DOI:
10.1039/D4TC01801J
(Review Article)
J. Mater. Chem. C, 2024,
12, 13745-13761
Recent advances in small-molecule organic fluorescent semiconductors
Received
2nd May 2024
, Accepted 7th August 2024
First published on 8th August 2024
Abstract
Organic fluorescent semiconductors are highly desired for the advancement of integrated optoelectronic devices, such as organic light-emitting transistors (OLETs), electrically pumped organic lasers (EPOLs), and so on. Thanks to the joint efforts of chemists and materials scientists, rapid developments of small-molecule organic fluorescent semiconductors have been witnessed in recent years. The optoelectronic properties have been greatly improved and several small-molecule organic fluorescent semiconductors with excellent comprehensive performances (mobility > 1.0 cm2 V−1 s−1 and photoluminescence quantum yield efficiency (PLQY) > 20%) have emerged. The material database has also been greatly enriched in recent years including anthracene derivatives, fluorene derivatives, thiophene/phenylene co-oligomers (TPCOs), distyrylbenzene derivatives, etc. Therefore, it is timely and of great significance to summarize the recent advances in small-molecule organic fluorescent semiconductors to offer some hints for researchers in this field. In this review, we first highlight the aggregation structures, different types, recent research progress and design strategies of small-molecule organic fluorescent semiconductors. Then, we will briefly introduce the recent achievements in optoelectronic applications of small-molecule organic fluorescent semiconductors. Finally, we will give a conclusion and outlook about the challenges and opportunities for the future development of small-molecule organic fluorescent semiconductors.

Jie Li
| Jie Li is an associate professor in the Institute of Molecular Aggregation Sciences, Tianjin University. She received her Bachelor's degree from Beijing Institute of Technology (BIT) in 2013 and PhD degree from Institute of Chemistry, Chinese Academy of Sciences (ICCAS), in 2018 (Supervisor: Prof. Wenping Hu). Her current research interests mainly focus on organic optoelectronic materials (organic emissive semiconductors and organic photoresponsive materials) and related device applications. |
1. Introduction
Since A. J. Heeger, A. G. Macdiarmid and H. Shirakawa synthesized the conductive polymer polyacetylene in 1977, revealing the fact that “organic polymers can be electrically conductive after proper doping”,1 numerous researchers have devoted their efforts to the development of organic semiconducting materials2–4 and organic conductive materials,5 and the field of “organic electronics” has also emerged. After decades of development, various high-performance organic semiconductors have been reported, and the comprehensive properties of some organic electronic materials have even exceeded those of polycrystalline silicon.6,7 Compared with their inorganic counterparts, organic semiconductors exhibit unique advantages, including intrinsic flexibility, light weight, solution processing, and good biocompatibility,8–11 showing great potential in various applications, such as organic flexible displays,12 organic circuits,13,14 organic sensors,15 and organic batteries.16,17
