
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogen-bonded π-conjugated supramolecular polymers
Pedro Ximenis
 ,
Daniel Martínez
,
Llorenç Rubert
and
Bartolome Soberats
*
,
Daniel Martínez
,
Llorenç Rubert
and
Bartolome Soberats
*
Department of Chemistry, Universitat de les Illes Balears, 07122 Palma de Mallorca, Spain. E-mail: b.soberats@uib.es
First published on 14th November 2025
Abstract
The self-assembly of π-conjugated molecules offers a promising route for designing advanced functional materials with tailored optical and electronic properties. Owing to their nature, organic π-conjugated scaffolds spontaneously assemble by π–π stacking, while the introduction of hydrogen-bonding (H-bonding) interactions in these systems has emerged as a key strategy to gain control over self-assembly processes and the resulting supramolecular assemblies. H-bonding provides both specificity and directionality in non-covalent interactions, facilitating the formation of well-ordered and stable structures, such as supramolecular polymers. This review examines recent advances in design strategies that leverage H-bonding chromophores to fine-tune self-assembly behavior in solution, discussing the impact of monomer design and the experimental conditions on molecular packing and the morphologies of the resulting assemblies. Along with the thermodynamic advantages of H-bonding, its impact on self-assembly kinetics is also discussed, highlighting phenomena such as pathway complexity and related concepts like living supramolecular polymerization, secondary nucleation and supramolecular polymorphism. By providing a comprehensive overview of the current state of the field, this work aims to guide future research efforts toward the rational design of hierarchically ordered π-conjugated supramolecular materials.
 Pedro Ximenis | Pedro Ximenis obtained his BSc Chemistry from the University of the Balearic Islands (UIB) and then his MSc Molecular Nanoscience and Nanotechnology from the Universitat de Valencia (UV), where he began his research in supramolecular assemblies under the supervision of Dr. Soberats. Now, he is currently a PhD student in the same group at the UIB. His main research topic is the design and study of non-conventional supramolecular assemblies based on dyes driven by Hydrogen Bonding. He is working on liquid crystalline systems and supramolecular polymers and copolymers |
 Daniel Martínez | Daniel Martínez received his BSc Chemistry and MSc Chemical Science and Technology from the University of the Balearic Islands (UIB) in which both bachelor's and master's theses were supervised by Dr Ángel García Raso. In 2022, he joined the Supramolecular Chemistry research group, at the same institution, as a support researcher, and in 2024 he started his PhD studies under the supervision of Dr Bartolomé Soberats. His research focuses on the study of supramolecular polymers and liquid crystals based on unconventionally assembled organic chromophores. |
 Llorenç Rubert | Llorenç Rubert is a PhD candidate under the supervision of Dr Bartolomé Soberats in the University of the Balearic Islands, Spain. His research focuses on design, study and control of supramolecular polymers and liquid crystals. He has worked on 2D-supramolecular polymers, pathway complexity and co-assembled luminescent liquid crystals. |
 Bartolome Soberats | Dr Bartolomé Soberats is an Associate Professor at the University of the Balearic Islands (UIB). He obtained his PhD from UIB and subsequently held postdoctoral positions at the University of Tokyo and the University of Würzburg. His research focuses on the design and development of supramolecular polymers and functional liquid crystals. He has contributed to the advancement of unconventional assemblies of liquid-crystalline dyes, dynamic covalent liquid crystals, and the exploration of pathway complexity and two-dimensional supramolecular polymers. |
1. Introduction
1.1. Prologue
The field of supramolecular chemistry has, over the past few decades, evolved from the early studies of host–guest interactions, to embrace more complex systems capable of dynamic, adaptive behaviour and hierarchical self-organization.1–6 In this context, the concept of supramolecular polymerization has gained increasing attention, not only as a model to study biological processes but also as a tool to develop innovative materials.7–10 Supramolecular polymers are large, dynamic assemblies formed through reversible non-covalent interactions (such as hydrogen bonding (H-bonding), π–π stacking, metal coordination, or hydrophobic effects) between monomeric units. These monomeric units can vary in structure and nature but typically consist of organic or organometallic components with suitable solubility in either aqueous or organic solvents. Unlike traditional covalent polymers,11 these materials exhibit adaptive properties, self-healing behaviour, and stimuli-responsiveness, making them ideal candidates for a wide range of applications.12–16Among the diverse molecular systems explored for supramolecular polymerization, extended π-conjugated organic scaffolds or dyes have emerged as versatile monomeric building blocks, not only for their intrinsic optoelectronic properties but also for their ability to promote directional π–π stacking between the aromatic surfaces.17–21 Furthermore, by tailoring the molecules and adding peripheral groups, it is possible to introduce additional interactions like H-bonding, electrostatic forces, or solvophobic effects, enhancing the self-assembly tendency. The combination of different non-covalent interactions has been shown to be an effective strategy for constructing π-conjugated supramolecular polymers with well-defined morphologies, tuneable photophysical properties, and dynamic behaviour. In this context, a key advantage of self-assembled π-conjugated materials lies in the fact that their properties emerge from the entire supramolecular architecture rather than solely from the molecular structures of the monomers, leading to emergent features such as exciton delocalization, aggregation-induced emission or quenching and enhanced charge mobility.22,23 Through rational design of the monomeric building blocks, the structure and properties of the polymers can be finely tuned, making such systems highly attractive for applications in organic electronics, photovoltaics, sensing, and bioimaging.24,25
The incorporation of H-bonding motifs into a chromophore scaffold offers several advantages for the construction of supramolecular polymers.26–28 The directionality in hydrogen bonds (H-bonds) guides hierarchical self-assembly, typically into one-dimensional polymers or more complex architectures, and provides additional stability to the resulting architecture. This strategy has been successfully applied to a variety of dye-systems, including perylene bisimides (PBIs),29 naphthalene diimides (NDIs),30 BODIPY,31 and diketopyrrolopyrroles (DPPs),32 among others. For instance, PBIs readily form stable, columnar stacks solely by π–π interactions,33 while the introduction of H-bonding amide units in the monomer design results in highly ordered supramolecular structures with excellent thermal and photostability.34 The introduction of H-bonds in supramolecular polymerization affects the thermodynamics of the process but it can also interfere in the kinetics, altering the energy landscape.35
This review aims to provide a focused overview of recent progress in the development of H-bonded π-conjugated polymers (Fig. 1). A special emphasis will be placed on how H-bonding is strategically implemented in the monomer structure to gain control over the self-assembly processes, molecular packing, and polymer morphology, as well as the photophysical properties. In this context, we aim to highlight not only the fundamental insights but also new avenues for the rational design of next-generation supramolecular materials.
 | ||
| Fig. 1 Outlook of the scope of this review covering different aspects of H-bonded supramolecular polymers based on π-conjugated systems. | ||
1.2. Supramolecular polymers and H-bonding
The concept of supramolecular polymers began to crystallize in the early 1990s, fuelled by the idea that non-covalent interactions could drive the formation of polymer-like structures with dynamic and reversible properties. A pivotal early example was published in 1988, when T. Aida and S. Inoue reported the formation of fibrous aggregates from the porphyrin derivative 1 (Fig. 2a).36 Their study revealed significant changes in the UV/vis absorption spectra by increasing concentration, which was related to an aggregation process, highlighting the effects of supramolecular polymerization on the chromophore properties. Interestingly, in this case, the assembly was stabilized by π–π interactions, with no participation of H-bonding. | ||
| Fig. 2 (a) Molecular structure of porphyrin 1 and the proposed assembly pattern. Molecular structure and H-bonding patterns of compounds (b) 2a,b and (c) 3. (d) General molecular structure of a BTA monomer and the representation of its self-assembly pattern via triple H-bonds (reproduced from ref. 39, with permission from the Royal Society of Chemistry, copyright 2012). (e) Different H-bonding motifs relevant for the development of supramolecular polymers. | ||
Around the same time, Jean-Marie Lehn introduced the concept of dynamic polymers capable of adapting and reorganizing in response to external stimuli due to the reversible nature of non-covalent interactions. Notably, they reported polymeric assemblies formed through triple H-bonds using bifunctional diamidopyridines and uracil derivatives (2a and 2b), highlighting the potential of H-bonding as a key driving force (Fig. 2b).37 H-bonds are directional and relatively strong non-covalent interactions with binding energies ranging from 5 to 40 kJ mol−1, depending on the chemical environment and the strength of the donor and acceptor.2 Compared to other noncovalent forces, H-bonds are stronger and more directional than van der Waals interactions (e.g., London dispersion forces), which are generally weak and non-directional. Unlike π–π stacking or hydrophobic interactions, which rely on electronic polarizability or solvent exclusion, respectively, H-bonds are highly specific and contribute significantly to conformational control and molecular alignment in supramolecular assemblies.
Accordingly, H-bonding emerged as a key motif in the design of supramolecular polymers. In this context, the works on H-bonded supramolecular polymers of Meijer and co-workers using ureidopyrimidinones (UPy)38 (Fig. 2c) and later benzene-1,3,5-tricarboxamides (BTAs)39 (Fig. 2d) proved to be especially influential. These systems have since served as model platforms for in-depth thermodynamic and mechanistic studies of supramolecular polymerization.7–16 In recent years, advances in molecular design, supported by synthetic chemistry, have been pivotal in expanding the scope and robustness of H-bonded supramolecular polymers. The incorporation of well-defined and highly directional motifs such as diaminotriazines-uracyl,40 bis(acylamino)triazines, barbiturates,41 ureas,42 and squaramides,43–45 (Fig. 2e) among others, has led to a versatile toolkit of H-bonding units capable of establishing predictable and tuneable interaction patterns. These motifs not only provide strong and reversible interactions but also enable fine control over the polymer architecture, stability, and responsiveness. Notably, many of these units, along with simpler functional groups like amides or amino acid sequences, can be seamlessly integrated into various organic scaffolds, including π-conjugated systems.17–25 This strategy allows for the creation of supramolecular polymers that combine superior structural order with electronic or optical functionality.
1.3. Self-assembly of dyes and electronic considerations
Historically, the interest in organic π-conjugated molecules can be traced back to their use in textile coloration in the 19th century, when synthetic dyes such as mauveine and indigo marked the beginning of industrial organic chemistry. These early dyes were valued for their chromatic properties, but only much later their potential in photonics, electronics, and sensing become fully recognized.46 The transition from simple colorants to functional molecular materials has paralleled developments in synthetic chemistry, which enabled to gain control to prepare complex molecules with the desired structure, planarity, and substitution patterns. This control has enabled the design of dyes not merely as isolated chromophores, but as building blocks capable of spontaneously assembling into higher-order architectures.47The delocalization of π-electrons across the π-conjugated organic systems imparts unique electronic properties to these molecules. This configuration defines the energy gap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), dictating the absorption and emission properties.48 The electronic characteristics of π-conjugated systems can be precisely tuned through molecular engineering strategies such as extending the conjugation length, introducing electron-donating or -withdrawing substituents, and incorporating heteroatoms into the π-framework. These structural modifications directly influence key parameters like energy levels and absorption/emission profiles.
Importantly, the photophysical behavior of dye molecules changes upon self-assembly due to the interaction between individual monomeric units. These monomer–monomer interactions are crucial in dictating the optical absorption, emission properties, and excited-state dynamics of the resulting assemblies. The first approximation to this phenomenon was studied in the molecular exciton theory,48–50 which was then extended to include vibronic coupling,51–55 and finally address short-range charge transfer interactions.52 Collectively, these methods offer a comprehensive description of how supramolecular packing modulates the electronic features of chromophore-based polymers.
Molecular exciton theory, originally formulated by Kasha and grounded in earlier work by Davydov on excitonic splitting, conceptualizes chromophores as point oscillators, so-called molecular oscillators, that interact via their transition dipole moments, described as the sum of position vectors of all particles with charge, characterized by their direction and probability.48–50 In this context, the simplest model involves a dimer of two interacting molecules, where each monomer is treated as an independent oscillator. When two molecules are brought close enough, their transition dipoles (µ) interact through coulombic dipole–dipole coupling. This interaction leads to a splitting of the excited-state energy levels into two excitonic states, one higher and one lower in energy than the monomeric excited state. Note that the splitting is not centered on the s1 level, because this is slightly displaced due to van der Waals interactions (ΔEvdW) (Fig. 3a).
 | ||
| Fig. 3 (a) Representation of the energetic levels for a monomer, an H-dimer and a J-dimer. (b) and (c) Representation of the energetic levels for dimers in different geometries: changing θ maintaining d constant (b) and changing α (c). (a)–(c) Green solid arrows represent the relative orientation of the molecular transition dipole moments of the allowed transitions; the global transition dipole moment of the dimer is represented with dashed green arrows; and purple solid arrows indicate the relative orientation of the molecular transition dipole moments of the forbidden transitions. (d) Representation of the vibrational energetic levels and absorption bands for a monomer and H or J-type aggregates with different levels of coulomb coupling and strong vibrational coupling. (e) The HOMO and LUMO of perylene. Hole transfer (th) and electron transfer (te) contributions within a π-stacked perylene dimer (3.5 Å stacking distance) as a function of short- and long-axis displacements. The product −te th(∝ JCT) and coulomb coupling as a function of short- and long-axis displacements. Red regions indicate a negative coupling (J-aggregation), and blue regions indicate a positive coupling (H-aggregation) (reproduced from ref. 52 with permission from the American Chemical Society, copyright 2018). | ||
The relative orientation of the dipoles determines the permitted and forbidden states, leading to what are commonly known as H-type interactions (hypsochromic, blue-shifted), where the permitted excitonic state is the one with higher energy, and J-type interactions (bathochromic, red-shifted), where the permitted excitonic state is the one with lower energy (Fig. 3a–c).56,57 A critical angle of θ = 54.7° indicates the transition point between the H-type and J-type coupling regimes (Fig. 3b). Other geometries may present non-aligned dipoles leading to mixed exciton states or partially allowed transitions as for example by relative rotation with the respect of the angle α (Fig. 3c).58,59 However, these approximations consist of idealized geometries (e.g. spherical dipoles or perfectly parallel arrangements) and may differ from real supramolecular structures.
The exciton model can be extended to higher-order aggregates (trimers, tetramers, etc.), treating each additional chromophore as a new dipolar oscillator in the assembly. This progression leads to band formation, exciton delocalization, and spectral changes including band narrowing and loss of vibronic fine structure. However, these extensions rely on high structural order and limited environmental perturbation, limiting their applicability to ideal systems. Accordingly, the classical exciton theory can be applied to systems with large transition dipole moments and negligible vibronic coupling, but fails for most organic chromophores, which exhibit significant vibrational coupling and/or charge transfer character.60
To overcome these limitations, vibronic coupling was incorporated into exciton theory, most notably by Spano and co-workers. The updated model considers both electronic coupling and coupling to molecular vibrations, characterized by the Franck–Condon principle and the Huang–Rhys factor, which correlates the electronic and molecular vibrations.51–55 In the weak coupling regime, the vibronic structure of aggregates can be described quantitatively using intensity ratios between successive vibronic peaks. The relative intensities depend on the interplay between the exciton coupling, vibrational energy, and the vibronic overlap functions. As shown in Fig. 3d, in H-aggregates, increasing exciton coupling shifts intensity from A1 to A2 levels and beyond, while in J-aggregates, intensity accumulates in the A1 level with minimal contribution from higher vibronic levels. This refined theory enables a more accurate interpretation of experimental spectra, particularly in systems where the vibrational fine structure is evident and traditional exciton theory fails to account for spectral intensities.61
A second major correction to exciton coupling theory involves the inclusion of short-range charge transfer interactions.52 These interactions arise from direct wavefunction overlap between adjacent chromophores, typically occurring at intermolecular distances below ∼4 Å, well within the range of typical supramolecular contacts. Charge transfer interactions are highly sensitive to geometry and molecular electronic structure as shown in Fig. 3e for perylene. Two principal contributions define charge transfer coupling: (i) hole transfer (th) integral (HOMO–HOMO overlap) and (ii) electron transfer (te) integral (LUMO–LUMO overlap). Their combined effect (−te·th) contributes to total coupling energy, which modifies the coulombic term (Fig. 3e) and must be considered jointly with vibrational coupling for accurate spectral predictions. Depending on the relative signs and magnitudes, coulomb and charge transfer contributions may reinforce or cancel each other, leading to cases where aggregate formation occurs without noticeable spectral shifts, known as null exciton coupling.62 This behavior has been observed in carefully designed systems, such as foldamers, where geometrical precision can control the electronic coupling geometry.63,64 More recently, the formation of aggregates with null exciton coupling interactions has been reported in a Z-shape PBI (see Section 4.1).65
In terms of emission properties, H-type dimers typically suppress fluorescence due to forbidden transitions from the lowest excitonic state, whereas J-type dimers enhance emission due to allowed transitions with increased oscillator strength. These differences underlie many observed trends in supramolecular chromophore assemblies, such as the pronounced fluorescence quenching in H-aggregates and enhanced brightness in J-aggregates.60,66,67
Understanding the interactions between chromophores is essential for designing supramolecular materials with tailored functions. These interactions directly influence key optical properties, making molecular-level control a powerful tool in material design.
2. Supramolecular polymerization and thermodynamics
The study of supramolecular polymers is predominantly conducted in solution, as they are dynamic species existing as supramolecular entities in solvated states in equilibrium with the monomers. In this review, we primarily focus on systems that assemble in organic solvents, as aqueous environments are considerably more complex and often weaken H-bonding due to competition with water molecules.10 Supramolecular polymerization processes in organic solvents are typically favoured by increasing the monomer concentration and decreasing the temperature. Moreover, the nature of solvent has a strong influence on the polymerization behaviour. For a given system, a good solvent is one that dissolves the monomer and prevents aggregation, while a bad solvent promotes self-assembly.68–74 Thus, in solvent mixtures, the ratio of good to poor solvent governs the polymerization tendency of the system.In this section, we will explore various aspects of the study of π-conjugated supramolecular polymers, considering mechanistic aspects of supramolecular polymerization, as well as the different methods applied to the characterization of supramolecular species.
2.1. Supramolecular polymerization mechanisms
A monomeric species can self-assemble into supramolecular polymers via different mechanisms depending, for example, on its molecular structure, the solvent and the intermolecular interactions involved in the self-assembly process.75 These mechanisms are typically classified into two main types: isodesmic and nucleation–elongation, and are studied considering the assembly into one-dimensional (1D) assemblies (Fig. 4). In studies of supramolecular polymerization mechanisms, the systems are typically treated as equilibria involving only two species: the monomer and the polymer. To evaluate the supramolecular polymerization mechanism of a given system, the parameter “degree of aggregation (αagg)” is used, which varies depending on the conditions (concentration, temperature and solvent composition) (see Section 2.2). The degree of aggregation is a numerical representation of the aggregated species in solution, where αagg = 1 indicates full aggregation and αagg = 0 corresponds to the monomeric state.76 This parameter can be experimentally calculated using eqn (1):
 | (1) |
 | ||
| Fig. 4 Schematic equilibria for the (a) and (b) isodesmic and the (d) and (e) nucleation-elongation aggregation mechanisms, where M is the monomer and Mn the nucleus. Schematic illustration of an (b) isodesmic and (e) nucleation–elongation processes represented as a function of concentration, rate of “bad solvent” and temperature. Evolution in Gibbs free energy (ΔG) for (c) isodesmic and (f) cooperative mechanisms along the polymerization process. The ΔG of the processes is represented as a function of the degree of polymerization (DP). ΔGe = Gibbs free energy for the elongation step; ΔGn = Gibbs free energy for the nucleation step (reproduced from ref. 76 and 96). | ||
It is noteworthy that while the isodesmic mechanism is common in simple π-conjugated supramolecular polymers formed solely by π–π stacking, it is rarely observed when H-bonding groups are introduced into the monomeric scaffold. One of the scarce examples of a H-bonded π-conjugated supramolecular polymers formed by an isodesmic mechanism was reported by Xiaoming He and coworkers.78 They found that an anthanthrene derivative (4) with amide-amide or amide-ester motifs at the molecular spacers forms H- or J-aggregates (Fig. 5a). In particular, the amide-ester motif promotes the formation of J-aggregates via an isodesmic mechanism, as it can be deduced from the sigmoidal trend of the variable temperature aggregation curve (Fig. 5a). The authors do not provide an explanation for the isodesmic nature of this system; however, we assume that the molecular structure is not fully planar, which partially disrupts the concomitant H-bonding and π–π interactions, thereby preventing cooperativity in the self-assembly process. Other relevant examples of isodesmic H-bonded supramolecular polymers can be found in the literature, although they remain scarce.79–81
The elongation-nucleation mechanism encompasses two sub-types: cooperative and anti-cooperative. To distinguish the cooperative and anti-cooperative mechanisms is useful to calculate the cooperativity factor (σ), defined as σ = Kn/Ke. For the cooperative mechanism σ value must be σ < 1, implying that the nucleation step is less favourable than the elongation one. On the other hand, if σ > 1, the nucleation step is more favourable, and the self-assembly mechanism can be stablished as anti-cooperative.83 In the case that σ = 1, an isodesmic mechanism operates.
In H-bonded π-conjugated supramolecular polymers, the most common mechanism is the cooperative one, characterized by a more favourable elongation step compared to the nucleation one. This mechanism often results in the formation of long supramolecular fibres. In a reference study, Würthner and coworkers presented a chiral PBI twisted scaffold (5) that self-assembles forming J-aggregates via H-bonding interactions between diimide groups following a cooperative mechanism (Fig. 5b). This fact could be demonstrated through concentration-dependent experiments.84 The corresponding aggregation curve, obtained by plotting αagg vs. KCT, describes a non-sigmoidal behaviour characterized by a sharp increase of αagg once reaching a critical concentration (Fig. 5b).
 | ||
| Fig. 5 (a) Molecular structure of compound 4, and the corresponding temperature dependent UV/vis absorption study and the aggregation curve fitted to an isodesmic mechanism. (reproduced from ref. 78, with permission of Wiley-VCH GmbH, copyright 2025) (a) Molecular structure of compound 5, and the corresponding concentration dependent UV/vis absorption study and the experimental aggregation curve (inset, black dots) (reproduced from ref. 84, with permission of the American Chemical Society, copyright 2009). The solid line shows the theoretical curve for an isodesmic mechanism, while the non-sigmoidal experimental data indicate the operation of a cooperative mechanism. (c) Molecular structure of compound 6, and the corresponding concentration dependent UV/vis absorption study and the experimental aggregation curve fitted to an anti-cooperative mechanism (reproduced from ref. 82 with permission of the Royal Society of Chemistry, copyright 2015). | ||
When the nucleation constant (Kn) is larger than the elongation constant (Ke), the mechanism becomes anti-cooperative. This results in the formation of short oligomers with narrow size distributions due to the less favourable elongation step.8,85 This mechanism was observed for the PBI 6 functionalized with two chiral amino acids with benzylamine groups and solubilizing pendants (Fig. 5c).82 For the first time an anti-cooperative supramolecular polymerization mechanism could be elucidated in detail from the theoretical point of view using a new K2–K model (Fig. 5c) derived from the previous model reported by R. F. Goldstein and L. Stryer.86 Following this work other examples of H-bonding chromophores were found to assemble via the anti-cooperative mechanism.83,87 This polymerization mechanism is relatively uncommon, but the resulting materials are of interest due to their short length, and inefficient packing, which in some cases has been found to be advantageous for preserving the emission properties of the monomer.87
As previously mentioned, the presence of H-bonding units in the monomer scaffold plays a critical role in the polymerization mechanism. Importantly, the interplay of two non-covalent interactions (H-bonding and π–π stacking) during the polymerization process seems to promote the cooperative mechanism. Indeed, various studies compared similar scaffolds with amide versus ester groups, consistently supporting this statement.78,88
2.2. Methodologies to determine supramolecular polymerization mechanisms
Under thermodynamic equilibrium, the monomer and aggregated species coexist in a constant ratio. Importantly, this ratio is highly sensitive to experimental conditions, and therefore the polymerization mechanism is studied by systematically varying the experimental conditions.8 Several methods are available to monitor these processes, which can be followed using various experimental techniques. The three most widely used approaches to experimentally determine the supramolecular polymerization mechanism are the concentration-dependent, solvent-dependent, and temperature-dependent methods.The experimental curves obtained from these experiments can be fitted by applying the corresponding mathematical model, which permits extracting the association constants and thermodynamic parameters for the system. In the last few years, several authors have laid the theoretical foundation for understanding the behaviour of supramolecular systems. The K2–K model described by Goldstein and Stryer86 and its extension to anti-cooperative mechanism by F. Würthner and coworkers,82 the nucleation-elongation model by Zhao and Moore89 or the van der Schoot model90 are just a few examples of the vast work performed in this field.91–93 Also it is worth remarking the work from ten Eikelder and coworkers in the study of more complex systems and coassemblies.94,95 Details about these mathematical models are out of the scope of this review.
