
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
[(TMEDA)Li(μ-PPh2)2K(TMEDA)(THF)]: a heterobimetallic molecular lithium–potassium phosphide complex†
Michelle H.
Crabbe
,
Danielle
O’Meara
,
Alan R.
Kennedy
 ,
Catherine E.
Weetman
* and
Robert E.
Mulvey
*
,
Catherine E.
Weetman
* and
Robert E.
Mulvey
*
Department of Pure and Applied Chemistry, University of Strathclyde, Glasgow, G1 1XL, UK. E-mail: catherine.weetman@strath.ac.uk; r.e.mulvey@strath.ac.uk
First published on 22nd May 2025
Abstract
Made by a co-complexation reaction, heterobimetallic [(TMEDA)Li(μ-PPh2)2K(TMEDA)(THF)] has been crystallographically and spectroscopically characterised, modelled by DFT calculations, and its performance compared against those of its homometallic phosphide components in preliminary studies of its application in catalytic hydrophosphination reactions of a representative alkene.
The beginning of organometallic compounds containing two distinct alkali metals can be traced back to Wittig's 1950s discovery of di-organo-lithium–sodium compounds (here, organo is n-Bu or Ph).1–3 Brønsted basicity is a feature of these compounds and also of related early bimetallics such as potassium hydride-butyllithium.4 However, it was when Lochmann5 and Schlosser,6 building on studies by Morton,7,8 independently introduced the n-butyllithium–potassium-t-butoxide “superbase” that the synthetic community had found its first widely utilised bi-alkali-metal reagent. Significantly, hitherto none of these aforementioned bimetallics has been unequivocally structurally characterised, though assiduous work by Klett has uncovered a complexity of various LixKyNpz(OtBu)x+y−z structures using neo-pentyl as an n-Bu substitute that almost certainly reveals the essential likeness of the Li–K superbase structure/s.9,10 Mixed alkali metal structures with organonitrogen ligands have also appeared such as guanidinide LiNa3[O
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P(NMe2)3]3[NC(NMe2)2]4,11 and the series of hexamethyldisilazides [(THF)xM1M2N(SiMe3)2],12 the first mixed bi-alkali-metal examples of a popular utility amide. Alkali metal amides have long been complementary useful reagents to their alkyl alkali metal counterparts, and they too have been incorporated into synergistically operative mixed alkali metal compounds,13 notably the LINK (nBuLi/KOtBu/TMP) system14 (TMP is 2,2,6,6-tetramethylpiperidide), as well as mixed mono-alkali-metal bimetallics such as the turbo-Grignard reagents and inverse crown complexes,15 each of which incorporates a non-alkali metal, most typically magnesium.
P(NMe2)3]3[NC(NMe2)2]4,11 and the series of hexamethyldisilazides [(THF)xM1M2N(SiMe3)2],12 the first mixed bi-alkali-metal examples of a popular utility amide. Alkali metal amides have long been complementary useful reagents to their alkyl alkali metal counterparts, and they too have been incorporated into synergistically operative mixed alkali metal compounds,13 notably the LINK (nBuLi/KOtBu/TMP) system14 (TMP is 2,2,6,6-tetramethylpiperidide), as well as mixed mono-alkali-metal bimetallics such as the turbo-Grignard reagents and inverse crown complexes,15 each of which incorporates a non-alkali metal, most typically magnesium.
In contrast to the sizable number of known organocarbon and organonitrogen mixed alkali metal compounds,16,17 organophosphorus examples are exceptionally rare. Westerhausen reported a phosphanediide example in [Li6K6Ba{P(SiMe3)}6(OSiiPr3)2],18 but to the best of our knowledge, hitherto there has been no secondary phosphide example. Here, we open this chemical space by the successful synthesis, NMR and X-ray crystallographic characterisation of the monomeric, dinuclear lithium–potassium phosphide, [(TMEDA)Li(μ-PPh2)2K(TMEDA)(THF)], 1 (TMEDA is N,N,N′,N′-tetramethylethylenediamine).