Organic fluorescent semiconductors, i.e. multifunctional organic semiconductors with a combination of charge transport and solid-state fluorescence, are the material basis of organic optoelectronic integrated devices (OLETs, EPOLs, organic flexible displays, etc.), which have attracted intensive attention in recent years. Organic light-emitting transistors (OLETs) are a kind of organic optoelectronic device that integrates the self-emission characteristics of organic light-emitting diodes (OLEDs) and the switching function of organic field-effect transistor (OFETs).18,19 On the one hand, it is an important device element for the next generation of transformative flexible displays and new organic laser technology, which is of great significance for the simplification of active matrix displays and the realization of EPOLs.20,21 On the other hand, high-performance organic single crystal light-emitting transistors based on organic fluorescent semiconductors could become the model device for us to directly observe and study the charge transport and recombination process of organic semiconductor carriers.22 However, although the first OLET device was reported as early as 2003,23 the research and application of OLETs have progressed very slowly. The design and synthesis of such materials are facing great challenges because of the contradictory requirements: high mobility organic semiconductors usually require closely packed molecules with strong intermolecular interactions, while strong intermolecular interactions tend to quench solid-state emission. For example, rubrene, as a star organic semiconductor molecule, has a single crystal mobility higher than 15 cm2 V−1 s−1,24,25 but its crystal fluorescence quantum yield is less than 1%; pentacene, as a classical transport material, has very weak solid-state emission.26,27 Thanks to the cooperative efforts devoted by chemists, materials scientists and theoretical scientists, impressive achievements in small-molecule organic fluorescent semiconductors have been made in recent years. The comprehensive optoelectronic properties have improved and several small-molecule organic fluorescent semiconductors with excellent comprehensive performances (mobility > 1.0 cm2 V−1 s−1 and PLQY > 20%) have emerged. The material database was also greatly enriched in recent years, with high-performance molecular systems being comprehensively studied, such as anthracene derivatives, fluorene derivatives, TPCOs, distyrylbenzene derivatives, etc. Moreover, the structure–property relationship is also deeply investigated and efficient molecular design strategy is proposed theoretically. Several excellent reviews have also been reported, including those outlining the key points of organic semiconductors for electronics and photonics or organic light-emitting transistors, and discussing the design strategies of organic emissive semiconductors.28–31 In this review, we will summarize the recent advances in small-molecule organic fluorescent semiconductors: (i) comprehensively review the aggregation structures, different materials types, recent research progress and design strategies of small-molecule organic fluorescent semiconductors; (ii) briefly summarize the recent achievements in optoelectronic applications based on small-molecule organic fluorescent semiconductors; and (iii) give a conclusion and outlook about the challenges and opportunities for the development of small-molecule organic fluorescent semiconductors (Scheme 1).
 |
| | Scheme 1 Schematic illustration of recent advances in small-molecule organic fluorescent semiconductors. | |
2. Aggregation structures