In concentration dependent experiments the αagg is plotted versus the concentration normalized with the elongation constant Ke,82 according to the appropriate mathematical model. Variable concentration experiments are technically demanding, due to the several solutions that must be prepared, which sometimes implies the lack of data points and poor fitting to the mathematical models.96 The concentration dependent experiments are essential to apply the Goldstein-Stryer K2–K mathematical model to describe anti-cooperative systems.86,97
In variable-temperature experiments, changes in the degree of polymerization (αagg) are monitored as a function of temperature. This is the most widely used procedure to assess the polymerization mechanisms due to its simplicity and the high numbers of data points that can be automatically obtained, leading to more reliable experimental curves.96 However, this methodology is sensible to kinetic effects. To mitigate these kinetic effects, the experiments can be carried out at relatively slow heating/cooling rates. The comparison of the heating and cooling curves is usually utilized to evaluate the magnitude of the kinetic effects in the system.8
Finally, solvent-dependent experiments are carried out at constant concentration and temperature in mixtures of two solvents, a “good solvent” and a “bad solvent”. Good solvents are those that solvate the monomer effectively, while bad solvents promote aggregation. In this context, in solvent dependent or “denaturation” experiments the degree of aggregation (αagg) is monitored as a function of the ratio of good/bad solvent and fitted to the appropriate model.68 The use of this technique has spread in the last few years due to its simplicity compared to the concentration dependent methodology and the higher reliability compared to the temperature dependent one which is sensible to kinetic effects and the sample decomposition at higher temperatures.98
2.3. Common techniques to study supramolecular polymers
A combination of complementary techniques is typically employed not only to characterize the polymerization processes itself, but also to elucidate the structural features and morphology of the resulting supramolecular polymers.3,99,100Nuclear magnetic resonance (NMR) spectroscopy provides detailed insights into binding interactions and the dynamics of reversible association.101 NMR enables the monitoring of polymerization processes as a function of temperature or concentration, typically at relatively high concentrations (>10−4 M). Moreover, by more complex NOESY/ROESY or DOSY experiments it is possible to extract information about the supramolecular interactions in the system and the size of the supramolecular polymers. Other spectroscopic methods, such as UV/vis absorption and fluorescence spectroscopy, have proven particularly powerful for tracking the aggregation behaviour at lower concentrations. Remarkably, these techniques are especially well suited for studying π-conjugated polymers due to their absorption/emission profiles in the UV, visible or near infrared (NIR) regions. Related spectroscopic techniques like circular dichroism (CD) and circularly polarized luminescence (CPL)102 can be used to study chiral systems. In addition to the techniques mentioned above, Fourier-transform infrared (FT-IR) spectroscopy is frequently used to monitor the formation and strength of H-bonds within the assemblies.103
Supramolecular polymerization processes can also be investigated using calorimetric techniques, including differential scanning calorimetry (DSC) and isothermal titration calorimetry (ITC), which provide quantitative data on the thermodynamics of the self-assembly processes.104
Light scattering,105 along with X-ray and neutron scattering,106 are widely used to determine the morphology and size of supramolecular assemblies in the solution state. However, the morphologies of the aggregates are often studied in the solid state by microscopic techniques after deposition of liquid samples on substrates and evaporation of the solvent. Atomic force microscopy (AFM)107 and transmission electron microscopy (TEM)108 provide direct visualization of nanoscale morphology and hierarchical organization at nanometre resolution.
The integration of theoretical calculations into the study of supramolecular polymers has proven increasingly valuable, offering a complementary perspective to experimental approaches and deepening our understanding of the underlying molecular processes.109 Computational methods such as density functional theory (DFT) allow for the precise modelling of non-covalent interactions at the atomic level, helping to elucidate the structural features and energetics of monomer association.110 More recently, Grimme's GFN2-xTB method,111 a semiempirical tight-binding approach optimized for large systems, has been increasingly used to model supramolecular assemblies. This method efficiently models non-covalent interactions, including dispersion forces, through advanced treatments of atomic multipole moments and self-consistent incorporation of the D4 dispersion model. It provides reliable geometries and interaction energies, making it well-suited for studying the structure and stability of supramolecular polymers. On the other hand, molecular dynamics (MD) simulations allow the exploration of the dynamic behaviour and structural evolution of supramolecular polymers over time, providing insight into assembly pathways, stability, and responsiveness to external stimuli at the molecular level.112–114
3. Monomer design in π-conjugated supramolecular polymers
Given the electronic factors that are involved in the self-assembly of π-conjugated molecules, precise control over aggregation processes is essential to obtain supramolecular polymers with the desired morphology, molecular packing, and functional properties. One of the most effective strategies for guiding self-assembly is rational monomer design. By carefully selecting the structural configuration and combining different functional groups within the monomer structure, it is possible to program its spontaneous assembly into well-defined supramolecular architectures. Importantly, rational monomer design does not only enable to gain control over the self-assembly under thermodynamic conditions but can also affect the kinetics of the process, generating in some cases pathway complexity (see Section 7).In the design of π-conjugated supramolecular polymers, the key structural element is the chromophore or π-conjugated scaffold, which provides the optical properties and promotes assembly through π–π stacking interactions. The nature of the π-conjugated core, its planarity, the presence of heteroatoms, and the extent of conjugation determine the electronic properties of the system, including its absorption and emission characteristics. Simple π-conjugated scaffolds typically exhibit poor solubility in water and in many organic solvents, which limits the study of their self-assembly behaviour in solution. To address this, π-conjugated monomers are commonly functionalized with solubilizing groups (Fig. 6), which vary depending on the solvent used. For example, alkyl chains are often introduced to improve solubility in low-polarity organic solvents such as methylcyclohexane (MCH), while oligoethylene glycol (OEG) chains provide solubility in polar media. In this context, the 3,4,5-tris(alkyloxy)benzene unit is a widely used solubilizing motif, known not only for enhancing solubility in organic solvents but also for imparting liquid-crystalline behaviour.29 To gain further control over self-assembly, H-bonding units are frequently incorporated. These are typically positioned either in the linker between the conjugated core and the solubilizing groups (Fig. 6) or, in some cases, are directly integrated into the core itself. Amides are the most employed H-bonding functionalities in the development of H-bonded π-conjugated polymers, although other groups such as ureas, imides, and related motifs have also been successfully utilized (Fig. 2e).
 | ||
| Fig. 6 Outlook on the general monomer design structure for developing H-bonded π-conjugated supramolecular polymers, and the possible molecular packing modes and morphologies of the resulting assembled structures. | ||
Monomer design influences the self-assembly process and consequently the molecular packing and morphology of the outcome supramolecular structure. As discussed before the molecular packing is critical for the material properties as for example in the formation of H- or J-aggregates (Fig. 6). However, the morphology of the aggregates is also relevant for their anisotropic features and their potential applicability. Most π-conjugated supramolecular polymers consist of 1D assemblies (Section 4), which are among the most accessible to characterize. Notably, regardless of monomer design, many systems also form particle-like aggregates ranging in size from nanometres to microns (Fig. 6). These aggregates are often ill-defined and are typically not classified strictly as supramolecular polymers due to their discrete nature, although they are frequently observed as kinetically trapped species (Section 7) and remain relevant to the self-assembly landscape. As a variation of 1D supramolecular polymers, helical structures have proven highly relevant for fundamental studies of chirality and for advanced functional applications (Section 5).8,102 In recent years, two-dimensional (2D) supramolecular polymers have also attracted increasing attention (Section 6), although their modes of assembly remain under active investigation.115 In contrast, far less is known about ordered three-dimensional (3D) supramolecular structures (Fig. 6).116 While these assemblies are relevant in liquid-crystalline states,117 their formation and stabilization in the solution state is probably hindered by challenges such as precise 3D assembly control and uncontrolled precipitation/crystallization of the final species.
Increasing attention has recently been directed not only toward single-component systems but also towards more complex, multicomponent assemblies, as copolymers, which offer new opportunities for understanding hierarchical organization and emergent functionalities.118 Multicomponent systems share a symbiotic relationship with other areas of supramolecular polymerization. For instance, they have proven to be essential in the discovery and development of the chirality amplification phenomenon (Section 5). Moreover, kinetic control and pathway complexity (Section 7) have enabled the formation of a wide variety of copolymers, particularly block copolymers,119–121 which were largely inaccessible through thermodynamic control, with only a few notable exceptions.122,123 Despite some specific examples, the design and development of supramolecular co-polymers will not be largely discussed in this review. For a more comprehensive overview of supramolecular copolymerization, readers are referred to several excellent and exhaustive reviews already available in the literature.124–128
4. 1D supramolecular polymers
In this section, we provide an overview of the various types of H-bonded π-conjugated 1D supramolecular polymers, highlighting key monomer design features that govern their formation and properties. In particular, we will discuss four distinct types of 1D assemblies: simple stacked, complex/tubular, main-chain, and stranded architectures.4.1. Simple stacked assemblies
Organic π-conjugated molecules spontaneously assemble into 1D polymers through π–π stacking interactions along the polymer axis. This type of assembly facilitates the overlap of the molecular orbitals and the corresponding excitonic coupling, providing in turn electron/hole transport. An early example of such 1D assemblies was reported by Aida for the amphiphilic porphyrin 1 (Fig. 2a) that forms H-type aggregates according to the UV/vis experiments.36 However, such π-stacked polymers are often weakly bonded and can exhibit highly dynamic behaviour, particularly in terms of rotational and translational order. The introduction of H-bonding units into the monomeric structure significantly strengthens the non-covalent interactions. In the common monomer design, the chromophore central core is surrounded by H-bonding units, and the corresponding supramolecular polymers are fibrillar structures where the aromatic cores align at the centre, stabilized by peripheral H-bonds along the fibre axis (Fig. 7, centre). The enhanced stability of these assemblies arises from the cooperative interplay between π–π stacking and H-bonding, both operating along the stacking direction, leading to reinforced 1D supramolecular polymers. As a result, these polymers typically exhibit cooperative self-assembly mechanisms, giving rise to long, stable fibrillar structures, which, depending on the conditions, can entangle and form gels.129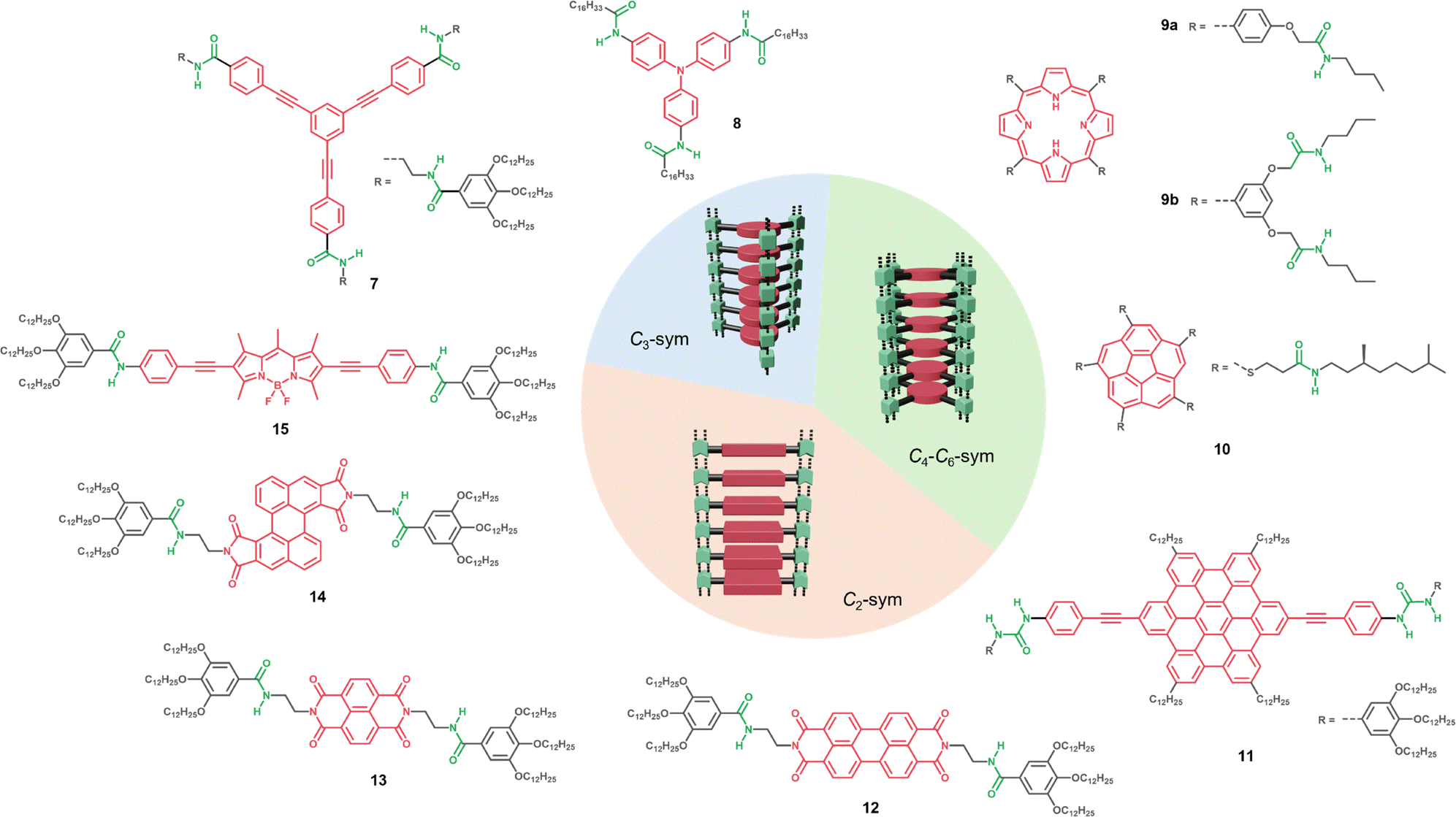 | ||
| Fig. 7 Schematic representation of simple-stacked 1D assembly structures depending on their symmetry (sym) and the molecular structures of monomers 7–15. C3-sym (7 and 8), C4–C6-sym (9a, 9b, 10 and 11) and C2-sym (11–15). | ||
Many examples of π-extended supramolecular polymers incorporating H-bonding units were inspired by BTA systems, which are well known for engaging in directional triple H-bonding and π–π stacking, leading to the formation of fibre-like assemblies (Fig. 7).130,131 For example, a C3-symmetric oligo phenylene ethynylene(OPE)-based tricarboxamide 7 was reported to self-assemble through multiple hydrogen bonds and π–π stacking interactions, forming 1D assemblies that further entangle to create gels.132 Importantly, in this example each of the three arms of the central core was functionalized with two amide groups reinforcing the interaction strength and cooperativity in the system. A similar behaviour was observed for the tris-amide triarylamine 8, which also forms 1D H-bonded assemblies.133 Remarkably, the triarylamine core provides the resulting polymer distinctive electronic, magnetic, and optical properties, indicating potential applications in organic electronics and spintronics.134
Other examples rely on disk-like π-conjugated monomers that typically exhibit a higher degree of symmetry, where the chromophore unit offers two to six functionalization sites for the incorporation of H-bonding motifs and solubilizing groups. A notable example of a supramolecular polymer formed from a monomer with C4 symmetry was reported by the Shinkai group.135 The porphyrin derivative 9a is functionalized with four amide groups and four lateral alkyl chains and forms organogels in different solvents based on fibrillar H-aggregates. Interestingly, when the number of amide groups is increased (9b), the new configuration leads to the formation of J-aggregates, maintaining the one-dimensional fibrillar structure. This represents a remarkable example of how closely related compounds, forming polymers with similar morphologies, can exhibit distinct molecular packing and optical properties. In another example, Aida reported a C5-symmetric corannulene 10, functionalized with five alkyl chains via amide linkages.136,137 This bowl-shape molecule presents different monomeric conformations (discussed in Section 7.2), but under the appropriate conditions self-assemble by multiple H-bonds forming a 1D stacked supramolecular polymer. Klaus Müllen and co-workers138 reported a hexabenzocoronene 11 functionalized with urea moieties at two of the six peripheral positions. This design leads to the self-assembly of the monomers via complementary H-bonding and π-stacking. The other four peripheral positions are functionalised with alkyl chains which provides solubility in organic solvents.
In many cases, the substitution pattern of the π-conjugated monomers is dictated by the shape of the chromophore as well as synthetic limitations. In this context, the design of linear π-conjugated monomers with C2 symmetry has been widely utilized in the field. Representative examples of C2-monomer designs leading to stacked 1D assemblies were reported using the rylene diimide derivatives, known for their highly conjugated aromatic cores and for their electron deficient nature.29,139 For example, Würthner34 and Shinkai140 reported PBI 12 and NDI 13, forming supramolecular polymers with excellent gelation properties in apolar solvents. More recently, Sánchez and coworkers reported the Z-shaped PBI 14.65 Remarkably, this unconventional molecular design leads to the formation of distinct aggregates, including a thermodynamically stable species that exhibits null exciton coupling. This so-called “null aggregate” is characterized by an absorption spectrum nearly identical to that of the monomer, but slightly red-shifted.
In another example based on a rod-like molecule, Fernández and coworkers reported the planar BODIPY dye 15 bearing two amide groups on both sides.88 In this case the molecular structure promotes 1D stacking via synergistic H-bonding and π–π interactions, as it was also observed in compounds 7–14 (Fig. 7). However, it should be highlighted that not all the C2-symmetry monomer designs lead to this type of 1D assembly.44 For instance, the similar molecular design of 15 but using a benzodithiophene core instead of BODIPY leads to the formation of nanoparticle-like H-bonded assemblies, in which 1D growth is hampered due to distortions in the aromatic backbone upon the polymerization process.83
4.2. Complex stacked and nanotubular assemblies
Section 4.1 discussed examples of π-conjugated supramolecular polymers formed through the cooperative interplay of two non-covalent interactions along the same dimension. However, in other cases, supramolecular polymers are built by two orthogonal non-covalent interactions that do not act cooperatively or along the same spatial dimension. This often leads to the formation of more complex architectures. In many of such systems, the monomers are carefully designed to first assemble via H-bonding into discrete supramolecular entities, such as dimers or macrocyclic oligomers composed of various units. These discrete entities can be considered a class of supramolecular monomers that subsequently undergo further self-assembly, through π–π stacking interactions or secondary H-bonding motifs, into polymeric structures. Depending on the nature of the initial supramolecular monomers, the resulting polymers can form either fibrillar or tubular structures (Fig. 8). | ||
| Fig. 8 Molecular design, supramolecular oligomer scheme and supramolecular polymer assembly of discrete 1D supramolecular assemblies. (a) PBI 16 assembly with melamine (reproduced from ref. 142 with permission of Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, copyright 1999). (b) Rosette assembly of OPV 17. (c) Dimer-based assembly of OPV 18. (d) Rosettes and nanotube assembly from 19. (e) Nanotoroidal assembly of bisazobenzene 20. (f) Nanotubes formed by cytosine and guanine units in 21. | ||
One of the first examples of this type of assembly was reported by Meijer, Würthner and coworkers.141,142 They described the self-organization of PBI 16 and melamine scaffolds through the formation of complementary H-bonds between the two units. The PBI was designed with phenoxy groups at the four bay positions, leaving the imidic positions free to enable H-bonding interactions with melamine. Although the exact supramolecular interaction modes were not clearly reported, it was proposed that a rosette-like structure is formed initially, which subsequently stacks into one-dimensional polymers. AFM experiments revealed the formation of fibrillar assemblies (Fig. 8a).
Another important relevant π-conjugated scaffold used to create this type of assembly is the oligo phenylene vinylene (OPV). Meijer and coworkers described an unsymmetrically functionalized OPV with a diamino triazine moiety and a wedge-shaped solubilizing group (17).143 This design enables the self-assembly of the molecules into hexameric rosettes that further organize into supramolecular tubules (Fig. 8b). Later, they designed a similar OPV, but instead of the diamino triazine moiety, they inserted an ureidotriazine unit (18).144,145 This modification led to the molecules to form dimeric H-bonded species that subsequently stack forming a 1D helical stack (Fig. 8c). In a following work, OPV 18 was combined with the PBI 16 to form trimeric discrete supramolecular units that co-assemble forming a chiral 1D supramolecular polymer via π–π stacking interactions.146 The three-component supramolecular entity was formed through complementary H-bonding between the imide groups of the PBI and the ureidotriazine units in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio. This system incorporates both donor and acceptor chromophores and was proposed for photovoltaic applications.
:2 ratio. This system incorporates both donor and acceptor chromophores and was proposed for photovoltaic applications.
Yagai's group reported a family of barbiturate functionalized π-conjugated systems forming different assembled structures.147–149 For example, the thienyl[oligo(hexylthiophene)]s 19 utilized the barbiturate unit to form H-bonded supramolecular rosettes composed of six molecular units.150 These discrete rosettes follow a second assembly process via π–π stacking, forming nanorods (Fig. 8d) that were proposed for photovoltaic applications. The same group also reported the formation of tubular assemblies using scissors-shaped molecules.151–153 This design is based on a bisazobenzene molecule functionalized with two amide groups and two chiral side chains (20). This molecule assembles through a combination of π–π stacking and H-bonding, forming nanotoroidal structures that were clearly observed by AFM. Remarkably, further assembly of these toroidal structures via additional π–π stacking interactions led to the formation of nanotubes (Fig. 8e).
A similar strategy was applied by the González group that exploited complementary interactions between nucleobases to develop tubular assemblies.154 For example, molecule 21 couples a 1,4-diethynylbenzene core with a cytosine and a guanine units, functionalized with benzylic wedges to enhance solubility in organic solvents (Fig. 8f).155 Importantly, an amide group was introduced into the alkyl chain linking the nucleobase and the dendron 21. This specific design enables a two-level hierarchical self-assembly process: first, a cyclization in which the monomers interact via H-bonding to form a cyclic tetramer; and second, an elongation step where these tetramers stack to form a tubular structure. In this second process, both π–π interactions and H-bonds between the amide groups stabilize the nanotubes along the stacking direction (Fig. 8f).156,157 These systems were shown to be useful for innovative host–guest functions.158,159
4.3. Main-chain assemblies
The π-conjugated supramolecular polymers described before are formed via the interplay of two non-covalent interactions, the H-bonds and the π–π stacking, resulting mainly in assemblies based on the stacking of the chromophores. In contrast, main-chain supramolecular polymers feature repeating units connected through end-to-end reversible interactions, such as H-bonds, giving rise to a chain-like supramolecular backbones (Fig. 9a).160,161 This design generally prevents the excitonic coupling between chromophores but still permit the charge transfer processes between adjacent units. | ||
| Fig. 9 (a) Schematic representation of a main-chain 1D assembly. (b) Early reported example of main-chain assembly between anthracenes 22 and 23. (c) Molecular structures of compounds 24–27 forming main-chain 1D assemblies. | ||
Earliest examples of H-bond-driven main-chain type supramolecular polymers were reported by Lehn37 and Meijer (Fig. 2, compounds 2 and 3).38,162 In the context of π-conjugated systems, one of the earliest examples was again reported by Lehn and co-workers. They designed two different anthracene derivatives 22 and 23,163,164 functionalised with two uracil and two 2,6-diacylaminopyridine motifs, respectively. The equimolar mixture of the two compounds leads to the formation of polymeric chains by triple H-bonding interactions (Fig. 9b).
Shifting focus to a different central chromophore, Würthner reported a cyanine design inspired by the Hamilton type receptor system described by Lehn in 2002.165 The cyanine molecule 24 features a cyanine scaffold functionalized with a barbituric acid moiety on one side and two 2,6-diamidopyridine groups on the other side, also known as Hamilton receptor groups (Fig. 9c).166 This design enables self-assembly through six directional H-bonds, resulting into a supramolecular polymer that forms a fluorescent organogel under the appropriate conditions.166
Meijer and co-workers designed a series of dyes bisfunctionalized with UPy moieties to promote the formation of 1D main-chain supramolecular polymers.167,168 They reported three distinct emissive monomers based on oligofluorene 25, (OPE) 26, and PBI 27 (Fig. 9c). Each of these chromophores self-assembles into linear supramolecular polymers via UPy-UPy quadruple H-bonding (Fig. 2c). Moreover, when mixed under appropriate conditions, the monomers co-assemble into random copolymers. These mixed assemblies exhibit efficient charge transfer along the polymer backbone, making them promising candidates for application in optoelectronic devices.