Co-complexation reactions, such as in the addition of two different monometallic complexes, have become a facile and efficient way of generating heterobimetallic complexes.19–21 To access new mixed alkali metal organophosphorus compounds, we followed co-complexation procedures. Equimolar quantities of LiPPh2 and KPPh2 in toluene solution formed an orange suspension after 1 h at room temperature, which on adding two molar equivalents of Lewis donor TMEDA followed by twelve equivalents of THF gave an orange solution (Scheme 1). Volatiles were removed under reduced pressure to yield an orange oil, which upon trituration with hexane gave an isolable orange powder. THF was required to solubilise 1 in d6-benzene for NMR characterisation. The 1H spectrum revealed the expected singlet resonances for TMEDA at 1.97 ppm and 2.01 ppm, and resonances in the aromatic range 6.77–7.82 ppm. Both the 7Li and 31P NMR spectra showed one singlet at 1.48 ppm and −21.2 ppm, respectively. 31P NMR analysis of solutions prepared of component monometallic complexes [LiPPh2(TMEDA)]2 [−26.0 ppm] and [KPPh2(TMEDA)]n [−15.7 ppm]22 (see ESI,† p. 14–17) under the same conditions show that the resonance for the product of the co-complexation reaction (−21.2 ppm) lies near the midpoint of those for these compounds, suggestive of the desired formation of the heterobimetallic complex. The 7Li NMR shift of [LiPPh2(TMEDA)]2 [1.16 ppm] is also downfield with respect to that of the mixed-metal solution. Heating the orange suspension in toluene/THF and cooling the mixture at −30 °C, afforded orange crystals. An X-ray diffraction study confirmed these to be heterobimetallic monomer 1 (Fig. 1) crystallised in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) .
.
 | ||
| Scheme 1 Co-complexation synthesis of the title compound 1. | ||
 | ||
| Fig. 1 Molecular structure of LiK(PPh2)2(TMEDA)2THF, 1. Thermal ellipsoids have been drawn at 50% probability, H atoms omitted and TMEDA ligands drawn in wireframe for clarity. Selected bond lengths (Å) and angles (°): Li1–P1 2.602(2); Li1–P2 2.599(3); K1–P1 3.4466(5); K1–P2 3.2419(4); Li1–N1 2.130(3); Li1–N2 2.126(3); K1–N3 2.839(2); K1–N4 2.906(10); K1–O1 2.6892(14); K1–C7 3.2983(14); K1–C8 3.1566(14); P2–Li1–P1 116.36(9); P2–K1–P1 82.649(11); K1–P1–Li1 77.99(6); K1–P2–Li1 81.99(6). | ||
The core feature of 1 is a four-membered LiPKP ring, which is near-planar (∑endocyclic bond angles, 359°), with an obtuse angle at Li [116.36(9)°], acute angle at K [82.649(11)°] and dissimilar endocyclic angles at the two P bridges [77.99(6)°; 81.99(6)°]. Li–P bond distances [2.602(2) Å and 2.599(2) Å] are within experimental error of each other, while more asymmetry exists in the K–P bonds [3.4466(5) Å and 3.2419(4) Å]. These Li–P and K–P distances are within the range of literature values.23 These metric differences reflect the greater coordinative asymmetry at K, whereas Li has a distorted tetrahedral geometry comprising two P and two (TMEDA) N atoms [Li1–N1 = 2.130(3) Å; Li1–N2 = 2.126(3) Å], the larger K atom has a coordination number of 7. This comprises two P, two (TMEDA) N and one (THF) O heteroatoms, with saturation completed by the π system of one Ph of the Ph2P ligand via a η2-C2 (ipso–ortho) edge [K1–C7 3.2983(14); K1–C8 3.1566(14)]. This π coordination accounts for the unique Ph2P ligand environments with P1 in a more distorted tetrahedral geometry than P2 (mean bond angles, 95.4° and 107.8°, respectively) (see Table S4 for selected bond metrics, ESI†). Here, applying the criteria of Alvarez24 for a geometric analysis of the hapticity of the aryl interactions in 1 (see ESI,† p. 19) predicts that K1 exhibits η2 coordination to the π system reinforcing the crystallographic metric data.
