
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
RAFT polymerisation of renewable terpene (meth)acrylates and the convergent synthesis of methacrylate–acrylate–methacrylate triblock copolymers†
Rachel L.
Atkinson
 a,
Olivia R.
Monaghan
a,
Matthew T.
Elsmore
b,
Paul D.
Topham
c,
Daniel T. W.
Toolan
d,
Matthew J.
Derry
c,
Vincenzo
Taresco
a,
Robert A.
Stockman
a,
Davide S. A.
De Focatiis
b,
Derek J.
Irvine
b and
Steven M.
Howdle
*a
a,
Olivia R.
Monaghan
a,
Matthew T.
Elsmore
b,
Paul D.
Topham
c,
Daniel T. W.
Toolan
d,
Matthew J.
Derry
c,
Vincenzo
Taresco
a,
Robert A.
Stockman
a,
Davide S. A.
De Focatiis
b,
Derek J.
Irvine
b and
Steven M.
Howdle
*a
aSchool of Chemistry, University of Nottingham, University Park, Nottingham NG7 2RD, Nottingham, UK. E-mail: steve.howdle@nottingham.ac.uk
bFaculty of Engineering, University Park, Nottingham NG7 2RD, Nottingham, UK
cAston Institute of Materials Research, Aston University, Aston Triangle, Birmingham, B4 7ET, UK
dDepartment of Chemistry, The University of Sheffield, Dainton Building, The University of Sheffield, Brook Hill, Sheffield S3 7HF, UK
First published on 5th May 2021
Abstract
Terpenes are ideal candidates for sustainable polymer feedstocks, due to their natural abundance and availability from existing waste streams. Previously, we have shown that a range of terpene(meth)acrylate monomers can be synthesised from the most commonly available terpenes (α-pinene, β-pinenene and limonene) and that these readily undergo radical polymerisation. We now report the synthesis of well-defined polymers and precise di- and multiblock copolymer architectures by use of RAFT control. A very wide range of Tg values are observed for the terpene (meth)acrylate homopolymers, from −3 °C for poly(limonene acrylate), up to +168 °C for poly(α-pinene methacrylate), and we exploit these to create renewably-sourced hard–soft block copolymers. We also report the synthesis of difunctional poly(α- and β-pinene methacrylate) macro-RAFT agents and the preparation of ABA triblock copolymers. Promising adhesive properties are observed for a triblock copolymer comprised of poly(α-pinene methacrylate) and poly(butyl acrylate) blocks. A range of fully terpene-based triblock copolymers containing poly(limonene acrylate) soft blocks are also reported.
Introduction
Growing awareness of the negative environmental impacts of fossil fuel usage has led to increased demand for naturally-sourced and renewable alternatives.1 Derived primarily from biomass, terpenes have been identified as a potential feedstock for sustainable polymer synthesis. These renewably-sourced hydrocarbons are an ideal monomer source, as many are available from existing waste streams.2Direct polymerisation of terpenes has been attempted using cationic polymerisation, with the most successful being the cationic polymerisation of β-pinene.2 The monoterpene that most readily undergoes direct radical polymerisation is myrcene, which was successfully polymerised via reversible addition–fragmentation chain transfer (RAFT) polymerisation up to 9000 g mol−1, though these reactions only reached low conversions after 3 days.3 Conversion was improved by increasing the temperature, but at the cost of increased branching and a broadening of the dispersity.4 RAFT copolymerisations of unmodified β-pinene with n-BuA,5 methyl acrylate6 and acrylonitrile7 have also been reported, as have those of both limonene and β-pinene with various maleimides.8 Isoprene monomer has also been polymerised via free-radical methods to produce oligomers in greater than 40% yield, in this study both RAFT and catalytic chain transfer polymerisation (CCTP) methods were successfully applied.9,10
In many cases, functionalisation of monoterpenes has been found to increase their potential as monomers. Approaches have included the synthesis of ring structures, which can undergo ring-opening polymerisation (ROP),11–14 or the addition of thiol groups to allow for polycondensation.15–17
Several terpene-based (meth)acrylate monomers have been reported, and were shown to undergo facile radical polymerisation.18 Sainz et al. described the addition of (meth)acrylate moieties to a terpene double bond via a two-step reaction, consisting of a hydroboration/oxidation step to produce an alcohol intermediate, followed by an esterification (Scheme 1). An alternative route to the synthesis of terpene-derived acrylates was also developed, involving acrylic acid rather than the toxic acryloyl chloride, improving the overall sustainability of the monomer synthesis. More recently, a variety of options for improving the sustainability of the esterification step, for a range of existing terpenoids, have been highlighted by Droesbeke et al.19 A further study by Castagnet et al. has also reported the sustainable, enzyme-catalysed syntheses of terpene (meth)acrylates, utilising microwave irradiation.20
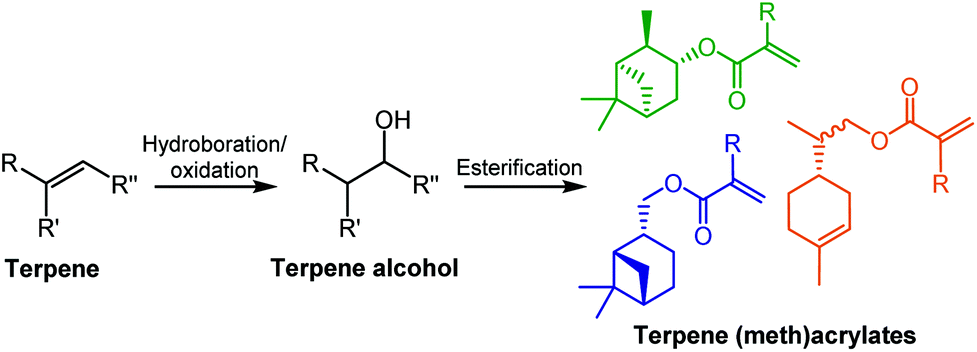 | ||
| Scheme 1 General monomer synthesis reaction scheme and chemical structures of six terpene-based monomers produced by Sainz et al.18 | ||
Sainz et al. also demonstrated that the polymerisation of these terpene (meth)acrylate monomers could be influenced by varying the quantity of a chain transfer agent (CTA), dodecanethiol.18 High conversions and molecular weights ranging from 5000–32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 were observed, depending on the CTA quantity. Each homopolymer exhibited a characteristic glass transition temperature (Tg) ranging from 0–145 °C at the molecular weights obtained.
000 g mol−1 were observed, depending on the CTA quantity. Each homopolymer exhibited a characteristic glass transition temperature (Tg) ranging from 0–145 °C at the molecular weights obtained.
The addition of (meth)acrylate functionality also facilitates the use of reversible deactivation radical polymerisation (RDRP) techniques, such as RAFT, without the need for additional comonomers. The major advantage of using such RDRP techniques is the possibility of forming more complicated polymer architectures, such as (multi)block and star copolymers.21,22
There have been a limited number of examples of the polymerisation of terpene (meth)acrylate homopolymers via RDRP. One approach is single-electron transfer living radical polymerisation (SET-LRP), used to polymerise both α-pinene acrylate23 and menthyl acrylate.24 More recently, RAFT polymerisation of terpene (meth)acrylates has also been investigated: Noppalit et al. reported the RAFT polymerisation of tetrahydrogeraniol acrylate25 and also the nitroxide mediated polymerisation (NMP) of tetrahydrogeraniol methacrylate.26 Additionally, Montanari et al. have produced a carvone-derived, tetraol, acrylate monomer, which can be polymerised via RAFT to produce a hydrophilic polymer.27 This was then used in the synthesis of an amphiphilic block copolymer, incorporating a poly(β-pinene acrylate) hydrophobic block.
In this paper, we have developed the polymerisation of the previously reported, terpene-based monomers to produce more specialised, bio-based materials, with a focus on di- and triblock copolymers. Hard–soft block copolymers were targeted, in order to exploit the wide range of Tgs exhibited by the poly[terpene (meth)acrylates]. Combining high Tg (hard) blocks with low Tg (soft) blocks should lead to a nanoscale, 3D network of hard and soft domains when phase separated. Such materials are desirable for self-healing,28 and shape-memory polymer applications,29 and are often used as additives to improve properties such as viscosity,30 and impact strength.31 ABA triblock copolymers, where A is a hard block and B is a soft block, have found uses as thermoplastic elastomers (TPEs)32 and pressure sensitive adhesives (PSAs),33 depending on their material properties.
Results and discussion
RAFT polymerisation of terpene (meth)acrylates
Initially, RAFT homopolymerisations of each of the six (meth)acrylate ((M)A) monomers were conducted using 2-cyano-2-propyl dodecyl trithiocarbonate (CPDT) as the RAFT agent (Scheme 2), which is known to be compatible with both acrylates and methacrylates.34 Homopolymers with molecular weights of 5000 and 30000 g mol−1 were targeted, to determine whether RAFT can be used to control the polymerisation at both low and high molecular weights.
