DOI:
10.1039/D5PY00203F
(Paper)
Polym. Chem., 2025,
16, 2751-2756
Synthesis of electron-deficient polymers based on sulfur-bridged dithienylboranes as a building block†
Received
28th February 2025
, Accepted 12th May 2025
First published on 15th May 2025
Abstract
π-Conjugated materials incorporating tricoordinate boron are known to possess relatively deep LUMO energy levels owing to the interaction between the empty p orbital of boron and the π* orbital. However, polymers with simple triarylborane structures do not have deep LUMO energy levels for applications in electron-accepting optoelectronic materials, such as n-type semiconductors. In this study, we synthesized new p–π* conjugated polymers by copolymerizing thiaborin units containing boron and sulfur atoms and diketopyrrolopyrrole (DPP), a well-known acceptor unit. The resulting polymers exhibited extended conjugation through the boron p orbital and strong absorption in the visible to near-infrared region. Although these polymers did not show n-type semiconductor behavior, they possessed deep LUMO energy levels lower than −3.6 eV. Furthermore, titration experiments using an amine base revealed that the polymer containing a sulfone unit has strong Lewis acidity, as evidenced by changes in the UV-vis absorption spectrum upon coordination of the base.
Introduction
π-Conjugated polymers are widely utilized in various applications, including organic electronic devices,1–3 and numerous conjugated polymers with diverse building blocks have been synthesized. Among the building blocks of π-conjugated polymers, electron-deficient units, such as benzothiadiazole (BT) and diketopyrrolopyrrole (DPP), are indispensable for the development of donor–acceptor (D–A) type polymers4,5 and n-type semiconducting polymers.6–8 Consequently, the synthesis of new electron-deficient units has been actively pursued. On the other hand, tricoordinate boron, which possesses an empty 2p orbital, generally exhibits electron-accepting characteristics.9–12 D–A type polymers employing tricoordinate boron as an acceptor have been extensively reported.13–20 For example, Jäkle and co-workers synthesized dithienylborane (FBDT, Fig. 1) with a bulky tris(2,4,6-trifluoromethylphenyl) (FMes) group and reported p–π* conjugated polymers that used FBDT as a building block.13 Helten and co-workers developed polymers by combining furan and tricoordinate boron.15,16 However, the electron-accepting ability of tricoordinate boron is inferior to that of typical π-conjugated acceptor units, such as BT and DPP (Fig. S1†). For example, we showed in our previous study that a D–A type polymer composed of FBDT and dithienosilole, a strong electron-donating unit, exhibited a relatively wide bandgap of 2.07 eV and weak dependence of its fluorescence spectra on solvent polarity.14 Furthermore, the two thiophene rings in the BDT framework demonstrated reduced coplanarity owing to steric repulsion caused by the internal β-hydrogens, potentially diminishing intermolecular interactions in the solid state. Although several n-type semiconductor materials based on p–π* conjugated systems have been reported, the drawbacks of the triarylborane units leave significant room for performance improvement.21 Inspired by the development of a new dithienylborane building block,22 we have previously synthesized FDTSB (Fig. 1), a sulfur-bridged dithienylborane with a FMes group on boron, and its polymer p(FDTSB-2T) (Fig. 1).19 This polymer exhibited significantly red-shifted absorption in the solid state compared to in solution, indicating that the introduction of sulfur and the improved coplanarity of the thiophene rings enhanced intermolecular interactions in the p–π* conjugated polymer. Additionally, we reported that DTSO2B (Fig. 1), obtained by oxidizing the sulfur atoms in the DTSB structure, showed a lowered LUMO energy level caused by the sulfur oxidation (Fig. S1†), making it a strong electron-acceptor unit.23 In this study, we synthesized new electron-deficient acceptor–acceptor (A–A) type p–π* conjugated polymers containing sulfur-bridged dithienylborane units (FDTSB and DTSO2B) copolymerized with DPP (Fig. 1). Electrochemical measurements revealed that these polymers possessed deep LUMO energy levels, demonstrating that the FDTSB and DTSO2B units worked effectively as acceptor units in electron-deficient polymers.
