are often used as photosensitizer in ruthenium nanodrugs, and the other components, such as CuS NP160 or graphene,316 are considered as photothermal conversion agents. It has also been reported17,317 that synthesized self-assembled RuNPs can be used as both photosensitizer and photothermal conversion agent.Tianming Yao and Shuo Shi et al.160 designed a multifunctional nanoparticle UiO-Ra-DOX-CuS based on Ru(II) polypyridyl alkyne complex Ra. Ra acts as a photosensitizer and forms a metal–organic framework (MOF) through click reaction. Doxorubicin (DOX) was incorporated into the porous MOFs during synthesis as a chemotherapy agent, and small CuS NPs as a photothermal conversion agent was loaded on the surface of nanoparticles by physical adsorption. This study is a typical example of Ru-mediated PDT, PTT and CT co-therapy.
Zong-Wan Mao et al.316 constructed rGO–Ru–PEG nanoparticles by using reduced graphene oxide (rGO) sheets and phosphorescent polyethylene glycol-modified Ru(II) complex (Ru–PEG) for combined PTT and PDT therapy. In this complex, Ru–PEG acts as photosensitizer and imaging agent and rGO acts as PTT agent. PTT and PDT under 808 nm and 450 nm laser irradiation, respectively, showed excellent antitumor therapeutic effects both in vivo and in vitro.
5.6 The combination of PACT and PDT
The combination of PACT and PDT is also a common strategy for using Ru complexes as antitumor agents. Dan Gibson and Gilles Gasser et al.235 designed a novel Pt(IV)–Ru(II) conjugation for PACT&PDT combined therapy. Upon entry into the cancer cell, the Pt(IV) center is reduced to Pt(II). Ru(II) polypyridine complex and phenylbutyric acid are released. In this complex, Pt(II) acts as a chemotherapeutic agent and Ru(II) complex acts as a photosensitizer of PDT. The synergistic effect of the combination therapy showed excellent cytotoxic activity in 2D and 3D cells.5.7 The combination of PTT, PDT and PACT
Bang-Ping Jiang et al.166 designed a hybrid ruthenium nanoaggregate of hyaluronic acid (HA-Ru NAs) to enhance cancer phototherapy through receptor-mediated targeting (RMT) and tumor microenvironment response (TMR). In this nanosystem, the HA component imparted RMT properties to the HA-Ru NAs, enabling them to selectively recognize CD44 overexpressed cancer cells, while the Ru nanocomponent imparted TMR therapeutic activity to the HA-Ru NAs. In particular, Ru nanocomponents exhibit near-infrared mediated photothermal and photodynamic functions, which can catalyze H2O2 to produce O2 in tumor tissues for alleviating tumor hypoxia and toxicity of ˙OH for chemodynamic therapy. Therefore, HA-Ru NAs can be used for PTT/PDT/PACT combined therapy, which effectively improves the specificity and efficacy of phototherapy, and also simplifies the therapeutic procedure.6. Conclusions
Ruthenium complexes such as NAMI-A, KP1019 and KP1339 have entered clinical studies as a replacement for platinum chemotherapy drugs. Recently, TLD1433 was also proved to be effective in phase II clinical trials in photodynamic therapy. All these reports greatly promote the research in the field. Many novel ruthenium complexes have been designed with various ligands, including bipyridine and aromatic ruthenium complexes, ligands from natural products and derivatives, such as β-carboline, quinoline/isoquinoline, curcumin, amino acids, flavones, sugars, etc. Many of them offer good antitumor activity both in vitro and in vivo. The structure of ligands affects the activity of complexes by lipophilicity, steric hindrance and charge carrying.Together with new therapeutic methods and technologies evolved in recent years, research activity in ruthenium antitumor drugs is not limited to synthesizing new ruthenium complexes, but includes combining ruthenium complexes with new therapeutic technologies. New trends in ruthenium research are emerging in two directions. One is the development of ruthenium complexes in combination with drug-delivery methods from single-molecule drugs to nanocomplexation, and further to self-assembled excipient-free nanomedicine. The other is the potential antitumor effect of ruthenium complexes in combined therapies, such as PDT, PTT, PACT and immunotherapy.
The ADME studies help us understand the mechanism of action and overcome the side effects of ruthenium complexes. Small ruthenium complexes are taken up by simple diffusion, assisted diffusion, active transport, or endocytosis. Due to their size, ruthenium nanoparticles are absorbed through endocytosis, especially receptor-mediated endocytosis, which also increases tumor targeting. At present, ruthenium complexes and their nanomaterials have been found to be distributed in the nucleus, mitochondria, lysosome, and endoplasmic reticulum, and exert antitumor effects through DNA binding, ROS generation and lysosomal degradation. The active hydrolysis of ruthenium in vivo has been generally accepted, and the targeted hydrolysis of chlorine ion or bipyridine is an effective method for designing ruthenium complexes for targeted delivery to reduce side effects.
With the rapid development of the field, a prospective future for ruthenium complexes as antitumor drugs can be anticipated. Excipient-free nanodrugs is also a field not only for ruthenium complexes. It is an effective method to reduce the use of excipients, especially in parenteral delivery when the choice of excipients is limited. In addition, ruthenium nanoparticles show photothermal, photodynamic, and chemodynamic activity. The use of these unique properties of Ru-nanodrugs will drastically simplify the design of drug-delivery vectors for combined therapy.
Author contributions
Conceptualization, Yu Lu and Di Zhu; resources, Wei Wang and Yuji Wang; data curation, Yu Lu and Wei Wang; writing—original draft preparation, Yu Lu, Wei Wang; writing—review and editing, Yu Lu, Quynh Le, Wei Wang; visualization, Yu Lu; supervision, Wei Wang and Yuji Wang; project administration, Yuji Wang; funding acquisition, Yuji Wang. All of the authors have read and approved the final manuscript.Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
This work was supported by the Beijing Municipal Colleges and Universities High Level Talents Introduction and Cultivate Project – Beijing Great Wall Scholar Program (CIT&TCD 20180332, China). Yu Lu thanks the China Scholarship Council (CSC) for financial support (File No. CSC No. 202008110286).References
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, CA-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- J. Ferlay, M. Colombet, I. Soerjomataram, D. M. Parkin, M. Piñeros, A. Znaor and F. Bray, Int. J. Cancer, 2021, 149, 778–789 CrossRef CAS PubMed.
- L. Huang, S. Zhao, F. Fang, T. Xu, M. Lan and J. Zhang, Biomaterials, 2021, 268, 120557 CrossRef CAS PubMed.
- A. G. Waks and E. P. Winer, J. Am. Med. Assoc., 2019, 3, 288–300 CrossRef PubMed.
- C. Moncharmont, P. Auberdiac, A. Mélis, S. Afqir, C. Pacaut, C. Chargari, Y. Merrouche and N. Magné, Bull. Cancer, 2011, 98, 164–175 CrossRef PubMed.
- Y. Lai, J. Lu, H. Hu and Y. Wang, Ther. Innov. Regul. Sci., 2015, 49, 181–193 CrossRef PubMed.
- Y. Zheng, K. Suntharalingam, T. C. Johnstone, H. Yoo, W. Lin, J. G. Brooks and S. J. Lippard, J. Am. Chem. Soc., 2014, 136, 8790–8798 CrossRef CAS PubMed.
- L. Qi, Q. Luo, Y. Zhang, F. Jia, Y. Zhao and F. Wang, Chem. Res. Toxicol., 2019, 32, 1469–1486 Search PubMed.
- S. Ghosh, Bioorg. Chem., 2019, 88, 102925 CrossRef CAS PubMed.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS PubMed.
- M. A. Pujante-Galián, S. A. Pérez, M. G. Montalbán, G. Carissimi, M. G. Fuster, G. Víllora and G. García, Molecules, 2020, 25, 5063 CrossRef PubMed.
- X. Zhang, D. Liu, F. Lv, B. Yu, Y. Shen and H. Cong, Colloids Surf., B, 2019, 182, 110373 CrossRef CAS PubMed.
- K. Lin, Z. Zhao, H. Bo, X. Hao and J. Wang, Front. Pharmacol., 2018, 9, 1323 CrossRef CAS PubMed.
- P. V. Simpson, N. M. Desai, I. Casari, M. Massi and M. Falasca, Future Med. Chem., 2019, 11, 119–135 CrossRef CAS PubMed.
- S. Y. Lee, C. Y. Kim and T. Nam, Drug Des., Dev. Ther., 2020, 14, 5375–5392 CrossRef CAS PubMed.