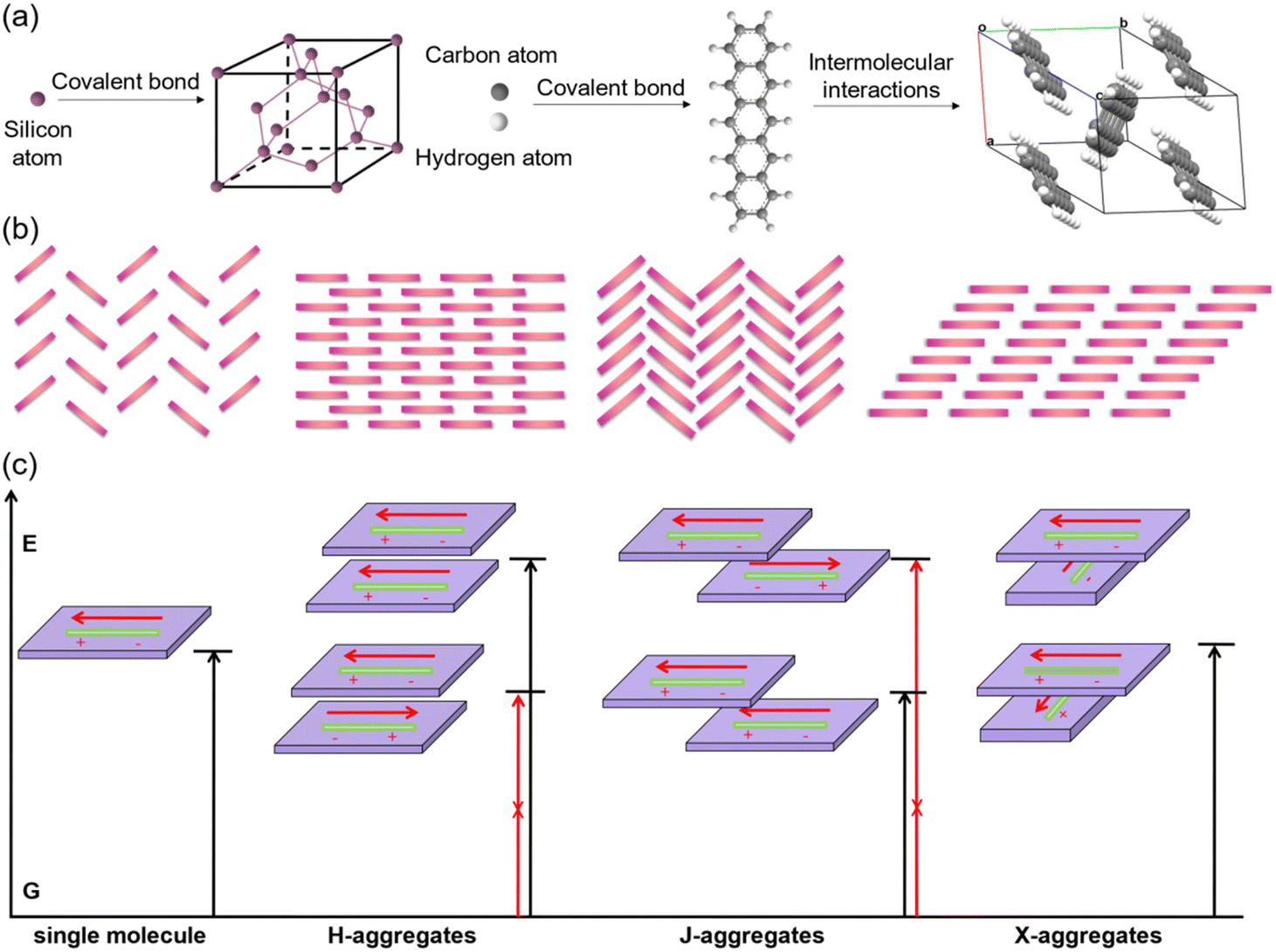
In comparison with inorganic semiconductors that are formed by atoms directly linked with covalent bonds, organic semiconductors composed of organic molecules are aggregated by weak intermolecular interactions (Fig. 1a). The energy of weak intermolecular interactions is usually small, so the interaction type, intensity, and orientation can be adjusted by molecular design, crystal engineering, etc., and thus organic semiconductors demonstrate specific molecular stacking together with designed electrical or optical properties.32 Generally, there are four typical organic semiconductor stacking motifs (Fig. 1b): (i) classical herringbone stacking, which mainly depends on the C–H⋯π interactions between the edges and faces of the aromatic ring, where there is no π⋯π overlap between adjacent molecules; (ii) coplanar herringbone stacking, which mainly depends on the face-to-face interactions between adjacent molecules; (iii) one-dimensional (1D) sliding stacking, in which molecules are mainly stacked along the π⋯π overlap direction; and (iv) two-dimensional layered stacking, where strong intermolecular interactions may produce two-dimensional (2D) network charge transport characteristics.33 Theoretically, the two key parameters of transfer integral and reorganization energy determine the electrical properties of organic semiconductors.34 Tight molecular stacking caused by strong intermolecular interactions, appropriate electronic structure and maximum orbital overlap are all very important to improve the carrier transport in high-performance organic semiconductors through rational molecular design. Two-dimensional layered stacking is believed to be the best molecular stacking motif to achieve high mobility organic semiconductors, as in the case of TIPS-PEN.27 For organic fluorescent semiconductors, the optical properties are also closely related to their aggregation structures,35,36 which is generally discussed according to the molecular exciton model, or in other words, based on the dipole–dipole interaction model. In theoretical research, the aggregates are usually simplified as dimers to discuss the effect of their transition dipoles on emission features (Fig. 1c). For dimers, the interaction between the excited states will generate two splitted energy level excited states and there are three representative cases that need to be discussed: (i) for H-aggregates, the dimers are face-to-face paralleled, and the lower excited state corresponds to the reversely parallel arrangement of the dipole or the wave functions, resulting in the optical forbidden transition between the lower excited state and the ground state. In this case, the total transition dipole reaches the maximum, and the radiation transition oscillator strength mainly concentrates on the higher excited state. The absorption of H-aggregates is hypsochromically shifted compared with that of the single molecule. According to Kasha's rule, the excited molecules need to relax to the lowest vibrational energy level of the excited state before transition to the ground state, so the H-aggregation is not conducive to emission. It is the universal law that the strong electronic coupling of H-aggregation will produce the largetransfer integral and is favorable for charge transport which will quench emission, except for the H-aggregation cases of DBTVB37 and Hex-4-TFPTA.38 (ii) For J-aggregates, the two