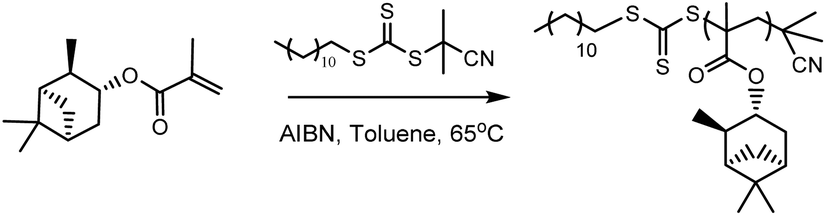 | ||
| Scheme 2 αPMA polymerisation in the presence of CPDT as a RAFT agent. | ||
RAFT polymerisation of the terpene methacrylate monomers resulted in polymers with low molecular weight distributions and Mn values close to those targeted (Table 1). The RAFT polymerisation of both αPMA and βPMA at 65 °C demonstrated good RAFT control, with Đ < 1.3 in all cases. A dispersity of less than 1.3 is indicative of a polymerisation proceeding via a controlled mechanism.35 For PLiMA, the refractive index GPC data showed a narrow peak, indicating good RAFT control. However, data from the light scattering detector showed the presence of a very small high molecular weight peak, (Fig. S1†) which could be due to PLiMA formed via uncontrolled free radical polymerisation, or a high molecular weight branched polymer indicating that the polymerisation was not fully controlled. A potential source of this branching is the cyclic double bond on the pendant group (Scheme 1).
| Entry | Monomer | Target Mn (g mol−1) | Conv.a (%) | M n(th)b (g mol−1) |
M
nc (g mol−1) |
M
wc (g mol−1) |
Đ |
|---|---|---|---|---|---|---|---|
| a Calculated by comparing integrals of monomer and polymer peaks in 1H NMR. b Calculated from conversion and target Mn. c Measured by GPC. Each reaction was carried out over 24 h and used CPDT as the RAFT agent (except entry 13 which used DDMAT). | |||||||
| 1 | αPMA | 5000 | 97 | 4850 | 6100 | 8000 | 1.28 |
| 2 | αPMA | 30000 |
81 | 24300 |
18800 |
21900 |
1.17 |
| 3 | βPMA | 5000 | 99 | 4950 | 6500 | 8000 | 1.22 |
| 4 | βPMA | 30000 |
93 | 27900 |
28600 |
30700 |
1.05 |
| 5 | LiMA | 5000 | 99 | 5000 | 5700 | 7200 | 1.14 |
| 6 | LiMA | 30000 |
97 | 29100 |
20700 |
26700 |
1.29 |
| 7 | αPA | 5000 | 85 | 4250 | 4300 | 4700 | 1.10 |
| 8 | αPA | 30000 |
99 | 29700 |
28200 |
30400 |
1.08 |
| 9 | βPA | 5000 | 91 | 4600 | 12400 |
20100 |
1.62 |
| 10 | βPA | 30000 |
82 | 24600 |
30100 |
40800 |
1.35 |
| 11 | LiA | 5000 | 90 | 4500 | 13800 |
38200 |
2.77 |
| 12 | LiA | 30000 |
78 | 23400 |
39500 |
100000 |
2.53 |
| 13 | LiA (DDMAT) | 30000 |
74 | 22200 |
58200 |
64700 |
3.20 |
The RAFT polymerisation of αPA (at Mn = 5000 and 30000 g mol−1) with CPDT exhibited excellent control, with low dispersities (<1.1) and the targeted Mns being achieved. βPA polymerisation exhibited acceptable RAFT control when targeting 30000 g mol−1, with Mn and Đ values slightly higher than desirable. But when targeting 5000 g mol−1, the PβPA product was found to have significantly increased Mn and Đ values.
Finally, the RAFT homopolymerisation of LiA gave less promising results, with a dispersities above 2.5 at both high and low molecular weights, indicating that some branching or minor crosslinking may have occurred. As CPDT is known to be a more appropriate RAFT agent for methacrylates than acrylates,34 a LiA polymerisation reaction was also carried out using the RAFT agent 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid (DDMAT), which has a less stable leaving group relative to CPDT and is therefore more suited to acrylate polymerisation. Surprisingly, we observed even poorer chain length control (Mn = 60600 g mol−1), a higher dispersity (Đ = 3.20) and a maximum conversion of 74% was reached, even when the reaction was allowed to continue for 74 hours. The lack of control over the LiA polymerisation may also be due to branching, as described earlier.
Terpene-derived hard–soft diblock copolymers
The major benefit of using RDRP techniques such as RAFT, is that they can provide a route to a wide variety of polymer architectures and functionalities. An investigation was therefore carried out to determine if the terpene monomers could be chain extended from a previously synthesised macro-RAFT agent, using the well-understood poly(methyl methacrylate) PMMA as the first block. PMMA macro-RAFT agents (ca. 15000 g mol−1, Đ < 1.2) were reinitiated in the presence of each of the six terpene-based monomers, (Scheme 3) with target Mn values of 15000 g mol−1 for the second block. All three methacrylate monomers were shown to have grown in a controlled manner, to produce diblock copolymers with molecular weights close to the targeted values and Đ < 1.3 (Table S1 and Fig. S2†). Additionally, it was found that αPA also produced a reasonably well-controlled diblock copolymer with Đ = 1.31. βPA exhibited an increased Đ = 1.48, caused by the low molecular weight shoulder peak which indicated the presence of some dead chains.
 | ||
| Scheme 3 General synthesis of a polymethacrylate-b-polyacrylate diblock copolymer using the RAFT agent CPDT. | ||
In the case of LiA, the use of a PMMA-macro RAFT agent led to a remarkable improvement in the RAFT control of the polymerisation (Fig. S2†). Chain extension of PMMA with LiA was well-controlled, resulting in a diblock copolymer with Đ = 1.28, though the conversion remained low at only 65%. The successful diblock copolymer syntheses confirmed that each of the terpene (meth)acrylate monomers has the potential to be used in the synthesis of block copolymers.
However, commercial MMA production is currently not considered to be sustainable,36 therefore the synthesis of fully renewable diblock copolymers is preferred. RAFT polymerisation has therefore been used to synthesise renewable, terpene-derived block copolymer architectures, with a focus on combining hard and soft blocks. The very wide range of Tgs observed for these poly[terpene (meth)acrylates] allows for the synthesis of fully terpene-derived, hard–soft diblock copolymers. The Tgs for each of the homopolymers measured by DMA are highlighted (Table 2).
| Polymer | Structure | T g (°C) | Polymer | Structure | T g (°C) |
|---|---|---|---|---|---|
| PαPMA |
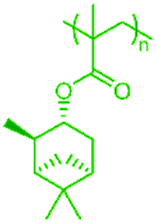
|
168 | PβPMA |
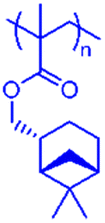
|
121 |
| PαPA |
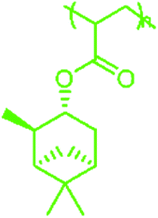
|
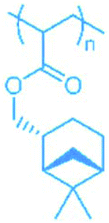
84 | PLiMA |
|
51 |
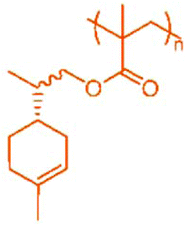
| PβPA |
|
41 | PLiA |
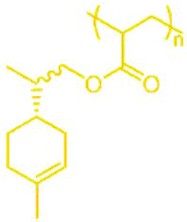
|
−3 |
Some of these Tg values are slightly higher than those reported by Sainz et al.18 This is because some of the polymers had higher molecular weights and additionally, the materials were analysed using dynamic mechanical analysis (DMA), which is known to slightly increase Tg compared to DSC.37 PαPMA and PβPMA are clear choices for a high Tg hard block with values of 168 and 121 °C respectively, while PLiA has the lowest Tg at −3 °C, so could be utilised as a soft block. PβPA exhibited a Tg of 41 °C at 27000 g mol−1, but it was reported in further work by Sainz,38 that for a lower Mn of 4200 g mol−1, the Tg was decreased to −8 °C,38 opening up the use of short lengths of PβPA as a soft block. The Tg of PLiMA was not previously reported but was found to be 51 °C at Mn = 29000 g mol−1, measured by DMA. PαPA and PLiMA were not considered for use in hard–soft block copolymers, as their Tgs are in the intermediate range, making them unsuitable as either hard or soft blocks. Thus, from our range of terpene-derived monomers, we have the potential for methacrylate hard blocks and acrylate soft blocks. The synthesis of methacrylate–acrylate block copolymers via RAFT polymerisation requires the synthesis of the methacrylate block first, followed by chain extension with the acrylate, and this is driven by the differing reactivities of the two monomers.39
To create these block copolymers, macro-RAFT agents of PαPMA and PβPMA were required. The previous PαPMA and PβPMA homopolymers were obtained after 24 h of polymerisation at 65 °C, when the reaction had reached full conversion. However, in order to retain chain-end fidelity and thus ensure that the polymer can be reinitiated and chain extended. It is preferable to stop the polymerisation at well below full conversion, before any significant chain termination has occurred.40 Some methods of RAFT polymerisation have been optimised to chain extend effectively up to 100% conversion,41,42 but for solution polymerisation of these previously untested monomers, it was found that taking 60% conversion as an upper limit was the most reliable method of ensuring chain end fidelity.
The βPMA polymerisation targeting an Mn of 30000 g mol−1 at 65 °C consistently reached 60% conversion, equating to 18000 g mol−1 in 4–6 hours, with low dispersities. For the equivalent αPMA polymerisation, reproducibility was an issue; initially, the αPMA polymerisation would reach 60% between 5–8 h, however some polymerisations would take much longer, exhibiting an extended initiation period of several hours, where no polymer was formed. It was found that increasing the reaction temperature to 75 °C eliminated this issue, and at this temperature, well-defined PαPMA homopolymers were reliably produced in 2.5–3 hours.