- A. Rilak Simović, R. Masnikosa, I. Bratsos and E. Alessio, Coord. Chem. Rev., 2019, 398, 113011 CrossRef.
- S. Zhao, X. Zhu, C. Cao, J. Sun and J. Liu, J. Colloid Interface Sci., 2018, 511, 325–334 CrossRef CAS PubMed.
- A. E. Graminha, J. Honorato, R. S. Correa, M. R. Cominetti, A. C. S. Menezesd and A. A. Batista, Dalton Trans., 2021, 323–335 RSC.
- M. Nišavić, M. Stoiljković, I. Crnolatac, M. Milošević, A. Rilak and R. Masnikosa, Arabian J. Chem., 2018, 11, 291–304 CrossRef.
- Y. Lu, D. Zhu, L. Gui, Y. Li, W. Wang, J. Liu and Y. Wang, J. Nanobiotechnol., 2021, 19, 115 CrossRef CAS PubMed.
- A. Bicek, I. Turel, M. Kanduser and D. Miklavcic, Bioelectrochemistry, 2007, 71, 113–117 CrossRef CAS PubMed.
- X. Zeng, Y. Wang, J. Han, W. Sun, H. J. Butt, X. J. Liang and S. Wu, Adv. Mater., 2020, 32, 2004766 CrossRef CAS PubMed.
- R. G. Teixeira, D. C. Belisario, X. Fontrodona, I. Romero, A. I. Tomaz, M. H. Garcia, C. Riganti and A. Valente, Inorg. Chem. Front., 2021, 1983–1996 RSC.
- S. Monro, K. L. Colón, H. Yin, J. Roque, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2018, 119, 797–828 CrossRef PubMed.
- X. Li, A. K. Gorle, M. K. Sundaraneedi, F. R. Keene and J. G. Collins, Coord. Chem. Rev., 2018, 375, 134–147 CrossRef CAS.
- J. M. Rademaker-Lakhai, D. van den Bongard, D. Pluim, J. H. Beijnen and J. H. Schellens, Clin. Cancer Res., 2004, 10, 3717–3727 CrossRef CAS PubMed.
- C. G. Hartinger, M. A. Jakupec, S. Zorbas-Seifried, M. Groessl, A. Egger, W. Berger, H. Zorbas, P. J. Dyson and B. K. Keppler, Chem. Biodivers., 2008, 5, 2140–2155 CrossRef CAS PubMed.
- R. Trondl, P. Heffeter, C. R. Kowol, M. A. Jakupec, W. Berger and B. K. Keppler, Chem. Sci., 2014, 5, 2925–2932 RSC.
- E. Alessio, Eur. J. Inorg. Chem., 2017, 1549–1560 CrossRef CAS.
- E. Ortega, G. Vigueras, F. J. Ballester and J. Ruiz, Coord. Chem. Rev., 2021, 446, 214129 CrossRef CAS.
- J. Liu, H. Lai, Z. Xiong, B. Chen and T. Chen, Chem. Commun., 2019, 55, 994–9914 Search PubMed.
- M. Mirrahimi, J. Beik, M. Mirrahimi, Z. Alamzadeh, S. Teymouri, V. P. Mahabadi, N. Eslahi, F. Ebrahimi Tazehmahalleh, H. Ghaznavi, A. Shakeri-Zadeh and C. Moustakis, Int. J. Biol. Macromol., 2020, 158, 617–626 CrossRef CAS PubMed.
- R. B. Mokhtari and T. S. H. Narges, Oncotarget, 2017, 23, 38022–38043 CrossRef PubMed.
- K. S. Saini and C. Twelves, Br. J. Cancer, 2021, 125, 155–163 CrossRef PubMed.
- L. M. Colli, M. J. Machiela, H. Zhang, T. A. Myers, L. Jessop, O. Delattre, K. Yu and S. J. Chanock, Cancer Res., 2017, 77, 3666–3671 CrossRef CAS PubMed.
- Q. Zhang and L. Li, J BUON, 2018, 3, 561–567 Search PubMed.
- F. Meric-Bernstam, J. Larkin, J. Tabernero and C. Bonini, Lancet, 2021, 397, 1010–1022 CrossRef CAS.
- S. Gurunathan, M. Kang, M. Qasim and J. Kim, Int. J. Mol. Sci., 2018, 19, 3264 CrossRef PubMed.
- M. Zhao, D. van Straten, M. L. D. Broekman, V. Préat and R. M. Schiffelers, Theranostics, 2020, 10, 1355–1372 CrossRef CAS PubMed.
- D. Ghosh, S. Nandi and S. Bhattacharjee, Clin. Transl. Med., 2018, 7, 33 Search PubMed.
- L. Zeng, P. Gupta, Y. Chen, E. Wang, L. Ji, H. Chao and Z. Chen, Chem. Soc. Rev., 2017, 46(19), 5771–5804 RSC.
- J. Shen, T. W. Rees, L. Ji and H. Chao, Coord. Chem. Rev., 2021, 443, 214016 CrossRef CAS.
- F. E. Poynton, S. A. Bright, S. Blasco, D. C. Williams, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2017, 46, 7706–7756 RSC.
- R. G. Kenny and C. J. Marmion, Chem. Rev., 2019, 119, 1058–1137 CrossRef CAS PubMed.
- G. Sava, S. Zorzet, C. Turrin, F. Vita, M. Soranzo, G. Zabucchi, M. Cocchietto, A. Bergamo, S. DiGiovine, G. Pezzoni, L. Sartor and S. Garbisa, Clin. Cancer Res., 2003, 1898–1905 CAS.
- A. Vacca, M. Bruno, A. Boccarelli, M. Coluccia, D. Ribatti, A. Bergamo, S. Garbisa, L. Sartor and G. Sava, Br. J. Cancer, 2002, 86, 993–998 CrossRef CAS PubMed.
- C. Pelillo, H. Mollica, J. A. Eble, J. Grosche, L. Herzog, B. Codan, G. Sava and A. Bergamo, J. Inorg. Biochem., 2016, 160, 225–235 CrossRef CAS PubMed.
- H. A. Burris, S. Bakewell, J. C. Bendell, J. Infante, S. F. Jones, D. R. Spigel, G. J. Weiss, R. K. Ramanathan, A. Ogden and D. Von Hoff, ESMO Open, 2016, 1, e154 CrossRef PubMed.
- G. Golbaghi and A. Castonguay, Molecules, 2020, 25, 265 CrossRef CAS PubMed.
- Q. Sun, Y. Li, H. Shi, Y. Wang, J. Zhang and Q. Zhang, Molecules, 2021, 26, 4389 CrossRef CAS PubMed.
- M. Savic, A. Arsenijevic, J. Milovanovic, B. Stojanovic, V. Stankovic, A. Rilak Simovic, D. Lazic, N. Arsenijevic and M. Milovanovic, Molecules, 2020, 25, 4699 CrossRef CAS PubMed.
- J. Shum, P. K. Leung and K. K. Lo, Inorg. Chem., 2019, 58, 2231–2247 CrossRef CAS PubMed.
- M. Allison, P. Caramés Méndez, C. M. Pask, R. M. Phillips, R. M. Lord and P. C. McGowan, Chem. – Eur. J., 2021, 27, 3737–3744 CrossRef CAS PubMed.
- S. Bu, G. Jiang, G. Jiang, X. Duan, J. Wang and X. Liao, J. Biol. Inorg. Chem., 2020, 747–757 CrossRef CAS PubMed.
- G. Jiang, W. Zhang, M. He, Y. Gu, L. Bai, Y. Wang, Q. Yi and F. Du, Spectrochim. Acta, Part A, 2020, 227, 117534 CrossRef CAS PubMed.
- S. Arlt, V. Petković, G. Ludwig, T. Eichhorn, H. Lang, T. Rüffer, S. Mijatović, D. Maksimović-Ivanić and G. N. Kaluđerović, Molecules, 2021, 26, 1860 CrossRef CAS PubMed.
- C. Sonkar, N. Malviya, N. Sinha, A. Mukherjee, S. Pakhira and S. Mukhopadhyay, BioMetals, 2021, 34, 795–812 CrossRef CAS PubMed.
- L. Biancalana, G. Pampaloni and F. Marchetti, Chimia, 2017, 71, 573 CrossRef CAS PubMed.
- L. A. Hager, S. Mokesch, C. Kieler, S. Alonso-de Castro, D. Baier, A. Roller, W. Kandioller, B. K. Keppler, W. Berger, L. Salassa and A. Terenzi, Dalton Trans., 2019, 48, 12040–12049 RSC.