molecules of the dimer slip along the molecular long axis, the radiation oscillator strength mainly concentrates on the lower excited state and the optical transition is allowed, which results in the red shift of the absorption compared with the single molecule. There is certain displacement along the molecular long axis and thus interactions between dipoles are weakened, and the lowest energy level transition is optically allowed. The intermolecular interactions, though weakened to some extent due to the molecular displacement, together with optically allowed transition make J-aggregation an excellent aggregation mode to achieve solid-state emission and charge transport simultaneously, as exemplified in cases such as 2,6-DPA,39 2,6-DNA,40 and NBTA.41 (iii) For X-aggregates (or cross-dipole stacking), one of the molecules rotates along its center and the adjacent molecules stack parallel with a certain cross angle. The interaction between the dimers decreases as the crossing angle increases, and the energy level splitting degree of the excited state also decreases. When the two molecules are vertically stacked, the energy level splitting disappears. The total transition dipoles correspond to the two excited states of cross-stacked dimers and the transitions are optically allowed, which favors emission.42–44 The chemical structures of X-aggregates generally feature a π-conjugated central core and large sterically hindered peripheral substituents. Due to the special requirements of the molecular structure, organic fluorescent semiconductors with an X-aggregation mode are difficult to be designed and quite rare, except for the cases of TES-DPA,44 BDPVA,45 and PQ-4ClP.46 As a representative case of X-aggregates, two types of C–H⋯π interactions (I and II) with distinctive distance and orientation drive the adjacent BDPVA molecules in the same molecular column to pack rotationally along the b axis to avoid the steric effect, i.e., to arrange in a cross-dipole stacking manner. Meanwhile, the adjacent molecular columns are connected by another class of C–H⋯π interactions (III) between the peripheral groups to form the molecular stacking network.
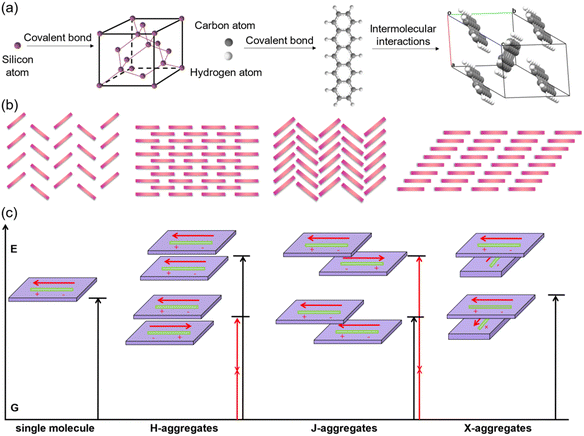 |
| | Fig. 1 (a) Schematic illustration of the formation of a single crystal unit cell of representative inorganic semiconductors (silicon) and organic semiconductors (pentacene). (b) The four classical packing motifs for organic semiconductors. (c) The energy splitting diagrams of organic aggregates. Reproduced with permission.35 Copyright 2021, John Wiley and Sons. | |
In addition to the dipole–dipole interaction model, quantum chemists have also conducted investigations to gain insights into the structure–property relationship of organic fluorescent semiconductors.47,48 Liao et al. proposed the excitonic effective mass (Me*) and charge effective mass (Mc*) to characterize the degree of luminescence quenching and the ability of carrier transport and found that the C–H⋯π interactions can induce heavy Me* and light Mc* to balance emission efficiency and carrier mobility. Shuai et al. proposed the four-state model which consists of two local Frenkel excitons and two intermolecular charge transfer (ICT) excitons aiming to clarify the relationship between mobility and luminescence in π-conjugated molecules, rendered a universal descriptor and discussed the relationship between the descriptor and the different packing structures.