 |
| | Fig. 1 Dithienylborane-based building blocks and their polymers. | |
Results and discussion
Synthesis
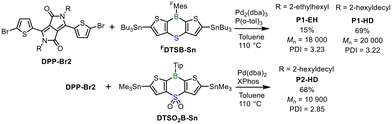
The synthetic route of the p–π* conjugated polymers is shown in Scheme 1. To enhance polymer solubility, a 2-hexyldecyl group was introduced onto the nitrogen atoms of the DPP unit. The DTSB and DTSO2B units had different aryl groups on the boron atom because the FMes group could not be introduced to the DTSO2B monomer.23 The A–A type p–π* conjugated polymers were synthesized via a palladium-catalyzed Stille cross-coupling reaction between the distannylated DTSB or DTSO2B monomer and the dibrominated DPP comonomer. It should be noted that FDTSB-Sn contains tributylstannyl groups, while DTSO2B-Sn contains trimethylstannyl groups. This is because the synthesis of these stannylated monomers has already been established.19,23 First, we synthesized copolymer P1-EH from DPP-Br2 bearing 2-ethylhexyl groups on the nitrogen atoms and FDTSB-Sn. However, P1-EH was poorly soluble, and the yield after the purification by Soxhlet extraction using chloroform was only 15%. To improve polymer solubility, the alkyl groups on the nitrogen atoms were replaced with 2-hexyldecyl groups, and polymers P1-HD and P2-HD were synthesized using the DPP monomer with 2-hexyldecyl groups. The crude polymers with the 2-hexyldecyl groups were purified by removing the palladium catalyst with a short silica pad, followed by reprecipitation from ethanol/chloroform. P1-HD and P2-HD were soluble in organic solvents, such as chloroform and THF, and their yields were good (Scheme 1). GPC analysis using polystyrene standards revealed number-average molecular weights (Mn) of 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 20000, and 10900 for P1-EH, P1-HD, and P2-HD, respectively, confirming the formation of sufficiently high molecular weight polymers. In the case of P1-EH and P1-HD, significant signal broadening was observed in the 1H NMR spectra, and signals corresponding to the thiophene rings and the FMes group were detected in the aromatic region (Fig. S2†). In the case of P2-HD, the 1H NMR spectrum was well resolved, and all peaks could be clearly assigned (Fig. S2†), which may be due to the lower molecular weight and higher solubility of P2-HD in CDCl3 compared to P1. In the 11B NMR spectrum of P2-HD, a broad peak was observed at 45 ppm (Fig. S3†), indicating that the boron center in the polymer is tricoordinate. In the 11B NMR spectra of P1-EH and P1-HD, a broad peak was observed at approximately 33 ppm (Fig. S3†), which appeared at a higher field than those of P2-HD and typical p–π* conjugated polymers.13,15,16 Although the reason for this is unclear, a similar observation has been reported in p–π* conjugated polymers based on the FDTSB unit.19 The 19F NMR spectrum of P1-HD displayed two signals attributable to the FMes group (Fig. S4†), suggesting that the FDTSB unit is preserved within the polymer structure. To further analyze the polymer structures, MALDI-TOF mass spectrometry was conducted (Fig. S5†). For P1-EH, most of the observed peaks could be assigned to the expected polymer structure, suggesting that structural defects were minimal. In contrast, the mass spectrum of P1-HD showed signals corresponding to the repeating units, similar to P1-EH, but also displayed additional peaks indicative of structural defects. Notably, molecular ion peaks corresponding to species containing greater numbers of DPP units than FDTSB units were observed, implying that homocoupling of DPP units occurred during the polymerization of P1-HD (Fig. S5†). Since homocoupling reactions are known to be promoted at high temperatures,24 it may be possible that in the case of the poorly soluble P1-EH, the polymer was precipitated from the reaction medium at an early stage, thereby suppressing further homocoupling. In the mass spectrum of P1-HD, butyl-terminated structures were also detected (Fig. S5†), which are likely formed through the transmetalation of butyl groups from tributyltin groups during the cross-coupling reaction. For P2-HD, although the precise structure of the end groups could not be verified from the mass spectrum, repeating units corresponding to DPP and DTSO2B units were observed (Fig. S5†), indicating the presence of an ideal structure in the polymer. Thermogravimetric (TG) analysis in air revealed that the 5% weight loss temperature (Td5) of P1-HD was 354 °C while that of P2-HD was 280 °C, indicating that P1-HD possesses higher thermal stability than P2-HD (Fig. S6†). This difference could be attributed to the difference of the aryl substituent on the boron atom.14 The Td5 of p(FDTSB-2T), which has an FDTSB unit like P1-HD, was 328 °C,19 demonstrating that copolymerization with the DPP unit slightly enhances the thermal stability of the p–π* conjugated polymer.
