
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSquare planar Pd(II)/oximate complexes as ‘metalloligands’ for the directional assembly of 4f-metal ions: a new family of {Ln2Pd} (Ln = lanthanide) clusters exhibiting slow magnetization relaxation†
Konstantinos N.
Pantelis
a,
Catherine P.
Raptopoulou
 b,
Vassilis
Psycharis
*b,
Jinkui
Tang
c and
Theocharis C.
Stamatatos
*ad
b,
Vassilis
Psycharis
*b,
Jinkui
Tang
c and
Theocharis C.
Stamatatos
*ad
aDepartment of Chemistry, University of Patras, 26504 Patras, Greece. E-mail: thstama@upatras.gr; Tel: +30-2610996730
bInstitute of Nanoscience and Nanotechnology, NCSR “Demokritos”, Aghia Paraskevi Attikis 15310, Greece. E-mail: v.psycharis@inn.demokritos.gr; Tel: +30-2106503346
cState Key Laboratory of Rare Earth Resource Utilization, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, P. R. China
dInstitute of Chemical Engineering Sciences, Foundation for Research and Technology – Hellas (FORTH/ICE – HT), Platani, P.O. Box 1414, 26504, Patras, Greece
First published on 25th November 2024
Abstract
A relatively unexplored approach in heterometallic chemistry of transition metals and lanthanides has been developed toward the controlled synthesis of a new family of linear heterotrinuclear Ln(III)–Pd(II)–Ln(III) complexes with the general formula [Ln2Pd(pao)2(NO3)6(MeOH)2(H2O)2]·[Pd(pao)2]4, where LnIII = DyIII (2), GdIII (3), ErIII (4) and YbIII (5). This strategy was based on the diamagnetic ‘metalloligand’ [Pd(pao)2] (1), where pao− is the anion of 2-pyridinealdoxime, containing two dangling oximate O-atoms which were trans to each other and available for binding with oxophilic lanthanide ions. Because of their trans-configuration, the [Pd(pao)2] ‘metalloligand’ was able to direct the binding of two {Ln(NO3)3(MeOH)(H2O)} units on opposite sites, thus yielding the reported trinuclear {Ln–Pd–Ln} clusters. Complexes 2–5 constitute a new family of trinuclear heterometallic {Ln2Pd} species, and they represent the first examples of a directional assembly approach towards the coordination of 4f-metal ions. Compounds 2 and 5 exhibit out-of-phase signals under applied dc fields of 300 and 2000 Oe, respectively, characteristics of the slow magnetization relaxation, albeit with very small energy barriers for the magnetization reversal. This was due to the combined onset of fast quantum tunneling and the weak crystal field effects induced by the coordinated ligands. The combined results highlight the potential of using the ‘metal complexes as ligands’ method to deliberately prepare heterometallic PdII–LnIII complexes with unique structural and interesting physicochemical (magnetic, optical, catalytic) properties.
1. Introduction
More than three decades ago, a significant breakthrough in the research fields of inorganic chemistry and molecular magnetism occurred when researchers discovered that a coordination complex known as {Mn12} could retain its magnetization for extended periods at liquid helium temperatures. This discovery marked the introduction of Single-Molecule Magnets (SMMs),1 a group of molecules characterized by a large ground state spin (S) and an Ising-type (or easy-axis) magnetoanisotropy, resulting from a negative zero-field splitting parameter (D). The combined presence of S and D leads to an effective energy barrier (Ueff) for magnetization reversal. SMMs are superparamagnetic-type materials, where magnetization relaxes slowly below a specific blocking temperature (TB), and the phenomenon can be observed through the appearance of out-of-phase ac magnetic susceptibility signals and magnetic hysteresis loops.2 Scientists are currently investigating SMMs due to their potential applications in quantum computing, spintronics, magnetic resonance imaging (MRI), and various other fields of science and technology related to information processing and data storage.3
ac magnetic susceptibility signals and magnetic hysteresis loops.2 Scientists are currently investigating SMMs due to their potential applications in quantum computing, spintronics, magnetic resonance imaging (MRI), and various other fields of science and technology related to information processing and data storage.3
Compelling evidence has highlighted the paramount importance of single-ion anisotropy in the design of SMMs with substantial energy barriers for magnetization reversal.4 A pivotal moment in this understanding occurred in 2003 when researchers recognized SMM behavior in mononuclear [LnIII(pc)2]− complexes featuring trivalent lanthanide ions (LnIII = TbIII, DyIII; pc2− = dianion of phthalocyanine).5 Subsequently, significant emphasis was placed on the synthesis and investigation of SMMs based on mononuclear, dinuclear, and polynuclear LnIII compounds.6 A groundbreaking recent development in this field resulted from the synthesis and study of [(CpiPr5)Dy(Cp*)]+ (CpiPr5 = penta-iso-propylcyclopentadienyl, Cp* = pentamethylcyclopentadienyl). This complex behaves as an SMM with a record blocking temperature of 80 K, which is above the landmark temperature of 77 K (the boiling point of liquid nitrogen).7 The SMM behavior observed in mononuclear LnIII complexes is attributed to the substantial anisotropy of individual ions, arising from strong spin–orbit coupling and the effects of the coordinated ligands on the overall crystal field.8 The DyIII-based SMMs dominate the field; the DyIII is a Kramers ion characterized by an odd number of 4f electrons and has always a bistable ground state.9 The intrinsic quadrupole moment approximation of f-orbitals classifies lanthanide ions into oblate, prolate, or spherical shapes based on their electron density distribution. This classification makes it easier to introduce anisotropy in these ions, particularly when compared to transition metals.6d Although f-electrons are deeply embedded in the core shell, the ligand field can still have a partial influence on their energies, affecting their magnetic properties. This means that by adjusting the ligand field, it is possible to explore the magnetic dynamics by modulating the f-electron cloud.10
In the pursuit of SMMs with enhanced properties, various attempts have focused on the synthesis of heterometallic compounds featuring both paramagnetic 4f- and diamagnetic 3d-metal ions, with a particular emphasis on ZnII and low-spin CoIII ions.11 The primary objective is the stabilization of the electronic ground state with the largest mJ and the increase of the energy gap between the ground and the first excited mJ states of the anisotropic lanthanide ion(s). The presence of diamagnetic metal ions influences the electron density distribution of coordinating ligands by inducing polarization effects, thus affecting the strength of the crystal field.11a Studies have shown that the presence of diamagnetic 3d-metal ions results in an increased negative charge on the oxygen donor atoms of the bridging ligands. This leads to larger energy gaps between the mJ states, resulting in higher Ueff values and the weakening (or even suppression) of quantum tunneling of magnetization (QTM) as a relaxation process.12
The focus has predominantly been on first-row transition metals (3d), primarily because they are more manageable, both experimentally and theoretically, compared to their 4d counterparts. A key distinction between 3d- and the heavier 4d-metal ions is the more extended valence orbitals of the latter. Among the 4d-block elements, it has been demonstrated that diamagnetic transition metal ions, such as the electron-deficient d8 PdII, can donate electron density to a lanthanide center, implying significant modifications on the magnetic properties of the resulting heterometallic PdII/LnIII compounds.13 Because of the tendency of the heavy PdII ion to form square planar geometries, the coordinated ligands should be able to satisfy these needs and facilitate the formation of stable species. A recent search in the Cambridge Crystallographic Data Centre (CCDC) revealed limited examples of non-organometallic PdII/LnIII coordination compounds. The majority of these involve carboxylate or thiocarboxylate ions as ligands, along with structures incorporating phosphines, arsenates, and derivatives of pyridine and pyrimidine.13,14 All of these complexes have been prepared via self-assembly synthetic routes using metal precursors and various ligand ‘blends’.