Initially, PαPMA macro-RAFT agents were chain extended with βPA and LiA, (Table 3). Chain extension of PαPMA with βPA produced a polymer with a Đ < 1.3 and the desired molecular weight; GPC data (Fig. 2a) show that the peak is slightly broadened relative to the PαPMA peak, suggesting some minor loss of control over the block copolymer synthesis.
| Code | Macro-RAFT (B1)a | M n (B1)b (g mol−1) | Monomer (B2) | Target Mn B2 (g mol−1) | Conv.c (%) | M n (th)d (g mol−1) |
M
nb (g mol−1) |
M
wb (g mol−1) |
Đ |
|---|---|---|---|---|---|---|---|---|---|
| a PαPMA and PβPMA macro-RAFT agent syntheses are detailed in Table S3.† b Measured by GPC. c Calculated by comparing integrals of monomer and polymer peaks in 1H NMR. d Calculated from conversion and target Mn. Each chain extension reaction was stopped after 24 h. More details on the macro-RAFT agent syntheses can be found in Table S3.† | |||||||||
| PαPMA-b-PβPA | PαPMA1 | 19000 |
βPA | 19000 |
77 | 32000 |
29500 |
38100 |
1.29 |
| PαPMA-b-PLiA1 | PαPMA2 | 18500 |
LiA | 18500 |
55 | 28700 |
33800 |
43000 |
1.27 |
| PαPMA-b-PLiA2 | PαPMA2 | 18500 |
LiA | 29200 |
63 | 36900 |
36700 |
59000 |
1.61 |
| PβPMA-b-PβPA | PβPMA1 | 22100 |
βPA | 16000 |
88 | 36200 |
38300 |
53900 |
1.41 |
| PβPMA-b-PLiA1 | PβPMA2 | 24100 |
LiA | 24100 |
54 | 37000 |
34400 |
43200 |
1.25 |
| PβPMA-b-PLiA2 | PβPMA2 | 24100 |
LiA | 42100 |
69 | 52100 |
56600 |
109800 |
1.94 |
LiA chain extension of ca. 10000 g mol−1 resulted in a well-defined diblock copolymer (PαPMA-b-PLiA1, Fig. 1b and Table 3). A longer LiA chain extension of 18000 g mol−1 showed increased dispersity, indicating chain growth was progressing a less controlled manner (PαPMA-b-PLiA2, Table 3), but Đ = 1.61 is still much lower than that obtained for LiA homopolymerisation.
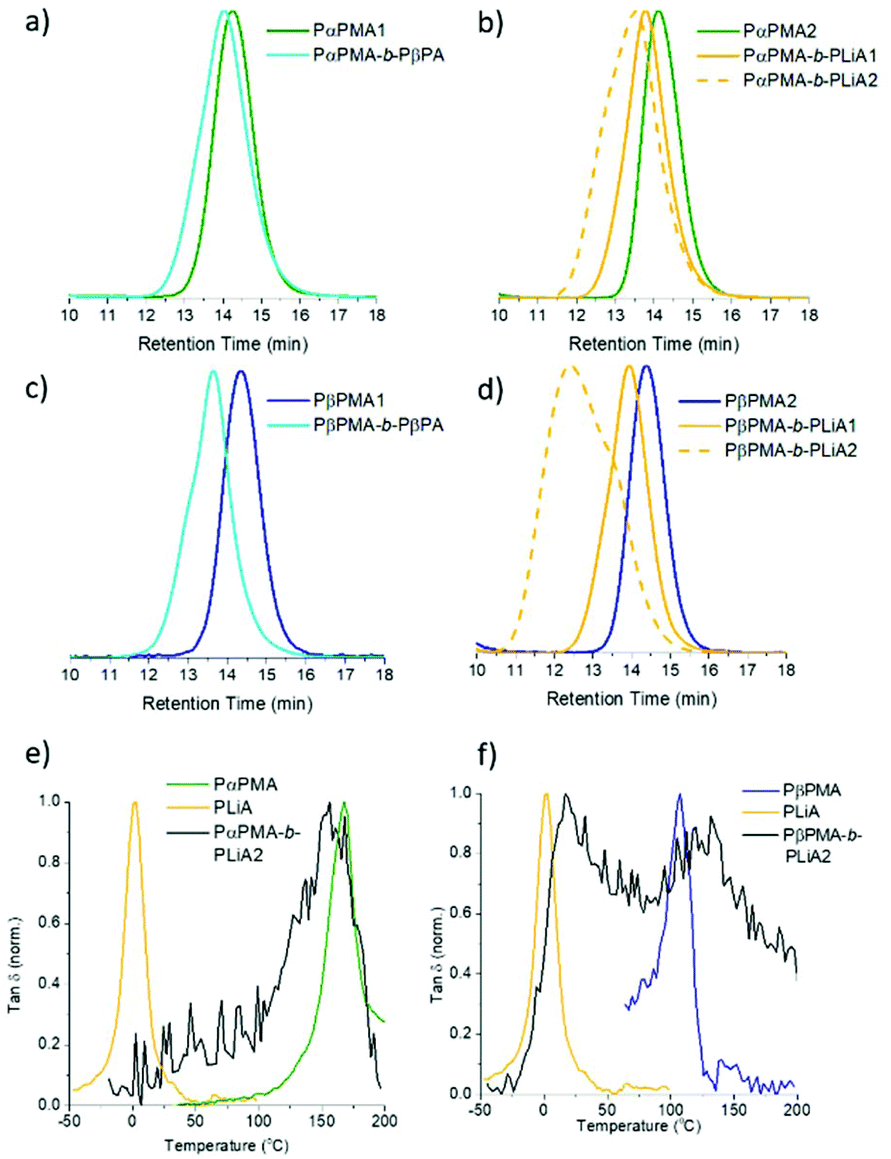 | ||
| Fig. 1 (a–d) GPC traces showing controlled chain extension of PαPMA with (a) βPA and (b) LiA, and of PβPMA with (c) βPA and (d) LiA. (e and f) DMA traces for (d) PαPMA-b-PLiA2 and (e) PβPMA-b-PLiA2, overlaid with the DMA traces of the relevant homopolymers (synthesised to full conversion). | ||
Subsequently, chain extension of the PβPMA macro-RAFT agent was investigated. The Tg of βPMA at 121 °C is lower than that of αPMA at 168 °C, and could offer lower processing temperatures which may be preferred for an application where a very high Tg is not required. Note that this value is still higher than the Tg of petroleum-derived polystyrene at 100 °C, a hugely popular polymer used in TPE synthesis. Polystyrene-based triblock copolymers such as polystyrene-block-polybutadiene-block-polystyrene (SBS), have dominated the TPE market for over 50 years.43
LiA and βPA were also chain extended from a PβPMA macro-RAFT agent. GPC results suggest that the chain extension of PβPMA with βPA was less well-controlled than for PαPMA. The dispersity was increased slightly to Đ = 1.41, though the targeted Mn was achieved. This can also be observed as a high molecular weight shoulder peak in the GPC trace (Fig. 1c). However, chain extension of PβPMA with LiA to ca. 13000 g mol−1 proceeded in a similarly well-controlled fashion as for PαPMA, with Đ = 1.25 (PβPMA-b-PLiA1, Table 3). Again, a longer chain extension with LiA (28000 g mol−1) did not occur in a well-controlled manner and only broad dispersity was achieved (PβPMA-b-PLiA2, Table 3).
DMA results were obtained for each of the diblock copolymers; for those containing a PβPA soft block only one peak was observed, indicating a lack of phase separation. This miscibility is likely due to the similarity in the pendant groups of the hard and soft blocks. However, some phase separation was observed for PαPMA-b-PLiA2 and PβPMA-b-PLiA2 (Fig. 1e and f). In the case of PβPMA-b-PLiA2, two peaks can clearly be seen in the DMA trace, relating to the Tgs of the hard and soft blocks. The DMA trace for PαPMA-b-PLiA shows a peak that clearly corresponds to the PαPMA hard block. Unusually, a low Tg peak is not seen, though there is a considerable amount of noise in that region. According to the Fox equation:44
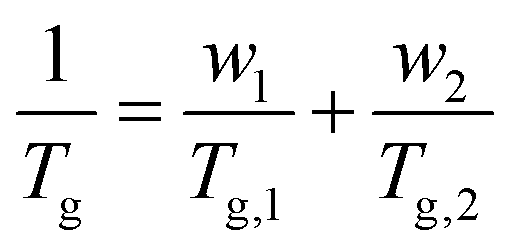 | (1) |
Overall, we have screened these monomers, which had not previously been polymerised via RAFT, to show that well defined polymers can be prepared for the majority of the terpene-based monomers. However, the lack of control over the PLiA homopolymerisation and higher molecular weight chain extensions limit its usefulness. Further work to improve the RAFT control of LiA is required if this is to be an easily accessible soft block.
ABA triblock copolymers
ABA triblock copolymers are a particularly useful form of hard–soft block copolymer, as they have the potential to exhibit elastomeric properties45 and could act as thermoplastic elastomers (TPEs). TPEs with an ABA triblock copolymer structure, incorporating hard methacrylate and soft acrylate blocks, are only effective in the form polymethacrylate-block-polyacrylate-block-polymethacrylate (i.e. hard–soft–hard), as the soft, central block must bridge two hard domains. This means that it is necessary to use a Z-connected, difunctional RAFT agent, which will synthesise the outer, polymethacrylate blocks first; a method known as a convergent chain growth.39 Unfortunately, there are not currently any commercially available RAFT agents of this type that exhibit good RAFT control over methacrylate polymerisation. S,S-Dibenzyl trithiocarbonate (DBTTC), the most commonly used difunctional RAFT agent, is not suitable for use with methacrylates, due to insufficient radical stabilisation of the leaving groups.46The difunctional RAFT-agent S,S’-bis(α,α′-dimethylacetic acid)trithiocarbonate (BDAT) is a Z-connected RAFT agent that is typically more suited to acrylate polymerisation than methacrylate, but it has been shown to provide some control over MMA polymerisation under certain conditions.47 The reported PMMA was synthesised with higher than expected molecular weights, due to an initial uncontrolled period, but after around 10 minutes the polymerisation became controlled and a linear proportionality for Mn as a function of conversion was observed. We have investigated this approach using the terpene methacrylate monomers, speculating that BDAT may provide sufficient control over the αPMA polymerisation to permit the synthesis of triblock copolymers. Initial studies focused on using PαPMA as the hard block (Scheme 4).