000, 20000, and 10900 for P1-EH, P1-HD, and P2-HD, respectively, confirming the formation of sufficiently high molecular weight polymers. In the case of P1-EH and P1-HD, significant signal broadening was observed in the 1H NMR spectra, and signals corresponding to the thiophene rings and the FMes group were detected in the aromatic region (Fig. S2†). In the case of P2-HD, the 1H NMR spectrum was well resolved, and all peaks could be clearly assigned (Fig. S2†), which may be due to the lower molecular weight and higher solubility of P2-HD in CDCl3 compared to P1. In the 11B NMR spectrum of P2-HD, a broad peak was observed at 45 ppm (Fig. S3†), indicating that the boron center in the polymer is tricoordinate. In the 11B NMR spectra of P1-EH and P1-HD, a broad peak was observed at approximately 33 ppm (Fig. S3†), which appeared at a higher field than those of P2-HD and typical p–π* conjugated polymers.13,15,16 Although the reason for this is unclear, a similar observation has been reported in p–π* conjugated polymers based on the FDTSB unit.19 The 19F NMR spectrum of P1-HD displayed two signals attributable to the FMes group (Fig. S4†), suggesting that the FDTSB unit is preserved within the polymer structure. To further analyze the polymer structures, MALDI-TOF mass spectrometry was conducted (Fig. S5†). For P1-EH, most of the observed peaks could be assigned to the expected polymer structure, suggesting that structural defects were minimal. In contrast, the mass spectrum of P1-HD showed signals corresponding to the repeating units, similar to P1-EH, but also displayed additional peaks indicative of structural defects. Notably, molecular ion peaks corresponding to species containing greater numbers of DPP units than FDTSB units were observed, implying that homocoupling of DPP units occurred during the polymerization of P1-HD (Fig. S5†). Since homocoupling reactions are known to be promoted at high temperatures,24 it may be possible that in the case of the poorly soluble P1-EH, the polymer was precipitated from the reaction medium at an early stage, thereby suppressing further homocoupling. In the mass spectrum of P1-HD, butyl-terminated structures were also detected (Fig. S5†), which are likely formed through the transmetalation of butyl groups from tributyltin groups during the cross-coupling reaction. For P2-HD, although the precise structure of the end groups could not be verified from the mass spectrum, repeating units corresponding to DPP and DTSO2B units were observed (Fig. S5†), indicating the presence of an ideal structure in the polymer. Thermogravimetric (TG) analysis in air revealed that the 5% weight loss temperature (Td5) of P1-HD was 354 °C while that of P2-HD was 280 °C, indicating that P1-HD possesses higher thermal stability than P2-HD (Fig. S6†). This difference could be attributed to the difference of the aryl substituent on the boron atom.14 The Td5 of p(FDTSB-2T), which has an FDTSB unit like P1-HD, was 328 °C,19 demonstrating that copolymerization with the DPP unit slightly enhances the thermal stability of the p–π* conjugated polymer.