On the other hand, the ‘metal complexes as ligands’ strategy (often referred to as ‘metalloligands’) has been frequently employed in heterometallic chemistry of various metal ions.15 This synthetic route offers distinct advantages when compared to the commonly used self-assembly method. It enables more precise control over the reaction's progress and the resulting products. This modular approach facilitates the synthesis of larger molecules by combining mononuclear or dinuclear complexes with suitable polynucleating ligands capable of binding with different metal centers. In this work, the aim was to obtain heterotrinuclear PdII/LnIII complexes with a particular Ln–Pd–Ln configuration (Fig. 1), using ‘end-capping’ ligands for the LnIII ions and a central ‘metalloligand’ with bridging capabilities.
 | ||
| Fig. 1 Schematic representation for the targeted synthesis of heterotrimetallic complexes with the Ln–Pd–Ln configuration by using the appropriate ‘metalloligands’ and ‘end-capping’ ligands. | ||
To this direction, mononuclear metal oximate complexes appear to be promising building blocks.16 Our research group and others have conducted extensive studies in the field of coordination chemistry with ligands belonging to the family of 2-pyridyl oximes. These ligands can be described as ‘compartmentalized’ due to their affinity for binding ‘soft’ 3d-metal ions, such as NiII and CuII, through their N-donor atoms, and ‘hard’ 4f-metal ions through their O-donor(s), following the HSAB (Hard and Soft Acids and Bases) model.16,17
In our quest to explore the PdII/LnIII chemistry with this group of ligands, we directed our synthetic efforts toward the simplest member of the 2-pyridyl oxime family, known as 2-pyridinealdoxime (paoH, Fig. 2). In addition to yielding a plethora of structurally and magnetically intriguing homometallic 3d- and heterometallic 3d/3d′- and 3d/4f-metal complexes through self-assembly processes,18 paoH has gained recognition for its exceptional utility as a part of ‘metalloligands’. Upon deprotonation, pao− ligation seems capable of stabilizing square planar complexes via the pyridine and oximate N-atoms, while the unbound, ‘free’ oximate O-atom would bridge neighboring metal ions.19,20 This observation made us to suspect that such {Pd(oximate)2} species might have the potential to act as ‘metalloligands’, exploiting the nucleophilicity of the oximate anion and its ability to coordinate with oxophilic LnIII ions.
 | ||
| Fig. 2 Structural formula and abbreviation of the organic ligand 2-pyridinealdoxime (paoH) used in this study. The red and blue coloration illustrates the ‘hard’ O- and the ‘soft’ N-donor atoms, respectively. | ||
We report herein the high-yield synthesis and use of the mononuclear [Pd(pao)2] (1) ‘metalloligand’, bearing two trans oximate O-atoms readily available for binding to 4f-metal ions on opposite sides, for the isolation of a new family of [Ln2Pd(pao)2(NO3)6(MeOH)2(H2O)2]·[Pd(pao)2]4 (LnIII = Dy (2), Gd (3), Er (4), Yb (5)) complexes with a linear Ln–Pd–Ln conformation and additional co-crystallized [Pd(pao)2] units forming supramolecular stacks. Complexes 2 and 5 exhibit slow magnetization relaxation upon application of an external dc field.
2. Experimental section
a. Materials, physical and spectroscopic measurements
All manipulations were performed under aerobic conditions using materials (reagent grade) and solvents as received unless otherwise noted. Elemental analyses (C, H, and N) were performed by the University of Patras microanalytical service. Infrared (IR) spectra (4000–400 cm−1) were recorded in the solid state using a PerkinElmer 16 PC spectrometer with samples prepared as KBr pellets. Electrospray ionization (ESI) mass spectra (MS) were taken on a Bruker HCT Ultra mass spectrometer from a solution of complex 1 prepared in MeCN. Variable-temperature direct and alternating current (dc and ac, respectively) magnetic susceptibility studies were performed at the temperature range 1.9–300 K using a Quantum Design MPMS XL-7 SQUID magnetometer equipped with a 7 T magnet. Diamagnetic corrections were applied to the observed paramagnetic susceptibility using Pascal's constants.21b. Synthesis of [Pd(pao)2] (1)
To a stirred, colorless solution of paoH (0.05 g, 0.30 mmol) and Et3N (42 μL, 0.30 mmol) in solvent MeOH (15 mL) was added solid PdCl2 (0.03 g, 0.15 mmol). The resulting yellow slurry was refluxed for 6 h, during which time all the PdCl2 solid dissolved and the color of the solution remained yellow. The solution was then filtered, and the filtrate was allowed to evaporate slowly at room temperature. After two days, X-ray quality well-formed yellow needle-shaped crystals of 1·2H2O appeared, and these were collected by filtration, and washed with cold MeOH (2 × 2 mL) and Et2O (2 × 5 mL). The yield was 90% (based on the organic chelate available). Upon dryness (under vacuum) the crystalline solid was analyzed as 1. Anal. calc. for C12H10PdN4O2 (found values in parentheses): C 41.34% (41.37%), H 2.89% (2.94%) and N 16.07% (16.01%). Selected IR data (KBr, cm−1): v = 1668 (w), 1603 (m), 1510 (s), 1478 (m), 1424 (w), 1334 (s), 1248 (s), 1177 (m), 1151 (w), 1037 (w), 895 (w) 835 (m), 777 (w), 681 (w), 656 (w), 573 (w), 525 (w), 486 (w), 432 (m).c. Synthesis of [Dy2Pd(pao)2(NO3)6(MeOH)2(H2O)2]·[Pd(pao)2]4 (2)
To a stirred, clear yellow solution of the [Pd(pao)2] precursor (0.03 g, 0.10 mmol) in a solvent mixture comprising MeOH/MeCN (15 mL, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 v/v) was added solid Dy(NO3)3·5H2O (0.09 g, 0.20 mmol). The resulting yellow slurry was refluxed for 4 h, during which time all solids were dissolved and the color of the solution turned light orange. The solution was then filtered, and the filtrate was allowed to evaporate slowly at room temperature. After four days, X-ray quality orange needle-shaped crystals of 2 appeared, and these were collected by filtration, washed with cold MeCN (2 × 2 mL) and Et2O (2 × 5 mL), and dried in air. The yield was 30% (based on the total available Dy). The crystalline solid was analyzed as 2. Anal. calc. for C62H62Dy2Pd5N26O32 (found values in parentheses): C 29.31% (29.42%), H 2.46% (2.53%) and N 14.34% (14.22%). Selected IR data (KBr, cm−1): v = 1608 (s), 1517 (m), 1487 (m), 1434 (m), 1384 (s), 1349 (m), 1249 (s), 1182 (m), 1156 (w), 1030 (w), 902 (w) 835 (w), 770 (m), 682 (w), 662 (w), 575 (w), 527 (w), 431 (w).
:2 v/v) was added solid Dy(NO3)3·5H2O (0.09 g, 0.20 mmol). The resulting yellow slurry was refluxed for 4 h, during which time all solids were dissolved and the color of the solution turned light orange. The solution was then filtered, and the filtrate was allowed to evaporate slowly at room temperature. After four days, X-ray quality orange needle-shaped crystals of 2 appeared, and these were collected by filtration, washed with cold MeCN (2 × 2 mL) and Et2O (2 × 5 mL), and dried in air. The yield was 30% (based on the total available Dy). The crystalline solid was analyzed as 2. Anal. calc. for C62H62Dy2Pd5N26O32 (found values in parentheses): C 29.31% (29.42%), H 2.46% (2.53%) and N 14.34% (14.22%). Selected IR data (KBr, cm−1): v = 1608 (s), 1517 (m), 1487 (m), 1434 (m), 1384 (s), 1349 (m), 1249 (s), 1182 (m), 1156 (w), 1030 (w), 902 (w) 835 (w), 770 (m), 682 (w), 662 (w), 575 (w), 527 (w), 431 (w).