To gain knowledge of the structure of 1 in solution, we studied it by 1H diffusion-ordered NMR spectroscopy (DOSY) in d8-toluene (see ESI,† p. 12). Excess THF was required to dissolve 1, since adding fewer equivalents, agitating or heating would cause 1 to precipitate from solution. The order of addition of reagents is also important, as when THF was added first, a mixed-metal species did not form and only the known polymeric [KPPh2]n was isolable.25 DOSY data imply that the LiK(PPh2)2 core is retained in the solution state. The diffusion coefficient D obtained for LiK(PPh2)2 (mean, 4.94 × 10−10 m2 s−1) predicts that the LiK species in solution has an 18% higher experimental MWDOSY (874 g mol−1) than the theoretical MW of crystalline LiK(PPh2)2(TMEDA)2(THF) (721 g mol−1). From our closest prediction to this theoretical MW in [(TMEDA)Li(μ-PPh2)2K(TMEDA)2(THF)3] (MWdiff −1%), one could speculate that the K–π interactions in the crystal are replaced by two extra K–O (THF) bonds, especially as extra THF was added to dissolve the crystals in d8-toluene and since only one Ph2P type ligand was seen in the 31P NMR spectrum at temperatures as low as –40 °C (see ESI,† Fig. S8, p. 7). However, given that both TMEDA and THF have estimated MWs higher than their theoretical MWs (by −49 and −12%, respectively), complex equilibria are likely to be in play making it difficult to know with any certainty the species present in solution.
The most common alkali metal based structures in the Cambridge Structural Database (CSD)26 are those of lithium compounds, with the greater interest in studying such structures stimulated mainly by the vast utility of organolithium compounds in synthesis. This detail holds true for alkali metal diorganophosphide compounds derived from secondary phosphines (R2PH), with lithium examples particularly useful as diorganophosphido transfer agents.27 In the case of diphenylphosphine, Ph2PH, the phosphine used in this study, there are only a handful of Ph2PLi structures in the database. Aligning well with the fundamental principles of and diversity within organolithium structural chemistry,28 this small collection of structures contains, a solvent-separated ion pair, a monomer, a discrete dimer, and three one-dimensional chain polymers (Fig. 2), with the degree of aggregation controlled by the Lewis base donor solvent stabilising the Lewis acidic metal centre, namely, 12-crown-4, PMDETA, TMEDA, THF (×2), Et2O, and DME, respectively. The key factor in this trend is clearly the denticity number of the Lewis base with the hexadentate, tridentate and bidentate donors dictating the solvent-separated ion pair, dimeric, and monomeric structures, respectively, while the weaker low denticity oxygen-based donors are not strong enough to cleave the infinite (P–Li) chain that presumably exists in donor-solvent free Ph2PLi, which due to its lack of solubility in non-donor solvents has evaded its crystallographic characterisation. Lithium derivatives of other diorganophosphide structures are known, such as those from Cy2PH,29t-Bu2PH,30,31 Dipp2PH,32 Mes2PH,32,33 (Dipp)MesPH,32 [(Me3Si)2CH]2PH (technically from [(Me3Si)2CH]2PCl),34 [(Me3Si)2CH](Tipp)PH,35 [(Me3Si)2CH](Dipp)PH,36 and [(Me3Si)2CH](Ph)PH.37 Noteworthy among these structures is [LiPt-Bu2(THF)]4,38 a discrete tetrameric molecule best described as a four Li–P rung ladder that is truncated by the presence of THF caps at the ladder end rungs. This structure is an early example of lithium organoelement structures that fit perfectly well into the ring-laddering concept39 developed initially through the study of lithium organonitrogen compounds.
 | ||
| Fig. 2 Selection of reported crystallographically-characterised donor adducts of Ph2PLi. | ||
A search of the CSD revealed only four hits for Ph2PK structures (Fig. 3). The larger coordination sphere of potassium40 generally gives rise to more complexity in these structures compared to that of its lithium counterparts. Donor-solvent-free Ph2PK,25 the product of an unplanned elimination reaction of Ph2P from metallation of HC(SiMe3)2(SiMe2PPh2), exists as a complicated supramolecular network structure containing connected KPKP and KPKPKP four- and six-membered rings, respectively, which propagate mainly via K–Ph π-contacts. The bis-solvate [{K(dioxane)2PPh2}∞]41 continues this 3-dimensional network connectivity through a combination of dioxane bridges, which have transoid type O donor centres and K–Ph π-contacts. In [{Ph2PK(PMDETA)}∞]42 the extra denticity provided by a tridentate PMDETA ligand reduces the supramolecular structure to a one-dimensional K–P bonded chain. Finally, the structure of [{(Ph2P)[K2(18-crown-6)2]+[PPh2]−}]43 is charge-separated comprising a Ph2P− anion sandwiched between two K(18-crown-6)+ cations as well as a second, separated Ph2P− anion. Potassium derivatives of other diorganophosphide structures are known including Dipp2PH,32t-Bu2PH,44 Ph(tBu)PH,45 and (Ter)iPrPH.46 Note that no mixed lithium–potassium diphenylphosphide structure was found in CSD searches.26 Evidence that such structural knowledge is more important than just for curiosity value comes from our recent report on catalytic hydrophosphination reactions of alkynes.47 Here, a clear correlation was found between the catalytic efficiency and the denticity of the donor and aggregation state of the NaPPh2·donor catalyst employed.