4.4. Stranded assemblies
It is noteworthy that in conventional main-chain π-conjugated supramolecular polymers, additional π–π interactions between different chains are typically absent. However, in some cases, both end-to-end H-bonding and π–π stacking interactions between π-conjugated monomers can occur simultaneously, leading to the formation of 1D stranded structures in which excitonic coupling emerges. In these assemblies, the chromophores first organize into H-bonded chains, which then further associate through π–π interactions into 1D structures composed of a variable number of strands (Fig. 10a). As a result, the π-surfaces of the chromophores align parallel to the fibrillar axis,169 an arrangement that differs from that observed in more conventional π -conjugated polymers (Fig. 7). The geometry of the interactions between chromophores, as well as the number of strands comprising each fibre, governs the nature and strength of the excitonic coupling, ultimately influencing the properties of the resulting supramolecular structure. | ||
| Fig. 10 (a) Schematic representation of the formation of stranded 1D assemblies by H-bonding and π–π interactions. (b) Earliest example of stranded 1D assembly based on PBI 28 (reproduced from ref. 84 with permission of American Chemical Society, Copyright 2009). (c) Molecular structures of compounds 29–32 self-assembled into stranded 1D supramolecular polymers. | ||
A remarkable example of stranded supramolecular polymers was reported by Frank Würthner and co-workers and consists of a PBI.170 PBI 28 is functionalized at the four bay positions with wedge-shaped groups to enhance solubility in organic solvents, while presents N–H groups at the imide positions (Fig. 10b). This design promotes the formation, in MCH, of double-stranded cable-like J-aggregates with high fluorescence quantum yields. The assembly is driven by H-bonds between the imide groups, which guide the formation of linear PBI chains that subsequently associate into double-stranded structures through slipped π–π interactions. In subsequent studies, it was demonstrated that the formation of these J-aggregates proceeds via a cooperative mechanism, and that supramolecular chirality can be induced by incorporating chiral side chains into the monomer design.84 These PBIs were widely studied as supramolecular materials beyond solution-phase behaviour.64 Notably, they have been reported to form hydrogels that exhibit temperature-dependent polymorphic behaviour, switching between H- and J-type assemblies.171 In addition, these systems were shown to form liquid-crystalline columnar J-aggregates in the solid state.172 Remarkably, solid-state X-ray studies enabled to reveal a correlation between the substitution pattern of the wedge-shaped groups and the number of strands composing the corresponding columnar assemblies.173
Similar molecular design was applied to develop NDI stranded supramolecular polymers. Ghosh and coworkers reported NDI 29a featuring two alkylamino substitutions at the naphthalene positions, while the imidic positions remain unsubstituted (Fig. 10c).174 This molecular design facilitates self-assembly by H-bonds, into J-type aggregates with a tubular morphology. More recently, it has been described a similar NDI derivative functionalized with wedge-shaped groups (29b) (Fig. 10c).175 This analogue formed fibrillar J-aggregates in MCH and exhibited liquid-crystalline behaviour in the bulk. Interestingly, wide angle X-ray scattering experiments determined the formation of four-stranded columnar liquid-crystalline phase in the solid state.
Such type of stranded assemblies was also achieved with other dyes incorporating lactam units in their core. DPP 30 features two lactam units to enable H-bonding, while the lateral positions are functionalized with wedge-shaped groups (Fig. 10c). The self-assembly process of this compounds follows a nucleation-elongation mechanism.176 The resulting fibre-like polymers were utilized to prepare anisotropic microfibres by an electrospinning process.177 The studies in bulk revealed the formation of a columnar liquid-crystalline phase based on a complex assembly mode consisting of the stack of DPP-dimers rotated by 90° forming a quadruple H-bonding motif. More recently, aryldipyrrolidone 31 forming fibrillar stranded assemblies in MCH as well as a liquid-crystalline columnar phase was reported (Fig. 10c).178 In all these examples, the realization of solid-state X-ray studies permitted more accurate characterization of the self-assembly mode of the stranded assemblies.
Another molecular design enabling stranded assemblies was reported by Chen and co-workers.179 They synthesized a BODIPY derivative functionalized at the equatorial position with a wedge-shaped group, while the axial positions bear two uracil moieties (32) (Fig. 10c). The self-assembly of this compound is driven by H-bonding and slipped π–π stacking interactions, resulting in J-type aggregates with a fibre/ribbon morphology. Steric effects induced by substituents at the meso-position promote the brick-like assembly of the H-bonded strands permitting the J-aggregate formation.
5. Chiral supramolecular polymers
Chirality is described as the property of an object that cannot be superimposed on its mirror image, and is ubiquitous in nature, appearing at various scales, from the macroscopic level (e.g., our hands, which are mirror images of each other; Fig. 11a) to the subatomic level (e.g., only left-handed helical neutrinos exist).180,181 In supramolecular chemistry, chirality is crucial for replicating the stereoselective interactions found in biological systems and for designing functional materials with properties such as CPL, enantioselective recognition, and asymmetric catalysis.2 | ||
| Fig. 11 (a) Mirror image of left and right hands as an example of chirality at the macroscopic scale. (b) Schematic representation asymmetry transfer in supramolecular polymers. (c) Schematic representation of circular dichroism (CD) phenomenon (reproduced from ref. 242, with permission of Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, copyright 2019). (d) Examples of point, axial, planar and helical chirality. | ||
In this line, the self-assembly of monomers endowed with one or more asymmetry (chiral) elements can lead to the formation of homochiral supramolecular polymers, through a process known as asymmetry transfer (Fig. 11b). In this process, the asymmetry is transmitted from the monomer to the supramolecular structure during the self-assembly. Although it is not necessary, it has been proved that the presence of H-bonding groups in the monomer structure has a positive influence on the transfer of asymmetry.102 Furthermore, π-conjugated systems are particularly attractive platforms to develop chiral supramolecular polymers due to their inherent electronic properties, which can be enhanced by the introduction of asymmetry.180 The absorption properties of π-conjugated scaffolds permit the study of supramolecular polymerization by CD, which measures the differential absorption of left- and right-circularly polarized light as a function of wavelength (Fig. 11c). Accordingly, CD spectroscopy emerged as a key tool for the study of chiral supramolecular polymers.
Remarkably, π-conjugated monomers can exhibit various forms of asymmetry (Fig. 11d), which can efficiently render chiral/helical supramolecular polymers. The most common strategy involves introducing point chirality into the monomers, typically by attaching chiral alkyl chains to an otherwise achiral π-conjugated core. Alternatively, monomers based on scaffolds with inherent asymmetry, such as axial or helical chirality, can also be used. Notably, the vast majority of chiral supramolecular polymers are 1D, forming helical fibres (Fig. 11b), although other types of hierarchical chiral assemblies have also been reported.102 In the following sections, we will examine representative monomer designs that give rise to chiral supramolecular polymers, along with the main approaches used to investigate asymmetry transfer in these systems.
5.1. π-Conjugated monomers incorporating point chirality
The incorporation of point chirality into the monomer structure is the most used strategy to assess chiral supramolecular assemblies. A general monomer design consists of a chromophore functionalized with a H-bonding groups and lateral chains with carbon stereocenters (Fig. 6). Usually one or more chiral centres are introduced in the solubilizing side alkyl chains137,151,182,183 or in alkyl spacers between the core and the side chains.118,184 In particular, aliphatic scaffolds of natural origin, like citronellol (3,7-dimethyl-6-octenol) (Fig. 11d) or amino acid derivatives, are utilized. Importantly, the transfer of the asymmetry in the systems is dependent on the number of chiral centres in the monomer, while the spatial distance between the stereocenters and the stacking core has also an influence.185Generally, π-conjugated monomers functionalized with chiral chains form simple 1D stacks with a rotational offset in between molecules, which leads to the induction of helicity (Fig. 11b and 12a, b). Importantly, the helicity of these 1D supramolecular polymers is dictated by the chirality in the alkyl chains (Fig. 11b). One of the first molecules utilized to study chirality in supramolecular polymers was again the BTA analogue (Fig. 2d).186,187 For example, BTA (S)-33 was functionalized with three bipyridine spacers linked to three wedge-shaped solubilizing groups. Importantly, these lateral solubilizing groups incorporated (S)-3,7-dimethyloctyl chains, and provided nine chiral centres to the monomer that effectively induced chiral supramolecular polymerization. More recently, L. Sánchez and coworkers studied the asymmetry amplification of other extended BTA analogues as 1,3,5-triphenylbenzenecarboxamides (see Fig. 7, compound 7), OPE-tricarboxamides188,189 and trisbiphenylamine-tricarboxiamides.190
 | ||
| Fig. 12 (a) Schematic representation of the asymmetry transfer in C3-symmetry scaffolds upon aggregation by point chirality and BTA (S)-33, triarylamines (S)-34 and (S)-35 and N-heterotriangulenes (S)-36 and (R)-36 chemical structures. (b) Schematic representation of C2-symmetry monomer chiral self-assembly and chemical structure for chiral PBI (S)-37 and chiral N-annulated perylenes (S)-38 and (R)-38 (c) Chemical structures of scissor-shaped monomers endowed with point chirality (R)-39 and (R)-40 employed by S. Yagai and coworkers and schematic representation of different supramolecular aggregates than can be observed in the supramolecular polymerization process (R)-39 depending on the molar fraction of the cross-linked specie, namely: helical ribbon, superhelical fibril, toroid and amoeba-like fibrils (reproduced from ref. 196, with permission of Springer Nature, copyright 2015). | ||
Other C3 N-centered triarylamine trisamides (S)-34 and (S)-35 functionalized with chiral alkyl chains were also shown to assemble into 1D helical stacks formed by the interplay of triple H-bonds and π–π stacking.191 In another design, L. Sánchez studied the self-assembly behaviour of chiral N-heterotriangulenes (S)-36 and (R)-36 in a MCH/toluene mixtures and in CCl4.192 Remarkably, molecules 34–36 presented pathway complexity (Section 7.1) in their self-assembly processes, forming different helical aggregates with different handedness depending on the experimental conditions. Furthermore, they demonstrated that the branched chiral side chains in (S)-36 induce significant kinetic retardation of the polymerization process, compared to the achiral analogue.
In addition to these examples based on C3-symmetric molecules, there are also systems constructed from C2-symmetric monomers. For example, F. Würthner and co-workers designed a PBI (S)-37 bearing chiral side chains and two amide groups (Fig. 12b).193 The authors stated that (S)-37 aggregates by π–π stacking and H-bonding forming helical supramolecular polymers. The steric crowding caused by the presence of terminal dimethyl groups in the lateral chains causes the formation of homochiral helical J-type aggregates, while the derivatives bearing simple lineal alkyl chains form achiral H-type aggregates.193 More recently, L. Sánchez and coworkers designed N-annulated perylene scaffolds bearing four amide groups, and chiral lateral chains, (S)- and (R)-38 (Fig. 12b).194 Upon self-assembly in organic media, these molecules form P- and M- type helix supramolecular polymers, respectively.
S. Yagai and co-workers have also implemented point chirality to their monomers,151,153,195–197 but in this case more complex hierarchical chiral supramolecular polymers were obtained instead of the simple stacked assemblies (Fig. 12c). For example, the scissor-schapped stilbene dyad (R)-39, bearing amide groups and two lateral 3,7-dimethyl-6-octyl chains, self-assembles by π–π stacking and H-bonding forming right-handed superhelical fibrils. Interestingly, upon UV light irradiation the stilbene units suffer a [2+2] photocycloaddition forming a cross-linked dyad which is unable to form well-defined nanoaggregates. However, in the presence of small amounts of these cross-linked species a whole new co-assembly pathway emerges. It is noteworthy to mention that high molar fractions of the cross-linked monomer with respect to 39 deteriorates the aggregation ability. For instance, the presence of only a 0.1 or 0.25 molar fraction of cross-linked monomers triggers the formation of superhelical fibrils with helix inversion, an 0.6 molar ratio produces amorphous amoeba-like fibrils coexisting with annular structures, and the presence of a molar fraction of 0.8 induces the formation of only amoeba-like fibrils.196 More recently, following a similar molecular design they prepared the scissor-shape dyad (R)-40 bearing two photoresponsive azobenzene units and they studied effects of photoirradiation and secondary nucleation (Section 7.3.3) on the resulting chiral supramolecular arquitectures.197
5.2. π-Conjugated monomers incorporating axial chirality
Axial chirality is defined as the stereoisomerism that arises from the 3D spatial arrangement of two pairs of substituents around a chiral axis.198 This type of asymmetry element can be embedded directly within specific π-conjugated scaffolds or introduced into the monomer structure by attaching a chiral axial unit as a pendant group. The synthesis and isolation of these pure chiral compounds is often nontrivial, frequently requiring enantioselective separation of the enantiomers via chiral high-performance liquid chromatography (HPLC).One of the most relevant examples of supramolecular polymers based on a monomer with axial chirality was reported by S. George and co-workers, combining two NDI units and a binaphthyl core.199,200 In this design, the main NDI chromophores are achiral, and the pendant binaphthyl unit provides axial chirality to the system (Fig. 13a). The self-assembly behaviour of compounds ((S) and (R)-41-42 and (S)-43) was studied under different conditions including via primary and secondary nucleation events, as well as living supramolecular polymerization (LSP) (Section 7.3). Importantly, despite the strong kinetic effects involved in the system, the pure compounds assemble into 1D stacks whose helicity is dictated by the axial chirality of the pedant binaphtyl group (Fig. 13a). A similar approach to design chiral monomers was applied by utilizing the trans-1,2-bis(amido)cyclohexane moiety linked also to two NDI units.123,201,202 For example, it has been reported compound (S,S)-44, with two electron rich NDI units, and compound (S,S)-45 with two electron poor NDI units (Fig. 13a). These compounds self-assemble into chiral stacks, and more interestingly the mixtures of the two compounds lead to the formation of block copolymers with controlled stereoselectivity under thermodynamic conditions.123
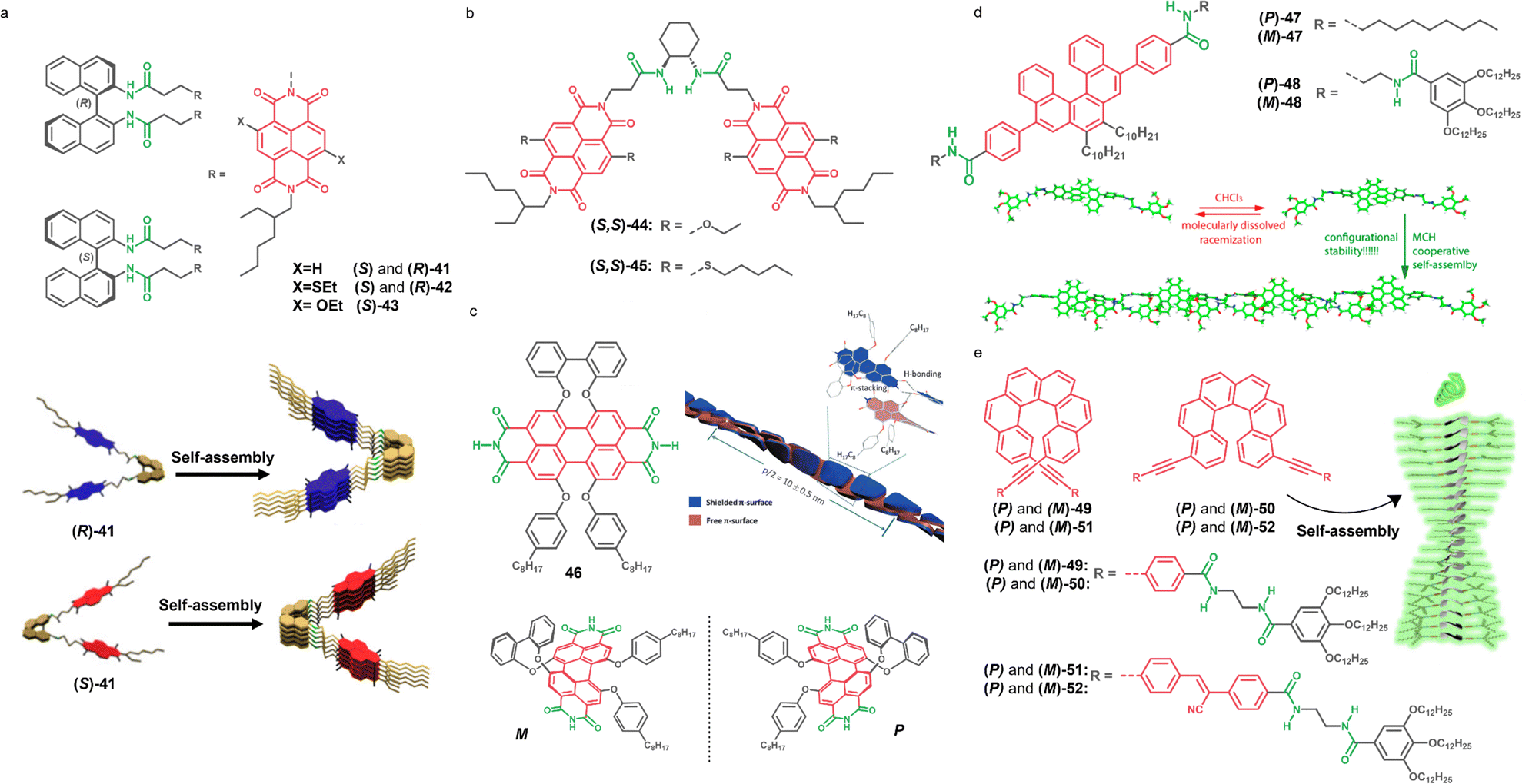 | ||
| Fig. 13 (a) Chemical structures of bichromophoric units with axially chiral binaphthyl units 41–43 and schematic representation of the self-assembly for bichromophoric units with binaphtyl axial pendants (S)- and (R)-41 (reproduced from ref. 199 with permission of Wiley-VCH GmbH, copyright 2020). (b) Chemical structure of bichromophoric units with axially chiral trans-1,2-bis(amido)cyclohexane moieties 44 and 45. (c) Chemical structure of core-substituted PBI 46 with a schematic representation of both M and P atropo-enantiomers M and P and a schematic representation for the aggregate of the enantiopure M atropo-enantiomer (reproduced from ref. 205 with permission of Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, copyright 2012). (d) Chemical structure of [5]helicenes 47 and 48 and a schematic representation of the aggregate of the former (M)-47 in MCH (reproduced from ref. 98 with permission of the American Chemical Society, copyright 2018).(e) Chemical structures of [6]helicenes 49, 50, 51, 52 and schematic representation of the self-assembled structure for 50 and 52 (reproduced from ref. 212 with permission from the Royal Society of Chemistry). | ||
In other examples, the axial chirality is introduced directly to the main chromophore presenting a permanent chiral axis. With the aim of preventing H-aggregation and favouring J-aggregation, F. Würthner and co-workers developed PBIs with a locked twisted-core by introducing bulky substituents at the bay potions and generating a chiral axis in the π-surface.203 This initial design was later improved by functionalizing the PBIs with a biphenyl bridge at one of the two bay positions, which facilitated the separation of the two atropoisomers by chiral HPLC.204 In this line, PBI 46 with N–H groups at the imide positions self-assembles into stranded 1D J-aggregates and its helicity is dictated by the axial chirality of the monomer (Fig. 13c).205 It is noteworthy that similar helical stranded PBI J-aggregates were also achieved by introducing point chirality in the molecule, specifically through the attachment of wedge-shaped groups bearing chiral alkyl chains at the bay positions (see Fig. 5b, compound 5).84
5.3. π-Conjugated monomers incorporating planar and helical chirality
There are various examples of chiral supramolecular polymers based on monomers presenting planar chirality (Fig. 11d),206–209 however, these monomers lack either H-bonding groups or chromophoric nature and therefore were not included in this review. In contrast, there are various examples of systems exploiting helical chirality (Fig. 11d). Helicenes are archetypical chiral molecules with strong fluorescence and CPL properties.210 L. Sánchez and co-workers reported in 2018 two 5,7,8,10-tetrasubstituted [5]helicenes, compounds (P)-47 and (M)-47 with two amide groups and compounds (P)-48 and (M)-48 with four. The corresponding enantiomers of these compounds could be successfully separated by chiral HPLC of the racemic mixtures, but when left in solution undergo racemization. They stated that (P) and (M)-47 did not form any supramolecular polymer, while (P)-48 and (M)-48 could polymerize by a cooperative mechanism due to the presence of the two extra amide groups, demonstrating the relevance of the H-bonding interaction in the supramolecular polymerization process. Interestingly, under the appropriate conditions, the polymerized systems do not suffer racemization. Due to the twisted shape of the helicene moiety, the π–π stacking is not possible, and therefore the resulting assemblies consist of chiral main-chain polymers formed by H-bonding (Fig. 13d).98More recent examples of helicenes in supramolecular polymers are the 2,15- and 4,13- disubstituted [6]-helicenes reported by L. Sánchez and co-workers in collaboration with J. Crassous (Fig. 13e).211,212 They utilized a 2,15-substituted scaffold to prepare (P) and (M)-49 and (P) and (M)-51, which assembles with a zig-zag arrangement by H-bonding, since the contorted shape of the molecule prevents the π–π stacking. In the following work they prepared (P) and (M)-50 and (P) and (M)-52.211,212 In the new compounds, due to the 4,13-disubstitution, the π-surface becomes accessible, resulting in a helical self-assembly by combination of the H-bonding and the π–π stacking interactions (Fig. 13e).
Besides the helical chirality, inherent chirality also has been utilized to prepare chiral supramolecular polymers. Worth to be mentioned, for example, are the corannulene scaffold studied by Aida and co-workers137 and the subphthalocyanines studied by D. González, T. Torres, E. Ortí and co-workers,213 among others.
5.4. Asymmetry amplification and symmetry breaking
In the previous section, we reviewed various examples of monomeric species capable of forming helical supramolecular polymers as a result of asymmetry transfer from the monomer to the polymer (Fig. 11b). Typical investigations of chiral supramolecular polymers focus on elucidating the mechanism and thermodynamics of the polymerization process, primarily using CD techniques. Additional studies often involve the study of mixtures of a chiral monomer with either an achiral analogue or the corresponding enantiomer. These approaches are valuable for understanding the principles of chirality transfer in supramolecular systems under different conditions. In this context, two of the most representative phenomena are asymmetry amplification and symmetry breaking.On the other hand, in the Majority rules experiment when monomers bearing opposite chirality (enantiomers) are mixed in varying ratios, asymmetry amplification may lead the predominant monomer to dictate its preferred helicity over the minority enantiomer.153,220 In both cases, a non-linear increase in the enantiomeric excess (ee) is distinctive of the existence of chiral amplification phenomena and can be influenced by different factors, such as the temperature,221 the number and the positions of stereogenic groups,189,220 or the shape of the molecule.222
A key issue in the asymmetry amplification phenomena is the geometrical complementarity of the different monomers and their energetically favoured tendency to co-assemble, if these two factors are not present, self-sorted homopolymers will be obtained and no asymmetry amplification will occur.102 The self-assembly mechanism and H-bonding have crucial roles in this process, as scarce examples of asymmetry amplification have been found in isodesmic or slightly cooperative supramolecular polymers.223,224
5.5. Supramolecular polymers exhibiting circularly polarized luminescence (CPL)
As mentioned at the beginning of this chapter, CD has become a fundamental tool to study chiral self-assembly processes of π-conjugated scaffolds. However, more recently, another phenomenon has gained increasing interest in the field due to its potential applications: CPL (Fig. 14a). CPL is the differential emission of left- and right-circularly polarized light from chiral luminophores (single molecule or supramolecular entity) in their excited state. This phenomenon has become one of the cornerstones in the development of functional materials due to its potential applications in several fields such as 3D displaying,229 encrypted storage and transmission of information,230,231 chiral sensing,232–234 or bioimaging.235,236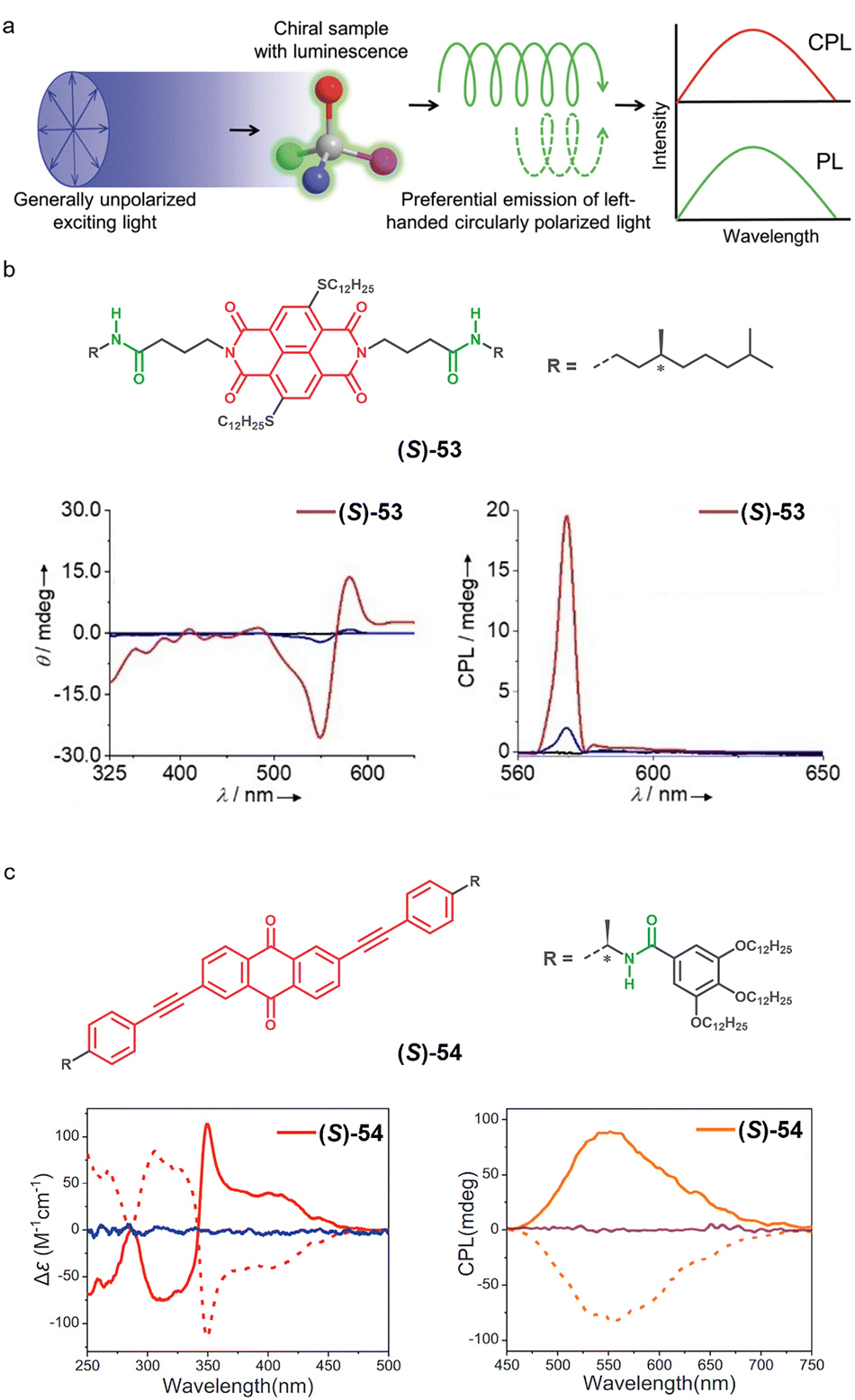 | ||
| Fig. 14 (a) Schematic representation of the circularly polarized luminescence (CPL) phenomenon (reproduced from ref. 242 with permission of Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, copyright 2019). (b) Chemical structure of chiral sulfur-substituted naphtalene-diimide (S)-53 along with its CD and CPL spectra (red lines) in decane/THF (9:1) (c = 0.1 mM) (reproduced from ref. 252 with permission of Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, copyright 2020). (c) Chemical structure of chiral anthraquinone derivative (S)-54 along with its CD (red line) and CPL (λεx = 365 nm) (orange line) spectra in MCH (c = 20 mM) (reproduced from ref. 239 with permission of Wiley-VCH GmbH, copyright 2025). | ||
One of the main focuses in this field has been to improve the dissymmetry factor (glum) and consequently the CPL brightness (BCPL), both of which are crucial for the performance of any CPL active material.237–239 The glum factor can be defined as the parameter that quantifies the difference in luminescence between two enantiomers or as 2(IL − IR)/(IL + IR), where IR and IL are the intensity of the right- and left-handed circularly polarized emissions (Fig. 14a), respectively, and its theoretical maximum value is 2. On the other hand, BCPL is expressed as BCPL = ελ × Φlum × |glum|/2, where ελ is the molar extinction and Φlum is the luminescence quantum yield.