d. Synthesis of [Gd2Pd(pao)2(NO3)6(MeOH)2(H2O)2]·[Pd(pao)2]4 (3), [Er2Pd(pao)2(NO3)6(MeOH)2(H2O)2]·[Pd(pao)2]4 (4) and [Yb2Pd(pao)2(NO3)6(MeOH)2(H2O)2]·[Pd(pao)2]4 (5)
X-ray quality crystals of complexes 3–5 were prepared in the same manner as complex 2 but using Gd(NO3)3·5H2O (0.09 g, 0.20 mmol), Er(NO3)3·5H2O (0.09 g, 0.20 mmol), and Yb(NO3)3·5H2O (0.09 g, 0.20 mmol) as the lanthanide sources. The crystals were collected by filtration, washed with cold MeCN (2 × 2 mL) and Et2O (2 × 5 mL), and dried under in air; the yields were: 28% (3), 32% (4), and 25% (5). We have further confirmed the identities of 3–5 by (i) IR spectroscopic comparison with the authentic, single-crystalline sample of 2 (Fig. S2†), and (ii) elemental analyses. Anal. calc. for 3 (found values in parentheses): C 29.44% (29.53%), H 2.47% (2.56%) and N 14.39% (14.28%). Anal. calc. for 4 (found values in parentheses): C 29.20% (29.11%), H 2.45% (2.32%) and N 14.28% (14.39%). Anal. calc. for 5 (found values in parentheses): C 29.07% (29.19%), H 2.44% (2.54%) and N 14.22% (14.11%).e. Single-crystal X-ray crystallography
Yellow single-crystals of complex 1 (0.32 × 0.19 × 0.05 mm) and orange single-crystals of the family of complexes 2–5 (0.41 × 0.19 × 0.14 mm for 2; 0.45 × 0.08 × 0.07 mm for 3; 0.40 × 0.08 × 0.08 mm for 4; 0.43 × 0.09 × 0.06 mm for 5), were taken directly from the mother liquor and immediately cooled at 160(2) K. X-ray diffraction data were collected on a Rigaku R-AXIS SPIDER Image Plate diffractometer using graphite-monochromated MoKα (λ = 0.71073 Å) radiation. Data collection (ω-scans) and processing (cell refinement, data reduction, and empirical absorption correction) were performed using the CrystalClear program package.22 The structures of 1–5 were solved by direct methods using SHELXS ver. 2013/1,23 and refined by full-matrix least-squares techniques on F2 with SHELXL ver. 2014/6.24 All H atoms in the structures of 1–5 were introduced at calculated positions and refined as riding on their respective bonded atoms. All non-H atoms were refined anisotropically. It is important to mention that, in all structures, the ligand pao− is disordered over two positions. Specifically in 1, the pao− ligand occupies two positions with occupancies 0.581(5) and 0.489(5), respectively. In the structures of 2–5, the pao− ligand in the {Ln2Pd} heterometallic unit also occupies two positions with occupancies 0.705(5) and 0.295(5) in 2, 0.707(8) and 0.293(8) in 3, 0.635(7) and 0.365(7) in 4, and 0.60(1) and 0.40(1) in 5. Moreover, in the structures of 2–5, the coordinated pao− ligand to one of the [Pd(pao)2] units, defined by Pd2, also occupies two positions with occupancies 0.544(4) and 0.456(5) in 2, 0.537(7) and 0.463(7) in 3, 0.560(6) and 0.440(6) in 4, and 0.553(9) and 0.447(9) in 5. The discussion of the above disordered parts of the structures 1–5 is important for the comprehensive structural analysis and description of the supramolecular properties of these complexes (vide infra).Various figures of all structures were created, using Diamond 3 and Mercury software packages.25 The unit cell parameters, structure solution, and refinement details of the monomeric 1 and the heterometallic complexes 2–5 are summarized in Table S1.† Further crystallographic details of compounds 1–5 can be found in the corresponding CIF files provided in the ESI.†
3. Results and discussion
a. Synthetic comments and IR spectra
The most challenging step towards the preparation of the targeted heterometallic compounds has been the synthesis of a suitable ‘metalloligand’ which would bear free O-binding sites and simultaneously retain its solid-state structure in solution. Square planar palladium(II) complexes (d8, diamagnetic) bearing chelating ligands are both thermodynamically stable and kinetically inert, rendering themselves as ideal ‘metalloligands’ and fruitful building blocks for the synthesis of new compounds with interesting architectures and properties. Hence, from the reaction between PdCl2, paoH and Et3N in a 1:2:2 molar ratio, in solvent MeOH, under refluxing conditions to enhance the solubility of the PdCl2 starting material, we came across the yellow crystalline mononuclear compound [Pd(pao)2] (1) in excellent yields (∼90%). The general formation of 1 is summarized by the following stoichiometric eqn (1): | (1) |
Positive (+) ion ES-MS studies were conducted to investigate the stability of 1 in MeCN, a solvent which is widely used for the synthesis of heterometallic transition metal/lanthanide complexes.11 Indeed, complex 1 proved to preserve its structure in solution with a single, high-intensity ion signal located at 349.6 m/z and corresponding to the single-charged [Pd(pao)(paoH)]+ species (Fig. 3). The trans-arrangement of the free oximate O-atoms prompted us to carry out structure-directing reactions of 1 with two equivalents of different lanthanide(III) precursors, aiming at the preparation of {Ln–Pd–Ln} compounds.
 | ||
| Fig. 3 Positive ion ES-MS spectrum of 1 in MeCN. | ||
Indeed, from the 1:2 general reaction between 1 and Ln(NO3)3·5H2O (LnIII = DyIII, GdIII, ErIII, YbIII) in MeOH/MeCN under refluxing conditions, an orange-colored solution was obtained, from which orange crystals of the mixed-complex [Ln2Pd(pao)2(NO3)6(MeOH)2(H2O)2]·[Pd(pao)2]4 (Ln = DyIII (2); GdIII (3); ErIII (4); YbIII (5)) were formed in yields ranging from 25–35% depending on the lanthanide ion. The general formation of complexes 2–5 is summarized by the following stoichiometric eqn (2).
 | (2) |
Complexes 2–5 are stable and crystalline solids at room temperature and non-sensitive toward air and moisture. It is noteworthy to mention that in our hands it has not been possible to isolate the same crystalline compounds 2–5 from one-pot reactions between PdX2 (X− = various anions), Ln(NO3)3·5H2O and ligand paoH, under different conditions, solvents and external bases. It appears that the heterometallic complexes 2–5 can only be prepared in pure and crystalline forms by using complex 1 as the ‘metalloligand’ and Ln(NO3)3·5H2O as a dual provider of LnIII ions and NO3− ancillary groups (vide infra). For the preparation of 2–5, the choice of the reaction solvent mixture has been proved crucial for their crystallization and the formation of single-crystals; in the absence of either MeOH or MeCN, we have been unable to identify any heterometallic compound, contrariwise homometallic [PdCl2(paoH)2] complex was readily formed.26 Surprisingly, the heterometallic complexes 2–5 co-crystallize with four monomeric [Pd(pao)2] units (vide infra); that has been chemically unexpected given the fact that the crystalline 2–5 can be obtained only through the 1:2 reactions between the corresponding ‘metalloligand’ and the lanthanide precursors. When the stoichiometric 5:2 reactions between [Pd(pao)2] and Ln(NO3)3·5H2O were carried out, the ‘metalloligand’ has been the sole isolated product in all cases and by all chemical means. It is likely that the supramolecular packing of [Pd(pao)2] units facilitates the stabilization of the crystal lattice and eventually the formation of the mixed heterometallic–homometallic complexes.
The IR spectra of complexes 1–5 are presented in Fig. S1 and S2.† Complexes 2–5 are isostructural, and therefore only the IR spectrum of 2 will be discussed as a representative example. The presence of coordinated aqua and methanol ligands is confirmed by the appearance of a broad IR band centered at 3382 cm−1; the broad character of this band is indicative of hydrogen bonding interactions, as it turned out later by crystallographic studies. The IR spectrum of 2 shows the characteristic bands of bidentate chelating nitrato ligands. The bands at 1487 and 1249 cm−1 are assigned to the ν1(A1)[ν(N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)] and ν5(B2)[νas(NO2)] modes,27 the former probably overlapped with an aromatic stretch. The large separation (238 cm−1) of the two stretching nitrato modes indicates the bidentate character of these groups.27 The spectrum exhibits a medium intensity band at 1384 cm−1, characteristic of the ν3(E′)[νd(NO)] mode of the ionic nitrate with D3h symmetry.27 Such species do not exist in the crystal structure of the neutral compound 2. This is not unusual in IR spectroscopy of coordination compounds, and it can be rationalized in terms of a partial replacement of nitrato groups by bromides from the excess KBr that was used for the preparation of the IR pellet, thus producing ionic nitrates (KNO3);28 this solid-state replacement is facilitated by the employed hydraulic pressure. The IR spectrum of complex 2 also revealed the in-plane deformation band of the 2-pyridyl ring of the pao− ligands in the range ∼770–682 cm−1, thus confirming the participation of the ring's nitrogen atom in coordination.29 Several bands appear in 1609 cm−1 and in the ∼1517–1349 cm−1 range, assigned to contributions from the v(CN) vibrations and the stretching vibrations of the aromatic rings of pao− groups, respectively. The ν(N–O) band of the oximate group is situated at 1156 cm−1 and can be attributed to the increased presence of double bond character (NO) in the electronic structure of the oximate group, resulting from the deprotonation and coordination of the oximate O-atom. As a result, the ν(NO) vibration shifts to higher wavenumbers in the complexes that include deprotonated and coordinated oximate groups in comparison to the unbound, neutral ligands.30
O)] and ν5(B2)[νas(NO2)] modes,27 the former probably overlapped with an aromatic stretch. The large separation (238 cm−1) of the two stretching nitrato modes indicates the bidentate character of these groups.27 The spectrum exhibits a medium intensity band at 1384 cm−1, characteristic of the ν3(E′)[νd(NO)] mode of the ionic nitrate with D3h symmetry.27 Such species do not exist in the crystal structure of the neutral compound 2. This is not unusual in IR spectroscopy of coordination compounds, and it can be rationalized in terms of a partial replacement of nitrato groups by bromides from the excess KBr that was used for the preparation of the IR pellet, thus producing ionic nitrates (KNO3);28 this solid-state replacement is facilitated by the employed hydraulic pressure. The IR spectrum of complex 2 also revealed the in-plane deformation band of the 2-pyridyl ring of the pao− ligands in the range ∼770–682 cm−1, thus confirming the participation of the ring's nitrogen atom in coordination.29 Several bands appear in 1609 cm−1 and in the ∼1517–1349 cm−1 range, assigned to contributions from the v(CN) vibrations and the stretching vibrations of the aromatic rings of pao− groups, respectively. The ν(N–O) band of the oximate group is situated at 1156 cm−1 and can be attributed to the increased presence of double bond character (NO) in the electronic structure of the oximate group, resulting from the deprotonation and coordination of the oximate O-atom. As a result, the ν(NO) vibration shifts to higher wavenumbers in the complexes that include deprotonated and coordinated oximate groups in comparison to the unbound, neutral ligands.30
b. Description of structures
Due to the similarities in the crystal structures of complexes 2–5, only the structural features of heterometallic 2 will be discussed in detail as a representative example. Selected interatomic distances and angles for complexes 1 and 2 are listed in Tables S2 and S3,† respectively. Complexes 2–5 are crystal lattice solvate-free, while compound 1 crystallizes with two H2O solvate molecules.Complex 1 crystallizes in the centrosymmetric space group P21/a. The asymmetric unit of 1 contains half of the [Pd(pao)2]·2H2O units. The complex possesses a center of symmetry and the Pd atoms are located at the center of symmetry (Fig. 4a). The pao− ligands are disordered over two orientations (Fig. S3†). The PdII ion is four-coordinate in a square planar environment consisting of four N-atoms derived from the two bidentate chelating pao− ligands (Fig. 4a), as confirmed by the PdII–N bond distances and the N–Pd–N bond angles (Table S2†). The two coordinated pao− ligands are trans to each other. Both disordered sites of the pao− ligands display the same coordination mode around the PdII center.