3. Materials and properties
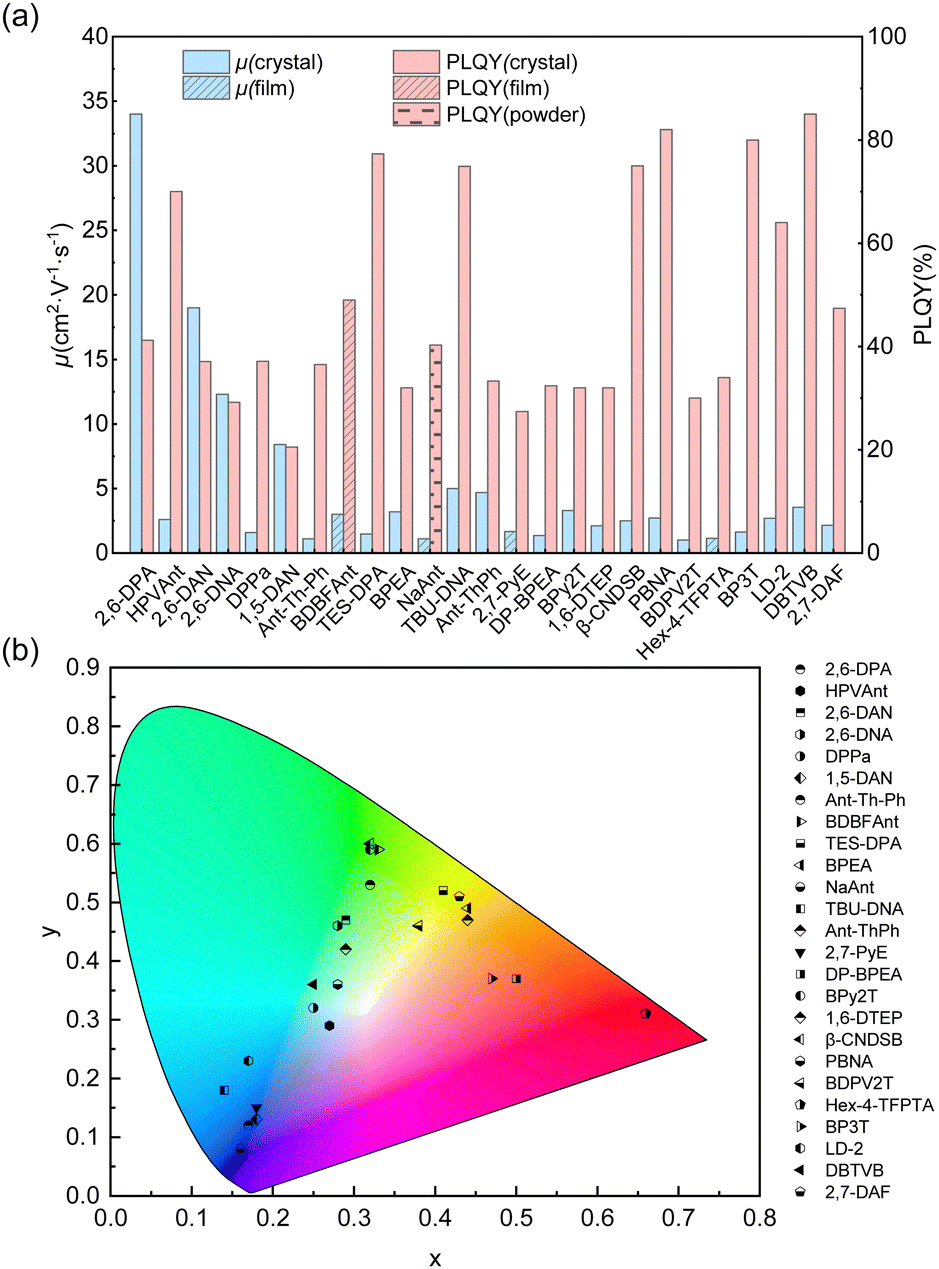
It is challenging to design molecules combining high carrier mobility with efficient solid-state emission because compact packing with strong and plentiful intermolecular interactions usually not only gives rise to excellent charge transport properties but also quenches the solid-state emission. Thanks to the joint efforts of chemists and materials scientists, great achievements in small-molecule organic fluorescent semiconductors have been made in recent years. The optoelectronic properties of organic fluorescent semiconductors have progressed and dozens of small-molecule organic fluorescent semiconductors with excellent comprehensive performances (μ > 1.0 cm2 V−1 s−1, PLQY > 20%, Fig. 2a) and diverse emission colours (Fig. 2b) emerged. The material database was also greatly enriched in recent years and excellent molecular systems, such as anthracene derivatives, fluorene derivatives, TPCOs, distyrylbenzene derivatives, have emerged, which will be discussed below.