Supramolecular polymerization of π-conjugated systems provides an excellent platform to explore CPL, especially when using chiral monomers with strong photoluminescence and tendency to form ordered chiral aggregates.240–242 In this context, the formation of low emissive H-aggregates (Section 1.3) is avoided, and instead, the authors have focused on the search of CPL-active J-type aggregates or either aggregation-induced emission (AIE)243 or aggregation-induced enhanced emission (AIEE).244
Luminescent scaffolds utilized to prepare CPL active chiral supramolecular polymers include cyanostilbenes74,245,246 helicenes,211,212 and bichromophoric units appended with chiral pendants as the trans-1,2-bis(amido)cyclohexane or binaphthyl moieties (see Sections 5.2 and 5.3).247–251 These systems have shown remarkable glum and BCPL values through the self-assembly of chiral luminophores.
In addition, S. Ghosh and co-workers designed a chiral sulfur-substituted naphthalene-diimide (S)-53 that exhibited H-bonding driven aggregation in decane (Fig. 14b). The cooperative supramolecular polymerization led to J-type aggregates with a fibrillar morphology, confirmed by UV/vis spectroscopy and AFM studies, respectively. Furthermore, CPL spectroscopy studies showed a glum value of 4.6 × 10−2 at the emission maxima, which is among the highest values reported for chiral supramolecular assemblies in solution.252 More recently, F. Wang and co-workers prepared an anthraquinone derivative with chiral amidic spacers (S)-54 featuring efficient J-aggregation by π–π stacking and H-bonding. CD spectroscopy corroborated the appearance of a bisignate Cotton signal upon cooling the solution, indicative of the formation of a chiral supramolecular polymer (Fig. 14c). CPL studies revealed a |glum| value of 0.013 and a BCPL value of 13.8 M−1 cm−1. Comparative studies with a methylated analogue of (S)-54 in the amide positions (blocking H-bonding) confirmed the essential role of H-bonds in the aggregation and CPL properties.239
Another interesting approach to produce CPL active systems involves the utilization of two components: a helical supramolecular polymer and an achiral emissive chromophore. Liu and co-workers reported the use of 1,3,5-benzenetricarbonyl L-glutamate diethyl ester that gelates forming hexagonal chiral nanotubes which can encapsulate a wide range of guest compounds.253 In this study, six different achiral luminophores were encapsulated, and in all the cases, the supramolecular polymer matrix was found to be able to transfer the chirality from the gel to the guest molecules, generating CPL-active materials with glum values ranging from ±0.2 × 10−3 and ±1.7 × 10−3.
6. 2D supramolecular polymers
Most supramolecular polymers based on H-bonded π-conjugated scaffolds have traditionally formed 1D architectures.7–10 However, probably due to the discovery of the potential of covalent 2D materials, most notably graphene, growing interest has emerged in developing 2D supramolecular polymers.254 These emerging materials are defined by a self-assembly process of monomers where the polymer is growing in two directions by the interplay of one or more non-covalent interactions. As a result of this growing process, diverse assemblies leading to different 2D morphologies such as nanosheets, spirals, and porous networks have been reported.255–259The earliest approaches to 2D supramolecular polymers were in aqueous environments and relied on amphiphilic monomers composed of hydrophobic π-conjugated cores and hydrophilic side chains.260–266 M. Lee and co-workers were pioneers in this area, reporting the aqueous self-assembly of an octa-p-phenylene derivative bearing flexible oligoether dendrons. This system formed extended 2D nanosheets in water, establishing a foundational platform that has been expanded with various aromatic cores and solubilizing side chains.260–266 In general, aqueous assemblies of organic amphiphiles are mainly dependent on the monomer design where the balance of the ratio of the lipophilic and hydrophilic parts defines the morphology of the aggregate. A notable example of hierarchical 2D self-assembly in water includes the work of Montenegro and co-workers, who functionalized a cyclic peptide with apolar residues to guide first the self-assembly into 1D nanotubes via H-bonding and π-stacking and then with directional hydrophobic interactions to form 2D networks.267
In organic solvents the obtention of 2D structures is more challenging since it requires a more careful monomer design with the capability to establish directional non-covalent interactions to enable the growth in two dimensions. In this context, hierarchical self-assembly of 1D structures has emerged as a key strategy to achieve 2D polymers. In these systems, monomers first form 1D assemblies, which later associate laterally via secondary non-covalent interactions to generate 2D architectures (Fig. 15a). The nature of these secondary interactions is crucial, as their directionality and strength determine the ability of the system to grow in the second dimension. For example, Sugiyasu, Takeuchi, and co-workers reported in a seminal example a family of porphyrins (55a,b) self-assembling by H-bonding and π-stacking along one axis and van der Waals interactions between peripheral alkyl chains along the second dimension (Fig. 15b).268,269 Porphyrins 55a and 55b form nanosheets in an organic solvent according to AFM experiments (Fig. 15a, right). Sánchez and co-workers observed a similar behaviour in the N-annulated PBI 56, achieving J-aggregated 1D stacks that further associate through sidechain van der Waals forces to yield metastable 2D sheets in MCH.270
 | ||
| Fig. 15 (a) Schematic representation of a hierarchical 2D assembly; from left to right: example of the monomer design, primary interactions, secondary interactions, construction of the 2D sheet and AFM image of the 2D hierarchical system of molecule 55a (reproduced from ref. 268, with permission of Springer Nature, copyright 2016). (b) Molecular structures of compounds 55–59 yielding 2D hierarchical assemblies. (c) Molecular structure of porphyrin 60 (reproduced from ref. 274, with permission of Springer Nature, copyright 2020) and representation of its self-assembly mode into 2D hierarchical spiral or concentric toroid assemblies. (right) Schematic representation and illustration of the formation of the concentric toroids and spirals and AFM images of the corresponding supramolecular assemblies of compound 60. (d) From left to right: Molecular structure of PBI 61, schematic representation of the formation of brick-like 2D assemblies and AFM image of the ribbons formed by PBI 61 (reproduced from ref. 278, with permission of Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, copyright 2020). | ||
Using more polar organic solvents, Fernández and co-workers utilized amphiphilic bispyridyl Pt(II) complex 57 (Fig. 15b) that also self-assembles into 2D nanostructures in THF/water.271 In this example, the 2D growth is directed by π–π interactions and orthogonal N–H⋯Cl H-bonding. In another example, Ghosh and co-workers reported an unsymmetrical NDI derivative 58, bearing a pendant carboxylic acid group at one of the imide positions, which forms hierarchical 2D assemblies. By modulating the monomer conformation with external additives, they achieved thermodynamically controlled supramolecular polymerization of 58, driven by synergistic H-bonding and π–π stacking interactions, resulting in the formation of ultra-thin 2D nanosheets.272 More recently, Tian and co-workers used triphenylamine 59, with three enaminone-linked pyridine hydrochloride salts at the periphery to obtain H-bonded based 2D assemblies.273 The resulting assemblies are porous and feature in-plane 1D conduits presenting electron transporting features, which enable a remarkable photocatalytic performance in hydrogen production via water splitting.
The hierarchical approximation to develop 2D polymers has also yielded unconventional spiral and toroidal assemblies (Fig. 15c). In non-polar solvents, porphyrin 60, developed by Sugiyasu and co-workers, self-assembles into a 1D supramolecular polymers that spontaneously coiled into an Archimedean spiral (Fig. 15c). These layered spirals are formed by a nucleation-elongation process and are stabilized by fluorophilic secondary interactions (Fig. 15c).274 Interestingly, at lower concentrations the same compound produces concentric toroids (Fig. 15c), which can further associate by weaker interactions to yield extended 2D aggregates.274 The morphology of these exotic spiral assemblies has been well characterized by microscopy experiments (see AFM images in Fig. 15c) when the samples are deposited on surfaces, while their stability in the solution state is not well known. In another example, Sánchez and Yagai reported the on-surface formation of 2D porous networks by the association of nanotoroidal structures based on a scissor-shaped azobenzene dyad.275
An alternative and scarcely explored strategy for constructing 2D supramolecular polymers involves the formation of brick-like structures using monomers capable of establishing slipped interactions through one or two non-covalent forces (Fig. 15d). For example, Shimizu and Soberats designed a bis-urea macrocycle that assembles into brick-like nanosheets via directional urea–urea hydrogen bonds.276 In a similar vein, Würthner and co-workers developed 2D supramolecular polymers from a bis(squaraine) derivative that self-assembles through slipped π–π interactions.277 This assembly mode has also been reported for a H-bonded π-conjugated system. PBI 61 (Fig. 15d), bearing NH groups at the imide positions, assembles into H-bonded chains in which all molecules adopt the same axial twist (either P or M) due to the flexibility of the molecular structure.278 The alternating association of P- and M-twisted PBI chains via π–π interactions leads to the formation of extended 2D nanosheets (Fig. 15d). Notably, the slipped stacking of the PBI cores in this arrangement results in the emergence of J-type coupling.
7. Pathway complexity and controlled supramolecular polymerization
7.1. Pathway complexity
Controlling the nanoscale organization of supramolecular polymers based on π-conjugated molecules has proven to be a key challenge for enhancing their optoelectronic properties.13,17,19 Over the past few decades, significant efforts have been devoted to developing diverse monomeric molecular designs and identifying the structural features that influence the assembly processes.8,76,279 Other factors such as solvent, temperature, and concentration have also been shown to play a critical role in the thermodynamics of supramolecular polymerization.8 However, the kinetic aspects of supramolecular polymerization were largely overlooked for many years, despite their crucial importance. Kinetically controlled supramolecular polymerization processes involve more complex scenarios, where intermediate metastable species delaying the progression toward the thermodynamically stable state appear.280–284 Pathway complexity refers to the coexistence and competition of multiple self-assembly routes that one monomer can follow. Despite this phenomenon was previously observed in various systems,285–287 the concept gained attention after Meijer and co-workers demonstrated, in 2012, that the self-assembly of the chiral OPV 18 (Fig. 8c) could lead to two competing helical forms: a thermodynamically stable M-type helix and a kinetically favoured P-type helix.145 Their findings showed that the early-formed P-type helix prevents the direct formation of the more stable M-type helix, which is slowly formed with time (Fig. 16a). | ||
| Fig. 16 (a) Schematic illustration of the self-assembly behaviour of chiral compound 18 characterized for two competing pathways (reproduced from ref. 145 with permission of Springer Nature, copyright 2012). Energy landscapes illustrating the pathway complexity for (b) a competitive process and (c) a consecutive process. The competitive process involves an off-pathway kinetically trapped and the consecutive process an on-pathway intermediate. M: monomer; A: kinetically trapped states; and B: thermodynamically stable state. | ||
Although the pathway complexity can be simply observed during conventional sample preparation, researchers employ various sample processing methods to access to kinetically trapped states.280–284 Common strategies to obtain kinetically trapped states include rapid cooling, which prevents equilibration and traps monomers or kinetic aggregates, and solvent-switching protocols, where mixing a good solvent with a poor one modulates the aggregation. Other methods involve adjusting the monomer concentration, using specific co-solvents or additives, or applying external stimuli such as ultrasonication, stirring, light irradiation, or seed addition.280–284,288
Time-dependent interconversion studies are typically used to study pathway complexity. The observation of pathway complexity in different systems has led to the identification of two representative scenarios.280–284 The consecutive route involves direct transformations between kinetic on-pathway species and thermodynamic species (Fig. 16b), whereas the competitive route involves the formation of an off-pathway intermediate that disassembles into monomers before forming the stable aggregates (Fig. 16c). These kinetically trapped species can be of monomeric (inactive/dormant) or of aggregated nature. The study of the mechanism in the kinetic transformation of supramolecular polymers is usually studied by time-dependent studies at different concentrations.280–288
7.2. The role of H-bonding in pathway complexity
Despite the investigations of different processing protocols that can be applied, not all the monomers are able to show pathway complexity and kinetic effects in their polymerization schemes, and some structural features in the monomer are required.280–284,289 In this regard, the presence of H-bonding groups in monomeric π-conjugated species has been shown to be a crucial feature. For example, intramolecular H-bonding can lead to the formation of kinetically trapped monomers under certain conditions.35,137,290–292 These trapped monomers (inactive or dormant monomers) are unable to undergo supramolecular polymerization, and only the disruption of these intramolecular bonds allows polymerization to proceed, typically through the interplay of H-bonding and π–π interactions. For instance, Würthner and co-workers reported that PBI 12 forms, after rapid cooling of the sample, a kinetically trapped monomeric state (closed form) through intramolecular H-bonds between the amide N–H and the carbonyl groups of the imide part (Fig. 17a).35 In another example, Aida and co-workers described the C5-symmetric corannulene derivative 10 with five amide-appended side chains, which forms a deeply trapped monomeric state stabilized by fivefold intramolecular H-bonds in nonpolar MCH (Fig. 17a).137 The concept of H-bond-stabilized inactive monomers has also been achieved though the folding of the monomeric species which has been demonstrated in the pyrene-substituted diamide 62 (Fig. 17a).290 The evolution of closed state inactive monomers into the extended forms enables the polymerization of the system. The disruption of the intramolecular H-bonds and the opening of the molecules into their extended form occur over time but can also be triggered by the application of external stimuli. | ||
| Fig. 17 (a) Molecular structures and the extended and closed forms of the H-bonding monomers 12, 10 and 62. (b) (left) Schematic representation of the self-assembly behaviour of compound 63 (reproduced from ref. 293 with permission of American Chemical Society, copyright 2015). (middle) Molecular structures of compounds 63 and 64. (right) Energy self-assembly landscape for compound 64 (reproduced from ref. 296 with permission of Wiley-VCH GmbH, copyright 2023). (c) Molecular structure of compounds 65 and 66 and the corresponding illustration of the different aggregates they form (reproduced from ref. 303 with permission of the Royal Society of Chemistry, copyright 2024). | ||
Alternatively, intramolecular H-bonding in monomeric π-conjugated species can also lead to the formation of kinetically trapped aggregates rather than inactive monomers (Fig. 17b). The tendency of these closed state monomers to form kinetically trapped aggregates is not generally driven by H-bonding interactions and instead arise from the interplay π–π stacking and solvophobic effects, while steric effects may also contribute. Such a case has been observed in various systems based on PBIs,293,294 N-annulated PBIs,295,296 N-annulated perylenes,297 N-heterotriangulenes,192 diketopyrrolopyrroles,298 and Pt(II) complexes,299,300 among others. Functionalizing π-conjugated molecules with various amide groups has been proven to be an effective strategy to promote intramolecular H-bonding patterns enabling the formation of kinetically trapped aggregates.192,293–300 For example, this behaviour has been observed in the bisdendronized PBI 63 bearing butyl spacers between the core and the wedge-shaped benzeneamide motifs (Fig. 17b).293 While the derivative 12, with shorter ethyl spacers, forms a kinetically trapped monomer upon rapid cooling of the solution (Fig. 17a),35 PBI 63 assembles into an off-pathway nanoparticle-like kinetic aggregate under similar conditions (Fig. 17b). This kinetic aggregate eventually evolves via a competitive process into fibrous aggregates over time. In another example, Sánchez and co-workers reported N-annulated perylene derivative 64 forming kinetically trapped J-aggregates based on intramolecularly H-bonded monomers (Fig. 17b).296 The kinetic J-aggregates evolved with time to the thermodynamic H-aggregates through a competitive mechanism. Interestingly, the alkyl spacer length between the amide groups and the core was tuned to modulate the kinetics and energy landscape of supramolecular polymerization (Fig. 17b).296
The formation of kinetically trapped aggregated species has also been observed in monomers capable of self-assembly via two or more distinct H-bonding patterns.301–305 This is the case of the pioneering example of the H-bonding porphyrin 65 reported by Sugiyasu, Takeuchi and coworkers (Fig. 17c).301,302 This type of porphyrins, functionalized with two dendronized amide arms, forms kinetically trapped J-aggregates by tilted H-bonds between amide groups, while this species evolve upon time into more stable H-aggregates characterized by coplanar interactions of the porphyrin cores and more lineal H-bonds (Fig. 17c). This system was utilized to gain control over supramolecular polymerization (see Section 7.3). In a more recent example, BOPHY 66 initially assembles upon cooling into a kinetically favoured H-type supramolecular polymer (66-AggA) in MCH, which gradually transforms into a thermodynamically stable structure (66-AggB) through a competing pathway (Fig. 17c).303 Theoretical studies suggest that the BF2 substituents create steric clashes within the antiparallel, face-to-face arrangement of 66-AggA, prompting the system to reorganize into the more stable polymer 66-AggB, where the monomer units are laterally offset. This new configuration reduces steric strain while preserving the H-bonds between the amide side groups.
7.3. Controlled supramolecular polymerization
Controlled supramolecular polymerization represents a significant conceptual advance in supramolecular chemistry, bridging the precision of covalent polymerization with the dynamic nature of non-covalent assembly.279 Unlike spontaneous supramolecular polymerization, which typically yields polydisperse structures through uncontrolled nucleation and growth, the controlled polymerization relies on suppressing uncontrolled aggregation by creating dormant species. This dormant species can then be activated, by a seed, external stimulus, or initiating species, to trigger a chain-growth mechanism, resulting in some cases in supramolecular polymers with predictable length, low dispersity, and in some cases, living characteristics.280–282 Recent advances in the controlled supramolecular polymerization of π-conjugated systems have enabled the formation of well-defined 1D and 2D nanostructures from small-molecule chromophores. | ||
| Fig. 18 (a) Schematic representation of the living supramolecular polymerization process and the molecular structure of PBI 67. The initiator refers to a seed or a single molecule. The trapped species can consist of kinetically trapped monomeric aggregated species. (b) Molecular structures of compounds 10 and 10-Me, and a schematic representation of the supramolecular polymerization process triggered by adding small amounts of 10-Me into a solution of 10 (reproduced from ref. 137 with permission of the American Association for the Advancement of Science, copyright 2015). (c) Molecular structure of compound 68 and the corresponding self-assembly energy landscape of the light controlled self-assembly process (reproduced from ref. 314 with permission of the American Chemical Society, copyright 2016). (d) Molecular structure of compounds 69a,b and the schematic illustration of the light controlled self-assembly process (reproduced from ref. 315 with permission of the American Chemical Society, copyright 2021). (e) Molecular structures of compounds 70, 71a and 71b, and the corresponding self-assembly energy landscape for the controlled supramolecular polymerization process (reproduced from ref. 317 with permission of the American Chemical Society, copyright 2023). | ||
To achieve LSP, the system may polymerize via the cooperative mechanism and present kinetic trapping of either monomers or aggregated species. Those trapped states serve as a reservoir of monomers for the controlled growing of supramolecular polymers from initiator species, which can be designed as single molecules or pre-formed seeds (Fig. 18a). In seeded LSP, pre-formed seeds are prepared via ultrasonication and are based on aggregates larger than the critical nucleus. These seeds always consist of the most thermodynamically stable aggregated form of the system. When the seeds (or the initiator) are added to a solution of the kinetically trapped states under specific conditions, the elongation process is directly activated, and the monomers are selectively added at the seed ends in a chain-growth fashion (Fig. 18a). True living behaviour is confirmed only if growth can be repeated in multiple cycles, with kinetic and structural evidence.
Pioneering example of LSP was reported by Sugiyasu and Takeuchi in a H-bonding porphyrin system.301 Porphyrin 65 (Fig. 17c) initially forms J-type aggregates as nanoparticles that, over time, evolve into thermodynamically stable fibrillar H-type aggregates (Fig. 17c). The kinetic transformation can be significantly accelerated by introducing seeds of the thermodynamically favoured H-type aggregates (Fig. 18a), prepared via sonication of preformed fibrillar structures. This process produces fibres with controlled length. Repeating this seeding process over several cycles yields supramolecular polymers with low polydispersity.301
An analogous seeding strategy was followed by the Würthner group in PBI systems.35,293,294 For example, PBI 67 forms a kinetically trapped H-aggregate based on the closed conformation of the monomer (Fig. 17a). Upon addition of seeds of the thermodynamic J-aggregate, the system undergoes LSP.294 These studies showed that LSP can be achieved in systems exhibiting off-pathway kinetically trapped aggregates,293,294 but also in systems presenting trapped monomer species in a closed H-bonded conformation (Fig. 17a).35 Interestingly, LSP was also applied for the development of PBI based copolymers with precise control over the supramolecular 1D blocky structures.120
Another relevant contribution in this area was made by Aida and co-workers in 2015 using a single molecule as the LSP initiator.137 In this system, the non-methylated corannulene derivative 10 (Fig. 18b) forms intramolecular pentafold H-bonds between amide groups (Fig. 17a), which effectively prevent self-assembly. In contrast, the N-methylated analogue 10-Me are incapable of forming intramolecular H-bonds or self-assemble into polymers. Interestingly, trace amounts of the methylated compound 10-Me were shown to act as initiators (Fig. 18a) for the LSP triggering the rapid supramolecular polymerization via H-bonds in 10 (Fig. 18b).
While the majority of examples of supramolecular polymerization target the obtention of 1D structures, the concept has also been extended to 2D polymers.268,308–310 For example, Sugiyasu and coworkers reported the controlled 2D supramolecular polymerization from a single porphyrin-based monomers 55a,b (Fig. 15b).268 Upon cooling the solution, the monomer first assembles into metastable J-aggregate nanoparticles via an isodesmic pathway, and then undergo time-dependent autocatalytic transformation into nanosheets through a short-slipping J-aggregation mode. The 2D growth is driven by lateral van der Waals interactions among hexyl side chains and reinforced by H-bonding. This pathway can be selectively activated and precisely controlled using seed fragments, enabling 2D LSP yielding nanosheets with a uniform thickness. More recently a similar approach was applied to grow 2D block copolymers.311
In another example, Feringa and coworkers reported the stilbene-bis-urea photoswitch 69 showing light-responsive aggregation behaviour (Fig. 18d).315 While the cis-isomer is an inactive monomer stabilized by an intramolecular H-bond, the trans-form self-assembles into H-bonded fibrous aggregates. Thus, the light controlled cis–trans photoisomerization of this compound enables the rapid, reversible polymerization and gelation. Unlike conventional light irradiation, vibrational strong coupling has recently been applied to direct supramolecular polymerization toward otherwise inaccessible structures. By coupling the C–H stretching vibration in NDI based polymers, vibrational strong coupling redirected the assembly pathway to form toroids instead of thick fibres, formed under conventional conditions.316
Other strategies to control supramolecular polymerization involves the use of chemical additives.317–320 In a pioneering example, Würthner and coworkers achieved an exquisite control over supramolecular polymerization of squaraine 70 by using a molecular chaperone analogue 71a (Fig. 18e).317 In this approach, squaraine monomers are temporarily inactivated by complexation with a tetralactam macrocycle 71a (chaperone analogue), preventing spontaneous polymerization. Upon addition of a high-affinity initiator (diketopiperazine 71b), the macrocycle is removed, releasing the squaraine monomers to polymerize via π–π stacking and H-bonding (Fig. 18e).