 | ||
| Fig. 3 Selection of reported crystallographically-characterised donor adducts of Ph2PK. | ||
DFT calculations were performed to support the experimental findings in this study. QTAIM calculations reveal bond paths and critical points around the central Li–P–K–P core. Negative charges on the phosphorus extend in the direction of Li; however, the values of ρ(r) and ∇2ρ(r) between Li–P and K–P bonds are consistent with the expected closed-shell ionic interactions in each case (see ESI,† Fig. S19). 31P NMR calculations were also performed and match the observed experimental trends, with the Li–K heterobimetallic 31P calculated signal found between the two homometallic resonances. Energies of formation of 1 also support facile exchange processes between the three metal complexes (see ESI,† for details).
A preliminary investigation of the catalytic capability of 1 in a hydrophosphination reaction of an alkene was undertaken (Table 1). To aid with comparison of other systems reported by us,47,48 the conditions used with the substrate 1,1-diphenylethylene were 10 mol% catalyst loading at room temperature in d8-toluene. The conversion was calculated using adamantane as an internal standard. With 1 as a catalyst, 94% conversion occurred after only 20 minutes, with quantitative conversion after 1.25 h. This is in stark contrast to monometallic [LiPPh2(TMEDA)]2, which under the same conditions could only reach 6% conversion after 1 h, and 21% even after 24 h. [KPPh2(TMEDA)]n was also tested, and quantitative conversion was reached after 0.5 h. This trend is reminiscent of that of nBuLi < nBuLi.KOtBu < nBuK found in Lochmann–Schlosser superbase chemistry.5
|
|
||
|---|---|---|
| Catalyst | Time (h) | Conversion (%) |
| a Contains 0.06 mL of THF. Adamantane (0.035 mmol) was added as an internal standard to calculate NMR conversion. | ||
| 1 | 0.1 | 81 |
| 1 | 1.25 | >99 |
| [LiPPh2(TMEDA)]2 | 1 | 6 |
| [LiPPh2(TMEDA)]2 | 24 | 21 |
| [LiPPh2(TMEDA)]2a | 6 | 56 |
| [KPPh2(TMEDA)]n | 0.1 | 88 |
| [KPPh2(TMEDA)]n | 0.5 | >99 |
| [KPPh2(TMEDA)]na | 0.5 | >99 |
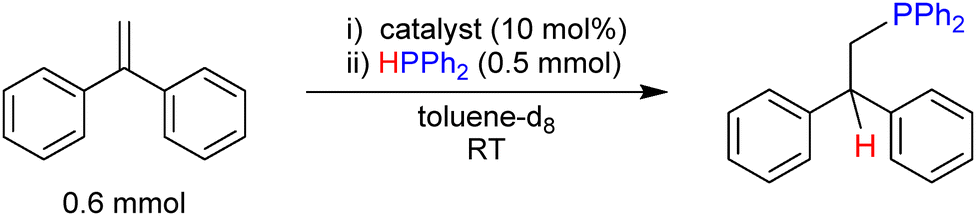
Note, the dehydrocoupling of phosphines is a known competing reaction,49,50 but when tested in our system 1 showed no evidence of it after 24 h. Importantly, 1 was retained in solution during the dehydrocoupling control reactions indicating that it does not dissociate into its monometallic counterparts during hydrophosphination catalysis. This work has established a unique mixed lithium–potassium phosphide complex derived from a common phosphine. New base metal compounds of this type, heterometallic and homometallic, may become increasingly important for the chemical community in its quest towards more sustainable practices that avoid the use of precious transition metals.
MHC, investigation, writing – original draft; DOM, investigation; ARK, validation; REM, conceptualisation, writing – review & editing; CW, formal analysis, writing – review & editing.