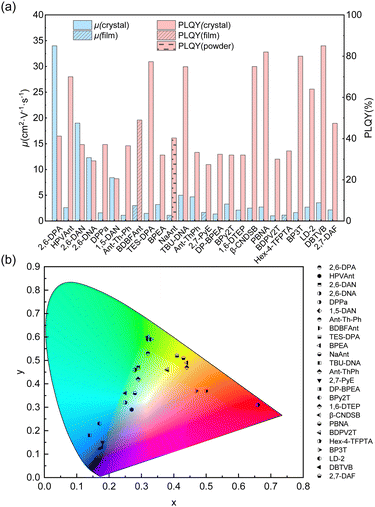 |
| | Fig. 2 Summary of the charge carrier mobility (μ), PLQY (a) and emission colours (b) of high performance (μmax > 1 cm2 V−1 s−1, PLQY > 20%) small-molecule organic fluorescent semiconductors. | |
3.1. Anthracene derivatives
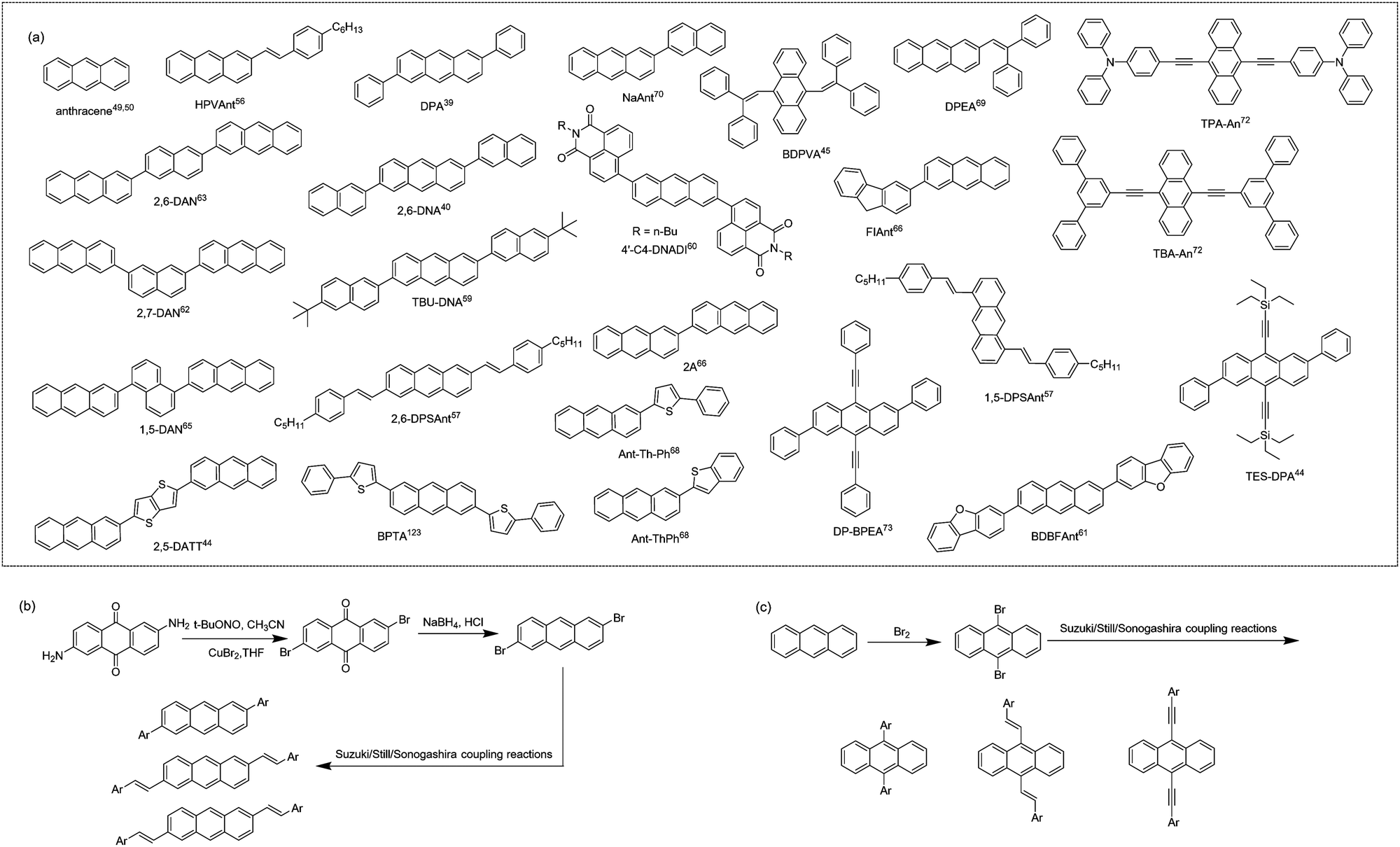
Among various functional organic molecules, anthracene, with three benzene rings linearly fused, is an organic molecule with excellent solid-state emission properties. The PLQYs of its single crystal, solution and powder are 64%, 28% and 18%, respectively.49,50 In the pursuit of high-performance anthracene-derived optoelectronic materials, the derivation of anthracene mainly focuses on 2,6-positions and/or 9,10-positions (Fig. 3a). As shown in Fig. 3b and c, the general synthetic routes for anthracene derivatives mainly include bromination and subsequent coupling reactions (Suzuki, Stille, Sonogashira coupling, etc.). For a long time, the optical and electrical properties of anthracene and its derivatives have been separately studied. In the design of anthracene-derived organic luminescent materials, research work mainly focuses on the introduction of steric groups to peri/end sites of anthracene to attenuate the intermolecular interactions to reduce exciton quenching and further to promote emission.51 For anthracene-based organic field-effect semiconductors, considering the moderate conjugated area of anthracene, researchers generally introduce conjugated groups at 2,6-positions and/or 9,10-positions of anthracene to increase the effective conjugated area and the packing density of the molecule, thereby increasing its charge transport properties.49,50,52,53 For example, Meng et al. synthesized DPVAnt by introducing the styryl group at the 2,6-positions of anthracene and the thin film and single crystal mobilities of which were 1.3 and 4.2 cm2 V−1 s−1, respectively. BPEA was designed by introducing the phenylethynyl group at 9,10-positions, and its α-phase single crystal mobility reached 0.64 cm2 V−1 s−1,54 which was higher than the mobility of the anthracene single crystal itself of 0.02 cm2 V−1 s−1.55 In 2012, Perepichka et al. designed and synthesized 2-(4-hexylstyryl) anthracene (HPVAnt),56 exhibiting PLQYs of 70% and 55% for the crystalline and solution states, respectively. Compared with other high-mobility acene-based organic semiconductors, such as tetracene, rubrene and pentacene, the better solid-state emission of HPVAnt is due to the fact that its S1 state energy (S1 = 2.94–3.15 eV) is less than twice the T1 state (T1 = 1.77 eV), thus resulting in the cessation of the singlet-fission process, which tends to cause solid-state fluorescence quenching. Due to the compact herringbone stacking feature of HPVAnt molecules, in which the anthracene cores of adjacent molecules are arranged perpendicularly to the long axis, the mobility