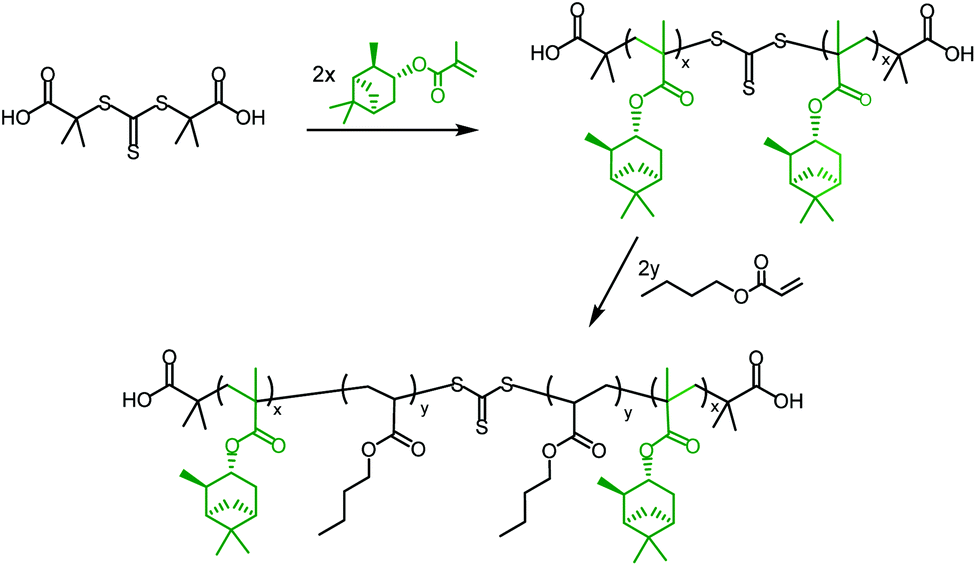 | ||
| Scheme 4 General synthesis of a PαPMA-b-PBuA-b-PαPMA triblock copolymer in two steps, using the RAFT agent BDAT. | ||
BDAT was used to synthesise difunctional macro-RAFT agents of PαPMA. Initial attempts resulted in Đ = 1.5–1.8, suggesting that these polymerisations were not fully RAFT-controlled. The molecular weight was also higher than expected in all cases, often reaching around double the predicted molecular weight, and the relationship to conversion was not consistent (Table S4†).
To assess the level of RAFT chain end fidelity in the PαPMA produced in this way, a chain extension reaction was carried out using PBuA, a very commonly used soft block for acrylic TPEs48–50 with Tg of ca. −50 °C. PαPMA was reinitiated in the presence of BuA and the polymerisation reached 95% conversion by 1H NMR. Somewhat surprisingly, GPC results showed that the chain extension occurred in a reasonably well-controlled manner, to produce a PαPMA-b-PBuA-b-PαPMA triblock copolymer, 74 kg mol−1, 23 wt% hard block (denoted as αBα-74-23). The polymer exhibited a minor low molecular weight shoulder peak, but no significant increase in dispersity was observed (Fig. 2a) suggesting reasonably good RAFT chain end fidelity of the PαPMA macro-RAFT agent.
 | ||
| Fig. 2 GPC traces for the chain extension of (a) PαPMA with BuA, (b) PβPMA with BuA, (c) PαPMA with LiA and (d) PβPMA with LiA. Details for the syntheses of each of the macro-RAFT agents can be found in Table S6.† | ||
Despite the apparent control issues during the macro-RAFT agent synthesis, it seems very possible that polymethacrylate-block-polyacrylate-block-polymethacrylate triblock copolymers can be produced successfully via this convergent RAFT method; the slight loss in molecular weight control in the first step being outweighed by the utility of this new approach. The good RAFT control and high conversion observed in the second step means that it is still possible to target specific molar ratios of blocks and should allow access to specific phase separated morphologies.
As discussed earlier, βPMA may also prove viable as a hard block, with a Tg of 121 °C. This lower value could allow significantly lower processing temperatures and may therefore be preferable for applications where extreme temperatures will not be required. A number of βPMA homopolymerisations were carried out using BDAT as the RAFT agent. As was seen for the equivalent αPMA polymerisations, higher than expected molecular weights were observed, as were dispersities above 1.4 (Table S5†)
Two PβPMA macro-RAFT agents were chain extended with PBuA to produce PβPMA-b-PBuA-b-PβPMA triblock copolymers: βBβ-67-25 (Fig. 2b and Table 4) and βBβ-38-23 (Fig. S3 and Table S7†). The targeted PBuA molecular weights for these block copolymers were 50000 and 30000 g mol−1 respectively, and both reactions reached high conversions. The GPC traces for each polymer clearly show that chain extension has occurred, but a low molecular weight shoulder peak can be observed in both cases, indicating the presence of some dead chains and causing a slight increase in the dispersity.
| Polymer | M n(block 1)/Đa (kg mol−1) | M n(triblock)/Đa (kg mol−1) | wt% (block 1) (NMR)b |
|---|---|---|---|
| a Measured by GPC. b Calculated from 1H NMR. c Taken from wt% reagents added as >96% conv. d Complete overlap of 1H NMR polymer peaks – GPC column calibration used as wt% could not be obtained. More detailed results for these polymerisations can be found in Tables S6 and S7.† | |||
| αBα-74-23 | 17/1.58 | 74/1.31 | 23 |
| βBβ-67-25 | 17/1.52 | 55/1.38 | 25c |
| αLα | 16/1.56 | 46/2.01 | 45 |
| βLβ | 9/1.47 | 21/3.21d | |
The limonene acrylate polymer has the lowest Tg of all the terpene acrylates, at −3 °C. PLiA was therefore most promising as a soft block for the synthesis of a fully terpene-derived triblock copolymer. However, LiA homopolymerisation was not well controlled via RAFT, but chain extension from a polymethacrylate macro-RAFT agent gave significantly improved results. To take advantage of this, macro-RAFT agents of PαPMA and PβPMA were chain extended with LiA, leading to hard–soft–hard, renewably sourced triblock copolymers. Although the LiA polymerisation step did not reach full conversion, stopping at 60–70% for both chain extensions, it was clear that the peak had shifted to lower retention times. In the case of αLα, the peak is broadened (Đ = 2.01, Table 4) compared to the macro-RAFT agent, but is still symmetrical. This suggests reasonably good chain end fidelity, as no significant low molecular weight tailing is observed (Fig. 2c), but a lack of control and potentially some branching occurs during the chain extension step. For βLβ, the peak has again shifted to lower retention times, but with significant low molecular weight tailing (Fig. 2d). This indicates a loss of both control over the LiA chain extension and RAFT chain end fidelity in the macro-RAFT agent which might introduce some homopolymer in the material and this could be detrimental to its elastic recovery. However, additional block A homopolymer (PβPMA) may be less of a concern than additional block B homopolymer (PLiA), since block B is the bridging chain between hard domains.
The chain extension with BuA has proved the most successful. While some LiA chain extension has occurred, the low conversions and high dispersities mean that PLiA needs further optimisation to be investigated fully for use in a TPE. However, we have shown that it is possible to synthesise polymethacrylate-block-polyacrylate-block-polymethacrylate triblock copolymers via a convergent RAFT polymerisation method, with some sacrifice of the dispersity and molecular weight control of the first methacrylate polymerisation. For the remainder of this work, we focus on the behaviour and potential applications of the PαPMA-b-PBuA-b-PαPMA (αBα) materials.
Morphological characterisation of PαPMA-b-BuA-b-PαPMA (αBα)
Four αBα triblock copolymers were synthesised, each with varying wt% of hard block (Table 5). No significant dispersity increase was observed for each BuA chain extension, though again, some minor low molecular weight shoulder peaks can be seen (Fig. S4†). For a series of polymers having the same hard and soft blocks we would expect an increase in the ratio of hard polymer to soft polymer to have a significant effect on the tensile properties of the material.51| Polymer | M n(PαPMA)/Đa (kg mol−1) | M n(triblock)/Đa (kg mol−1) | wt% (PαPMA) (1H NMR)b | D | Morphologyc |
|---|---|---|---|---|---|
| a Measured by GPC. b Calculated from 1H NMR data. c Measured by AFM. Additional data for these polymerisations can be found in Table S7, ESI.† | |||||
| αBα-74-23 | 17/1.58 | 74/1.31 | 23 | 33 | Sphere/cylinder |
| αBα-64-28 | 18/1.60 | 64/1.35 | 28 | 41 | LAM |
| αBα-60-33 | 18/1.60 | 60/1.33 | 33 | 38 | LAM/inv. cylinder |
| αBα-91-42 | 42/1.66 | 91/1.63 | 42 | NA | NA |
AFM studies have been performed on each αBα triblock copolymer to explore the phase-separated morphologies (Fig. 3). αBα-74-23 displayed a morphology consisting of small, hard domains with the soft block as the matrix phase. Such a morphology is indicative of being within the spherical or cylindrical regions of the thermodynamic phase diagram. Such morphologies are desirable for TPEs, as the small domains act as physical crosslinks, while still allowing the soft PBuA matrix to be reasonably mobile.45 A dominant length scale of 33 nm was determined for αBα-74-23, by taking the Fast Fourier Transform (FFT) of the AFM image (Fig. 3). Other samples, αBα-64-28 and αBα-60-33, also showed well-defined, phase separated morphologies with dominant length scales of 41 and 38 nm, respectively. αBα-64-28 appears to have adopted a lamellar type morphology, which may be attributed to either lamellae perpendicularly orientated to the substrate surface or cylinders orientated parallel to the substrate surface. αBα-60-33 showed a similar morphology, but the increased hard polymer content is apparent in the AFM image. Some areas appear to show an inverse cylindrical-type morphology, where soft domains are dispersed in a hard polymer matrix; the opposite of the desired morphology. Finally, αBα-91-42 showed the least well-defined morphology of the four αBα triblock copolymers, and appears to be fully comprised of large soft domains within a hard matrix.