- M. Santi, A. K. Mapanao, L. Biancalana, F. Marchetti and V. Voliani, Eur. J. Med. Chem., 2021, 212, 113143 CrossRef CAS PubMed.
- F. Rinaldi-Neto, A. B. Ribeiro, N. H. Ferreira, I. S. Squarisi, K. M. Oliveira, R. P. Orenha, R. L. T. Parreira, A. A. Batista and D. C. Tavares, J. Inorg. Biochem., 2021, 222, 111497 CrossRef CAS PubMed.
- B. Chen, J. Lu, N. Jiang, X. Ma, R. Li and R. Ye, JBIC, J. Biol. Inorg. Chem., 2021, 26, 909–918 CrossRef CAS PubMed.
- S. Li, G. Xu, Y. Zhu, J. Zhao and S. Gou, Dalton Trans., 2020, 49, 9454–9463 RSC.
- F. Caruso, M. Rossi, A. Benson, C. Opazo, D. Freedman, E. Monti, M. B. Gariboldi, J. Shaulky, F. Marchetti, R. Pettinari and C. Pettinari, J. Med. Chem., 2012, 55, 1072–1081 CrossRef CAS PubMed.
- W. Su, X. Wang, X. Lei, Q. Xiao, S. Huang and P. Li, J. Organomet. Chem., 2017, 833, 54–60 CrossRef CAS.
- F. Caruso, R. Pettinari, M. Rossi, E. Monti, M. B. Gariboldi, F. Marchetti, C. Pettinari, A. Caruso, M. V. Ramani and G. V. Subbaraju, J. Inorg. Biochem., 2016, 162, 44–51 CrossRef CAS PubMed.
- A. Antonyan, A. De, L. A. Vitali, R. Pettinari, F. Marchetti, M. R. Gigliobianco, C. Pettinari, E. Camaioni and G. Lupidi, Biochimie, 2014, 99, 146–152 CrossRef CAS PubMed.
- X. Lei, W. Su, P. Li, Q. Xiao, S. Huang, Q. Qian, C. Huang, D. Qin and H. Lan, Polyhedron, 2014, 81, 614–618 CrossRef CAS.
- P. Ramadevi, R. Singh, S. S. Jana, R. Devkar and D. Chakraborty, J. Organomet. Chem., 2017, 833, 80–87 CrossRef CAS.
- M. Kubanik, J. K. Y. Tu, T. Söhnel, M. Hejl, M. Jakupec, W. Kandioller, B. Keppler and C. G. Hartinger, Metallodrugs, 2015, 1, 24–35 Search PubMed.
- A. Kurzwernhart, W. Kandioller, É. A. Enyedy, M. Novak, M. A. Jakupec, B. K. Keppler and C. G. Hartinger, Dalton Trans., 2013, 42(17), 6193–6202 RSC.
- A. Pettenuzzo, R. Pigot and L. Ronconi, Metallodrugs, 2015, 1, 36–61 Search PubMed.
- A. C. Fernandes, Curr. Med. Chem., 2019, 35, 6412–6437 CrossRef PubMed.
- C. Tan, S. Lai, S. Wu, S. Hu, L. Zhou, Y. Chen, M. Wang, Y. Zhu, W. Lian, W. Peng, L. Ji and A. Xu, J. Med. Chem., 2010, 53, 7613–7624 CrossRef CAS PubMed.
- J. Chen, Y. Deng, J. Wang, S. Chen, F. Peng, X. He, M. Liu, H. Luo, J. Zhang and L. Chen, JBIC, J. Biol. Inorg. Chem., 2021, 26, 793–808 CrossRef CAS PubMed.
- J. Chen, F. Peng, Y. Zhang, B. Li, J. She, X. Jie, Z. Zou, M. Chen and L. Chen, Eur. J. Med. Chem., 2017, 140, 104–117 CrossRef CAS PubMed.
- Y. Chen, M. Qin, J. Wu, L. Wang, H. Chao, L. Ji and A. Xu, Eur. J. Med. Chem., 2013, 70, 120–129 CrossRef CAS PubMed.
- Y. Chen, M. Qin, L. Wang, H. Chao, L. Ji and A. Xu, Biochimie, 2013, 95, 2050–2059 CrossRef CAS PubMed.
- J. Chen, J. Wang, Y. Deng, B. Li, C. Li, Y. Lin, D. Yang, H. Zhang, L. Chen and T. Wang, Eur. J. Med. Chem., 2020, 203, 112562 CrossRef CAS PubMed.
- D. Havrylyuk, B. S. Howerton, L. Nease, S. Parkin, D. K. Heidary and E. C. Glazer, Eur. J. Med. Chem., 2018, 156, 790–799 CrossRef CAS PubMed.
- S. Chatterjee, S. Kundu, A. Bhattacharyya, C. G. Hartinger and P. J. Dyson, JBIC, J. Biol. Inorg. Chem., 2008, 13, 1149–1155 CrossRef CAS PubMed.
- A. Weiss, R. H. Berndsen, M. Dubois, C. Müller, R. Schibli, A. W. Griffioen, P. J. Dyson and P. Nowak-Sliwinska, Chem. Sci., 2014, 5, 4742–4748 RSC.
- L. He, S. Liao, C. Tan, R. Ye, Y. Xu, M. Zhao, L. Ji and Z. Mao, Chem. – Eur. J., 2013, 19, 12152–12160 CrossRef CAS PubMed.
- M. P. Chelopo, S. A. Pawar, M. K. Sokhela, T. Govender, H. G. Kruger and G. E. M. Maguire, Eur. J. Med. Chem., 2013, 66, 407–414 CrossRef CAS PubMed.
- B. A. Lakshmi, A. S. Reddy, R. Sangubotla, J. W. Hong and S. Kim, Colloids Surf., B, 2021, 204, 111773 CrossRef CAS PubMed.
- J. Karges, J. Li, L. Zeng, H. Chao and G. Gasser, ACS Appl. Mater. Interfaces, 2020, 12, 54433–54444 CrossRef CAS PubMed.
- Y. Dou, Y. Shang, X. He, W. Li, Y. Li and Y. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 13954–13963 CrossRef CAS PubMed.
- C. Y. Wu and W. Wang, Pharmaceutics, 2022, 14, 819 CrossRef CAS PubMed.
- Y. Shi, R. van der Meel, X. Chen and T. Lammers, Theranostics, 2020, 10, 7921–7924 CrossRef PubMed.
- J. Park, Y. Choi, H. Chang, W. Um, J. H. Ryu and I. C. Kwon, Theranostics, 2019, 9, 8073–8090 CrossRef CAS PubMed.
- J. Fang, W. Islam and H. Maeda, Adv. Drug Delivery Rev., 2020, 157, 142–160 CrossRef CAS PubMed.
- M. A. Subhan, S. S. K. Yalamarty, N. Filipczak, F. Parveen and V. P. Torchilin, J. Pers. Med., 2021, 11, 571 CrossRef PubMed.
- J. Medler and H. Wajant, Expert Opin. Ther. Targets, 2019, 23, 295–307 CrossRef CAS PubMed.
- E. Crescenzi, A. Leonardi and F. Pacifico, Int. J. Mol. Sci., 2021, 22, 12333 CrossRef CAS PubMed.
- X. Song, Q. Si, R. Qi, W. Liu, M. Li, M. Guo, L. Wei and Z. Yao, Front. Immunol., 2021, 12, 800630 CrossRef CAS PubMed.
- M. Alavi and M. Hamidi, Drug Metab. Pers. Ther., 2019, 1, 20180032 Search PubMed.
- Y. Sakurai and H. Harashima, Expert Opin. Drug Delivery, 2019, 16, 915–936 CrossRef CAS PubMed.
- A. Shafei, W. El-Bakly, A. Sobhy, O. Wagdy, A. Reda, O. Aboelenin, A. Marzouk, K. El Habak, R. Mostafa, M. A. Ali and M. Ellithy, Biomed. Pharmacother., 2017, 95, 1209–1218 CrossRef CAS PubMed.
- N. Amreddy, A. Babu, R. Muralidharan, P. Janani, A. Srivastava, R. Ahmed, M. Mehta, M. Anupama and R. Ramesh, Adv. Cancer Res., 2018, 115–170 CrossRef CAS PubMed.