of the thin film and single crystal field-effect transistors based on HPVAnt reached 1.5 cm2 V−1 s−1and 2.6 cm2 V−1 s−1, respectively. Subsequently, three organic molecules, 2,6-DPSAnt, 1,5-DPSAnt and 9,10-DPSAnt, were designed by introducing the 4-pentylstyryl group at the 2,6-positions, 1,5-positions and 9,10-positions of anthracene, and substitution site effects of anthracene derivatives on the fluorescence properties and charge transport properties were also studied.57 The single crystal structure reveals that there is certain twisted angle between the styrene group and the anthracene core: 2,6-DPSAnt (15°) < 1,5-DPSAnt (30°) < 9,10-DPSAnt (60°). The smaller twisted angle makes 2,6-DPSAnt still maintain herringbone stacking and relatively stronger intermolecular interactions, while 1,5-DPSAnt and 9,10-DPSAnt exhibit one-dimensional stacking. The larger slip leads to no π–π overlap between anthracene cores. This stacking motif leads to the result that 2,6-DSPAnt demonstrates the best thin film charge transport performance with a μmax of 0.75 cm2 V−1 s−1, while 9,10-DPSAnt has no charge transport ability due to weak intermolecular interactions and poor film formation. In terms of luminous efficiency, the order is 1,5-DPSAnt (4–9%) < 2,6-DPSAnt (14%) < 9,10-DPSAnt (18–22%). The results indicate that it is easier to achieve a balance between high mobility and strong solid-state emission by conjugation extension at the 2,6-positions of anthracene. Enlightened by these results, by introducing a phenyl group at the 2,6-positions of anthracene with the carbon–carbon single bond, Hu et al. reported a high mobility organic fluorescent semiconductor, 2,6-diphenylanthracene (2,6-DPA).39,58 There is a slight twisted angle (∼20°) between the benzene ring and the anthracene core of 2,6-DPA, and the molecules adopt a herringbone stacking mode. The large redshift of the absorption spectrum in the aggregated state in contrast with the solution state indicates its J-aggregation mode. According to the dipole–dipole interaction model, such kind of aggregation mode can effectively reduce the quenching of fluorescence, which further leads to the higher luminescence quantum yield of 2,6-DPA (single crystal, PLQY = 41.2%). The tight herringbone stacking and the multiple C–H⋯π interactions between adjacent 2,6-DPA molecules are responsible for the record high single crystal mobility of 34 cm2 V−1 s−1 along the b axis and 23 cm2 V−1 s−1 along the c axis were achieved. Later, 2,6-DNA was developed by further aromatic extension at the 2,6-positions of anthracene, and also displayed classical herringbone packing and J-aggregation modes, with a μmax (single crystal) of up to 12.3 cm2 V−1 s−1 and a PLQY of 29.2% for single crystals.40 By introducing tert-butyl to the terminal site of 2,6-DNA, the high performance organic fluorescent semiconductor, TBU-DNA, was also