 | ||
| Fig. 3 AFM images for each of the synthesised αBα triblock copolymers (height images), with FFTs shown as an insert. 1 × 1 μm area shown. Lighter areas correspond to the PαPMA domains and darker regions correspond to the PBuA domains. | ||
AFM analysis of the phase separated morphologies of the four αBα triblock copolymers are highly surface sensitive, due to both the nature of AFM measurements and the intricacies of thin-film processing in determining morphological evolution towards thermodynamic equilibrium. Complementary small-angle X-ray scattering (SAXS) experiments were performed on bulk, solvent cast samples to provide further insights into the nanoscale behaviour of the αBα triblock copolymers (Fig. S5†). The SAXS patterns clearly indicate microphase separation in each of the triblock copolymers, but there is insufficient order to be conclusive about the morphology assignment. Tentatively, the αBα-74-23 triblock copolymer appears to show lamellar morphology (a second peak is observed at 2q*, where q* is the primary scattered peak). However, this assignment does not match with that expected for a block copolymer containing 23 wt% of the minor component, where a spherical or cylindrical morphology would be expected (as shown in the AFM), and we cannot rule out the presence of additional peaks (e.g. at √2 and √3 for bcc spheres or √3 for hexagonally packed cylinders) between the relatively broad peaks that are clearly observed. We can conclude that both AFM and SAXS demonstrate that the αBα-[74-23, 64-28 and 91-42] triblock copolymers form phase separated nanomorphologies. Clearly, contributions of both sample processing and the specificity of the morphological technique employed (e.g. surface vs. bulk) indicate that these polymers exhibit complex phase behaviour and these will be the subject of future detailed investigations.
Tensile and adhesive properties of an α-pinene-based pressure sensitive adhesive (PSA)
The previous AFM measurements have demonstrated that αBα-74-23 is capable of forming a desirable morphology for use as a TPE: discrete hard domains dispersed within a soft matrix, so this material was taken forward for mechanical characterisations. Tensile measurements were carried out with engineering stress (σ) and strain (ε) being reported for ease of comparison with tensile data from other sources (Table 6). The averaged tensile data (Fig. 4 – circles) shows up to ε = 300% for 8 specimens of αBα-74-23, with an average thickness of 0.623 ± 0.028 mm, where ± represents one standard error. Excellent repeatability between specimens was found up to ∼100% strain, above which some deviation occurs. An average ultimate tensile strain (εUTS), defined as the strain at the ultimate tensile stress (σUTS), exceeding 500% was measured for αBα-74-23 (Table 6) at a σUTS of 722 kPa. This behaviour is typical of polyacrylate materials which generally exhibit a higher chain entanglement length than some other typically used soft blocks (such as polybutadiene or polyisoprene), which in turn reduces the density of entanglements in the soft matrix.52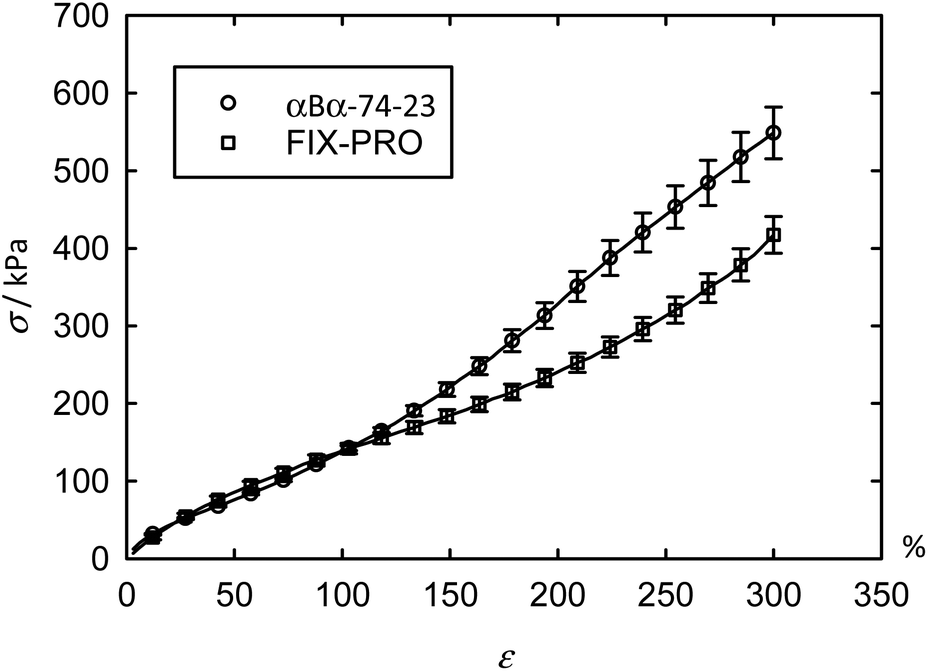 | ||
| Fig. 4 Average tensile performance for αBα-74-23 and FIX-PRO® are given as squares and circles respectively, showing similar tensile performance up to 120% strain. Error bars denoting ±one standard error at symbols are given for clarity, with lines through data representing the full curve in each case. | ||
| Material | σ UTS (kPa) | ε UTS (%) | E s at 1% (kPa) | E s at 10% (kPa) | E s at 100% (kPa) | E s at 300% (kPa) | G′ at 1 Hz (kPa) |
|---|---|---|---|---|---|---|---|
| αBα-74-23 | 722.0 ± 15.0 | 504.1 ± 38.4 | 454.1 ± 43.2 | 282.9 ± 6.4 | 138.2 ± 3.0 | 182.9 ± 11.1 | 906.6 |
| FIX-PRO® | 1762.5 ± 165.1 | 463.2 ± 4.4 | 230.3 ± 46.2 | 230.4 ± 27.7 | 138.7 ± 6.5 | 139.1 ± 7.9 | 158.4 |
The films of αBα-74-23 used in the tensile measurements were observed to adhere well to many substrates, and the tack was observed to be lasting. This is not surprising as this type of soft, acrylic TPE material is well-suited for use as a PSA, and can offer improvement over some of the more common types. It is known that soft, acrylic polymers behave well as PSAs,56–58 however these typically require a cross-linked chemical structure to prevent creep, making them difficult to reprocess and recycle. A number of materials of this type have been synthesised from terpene (meth)acrylate monomers. Examples of this include tetrahydrogeraniol acrylate-based polymers from Baek et al., one combined with menthyl acrylate,59 and a separate combined with isobornyl methacrylate.60 A recent paper by Droesbeke et al. has utilised tetrahydrogeraniol, citronellol, menthol and isoborneol (meth)acrylate monomers, in the emulsion polymerisation of waterborne PSAs.61
TPE triblock copolymers such as SBS offer a recyclable alternative, however, for these materials to be useful as PSAs, they require the addition of a tackifier to induce sufficient wettability to a substrate and improve adhesive performance.62
Alternatively, the polyterpene acrylic TPE we have developed can be effectively melted or dissolved and reprocessed, and the low modulus and high wettability of the acrylic polymers eliminate the need for any additional tackifier. Furthermore, polyterpene resins are desirable for use in adhesives due to their intrinsic tackiness and have found application as tack modifiers in hot melt and pressure sensitive adhesives for this reason.63
To assess the potential application as a PSA, the mechanical properties of the novel material were compared to those of a commercial PSA, FIX-PRO®. 7 specimens of FIX-PRO® with an average thickness of 0.923 ± 0.038 mm were tested in the same way for comparison, showing comparable performance to the terpene material below 120% strain (Fig. 4 – squares). Tensile data for the individual specimens can be found in Fig. S6 and S7,† for αBα-74-23 and FIX-PRO® respectively. On average, αBα-74-23 exhibited a lower σUTS than the commercial elastomer by a factor of 2.44, but showed a slightly greater εUTS, by a factor of 1.09. Owing to the non-linear nature of the tensile data for these elastomeric materials, a secant modulus (Es) has been determined at 1%, 10%, 100% and 300% strain for the purpose of direct comparison between the stiffnesses of αBα-74-23 and FIX-PRO®, as provided in Table 6. Es at 10% and 100% strains were found to be very similar in magnitude. At large strains, where ε = 300%, a difference in stiffness of 23.9% was observed, with the αBα-74-23 being stiffer than the FIX-PRO®. Based on these results, it can be concluded that under the degree of tensile deformations typically experienced during removal from a backing film and application to a surface, where strains are likely to be small; of the order of <100%, the terpene-based elastomer behaves sufficiently similarly to the benchmark commercial material to infer suitability to this target application.
At large strains, αBα-74-23 exhibits a small degree of strain hardening, whereas the stiffness of FIX-PRO® remains approximately constant. The behaviour of both elastomers is in line with similar amorphous polymer systems.53,54 In order to better understand the large strain regime, a simple neo-Hookean model with finite extensibility, here implemented as a Gent model,55 was fitted to the data in parallel with a constant flow stress.