- L. Tang, A. Zhang, Y. Mei, Q. Xiao, X. Xu and W. Wang, Pharmaceutics, 2021, 13, 2145 CrossRef CAS PubMed.
- N. Huang, X. Chen, X. Zhu, M. Xu and J. Liu, Biomaterials, 2017, 141, 296–313 CrossRef CAS PubMed.
- Y. Liu, L. Ma, H. Zhou, X. Zhu, Q. Yu, X. Chen, Y. Zhao and J. Liu, J. Mater. Chem. B, 2018, 6, 3497–3514 RSC.
- L. Zhu, Z. Kuang, P. Song, W. Li, L. Gui, K. Yang, F. Ge, Y. Tao and W. Zhang, Nanotechnology, 2021, 32, 455103 CrossRef CAS PubMed.
- H. Shi, J. Lou, S. Lin, Y. Wang, Y. Hu, P. Zhang, Y. Liu and Q. Zhang, J. Inorg. Biochem., 2021, 221, 111489 CrossRef CAS PubMed.
- C. Liu, D. Yu, F. Ge, L. Yang and Q. Wang, Anal. Bioanal. Chem., 2019, 411, 4193–4202 CrossRef CAS PubMed.
- X. Fan, S. Wang, H. Liu, Z. Li, Q. Sun, Y. Wang and X. Fan, Talanta, 2022, 236, 122830 CrossRef CAS PubMed.
- P. Zhang, H. Huang, J. Huang, H. Chen, J. Wang, K. Qiu, D. Zhao, L. Ji and H. Chao, ACS Appl. Mater. Interfaces, 2015, 7, 23278–23290 CrossRef CAS PubMed.
- N. Soliman, G. Gasser and C. M. Thomas, Adv. Mater., 2020, 32, 2003294 CrossRef CAS PubMed.
- M. Yang, R. Zhao, Y. Fang and J. Shao, Int. J. Pharm., 2019, 570, 118663 CrossRef CAS PubMed.
- P. Xing and Y. Zhao, Adv. Mater., 2016, 28, 7304–7339 CrossRef CAS PubMed.
- W. Li, Y. Yang, C. Wang, Z. Liu, X. Zhang, F. An, X. Diao, X. Hao and X. Zhang, Chem. Commun., 2012, 48, 8120 RSC.
- M. Zhou, X. Zhang, Y. Yang, Z. Liu, B. Tian, J. Jie and X. Zhang, Biomaterials, 2013, 34, 8960–8967 CrossRef CAS PubMed.
- Y. Shen, E. Jin, B. Zhang, C. J. Murphy, M. Sui, J. Zhao, J. Wang, J. Tang, M. Fan, E. Van Kirk and W. J. Murdoch, J. Am. Chem. Soc., 2010, 132, 4259–4265 CrossRef CAS PubMed.
- J. Fan, Y. Cheng and M. Sun, Chem. Rec., 2020, 20, 1474–1504 CrossRef CAS PubMed.
- Y. Liu, B. M. Crawford and T. Vo-Dinh, Immunotherapy, 2018, 10, 1175–1188 CrossRef CAS PubMed.
- P. Singh, S. Pandit, V. R. S. S. Mokkapati, A. Garg, V. Ravikumar and I. Mijakovic, Int. J. Mol. Sci., 2018, 19, 1979 CrossRef PubMed.
- M. Martinez-Calvo, K. N. Orange, R. B. Elmes, P. B. la Cour, D. C. Williams and T. Gunnlaugsson, Nanoscale, 2016, 8, 563–574 RSC.
- S. Estalayo-Adrián, G. J. McManus, H. L. Dalton, A. J. Savyasachi, J. M. Kelly and T. Gunnlaugsson, Dalton Trans., 2020, 49, 14158–14168 RSC.
- D. Gopalakrishnan, S. Saravanan, R. Merckx, K. A. Madan, T. Khamrang, M. Velusamy, K. Vasanth, S. Sunitha, R. Hoogenboom, S. Maji and M. Ganeshpandian, Dalton Trans., 2021, 50, 8232–8242 RSC.
- F. Limosani, H. Remita, P. Tagliatesta, E. M. Bauer, A. Leoni and M. Carbone, Materials, 2022, 15, 1207 CrossRef CAS PubMed.
- A. S. C. Gonçalves, C. F. Rodrigues, A. F. Moreira and I. J. Correia, Acta Biomater., 2020, 116, 105–137 CrossRef PubMed.
- J. Zheng, X. Cheng, H. Zhang, X. Bai, R. Ai, L. Shao and J. Wang, Chem. Rev., 2021, 121, 13342–13453 CrossRef CAS PubMed.
- C. R. Mayer, E. Dumas, F. Miomandre, R. Méallet-Renault, F. Warmont, J. Vigneron, R. Pansu, A. Etcheberry and F. Sécheresse, New J. Chem., 2006, 30, 1628–1637 RSC.
- C. R. Mayer, E. Dumas and F. Sécheresse, J. Colloid Interface Sci., 2008, 328, 452–457 CrossRef CAS PubMed.
- P. Pramod, P. K. Sudeep, K. G. Thomas and P. V. Kamat, J. Phys. Chem. B, 2006, 42, 20737–20741 CrossRef PubMed.
- Y. Yu, M. Zhou and H. Cui, J. Mater. Chem., 2011, 21, 12622 RSC.
- M. Jebb, P. K. Sudeep, P. Pramod, K. George Thomas and P. V. Kamat, J. Phys. Chem. B, 2007, 111, 6839–6844 CrossRef CAS PubMed.
- N. J. Rogers, S. Claire, R. M. Harris, S. Farabi, G. Zikeli, I. B. Styles, N. J. Hodges and Z. Pikramenou, Chem. Commun., 2014, 50, 617–619 RSC.
- R. B. P. Elmes, K. N. Orange, S. M. Cloonan, D. C. Williams and T. Gunnlaugsson, J. Am. Chem. Soc., 2011, 133, 15862–15865 CrossRef CAS PubMed.
- P. Zhang, J. Wang, H. Huang, H. Chen, R. Guan, Y. Chen, L. Ji and H. Chao, Biomaterials, 2014, 35, 9003–9011 CrossRef CAS PubMed.
- P. Zhang, J. Wang, H. Huang, B. Yu, K. Qiu, J. Huang, S. Wang, L. Jiang, G. Gasser, L. Ji and H. Chao, Biomaterials, 2015, 63, 102–114 CrossRef CAS PubMed.
- P. Zhang, J. Wang, H. Huang, K. Qiu, J. Huang, L. Ji and H. Chao, J. Mater. Chem. B, 2017, 5, 671–678 RSC.
- N. Thangavel, I. Jayakumar, M. Ravichandran, V. Vaidyanathan Ganesan and B. U. Nair, Spectrochim. Acta, Part A, 2019, 215, 196–202 CrossRef CAS PubMed.
- G. Mangiapia, G. Vitiello, C. Irace, R. Santamaria, A. Colonna, R. Angelico, A. Radulescu, G. D'Errico, D. Montesarchio and L. Paduano, Biomacromolecules, 2013, 14, 2549–2560 CrossRef CAS PubMed.
- G. Vitiello, A. Luchini, G. D'Errico, R. Santamaria, A. Capuozzo, C. Irace, D. Montesarchio and L. Paduano, J. Mater. Chem. B, 2015, 3, 3011–3023 RSC.
- J. A. Lebrón, F. J. Ostos, M. López-López, M. L. Moyá, C. Sales, E. García, C. B. García-Calderón, M. García-Calderón, M. J. Peña-Gómez, I. V. Rosado, F. R. Balestra, P. Huertas and P. López-Cornejo, Pharmaceutics, 2020, 12, 482 CrossRef PubMed.
- H. Zhou, M. Chen, Y. Liu and S. Wu, Macromol. Rapid Commun., 2018, 39, 1800372 CrossRef PubMed.
- G. Lv, L. Qiu, G. Liu, W. Wang, K. Li, X. Zhao and J. Lin, Dalton Trans., 2016, 15, 18147–18155 RSC.
- M. Mladenović, I. Morgan, N. Ilić, M. Saoud, M. V. Pergal, G. N. Kaluđerović and N. Ž. Knežević, Pharmaceutics, 2021, 13, 460 CrossRef PubMed.
- M. J. Ramalho, S. Andrade, J. A. Loureiro and M. Do Carmo Pereira, Drug Delivery Transl. Res., 2020, 10, 380–402 CrossRef PubMed.