The award of a George Fraser Scholarship (PhD studentship to M. H. C.) is gratefully appreciated. Craig Irving is also thanked for help with NMR. Results were obtained using the ARCHIE-WeSt High Performance Computer (https://www.archie-west.ac.uk) based at Strathclyde (Grant code EP/K000586/1).
Data availability
NMR data, crystallographic data and XYZ files are available from the University of Strathclyde Knowledge Base at https://doi.org/10.15129/a7b30f49-5910-417c-87ae-409ca2181178.Conflicts of interest
The authors have no conflicts of interest to declare.Notes and references
- G. Wittig, R. Ludwig and R. Polster, Chem. Ber., 1955, 88, 294–301 CrossRef CAS.
- G. Wittig and F. Bickelhaupt, Chem. Ber., 1958, 91, 865–872 CrossRef CAS.
- G. Wittig and E. Benz, Chem. Ber., 1958, 91, 873–882 Search PubMed.
- G. G. Eberhardt, J. Org. Chem., 1964, 29, 643–645 CrossRef CAS.
- L. Lochmann and M. Janata, Cent. Eur. J. Chem., 2014, 12, 537–548 CAS.
- M. Schlosser, Organometallics in Synthesis: A Manual, John Wiley & Sons, Ltd, 2001, pp. 1–352 Search PubMed.
- A. A. Morton, E. E. Magat and R. L. Letsinger, J. Am. Chem. Soc., 1947, 69, 950–961 CrossRef CAS.
- A. A. Morton and M. E. T. Holden, J. Am. Chem. Soc., 1947, 69, 1675–1681 CrossRef CAS PubMed.
- P. Benrath, M. Kaiser, T. Limbach, M. Mondeshki and J. Klett, Angew. Chem., Int. Ed., 2016, 55, 10886–10889 CrossRef CAS PubMed.
- B. Jennewein, S. Kimpel, D. Thalheim and J. Klett, Chem. – Eur. J., 2018, 24, 7605–7609 CrossRef CAS PubMed.
- W. Clegg, R. E. Mulvey, R. Snaith, G. E. Toogood and K. Wade, J. Chem. Soc., Chem. Commun., 1986, 1740–1742 RSC.
- P. G. Williard and M. A. Nichols, J. Am. Chem. Soc., 1991, 113, 9671–9673 CrossRef CAS.
- D. R. Armstrong, A. R. Kennedy, R. E. Mulvey and S. D. Robertson, Chem. – Eur. J., 2011, 17, 8820–8831 CrossRef CAS PubMed.
- P. Fleming and D. F. O’Shea, J. Am. Chem. Soc., 2011, 133, 1698–1701 CrossRef CAS PubMed.
- G. W. Honeyman, D. R. Armstrong, W. Clegg, E. Hevia, A. R. Kennedy, R. McLellan, S. A. Orr, J. A. Parkinson, D. L. Ramsay, S. D. Robertson, S. Towie and R. E. Mulvey, Chem. Sci., 2020, 11, 6510–6520 RSC.
- D. Barr, W. Clegg, R. E. Mulvey and R. Snaith, J. Chem. Soc., Chem. Commun., 1989, 57–58 RSC.
- N. D. R. Barnett, R. E. Mulvey, W. Clegg and P. A. O’Neil, Polyhedron, 1992, 11, 2809–2812 CrossRef CAS.
- M. Westerhausen, S. Weinrich, G. Kramer and H. Piotrowski, Inorg. Chem., 2002, 41, 7072–7076 CrossRef CAS PubMed.
- R. Campbell, E. Crosbie, A. R. Kennedy, R. E. Mulvey, R. A. Naismith and S. D. Robertson, Aust. J. Chem., 2013, 66, 1189–1201 CrossRef CAS.
- D. R. Armstrong, E. V. Brouillet, A. R. Kennedy, J. A. Garden, M. Granitzka, R. E. Mulvey and J. J. Trivett, Dalton Trans., 2014, 43, 14409–14423 RSC.
- N. Jin, A. Logallo and E. Hevia, Organometallics, 2025, 44, 197–206 CrossRef CAS.
- R. E. Mulvey, K. Wade, D. R. Armstrong, G. T. Walker, R. Snaith, W. Clegg and D. Reed, Polyhedron, 1987, 6, 987–993 CrossRef CAS.
- As compared to all Li–P and K–P containing structures deposited with the Cambridge Structural Database (CSD).