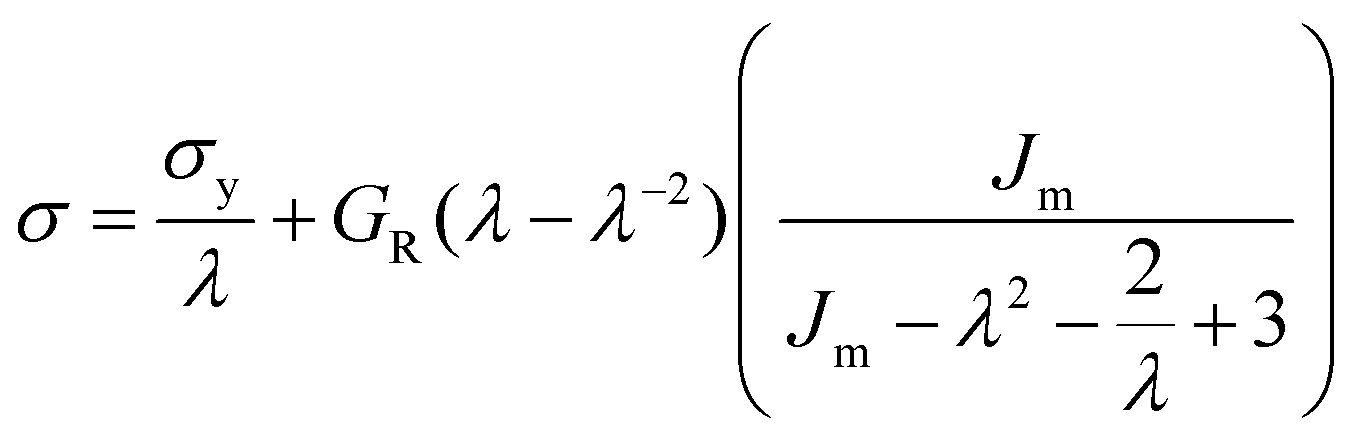 | (2) |
| Material | G R (kPa) | σ y (kPa) | J m | λ max | R 2 |
|---|---|---|---|---|---|
| αBα-74-23 | 68.3 ± 1.4 | 6.4 ± 1.0 | 16.9 ± 1.2 | 4.4 ± 0.1 | 0.9979 ± 0.0006 |
| FIX-PRO® | 66.8 ± 3.2 | 13.6 ± 1.8 | 35.7 ± 0.8 | 6.2 ± 0.1 | 0.9987 ± 0.0002 |
Probe tack adhesion testing was carried out for 3 specimens of αBα-74-23 and 5 specimens of FIX-PRO®, the results of which are presented in Table 6. Fig. 5 shows measurements of force during probe separation as a function of the displacement normalised by thickness at 0 N loading for αBα-74-23 and FIX-PRO®. A peak adhesive force of 4.33 ± 0.21 N was observed for αBα-74-23, which is comparable to that observed for the commercial material, with an average peak force of 4.49 ± 0.13 N. The actual displacement at peak force was found to be lower for αBα-74-23 than for FIX-PRO®, but when normalised by the specimen thickness, both materials exhibit similar adhesion behaviour. Representative force-displacement results for single specimens of αBα-74-23 and FIX-PRO® are also provided to full detachment in Fig. 6, showing complete failure of the adhesive bond. The work of adhesion was determined via numerical integration as W = 69.3 and 39.5 kJ m−3 respectively for αBα-74-23 and FIX-PRO®. This indicates that almost double the energy is required to cause failure of the adhesive bond for the novel elastomer compared with the commercial material.
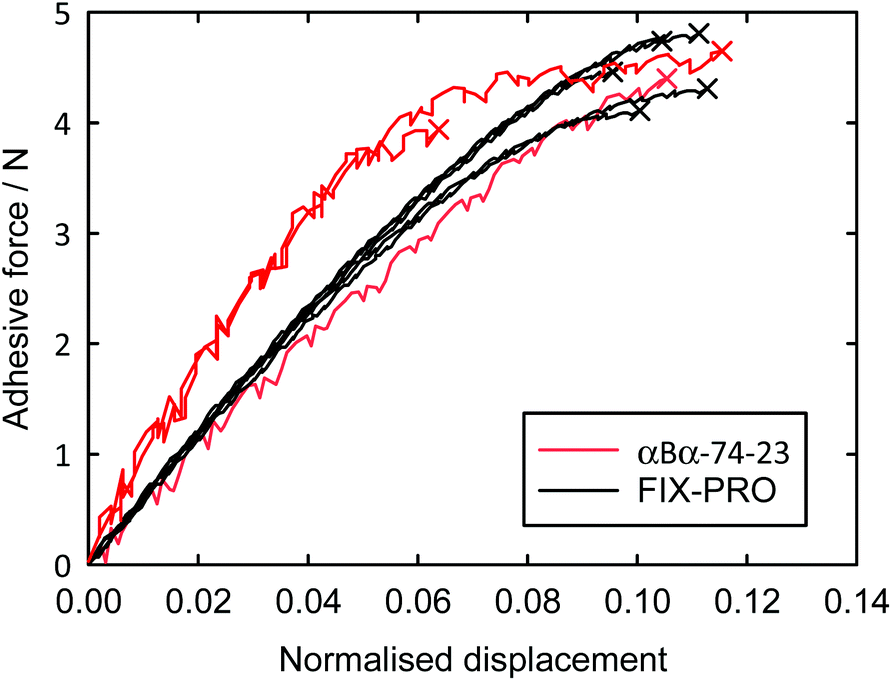 | ||
| Fig. 5 Adhesive probe tack force as a function of displacement normalised by specimen thickness, for 3 specimens of αBα-74-23 and for 5 specimens of FIX-PRO®. Crosses indicate failure properties for both materials. | ||
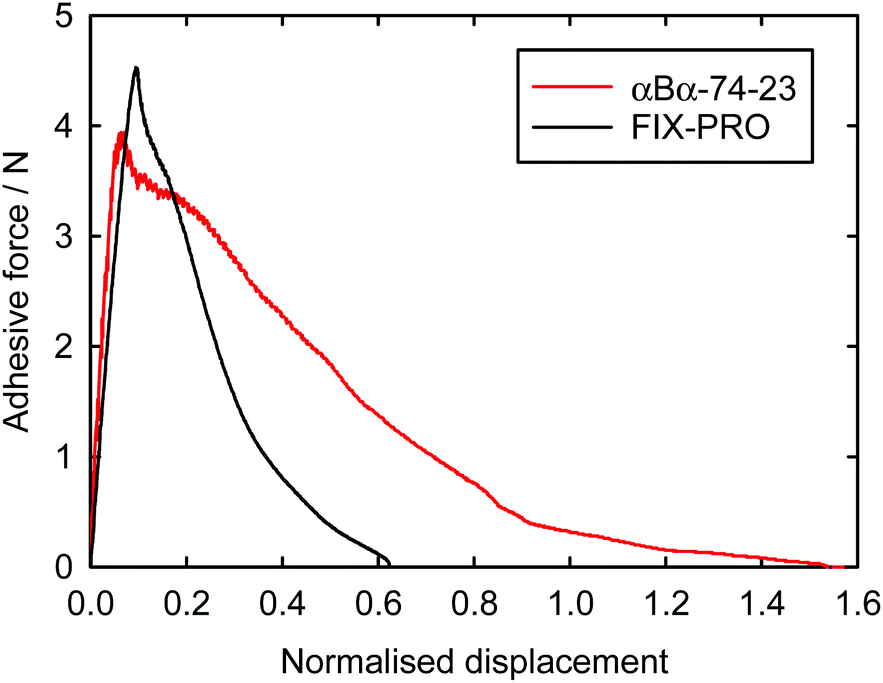 | ||
| Fig. 6 Representative probe adhesion results for single specimens of αBα-74-23 and FIX-PRO® to full detachment. W = 69.3 kJ m−3 for αBα-74-23, and 39.5 kJ m−3 for FIX-PRO® were determined from the areas under the curves. | ||
Dynamic shear measurements performed at 25 °C on both materials showed no presence of an intersection in the elastic and viscous shear moduli within a frequency range of 0.01–10 Hz, indicating that structural relaxation does not occur at typical adhesive application timescales when operating under ambient conditions. The elastic shear modulus (G′) was found to be greater for αBα-74-23 than for FIX-PRO® by a factor of 5.72 at an oscillation frequency of 1 Hz (Table 6), showing some difference in the small-strain behaviour as indicated by the tensile measurements.
Whilst further investigation into the influence of adhesive dwell time, application force and substrate conditions are required to verify specific application suitability, the findings based on these preliminary assessments are promising. The similarity of both tensile and adhesive properties between the terpene-derived material and a commercial PSA highlight the potential for these novel copolymers as renewable alternatives to existing oil-based adhesives.
Conclusions
A range of homopolymers and block copolymers of terpene-derived (meth)acrylates have been synthesised via RAFT polymerisation. The pinene methacrylates were shown to polymerise well and act as macro-RAFT agents, while LiMA and the terpene acrylates were polymerised with varying degrees of RAFT control. Chain extension of these macro-RAFT agents with PβPA and PLiA (extensions of 10–18000 g mol−1) were successful, with targeted molecular weights reached and reasonably low dispersities observed. Some loss of control was observed for longer PLiA chain extensions, and further work is required to establish full RAFT control. The difunctional, Z-connected RAFT agent, BDAT, was then employed to synthesise ABA triblock copolymers with the potential to act as TPEs and PSAs. This method was successfully used to synthesise PαPMA-b-PBuA-b-PαPMA triblock copolymers with reasonable dispersities in two steps. We have also begun to extend this method to include the additional monomers βPMA and LiA, creating partially and fully renewable, terpene (meth)acrylate polymers, with varying levels of RAFT control achieved.
The morphologies of a number of PαPMA-b-PBuA-b-PαPMA triblock copolymers, at varying hard:soft polymer ratios, have been investigated. The material with the most promising structure was taken forward for further measurements, and was found to demonstrate good tensile properties and adhesive behaviour, comparable to a commercial PSA.