- S. Karaosmanoglu, M. Zhou, B. Shi, X. Zhang, G. R. Williams and X. Chen, J. Controlled Release, 2021, 329, 805–832 CrossRef CAS PubMed.
- O. A. Kuchur, S. A. Tsymbal, M. V. Shestovskaya, N. S. Serov, M. S. Dukhinova and A. A. Shtil, J. Inorg. Biochem., 2020, 209, 111117 CrossRef CAS PubMed.
- E. M. See, C. Tossi, L. Hällström and I. Tittonen, ACS Omega, 2020, 5, 10671–10679 CrossRef CAS PubMed.
- Y. Zhang, J. Yu, H. Niu and H. Liu, J. Colloid Interface Sci., 2007, 313, 503–510 CrossRef CAS PubMed.
- G. Chen, M. Xu, S. Zhao, J. Sun, Q. Yu and J. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 33645–33659 CrossRef CAS PubMed.
- S. Zhu, X. Lin, P. Ran, Q. Xia, C. Yang, J. Ma and Y. Fu, Biosens. Bioelectron., 2017, 91, 436–440 CrossRef CAS PubMed.
- H. Zhou, Y. Gong, Y. Liu, A. Huang, X. Zhu, J. Liu, G. Yuan, L. Zhang, J. Wei and J. Liu, Biomaterials, 2020, 237, 119822 CrossRef CAS PubMed.
- S. Mandal, J. Dinda, A. Mahapatra, W. Hwang, G. Roymahapatra and A. Mishra, J. Cancer Res. Ther., 2015, 11, 105 CrossRef PubMed.
- H. Wang, Y. Yuan, Y. Zhuo, Y. Chai and R. Yuan, Anal. Chem., 2016, 4, 2258–2265 CrossRef PubMed.
- S. Ramasamy, D. Benneta and S. Kim, RSC Adv., 2015, 79616–79623 RSC.
- M. Xu, Y. Wen, Y. Liu, X. Tan, X. Chen, X. Zhu, C. Wei, L. Chen, Z. Wang and J. Liu, Nanoscale, 2019, 19, 9661–9678 RSC.
- R. Zhang, X. Fan, Z. Meng, H. Lin, Q. Jin, F. Gong, Z. Dong, Y. Li, Q. Chen, Z. Liu and L. Cheng, Theranostics, 2019, 9, 8266–8276 CrossRef CAS PubMed.
- X. Zhu, H. Zhou, Y. Liu, Y. Wen, C. Wei, Q. Yu and J. Liu, Acta Biomater., 2018, 82, 143–157 CrossRef CAS PubMed.
- S. Zhang, L. Li, S. Zhao, Z. Sun, M. Hong and J. Luo, J. Mater. Chem. A, 2015, 3, 15764–15768 RSC.
- S. Y. Lee, C. Y. Kim and T. Nam, Drug Des., Dev. Ther., 2020, 14, 5375–5392 CrossRef CAS PubMed.
- Y. Zhou, Q. Yu, X. Qin, D. Bhavsar, L. Yang, Q. Chen, W. Zheng, L. Chen and J. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 15000–15012 CrossRef CAS PubMed.
- C. Yin, Z. Wang, X. Ding, X. Chen, J. Wang, E. Yang, W. Wang, L. L. Martin and D. Sun, J. Mater. Chem. B, 2021, 3808–3825 RSC.
- H. Vardhan, A. Nafady, A. M. Al-Enizi, K. Khandker, H. M. El-Sagher, G. Verma, M. Acevedo-Duncan, T. M. Alotaibi and S. Ma, Molecules, 2019, 24, 2284 CrossRef CAS PubMed.
- X. Zhao, H. Zhao, S. Wang, Z. Fan, Y. Ma, Y. Yin, W. Wang, R. Xi and M. Meng, J. Am. Chem. Soc., 2021, 143, 20828–20836 CrossRef CAS PubMed.
- X. Hu, Y. Lu, C. Dong, W. Zhao, X. Wu, L. Zhou, L. Chen, T. Yao and S. Shi, Chem. – Eur. J., 2020, 26, 1668–1675 CrossRef CAS PubMed.
- J. A. Johnson, Y. Y. Lu, A. O. Burts, Y. Xia, A. C. Durrell, D. A. Tirrell and R. H. Grubbs, Macromolecules, 2010, 43, 10326–10335 CrossRef CAS PubMed.
- S. Xue, C. Tan, M. Chen, J. Cao, D. Zhang, R.-R. Ye, L. N. Jin and Z. W. Mao, Chem. Commun., 2017, 53, 842–845 RSC.
- H. Fu, Y. Chen, Q. Yu and Y. Liu, Chem. Commun., 2019, 21, 3148–3151 RSC.
- Z. Zhao, X. Zhang, C. Li and T. Chen, Biomaterials, 2019, 192, 579–589 CrossRef CAS PubMed.
- K. Adamson, C. Dolan, N. Moran, R. J. Forster and T. E. Keyes, Bioconjugate Chem., 2014, 25, 928–944 CrossRef CAS PubMed.
- W. Wang, Z. Guo, Y. Lu, X. Shen, T. Chen, R. Huang, B. Zhou, C. Wen, H. Liang and B. Jiang, ACS Appl. Mater. Interfaces, 2019, 11, 17294–17305 CrossRef CAS PubMed.
- J. Zhang, V. Ramu, X. Zhou, C. Frias, D. Ruiz-Molina, S. Bonnet, C. Roscini and F. Novio, Nanomaterials, 2021, 11, 3089 CrossRef CAS PubMed.
- B. Tian, S. Hua and J. Liu, Carbohydr. Polym., 2020, 232, 115805 CrossRef CAS PubMed.
- D. Zhang, P. Lv, C. Zhou, Y. Zhao, X. Liao and B. Yang, Mater. Sci. Eng., C, 2019, 96, 872–886 CrossRef CAS PubMed.
- Y. Zhou, M. Zhao, Z. Mao and L. Ji, Chem. – Eur. J., 2008, 14, 7193–7201 CrossRef CAS PubMed.
- S. Lee, C. Lee, B. Kim, L. Q. Thao, E. S. Lee, J. O. Kim, K. T. Oh, H. Choi and Y. S. Youn, Colloids Surf., B, 2016, 147, 281–290 CrossRef CAS PubMed.
- X. Yuan, Z. Jia, J. Li, Y. Liu, Y. Huang, Y. Gong, X. Guo, X. Chen, J. Cen and J. Liu, J. Mater. Chem. B, 2021, 9, 7835–7847 RSC.
- I. H. El-Sayed, X. Huang and M. A. El-Sayed, Nano Lett., 2005, 5, 829–834 CrossRef CAS PubMed.
- J. H. W. Pahl, J. Koch, J. Götz, A. Arnold, U. Reusch, T. Gantke, E. Rajkovic, M. Treder and A. Cerwenka, Cancer Immunol. Res., 2018, 6, 517–527 CrossRef CAS PubMed.
- H. Ataee-Esfahani, J. Liu, M. Hu, N. Miyamoto, S. Tominaka, K. C. W. Wu and Y. Yamauchi, Small, 2013, 9, 1047–1051 CrossRef CAS PubMed.
- J. Li, C. Zhou, B. Dong, H. Zhong, S. Chen, Q. Li and Z. Wang, Cancer Biol. Ther., 2016, 17, 1231–1239 CrossRef CAS PubMed.
- M. Ravera, E. Gabano, M. J. McGlinchey and D. Osella, Dalton Trans., 2022, 2121–2134 RSC.
- D. Gibson, ChemMedChem, 2021, 16, 2188–2191 CrossRef CAS PubMed.
- D. Gibson, J. Inorg. Biochem., 2019, 191, 77–84 CrossRef CAS PubMed.
- X. Li, Y. Liu and H. Tian, Bioinorg. Chem. Appl., 2018, 2018, 1–18 CrossRef PubMed.
- Y. Zheng, D. Y. Zhang, H. Zhang, J. J. Cao, C. P. Tan, L. N. Ji and Z. W. Mao, Chem. – Eur. J., 2018, 24, 18971–18980 CrossRef CAS PubMed.
- L. Ma, R. Ma, Z. Wang, S. Yiu and G. Zhu, Chem. Commun., 2016, 52, 10735–10738 RSC.
- L. Ma, X. Lin, C. Li, Z. Xu, C. Chan, M. Tse, P. Shi and G. Zhu, Inorg. Chem., 2018, 57, 2917–2924 CrossRef CAS PubMed.