 | ||
| Fig. 4 Partially labeled representation of the molecular structures of (a) the ‘metalloligand’ 1 and (b) the heterotrinuclear {Dy2Pd(pao)2(NO3)6(MeOH)2(H2O)2} unit of complex 2. All H atoms are omitted for clarity. Color scheme: PdII, magenta; DyIII, yellow; O, red; N, blue; C, gray. Symmetry operation for the primed atoms: (′): −x, 2 − y, 2 − z for 1 and (′): −x, −y, 1 − z for 2. | ||
In the structure of 1·2H2O, intramolecular and intermolecular hydrogen bonds are observed (Fig. S3 and Table S5†) for both orientations A and B of the coordinated pao− ligands. The [Pd(pao)2]·2H2O complexes with pao− ligands in orientation A form chains along c axis through C5A–H5A⋯O1Aii hydrogen bonding interactions (Fig. S4a, Table S5,† symmetry code (ii): x, y, z − 1), while those in orientation B form similar H bonds through the C1B–H1B⋯O1Biii interactions (Fig. S4b, Table S5,† symmetry code (iii): −x, −y + 2, −z + 1). The stacking of coordination compounds through phenyl-chelate ring interactions is observed in several square planar transition metal complexes.31 Fig. S5† represents the stacking of complexes along a-axis with the distance of the average planes of neighboring complexes being 3.37(1) Å for orientation A, a result that indicates that the pyridine rings favor also this type of stacking. Hence, the [Pd(pao)2] square planar complexes belong to fraction-2 of five-member chelating ring with fused 6-membered aromatic rings, i.e. the pyridine rings in this case.31c A parameter used to characterize this type of stacking is the difference of the distance of the centroids of the two pyridine rings with the distance of the centroid of the pyridine ring with the centroid of the chelate ring, represented by the symbol Δ (Δ = 0.736(1) Å). This positive value of Δ indicates a phenyl-chelate interaction type;31c however, in the case of 1 this interaction is a phenyl-metal type (inset of Fig. S5,† for orientations A and B). For orientation B, the distance of the average planes is 3.62(7) Å and Δ value is 0.912(1) Å. The complexes stacked along a-axis interact through C5A–H5A⋯O1A or C1B–H1B⋯O1B hydrogen bonds, thus forming layers parallel to the [010] planes which lie at the bottom or top (and in the middle) of the cell (Fig. S6†). Among these layers, zig-zag chains of lattice water molecules are formed through O1W–H1WB⋯O1Wvi hydrogen bonds (Table S5,† symmetry code: (vi): x + 1/2, −y + 3/2, z), which also assist in building the 3-D architecture of the structure; these interaction serve to further link complexes belonging to neighboring layers through the O1W–H1WA⋯O1A (or C1B–H1B⋯O1B) hydrogen bonds (Table S5†).
Compounds 2–5 crystalize in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) centrosymmetric space group; given that all four structures are isomorphous, only the structure of 2 (Fig. 4b) will be described in detail. The asymmetric unit of the cell contains half of the formula unit {[Dy2Pd(pao)2(NO3)6(MeOH)2(H2O)2]·[Pd*(pao)2]2·[Pd**(pao)2]2}, where Pd, Pd* and Pd** represent symmetry-independent PdII atoms and they correspond to the atoms labeled as Pd1, Pd2 and Pd3, respectively, in the following discussion. Thus, the structure of 2 included three types of complexes and they will be described separately. The structure of the complex [Dy2Pd(pao)2(NO3)6(MeOH)2(H2O)2] (2A) is shown in Fig. 4b and S7a.† Pd1 atom is located at a center of symmetry and the pao− ligands are also disordered as in 1. Pd2 and Pd3 occupy the same coordination environment as in 1, thus forming square planar co-complexes 2B and 2C, respectively, within the structure of 2. Although, Pd2 occupies a general position, the two symmetry-independent pao− chelates are disordered over positions “A” and “B” (Fig. S7b†). On the other hand, Pd3 also occupies a general position albeit in this case the two symmetry-independent pao− ligands are ordered (Fig. S7c†).
centrosymmetric space group; given that all four structures are isomorphous, only the structure of 2 (Fig. 4b) will be described in detail. The asymmetric unit of the cell contains half of the formula unit {[Dy2Pd(pao)2(NO3)6(MeOH)2(H2O)2]·[Pd*(pao)2]2·[Pd**(pao)2]2}, where Pd, Pd* and Pd** represent symmetry-independent PdII atoms and they correspond to the atoms labeled as Pd1, Pd2 and Pd3, respectively, in the following discussion. Thus, the structure of 2 included three types of complexes and they will be described separately. The structure of the complex [Dy2Pd(pao)2(NO3)6(MeOH)2(H2O)2] (2A) is shown in Fig. 4b and S7a.† Pd1 atom is located at a center of symmetry and the pao− ligands are also disordered as in 1. Pd2 and Pd3 occupy the same coordination environment as in 1, thus forming square planar co-complexes 2B and 2C, respectively, within the structure of 2. Although, Pd2 occupies a general position, the two symmetry-independent pao− chelates are disordered over positions “A” and “B” (Fig. S7b†). On the other hand, Pd3 also occupies a general position albeit in this case the two symmetry-independent pao− ligands are ordered (Fig. S7c†).
The heterotrinuclear centrosymmetric {Dy2Pd} unit can be described as a perfectly linear array of two external DyIII (Dy1, Dy1′) and a central PdII ions (Dy1–Pd1–Dy1′ angle = 180°), linked together through the oximate O-atoms (O1A, O1A′) of the {Pd(pao)2} ‘metalloligand’. Thus, the pao− ligands act as tridentate chelating and bridging, adopting the η1:η1:η1:μ coordination mode, with the ‘hard’ oximate O-atoms binding to the oxophilic DyIII atoms. Peripheral ligation about the two symmetry-related DyIII atoms is provided by six bidentate chelating NO3− groups, and two terminally bound aqua (O1W, O1W′), and two monodentate MeOH (O1M, O1M′) solvate molecules. The intramolecular Dy1⋯Dy1′ separation in 2 is 10.560 Å, presaging negligible magnetic interactions between the paramagnetic centers.
The Dy1 (and its symmetry-related Dy1′) atom is bound to nine O-donor atoms. One of them (O1A) belongs to the deprotonated oximate groups of the {Pd(pao)2} subunit, six oxygen atoms (O2, O3, O5, O6, O8, O9) belong to the three bidentate chelating nitrato groups, while the remaining two oxygen atoms belong to the terminal MeOH (O1M) and H2O (O1W) molecules. The Dy–O1A bond (2.282 Å) is the shortest among all Dy–O bond lengths (Table S3†), in accordance with similar separations in Dy–Ooximate complexes.30 The coordination geometry of Dy1 center was determined by the Continuous Shape Measures (CShM) approach of the SHAPE program,32 which allows one to numerically evaluate by how much a particular polyhedron deviates from the ideal shape (Table S4†). Thus, the coordination geometry of Dy1 (and Dy1′) in 2 (Fig. 5) can be described as distorted muffin (CShM value = 2.43) with the second closest polyhedron being that of capped square antiprism (CShM value = 2.57). The basal trigonal plane of the muffin is formed by O5, O6, and O8 atoms, while the equatorial pentagonal plane is made of O1M, O2, O9, O1W, and O3, and the oximate O1A atom occupies the vertex of the muffin.
 | ||
| Fig. 5 The distorted muffin coordination polyhedron of Dy1 (and Dy1′) in 2 with the corresponding atom labeling. The white thin lines define the vertices of the ideal polyhedron. Color scheme: DyIII, yellow; O, red. | ||
From a supramolecular perspective, there are hydrogen bonding interactions between the dangling oximate O-atoms as acceptors (O21A and O41) of the [Pd(pao)2] co-crystallized units and the O–H groups of the coordinated MeOH and H2O molecules as donors. Their dimensions are: O1W⋯O21A = 2.629 Å, O1W⋯O41 = 2.598 Å and O1M⋯O11A = 2.603 Å. These interactions serve to hold together the trinuclear {Dy2Pd} cluster with the [Pd(pao)2] units. Further intermolecular hydrogen bonds between molecules belonging to neighboring stacks of pentads stabilize the 3-D supramolecular structure of 2.