Other approaches to control supramolecular polymerization, not analysed here, include liquid–liquid phase separation events,321 the use of adaptive systems,322 or crowding agents.319,323
 | ||
| Fig. 19 (a) Schematic representation of the supramolecular polymerization process composed of a primary nucleation, an elongation and a secondary nucleation step (reproduced from ref. 325 with permission of Springer Nature, copyright 2025). (b) Molecular structure of compound 72 and the corresponding illustration of its the self-assembly behaviour forming nanotoroids and catenane structures via secondary nucleation (reproduced from ref. 325 with permission of Springer Nature, copyright 2025). Inset shows the AFM image of a nano-poly[5]catenane (reproduced from ref. 328 with permission of Springer Nature, copyright 2020). (c) Schematic representation of the self-assembly behaviour of porphyrin 60, leading to the formation of concentric toroidal assemblies via secondary nucleation (reproduced from ref. 274, with permission of Springer Nature, copyright 2020). (d) Molecular structure of compound (S)-34 and the corresponding illustration of its self-assembly behaviour forming chiral helical fibrils by primary nucleation and superhelical assemblies via secondary nucleation (reproduced from ref. 330 with permission of Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, copyright 2019). | ||
While secondary nucleation in supramolecular polymerization was observed by Bert Meijer and co-workers in 2008 in chiral oligothiophenes,326 the phenomenon was not clearly demonstrated until recently. The most exhaustive studies of secondary nucleation were reported by the Yagai group.325 For example, the barbituric acid-functionalized π-conjugated compound 72 self-assembles into toroidal nanostructures through hierarchical supramolecular polymerization (Fig. 19b).327,328 Each toroid originates from H-bonded rosettes formed by approximately 600 molecules. Remarkably, secondary nucleation on the inner surfaces of these toroids drives the formation of complex interlocked topologies known as nano-poly[n]catenanes, including [2]- to [22]-catenanes. The AFM image of a nano-poly[5]catenane is shown in Fig. 19b. van der Waals interactions between monomers and toroid surfaces facilitate this hierarchical growth.327 The utilization of this type of molecules and secondary nucleation provided new tools for the creation of exotic supramolecular topologies.
Sugiyasu and co-workers demonstrated a sophisticated example of secondary nucleation-controlled supramolecular polymerization through the formation of supramolecular concentric toroids using fluoroalkyl-substituted metalloporphyrins 60 (Fig. 15c).274,329 Upon cooling, primary toroids are formed under dilute conditions through ring closure of curved supramolecular 1D stacks. These toroids acted as seeds for secondary nucleation along their fluorinated inner surfaces, enabling the growth of larger, concentric toroidal structures (Fig. 19c).329 A solvent-mixing protocol allowed for kinetically controlled toroid synthesis with narrow polydispersity, and monomer addition enabled further size modulation without loss of uniformity.274 In a second work, site-selective secondary nucleation of different porphyrin monomers led to the construction of 2D block architectures with high sequence control. Notably, selective depolymerization of inner toroids using 4-dimethylaminopyridine revealed reactivity differences between the inner and outer surfaces, attributed to their distinct curvature and side-chain composition.
The secondary nucleation approach has also been employed to construct hierarchical supramolecular polymers from chiral π-conjugated monomers.200,330–332 Giuseppone and co-workers demonstrated this using a triphenylamine-based monomer (S)-34, where the occurrence of secondary nucleation could be finely tuned by varying the concentration and cooling rate.330 At low concentrations and under fast cooling, P-helical fibrils are formed (Fig. 19d). In contrast, higher concentrations or slower cooling rates promote the secondary nucleation of these fibrils into larger M-helical superstructures, showcasing a clear transition from primary to hierarchical organization (Fig. 19d).
7.4. Supramolecular polymorphism
Polymorphism refers to the ability of a molecule or polymer to adopt distinct structural arrangements depending on factors such as solvent, temperature, concentration, or processing conditions.334 In chemical biology, polymorphism manifest in the tertiary and quaternary structures of amyloid aggregates, with different forms often linked to specific biological functions or pathological effects.335 In the pharmaceutical industry, crystal polymorphism significantly influences drug solubility, stability, and bioavailability.336 Likewise, in organic electronics, molecular packing variations directly impact charge transport, optical properties, and overall device performance.337 Controlling polymorphism in π-conjugated supramolecular polymers can therefore enable access to diverse supramolecular structures with tailored packing, morphology, and properties.Systems presenting supramolecular polymorphism are characterized for presenting two or more distinct stable aggregated states.44,195,278,280,281,338–348 Fig. 20a shows the energy landscape for PBI 73 reported by the Würthner group, which exhibits three distinct one-dimensional supramolecular polymorphs (73-Agg1–3) in the same solvent, at the same concentration, and at room temperature.339 73-Agg2 and 73-Agg3 were formed from kinetically trapped 73-Agg1 by applying ultrasonication (physical trigger) or seeding (chemical trigger). The three distinct aggregates differ in their π–π stacking arrangements and H-bonding patterns as shown in Fig. 20a. Supramolecular polymorphism was also observed for PBI 61 (Fig. 15d), which features bay-position acyloxy groups and NH at the imide positons.278 This compound forms either fibrilar or lamellar polymorphs depending on the cooling rate (Fig. 20b). These polymorphic structures arise from homo- or heterochiral self-assembly of atropoenantiomeric PBIs. Fast cooling promotes narcissistic self-sorting into the homochiral form, whereas slower cooling enables a transformation into heterochiral sheet-like assemblies.
 | ||
| Fig. 20 (a) Molecular structure of compound 73 and the corresponding self-assembly energy landscape leading to the formation of Agg1, Agg2 and Agg3. The assembly mode of the three aggregates is schematically depicted in the inset (reproduced from ref. 339 with permission of the American Chemical Society, copyright 2019). (b) Scheme of the polymorphic behaviour of compound 61 forming fibres or sheet structures depending on the thermal treatment of the sample (reproduced from ref. 278 with permission of Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, copyright 2020). (c) Molecular structure of compound 74 and the scheme of its self-assembly behaviour forming polymorph A via pseudo-parallel interactions or polymorph B via slipped interactions depending on the thermal treatment of the sample (reproduced from ref. 340 with permission of American Chemical Society, copyright 2019). (d) Molecular structure of compounds (S)-75, (R)-75 and (A)-75 and a schematic illustration of the corresponding polymorphs I and II providing distinct chiral amplification behaviour (reproduced from ref. 349 with permission of Wiley-VCH GmbH, copyright 2025). (e) Molecular structure of compounds 76a–d and a schematic illustration of the corresponding self-assembly behaviour into planar or convex rosettes and the subsequently into toroidal and fibrillar aggregates (reproduced from ref. 342 with permission of the Royal Society of Chemistry, copyright 2023). (f) Molecular structure of compound 77 and a scheme of its self-assembly behaviour including the light modulation of supramolecular polymorphism (reproduced from ref. 195 with permission of the Royal Society of Chemistry, copyright 2022). | ||
Supramolecular polymorphism has also been reported for OPE-based Pt(II) complexes.340,341 The Fernández group reported the Pt(II) complex 74 that forms two stable polymorphic structures (polymorphs A and B) simultaneously, depending on assembly conditions in MCH.340 Polymorph 74-A forms under fast cooling conditions (2 K min−1) via an isodesmic mechanism and features a slipped molecular arrangement stabilized by N–H⋯Cl–Pt interactions (Fig. 20c). In contrast, polymorph 74-B emerges through a cooperative mechanism under slow cooling (0.1 K min−1), displaying pseudoparallel stacking driven by N–H⋯O(alkoxy) H-bonds (Fig. 20c). Conversion from polymorphs 74-A to 74-B is only possible by thermal annealing, where aggregate 74-A must fully disassemble into monomers before reassembling into the 74-B polymorph. In a recent example, the G. Fernández group has demonstrated the relevance of polymorphism in the control of the amplification of asymmetry using chiral Pt(II) complexes, (S)-75 and (R)-75, alongside an achiral model compound (A)-75 (Fig. 20d).349 The chiral monomers form two stable polymorphs in MCH at room temperature (AggI and AggII) distinguished by their defined rotational offset during supramolecular polymerization, leading to distinct morphologies and photophysical properties (Fig. 20d). In the asymmetry amplification experiments, both polymorphs showed amplification effects, while AggII (with more twisted packing and stronger H-bonding) shows superior performance in SaS experiments with achiral compound (A)-75. Remarkably, coassembly of (A)-75 and (S)-75 into AggII results in CPL, an emergent property absent in the supramolecular polymers of the individual components.349
Other examples of supramolecular polymorphism in π-conjugated systems have mainly focused on morphological aspects, even when the molecular packing remains barely altered.195,319,342,343 Yagai and co-workers reported supramolecular polymorphism in the barbiturate conformers 76a–d that present different molecular conformations due to the flexible double bond connecting the two naphthalene units (Fig. 20e).342 These compounds self-assemble into H-bonded cyclic hexamers (rosettes) (Fig. 8d) that subsequently yield toroidal or straight fibrous structures depending on the used conformer. Differences in the rosettes’ planarity determine the stacking mode: translationally displaced stacking leads to curved nanorings, while aligned stacking produces straight nanorods (Fig. 20e). The same group also reported the scissor-shaped azobenzene dyad 78 forming nanotoroids as kinetic products and gradually transitions to thermodynamically stable fibres upon cooling (Fig. 20f).195 Remarkably, the polymorphic behaviour can be controlled by light at constant temperature. Strong visible light irradiation of the cis-77 induces the formation of nanotoroids, while weaker light favours fibre formation (Fig. 20d). This is the first demonstration of light-driven modulation of supramolecular polymorphism, enabling the selective formation of distinct nanostructures without changing temperature.
8. Applications of hydrogen bonded π-conjugated supramolecular polymers
As we have seen, depending on their monomer structure and supramolecular organization, π-conjugated supramolecular polymers can exhibit a broad spectrum of properties. Still, the real-world application of supramolecular polymers remains limited, and no truly groundbreaking product has yet reached the market. Nevertheless, their potential is undeniable, and it is likely that supramolecular polymers will play a key role in the development of next-generation materials.15,350 While applications are not the main focus of this review, this section offers a brief overview of the key characteristics of π-conjugated H-bonded supramolecular polymers that highlight their potential for future applications.8.1. Photonic and optoelectronic applications
Owing to the intrinsic photophysical properties of π-conjugated scaffolds, the supramolecular polymer derivatives are inherently attractive for photonic and optoelectronic applications. In the field of photonics, J-aggregates are of great relevance due to their sharp red-shifted absorption, strong exciton coupling, and efficient light-harvesting properties.66 For example, J-aggregated cyanines have been used to construct supramolecular structures with light harvesting properties,59,351,352 or platinum acetylides were found to be good candidates to be used in optical waveguides.353PBI supramolecular polymers often form J-aggregates characterized by strong excitonic coupling and fluorescence properties.29,64,354 For example, the tetraphenoxy-substituted PBI modified with thiophene side arms (78) self-assembles into core–shell columnar structures made of up to seven strands (Fig. 21a).355,356 The presence of separated electron-accepting and an electron-donating regions allows independent pathways for electrons and holes, enabling efficient charge transport after light excitation. As a result, the material shows strong anisotropic photoconductivity with fast on–off switching under light irradiation (Fig. 21a). PBI J-aggregates have generally been applied in solar cells,357 thin-film transistors,358 optical waveguides,359 and optical microcavities,360 among others.
 | ||
| Fig. 21 (a) Molecular structure of 78 and its self-assembled core-shell structure with charge transport properties (reproduced from ref. 356 with permission of Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, copyright 2019). (b) Molecular structure of 79 and the corresponding representation of its 1D assembly (reproduced from ref. 361 with permission of the American Chemical Society, copyright 2020). (c) Molecular structure of 80, the self-assembly mode and the representation of the controlled supramolecular polymerization of 80 under the application of an electric field (reproduced from ref. 368 with permission of Springer Nature, copyright 2012). (d) Molecular structure of 81 and scheme of detection of nitrocompounds (reproduced from ref. 377 with permission of Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, copyright 2014). (e) Representation of the structure of a BTA supramolecular polymer catalyst in which the helicity is induced by adjusting the amount of BTA (S)- and (R)-82 (reproduced from ref. 390 with permission of Springer Nature, copyright 2024). (f) Molecular structure of photocatalyst 83 and its corresponding 2D assembly (reproduced from ref. 394 with permission of Springer Nature, copyright 2014). (g) Closed and open states of the photoswitch 84 (reproduced from ref. 419 with permission of Springer Nature, copyright 2018). (h) Molecular structure of BODIPY 85 and the representation of the formation of spherical aggregates of different sizes depending on the alkyl chain length. Scheme of its function for in vivo bioimaging (reproduced from ref. 416 with permission of Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, copyright 2025). | ||
H-aggregates have also been exploited for photonic applications, as for example the carbonyl-bridge tryarylamines.361 Compound 79 self-assembles into fibrillar aggregates with pronounced electronic coupling, enabling coherent exciton transport over distances exceeding 4 µm, limited only by the length of the supramolecular fiber (Fig. 21b). Interestingly, the transport properties of these tryarylamine family can be tuned by changing the peripherical groups.362–364
In a prominent example, Aida and coworkers reported amphiphilic hexabenzocoronenes self-assembling into micrometre-scale, graphite-like nanotubes with a uniform wall thickness.365 Interestingly, these tubular structures supported anisotropic charge transport.366 In the same context, Giuseppone reported a family of triarylamine scaffolds forming supramolecular 1D structures under different conditions.134,367 Light-triggered supramolecular polymerization under an electric field enabled the in situ growth of 80 nanowires that bridge electrode gaps in a highly ordered fashion (Fig. 21c).368 These self-assembled organic interconnects exhibit remarkable electronic properties, with an ohmic conductivity of 5 × 103 S m−1, and current densities up to 2 × 106 A cm−2. At cryogenic temperatures, the nanowires show metallic behavior, underscoring their exceptional performance compared to typical organic materials. In another example, urea-functionalized bithiophene derivatives were shown to assemble into nanofibres that support highly mobile charge carriers, as revealed by pulse radiolysis.369 Similar behaviour was reported for oligothiophene-based chromophores that allows the formation of long-lived photocharges.370
These examples underscore the potential of π-conjugated supramolecular polymers in key optoelectronic devices,29,59,64,351,361,366,369,371,372 including organic field-effect transistors (OFETs),373 organic light-emitting diodes (OLEDs),374 and organic photovoltaics.375
8.2. Sensing and separation
π-Conjugated supramolecular polymers have also emerged as promising platforms for sensing, exploiting their inherent stimuli-responsive properties. For instance, it has been demonstrated that they can be applied to efficiently detect explosive nitroaromatic compounds,376,377 through fluorescence quenching mechanisms.17,378 One example of the sensing of explosive nitroaromatic compounds consists of pyrene 81, forming supramolecular polymers via π-stacking and H-bonds (Fig. 21d).377 The resulting supramolecular structure allows detection due to donor–acceptor interactions and H-bonding between the polymer and the nitrocompound. In the same line, fluorescence quenching mechanisms were used for the detection of metal ions using the bispillar[5]arene-based supramolecular polymer designed by Tai-Bao Wei.379 This system exhibited AIE, and was successfully applied for the fluorescence detection and removal of mercury(II) through host–guest interactions.Supramolecular assemblies also have great potential not only for the sensing of chiral compounds, but also for their separation.380 For example, chiral and achiral hydrazide foldamers were shown to bind selectively to saccharides adopting well-defined supramolecular helical assemblies or forming host–guest foldamers whose conformation can be monitored by different techniques.381,382 While still is a growing field, supramolecular assemblies have also been proposed for separation and purification applications, as for example as components in separation membranes.383–385
8.3. Catalysis
Another emergent application of supramolecular polymers lies in the field of catalysis.386–388 For instance, a water-soluble BTA derivative functionalized with L-proline co-assembles into homochiral helical supramolecular polymers that exhibit catalytic activity and selectivity in aldol reactions involving p-nitrobenzaldehyde and cyclohexanone.389 More recently, BTA-based supramolecular polymers have been employed to develop a switchable asymmetric copper catalyst that enables selective access to different stereoisomers of an amino alcohol through a hydrosilylation–hydroamination cascade.390 This was achieved using helical polymers composed of different BTA monomers, where the handedness of the helices was tuned by adding a chiral BTA co-monomer, (S) or (R)-82 (Fig. 21e). The helicity of the polymers dictates the outcome of the catalytic transformation of an amino alcohol.Supramolecular π-conjugated polymers are particularly relevant in photocatalysis, where the chromophore plays a central role in light absorption and energy transfer within the system.391 Photocatalityc supramolecular polymers are mainly used in aqueous systems, but not always H-bonds are involved. One example of a photocatalytic system consists of a platinum(II) metallacycle with AIE-active TPE units acting as efficient light-harvesting antennas in water.392 The monomer self-assembles into nanospheres, which enable a highly efficient two-step energy transfer cascade, and the photocatalytic C–H alkylation in water. Other catalytic processes include water splitting and CO2 reduction.393 Stupp and coworkers reported a hydrogel scaffold formed through the self-assembly of a perylene monoimide amphiphile 83 that acts as both (Fig. 21f) a light-harvesting platform and a host for a nickel-based catalyst.394,395 This supramolecular material promotes efficient electronic coupling between chromophores and supports photocatalytic hydrogen generation within the three-dimensional hydrogel matrix. Furthermore, it can be easily deployed on surfaces or embedded in porous supports, offering a versatile platform for artificial photosynthesis. In another example, a spherical chromatophore-like nanomicelle system was developed by self-assembly of Zn–porphyrin amphiphiles with a Co catalyst in water.396 The resulting hierarchical organization generates an antenna effect that promotes CO2-to-CH4 conversion with remarkable performance (TON > 6600, 89% selectivity, 15% solar-to-fuel efficiency).
8.4. Biomedical applications
Supramolecular polymers likely exhibit the greatest potential for biomedical applications.397–400 However, these supramolecular polymers do not always consist of chromophores or involve the participation of H-bonding systems. It should be highlighted the pioneering work of the Stupp group, demonstrating the use of supramolecular polymers in regenerative medicine,401 muscle actuators,402 and therapeutics,403 among others. In addition, numerous studies have shown the potential of supramolecular polymers in drug delivery systems.404,405π-Conjugated supramolecular polymers have been exploited in biomedical applications for their unique electronic and optical properties.406,407 Various dye-based assemblies have been employed for phototherapy408–413 and bioimaging,414–417 enabling both therapeutic and diagnostic functionalities. Chen and Wang reported supramolecular nanoparticles based on a Pt(IV)-modified β-cyclodextrin–ferrocene monomer.418 In tumors, the nanoparticles dissociate in response to H2O2, releasing hydroxyl radicals and Pt(IV) prodrugs, which convert to cis-platin and amplify H2O2 generation. This self-augmented cascade enhances chemodynamic therapy while enabling renal clearance for low systemic toxicity. Other systems rely on light stimuli to modulate its function. In this context, Ding, Tang and coworkers developed a smart organic nanoparticle based on the photoswitch 84, useful for photoacoustic imaging and photodynamic therapy.419 By light-triggered switching between ring-closed and ring-opened states (Fig. 21g), the nanoparticle directs absorbed energy either to thermal deactivation for photoacoustic imaging or to fluorescence and ROS generation for photodynamic therapy.
More recently, a series of amphiphilic aza-BODIPY dyes (85) were shown to self-assemble in water into nanostructures with tunable size and photophysical properties.416 Smaller assemblies (C1) show weak emission, while longer-chain derivatives (C8–C16) form J-aggregates with strong NIR fluorescence and photoacoustic signals (Fig. 21h). These dye assemblies demonstrate excellent biocompatibility, size-dependent biodistribution, and effective in vitro and in vivo imaging, highlighting the potential of self-assembled dyes as versatile contrast agents for biomedical applications.
9. Conclusions and future outlook
Hydrogen bonding has proven to be one of the most powerful and versatile noncovalent interactions for directing the self-assembly into well-defined supramolecular architectures. The inherent directionality, tunable strength, and reversibility of hydrogen bonds make them ideally suited for programming order from the molecular levels to macroscopic domains. Throughout this review, we have highlighted how hydrogen-bonding motifs can be rationally combined with π-conjugated scaffolds allowing a certain level of control over the self-assembly mechanism, molecular packing, and aggregate morphology, as well as the induction of pathway complexity and tuning of the photophysical properties.Considering the electronic implications and exciton coupling arising from the interactions between chromophores, the directionality and strength of hydrogen bonds play a pivotal role in guiding the assembly of π-conjugated systems. The general monomer design strategy for developing hydrogen-bonded π-conjugated supramolecular polymers involves the functionalization of the selected chromophoric unit with hydrogen-bonding motifs and the appropriate solubilizing groups. Through careful molecular engineering of these monomeric units, it is possible to achieve a high degree of control over the self-assembly process, guiding the formation of one-dimensional, two-dimensional, and/or chiral supramolecular architectures. The molecular packing within these architectures is critical, as it directly influences exciton coupling and determines whether H-type or J-type aggregates are formed, factors that are essential for tuning the optical, electronic, and functional properties of the resulting materials.
Importantly, the incorporation of hydrogen-bonding motifs and flexible linkers into π-conjugated scaffolds provides an additional versatility and tunability regarding the interaction's possibilities. Hydrogen-bonding units can establish diverse intra- and intermolecular interaction patterns, acting either synergistically or in competition with other noncovalent forces. This interplay gives rise to complex self-assembly landscapes, featuring multiple monomeric or aggregated species that may exist as kinetically trapped intermediates or thermodynamically stable structures. Kinetically trapped species, including dormant monomers or metastable aggregates, have proven particularly valuable for exerting control over the self-assembly process through mechanisms such as living supramolecular polymerization or secondary nucleation.
The integration of hydrogen-bonding units into π-conjugated systems has also enabled the development of adaptive and stimuli-responsive materials. External stimuli such as ultrasonication, light, or chemical additives can modulate the self-assembly processes, in terms of molecular packing, morphology, and optical or electronic behaviour. In this context, supramolecular polymorphism has emerged as a key concept, enabling the formation of two or more stable aggregated states from the same monomeric building blocks.
Despite significant progress, several challenges and opportunities remain in the field of hydrogen-bonded π-conjugated supramolecular assemblies. A more predictive understanding of how subtle variations in hydrogen-bond donor/acceptor patterns, molecular geometry, and solvent environment influence supramolecular outcomes is still lacking. This will be relevant for the development of three-dimensional architectures, but also in relation to pathway complexity, where the critical aspects of monomer design are not yet fully understood. In this context, developing new strategies to control supramolecular polymerization and harness supramolecular polymorphism remains a demanding but promising direction. Moreover, a deeper exploration of stimuli-responsive systems could not only offer new tools to control the kinetics and mechanisms of supramolecular polymerization but also provide a means to modulate in real-time the morphology and properties of the resulting materials.
Looking ahead, the utilization of new tools such as machine learning, molecular simulations, and advanced in situ characterization techniques (e.g., cryo-TEM, time-resolved spectroscopy, single-molecule methods, and small-angle X-ray scattering) will be essential to decipher the complex kinetics and thermodynamics of supramolecular assembly processes. These approaches may eventually allow us to design supramolecular systems with predictive control over structure, function, and dynamics, akin to how nature programs molecular machines and complex assemblies via hydrogen-bonding in proteins and nucleic acids. Although this review has primarily focused on single-component systems, the rational design of multicomponent supramolecular assemblies with precise sequence and spatial control remains a significant challenge. Gaining control over multicomponent assembly is crucial for progressing toward more complex, multifunctional, and life-like supramolecular systems with advanced levels of structural and functional sophistication.
Owing to their unique electronic properties, π-conjugated supramolecular polymers hold significant potential for applications in optoelectronics, photonics, catalysis, sensing, and biomedicine. However, translating these fundamental properties into practical applications remains a major challenge, largely due to issues related to structural stability, processability, and precise control over molecular organization at larger scales, or in multicomponent environments. Their dynamic, noncovalent nature, while advantageous for self-healing and adaptability, can compromise long-term stability and reproducibility. Bridging the gap between molecular design and application performance will require advances in processing methods, and a deeper understanding of structure–function relationships under operational conditions.
In conclusion, hydrogen-bonding modulation in the self-assembly of π-conjugated systems is not only a powerful strategy to develop functional materials but also a conceptual framework for advancing the field of supramolecular chemistry. As we move toward the development of programmable, adaptive, and responsive supramolecular systems, hydrogen bonding will undoubtedly remain at the heart of molecular design, enabling new frontiers in soft matter, materials science, and nanotechnology.
Author contributions
D. Martínez and L. Rubert: literature search, data collection and analysis, and writing. P. Ximenis: literature search, data collection and analysis, and writing and editing. B. Soberats: conceiving the topic, literature search, data collection, discussion, writing and editing, and overall supervision.Conflicts of interest
There are no conflicts to declareData availability
This review does not include any original research results, software, or code, nor were any new data generated or analyzed. Additional information about the review is available upon request from the authors.Acknowledgements
We acknowledge the grants CNS2022-135945, PID2022-142168NB-I00, TED2021-130946B-100, and PID2019-107779GA-I00, funded by MICIU/AEI/10.13039/501100011033 and by “ERDF/EU”, and by the “European Union NextGenerationEU/PRTR”. P. Ximenis and L. Rubert are thankful to Govern de les Illes Balears for their PhD fellowships. D. Martinez thanks the MICIN for the predoctoral FPI scholarship (PREP2022-000239).Notes and references
- J.-M. Lehn, Angew. Chem., Int. Ed. Engl., 1988, 27, 89–112 CrossRef.