- J. Karges, T. Yempala, M. Tharaud, D. Gibson and G. Gasser, Angew. Chem., Int. Ed., 2020, 59, 7069–7075 CrossRef CAS PubMed.
- J. van den Anker, M. D. Reed, K. Allegaert and G. L. Kearns, J. Clin. Pharmacol., 2018, 58, S10–S25 CrossRef CAS PubMed.
- S. Ravindran, J. K. Suthar, R. Rokade, P. Deshpande, P. Singh, A. Pratinidhi, R. Khambadkhar and S. Utekar, Curr. Drug Metab., 2018, 19, 327–334 CrossRef CAS PubMed.
- F. Lentz, A. Drescher, A. Lindauer, M. Henke, R. A. Hilger, C. G. Hartinger, M. E. Scheulen, C. Dittrich, B. K. Keppler and U. Jaehde, Anti-Cancer Drugs, 2009, 20, 97–103 CrossRef CAS PubMed.
- V. Novohradsky, A. Bergamo, M. Cocchietto, J. Zajac, V. Brabec, G. Mestroni and G. Sava, Dalton Trans., 2015, 44, 1905–1913 RSC.
- S. Mehanna, K. Bodman-Smith, C. F. Dahera and R. S. Khnayzer, Anal. Methods, 2020, 4517–4525 RSC.
- M. R. Gill and J. A. Thomas, Chem. Soc. Rev., 2012, 41, 3179–3192 RSC.
- S. Mayor and R. E. Pagano, Nat. Rev. Mol. Cell Biol., 2007, 8, 603–612 CrossRef CAS PubMed.
- Y. Lu, Z. Peng, D. Zhu, Y. Jia, A. Taledaohan, Y. Li, J. Liu, Y. Wang and Y. Wang, Int. J. Nanomed., 2021, 16, 5565–5580 CrossRef PubMed.
- Y. Sun, Y. Liang, W. Dai, X. Wang, J. Wang and S. Huang, Nano Lett., 2019, 5, 3229–3237 CrossRef PubMed.
- C. A. Puckett and J. K. Barton, J. Am. Chem. Soc., 2007, 129, 46–47 CrossRef CAS PubMed.
- C. A. Puckett and J. K. Barton, Biochemistry, 2008, 47, 11711–11716 CrossRef CAS PubMed.
- S. Park, J. L. Gray, S. D. Altman, A. R. Hairston, B. T. Beswick, Y. Kim and E. T. Papish, J. Inorg. Biochem., 2020, 203, 110922 CrossRef CAS PubMed.
- Y. Sun, Y. Liang, W. Dai, B. He, H. Zhang, X. Wang, J. Wang, S. Huang and Q. Zhang, Nano Lett., 2019, 19, 3229–3237 CrossRef CAS PubMed.
- J. J. Rennick, A. P. R. Johnston and R. G. Parton, Nat. Nanotechnol., 2021, 16, 266–276 CrossRef CAS PubMed.
- J. F. Machado, J. D. G. Correia and T. S. Morais, Molecules, 2021, 26, 3153 CrossRef CAS PubMed.
- W. Guo, W. Zheng, Q. Luo, X. Li, Y. Zhao, S. Xiong and F. Wang, Inorg. Chem., 2013, 52, 5328–5338 CrossRef CAS PubMed.
- M. A. Naves, A. E. Graminha, L. C. Vegas, L. Luna-Dulcey, J. Honorato, A. C. S. Menezes, A. A. Batista and M. R. Cominetti, Mol. Pharmaceutics, 2019, 16, 1167–1183 CrossRef CAS PubMed.
- L. M. Bareford and P. W. Swaan, Adv. Drug Delivery Rev., 2007, 8, 748–758 CrossRef PubMed.
- A. K. Bytzek, G. Koellensperger, B. K. Keppler and C. G. Hartinger, J. Inorg. Biochem., 2016, 160, 250–255 CrossRef CAS PubMed.
- M. Sulyok, S. Hann, C. G. Hartinger, B. K. Keppler, G. Stingeder and G. Koellensperger, J. Anal. At. Spectrom., 2005, 20, 856 RSC.
- J. Mo, N. P. Mai Le and R. Priefer, Eur. J. Med. Chem., 2021, 225, 113770 CrossRef CAS PubMed.
- M. Groessl, O. Zava and P. J. Dyson, Metallomics, 2011, 6, 591–599 CrossRef PubMed.
- K. Kardani, A. Milani, S. H. Shabani and A. Bolhassani, Expert Opin. Drug Delivery, 2019, 16, 1227–1258 CrossRef CAS PubMed.
- G. Guidotti, L. Brambilla and D. Rossi, Trends Pharmacol. Sci., 2017, 38, 406–424 CrossRef CAS PubMed.
- C. S. Burke, A. Byrne and T. E. Keyes, J. Am. Chem. Soc., 2018, 140, 6945–6955 CrossRef CAS PubMed.
- A. Byrne, C. S. Burke and T. E. Keyes, Chem. Sci., 2016, 7, 6551–6562 RSC.
- C. A. Puckett and J. K. Barton, J. Am. Chem. Soc., 2009, 131, 8738–8739 CrossRef CAS PubMed.
- C. A. Puckett and J. K. Barton, Bioorg. Med. Chem., 2010, 18, 3564–3569 CrossRef CAS PubMed.
- H. Cho, Y. Cho, M. S. Shim, J. Y. Lee, H. S. Lee and H. C. Kang, Biochim. Biophys. Acta, Mol. Basis Dis., 2020, 1866, 165808 CrossRef CAS PubMed.
- F. Wang, M. A. Ogasawara and P. Huang, Mol. Aspects Med., 2010, 31, 75–92 CrossRef CAS PubMed.
- P. Lu, B. J. Bruno, M. Rabenau and C. S. Lim, J. Controlled Release, 2016, 240, 38–51 CrossRef CAS PubMed.
- J. Liu, X. Liao, K. Xiong, S. Kuang, C. Jin, L. Ji and H. Chao, Chem. Commun., 2020, 56, 5839–5842 RSC.
- Q. Yu, Y. Liu, L. Xu, C. Zheng, F. Le, X. Qin, Y. Liu and J. Liu, Eur. J. Med. Chem., 2014, 82, 82–95 CrossRef CAS PubMed.
- E. M. De Francesco, B. Ózsvári, F. Sotgia and M. P. Lisanti, Front. Oncol., 2019, 9, 615 CrossRef PubMed.
- J. Liu, Y. Chen, G. Li, P. Zhang, C. Jin, L. Zeng, L. Ji and H. Chao, Biomaterials, 2015, 56, 140–153 CrossRef CAS PubMed.
- C. S. Burke, A. Byrne and T. E. Keyes, Angew. Chem., Int. Ed., 2018, 38, 12420–12424 CrossRef PubMed.
- D. Cullinane, K. S. Gkika, A. Byrne and T. E. Keyes, J. Inorg. Biochem., 2020, 207, 111032 CrossRef CAS PubMed.
- A. Martin, A. Byrne, C. S. Burke, R. J. Forster and T. E. Keyes, J. Am. Chem. Soc., 2014, 136, 15300–15309 CrossRef CAS PubMed.
- L. He, Y. Li, C. Tan, R. Ye, M. Chen, J. Cao, L. Ji and Z. Mao, Chem. Sci., 2015, 6, 5409–5418 RSC.
- B. Purushothaman, J. Lee, S. Hong and J. M. Song, J. Nanobiotechnol., 2020, 18, 102 CrossRef CAS PubMed.
- D. Zhang, Y. Zheng and H. Zhang, Nanoscale, 2017, 18966–18976 RSC.
- P. Srivastava, M. Verma, A. Kumar, P. Srivastava, R. Mishra, S. Sivakumar and A. K. Patra, Dalton Trans., 2021, 50, 3629–3640 RSC.
- J. Li, Z. Tian, Z. Xu, S. Zhang, Y. Feng, L. Zhang and Z. Liu, Dalton Trans., 2018, 47, 15772–15782 RSC.
- M. Yang, H. Zhao, Z. Zhang, Q. Yuan, Q. Feng, X. Duan, S. Wang and Y. Tang, Chem. Sci., 2021, 12, 11515–11524 RSC.
- H. Huang, B. Yu, P. Zhang, J. Huang, Y. Chen, G. Gasser, L. Ji and H. Chao, Angew. Chem., Int. Ed., 2015, 54, 14049–14052 CrossRef CAS PubMed.
- B. Liu, Y. Gao, M. A. Jabed, S. Kilina, G. Liu and W. Sun, ACS Appl. Bio Mater., 2020, 3, 6025–6038 CrossRef CAS PubMed.