In the crystal lattice, complexes 2A, 2B and 2C are stacked in groups of five as shown in Fig. 6, along the [101] direction. Complex 2A is located at the middle of the stack and since the {Dy2Pd} unit possesses a center of symmetry, 2B and 2C, which are placed below and above the central unit, are also related by center of symmetry and indicated by Pd2, Pd3 and Pd2′, Pd3′ in Fig. 6. The stacking of complexes extends along both directions of [101] through their overlap with Pd3′′ and Pd3′′′ containing complexes that belong to neighboring pentads (Fig. 6). The planar {Pd(pao)2} units within 2 belong also to fraction-2 as in the case of 1. The type of stacking between 2A and 2B, and 2B and 2C, are of phenyl-chelate interaction (Fig. 6, right) with Δ values of 0.433(1) and 0.906(1) Å, respectively.31c The angle between the planes of 2A and 2B is 1.3(3)° and between 2B and 2C is 2.0(2)°. The distance of Pd1 from the mean plane of 2B is 3.297(1) Å and that of Pd2 from 2C is 3.264(1) Å. Complexes containing Pd3 and Pd3′′′ atoms are centrosymmetrically-related, and they overlap in a pyridine-metal interaction with a Δ value of 1.061(1) Å and a distance of 3.351(4) Å along the planes.
 | ||
| Fig. 6 (left) Group of five complexes featuring Pd1, Pd2, Pd3 and their centrosymmetric analogues, Pd2′ and Pd3′, stacked along the [101] crystallographic direction. (right) Representations of the overlap of complexes 2A, 2b and 2C with each other. Symmetry codes (′): −x, −y, 1 − z; (′′): 1 + x, y, 1 + z; (′′′): −1 − x, −y, −z. | ||
Finally, complexes 2–5 represent rare examples of discrete, non-polymeric, heterometallic compounds with a LnIII–MII–LnIII linear conformation,33 and they constitute the only family of linear {Ln2Pd} complexes reported to date. The only other related example is the complex [Dy2Pd{(py)2CNO}2(hfac)6], where (py)2CNOH is the ligand di-2-pyridyl ketoxime and hfacH is 1,1,1,5,5,5-hexafluoropentane-2,4-dione, reported by Okazawa, Ishida and coworkers.34 In that case, the terminal DyIII ions are 8-coordinate with distorted square antiprismatic geometries.
c. Magnetic studies
Direct current (dc) magnetic susceptibility (χM) data on analytically pure, microcrystalline samples of 2–5 were collected in the 2–300 K temperature range under an applied magnetic field of 0.1 T. The data are plotted as χMT products vs. T in Fig. 7. From a magnetism perspective, complexes 2–5 can be viewed as “homodinuclear” lanthanide(III) species, provided that square planar Pd(II) is diamagnetic. Note that the very long intramolecular Ln⋯Ln distances in 2–5 preclude the consideration of any significant magnetic interactions between the metal centers. The room temperature χMT values for compounds 2 (28.32 cm3 K mol−1), 3 (15.38 cm3 K mol−1), 4 (22.31 cm3 K mol−1) and 5 (5.05 cm3 K mol−1) are in excellent agreement with the expected values of 28.34, 15.76, 22.96 and 5.14 cm3 K mol−1, for two DyIII (6H15/2, free ion; S = 5/2, L = 5, gJ = 4/3; χMT = 14.17 cm3 K mol−1), GdIII (8S7/2, free ion; S = 7/2, L = 0, gJ = 2; χMT = 7.88 cm3 K mol−1), ErIII (4I15/2, free ion; S = 3/2, L = 6, gJ = 6/5; χMT = 11.48 cm3 K mol−1) and YbIII (2F7/2, free ion; S = 1/2, L = 3, gJ = 8/7; χMT = 2.57 cm3 K mol−1) noninteracting ions, respectively.35 Upon cooling, the χMT products of complexes 2, 4 and 5 decrease slowly in the 300–50 K temperature range and then more rapidly reaching the values of 23.39 (2), 13.31 (4) and 3.22 (5) cm3 K mol−1 at 2.0 K, while the χMT product of the isotropic complex 3 remains almost constant at the entire temperature regime. The observed decrease of the χMT products is mostly due to the depopulation of the mJ sublevels of the ground J state of the anisotropic LnIII ions.36 The χMT versus T data for 3 were fit to an isotropic 1-J model that accounts for the long pathway Gd⋯Gd interaction (solid line in Fig. 7). The best-fit parameters were: J = −0.0012(3) cm−1 and g = 1.98(2), thus corroborating the negligible magnetic interactions between the metal centers. | ||
| Fig. 7 Plots of χMT vs. T for complexes 2–5 in a 0.1 T dc field. The solid pink line is the fit of the data for the isotropic GdIII-analogue; see the text for the fit parameters. | ||
The field dependence of the magnetization at 1.9, 3 and 5 K, for the magnetically more prominent complexes 2, 4 and 5, shows a relatively rapid increase at low fields without reaching saturation at ∼7 T, which indicates the presence of magnetic anisotropy (Fig. 8). Furthermore, the magnetization values of 2, 4 and 5 at 7 T are ∼10, 12 and 2.5NμB, respectively, much lower than the expected values for two (n = 2) DyIII (MS/NμB = ngJJ = 20NμB), ErIII (MS/NμB = ngJJ = 18NμB) and YbIII (MS/NμB = ngJJ = 8NμB) ions, which is due to crystal field effects that induce magnetic anisotropy. For complex 3, the magnetization value at 7 T is 13.7NμB, very close to the theoretical value for two isotropic GdIII ions (MS/NμB = ngJJ = 14NμB) (Fig. S8†).
 | ||
| Fig. 8 Plots of magnetization (M) vs. field (H) for complexes 2, 4 and 5 at three different low temperatures. The solid lines are guides for the eye only. | ||
The presence of anisotropic LnIII atoms prompted us to explore the magnetic dynamics of compounds 2, 4 and 5, which comprise either oblate (DyIII) or prolate (ErIII, YbIII) metal ions with dissimilar distribution of the f-electron density. Our goal was to determine whether these compounds exhibit (or not) slow relaxation of magnetization due to the electrostatic interactions of the crystal field (CF) with the oblate/prolate type electron density of the lanthanide ions. According to this electrostatic model, the oblate ions, having a strong axial CF below and above the xy basal plane, stabilize the largest mJ and maximize the uniaxial anisotropy. However, the prolate ions favor the equatorial coordination geometry to minimize the charge repulsion with the axial electron density.
Hence, we initially conducted alternating current (ac) magnetic susceptibility measurements without an applied dc field, using a 3.5 G ac field oscillating at various frequencies within the temperature range of 2–40 K. One common observation across all compounds was the total absence of frequency dependent signals in the out-of-phase 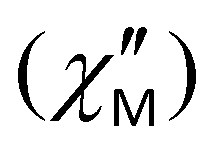 susceptibilities vs. T plots. However, by analyzing the dc field dependence of the
susceptibilities vs. T plots. However, by analyzing the dc field dependence of the  at 997 Hz and 1.9 K for compounds 2 (Fig. S9†) and 5 (Fig. S10†), we were able to identify dc fields of 300 and 2000 Oe, which could be suitable (optimum fields) for quenching the operative quantum tunnelling of the magnetization (QTM), a relaxation pathway responsible for the fast magnetization reversal of SMMs at low temperatures. Therefore, we carried out ac studies in the presence of the external dc fields.
at 997 Hz and 1.9 K for compounds 2 (Fig. S9†) and 5 (Fig. S10†), we were able to identify dc fields of 300 and 2000 Oe, which could be suitable (optimum fields) for quenching the operative quantum tunnelling of the magnetization (QTM), a relaxation pathway responsible for the fast magnetization reversal of SMMs at low temperatures. Therefore, we carried out ac studies in the presence of the external dc fields.