- J. W. Steed and J. L. Atwood, Supramolecular Chemistry, John Wiley Sons, Chichester, UK, 2009 Search PubMed.
- C. A. Schalley, Analytical Methods in Supramolecular Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2nd edn, 2012 Search PubMed.
- J. M. Lehn, Chem. Soc. Rev., 2017, 46, 2378–2379 RSC.
- J. M. Lehn, Eur. Rev., 2009, 17, 263–280 CrossRef.
- E. V. Anslyn and D. A. Dougherty, Modern Physical Organic Chemistry, University Science Books, California, USA, 2006 Search PubMed.
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4097 CrossRef CAS.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS.
- L. Yang, X. Tan, Z. Wang and X. Zhang, Chem. Rev., 2015, 115, 7196–7239 CrossRef CAS.
- E. Krieg, M. M. C. Bastings, P. Besenius and B. Rybtchinski, Chem. Rev., 2016, 116, 2414–2477 CrossRef CAS.
- G. Wypych, Handbook of Polymers, ChemTec Publishing, Ontario, Canada, 3rd edn, 2022 Search PubMed.
- A. W. Bosman, R. P. Sijbesma and E. W. Meijer, Mater. Today, 2004, 7, 34–39 CrossRef CAS.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS.
- D. B. Amabilino, D. K. Smith and J. W. Steed, Chem. Soc. Rev., 2017, 46, 2404–2420 RSC.
- T. D. Clemons and S. I. Stupp, Prog. Polym. Sci., 2020, 111, 101310 CrossRef CAS PubMed.
- Y. Xue, Y. Jin, Y. Zhang, F. Han and F. Wang, Chem. Mater., 2024, 36, 6347–6369 CrossRef CAS.
- H. Wang, X. Ji, Z. Li and F. Huang, Adv. Mater., 2017, 29, 1606117 CrossRef PubMed.
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS PubMed.
- D. González-Rodríguez and A. P. H. J. Schenning, Chem. Mater., 2011, 23, 310–325 CrossRef.
- A. Khasbaatar, Z. Xu, J. H. Lee, G. Campillo-Alvarado, C. Hwang, B. N. Onusaitis and Y. Diao, Chem. Rev., 2023, 123, 8395–8487 CrossRef CAS.
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- F. Würthner, Angew. Chem., Int. Ed., 2020, 59, 14192–14196 CrossRef PubMed.
- H. Chen and J. F. Stoddart, Nat. Rev. Mater., 2021, 6, 804–828 CrossRef CAS.
- A. Jain and S. J. George, Mater. Today, 2015, 18, 206–214 CrossRef CAS.
- Z.-T. Li and L.-Z. Wu, Hydrogen bonded supramolecular structures, Springer, Berlin Heidelberg, Berlin, Germany, 2015 Search PubMed.
- Q. Wan and B. C. Thompson, Adv. Sci., 2024, 11, 2305356 CrossRef CAS.
- E. D. Głowacki, M. Irimia-Vladu, S. Bauer and N. S. Sariciftci, J. Mater. Chem. B, 2013, 1, 3742–3753 RSC.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef.
- A. Das and S. Ghosh, Chem. Commun., 2016, 52, 6860–6872 RSC.
- B. Matarranz and G. Fernández, Chem. Phys. Rev., 2021, 2, 041304 CrossRef CAS.
- A. Ruiz-Carretero, N. R. Á. Rovelo, S. Militzer and P. J. Mésini, J. Mater. Chem. A, 2019, 7, 23451–23475 RSC.
- Z. Chen, B. Fimmel and F. Würthner, Org. Biomol. Chem., 2012, 10, 5845–5855 RSC.
- X. Q. Li, V. Stepanenko, Z. Chen, P. Prins, L. D. A. Siebbeles and F. Würthner, Chem. Commun., 2006, 3871–3873 RSC.
- S. Ogi, V. Stepanenko, K. Sugiyasu, M. Takeuchi and F. Würthner, J. Am. Chem. Soc., 2015, 137, 3300–3307 CrossRef CAS.
- T. Aida, A. Takemura, M. Fuse and S. Inoue, J. Chem. Soc. Chem. Commun., 1988, 391–393 RSC.
- C. Fouquey, J.-M.-M. Lehn and A.-M.-M. Levelut, Adv. Mater., 1990, 2, 254–257 CrossRef CAS.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. K. Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS PubMed.
- S. Cantekin, T. F. A. de Greef and A. R. A. Palmans, Chem. Soc. Rev., 2012, 41, 6125–6137 RSC.
- F. H. Beijer, R. P. Sijbesma, J. A. J. M. Vekemans, E. W. Meijer, H. Kooijman and A. L. Spek, J. Org. Chem., 1996, 61, 6371–6380 CrossRef CAS PubMed.
- S. Yagai, Y. Kitamoto, S. Datta and B. Adhikari, Acc. Chem. Res., 2019, 52, 1325–1335 CrossRef CAS PubMed.
- M. Yokoya, S. Kimura and M. Yamanaka, Chem. Eur. J., 2021, 27, 5601–5614 CrossRef CAS PubMed.
- V. Saez Talens, P. Englebienne, T. T. Trinh, W. E. M. Noteborn, I. K. Voets and R. E. Kieltyka, Angew. Chem., Int. Ed., 2015, 54, 10502–10506 CrossRef CAS PubMed.
- S. Bujosa, A. Doncel-Giménez, N. Bäumer, G. Fernández, E. Ortí, A. Costa, C. Rotger, J. Aragó and B. Soberats, Angew. Chem., Int. Ed., 2022, 61, e202213345 CrossRef CAS PubMed.
- S. Bujosa, L. Rubert, C. Rotger and B. Soberats, Commun. Chem., 2024, 7, 1–11 CrossRef PubMed.
- S. Yadav, K. S. Tiwari, C. Gupta, M. K. Tiwari, A. Khan and S. P. Sonkar, Results Chem., 2023, 5, 100733 CrossRef CAS.
- M. Gsänger, D. Bialas, L. Huang, M. Stolte and F. Würthner, Adv. Mater., 2016, 28, 3615–3645 CrossRef PubMed.
- M. Kasha, H. R. Rawls and M. A. El-Bayoumi, Pure Appl. Chem, 1965, 11, 371–392 CAS.
- A. S. Davydov, Sov. Phys. Uspekhi, 1964, 7, 145 CrossRef.
- Transition moment in IUPAC Compendium of Chemical Terminology, 5th edition, International Union of Pure and Applied Chemistry, 2025.
- F. C. Spano, Acc. Chem. Res., 2010, 43, 429–439 CrossRef CAS PubMed.
- N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069–7163 CrossRef CAS PubMed.
- J. Franck and E. G. Dymond, Trans. Faraday Soc., 1926, 21, 536–542 RSC.
- E. Condon, Phys. Rev., 1926, 28, 1182 CrossRef CAS.
- K. Huang, A. Rhys and N. F. Mott, Proc. R. Soc. Lond. Ser. Math. Phys. Sci., 1997, 204, 406–423 Search PubMed.
- F. Würthner, S. Yao, T. Debaerdemaeker and R. Wortmann, J. Am. Chem. Soc., 2002, 124, 9431–9447 CrossRef PubMed.
- J. Demuth, S. Bednarik, R. Machan, I. Mocak, T. Malinsky, M. A. E. Dahabova, J. Holcak, M. Miletin, J. Labuta, V. Novakova and P. Zimcik, Inorg. Chem. Front., 2025, 12, 1590–1608 RSC.
- A. Zitzler-Kunkel, E. Kirchner, D. Bialas, C. Simon and F. Würthner, Chem. Eur. J., 2015, 21, 14851–14861 CrossRef CAS PubMed.
- F. Würthner, Acc. Chem. Res., 2016, 49, 868–876 CrossRef PubMed.
- J. L. Bricks, Y. L. Slominskii, I. D. Panas and A. P. Demchenko, Methods Appl. Fluoresc., 2017, 6, 012001 CrossRef PubMed.
- D. Bialas, E. Kirchner, M. I. S. Röhr and F. Würthner, J. Am. Chem. Soc., 2021, 143, 4500–4518 CrossRef CAS PubMed.
- E. Sebastian and M. Hariharan, J. Am. Chem. Soc., 2021, 143, 13769–13781 CrossRef CAS PubMed.
- C. Kaufmann, D. Bialas, M. Stolte and F. Würthner, J. Am. Chem. Soc., 2018, 140, 9986–9995 CrossRef CAS PubMed.
- M. Hecht and F. Würthner, Acc. Chem. Res., 2021, 54, 2 CrossRef PubMed.
- A. J. Schwalb, C. Naranjo, A. Fernández-Alarcón, F. García, E. Ortí, J. Aragó and L. Sánchez, J. Am. Chem. Soc., 2025, 147(28), 25024–25034 CrossRef CAS PubMed.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef PubMed.
- J. Xu, M. Huang, H. Pang, Z. Weng, G. Hu, S. Zhang, Q. Yang and Q. Wu, Aggregate, 2024, 5, e546 CrossRef CAS.
- P. A. Korevaar, C. Schaefer, T. F. A. D. Greef and E. W. Meijer, J. Am. Chem. Soc., 2012, 134, 13482–13491 CrossRef CAS PubMed.
- G. Vantomme, G. M. ter Huurne, C. Kulkarni, H. M. M. ten Eikelder, A. J. Markvoort, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2019, 141, 18278–18285 CrossRef CAS PubMed.
- C. C. Lee, C. Grenier, E. W. Meijer and A. P. H. J. Schenning, Chem. Soc. Rev., 2009, 38, 671–683 RSC.
- L. Borsdorf, L. Herkert, N. Bäumer, L. Rubert, B. Soberats, P. A. Korevaar, C. Bourque, C. Gatsogiannis and G. Fernández, J. Am. Chem. Soc., 2023, 145, 8882–8895 CrossRef CAS PubMed.
- D. Basak, D. S. Pal, T. Sakurai, S. Yoneda, S. Seki and S. Ghosh, Phys. Chem. Chem. Phys., 2017, 19, 31024–31029 RSC.
- Y. Dorca, C. Naranjo, G. Ghosh, B. Soberats, J. Calbo, E. Ortí, G. Fernández and L. Sánchez, Chem. Sci., 2021, 13, 81–89 RSC.
- L. López-Gandul, C. Naranjo, C. Sánchez, R. Rodríguez, R. Gómez, J. Crassous and L. Sánchez, Chem. Sci., 2022, 13, 11577–11584 RSC.
- T. Seki, S. Yagai, T. Karatsu and A. Kitamura, J. Org. Chem., 2008, 73, 3328–3335 CrossRef CAS PubMed.
- M. Hartlieb, E. D. H. Mansfield and S. Perrier, Polym. Chem., 2020, 11, 1083–1110 RSC.
- C. Kulkarni, S. Balasubramanian and S. J. George, ChemPhysChem, 2013, 14, 661–673 CrossRef CAS PubMed.
- H. Miao, F. Yang, Y. Zhang, H. Li and X. He, Chem. Eur. J., 2025, e202404243 CrossRef CAS PubMed.
- T. Yamaguchi, Y. Naito, C. Kitamura, H. Takeshita, S. Ida, T. Ishi-i, K. Suzuki, T. Matsumoto, Y. Shiota, D. Suzuki, Y. Imai, T. Ikeda and S. Kato, Chem. Asian J., 2024, 19, e202400829 CrossRef CAS PubMed.
- W. T. Gallonde, M. Shekar, V. Dorcet, S. Rigaut and O. Galangau, Chem. Eur. J., 2025, 31, e202500443 CrossRef CAS PubMed.
- B. Narayan, C. Kulkarni and S. J. George, J. Mater. Chem. C, 2012, 1, 626–629 RSC.
- J. Gershberg, F. Fennel, T. H. Rehm, S. Lochbrunner and F. Würthner, Chem. Sci., 2016, 7, 1729–1737 RSC.
- Y. Dorca, C. Naranjo, G. Ghosh, B. Soberats, J. Calbo, E. Ortí, G. Fernández and L. Sánchez, Chem. Sci., 2021, 13, 81–89 RSC.
- T. E. Kaiser, V. Stepanenko and F. Würthner, J. Am. Chem. Soc., 2009, 131, 6719–6732 CrossRef CAS PubMed.
- R. R. van der Weegen, P. A. Korevaar, P. Voudouris, I. K. Voets, T. F. A. de Greef, J. A. J. M. Vekemans and E. W. Meijer, Chem. Commun., 2013, 49, 5532–5534 RSC.
- R. F. Goldstein and L. Stryer, Biophys. J., 1986, 50, 583–599 CrossRef CAS PubMed.
- I. Helmers, M. S. Hossain, N. Bäumer, P. Wesarg, B. Soberats, L. S. Shimizu and G. Fernández, Angew. Chem., Int. Ed., 2022, 61, e202200390 CrossRef CAS PubMed.
- A. Rödle, B. Ritschel, C. Mück-Lichtenfeld, V. Stepanenko and G. Fernández, Chem. Eur. J., 2016, 22, 15772–15777 CrossRef PubMed.
- D. Zhao and J. S. Moore, Org. Biomol. Chem., 2003, 1, 3471–3491 RSC.
- P. van der Schoot, Supramolecular Polymers. Theroy of Supramolecular Polymerization, CRC Press, Netherlands, 2nd edn, 2005 Search PubMed.
- G. Ercolani, J. Am. Chem. Soc., 2003, 125, 16097–16103 CrossRef CAS PubMed.
- J. F. Douglas, J. Dudowicz and K. F. Freed, J. Chem. Phys, 2008, 128, 35 CrossRef PubMed.
- G. Fernández, M. Stolte, V. Stepanenko and F. Würthner, Chem. Eur. J., 2013, 19, 206–217 CrossRef PubMed.
- H. M. M. T. Eikelder, B. Adelizzi, A. R. A. Palmans and A. J. Markvoort, J. Phys. Chem. B, 2019, 123, 6627–6642 CrossRef PubMed.
- H. M. M. T. Eikelder, A. J. Markvoort, T. F. A. D. Greef and P. A. J. Hilbers, J. Phys. Chem. B, 2012, 116, 5291–5301 CrossRef PubMed.
- M. M. J. Smulders, M. M. L. Nieuwenhuizen, T. F. A. D. Greef, P. V. D. Schoot, A. P. H. J. Schenning and E. W. Meijer, Chem. Eur. J., 2010, 16, 362–367 CrossRef CAS PubMed.
- B. Matarranz, S. Díaz-Cabrera, G. Ghosh, I. Carreira-Barral, B. Soberats, M. García-Valverde, R. Quesada and G. Fernández, Angew. Chem., Int. Ed., 2023, 62, e202218555 CrossRef CAS PubMed.
- J. S. Valera, R. Gómez and L. Sánchez, Org. Lett., 2018, 20, 2020–2023 CrossRef CAS PubMed.
- Y. Liu, Z. Wang and X. Zhang, Chem. Soc. Rev., 2012, 41, 5922–5932 RSC.
- R. Vill, J. Gülcher, P. Khalatur, P. Wintergerst, A. Stoll, A. Mourran and U. Ziener, Nanoscale, 2019, 11, 663–674 RSC.
- M. Pons and O. Millet, Prog. Nucl. Magn. Reson. Spectrosc., 2001, 38, 267–324 CrossRef CAS.
- F. García, R. Gómez and L. Sánchez, Chem. Soc. Rev., 2023, 52, 7524–7548 RSC.
- Y. Marechal and A. Witkowski, J. Chem. Phys, 1968, 48, 3697–3705 CrossRef CAS.
- A. Arnaud and L. Bouteiller, Langmuir, 2004, 20, 6858–6863 CrossRef CAS PubMed.
- S. K. Brar and M. Verma, TrAC Trends Anal. Chem., 2011, 30, 4–17 CrossRef CAS.
- C. M. Jeffries, J. Ilavsky, A. Martel, S. Hinrichs, A. Meyer, J. S. Pedersen, A. V. Sokolova and D. I. Svergun, Nat. Rev. Methods Primer, 2021, 1, 70 CrossRef.
- F. J. Giessibl, Rev. Mod. Phys., 2003, 75, 949 CrossRef.
- D. J. Smith, Characterization of Nanomaterials Using Transmission Electron Microscopy, Nanoscience and Nanotechnology Series, Royal Society of Chemistry, Cambridge, UK, 2015 Search PubMed.
- D. Bochicchio and G. M. Pavan, Adv. Phys. X, 2018, 3, 1436408 Search PubMed.
- I. A. W. Filot, A. R. A. Palmans, P. A. J. Hilbers, R. A. V. Santen, E. A. Pidko and T. F. A. D. Greef, J. Phys. Chem. B, 2010, 114, 13667–13674 CrossRef PubMed.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef PubMed.
- D. C. Rapaport, J. E. Johnson and J. Skolnick, Comput. Phys. Commun., 1999, 121–122, 231–235 CrossRef.
- D. Bochicchio, M. Salvalaglio and G. M. Pavan, Nat. Commun., 2017, 8, 147 CrossRef PubMed.
- M. Crippa, C. Perego, A. L. de Marco and G. M. Pavan, Nat. Commun., 2022, 13, 2162 CrossRef PubMed.
- C. M. Atienza and L. Sánchez, Chem. Eur. J., 2024, 30, e202400379 CrossRef PubMed.
- X. Xiao, H. Chen, X. Dong, J. Zhang, J. Zhang, H. Zhang, D. Wang and W. Tian, Mater. Chem. Front., 2022, 6, 3261–3270 RSC.
- J. Uchida, B. Soberats, M. Gupta and T. Kato, Adv. Mater., 2022, 34, 2109063 CrossRef PubMed.
- R. Liao, F. Wang, Y. Guo, Y. Han and F. Wang, J. Am. Chem. Soc., 2022, 144, 9775–9784 CrossRef PubMed.
- S. H. Jung, D. Bochicchio, G. M. Pavan, M. Takeuchi and K. Sugiyasu, J. Am. Chem. Soc., 2018, 140, 10570–10577 CrossRef PubMed.
- W. Wagner, M. Wehner, V. Stepanenko and F. Würthner, J. Am. Chem. Soc., 2019, 141, 12044–12054 CrossRef PubMed.
- S. Sarkar, A. Sarkar, A. Som, S. S. Agasti and S. J. George, J. Am. Chem. Soc., 2021, 143, 11777–11787 CrossRef PubMed.
- B. Adelizzi, A. Aloi, A. J. Markvoort, H. M. M. Ten Eikelder, I. K. Voets, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2018, 140, 7168–7175 CrossRef CAS PubMed.
- A. Sarkar, T. Behera, R. Sasmal, R. Capelli, C. Empereur-mot, J. Mahato, S. S. Agasti, G. M. Pavan, A. Chowdhury and S. J. George, J. Am. Chem. Soc., 2020, 142, 11528–11539 CrossRef CAS PubMed.
- M. M. Safont-Sempere, G. Fernández and F. Würthner, Chem. Rev., 2011, 111, 5784–5814 CrossRef CAS PubMed.
- A. Das and S. Ghosh, Angew. Chem., Int. Ed., 2014, 53, 2038–2054 CrossRef CAS PubMed.
- B. Adelizzi, N. J. Van Zee, L. N. J. de Windt, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2019, 141, 6110–6121 CrossRef CAS PubMed.
- Y. Chen and C. Liu, ChemPlusChem, 2024, 89, e202300623 CrossRef CAS PubMed.
- M. J. Mayoral, C. Rest, J. Schellheimer, V. Stepanenko and G. Fernández, Chem. Eur. J., 2012, 18, 15607–15611 CrossRef CAS PubMed.
- X. Huang, R. Li, Z. Duan, F. Xu and H. Li, Soft Matter, 2022, 18, 3828–3844 RSC.
- Y. Yasuda, E. Iishi, H. Inada and Y. Shirota, Chem. Lett, 1996, 25, 575–576 CrossRef.
- K. Hanabusa, C. Koto, M. Kimura, H. Shirai and A. Kakehi, Chem. Lett, 1997, 26, 429–430 CrossRef.
- F. Aparicio, F. García and L. Sánchez, Chem. Eur. J., 2013, 19, 3239–3248 CrossRef CAS PubMed.
- J. J. I. Armao, M. Maaloum, T. Ellis, G. Fuks, M. Rawiso, E. Moulin and N. Giuseppone, J. Am. Chem. Soc., 2014, 136, 11382–11388 CrossRef CAS PubMed.
- E. Moulin, J. J. I. Armao and N. Giuseppone, Acc. Chem. Res., 2019, 52, 975–983 CrossRef CAS PubMed.
- M. Shirakawa, S. Kawano, N. Fujita, K. Sada and S. Shinkai, J. Org. Chem., 2003, 68, 5037–5044 CrossRef CAS PubMed.
- J. Kang, D. Miyajima, Y. Itoh, T. Mori, H. Tanaka, M. Yamauchi, Y. Inoue, S. Harada and T. Aida, J. Am. Chem. Soc., 2014, 136, 10640–10644 CrossRef CAS PubMed.
- J. Kang, D. Miyajima, T. Mori, Y. Inoue, Y. Itoh and T. Aida, Science, 2015, 347, 646–651 CrossRef CAS PubMed.
- X. Dou, W. Pisula, J. Wu, G. J. Bodwell and K. Müllen, Chem. Eur. J., 2008, 14, 240–249 CrossRef CAS PubMed.
- S. V. Bhosale, M. A. Kobaisi, R. W. Jadhav, P. P. Morajkar, L. A. Jones and S. George, Chem. Soc. Rev., 2021, 50, 9845–9998 RSC.
- P. Mukhopadhyay, Y. Iwashita, M. Shirakawa, S. Kawano, N. Fujita and S. Shinkai, Angew. Chem., Int. Ed., 2006, 45, 1592–1595 CrossRef CAS PubMed.
- F. Würthner, C. Thalacker and A. Sautter, Adv. Mater., 1999, 11, 754–758 CrossRef.
- F. Würthner, C. Thalacker, A. Sautter, W. Schärtl, W. Ibach and O. Hollricher, Chem. Eur. J., 2000, 6, 3871–3886 CrossRef.
- P. Jonkheijm, A. Miura, M. Zdanowska, F. J. M. Hoeben, S. De Feyter, A. P. H. J. Schenning, F. C. De Schryver and E. W. Meijer, Angew. Chem., Int. Ed., 2004, 43, 74–78 CrossRef PubMed.
- S. J. George, Ž. Tomović, M. M. J. Smulders, T. F. A. de Greef, P. E. L. G. Leclère, E. W. Meijer and A. P. H. J. Schenning, Angew. Chem., Int. Ed., 2007, 46, 8206–8211 CrossRef CAS PubMed.
- P. A. Korevaar, S. J. George, A. J. Markvoort, M. M. J. Smulders, P. A. J. Hilbers, A. P. H. J. Schenning, T. F. A. De Greef and E. W. Meijer, Nature, 2012, 481, 492–496 CrossRef CAS PubMed.
- A. P. H. J. Schenning, J. v Herrikhuyzen, P. Jonkheijm, Z. Chen, F. Würthner and E. W. Meijer, J. Am. Chem. Soc., 2002, 124, 10252–10253 CrossRef CAS PubMed.
- B. Adhikari, X. Lin, M. Yamauchi, H. Ouchi, K. Aratsu and S. Yagai, Chem. Commun., 2017, 53, 9663–9683 RSC.
- H. Ouchi, X. Lin and S. Yagai, Chem. Lett., 2019, 48, 1009–1018 CrossRef CAS.
- K. Tashiro, T. Saito, H. Arima, N. Suda, B. Vedhanarayanan and S. Yagai, Chem. Rec., 2022, 22, e202100252 CrossRef CAS PubMed.
- S. Yagai, M. Suzuki, X. Lin, M. Gushiken, T. Noguchi, T. Karatsu, A. Kitamura, A. Saeki, S. Seki, Y. Kikkawa, Y. Tani and K. Nakayama, Chem. Eur. J., 2014, 20, 16128–16137 CrossRef CAS PubMed.
- S. Yagai, M. Yamauchi, A. Kobayashi, T. Karatsu, A. Kitamura, T. Ohba and Y. Kikkawa, J. Am. Chem. Soc., 2012, 134, 18205–18208 CrossRef CAS PubMed.
- T. Saito and S. Yagai, Eur. J. Org. Chem., 2020, 2475–2478 CrossRef CAS.
- T. Saito, T. Kajitani and S. Yagai, J. Am. Chem. Soc., 2023, 145, 443–454 CrossRef CAS PubMed.
- A. del Prado, D. González-Rodríguez and Y.-L. Wu, ChemistryOpen, 2020, 9, 409–430 CrossRef CAS PubMed.
- V. Vázquez-González, M. J. Mayoral, R. Chamorro, M. M. R. M. Hendrix, I. K. Voets and D. González-Rodríguez, J. Am. Chem. Soc., 2019, 141, 16432–16438 CrossRef PubMed.
- V. Vázquez-González, M. J. Mayoral, F. Aparicio, P. Martínez-Arjona and D. González-Rodríguez, ChemPlusChem, 2021, 86, 1087–1096 CrossRef PubMed.
- M. González-Sánchez, J. S. Valera, J. Veiga-Herrero, P. B. Chamorro, F. Aparicio and D. González-Rodríguez, Chem. Soc. Rev., 2025, 54, 5657–5697 RSC.