 |
| | Scheme 1 Synthetic scheme of DTSB/DTSO2B-containing polymers. | |
Optical properties
Next, the UV-vis absorption spectra of the p–π* polymers were measured in toluene (Fig. 2). The maximum absorption wavelengths of P1-EH, P1-HD, and P2-HD were observed at 722, 721, and 719 nm, respectively (Table 1), representing a redshift of over 280 nm compared to p(FDTSB-2T) (λmax = 433 nm in toluene).19 This redshift is attributed to the effective conjugation of the DPP unit. The maximum absorption wavelength of p(FBDT-DPP), a DPP-based p–π* conjugated polymer without sulfur bridging (Fig. 2a), was observed at 594 nm in chlorobenzene.21 This demonstrates that sulfur bridging improves the coplanarity of the thiophene rings and reduces the HOMO–LUMO energy gap. On the other hand, the maximum absorption wavelength of p(4T-DPP), a fully conjugated polymer composed of DPP and quaterthiophene (Fig. 2a), was 777 nm in chloroform,25 indicating that the conjugation through thiaborin rings causes a slight widening of the band gap. Although P1-EH and P1-HD share the same structure in the main chain, differing only in the alkyl group on the nitrogen atoms, their spectra markedly differed (Fig. 2b). The absorption spectrum of P1-EH exhibited apparent vibrational features for the 0–0 and 0–1 transitions, suggesting a rigid main-chain structure. On the other hand, the absorption spectrum of P1-HD showed a maximum at a similar wavelength and significantly broadened shoulder peaks in the longer wavelength region. The appearance of the shoulder peaks can be attributed to two possible reasons. One is the structural defects, including the homocoupled DPP units, as suggested by the MALDI-TOF mass spectrum (Fig. S5†). The other reason is the potential aggregation of the polymer even in solution.26 The absorption bands of P2-HD appeared in a similar region to those of P1-EH, suggesting that the oxidation of sulfur does not significantly affect the HOMO–LUMO energy gap in these polymers. The optical band gaps (Eoptg) of the polymers were determined from the absorption edges (Table 1). In toluene, the Eoptg values for P1-EH, P1-HD, and P2-HD were 1.46, 1.31, and 1.48 eV, respectively, significantly smaller than that of p(FBDT-DPP) (1.83 eV).21 Notably, the Eoptg of P2-HD was comparable to that of fully conjugated polymer p(4T-DPP) (1.2 eV).25 To investigate the influence of solvent polarity, the UV-vis absorption spectra of these p–π* conjugated polymers were measured in THF and dichloromethane in addition to in toluene (Fig. S7†). The absorption spectra of P1-EH and P1-HD demonstrated negligible changes across different solvents, indicating minimal dependence on solvent polarity in the ground state. On the other hand, the absorption spectra of P2-HD exhibited a slight blue shift in dichloromethane and THF compared to that in toluene. This shift may be attributed to aggregation, possibly because of the low solubility of P2-HD in toluene (vide infra).
 |
| | Fig. 2 Chemical structures of previously reported DPP polymers (a) and UV-vis absorption spectra of DTSB/DTSO2B-containing polymers in toluene and as film (b). | |
Table 1 Optical and electrochemical properties of DTSB/DTSO2B-containing polymers
| Polymer |
λ
abs (film)a/nm |
E
optg (film)b/eV |
V
redc/V |
V
oxd/V |
LUMOe/eV |
HOMOf/eV |
E
CVgg/eV |
|
Absorption maximum in toluene and in film.
Band gap estimated from the absorption edge in toluene and in film.
Onset potential of the reduction wave of film.
Onset potential of the oxidation wave of film.
LUMO = –4.8 − Vred.
HOMO = –4.8 − Vox.