Indeed, the appearance of frequency dependent tails of ac signals, in the out-of-phase  susceptibilities vs. T plots for 2 (Fig. 9a) under a 300 Oe applied dc field, at temperatures below ∼6 K, indicates the onset of magnetization relaxation and weak SMM behavior. These tails of
susceptibilities vs. T plots for 2 (Fig. 9a) under a 300 Oe applied dc field, at temperatures below ∼6 K, indicates the onset of magnetization relaxation and weak SMM behavior. These tails of  signals suggest that QTM is still the dominant relaxation mechanism, implying a small effective energy barrier (Ueff). Given the absence of peaks in the
signals suggest that QTM is still the dominant relaxation mechanism, implying a small effective energy barrier (Ueff). Given the absence of peaks in the  vs. T plots, we determined the SMM parameters by assuming that the magnetization relaxation follows a Debye process with only one characteristic time, allowing us to apply the Kramers–Kronig equations,37,38 resulting in the combined eqn (3), where ω is the angular frequency, τ0 is the pre-exponential factor, and kB is the Boltzmann's constant.
vs. T plots, we determined the SMM parameters by assuming that the magnetization relaxation follows a Debye process with only one characteristic time, allowing us to apply the Kramers–Kronig equations,37,38 resulting in the combined eqn (3), where ω is the angular frequency, τ0 is the pre-exponential factor, and kB is the Boltzmann's constant.
| ln(χ′′/χ′) = ln(ωτ0) + Ueff/kBT | (3) |
 | ||
Fig. 9 (a) Temperature dependence of the out-of-phase 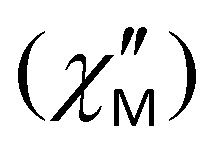 ac magnetic susceptibility under a 300 Oe applied dc field for complex 2, measured in a 3.5 G ac field oscillating at the indicated frequencies. The solid lines are guides only. (b) Debye plots of complex 2 for the indicated ac frequencies. The solid lines correspond to the fit of the data by applying the Kramers–Kronig eqn (3). ac magnetic susceptibility under a 300 Oe applied dc field for complex 2, measured in a 3.5 G ac field oscillating at the indicated frequencies. The solid lines are guides only. (b) Debye plots of complex 2 for the indicated ac frequencies. The solid lines correspond to the fit of the data by applying the Kramers–Kronig eqn (3). | ||
Using eqn (3), we derived the best-fit parameters for compound 2 (Fig. 9b), which gave: Ueff = ∼6.9(1) K and τ0 = 1.6(1) × 10−6 s. These values are consistent with the expected properties of a fast-relaxing SMM.11 The resulting energy barrier is quite small, indicating that a thermally assisted Orbach process is unlikely to be the operative mechanism for the magnetization reversal in 2.39
Under a 2000 Oe applied dc field, complex 5 displays frequency dependent in-phase  (Fig. S11†) and out-of-phase
(Fig. S11†) and out-of-phase 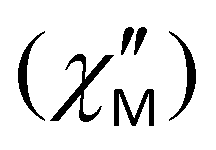 (Fig. 10a) signals below 3.0 K, thus suggesting the presence of slow relaxation of the magnetization consistent with a field-induced SMM behavior.40 The Cole–Cole plots of 5 were fitted at the low temperature regime using a generalized Debye model (Fig. S12†), and the obtained α parameters were found in the range of 0.08–0.13, indicating a relatively narrow distribution of relaxation times, which agrees with the presence of one predominant relaxation processes.38 To determine the temperature dependence of relaxation times (τ) and construct an Arrhenius-type plot, we fitted the data including only the Orbach relaxation mechanism (Fig. 10b); no reasonable values were obtained by fitting the data including Raman and QTM relaxation processes. The data in the temperature range of 1.9 to 3.0 K were analyzed using the following eqn (4):
(Fig. 10a) signals below 3.0 K, thus suggesting the presence of slow relaxation of the magnetization consistent with a field-induced SMM behavior.40 The Cole–Cole plots of 5 were fitted at the low temperature regime using a generalized Debye model (Fig. S12†), and the obtained α parameters were found in the range of 0.08–0.13, indicating a relatively narrow distribution of relaxation times, which agrees with the presence of one predominant relaxation processes.38 To determine the temperature dependence of relaxation times (τ) and construct an Arrhenius-type plot, we fitted the data including only the Orbach relaxation mechanism (Fig. 10b); no reasonable values were obtained by fitting the data including Raman and QTM relaxation processes. The data in the temperature range of 1.9 to 3.0 K were analyzed using the following eqn (4):
| τ−1 = τ0−1exp(−Ueff/kBT) | (4) |
exp(−Ueff/kBT) corresponds to the Orbach process, in which τ0 is the pre-exponential factor, kBT is the thermal energy, and Ueff is the effective energy barrier for the magnetization reversal. As the thermally assisted Orbach process dominates, the relaxation time has an exponential dependence on temperature (linear region), giving a Ueff value of ∼6.0 K as well as a τ0 value of 1.9 × 10−5 s.
 | ||
Fig. 10 (a) Frequency dependence of the out-of-phase 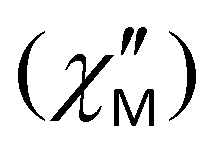 ac magnetic susceptibility under a 2000 Oe applied dc field for complex 5, measured in a 3.5 G ac field over the temperature range 1.9–3.5 K. Solid lines represent fits to the data in the frequency range 10–1500 Hz. (b) Arrhenius-type plot of ln(τ) vs. 1/T for complex 5, under a 2000 Oe applied dc field. The solid red line corresponds to the fit of the data by applying eqn (4). ac magnetic susceptibility under a 2000 Oe applied dc field for complex 5, measured in a 3.5 G ac field over the temperature range 1.9–3.5 K. Solid lines represent fits to the data in the frequency range 10–1500 Hz. (b) Arrhenius-type plot of ln(τ) vs. 1/T for complex 5, under a 2000 Oe applied dc field. The solid red line corresponds to the fit of the data by applying eqn (4). | ||
Although there are many {Yb2} complexes exhibiting field-induced relaxation of magnetization,41 complex 5 is the first example of a slow-relaxing magnetic species in which the YbIII ions are separated by such a long-distance spacer, i.e. the ‘metalloligand’ {Pd(pao)2}.
4. Conclusions
In summary, this work has demonstrated the effectiveness of the ‘metal complexes as ligands’ approach as a successful route for the controlled synthesis of new heterometallic PdII/LnIII complexes with targeted nuclearities and topologies. By utilizing the [Pd(pao)2] building block (pao− = anion of 2-pyridinealdoxime), we unveiled the coordination of the two dangling, trans oximate, O-atoms of pao− ligands with various LnIII ions, which led to a new family of linear compounds with the general formula [Ln2Pd(pao)2(NO3)6(MeOH)2(H2O)2]·[Pd(pao)2]4, where LnIII = Dy (2), Gd (3), Er (4), Yb (5). Compounds 2–5 constitute the only family of {Ln2Pd} molecular species reported to date. Apart from their structural (and supramolecular) interest, compounds 2 and 5 also display field-induced slow magnetization relaxation under applied dc fields of 300 and 2000 Oe, respectively. However, this is accompanied by relatively low energy barriers for the magnetization reversal due to the fast tunneling mechanism and the weak crystal field effects from the coordinated ligands which constitute the ‘muffin’-like polyhedron of the LnIII ions.We are currently trying to expand this synthetic route to various {Pd(oximate)2} ‘metalloligands’ with either trans or cis unbound oximate O-atoms in an attempt to direct not only the nuclearity (trimers or dimers) and dimensionality (0-D or 1/2-D) of the resulting heterometallic products but also the coordination of the negatively charged oximate O-atoms towards the desired axial or equatorial site (depending on the oblate or prolate LnIII ion).
Data availability
The data supporting this article has been included as part of the ESI.† Crystallographic data for 1–5 have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under 2385399 (1), 2385400 (2), 2385401 (3), 2385402 (4), and 2385403 (5).†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The research work was supported by the Hellenic Foundation for Research and Innovation (HFRI) under the 3rd Call for HFRI PhD Fellowships (Fellowship Number: 5618).References
- R. Sessoli, H. Tsai, A. R. Schake, S. Wang, J. Vincent, K. Folting, D. Gatteschi, G. Christou and D. N. Hendrickson, J. Am. Chem. Soc., 1993, 115, 1804 CrossRef CAS.
- R. Sessoli, D. Gatteschi and J. Villain, in Molecular Nanomagnets, Oxford University Press, Oxford, UK, 2006 Search PubMed.