- N. Bilbao, I. Destoop, S. De Feyter and D. González-Rodríguez, Angew. Chem., Int. Ed., 2016, 55, 659–663 CrossRef CAS PubMed.
- I. Sancho-Casado, F. Aparicio, M. González-Sánchez, R. Chamorro, V. Vega-Mayoral, J. Cabanillas-González and D. González-Rodríguez, Angew. Chem., Int. Ed., 2025, e202509156 CAS.
- L. S. Shimizu, Polym. Int., 2007, 56, 444–452 CrossRef CAS.
- J. D. Fox and S. J. Rowan, Macromolecules, 2009, 42, 6823–6835 CrossRef CAS.
- S. H. M. Söntjens, R. P. Sijbesma, M. H. P. van Genderen and E. W. Meijer, J. Am. Chem. Soc., 2000, 122, 7487–7493 CrossRef.
- M. Kotera, J.-M. Lehn and J.-P. Vigneron, Tetrahedron, 1995, 51, 1953–1972 CrossRef CAS.
- M. Kotera, J.-M. Lehn and J.-P. Vigneron, J. Chem. Soc. Chem. Commun., 1994, 197–199 RSC.
- V. Berl, M. Schmutz, M. J. Krische, R. G. Khoury and J.-M. Lehn, Chem. Eur. J., 2002, 8, 1227–1244 CrossRef CAS PubMed.
- R. Schmidt, M. Stolte, M. Grüne and F. Würthner, Macromolecules, 2011, 44, 3766–3776 CrossRef CAS.
- S. P. Dudek, M. Pouderoijen, R. Abbel, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2005, 127, 11763–11768 CrossRef CAS PubMed.
- R. Abbel, C. Grenier, M. J. Pouderoijen, J. W. Stouwdam, P. E. L. G. Leclère, R. P. Sijbesma, E. W. Meijer and A. P. H. J. Schenning, J. Am. Chem. Soc., 2009, 131, 833–843 CrossRef CAS PubMed.
- A. Tsuda, Md. A. Alam, T. Harada, T. Yamaguchi, N. Ishii and T. Aida, Angew. Chem., Int. Ed., 2007, 46, 8198–8202 CrossRef CAS PubMed.
- T. E. Kaiser, H. Wang, V. Stepanenko and F. Würthner, Angew. Chem., Int. Ed., 2007, 46, 5541–5544 CrossRef CAS PubMed.
- V. Grande, B. Soberats, S. Herbst, V. Stepanenko and F. Würthner, Chem. Sci., 2018, 9, 6904–6911 RSC.
- S. Herbst, B. Soberats, P. Leowanawat, M. Lehmann and F. Würthner, Angew. Chem., Int. Ed., 2017, 56, 2162–2165 CrossRef CAS PubMed.
- S. Herbst, B. Soberats, P. Leowanawat, M. Stolte, M. Lehmann and F. Würthner, Nat. Commun., 2018, 9, 2646 CrossRef PubMed.
- H. Kar, D. W. Gehrig, N. K. Allampally, G. Fernández, F. Laquai and S. Ghosh, Chem. Sci., 2016, 7, 1115–1120 RSC.
- E. Castellanos, R. M. Gomila, R. Manha, G. Fernández, A. Frontera and B. Soberats, J. Mater. Chem. C, 2023, 11, 10884–10892 RSC.
- B. Soberats, M. Hecht and F. Würthner, Angew. Chem., Int. Ed., 2017, 56, 10771–10774 CrossRef CAS PubMed.
- M. Hecht, B. Soberats, J. Zhu, V. Stepanenko, S. Agarwal, A. Greiner and F. Würthner, Nanoscale Horiz., 2018, 4, 169–174 RSC.
- P. Ximenis, L. Rubert, H. M. A. Ehmann and B. Soberats, Org. Chem. Front., 2025, 12, 430–436 RSC.
- Y. Zhang, P. Liu, H. Pan, H. Dai, X.-K. Ren and Z. Chen, Chem. Commun., 2020, 56, 12069–12072 RSC.
- M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS PubMed.
- R. A. Hegstrom and D. K. Kondepudi, Sci. Am., 1990, 262, 108–115 CrossRef.
- T. Seki, A. Asano, S. Seki, Y. Kikkawa, H. Murayama, T. Karatsu, A. Kitamura and S. Yagai, Chem. Eur. J., 2011, 17, 3598–3608 CrossRef CAS PubMed.
- F. Helmich, C. C. Lee, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2010, 132, 16753–16755 CrossRef CAS PubMed.
- M. Wehner, M. I. S. Röhr, V. Stepanenko and F. Würthner, Nat. Commun., 2020, 11, 5460 CrossRef CAS PubMed.
- M. A. Martínez, A. Doncel-Giménez, J. Cerdá, J. Calbo, R. Rodríguez, J. Aragó, J. Crassous, E. Ortí and L. Sánchez, J. Am. Chem. Soc., 2021, 143, 13281–13291 CrossRef PubMed.
- A. R. A. Palmans, J. A. J. M. Vekemans, E. E. Havinga and E. W. Meijer, Angew. Chem., Int. Ed. Engl., 1997, 36, 2648–2651 CrossRef CAS.
- A. J. Wilson, J. V. Gestel, R. P. Sijbesma and E. W. Meijer, Chem. Commun., 2006, 4404–4406 RSC.
- Y. Dorca, R. Sánchez-Naya, J. Cerdá, J. Calbo, J. Aragó, R. Gómez, E. Ortí and L. Sánchez, Chem. Eur. J., 2020, 26, 14700–14707 CrossRef CAS.
- E. E. Greciano, J. Calbo, J. Buendía, J. Cerdá, J. Aragó, E. Ortí and L. Sánchez, J. Am. Chem. Soc., 2019, 141, 7463–7472 CrossRef CAS PubMed.
- Y. Dorca, C. Naranjo, G. Ghosh, R. Gómez, G. Fernández and L. Sánchez, Org. Mater., 2020, 2, 41–46 CrossRef CAS.
- B. Adelizzi, I. A. W. Filot, A. R. A. Palmans and E. W. Meijer, Chem. Eur. J., 2017, 23, 6103–6110 CrossRef CAS PubMed.
- J. S. Valera, R. Gómez and L. Sánchez, Small, 2018, 14, 1702437 CrossRef PubMed.
- S. Ghosh, X.-Q. Li, V. Stepanenko and F. Würthner, Chem. Eur. J., 2008, 14, 11343–11357 CrossRef CAS PubMed.
- E. E. Greciano, R. Rodríguez, K. Maeda and L. Sánchez, Chem. Commun., 2020, 56, 2244–2247 RSC.
- N. Suda, T. Saito, H. Arima and S. Yagai, Chem. Sci., 2022, 13, 3249–3255 RSC.
- M. Yamauchi, T. Ohba, T. Karatsu and S. Yagai, Nat. Commun., 2015, 6, 8936 CrossRef CAS PubMed.
- T. Saito, D. Inoue, Y. Kitamoto, H. Hanayama, T. Fujita, Y. Watanabe, M. Suda, T. Hirose, T. Kajitani and S. Yagai, Nat. Nanotechnol., 2025, 20, 825–834 CrossRef CAS PubMed.
- M. Nic, L. Hovorka, J. JIrat, B. Kosata and J. Znamenacek, IUPAC compendium of chemical terminology-the gold book, International Union of Pure and Applied Chemistry, 2nd edn, 2005 Search PubMed.
- S. Sarkar, A. Sarkar and S. J. George, Angew. Chem., Int. Ed., 2020, 59, 19841–19845 CrossRef CAS PubMed.
- S. Sarkar, A. Sarkar, A. Som, S. S. Agasti and S. J. George, J. Am. Chem. Soc., 2021, 143, 11777–11787 CrossRef CAS PubMed.
- B. Narayan, K. K. Bejagam, S. Balasubramanian and S. J. George, Angew. Chem., Int. Ed., 2015, 54, 13053–13057 CrossRef CAS PubMed.
- A. Sarkar, S. Dhiman, A. Chalishazar and S. J. George, Angew. Chem., Int. Ed., 2017, 56, 13767–13771 CrossRef CAS.
- P. Osswald and F. Würthner, J. Am. Chem. Soc., 2007, 129, 14319–14326 CrossRef CAS PubMed.
- Z. Xie and F. Würthner, Org. Lett., 2010, 12, 3204–3207 CrossRef CAS.
- Z. Xie, V. Stepanenko, K. Radacki and F. Würthner, Chem. Eur. J., 2012, 18, 7060–7070 CrossRef CAS PubMed.
- T. Ogoshi, T. Furuta and T. A. Yamagishi, Chem. Commun., 2016, 52, 10775–10778 RSC.
- D. E. Fagnani, M. J. Meese Jr., K. A. Abboud and R. K. Castellano, Angew. Chem., Int. Ed., 2016, 55, 10726–10731 CrossRef CAS PubMed.
- W. R. Henderson, Y. Zhu, D. E. Fagnani, G. Liu, K. A. Abboud and R. K. Castellano, J. Org. Chem., 2020, 85, 1158–1167 CrossRef CAS.
- W. R. Henderson, A. Kumar, K. A. Abboud and R. K. Castellano, Chem. Eur. J., 2020, 26, 17588–17597 CrossRef CAS PubMed.
- T. Hirose, N. Ito, H. Kubo, T. Sato and K. Matsuda, J. Mater. Chem. C, 2016, 4, 2811–2819 RSC.
- R. Rodríguez, C. Naranjo, A. Kumar, P. Matozzo, T. K. Das, Q. Zhu, N. Vanthuyne, R. Gómez, R. Naaman, L. Sánchez and J. Crassous, J. Am. Chem. Soc., 2022, 144, 7709–7719 CrossRef PubMed.
- L. López-Gandul, R. Rodríguez, N. Vanthuyne, J. Crassous and L. Sánchez, Nanoscale, 2024, 16, 13041–13049 RSC.
- M. J. Mayoral, J. Guilleme, J. Calbo, J. Aragó, F. Aparicio, E. Ortí, T. Torres and D. González-Rodríguez, J. Am. Chem. Soc., 2020, 142, 21017–21031 CrossRef CAS.
- M. M. Green, M. P. Reidy, R. J. Johnson, G. Darling, D. J. O’Leary and G. Willson, J. Am. Chem. Soc., 1989, 111, 6452–6454 CrossRef.
- H. Gu, Y. Nakamura, T. Sato, A. Teramoto, M. M. Green, S. K. Jha, C. Andreola and M. P. Reidy, Macromolecules, 1998, 31, 6362–6368 CrossRef CAS.
- M. M. Green, B. A. Garetz, B. Munoz, H. P. Chang, S. Hoke and R. G. Cooks, J. Am. Chem. Soc., 1995, 117, 4181–4182 CrossRef CAS.
- J. V. Gestel, A. R. A. Palmans, B. Titulaer, J. A. J. M. Vekemans and E. W. Meijer, J. Am. Chem. Soc., 2005, 127, 5490–5494 CrossRef PubMed.
- T. Kim, T. Mori, T. Aida and D. Miyajima, Chem. Sci., 2016, 7, 6689–6694 RSC.
- A. Das, S. Ghosh and S. J. George, Angew. Chem., Int. Ed., 2023, 62, e202308281 CrossRef CAS.
- M. M. J. Smulders, P. J. M. Stals, T. Mes, T. F. E. Paffen, A. P. H. J. Schenning, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2010, 132, 620–626 CrossRef CAS.
- M. M. J. Smulders, I. A. W. Filot, J. M. A. Leenders, P. V. D. Schoot, A. R. A. Palmans, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2010, 132, 611–619 CrossRef CAS PubMed.
- Y. Jin, H. Zhao, F. Han, S. Jiang, Y. Xue and F. Wang, ACS Macro Lett., 2025, 14, 781–787 CrossRef CAS PubMed.
- J. Buendía, J. Calbo, F. García, J. Aragó, P. M. Viruela, E. Ortí and L. Sánchez, Chem. Commun., 2016, 52, 6907–6910 RSC.
- T. Metzroth, A. Hoffmann, R. Martín-Rapún, M. M. J. Smulders, K. Pieterse, A. R. A. Palmans, J. A. J. M. Vekemans, E. W. Meijer, H. W. Spiess and J. Gauss, Chem. Sci., 2010, 2, 69–76 RSC.
- B. Isare, M. Linares, L. Zargarian, S. Fermandjian, M. Miura, S. Motohashi, N. Vanthuyne, R. Lazzaroni and L. Bouteiller, Chem. Eur. J., 2010, 16, 173–177 CrossRef CAS PubMed.
- V. Stepanenko, X. Q. Li, J. Gershberg and F. Würthner, Chem. Eur. J., 2013, 19, 4176–4183 CrossRef CAS PubMed.
- J. Kim, J. Lee, W. Y. Kim, H. Kim, S. Lee, H. C. Lee, Y. S. Lee, M. Seo and S. Y. Kim, Nat. Commun., 2015, 6, 6959 CrossRef CAS PubMed.
- J. S. Kang, S. Kang, J. M. Suh, S. M. Park, D. K. Yoon, M. H. Lim, W. Y. Kim and M. Seo, J. Am. Chem. Soc., 2022, 144, 2657–2666 CrossRef CAS PubMed.
- K. Fu and G. Liu, ACS Nano, 2024, 18, 2279–2289 CrossRef CAS PubMed.
- C. Wagenknecht, C. M. Li, A. Reingruber, X. H. Bao, A. Goebel, Y. A. Chen, Q. Zhang, K. Chen and J. W. Pan, Nat. Photonics, 2010, 4, 549–552 CrossRef CAS.
- Q. Wang, H. Xu, Z. Qi, J. Mei, H. Tian and D. H. Qu, Angew. Chem., Int. Ed., 2024, 63, e202407385 CrossRef CAS PubMed.
- T. Zhao, J. Han, P. Duan and M. Liu, Acc. Chem. Res., 2020, 53, 1279–1292 CrossRef CAS PubMed.
- Y. Yang, R. C. D. Costa, M. J. Fuchter and A. J. Campbell, Nat. Photonics, 2013, 7, 634–638 CrossRef CAS.
- G. Liu, M. G. Humphrey, C. Zhang and Y. Zhao, Chem. Soc. Rev., 2023, 52, 4443–4487 RSC.
- G. Muller, Dalton Trans., 2009, 9692–9707 RSC.
- F. Song, G. Wei, X. Jiang, F. Li, C. Zhu and Y. Cheng, Chem. Commun., 2013, 49, 5772–5774 RSC.
- L. Arrico, L. D. Bari and F. Zinna, Chem. Eur. J., 2021, 27, 2920–2934 CrossRef CAS PubMed.
- Y. Nagata and T. Mori, Front. Chem., 2020, 8, 535258 Search PubMed.
- L. Gao, R. Liao, L. Ao, Y. Zhang, J. Jin and F. Wang, Angew. Chem., Int. Ed., 2025, 64, e202505776 CrossRef CAS PubMed.
- Y. Zhang, W. Yu, H. Li, W. Zheng and Y. Cheng, Chem. Eur. J., 2023, 29, e202204039 CrossRef CAS PubMed.
- C. Li, X. Jin, J. Han, T. Zhao and P. Duan, J. Phys. Chem. Lett., 2021, 12, 8566–8574 CrossRef CAS PubMed.
- Y. Sang, J. Han, T. Zhao, P. Duan, M. Liu, Y. Sang, M. Liu, T. Zhao, P. Duan and J. Han, Adv. Mater., 2020, 32, 1900110 CrossRef CAS PubMed.
- J. Luo, Z. Xie, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- B. K. An, S. K. Kwon, S. D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410–14415 CrossRef CAS PubMed.
- Y. Yin, Z. Chen, Y. Han, R. Liao and F. Wang, Org. Chem. Front., 2021, 8, 4986 RSC.
- L. López-Gandul, C. Naranjo, C. Sánchez, R. Rodríguez, R. Gómez, J. Crassous and L. Sánchez, Chem. Sci., 2022, 13, 11577–11584 RSC.
- J. Kumar, T. Nakashima, H. Tsumatori, M. Mori, M. Naito and T. Kawai, Chem. Eur. J., 2013, 19, 14090–14097 CrossRef CAS PubMed.
- J. A. Panis, M. Louis, A. Brosseau, S. Katao, F. de los Reyes, T. Nakashima, R. Métivier, C. Allain and T. Kawai, Chem. Eur. J., 2022, 28, e202201012 CrossRef CAS PubMed.
- S. Hu, L. Hu, X. Zhu, Y. Wang and M. Liu, Angew. Chem., Int. Ed., 2021, 60, 19451–19457 CrossRef CAS PubMed.
- M. J. Álvaro-Martins, C. Billiaux, P. Godard, R. Oda, G. Raffy and D. M. Bassani, Chem. Commun., 2023, 59, 7963–7966 RSC.
- S. Ghorai, S. Show and A. Das, Angew. Chem., Int. Ed., 2025, 64, e202500879 CrossRef CAS PubMed.
- A. Mukherjee and S. Ghosh, Chem. Eur. J., 2020, 26, 12874–12881 CrossRef CAS PubMed.
- H. Cao, P. Duan, X. Zhu, J. Jiang and M. Liu, Chem. Eur. J., 2012, 18, 5546–5550 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- I. S. Choi, N. Bowden and G. M. Whitesides, Angew. Chem., Int. Ed., 1999, 38, 3078–3081 CrossRef CAS PubMed.
- C. M. Atienza and L. Sánchez, Chem. Eur. J., 2024, 30, e202400379 CrossRef CAS PubMed.
- L. Gallego, J. F. Woods and M. Rickhaus, Org. Mater., 2022, 4, 137–145 CrossRef CAS.
- I. Insua, J. Bergueiro, A. Méndez-Ardoy, I. Lostalé-Seijo and J. Montenegro, Chem. Sci., 2022, 13, 3057–3068 RSC.
- X. He, M.-S. Hsiao, C. E. Boott, R. L. Harniman, A. Nazemi, X. Li, M. A. Winnik and I. Manners, Nat. Mater., 2017, 16, 481–488 CrossRef CAS PubMed.
- Y.-S. Yoo, J.-H. Choi, J.-H. Song, N.-K. Oh, W.-C. Zin, S. Park, T. Chang and M. Lee, J. Am. Chem. Soc., 2004, 126, 6294–6300 CrossRef CAS PubMed.
- J.-K. Kim, E. Lee, Y.-H. Jeong, J.-K. Lee, W.-C. Zin and M. Lee, J. Am. Chem. Soc., 2007, 129, 6082–6083 CrossRef CAS PubMed.
- E. Lee, Y.-H. Jeong, J.-K. Kim and M. Lee, Macromolecules, 2007, 40, 8355–8360 CrossRef CAS.
- S. Shin, S. Lim, Y. Kim, T. Kim, T.-L. Choi and M. Lee, J. Am. Chem. Soc., 2013, 135, 2156–2159 CrossRef CAS PubMed.
- Y. Kim, S. Shin, T. Kim, D. Lee, C. Seok and M. Lee, Angew. Chem., Int. Ed., 2013, 52, 6426–6429 CrossRef CAS PubMed.
- B. Sun, B. Shen, A. Urushima, X. Liu, X. Feng, E. Yashima and M. Lee, Angew. Chem., Int. Ed., 2020, 59, 22690–22696 CrossRef PubMed.
- B. Sun, Y. Kim, Y. Wang, H. Wang, J. Kim, X. Liu and M. Lee, Nat. Mater., 2018, 17, 599–604 CrossRef PubMed.
- I. Insua and J. Montenegro, J. Am. Chem. Soc., 2020, 142, 300–307 CrossRef PubMed.
- T. Fukui, S. Kawai, S. Fujinuma, Y. Matsushita, T. Yasuda, T. Sakurai, S. Seki, M. Takeuchi and K. Sugiyasu, Nat. Chem., 2017, 9, 493–499 CrossRef PubMed.
- N. Sasaki, J. Yuan, T. Fukui, M. Takeuchi and K. Sugiyasu, Chem. Eur. J., 2020, 26, 7840–7846 CrossRef PubMed.
- E. E. Greciano, J. Calbo, E. Ortí and L. Sánchez, Angew. Chem., Int. Ed., 2020, 59, 17517–17524 CrossRef PubMed.
- N. Bäumer, K. K. Kartha, J. P. Palakkal and G. Fernández, Soft Matter, 2020, 16, 6834–6840 RSC.
- A. Chakraborty, G. Ghosh, D. S. Pal, S. Varghese and S. Ghosh, Chem. Sci., 2019, 10, 7345–7351 RSC.
- J.-A. Zhang, X. Xiao, J. Wang, S. Luo, Y. Lu, Y.-Y. Pang and W. Tian, J. Am. Chem. Soc., 2025, 147, 13447–13460 CrossRef PubMed.
- N. Sasaki, M. F. J. Mabesoone, J. Kikkawa, T. Fukui, N. Shioya, T. Shimoaka, T. Hasegawa, H. Takagi, R. Haruki, N. Shimizu, S. Adachi, E. W. Meijer, M. Takeuchi and K. Sugiyasu, Nat. Commun., 2020, 11, 3578 Search PubMed.
- J. S. Valera, H. Arima, C. Naranjo, T. Saito, N. Suda, R. Gómez, S. Yagai and L. Sánchez, Angew. Chem., Int. Ed., 2022, 61, e202114290 CrossRef PubMed.
- L. Rubert, M. F. Islam, A. B. Greytak, R. Prakash, M. D. Smith, R. M. Gomila, A. Frontera, L. S. Shimizu and B. Soberats, Angew. Chem., Int. Ed., 2023, 62, e202312223 CrossRef PubMed.
- C.-A. Shen, D. Bialas, M. Hecht, V. Stepanenko, K. Sugiyasu and F. Würthner, Angew. Chem., Int. Ed., 2021, 60, 11949–11958 CrossRef PubMed.
- M. Hecht, P. Leowanawat, T. Gerlach, V. Stepanenko, M. Stolte, M. Lehmann and F. Würthner, Angew. Chem., Int. Ed., 2020, 59, 17084–17090 CrossRef PubMed.
- G. Ghosh, P. Dey and S. Ghosh, Chem. Commun., 2020, 56, 6757–6769 RSC.
- J. Matern, Y. Dorca, L. Sánchez and G. Fernández, Angew. Chem., Int. Ed., 2019, 58, 16730–16740 CrossRef PubMed.
- M. Wehner and F. Würthner, Nat. Rev. Chem., 2020, 4, 38–53 CrossRef.
- G. Ghosh, Giant, 2023, 14, 100160 CrossRef.
- P. A. Korevaar, T. F. A. de Greef and E. W. Meijer, Chem. Mater., 2014, 26, 576–586 CrossRef.
- T. Aida and E. w Meijer, Isr. J. Chem., 2020, 60, 33–47 CrossRef.
- A. Lohr and F. Würthner, Angew. Chem., Int. Ed., 2008, 47, 1232–1236 CrossRef PubMed.
- M. M. Bouman and E. W. Meijer, Adv. Mater., 1995, 7, 385–387 CrossRef.
- J.-H. Ryu and M. Lee, J. Am. Chem. Soc., 2005, 127, 14170–14171 CrossRef PubMed.
- S. Dhiman and S. J. George, Bull. Chem. Soc. Jpn, 2018, 91, 687–699 CrossRef.
- K. Sugiyasu, Polym. J., 2021, 53, 865–875 CrossRef.
- S. Ogi, K. Matsumoto and S. Yamaguchi, Angew. Chem., Int. Ed., 2018, 57, 2339–2343 CrossRef PubMed.
- J. Matern, Z. Fernández, N. Bäumer and G. Fernández, Angew. Chem., Int. Ed., 2022, 61, e202203783 CrossRef PubMed.
- S. Patra, S. Dhiman and S. J. George, Chem. Mater., 2024, 36, 9460–9468 CrossRef.
- S. Ogi, V. Stepanenko, J. Thein and F. Würthner, J. Am. Chem. Soc., 2016, 138, 670–678 CrossRef PubMed.
- W. Wagner, M. Wehner, V. Stepanenko, S. Ogi and F. Würthner, Angew. Chem., Int. Ed., 2017, 56, 16008–16012 CrossRef PubMed.
- E. E. Greciano, J. Calbo, E. Ortí and L. Sánchez, Angew. Chem., Int. Ed., 2020, 59, 17517–17524 CrossRef PubMed.
- C. Naranjo, S. Adalid, R. Gómez and L. Sánchez, Angew. Chem., Int. Ed., 2023, 62, e202218572 CrossRef CAS PubMed.
- E. E. Greciano, S. Alsina, G. Ghosh, G. Fernández and L. Sánchez, Small Methods, 2020, 4, 1900715 CrossRef CAS.
- N. Fukaya, S. Ogi, H. Sotome, K. J. Fujimoto, T. Yanai, N. Bäumer, G. Fernández, H. Miyasaka and S. Yamaguchi, J. Am. Chem. Soc., 2022, 144, 22479–22492 CrossRef CAS PubMed.
- J. Matern, I. Maisuls, C. A. Strassert and G. Fernández, Angew. Chem., Int. Ed., 2022, 61, e202208436 CrossRef CAS PubMed.
- J. Matern, Z. Fernández and G. Fernández, Chem. Commun., 2022, 58, 12309–12312 RSC.