Experimental
Materials
Monomers αPMA and αPA were obtained from Cornelius Specialities and the inhibitor was removed from both via an alumina column prior to use. β-Pinene and limonene-based monomers were synthesised following a literature method.18 For monomers methyl methacrylate (MMA, 99% ProSciTech) butyl acrylate (BuA, 99% BASF), the inhibitor was removed via an alumina column prior to use. The RAFT agent, S,S′-bis(α,α′-dimethyl-α′′-acetic acid)-trithiocarbonate (BDAT), was synthesised following a literature method.64 2,2-Azobis(2-methylpropionitrile) (AIBN, 98% Sigma Aldrich) was recrystallised twice before use. All other reagents were purchased from Sigma-Aldrich and used as purchased. Details can be found in the ESI.†Characterisation
Nuclear Magnetic Resonance (1H NMR) spectra were obtained on a Bruker Avance III HD 400 MHz. Samples were dissolved in deuterated chloroform (CDCl3).Gel permeation chromatography
Gel permeation chromatography (GPC) was used to acquire molecular weight and dispersity data for the polymers. An Agilent 1260 infinity multidetector SEC system, with a multi angle light scattering detector (MALS, Wyatt, Optilab Dawn 8+), was coupled to a Viscometer (Wyatt, ViscoStar-2) and a differential refractometer (DRI, Agilent 1260) for sample detection. The columns used were 2 x Agilent PLGEL 5 μm Mixed D (7.5 mm × 300 mm) and a PLGEL 5 μm guard column (7.5 mm × 50 mm). The mobile phase was THF at 1 mL min−1 at 40 °C. A known refractive index increment (dn/dc) value of 0.067 mL g−1 was used for PBuA.65dn/dc values were measured for the poly(terpene(meth)acrylates) and were as follows: PαPMA – 0.106 g mol−1, PβPMA – 0.107 mL g−1, PLiA – 0.085 mL g−1, PBuA – 0.084 mL g−1. For block copolymers, the dn/dc was determined using the following equation:26
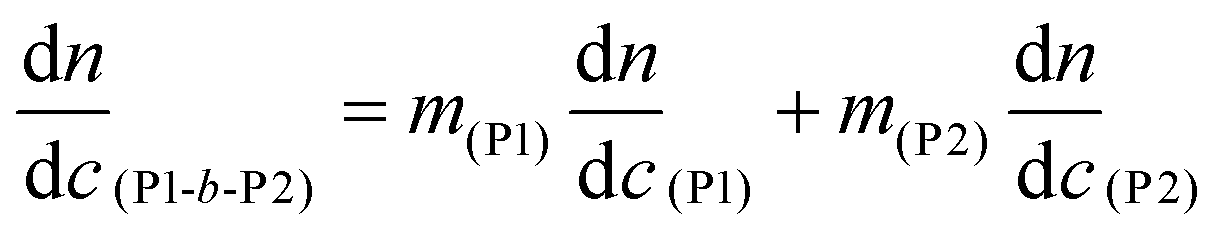 | (3) |
A standard column calibration (12 × PMMA standards across a range of Mn = 1000–400000 g mol−1) was used to calculate the molecular weights of PMMA and PMMA-based homopolymers. The dn/dc value of LiMA was not found, so PLiMA was also compared to PMMA standards. All polymers below 10000 g mol−1 were compared to PMMA standards, as below this length the light scattering data are less accurate. PαPMA homopolymers were measured by comparison with PMMA standards, as the values obtained from light scattering were inconsistent.
Dynamic Mechanical Analysis (DMA) was carried out on a Triton Technology DMA (TTDMA) (now Mettler Toledo DMA1) using the powder pocket accessory. Approximately 40 mg of sampled was added to the powder pocket and measured at 1 and 10 Hz in single cantilever bending geometry between −100 to 250 °C, or a narrower temperature window, depending on the region of interest. Tg values were taken from the peak in the tanδ curve at 1 Hz.
Atomic Force Microscopy (AFM) measurements were conducted on a Dimension FastScan AFM (Bruker Corporation), working in PeakForce quantitative nanomechanical property (PF-QNM) mode in air with an RTESPA-150 silicon probe (spring constant = 2.44 N m−1) Samples were prepared by dissolving 30 mg of polymer in 1 mL of toluene, then spin coating the solution onto a silicon wafer at 1500 rpm for 30 s. The resulting thin films were annealed at 180 °C for 24 hours, after which the oven was turned off and allowed to cool slowly to room temperature.
Free Polymer Films were prepared by solution casting circa 20 wt% solutions of polymer in THF onto an ETFE frame. An amount of approximately 1 g of polymer was used per 10 cm2 of the ETFE frame, as this was found to give a film thickness of around 0.5 mm, which was suitable for mechanical testing. The frame and solution were covered with foil, and the solvent was allowed to evaporate over 24 hours. The frame and polymer were placed in a vacuum oven at 25 °C for at least 3 days to remove all traces of solvent.
Small-Angle X-ray Scattering (SAXS) analyses were performed on copolymer films prepared via solvent casting from solutions (20 wt%) in THF. SAXS patterns were recorded at a synchrotron source (Diamond Light Source, station I22, Didcot, UK) using monochromatic X-ray radiation (X-ray wavelength λ = 0.999 Å, with scattering vector q ranging from 0.003 to 0.25 Å−1, where q = 4πsinθ/λ and θ is one-half of the scattering angle) and a 2D Pilatus 2 M pixel detector (Dectris, Switzerland). Scattering data were reduced utilising standard routines available at the beamline.66
Mechanical testing
The dried films were peeled off the ETFE sheet, and cut into strips of 5 × 25 mm2 using a pair of parallel razor blades for the purpose of tensile testing, or into an 8 mm diameter disc using a steel punch for the purpose of probe adhesion testing or rheometry. Specimens dimensions were determined using a combination of thickness gauge measurements and optical scanning followed by ImageJ analysis. All mechanical and rheological testing was carried out using an MCR302 Anton Paar rheometer.Dynamic shear rheometry was conducted using an 8 mm diameter parallel plate measuring system and temperature-controlled CTD450 oven coupled to a water recirculation cooling unit. Specimens were subjected to a compressive normal force of 25 N for 5 minutes to ensure full contact with the measuring plates, prior to commencing an isothermal frequency scan across 0.01–10 Hz at a strain amplitude of 0.1% under fixed gap conditions, maintaining contact between the specimen and plates.
For the purpose of tensile testing, a twin-drum Sentmanat Extensional Rheometry (SER) fixture with a fixed distance of 12.72 mm between the drums was employed in conjunction with the rheometer, applying a true strain rate of 0.03 s−1 until specimen failure. For adhesion probe experiments, an Anton Paar disposable aluminium rheometry plate with an average surface roughness of Ra = 0.8 μm was machined to a diameter of 5.8 mm. The probe was fixed to the rheometer measuring system and brought into contact with the elastomer at a speed of 10 μm s−1 until a normal force of 10 N was registered. The 10 N force was maintained for 1 minute, then the probe was retracted at a speed of 1.25 μm s−1 until the peak adhesive force was achieved. All mechanical testing was carried out at room temperature (22.56 ± 0.36 °C).
RAFT homopolymerisations
The RAFT agent, 2-cyano-2-propyl dodecyl trithiocarbonate (CPDT) (varying mol%) and AIBN (0.2 mol% of CPDT) were completely dissolved in toluene (1 mL), with the required monomer (1 g). The oxygen was removed from the reaction mixture via 3 freeze–pump–thaw cycles. The reaction mixture was then heated to 65 °C (75 °C for αPMA polymerisation where stated) for 24 h or until the desired conversion was reached. The reaction was terminated by exposure to air and the resultant polymers were purified by dissolving the reaction mixture in a minimum of THF and precipitating into ice cold methanol, filtered using Buchner filtration and dried in a vacuum oven. The reaction was monitored via1H NMR, and the resultant polymers were analysed by 1H NMR, GPC and DMA.Block copolymer synthesis via RAFT
A series of macro-RAFT agents were synthesised via the homopolymerisation method described above. The macro-RAFT agent polymerisations were terminated below 60% conversion to retain chain end fidelity. The reactions were monitored using 1H NMR and when nearing 60% conversion, the reaction mixture was exposed to air, quenching the radicals and terminating the reaction. Ratios of macro-RAFT agent to monomer were determined by the required molecular weight of the second block. A representative procedure was as follows: limonene acrylate (0.125 g, 0.6 mmol), PαPMA macro-RAFT agent (0.5 g, 0.0179 mmol) and AIBN (0.9 mg, 0.00357 mmol) were dissolved in toluene (3 mL) and transferred to a Schlenk tube. The reaction mixture underwent 3× freeze–pump–thaw cycles to remove any oxygen and was then heated to 65 °C. The reaction was monitored with 1H NMR and terminated at >95% conversion. The resultant PαPMA-b-PLiA block copolymer was purified via precipitation from THF into ice cold methanol.When synthesising triblock copolymers, the macro-RAFT agent synthesis was carried out on a ×5 scale, using BDAT as a difunctional RAFT agent. As a result of this scale up 2–3 precipitation steps were required to remove all unreacted monomer. The chain extensions were then carried out as for the diblock copolymers. The final triblock copolymers were analysed with 1H NMR, GPC and DMA, and in some cases AFM and SAXS.
Conflicts of interest
There are no conflicts to declare.References
- R. A. Sheldon, Green Chem., 2014, 16, 950–963 RSC.
- M. R. Thomsett, R. A. Stockman, T. E. Storr, S. M. Howdle and O. R. Monaghan, Green Mater., 2016, 4, 115–134 CrossRef.
- J. Hilschmann and G. Kali, Eur. Polym. J., 2015, 73, 363–373 CrossRef CAS.
- N. Bauer, J. Brunke and G. Kali, ACS Sustainable Chem. Eng., 2017, 5, 10084–10092 CrossRef CAS.
- A. L. Li, X. Y. Wang, H. Liang and J. Lu, React. Funct. Polym., 2007, 67, 481–488 CrossRef CAS.
- Y. Wang, A.-L. Li, H. Liang and J. Lu, Eur. Polym. J., 2006, 42, 2695–2702 CrossRef CAS.