- J. Chen, Q. Tao, J. Wu, M. Wang, Z. Su, Y. Qian, T. Yu, Y. Wang, X. Xue and H. Liu, J. Inorg. Biochem., 2020, 210, 111132 CrossRef CAS PubMed.
- F. Zhao, W. Wang and W. Wu, Dalton Trans., 2021, 50, 3536–3541 RSC.
- L. Xu, P. Zhang, X. Fang, Y. Liu, J. Wang, H. Zhou, S. Chen and H. Chao, J. Inorg. Biochem., 2019, 191, 126–134 CrossRef CAS PubMed.
- C. Gaiddon, I. Gross, X. Meng, M. Sidhoum, G. Mellitzer, B. Romain, J. Delhorme, A. Venkatasamy, A. C. Jung and M. Pfeffer, Molecules, 2021, 26, 5386 CrossRef CAS PubMed.
- J. Karges, T. Yempala, M. Tharaud, D. Gibson and G. Gasser, Angew. Chem., Int. Ed., 2020, 59, 7069–7075 CrossRef CAS PubMed.
- R. S. Erdmann, H. Takakura, A. D. Thompson, F. Rivera-Molina, E. S. Allgeyer, J. Bewersdorf, D. Toomre and A. Schepartz, Angew. Chem., Int. Ed., 2014, 53, 10242–10246 CrossRef CAS PubMed.
- J. Zhang, H. Li, C. Chan, R. Lan, W. Chan, G. Law, W. Wong and K. Wong, Chem. Commun., 2012, 48, 9646 RSC.
- P. Štarha and Z. Trávníček, Coord. Chem. Rev., 2019, 395, 130–145 CrossRef.
- D. S. Goodsell, Stem Cells, 2006, 514–515 CrossRef PubMed.
- S. Dasari and P. Bernard Tchounwou, Eur. J. Pharmacol., 2014, 740, 364–378 CrossRef CAS PubMed.
- T. Minervini, B. Cardey, S. Foley, C. Ramseyer and M. Enescu, Metallomics, 2019, 11, 833–844 CrossRef CAS PubMed.
- T. Yotsuyanagi, M. Usami, Y. Noda and M. Nagata, Int. J. Pharm., 2002, 246, 95–104 CrossRef CAS PubMed.
- C. Scolaro, C. G. Hartinger, C. S. Allardyce, B. K. Keppler and P. J. Dyson, J. Inorg. Biochem., 2008, 102, 1743–1748 CrossRef CAS PubMed.
- M. Bacac, A. C. G. Hotze, K. V. D. Schilden, J. G. Haasnoot, S. Pacor, E. Alessio, G. Sava and J. Reedijk, J. Inorg. Biochem., 2004, 98, 402–412 CrossRef CAS PubMed.
- B. G. Dwyer, E. Johnson, E. Cazares, K. L. McFarlane Holman and S. R. Kirk, J. Inorg. Biochem., 2018, 182, 177–183 CrossRef CAS PubMed.
- J. Chen, L. Chen, S. Liao, K. Zheng and L. Ji, J. Phys. Chem. B, 2007, 111, 7862–7869 CrossRef CAS PubMed.
- J. Chatlas, R. van Eldik and B. K. Keppler, Inorg. Chim. Acta, 1995, 233, 59–63 CrossRef CAS.
- C. G. Hartinger, S. Zorbas-Seifried, M. A. Jakupec, B. Kynast, H. Zorbas and B. K. Keppler, J. Inorg. Biochem., 2006, 100, 891–904 CrossRef CAS PubMed.
- I. Romero-Canelón, L. Salassa and P. J. Sadler, J. Med. Chem., 2013, 56, 1291–1300 CrossRef PubMed.
- K. Pelivan, W. Miklos, S. van Schoonhoven, G. Koellensperger, L. Gille, W. Berger, P. Heffeter, C. R. Kowol and B. K. Keppler, J. Inorg. Biochem., 2016, 160, 61–69 CrossRef CAS PubMed.
- D. Truhn, C. K. Kuhl, A. Ciritsis, A. Barabasch and N. A. Kraemer, Eur. Radiol., 2019, 29, 383–391 CrossRef PubMed.
- A. Yadav, T. Janaratne, A. Krishnan, S. S. Singhal, S. Yadav, A. S. Dayoub, D. L. Hawkins, S. Awasthi and F. M. MacDonnell, Mol. Cancer Ther., 2013, 12, 643–653 CrossRef CAS PubMed.
- M. Zhang, L. Wang, L. Heng, Z. Wang, W. Feng, H. Jin, S. Liu, S. Lan, Y. Liu and H. Zhang, ACS Appl. Bio Mater., 2022, 16, 2365–2376 CrossRef PubMed.
- Z. Lam, G. Balasundaram, K. V. Kong, B. Y. Chor, D. Goh, B. Khezri, R. D. Webster, W. K. Leong and M. Olivo, J. Mater. Chem. B, 2016, 4, 3886–3891 RSC.
- D. Sun, Y. Liu, Q. Yu, X. Qin, L. Yang, Y. Zhou, L. Chen and J. Liu, Biomaterials, 2014, 35, 1572–1583 CrossRef CAS PubMed.
- X. Ge, L. Sun, B. Ma, D. Jin, L. Dong, L. Shi, N. Li, H. Chen and W. Huang, Nanoscale, 2015, 7, 13877–13887 RSC.
- P. Kalomoiri, C. Rodríguez Rodríguez, K. K. Sørensen, M. Bergamo, K. Saatchi, U. O. Häfeli and K. J. Jensen, ChemBioChem, 2020, 21, 3338–3348 CrossRef CAS PubMed.
- Q. Zhang and L. Li, J BUON, 2018, 3, 561–567 Search PubMed.
- J. Hong and C. Yun, Curr. Cancer Drug Targets, 2018, 18, 139 CrossRef CAS PubMed.
- Y. Lu, J. Hua, F. Yan, C. Jiang, Y. Piao, Z. Ye, Z. Fu, H. Jiang, F. Wang and Y. Jiang, Medicine, 2021, 100, e26629 CrossRef CAS PubMed.
- W. Rao, Z. Deng and J. Liu, Crit. Rev. Biomed. Eng., 2010, 38, 101 CrossRef PubMed.
- Z. Dembic, Molecules, 2020, 25, 5776 CrossRef CAS PubMed.
- P. Sarbadhikary, B. P. George and H. Abrahamse, Theranostics, 2021, 11, 9054–9088 CrossRef CAS PubMed.
- P. D. Karkos, I. Koskinas, M. Stavrakas, S. Triaridis and J. Constantinidis, Ear Nose Throat J., 2021, 100, 19S–23S CrossRef PubMed.
- W. Chen, Y. Yuan and X. Jiang, J. Controlled Release, 2020, 328, 395–406 CrossRef CAS PubMed.
- U. Hafeez, S. Parakh, H. K. Gan and A. M. Scott, Molecules, 2020, 25, 4764 CrossRef CAS PubMed.
- M. A. Zaimy, N. Saffarzadeh, A. Mohammadi, H. Pourghadamyari, P. Izadi, A. Sarli, L. K. Moghaddam, S. R. Paschepari, H. Azizi, S. Torkamandi and J. Tavakkoly-Bazzaz, Cancer Gene Ther., 2017, 24, 233–243 CrossRef CAS PubMed.
- C. M. Hu, S. Aryal and L. Zhang, Ther. Delivery, 2010, 1, 323–334 CrossRef CAS PubMed.
- J. M. Dabrowski and L. G. Arnaut, Photochem. Photobiol. Sci., 2015, 14, 1765–1780 CrossRef CAS PubMed.
- C. A. Robertson, D. H. Evans and H. Abrahamse, J. Photochem. Photobiol., B, 2009, 96, 1–8 CrossRef CAS PubMed.
- J. Hess, H. Huang, A. Kaiser, V. Pierroz, O. Blacque, H. Chao and G. Gasser, Chem. – Eur. J., 2017, 23, 9888–9896 CrossRef CAS PubMed.
- F. Heinemann, J. Karges and G. Gasser, Acc. Chem. Res., 2017, 50, 2727–2736 CrossRef CAS PubMed.
- J. Mo, N. P. Mai Le and R. Priefer, Eur. J. Med. Chem., 2021, 225, 113770 CrossRef CAS PubMed.