Band gap estimated from Vox and Vred.
|
|
P1-EH
|
722 (712) |
1.46 (1.46) |
–1.23 |
0.50 |
–3.57 |
–5.30 |
1.73 |
|
P1-HD
|
721 (714) |
1.31 (1.29) |
–1.23 |
0.54 |
–3.57 |
–5.34 |
1.77 |
|
P2-HD
|
719 (728) |
1.48 (1.41) |
–1.19 |
0.72 |
–3.61 |
–5.52 |
1.91 |
Next, the UV-vis absorption spectra of the spin-coated thin films were measured (Fig. 2b). The absorption spectrum of P1-EH in the film was nearly identical to that in solution. In contrast, the absorption spectrum of P2-HD in the film showed a slight redshift compared to that in solution, suggesting the presence of π–π interactions in the solid state. The absorption spectrum of P1-HD, similar to that of P1-EH, exhibited minimal differences between the film and the solution; however, the prominent shoulder peaks may suggest aggregation even in solution for P1-HD. To investigate this, temperature-dependent UV-vis absorption spectra were measured in toluene (Fig. S8†). For P1-EH, only a slight decrease in the absorption band corresponding to the 0–0 transition was observed with increasing temperature. On the other hand, P1-HD exhibited an obvious decrease in the shoulder peaks with increasing temperature, suggesting that these shoulder peaks partially originate from aggregation.26 To further examine the aggregation behavior, UV-vis absorption spectra of P1-HD were measured at various concentrations. However, the intensity of the shoulder peaks remained unchanged even at low concentrations, indicating the no concentration dependence (Fig. S9†). These results suggest that P1-HD may form aggregates even at low concentrations, or that the shoulder peaks are largely attributable not only to aggregate but also to the structural defects arising from the homocoupled DPP units. The impact of the structural defects on the UV-vis absorption spectra is discussed in detail in the section on DFT calculations. P2-HD showed a blue shift in its absorption band with increasing temperature (Fig. S8c†), suggesting partial aggregation in toluene at room temperature. The shoulder peaks in the absorption spectrum of P1-HD and the redshifted absorption of P2-HD in the film relative to that in solution may indicate the presence of strong intermolecular interactions in the solid state, which are usually not observed in p–π* conjugated polymers, including p(FBDT-DPP).21 These results suggest that sulfur-atom bridging can enhance intermolecular interactions in p–π* conjugated polymers.
DFT calculations
To analyze the electronic structures of the polymers, DFT calculations were performed on oligomers with a degree of polymerization n of 2.5, namely, O1-Me and O2-Me, as models for the respective polymers (Fig. 3). For comparison of structures, similar calculations were also conducted on a model of p(4T-DPP), o(4T-DPP), and a p–π* conjugated oligomer without sulfur bridges, o(FBDT-DPP). Fig. 3 shows the optimized structures and the frontier orbitals of these oligomers. In o(4T-DPP), all the π-conjugated units were perfectly coplanar. Similarly, the main chains of thiaborin-based p–π* conjugated oligomers, O1-Me and O2-Me, were nearly coplanar. In contrast, o(FBDT-DPP), lacking the sulfur bridges, exhibited significant twisting between the two thiophene rings within the FBDT unit, with a dihedral angle of 36.5°. This is caused by the steric hindrance from β-hydrogen atoms on the thiophene rings. The HOMO is delocalized across the entire o(4T-DPP) molecule, while in p–π* conjugated oligomers, it is localized on the DPP unit owing to the interruption of conjugation at the boron bridge. Consequently, the HOMO energy levels of p–π* conjugated systems are much deeper than that of o(4T-DPP) (Fig. 3). The LUMO in all molecules is delocalized across the central DPP and peripheral oligothiophene units, with a clear contribution from the p orbital of boron in p–π* conjugated oligomers. Similar to the HOMO energy levels, the LUMO energy levels of the p–π* conjugated oligomers are also much deeper than that of o(4T-DPP) (Fig. 3). Notably, the LUMO energy level of the sulfone-containing O2-Me is the lowest among these oligomers, at −3.36 eV.