- (a) B. Zhou, R. Tao, S. Q. Shen and J. Q. Liang, Phys. Rev. A, 2002, 66, 010301 CrossRef; (b) J. Tejada, E. M. Chudnovsky, E. del Barco, J. M. Hernandez and T. P. Spiller, Nanotechnology, 2001, 12, 181–186 CrossRef CAS; (c) M. Mannini, F. Pineider, C. Danieli, F. Totti, L. Sorace, P. Sainctavit, M. A. Arrio, E. Otero, L. Joly, J. C. Cezar, A. Cornia and R. Sessoli, Nature, 2010, 468, 417–421 CrossRef CAS PubMed; (d) M. Bottrill, L. Kwok and N. J. Long, Chem. Soc. Rev., 2006, 35, 557–571 RSC.
- M. Nakano and H. Oshio, Chem. Soc. Rev., 2011, 40, 3239 RSC.
- N. Ishikawa, M. Sugita, T. Ishikawa, S. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694 CrossRef CAS PubMed.
- (a) D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110 CrossRef CAS; (b) P. Zhang, Y. N. Guo and J. Tang, Coord. Chem. Rev., 2013, 257, 1728 CrossRef CAS; (c) P. Zhang, L. Zhang and J. Tang, Dalton Trans., 2015, 44, 3923 RSC; (d) S. T. Liddle and J. van Slageren, Chem. Soc. Rev., 2015, 44, 6655 RSC.
- F. S. Guo, B. M. Day, Y. C. Chen, M. L. Tong, A. Mansikkamäki and R. A. Layfield, Science, 2018, 362, 1400–1403 CrossRef CAS.
- L. Sorace, C. Benelli and D. Gatteschi, Chem. Soc. Rev., 2011, 40, 3092 RSC.
- Y. N. Guo, G. F. Xu, Y. Guo and J. Tang, Dalton Trans., 2011, 40, 9953 RSC.
- N. Ishikawa, M. Sugita, T. Okubo, N. Takana, T. Iino and Y. Kaizu, Inorg. Chem., 2003, 42, 2440–2446 CrossRef CAS PubMed.
- (a) A. Chakraborty, J. Goura, P. Kalita, A. Swain, G. Rajaraman and V. Chandrasekhar, Dalton Trans., 2018, 47, 8841–8864 RSC; (b) N. C. Anastasiadis, C. D. Polyzou, G. E. Kostakis, V. Bekiari, Y. Lan, S. P. Perlepes, K. F. Konidaris and A. K. Powell, Dalton Trans., 2015, 44, 19791–19795 RSC; (c) A. S. Armenis, V. Vipanchi, K. N. Pantelis, L. Cunha-Silva, K. R. Vignesh, D. I. Alexandropoulos and Th. C. Stamatatos, Chem. – Eur. J., 2023, e202302337 CrossRef CAS.
- (a) A. Upadhyay, S. K. Singh, C. Das, R. Mondol, S. K. Langley, K. S. Murray, G. Rajaraman and M. Shanmugam, Chem. Commun., 2014, 50, 8838–8841 RSC; (b) I. Oyarzabal, J. Ruiz, J. M. Seco, M. Evangelisti, A. Camon, E. Ruiz, D. Aravena and E. Colacio, Chem. – Eur. J., 2014, 20, 14262–14269 CrossRef CAS PubMed; (c) A. Upadhyay, C. Das, S. Vaidya, S. K. Singh, T. Gupta, R. Mondol, S. K. Langley, K. S. Murray, G. Rajaraman and M. Shanmugam, Chem. – Eur. J., 2017, 23, 4903–4916 CrossRef CAS.
- (a) M. A. Sørensen, H. Weihe, M. G. Vinum, J. S. Mortensen, L. H. Doerrerb and J. Bendix, Chem. Sci., 2017, 8, 3566 RSC; (b) D. C. Izuogu, T. Yoshida, H. Zhang, G. Cosquer, K. Katoh, S. Ogata, M. Hasegawa, H. Nojiri, M. Damjanovic, W. Wernsdorfer, T. Uruga, T. Ina, B. K. Breedlove and M. Yamashita, Chem. – Eur. J., 2018, 24, 9285–9294 CrossRef CAS PubMed; (c) D. Chukwuma Izuogu, T. Yoshida, G. Cosquer, J. N. Asegbeloyin, H. Zhang, A. J. W. Thom and M. Yamashita, Chem. – Eur. J., 2020, 26, 6036–6049 CrossRef; (d) M. Karbowiak, C. Rudowicz and T. Ishida, Inorg. Chem., 2013, 52, 13199–13206 CrossRef CAS PubMed.
- (a) D. Osypiuk, B. Cristóvão and L. Mazur, J. Mol. Struct., 2022, 1261, 132924 CrossRef CAS; (b) Z. G. Lin, L. Y. Fan, A. Kondinski, N. Vankova, T. Heine, B. K. Chen, A. Haider, B. Wang, U. Kortz and C. W. Hu, Inorg. Chem., 2016, 55, 7811–7813 CrossRef CAS PubMed; (c) S. L. Huang, A. Q. Jiab and G. X. Jin, Chem. Commun., 2013, 49, 2403–2405 RSC; (d) M. Barsukova, N. V. Izarova, R. N. Biboum, B. Keita, L. Nadjo, V. Ramachandran, N. S. Dalal, N. S. Antonova, J. J. Carb, J. M. Poblet and U. Kortz, Chem. – Eur. J., 2010, 16, 9076–9085 CrossRef CAS; (e) B. Miroslaw, B. Cristóvão and Z. Hnatejko, Molecules, 2018, 23, 2423 CrossRef; (f) S. E. Nefedov, N. Y. Kozitsyna, M. N. Vargaftik and I. I. Moiseev, Polyhedron, 2009, 28, 172–180 CrossRef CAS; (g) Z. Wang, L. P. Zhou, T. H. Zhao, L. X. Cai, X. Q. Guo, P. F. Duan and Q. F. Sun, Inorg. Chem., 2018, 57, 7982–7992 CrossRef CAS PubMed; (h) M. B. Stuckart, N. V. Izarova, R. A. Barrett, Z. Wang, J. van Tol, H. W. Kroto, N. S. Dalal, P. J. Lozano, J. J. Carbó, J. M. Poblet, M. S. von Gernler, T. Drewello, P. de Oliveira, B. Keita and U. Kortz, Inorg. Chem., 2012, 51, 13214–13228 CrossRef; (i) L. You, W. Zhu, S. Wang, G. Xiong, F. Ding, B. Ren, I. Dragutan, V. Dragutan and Y. Sun, Polyhedron, 2016, 115, 47–53 CrossRef CAS; (j) F. Volcker, F. M. Muck, K. D. Vogiatzis, K. Fink and P. W. Roesky, Chem. Commun., 2015, 51, 11761 RSC; (k) J. A. R. Navarro and J. M. Salas, Chem. Commun., 2000, 235–236 RSC; (l) L. X. You, L. X. Cui, B. B. Zhao, G. Xiong, F. Ding, B. Y. Ren, Z. L. Shi, I. Dragutan, V. Dragutan and Y. G. Sun, Dalton Trans., 2018, 47, 8755 RSC; (m) Z. Lang, P. Yang, Z. Lin, L. Yan, M. X. Li, J. J. Carbo, U. Kortz and J. M. Poblet, Chem. Sci., 2017, 8, 7862 RSC.
- G. Kumar and R. Gupta, Chem. Soc. Rev., 2013, 42, 9403 RSC.
- P. Chaudhuri, Coord. Chem. Rev., 2003, 243, 143 CrossRef CAS.
- C. J. Milios, Th. C. Stamatatos and S. P. Perlepes, Polyhedron, 2006, 25, 134–194 CrossRef CAS.
- (a) M. Murugesu, K. A. Abboud and G. Christou, Polyhedron, 2004, 23, 2779–2788 CrossRef CAS; (b) C. D. Polyzou, Z. G. Lada, A. Terzis, C. P. Raptopoulou, V. Psycharis and S. P. Perlepes, Polyhedron, 2014, 79, 29–36 CrossRef CAS; (c) Th. C. Stamatatos, K. A. Abboud, S. P. Perlepes and G. Christou, Dalton Trans., 2007, 3861–3863 RSC; (d) K. F. Konidaris, E. Katsoulakou, M. Kaplanis, V. Bekiari, A. Terzis, C. P. Raptopoulou, E. M. Zoupa and S. P. Perlepes, Dalton Trans., 2010, 39, 4492–4494 RSC; (e) S. Zhang, L. Zhen, B. Xu, R. Inglis, K. Li, W. Chen, Y. Zhang, K. F. Konidaris, S. P. Perlepes, E. K. Brechin and Y. Li, Dalton Trans., 2010, 39, 3563–3571 RSC; (f) C. M. Liu, D. Q. Zhang and D. B. Zhu, RSC Adv., 2014, 4, 53870 RSC; (g) D. I. Alexandropoulos, C. Papatriantafyllopoulou, G. Aromi, O. Roubeau, S. J. Teat, S. P. Perlepes, G. Christou and Th. C. Stamatatos, Inorg. Chem., 2010, 49, 3962–3964 CrossRef CAS; (h) C. Papatriantafyllopoulou, Th. C. Stamatatos, C. G. Efthymiou, L. C. Silva, F. A. A. Paz, S. P. Perlepes and G. Christou, Inorg. Chem., 2010, 49, 9743–9745 CrossRef CAS.