- A. Falceto, E. Carmona and S. Alvarez, Organometallics, 2014, 33, 6660–6668 CrossRef CAS.
- A. G. Avent, D. Bonafoux, C. Eaborn, M. S. Hill, P. B. Hitchcock and J. D. Smith, J. Chem. Soc., Dalton Trans., 2000, 2183–2190 RSC.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- K. Issleib and E. Wenschuh, Chem. Ber., 1964, 97, 715–720 CrossRef CAS.
- V. H. Gessner, C. Däschlein and C. Strohmann, Chem. – Eur. J., 2009, 15, 3320–3334 CrossRef CAS PubMed.
- R. A. Bartlett, M. M. Olmstead and P. P. Power, Inorg. Chem., 1986, 25, 1243–1247 CrossRef CAS.
- G. Rabe, J. Riede and A. Schier, Acta Crystallogr. C, 1996, 52, 1350–1352 CrossRef.
- G. Stieglitz, B. Neumüller and K. Dehnicke, Z. Naturforsch., B, 1993, 48b, 156–160 CrossRef.
- K. Izod, P. Evans and P. G. Waddell, Dalton Trans., 2017, 46, 13824–13834 RSC.
- R. A. Bartlett, M. M. Olmstead, P. P. Power and G. A. Sigel, Inorg. Chem., 1987, 26, 1941–1946 CrossRef CAS.
- P. B. Hitchcock, M. F. Lappert, P. P. Power and S. J. Smith, J. Chem. Soc., Chem. Commun., 1984, 1669–1670 RSC.
- W. Clegg and R. W. Harrington, CSD Commun., 2021 DOI:10.5517/ccdc.csd.cc2736sc.
- K. Izod, P. Evans and P. G. Waddell, Inorg. Chem., 2020, 59, 863–874 CrossRef CAS PubMed.
- K. Izod, J. Stewart, E. R. Clark, W. Clegg and R. W. Harrington, Inorg. Chem., 2010, 49, 4698–4707 CrossRef CAS PubMed.
- R. A. Jones, A. L. Stuart and T. C. Wright, J. Am. Chem. Soc., 1983, 105, 7459–7460 CrossRef CAS.
- R. E. Mulvey, Chem. Soc. Rev., 1991, 20, 167–209 RSC.
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 186–197 Search PubMed.
- O. Kühl, J. Sieler and E. Hey-Hawkins, Z. Kristallogr. – Cryst. Mater., 1999, 214, 496–499 Search PubMed.
- K. Izod, W. McFarlane, B. V. Tyson, W. Clegg, R. W. Harrington and S. T. Liddle, Organometallics, 2003, 22, 3684–3690 Search PubMed.
- F. Dornhaus, M. Bolte, H. W. Lerner and M. Wagner, Eur. J. Inorg. Chem., 2006, 1777–1785 CrossRef CAS.
- M. Xu, A. R. Jupp and D. W. Stephan, Angew. Chem., Int. Ed., 2019, 58, 3548–3552 CrossRef CAS PubMed.
- G. W. Rabe, S. Kheradmandan, H. Heise, I. A. Guzei, L. M. Liable-Sands and A. L. Rheingold, Main Group Chem., 1998, 2, 221–228 CrossRef CAS.
- J. Bresien, Y. Pilopp, A. Schulz, L. S. Szych, A. Villinger and R. Wustrack, Inorg. Chem., 2020, 59, 13561–13571 CrossRef CAS PubMed.
- M. T. Whitelaw, S. Banerjee, A. R. Kennedy, A. van Teijlingen, T. Tuttle and R. E. Mulvey, Cell Rep. Phys. Sci., 2022, 3, 100942 CrossRef CAS.
- V. A. Pollard, A. Young, R. McLellan, A. R. Kennedy, T. Tuttle and R. E. Mulvey, Angew. Chem., Int. Ed., 2019, 58, 12291–12296 CrossRef CAS PubMed.
- V. Naseri, R. J. Less, R. E. Mulvey, M. McPartlin and D. S. Wright, Chem. Commun., 2010, 46, 5000–5002 RSC.
- S. Molitor, J. Becker and V. H. Gessner, J. Am. Chem. Soc., 2014, 136, 15517–15520 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2434958. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc02154e |
| This journal is © The Royal Society of Chemistry 2025 |