 |
| | Fig. 3 DFT-optimized structures and frontier orbitals of DPP-containing model oligomers at the B3LYP-D3BJ/6-31G(d) level. | |
To further investigate the impact of the structural defects observed in P1-HD, a geometry optimization was performed for an oligomer model O1-Me′, in which the connectivity of one FDTSB-DPP unit in O1-Me was reversed. Subsequent TD-DFT calculations were performed to evaluate the S0 → S1 electronic transitions (Tables S1 and S2†). Despite no significant difference in planarity between O1-Me and O1-Me′, the calculated S0 → S1 transition wavelength was 759 nm for O1-Me and 838 nm for O1-Me′ (Fig. S10†), indicating a substantial redshift in absorption induced by the presence of the DPP homocoupling unit. This result supports that the shoulder peaks observed in the UV-vis absorption spectra of P1-HD are significantly contributed from the structural defects.
Cyclic voltammetry and evaluation of carrier-transport properties
To investigate the redox properties of the p–π* conjugated polymers, cyclic voltammetry (CV) measurements were conducted on the polymer films (Fig. 4). The frontier orbital energy levels were calculated from the onset potentials of the first oxidation and reduction waves. The LUMO energy levels of P1-HD and P2-HD were determined to be −3.57 eV and −3.61 eV, respectively (Table 1), indicating deep LUMO energy levels. In particular, P2-HD exhibited a slightly deeper LUMO energy level than P1-HD, consistent with the results of DFT calculations. This indicates that the sulfone structure effectively contributes to lowering the LUMO energy level. The LUMO energy level of the non-sulfur-bridged polymer p(FBDT-DPP) was reported to be −3.58 eV,21 and the thiaborin-based polymers exhibited comparable or even deeper LUMO energy levels. These observations confirm that introducing thiaborin structures is an effective strategy for lowering the LUMO energy level in p–π* conjugated materials. Motivated by the extended conjugation and the deep LUMO energy levels of these p–π* conjugated polymers, we fabricated organic field-effect transistors (OFETs) using these polymers to investigate their p- and n-type semiconducting properties. Although these polymers showed good film-forming properties, attempts to evaluate the transistor characteristics of spin-coated films did not yield clear evidence of semiconducting behavior (Fig. S11†). The UV-vis absorption spectra suggested strong intermolecular interactions in these p–π* conjugated polymers (vide supra), which are generally advantageous for carrier transport. Nonetheless, one possible reason for the absence of semiconducting properties is that the bulky aryl groups attached to boron increased the intermolecular distance in the film, hindering carrier transport.
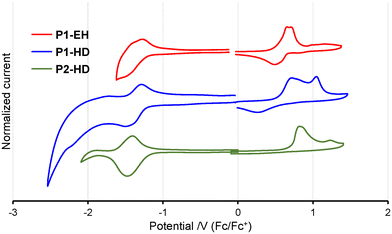 |
| | Fig. 4 Cyclic voltammograms of DTSB/DTSO2B-containing polymer films in acetonitrile with 0.1 M TBAPF6 at a scan rate of 100 mV s−1. | |
Optical responses to Lewis bases
Because p–π* conjugated polymers contain Lewis acidic tricoordinate boron, we investigated their response to Lewis bases. Our previous study has demonstrated that the DTSO2B unit, despite having a bulky Tip group on the boron atom, can still form a coordination bond with organic bases, such as 4-dimethylanimopyridine (DMAP).23 As the amount of DMAP added to a solution of P2-HD increased, the long-wavelength absorption band gradually decreased, and a new absorption band appeared around 630 nm (Fig. 5). This is believed to result from the disappearance of the p–π* conjugation upon the coordination of DMAP with boron. In contrast, no change was observed in the absorption spectrum of P1-HD upon adding DMAP (Fig. S12†), suggesting no coordination with boron. The difference in response to DMAP is thought to be due to the increased Lewis acidity of boron in the DTSO2B unit23 due to the strong electron-withdrawing properties of sulfone.