- S. Ogi, K. Sugiyasu, S. Manna, S. Samitsu and M. Takeuchi, Nat. Chem., 2014, 6, 188–195 CrossRef CAS PubMed.
- S. Ogi, T. Fukui, M. L. Jue, M. Takeuchi and K. Sugiyasu, Angew. Chem., Int. Ed., 2014, 53, 14363–14367 CrossRef CAS PubMed.
- R. M. Veedu, Z. Fernández, N. Bäumer, A. Albers and G. Fernández, Chem. Sci., 2024, 15, 10745–10752 RSC.
- H. Kar, G. Ghosh and S. Ghosh, Chem. Eur. J., 2017, 23, 10536–10542 CrossRef CAS PubMed.
- Y. Huang, R. Zhang, C. Shi, C. Wang, R. Liao, C. Zou, F. Wang and N. Huang, Chem. Eur. J., 2025, 31, e202500120 CrossRef CAS PubMed.
- T. Gädt, N. S. Ieong, G. Cambridge, M. A. Winnik and I. Manners, Nat. Mater., 2009, 8, 144–150 CrossRef PubMed.
- J. B. Gilroy, T. Gädt, G. R. Whittell, L. Chabanne, J. M. Mitchels, R. M. Richardson, M. A. Winnik and I. Manners, Nat. Chem., 2010, 2, 566–570 CrossRef CAS PubMed.
- Z. Jin, N. Sasaki, N. Kishida, M. Takeuchi, Y. Wakayama and K. Sugiyasu, Chem. Eur. J., 2023, 29, e202302181 CrossRef CAS PubMed.
- L. Kleine-Kleffmann, A. Schulz, V. Stepanenko and F. Würthner, Angew. Chem., Int. Ed., 2023, 62, e202314667 CrossRef CAS PubMed.
- P. Khanra, P. Rajdev and A. Das, Angew. Chem., Int. Ed., 2024, 63, e202400486 CrossRef CAS PubMed.
- Z. Jin, M. Takeuchi, Y. Wakayama and K. Sugiyasu, Chem. Eur. J., 2023, 29, e202302181 CrossRef CAS PubMed.
- S. Yagai and A. Kitamura, Chem. Soc. Rev., 2008, 37, 1520–1529 RSC.
- E. Fuentes, M. Gerth, J. A. Berrocal, C. Matera, P. Gorostiza, I. K. Voets, S. Pujals and L. Albertazzi, J. Am. Chem. Soc., 2020, 142, 10069–10078 CrossRef CAS PubMed.
- M. Endo, T. Fukui, S. H. Jung, S. Yagai, M. Takeuchi and K. Sugiyasu, J. Am. Chem. Soc., 2016, 138, 14347–14353 CrossRef CAS PubMed.
- F. Xu, L. Pfeifer, S. Crespi, F. K.-C. Leung, M. C. A. Stuart, S. J. Wezenberg and B. L. Feringa, J. Am. Chem. Soc., 2021, 143, 5990–5997 CrossRef CAS PubMed.
- S. Imai, T. Hamada, M. Nozaki, T. Fujita, M. Takahashi, Y. Fujita, K. Harano, H. Uji-i, A. Takai and K. Hirai, J. Am. Chem. Soc., 2025, 147, 23528–23535 CrossRef CAS PubMed.
- L. Kleine-Kleffmann, V. Stepanenko, K. Shoyama, M. Wehner and F. Würthner, J. Am. Chem. Soc., 2023, 145, 9144–9151 CrossRef CAS PubMed.
- C. Naranjo, Y. Dorca, G. Ghosh, R. Gómez, G. Fernández and L. Sánchez, Chem. Commun., 2021, 57, 4500–4503 RSC.
- N. Bäumer, E. Castellanos, B. Soberats and G. Fernández, Nat. Commun., 2023, 14, 1084 CrossRef PubMed.
- T. Fukui, N. Sasaki, M. Takeuchi and K. Sugiyasu, Chem. Sci., 2019, 10, 6770–6776 RSC.
- H. Fu, J. Huang, J. J. B. van der Tol, L. Su, Y. Wang, S. Dey, P. Zijlstra, G. Fytas, G. Vantomme, P. Y. W. Dankers and E. W. Meijer, Nature, 2024, 626, 1011–1018 CrossRef CAS PubMed.
- T. Dünnebacke, N. Niemeyer, S. Baumert, S. Hochstädt, L. Borsdorf, M. R. Hansen, J. Neugebauer and G. Fernández, Nat. Commun., 2024, 15, 5695 CrossRef PubMed.
- J. J. B. V. D. Tol, M. M. J. Dekker, Á. Müller, P. Springintveld, E. W. Meijer and G. VantommeAngew. Chem., Int. Ed., 2025, e202512216 Search PubMed.
- S. I. A. Cohen, S. Linse, L. M. Luheshi, E. Hellstrand, D. A. White, L. Rajah, D. E. Otzen, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9758–9763 CrossRef CAS PubMed.
- S. Datta, H. Itabashi, T. Saito and S. Yagai, Nat. Chem., 2025, 17, 477–492 CrossRef CAS PubMed.
- M. Wolffs, P. A. Korevaar, P. Jonkheijm, O. Henze, W. James Feast, A. P. H. J. Schenning and E. W. Meijer, Chem. Commun., 2008, 4613–4615 RSC.
- S. Datta, S. Takahashi and S. Yagai, Acc. Mater. Res., 2022, 3, 259–271 CrossRef CAS.
- S. Datta, Y. Kato, S. Higashiharaguchi, K. Aratsu, A. Isobe, T. Saito, D. D. Prabhu, Y. Kitamoto, M. J. Hollamby, A. J. Smith, R. Dalgliesh, N. Mahmoudi, L. Pesce, C. Perego, G. M. Pavan and S. Yagai, Nature, 2020, 583, 400–405 CrossRef CAS PubMed.
- N. Sasaki, J. Kikkawa, Y. Ishii, T. Uchihashi, H. Imamura, M. Takeuchi and K. Sugiyasu, Nat. Chem., 2023, 15, 922–929 CrossRef CAS PubMed.
- A. Osypenko, E. Moulin, O. Gavat, G. Fuks, M. Maaloum, M. A. J. Koenis, W. J. Buma and N. Giuseppone, Chem. Eur. J., 2019, 25, 13008–13016 CrossRef CAS PubMed.
- R. Laishram, S. Sarkar, I. Seth, N. Khatun, V. K. Aswal, U. Maitra and S. J. George, J. Am. Chem. Soc., 2022, 144, 11306–11315 CrossRef CAS PubMed.
- S. Sarkar, R. Laishram, D. Deb and S. J. George, J. Am. Chem. Soc., 2023, 145, 22009–22018 CrossRef CAS PubMed.
- S. R. Pramatha, D. Srideep, U. Pattnaik, R. Sahu, D. I. Suresh, A. C. Yadav, C. Muduli, S. K. Reddy, S. P. Senanayak and K. Venkata Rao, Nat. Commun., 2024, 15, 10808 CrossRef PubMed.
- J.-P. Brog, C.-L. Chanez, A. Crochet and K. M. Fromm, RSC Adv, 2013, 3, 16905–16931 RSC.
- R. Tycko, Neuron, 2015, 86, 632–645 CrossRef CAS PubMed.
- Q. Shi, H. Chen, Y. Wang, J. Xu, Z. Liu and C. Zhang, Int. J. Pharm., 2022, 611, 121320 CrossRef CAS PubMed.
- H. Chung and Y. Diao, J. Mater. Chem. C, 2016, 4, 3915–3933 RSC.
- N. M. Matsumoto, R. P. M. Lafleur, X. Lou, K.-C. Shih, S. P. W. Wijnands, C. Guibert, J. W. A. M. van Rosendaal, I. K. Voets, A. R. A. Palmans, Y. Lin and E. W. Meijer, J. Am. Chem. Soc., 2018, 140, 13308–13316 CrossRef CAS PubMed.
- M. Wehner, M. I. S. Röhr, M. Bühler, V. Stepanenko, W. Wagner and F. Würthner, J. Am. Chem. Soc., 2019, 141, 6092–6107 CrossRef CAS PubMed.
- A. Langenstroer, K. K. Kartha, Y. Dorca, J. Droste, V. Stepanenko, R. Q. Albuquerque, M. R. Hansen, L. Sánchez and G. Fernández, J. Am. Chem. Soc., 2019, 141, 5192–5200 CrossRef CAS PubMed.
- J. Matern, K. K. Kartha, L. Sánchez and G. Fernández, Chem. Sci., 2020, 11, 6780–6788 RSC.
- C. Otsuka, S. Imai, T. Ohkubo and S. Yagai, Chem. Commun., 2024, 60, 1108–1111 RSC.
- C. Otsuka, S. Takahashi, A. Isobe, T. Saito, T. Aizawa, R. Tsuchida, S. Yamashita, K. Harano, H. Hanayama, N. Shimizu, H. Takagi, R. Haruki, L. Liu, M. J. Hollamby, T. Ohkubo and S. Yagai, J. Am. Chem. Soc., 2023, 145, 22563–22576 CrossRef CAS PubMed.
- L. Rubert, H. M. A. Ehmann and B. Soberats, Angew. Chem., Int. Ed., 2025, 64, e202415774 CrossRef CAS PubMed.
- I. Debnath, T. Roy, J. Matern, S. A. H. Jansen, G. Fernández and K. Mahata, Org. Chem. Front., 2021, 8, 5432–5439 RSC.
- S. Datta and D. Chaudhuri, Angew. Chem., Int. Ed., 2022, 61, e202201956 CrossRef CAS PubMed.
- Y. Hong, Y. Jiang, C. Yuan, D. Wang, Y. Sun and J. Jiang, Chin. Chem. Lett., 2024, 35, 109909 CrossRef CAS.
- Y. Kasahara, T. Takeda, S. Dekura, Y. Ishii, H. Anetai, A. Takai, I. Hisaki, M. Takeuchi and T. Akutagawa, J. Am. Chem. Soc., 2025, 147, 18783–18795 CrossRef CAS PubMed.
- C. Montañez-Moyano, Y. Xue, M. V. Cappellari, A. Martínez-Manjarrés, L. Borsdorf, C. A. Strassert and G. Fernández, Angew. Chem., Int. Ed., 2025, e202509031 Search PubMed.
- J. Li, J. Wang, H. Li, N. Song, D. Wang and B. Z. Tang, Chem. Soc. Rev., 2020, 49, 1144–1172 RSC.
- D. M. Eisele, J. Knoester, S. Kirstein, J. P. Rabe and D. A. V. Bout, Nat. Nanotechnol., 2009, 4, 658–663 CrossRef CAS PubMed.
- C. Villegas, E. Krokos, P.-A. Bouit, J. L. Delgado, D. M. Guldi and N. Martín, Energy Environ. Sci., 2011, 4, 679–684 RSC.
- X. Wang, Y. Han, Y. Liu, G. Zou, Z. Gao and F. Wang, Angew. Chem., Int. Ed., 2017, 56, 12466–12470 CrossRef CAS PubMed.
- C. Rehhagen, M. Stolte, S. Herbst, M. Hecht, S. Lochbrunner, F. Würthner and F. Fennel, J. Phys. Chem. Lett., 2020, 11, 6612–6617 CrossRef CAS PubMed.
- M. Hecht, T. Schlossarek, S. Ghosh, Y. Tsutsui, A. Schmiedel, M. Holzapfel, M. Stolte, C. Lambert, S. Seki, M. Lehmann and F. Würthner, ACS Appl. Nano Mater., 2020, 3, 10234–10245 CrossRef CAS.
- M. Hecht, T. Schlossarek, M. Stolte, M. Lehmann and F. Würthner, Angew. Chem., Int. Ed., 2019, 58, 12979–12983 CrossRef CAS PubMed.
- Z. Xie, B. Xiao, Z. He, W. Zhang, X. Wu, H. Wu, F. Würthner, C. Wang, F. Xie, L. Liu, Y. Ma, W.-Y. Wong and Y. Cao, Mater. Horiz., 2015, 2, 514–518 RSC.
- M. Gsänger, J. H. Oh, M. Könemann, H. W. Höffken, A.-M. Krause, Z. Bao and F. Würthner, Angew. Chem., Int. Ed., 2010, 49, 740–743 CrossRef PubMed.
- H. Liu, X. Cao, Y. Wu, Q. Liao, Á. J. Jiménez, F. Würthner and H. Fu, Chem. Commun., 2014, 50, 4620–4623 RSC.
- P. Lova, V. Grande, G. Manfredi, M. Patrini, S. Herbst, F. Würthner and D. Comoretto, Adv. Opt. Mater., 2017, 5, 1700523 CrossRef.
- A. T. Haedler, K. Kreger, A. Issac, B. Wittmann, M. Kivala, N. Hammer, J. Köhler, H. W. Schmidt and R. Hildner, Nature, 2015, 523, 196–199 CrossRef CAS PubMed.
- A. T. Haedler, S. R. Beyer, N. Hammer, R. Hildner, M. Kivala, J. Köhler and H.-W. Schmidt, Chem. Eur. J., 2014, 20, 11708–11718 CrossRef CAS PubMed.
- B. Wittmann, F. A. Wenzel, S. Wiesneth, A. T. Haedler, M. Drechsler, K. Kreger, J. Köhler, E. W. Meijer, H.-W. Schmidt and R. Hildner, J. Am. Chem. Soc., 2020, 142, 8323–8330 CrossRef CAS PubMed.
- A. V. Gorbunov, A. T. Haedler, T. Putzeys, R. H. Zha, H.-W. Schmidt, M. Kivala, I. Urbanaviciutė, M. Wübbenhorst, E. W. Meijer and M. Kemerink, ACS Appl. Mater. Interfaces, 2016, 8, 15535–15542 CrossRef CAS PubMed.
- Y. Yamamoto, T. Fukushima, Y. Suna, N. Ishii, A. Saeki, S. Seki, S. Tagawa, M. Taniguchi, T. Kawai and T. Aida, Science, 2006, 314, 1761–1764 CrossRef CAS PubMed.
- Y. Yamamoto, T. Fukushima, W. Jin, A. Kosaka, T. Hara, T. Nakamura, A. Saeki, S. Seki, S. Tagawa and T. Aida, Adv. Mater., 2006, 18, 1297–1300 CrossRef CAS.
- J. J. I. Armao, I. Nyrkova, G. Fuks, A. Osypenko, M. Maaloum, E. Moulin, R. Arenal, O. Gavat, A. Semenov and N. Giuseppone, J. Am. Chem. Soc., 2017, 139, 2345–2350 CrossRef PubMed.
- V. Faramarzi, F. Niess, E. Moulin, M. Maaloum, J.-F. Dayen, J.-B. Beaufrand, S. Zanettini, B. Doudin and N. Giuseppone, Nat. Chem., 2012, 4, 485–490 CrossRef CAS PubMed.
- F. S. Schoonbeek, J. H. V. Esch, B. Wegewijs, D. B. A. Rep, M. P. D. Haas, T. M. Klapwijk, R. M. Kellogg and B. L. Feringa, Angew. Chem., Int. Ed., 1999, 38, 1393–1397 CrossRef PubMed.
- R. J. Hafner, D. Görl, A. Sienkiewicz, S. Balog and H. Frauenrath, Chem. Eur. J., 2020, 26, 9506–9517 CrossRef PubMed.
- J. P. Hill, W. Jin, A. Kosaka, T. Fukushima, H. Ichihara, T. Shimomura, K. Ito, T. Hashizume, N. Ishii and T. Aida, Science, 2004, 304, 1481–1483 CrossRef PubMed.
- M. Sofos, J. Goldberger, D. A. Stone, J. E. Allen, Q. Ma, D. J. Herman, W. W. Tsai, L. J. Lauhon and S. I. Stupp, Nat. Mater., 2009, 8, 68–75 CrossRef PubMed.
- Y. Guo, G. Yu and Y. Liu, Adv. Mater., 2010, 22, 4427–4447 CrossRef PubMed.
- G. Hong, X. Gan, C. Leonhardt, Z. Zhang, J. Seibert, J. M. Busch and S. Bräse, Adv. Mater., 2021, 33, 2005630 CrossRef PubMed.
- K. A. Mazzio and C. K. Luscombe, Chem. Soc. Rev., 2014, 44, 78–90 RSC.
- S. Shanmugaraju, H. Jadhav, R. Karthik and P. S. Mukherjee, RSC Adv., 2013, 3, 4940–4950 RSC.
- B. Gole, W. Song, M. Lackinger and P. S. Mukherjee, Chem. Eur. J., 2014, 20, 13662–13680 CrossRef PubMed.
- T. L. Mako, J. M. Racicot and M. Levine, Chem. Rev., 2019, 119, 322–477 CrossRef PubMed.
- Q. Lin, X.-M. Jiang, X.-Q. Ma, J. Liu, H. Yao, Y.-M. Zhang and T.-B. Wei, Sens. Actuators B Chem., 2018, 272, 139–145 CrossRef.
- Z. Chen, Q. Wang, X. Wu, Z. Li and Y.-B. Jiang, Chem. Soc. Rev., 2015, 44, 4249–4263 RSC.
- D.-W. Zhang, X. Zhao and Z.-T. Li, Acc. Chem. Res., 2014, 47, 1961–1970 CrossRef PubMed.
- C. Li, G.-T. Wang, H.-P. Yi, X.-K. Jiang, Z.-T. Li and R.-X. Wang, Org. Lett., 2007, 9, 1797–1800 CrossRef PubMed.
- F. Nabeel, T. Rasheed, M. Bilal, C. Li, C. Yu and H. M. N. Iqbal, Sep. Purif. Rev., 2020, 49, 20–36 CrossRef.
- E. Cohen, H. Weissman, E. Shimoni, I. Kaplan-Ashiri, K. Werle, W. Wohlleben and B. Rybtchinski, Angew. Chem., Int. Ed., 2017, 56, 2203–2207 CrossRef CAS PubMed.
- E. Krieg, H. Weissman, E. Shirman, E. Shimoni and B. Rybtchinski, Nat. Nanotechnol., 2011, 6, 141–146 CrossRef CAS PubMed.
- L. Bouteiller and M. Raynal, Supramol. Catal. New Dir. Dev., 2021, 93–106 Search PubMed.
- M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. V. Leeuwen, Chem. Soc. Rev., 2014, 43, 1660–1733 RSC.
- M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. V. Leeuwen, Chem. Soc. Rev., 2014, 43, 1734–1787 RSC.
- L. N. Neumann, M. B. Baker, C. M. A. Leenders, I. K. Voets, R. P. M. Lafleur, A. R. A. Palmans and E. W. Meijer, Org. Biomol. Chem., 2015, 13, 7711–7719 RSC.
- R. Chen, A. Hammoud, P. Aoun, M. A. Martínez-Aguirre, N. Vanthuyne, R. Maruchenko, P. Brocorens, L. Bouteiller and M. Raynal, Nat. Commun., 2024, 15, 4116 CrossRef CAS PubMed.
- X.-G. Tan, P. Jeyakkumar, Y.-X. Zhang, L.-W. Chu, Y.-R. Hao, S.-K. Li, K.-Y. Wang and X.-Y. Hu, cMat, 2024, 1, e31 CrossRef.
- D. Zhang, W. Yu, S. Li, Y. Xia, X. Li, Y. Li and T. Yi, J. Am. Chem. Soc., 2021, 143, 1313–1317 CrossRef CAS PubMed.
- J. Tian, J. Yu, Q. Tang, J. Zhang, D. Ma, Y. Lei and Z.-T. Li, Mater. Futur., 2022, 1, 042104 CrossRef CAS.
- A. S. Weingarten, R. V. Kazantsev, L. C. Palmer, M. McClendon, A. R. Koltonow, A. P. S. Samuel, D. J. Kiebala, M. R. Wasielewski and S. I. Stupp, Nat. Chem., 2014, 6, 964–970 CrossRef CAS PubMed.
- A. S. Weingarten, R. V. Kazantsev, L. C. Palmer, D. J. Fairfield, A. R. Koltonow and S. I. Stupp, J. Am. Chem. Soc., 2015, 137, 15241–15246 CrossRef CAS.
- J. Yu, L. Huang, Q. Tang, S.-B. Yu, Q.-Y. Qi, J. Zhang, D. Ma, Y. Lei, J. Su, Y. Song, J.-C. Eloi, R. L. Harniman, U. Borucu, L. Zhang, M. Zhu, F. Tian, L. Du, D. L. Phillips, I. Manners, R. Ye and J. Tian, Nat. Catal., 2023, 6, 464–475 CrossRef CAS.
- R. Dong, Y. Zhou, X. Huang, X. Zhu, Y. Lu and J. Shen, Adv. Mater., 2015, 27, 498–526 CrossRef CAS PubMed.
- D. Wang, G. Tong, R. Dong, Y. Zhou, J. Shen and X. Zhu, Chem. Commun., 2014, 50, 11994–12017 RSC.
- R. Dong, Y. Pang, Y. Su and X. Zhu, Biomater. Sci., 2015, 3, 937–954 RSC.
- N. R. B. Boase, E. R. Gillies, R. Goh, R. E. Kieltyka, J. B. Matson, F. Meng, A. Sanyal and O. Sedláček, Biomacromolecules, 2024, 25, 5417–5436 CrossRef CAS PubMed.
- J. B. Matson and S. I. Stupp, Chem. Commun., 2011, 48, 26–33 RSC.
- S. M. Chin, C. V. Synatschke, S. Liu, R. J. Nap, N. A. Sather, Q. Wang, Z. Álvarez, A. N. Edelbrock, T. Fyrner, L. C. Palmer, I. Szleifer, M. O. D. L. Cruz and S. I. Stupp, Nat. Commun., 2018, 9, 1–11 CrossRef PubMed.
- K. Sato, M. P. Hendricks, L. C. Palmer and S. I. Stupp, Chem. Soc. Rev., 2018, 47, 7539–7551 RSC.
- M. J. Webber and R. Langer, Chem. Soc. Rev., 2017, 46, 6600–6620 RSC.
- A. L. Schmitt, S. H.-J. Yoon, W.-D. Jang and H.-J. Yoon, J. Mater. Chem., 2009, 20, 211–222 Search PubMed.
- T. Jin, C. Huang, M. Cui, Y. Yang, Z. Wang, W. Zhu and X. Qian, J. Mater. Chem. B, 2020, 8, 10686–10699 RSC.
- Z. Shi, X. Han, W. Hu, H. Bai, B. Peng, L. Ji, Q. Fan, L. Li and W. Huang, Chem. Soc. Rev., 2020, 49, 7533–7567 RSC.
- Y. Chen Liu, G. Jian Liu, W. Zhou, G. Li Feng, Q. Yu Ma, Y. Zhang and G. Wen Xing, Angew. Chem., Int. Ed., 2023, 62, e202309786 CrossRef PubMed.
- Y. Liu, H. Wang, S. Li, C. Chen, L. Xu, P. Huang, F. Liu, Y. Su, M. Qi, C. Yu and Y. Zhou, Nat. Commun., 2020, 11, 1724 CrossRef CAS PubMed.
- Y. Dai, J. Sun, X. Zhang, J. Zhao, W. Yang, J. Zhou, Z. Gao, Q. Wang, F. Yu and B. Wang, Coord. Chem. Rev., 2024, 517, 216054 CrossRef CAS.
- Y. Chen, X. H. Zhang, D. B. Cheng, Y. Zhang, Y. Liu, L. Ji, R. Guo, H. Chen, X. K. Ren, Z. Chen, Z. Y. Qiao and H. Wang, ACS Appl. Mater. Interfaces, 2020, 14, 3650 Search PubMed.
- N. Kwon, H. Kim, X. Li and J. Yoon, Chem. Sci., 2021, 12, 7248–7268 RSC.
- X.-Y. Lou, G. Zhang, N. Song and Y.-W. Yang, Biomaterials, 2022, 286, 121595 CrossRef CAS PubMed.
- N. Mehwish, X. Dou, Y. Zhao and C. L. Feng, Mater. Horiz., 2019, 6, 14–44 RSC.
- G. Fan, Y. X. Lin, L. Yang, F. P. Gao, Y. X. Zhao, Z. Y. Qiao, Q. Zhao, Y. S. Fan, Z. Chen and H. Wang, Chem. Commun., 2015, 51, 12447–12450 RSC.
- A. Albers, S. Kuberasivakumaran, Z. Fernández, C. G. Daniliuc, Y. Li, M. Lee, C. Geyer, E. Hoffmann, C. Faber, A. Helfen, C. Grashoff, M. Masthoff and G. Fernández, Angew. Chem., Int. Ed., 2025, 64, e202500144 CrossRef CAS PubMed.
- V. S. Talens, G. Arias-Alpizar, D. M. M. Makurat, J. Davis, J. Bussmann, A. Kros and R. E. Kieltyka, Biomacromolecules, 2020, 21, 1060–1068 CrossRef PubMed.
- K. Yang, G. Yu, Z. Yang, L. Yue, X. Zhang, C. Sun, J. Wei, L. Rao, X. Chen and R. Wang, Angew. Chem., Int. Ed., 2021, 60, 17570–17578 CrossRef CAS PubMed.
- J. Qi, C. Chen, X. Zhang, X. Hu, S. Ji, R. T. K. Kwok, J. W. Y. Lam, D. Ding and B. Z. Tang, Nat. Commun., 2018, 9, 1848 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2025 |