- A. L. Li, X. Y. Wang, H. Liang and J. Lu, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5954–5966 CrossRef.
- M. Matsuda, K. Satoh and M. Kamigaito, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1774–1785 CrossRef CAS.
- J. Li, J. El Harfi, S. M. Howdle, K. Carmichael and D. J. Irvine, Polym. Chem., 2012, 3, 1495–1501 RSC.
- K. Adlington, J. El Harfi, J. Li, K. Carmichael, J. A. Guderian, C. B. Fox and D. J. Irvine, Biomacromolecules, 2016, 17, 165–172 CrossRef CAS PubMed.
- H. C. Quilter, M. Hutchby, M. G. Davidson and M. D. Jones, Polym. Chem., 2017, 8, 833–837 RSC.
- J. R. Lowe, M. T. Martello, W. B. Tolman and M. A. Hillmyer, Polym. Chem., 2011, 2, 702–708 RSC.
- N. Oberleitner, A. K. Ressmann, K. Bica, P. Gärtner, M. W. Fraaije, U. T. Bornscheuer, F. Rudroff and M. D. Mihovilovic, Green Chem., 2017, 19, 1–5 RSC.
- O. Hauenstein, M. Reiter, S. Agarwal, B. Rieger and A. Greiner, Green Chem., 2016, 18, 760–770 RSC.
- M. Claudino, J.-M. Mathevet, M. Jonsson and M. Johansson, Polym. Chem., 2014, 5, 3245–3260 RSC.
- M. Firdaus, L. Montero De Espinosa and M. A. R. Meier, Macromolecules, 2011, 44, 7253–7262 CrossRef CAS.
- M. Firdaus and M. A. R. Meier, Green Chem., 2013, 15, 370–380 RSC.
- M. F. Sainz, J. A. Souto, D. Regentova, M. K. G. Johansson, S. T. Timhagen, D. J. Irvine, P. Buijsen, C. E. Koning, R. A. Stockman and S. M. Howdle, Polym. Chem., 2016, 7, 2882–2887 RSC.
- M. A. Droesbeke and F. E. Du Prez, ACS Sustainable Chem. Eng., 2019, 7, 11633–11639 CrossRef CAS.
- T. Castagnet, G. Aguirre, J. M. Asua and L. Billon, ACS Sustainable Chem. Eng., 2020, 8, 7503–7512 CrossRef CAS.
- D. J. Keddie, Chem. Soc. Rev., 2014, 43, 496–505 RSC.
- R. T. A. Mayadunne, J. Jeffery, G. Moad and E. Rizzardo, Macromolecules, 2003, 36, 1505–1513 CrossRef CAS.
- N. Bensabeh, A. Moreno, A. Roig, O. R. Monaghan, J. C. Ronda, V. Cádiz, M. Galià, S. M. Howdle, G. Lligadas and V. Percec, Biomacromolecules, 2019, 20, 2135–2147 CrossRef CAS.
- N. Bensabeh, J. C. Ronda, M. Galià, V. Cádiz, G. Lligadas and V. Percec, Biomacromolecules, 2018, 19, 1256–1268 CrossRef CAS PubMed.
- S. Noppalit, A. Simula, N. Ballard, X. Callies, J. M. Asua and L. Billon, Biomacromolecules, 2019, 20, 2241–2251 CrossRef CAS PubMed.
- S. Noppalit, A. Simula, L. Billon and J. M. Asua, Polym. Chem., 2020, 11, 1151–1160 RSC.
- U. Montanari, V. Taresco, A. Liguori, C. Gualandi and S. M. Howdle, Polym. Int., 2021, 70, 499–505 CrossRef CAS.
- R. K. Bose, M. Enke, A. M. Grande, S. Zechel, F. H. Schacher, M. D. Hager, S. J. Garcia, U. S. Schubert and S. van der Zwaag, Eur. Polym. J., 2017, 93, 417–427 CrossRef CAS.
- J. H. Yang, B. C. Chun, Y. C. Chung and J. H. Cho, Polymer, 2003, 44, 3251–3258 CrossRef CAS.
- S. Keki, C. Bogacs, L. Bogacs, L. Daroczi and M. Zsuga, Angew. Makromol. Chemie, 1997, 245, 183–191 CrossRef CAS.
- J. Markarian, Plast. Addit. Compd., 2004, 6, 46–49 CrossRef CAS.
- W. Wang, W. Lu, A. Goodwin, H. Wang, P. Yin, N. G. Kang, K. Hong and J. W. Mays, Prog. Polym. Sci., 2019, 95, 1–31 CrossRef CAS.
- J. Shin, M. T. Martello, M. Shrestha, J. E. Wissinger, W. B. Tolman and M. A. Hillmyer, Macromolecules, 2011, 44, 87–94 CrossRef CAS.
- RAFT: Choosing the right agent to achieve controlled polymerization, Sigma Aldrich, http://www.sigmaaldrich.com/technical-documents/articles/materials-science/polymer-science/raft-polymerization.html, (accessed June 2020).
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- M. J. Darabi Mahboub, J. L. Dubois, F. Cavani, M. Rostamizadeh and G. S. Patience, Chem. Soc. Rev., 2018, 47, 7703–7738 RSC.
- D. M. O'Brien, R. L. Atkinson, R. Cavanagh, A. A. C. Pacheco, R. Larder, E. Krumins, A. J. Haddleton, C. Alexander, R. A. Stockman, S. M. Howdle and V. Taresco, Eur. Polym. J., 2020, 125, 109516 CrossRef.
- M. F. Sainz, PhD thesis, University of Nottingham, 2016.
- D. J. Keddie, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2012, 45, 5321–5342 CrossRef CAS.
- D. J. Keddie, Chem. Soc. Rev., 2014, 43, 496–505 RSC.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Nat. Commun., 2013, 4, 2505 CrossRef PubMed.
- G. Gody, T. Maschmeyer and P. B. Zetterlund, Macromolecules, 2014, 47, 639–649 CrossRef CAS.
- A.-V. Ruzette and L. Leibler, Nat. Mater., 2005, 4, 19–31 CrossRef CAS PubMed.
- T. G. Fox, Bull. Am. Phys. Soc., 1956, 1, 123 CAS.
- R. J. Spontak and N. P. Patel, Curr. Opin. Colloid Interface Sci., 2000, 5, 333–340 CrossRef.
- L. G. Williams, K. Sullivan and J. Tsanaktsidis, Select RAFT Agents for Making Well-Defined Functionalized Polymers, https://www.sigmaaldrich.com/technical-documents/articles/technology-spotlights/raft-agents-for-functionalized-polymers.html (accessed March 2021) Search PubMed.
- J. Ma and H. Zhang, Macromol. Res., 2014, 23, 67–73 CrossRef.
- Y. Luo, X. Wang, Y. Zhu, B. G. Li and S. Zhu, Macromolecules, 2010, 43, 7472–7481 CrossRef CAS.
- S. Wang, L. Shuai, B. Saha, D. G. Vlachos and T. H. Epps, ACS Cent. Sci., 2018, 4, 701–708 CrossRef CAS PubMed.
- X. Fu, Y. Yuan, X. Chen, Y. Xiao, J. Wang, C. Zhou and J. Lei, J. Appl. Polym. Sci., 2017, 134, 1–7 Search PubMed.
- G. Holden, in Encyclopedia of Polymer Science and Technology, John Wiley & Sons, Inc., Hoboken, NJ, United States, 2010 Search PubMed.
- W. Lu, Y. Wang, W. Wang, S. Cheng, J. Zhu, Y. Xu, K. Hong, N. G. Kang and J. Mays, Polym. Chem., 2017, 8, 5741–5748 RSC.
- C. C. W. J. Clarijs and L. E. Govaert, J. Polym. Sci., Part B: Polym. Phys., 2019, 57, 1001–1013 CrossRef CAS.
- S. Bahadur, Polym. Eng. Sci., 1973, 13, 266–272 CrossRef CAS.
- A. N. Gent, Rubber Chem. Technol., 1996, 69, 59–61 CrossRef CAS.
- R. Vendamme, N. Schüwer and W. Eevers, J. Appl. Polym. Sci., 2014, 131, 8379–8394 CrossRef.
- N. Karyu, M. Noda, S. Fujii, Y. Nakamura and Y. Urahama, J. Appl. Polym. Sci., 2016, 43639, 1–11 Search PubMed.
- X. Zhang, H. Liu, L. Yue, Y. Bai and J. He, Polym. Bull., 2019, 76, 3093–3112 CrossRef CAS.
- S. S. Baek and S. H. Hwang, Eur. Polym. J., 2017, 92, 97–104 CrossRef CAS.
- K. Y. Cho, A. Cho, H. J. Kim, S. H. Park, C. M. Koo, Y. J. Kwark, H. G. Yoon, S. S. Hwang and K. Y. Baek, Polym. Chem., 2016, 7, 7391–7399 RSC.
- M. A. Droesbeke, A. Simula, J. M. Asua and F. E. Du Prez, Green Chem., 2020, 22, 4561–4569 RSC.
- S. C. Temin, in Handbook of Adhesives, ed. I. Skeist, Springer US, Boston, MA, 3rd edn, 1990, pp. 641–633 Search PubMed.
- NPCS Board of Consultants & Engineers, in The Complete Book on Adhesives, Glues & Resins Technology, 2016, pp. 146–175 Search PubMed.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754–6756 CrossRef CAS.
- S. Mori and H. G. Barth, Size Exclusion Chromatography, Springer Berlin Heidelberg, New York, 1999 Search PubMed.
- B. R. Pauw, A. J. Smith, T. Snow, N. J. Terrill and A. F. Thünemann, J. Appl. Crystallogr., 2017, 50, 1800–1811 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py00326g |
| This journal is © The Royal Society of Chemistry 2021 |