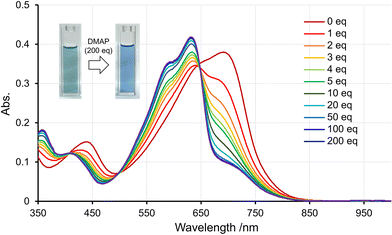 |
| | Fig. 5 UV-vis absorption data obtained from titrations of P2-HD with DMAP aliquots in CHCl3 at the concentration of 0.01 mg mL−1. Insets show the response of P1-HD and P2-HD to the addition of DMAP. | |
Finally, to elucidate the influence of aryl substituents on the boron atom, fluoride ion affinity (FIA) calculations were performed (Fig. S13†).27 Our previous study has shown that oxidation of sulfur significantly enhances the Lewis acidity of the boron atom, and the FIA value of the sulfone compound DTSO2B bearing a Tip group on boron is more than 50 kJ mol−1 higher than that of its sulfide counterpart, DTSB.23 When the Tip group was replaced with an FMes group, the calculated FIA values of FDTSB and FDTSO2B were found to be even higher than those of their corresponding Tip analogs. Despite the increased steric bulkiness of the FMes group, this increase of the Lewis acidity is attributed to the strong electron-withdrawing properties of the CF3 groups on the FMes group. Moreover, regardless of the aryl substituent, sulfone derivatives consistently exhibited higher FIA values than their corresponding sulfide analogs. Consequently, although O1-Me and O2-Me possess FMes and Tip groups on the boron atoms, respectively, O2-Me exhibited a higher FIA value than O1-Me. This result is consistent with experimental observations showing that only P2-HD formed a complex with DMAP.
Conclusions
We synthesized new electron-deficient polymers by copolymerizing thiaborin-based p–π* conjugated building blocks with a DPP unit. These polymers exhibited an extended π-conjugation through the boron p orbital and possessed a small bandgap. Cyclic voltammetry measurements revealed that the polymers had deeper LUMO energy levels than previously reported p–π* conjugated polymers, demonstrating that the combination of thiaborin and DPP units effectively enhanced the electron-deficient character of the polymer. Unfortunately, these polymers did not exhibit the expected n-type semiconductor characteristics. However, their deep LUMO energy levels suggested that incorporating alternative molecular designs to enhance intermolecular interactions within these polymer backbones could lead to the development of n-type semiconductor materials. Furthermore, titration experiments with DMAP revealed that P2-HD retained high Lewis acidity even in the polymer. This finding demonstrates that p–π* conjugated polymers incorporating the DTSO2B unit can be utilized as sensor materials for detecting Lewis bases.
Author contributions
Yohei Adachi: conceptualization; funding acquisition; project administration; supervision; writing – original draft; writing – review & editing. Ryuji Matsuura: investigation; visualization. Joji Ohshita: supervision; writing – review & editing.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers JP22K14666 and JP23K23400.
References
- Y. Li, Acc. Chem. Res., 2012, 45, 723 CrossRef CAS PubMed.
- A. Facchetti, Chem. Mater., 2011, 23, 733 CrossRef CAS.
- X. Guo, M. Baumgarten and K. Müllen, Prog. Polym. Sci., 2013, 38, 1832 CrossRef CAS.
- H. Zhou, L. Yang and W. You, Macromolecules, 2012, 45, 607 CrossRef CAS.
- C. Liu, K. Wang, X. Gong and A. J. Heeger, Chem. Soc. Rev., 2016, 45, 4825 RSC.
- H. Sun, X. Guo and A. Facchetti, Chem, 2020, 6, 1310 Search PubMed.
- Y. Lu, J.-Y. Wang and J. Pei, Chem. Mater., 2019, 31, 6412 CrossRef CAS.