- L. Wang, H. Yin, M. A. Jabed, M. Hetu, C. Wang, S. Monro, X. Zhu, S. Kilina, S. A. McFarland and W. Sun, Inorg. Chem., 2017, 56, 3245–3259 CrossRef CAS PubMed.
- M. Lin, S. Zou, X. Liao, Y. Chen, D. Luo, L. Ji and H. Chao, Chem. Commun., 2021, 57, 4408–4411 RSC.
- S. Li, J. Zhao, X. Wang, G. Xu, S. Gou and Q. Zhao, Inorg. Chem., 2020, 59, 11193–11204 CrossRef CAS PubMed.
- M. H. Kaulage, B. Maji, S. Pasadi, S. Bhattacharya and K. Muniyappa, Eur. J. Med. Chem., 2017, 139, 1016–1029 CrossRef CAS PubMed.
- J. Karges, S. Kuang, Y. C. Ong, H. Chao and G. Gasser, Chem. – Eur. J., 2021, 27, 362–370 CrossRef CAS PubMed.
- Y. Liu, P. Bhattarai, Z. Daib and X. Chen, Chem. Soc. Rev., 2019, 7, 2053–2108 RSC.
- X. Huang, X. Sun, W. Wang, Q. Shen, Q. Shen, X. Tang and J. Shao, J. Mater. Chem. B, 2021, 9, 3756–3777 RSC.
- G. Zhao, H. Wu, R. Feng, D. Wang, P. Xu, P. Jiang, K. Yang, H. Wang, Z. Guo and Q. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 3295–3304 CrossRef CAS PubMed.
- T. Liu, M. Zhang, W. Liu, X. Zeng, X. Song, X. Yang, X. Zhang and J. Feng, ACS Nano, 2018, 12, 3917–3927 CrossRef CAS PubMed.
- Z. Liu, K. Qiu, X. Liao, T. W. Rees, Y. Chen, Z. Zhao, L. Ji and H. Chao, Chem. Commun., 2020, 56, 3019–3022 RSC.
- H. Lu, S. He, Q. Zhang, X. Li, Z. Xie, Z. Wang, Y. Qi and Y. Huang, Biomater. Sci., 2021, 9, 7115–7123 RSC.
- C. Matera, A. M. J. Gomila, N. Camarero, M. Libergoli, C. Soler and P. Gorostiza, J. Am. Chem. Soc., 2018, 140, 15764–15773 CrossRef CAS PubMed.
- W. Fan, P. Huang and X. Chen, Chem. Soc. Rev., 2016, 6488–6519 RSC.
- C. Zhang, X. Guo, X. Da, Y. Yao, H. Xiao, X. Wang and Q. Zhou, Dalton Trans., 2021, 50, 7715–7724 RSC.
- C. Zhang, X. Guo, X. Da, Z. Wang, X. Wang and Q. Zhou, Dalton Trans., 2021, 50, 10845–10852 RSC.
- N. Tian, Y. Feng, W. Sun, J. Lu, S. Lu, Y. Yao, C. Li, X. Wang and Q. Zhou, Dalton Trans., 2019, 48, 6492–6500 RSC.
- M. G. Elias, S. Mehanna, E. Elias, R. S. Khnayzer and C. F. Daher, Chem.-Biol. Interact., 2021, 348, 109644 CrossRef CAS PubMed.
- A. K. Renfrew, J. Karges, R. Scopelliti, F. D. Bobbink, P. Nowak Sliwinska, G. Gasser and P. J. Dyson, ChemBioChem, 2019, 20, 2876–2882 CrossRef CAS PubMed.
- Y. Chen, L. Bai, P. Zhang, H. Zhao and Q. Zhou, Molecules, 2021, 26, 5679 CrossRef CAS PubMed.
- Z. Jin, S. Qi, X. Guo, Y. Jian, Y. Hou, C. Li, X. Wang and Q. Zhou, Chem. Commun., 2021, 57, 3259–3262 RSC.
- M. Abbott and Y. Ustoyev, Semin. Oncol. Nurs., 2019, 35, 150923 CrossRef PubMed.
- D. Goldberg, Crit. Rev. Clin. Lab. Sci., 2010, 47, 1–4 CrossRef PubMed.
- R. S. Riley, C. H. June, R. Langer and M. J. Mitchell, Nat. Rev. Drug Discovery, 2019, 18, 175–196 CrossRef CAS PubMed.
- V. Velcheti and K. Schalper, Am. Soc. Clin. Oncol. Educ. Book, 2016, 35, 298–308 CrossRef PubMed.
- A. Iscaro, N. F. Howard and M. Muthana, Curr. Pharm. Des., 2019, 17, 1962–1979 CrossRef PubMed.
- A. Chauhan, T. Khan and A. Omri, Int. J. Mol. Sci., 2021, 22, 8037 CrossRef CAS PubMed.
- K. S. Kapitanova, V. A. Naumenko, A. S. Garanina, P. A. Melnikov, M. A. Abakumov and I. B. Alieva, Biochemistry, 2019, 84, 729–745 CAS.
- X. Duan, C. Chan and W. Lin, Angew. Chem., Int. Ed., 2019, 58, 670–680 CrossRef CAS PubMed.
- S. Chung, R. A. Revia and M. Zhang, Nanoscale Horiz., 2021, 6, 696–717 RSC.
- D. J. Irvine and E. L. Dane, Nat. Rev. Immunol., 2020, 20, 321–334 CrossRef CAS PubMed.
- R. A. Odion, Y. Liu and T. Vo-Dinh, IEEE J. Sel. Top. Quantum Electron., 2021, 5, 4800109 Search PubMed.
- L. A. Dykman and N. G. Khlebtsov, Biomed. Opt. Express, 2019, 10, 3152 CrossRef CAS PubMed.
- D. V. Krysko, A. D. Garg, A. Kaczmarek, O. Krysko, P. Agostinis and P. Vandenabeele, Nat. Rev. Cancer, 2012, 12, 860–875 CrossRef CAS PubMed.
- S. Wang, Y. Bai, D. Wang, Y. Zhai, Y. Qiao, X. Zhao, Y. Yin, R. Xi, W. Wang, W. Zhao and M. Meng, Chem. Eng. J., 2022, 442, 136322 CrossRef CAS.
- L. Galluzzi, A. Buqué, O. Kepp, L. Zitvogel and G. Kroemer, Nat. Rev. Immunol., 2017, 17, 97–111 CrossRef CAS PubMed.
- D. Wernitznig, K. Kiakos, F. G. Del, N. Harrer, H. Machat, A. Osswald, M. A. Jakupec, A. Wernitznig, W. Sommergruber and B. K. Keppler, Metallomics, 2019, 11, 1044–1048 CrossRef CAS PubMed.
- P. Konda, I. J. Roque, L. M. Lifshits, A. Alcos, E. Azzam, G. Shi, C. G. Cameron, S. A. McFarland and S. Gujar, Am. J. Cancer Res., 2022, 12, 210–228 CAS.
- Y. Liu, Y. Wen, X. Chen, X. Zhu, Q. Yu, Y. Gong, G. Yuan, J. Liu and X. Qin, J. Mater. Chem. B, 2019, 7, 6210–6223 RSC.
- N. Toupin, M. K. Herroon, R. P. Thummel, C. Turro, I. Podgorski, H. Gibson and J. J. Kodanko, Chem. – Eur. J., 2022, 28, e202104430 CrossRef CAS PubMed.
- Q. Chen, L. He, X. Li, L. Xu and T. Chen, Biomaterials, 2022, 281, 121371 CrossRef CAS PubMed.
- H. Lai, D. Zeng, C. Liu, Q. Zhang, X. Wang and T. Chen, Biomaterials, 2019, 219, 119377 CrossRef CAS PubMed.
- F. Zhang, F. Chen, C. Yang, L. Wang, H. Hu, X. Li, X. Zheng, Z. Wang, Z. Chang, T. Li, L. Li, M. Ge, J. Du, W. Sun, W. F. Dong and D. Shao, Small, 2021, 17, 2100006 CrossRef CAS PubMed.
- D. Zhang, Y. Zheng, C. Tan, J. Sun, W. Zhang, L. Ji and Z. Mao, ACS Appl. Mater. Interfaces, 2017, 9, 6761–6771 CrossRef CAS PubMed.
- X. Huang, G. Chen, J. Pan, X. Chen, N. Huang, X. Wang and J. Liu, J. Mater. Chem. B, 2016, 4, 6258–6270 RSC.
| This journal is © The Royal Society of Chemistry 2022 |