- (a) S. Khanra, B. Biswas, C. Golze, B. Buchner, V. Kataev, T. Weyhermuller and P. Chaudhuri, Dalton Trans., 2007, 481–487 RSC; (b) C. D. Polyzou, E. S. Koumousi, Z. G. Lada, C. P. Raptopoulou, V. Psycharis, M. Rouzières, A. C. Tsipis, C. Mathonière, R. Clérac and S. P. Perlepes, Dalton Trans., 2017, 46, 14812 RSC; (c) P. Chaudhuri, T. Weyhermüller, R. Wagner, S. Khanra, B. Biswas, E. Bothe and E. Bill, Inorg. Chem., 2007, 46, 9003–9016 CrossRef CAS.
- (a) K. W. Nordquest, D. W. Phelps, W. F. Little and D. J. Hodgson, J. Am. Chem. Soc., 1976, 98(5), 1104–1107 CrossRef CAS; (b) A. A. Torabi, A. SoudoziI and R. Welter, Z. Kristallogr. - New Cryst. Struct., 2007, 222, 197–198 CAS; (c) K. Ha, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, m176–m177 CrossRef CAS PubMed.
- G. A. Bain and J. F. J. Berry, Chem. Educ., 2008, 85, 532 CrossRef CAS.
- CrystalClear, Rigaku/MSC Inc., The Woodlands, Texas, 2005 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
- (a) K. Brandenburg, DIAMOND, Release 3. 1f, Crystal Impact GbR, Bonn, Germany, 2008, p. 389 Search PubMed; (b) I. J. Bruno, J. C. Cole, P. R. Edgington, M. K. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, MERCURY, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389 CrossRef PubMed.
- K. Ha, Crystal structure of dichloro(2-pyridinealdoxime-κ2N,N′)palladium(II), PdCl2(C6H6N2O), Z. Kristallogr., 2011, 226, 525–526 CAS.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Wiley, New York, 4th edn, 1986, pp. 254–257 Search PubMed.
- G. J. Kleywegt, W. G. R. Wiesmeijer, G. J. van Driel, W. L. Driessen, J. Reedijk and J. H. Noordik, J. Chem. Soc., Dalton Trans., 1985, 2177 RSC.
- A. B. P. Lever and E. Mantovani, Inorg. Chem., 1971, 10, 817 CrossRef CAS.
- (a) C. D. Polyzou, H. Nikolaou, C. Papatriantafyllopoulou, V. Phycharis, A. Terzis, C. P. Raptopoulou, A. Escuer and S. P. Perlepes, Dalton Trans., 2012, 41, 13755 RSC; (b) Th. C. Stamatatos, E. Katsoulakou, A. Terzis, C. P. Raptopoulou, R. E. P. Winpenny and S. P. Perlepes, Polyhedron, 2009, 28, 1638 CrossRef CAS; (c) Th. C. Stamatatos, A. Escuer, K. A. Abboud, C. P. Raptopoulou, S. P. Perlepes and G. Christou, Inorg. Chem., 2008, 47, 11825–11838 CrossRef CAS PubMed.
- (a) D. N. Sredojevic, Z. D. Tomic and S. D. Zaric, Cent. Eur. J. Chem., 2007, 5, 20–31 CAS; (b) Z. D. Tomic, D. N. Sredojevic and S. D. Zaric, Cryst. Growth Des., 2006, 6, 29–31 CrossRef CAS; (c) D. N. Sredojevic, D. Z. Vojislavljevic, Z. D. Tomic and S. D. Zaric, Acta Crystallogr., Sect. B: Struct. Sci., 2012, 68, 261–265 CrossRef CAS.
- M. Llunell, D. Casanova, J. Girera, P. Alemany and S. Alvarez, SHAPE, Version 2.0, Universitat de Barcelona, Barcelona, Spain, 2010 Search PubMed.
- (a) F. Mori, T. Nyui, T. Ishida, T. Nogami, K.-Y. Choi and H. Nojiri, J. Am. Chem. Soc., 2006, 128, 1440–1441 CrossRef CAS PubMed; (b) A. Okazawa, R. Watanabe, M. Nezu, T. Shimada, S. Yoshii, H. Nojiri and T. Ishida, Chem. Lett., 2010, 39, 1331–1332 CrossRef CAS; (c) J. McGuire, B. Wilson, J. McAllister, H. N. Miras, C. Wilson, S. Sproules and J. H. Farnaby, Dalton Trans., 2019, 48, 5491–5495 RSC; (d) X. Zhu, W.-K. Wong, J. Guo, W.-Y. Wong and J.-P. Zhang, Eur. J. Inorg. Chem., 2008, 3515–3523 CrossRef CAS; (e) T. Sanada, T. Suzuki and S. Kaizaki, J. Chem. Soc., Dalton Trans., 1998, 959–965 RSC.
- A. Okazawa, H. Nojiri, T. Ishida and N. Kojima, Polyhedron, 2011, 30, 3140–3144 CrossRef CAS.
- G. G. Morgan and I. A. Kuhne, Practical Approaches to Biological Inorganic Chemistry (Second Edition): Molecular magnetochemistry, Chapter 3, School of Chemistry, University College Dublin, Dublin, Ireland, 2020 Search PubMed.
- J. L. Liu, Y. C. Chen and M. L. Tong, Chem. Soc. Rev., 2018, 47, 2431 RSC.
- (a) M. Grahl, J. Kotzler and I. Sessler, J. Magn. Magn. Mater., 1990, 90–91, 187–188 CrossRef CAS; (b) J. Bartolomé, G. Filoti, V. Kuncser, G. Schinteie, V. Mereacre, C. E. Anson, A. K. Powell, D. Prodius and C. Turta, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 014430–014446 CrossRef.
- K. S. Cole and R. H. Cole, J. Chem. Phys., 1941, 9, 341–351 CrossRef CAS.
- Y. N. Guo, G. F. Xu, Y. Guo and J. Tang, Dalton Trans., 2011, 40, 9953–9963 RSC.
- S. T. Liddle and J. van Slageren, Chem. Soc. Rev., 2015, 44, 6655–6669 RSC.
- (a) P.-H. Lin, W.-B. Sun, Y.-M. Tian, P.-F. Yan, L. Ungur, L. F. Chibotaru and M. Murugesu, Dalton Trans., 2012, 41, 12349–12352 RSC; (b) W.-B. Chen, L. Zhong, Y.-J. Zhong, Y.-Q. Zhang, S. Gao and W. Dong, Inorg. Chem. Front., 2020, 7, 3136–3145 RSC; (c) A. Mondal and S. Konar, Chem. – Eur. J., 2021, 27, 3449–3456 CrossRef CAS PubMed; (d) B. Lefeuvre, J. Flores Gonzalez, F. Gendron, V. Dorcet, F. Riobé, V. Cherkasov, O. Maury, B. Le Guennic, O. Cador, V. Kuropatov and F. Pointillart, Molecules, 2020, 25, 492 CrossRef CAS; (e) A. Gorczyński, D. Marcinkowski, M. Kubicki, M. Löffler, M. Korabik, M. Karbowiak, P. Wiśniewski, C. Rudowicz and V. Patroniak, Inorg. Chem. Front., 2018, 5, 605–618 RSC; (f) T.-Q. Liu, P.-F. Yan, F. Luan, Y.-X. Li, J.-W. Sun, C. Chen, F. Yang, H. Chen, X.-Y. Zou and G.-M. Li, Inorg. Chem., 2015, 54, 221–228 CrossRef CAS PubMed; (g) G. Brunet, R. Marin, M.-J. Monk, U. Resch-Genger, D. A. Gálico, F. A. Sigoli, E. A. Suturina, E. Hemmer and M. Murugesu, Chem. Sci., 2019, 10, 6799–6808 RSC; (h) B. Lefeuvre, J. Flores Gonzalez, C. A. Mattei, V. Dorcet, O. Cador and F. Pointillart, Inorganics, 2021, 9, 50 CrossRef CAS; (i) A. Mondal and S. Konar, Dalton Trans., 2021, 50, 13666–13670 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal data and refinement parameters, structural data (tables and figures), spectroscopic and additional magnetism figures for complexes 1–5. CCDC 2385399 (1), 2385400 (2), 2385401 (3), 2385402 (4), and 2385403 (5). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02691h |
| This journal is © The Royal Society of Chemistry 2025 |