- S. Griggs, A. Marks, H. Bristow and I. McCulloch, J. Mater. Chem. C, 2021, 9, 8099 Search PubMed.
- L. Ji, S. Griesbeck and T. B. Marder, Chem. Sci., 2017, 8, 846 RSC.
- X. Yin, J. Liu and F. Jäkle, Chem. – Eur. J., 2021, 27, 2973 CrossRef CAS PubMed.
- K. Tanaka and Y. Chujo, Macromol. Rapid Commun., 2012, 33, 1235 CrossRef CAS PubMed.
- H. Helten, Chem. – Asian J., 2019, 14, 919 CrossRef CAS PubMed.
- X. Yin, F. Guo, R. A. Lalancette and F. Jäkle, Macromolecules, 2016, 49, 537 Search PubMed.
- Y. Adachi, Y. Ooyama, Y. Ren, X. Yin, F. Jäkle and J. Ohshita, Polym. Chem., 2018, 9, 291 RSC.
- A. Lik, S. Jenthra, L. Fritze, L. Müller, K.-N. Truong and H. Helten, Chem. – Eur. J., 2018, 24, 11961 CrossRef CAS PubMed.
- L. Fritze, M. Fest, A. Helbig, T. Bischof, I. Krummenacher, H. Braunschweig, M. Finze and H. Helten, Macromolecules, 2021, 54, 7653 Search PubMed.
- C. Xue, M. Peng, Z. Zhang, X. Han, Q. Wang, C. Li, H. Liu, T. Li, N. Yu and Y. Ren, Macromolecules, 2022, 55, 3850 CrossRef CAS.
- X. Han, C. Xue, Z. Zhao, M. Peng, Q. Wang, H. Liu, N. Yu, C. Pu and Y. Ren, ACS Macro Lett., 2023, 12, 961 CrossRef CAS PubMed.
- Y. Adachi, M. Sakabe, T. Nomura and J. Ohshita, Polym. J., 2023, 55, 489 CrossRef CAS.
- A. F. Alahmadi, X. Yin, R. A. Lalancette and F. Jäkle, Chem. – Eur. J., 2023, 29, e202203619 Search PubMed.
- B. Meng, Y. Ren, J. Liu, F. Jäkle and L. Wang, Angew. Chem., Int. Ed., 2018, 57, 2183 CrossRef CAS PubMed.
- Y. Yan, Z. Sun, C. Li, J. Zhang, L. Lv, X. Liu and X. Liu, Asian J. Org. Chem., 2017, 6, 496 Search PubMed.
- Y. Adachi, R. Matsuura and J. Ohshita, Organometallics, 2024, 43, 829 Search PubMed.
- B. Ma, Q. Shi, X. Ma, Y. Li, H. Chen, K. Wen, R. Zhao, F. Zhang, Y. Lin, Z. Wang and H. Huang, Angew. Chem., Int. Ed., 2022, 61, e202115969 CrossRef CAS PubMed.
- Y. Li, P. Sonar, S. P. Singh, M. S. Soh, M. van Meurs and J. Tan, J. Am. Chem. Soc., 2011, 133, 2198 CrossRef CAS PubMed.
- R. Heuvel, F. J. M. Colberts, J. Li, M. M. Wienk and R. A. J. Janssen, J. Mater. Chem. A, 2018, 6, 20904 Search PubMed.
- P. Erdmann, J. Leitner, J. Schwarz and L. Greb, ChemPhysChem, 2020, 21, 987 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details with synthesis, characterization; absorption spectra; DFT computation results; NMR/mass spectra. See DOI: https://doi.org/10.1039/d5py00203f |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Ryuji
Matsuura
a,
Mitsuru
Sakabe
a,
Hiroki
Tobita
a,
Hideki
Murakami
b and
Joji
Ohshita
*a,
Ryuji
Matsuura
a,
Mitsuru
Sakabe
a,
Hiroki
Tobita
a,
Hideki
Murakami
b and
Joji
Ohshita