acquired.59 The suitable size of the terminal substituted group triggers the unique “slipped herringbone packing” of TBU-DNA, which further leads to the integrated high single crystal charge transport mobility (μmax = 5.0 cm2 V−1 s−1) and enhanced solid-state emission (single crystals, PLQY = 74.9%). By introducing naphthalene monoimide (NMI) acceptors at 2,6-positions of anthracene, 4′-C4-DNADI was recently reported, manifesting the electron transport characteristics (single crystal device, μ = 0.02 cm2 V−1 s−1) together with hot-exciton induced delayed fluorescence (PLQY = 26%).60 Meng et al. also synthesized 2,6-bis(dibenzo[b,d]furan-3-yl)anthracene (BDBFAnt) by introducing dibenzo[b,d]furan-3-yl at the 2,6-positions of anthracene, exhibiting a mobility of 3.0 cm2 V−1 s−1 and a PLQY of 49% for its film.61 Anthracene derivatives with the 2-(anthracen-2-yl) group as the substituent were also reported, among which the naphthalene-cored anthracene derivative, 2,6-DAN, exhibited the highest single crystal mobility of 19 cm2 V−1 s−1 and fluorescence emission efficiency of 37.09% (crystal powder).62–65 By introducing the conjugated unit at the 2-position of anthracene, asymmetric anthracene derivatives are also investigated.66–70 Meng et al. designed and synthesized two similar molecules, 2-fluorenyl-2-anthracene (FlAnt) and 2-anthryl-2-anthracence (2A), both of which show similar classical herringbone packing mode.66 The thin film mobility of FlAnt and 2A reaches 0.22 cm2 V−1 s−1 and 3.19 cm2 V−1 s−1, respectively, together with thin film emission efficiencies of 15.7% and 13.9%. To study the impact of different conjugated ways on the optoelectronic properties, 2-(anthracen-2-yl)-5-phenylthiophene (Ant-Th-Ph) and 2-(anthracen-2-yl)benzo[b]thiophene and (Ant-ThPh) were designed. High single crystal mobilities of 1.1 cm2 V−1 s−1 and 4.7 cm2 V−1 s−1 were obtained together with strong emission efficiencies of 36.52% and 33.32% for single crystals.68 In addition to modification at the 2,6-positions of anthracene, the introduction of specific functional groups at the 9,10-positions of anthracene was also studied. Tian et al. designed and synthesized the butterfly-shaped molecule, 9,10-bis(2,2-diphenylvinyl)anthracene (BDPVA), the solid-state emission efficiency of which reaches 60% for single crystals.45 OLETs based on the BDPVA single crystal showed ambipolar transport mobilities of μh = 0.08 cm2 V−1 s−1 and μe = 0.06 cm2 V−1 s−1, respectively.71 9,10-Bis([N,N-diphenyl]-4′-phenylethynyl)anthracene (TPA-An) and 9,10-bis([1′,3′-diphenyl]-5′-phenylethynyl)anthracene (TBA-An) were further synthesized,72 exhibiting remarkably high emission efficiencies of 98% and 99% at room temperature for single crystals, which are the highest PLQY values reported for small-molecule organic fluorescent semiconductors to date. Considerably high hole mobilities of 0.45 and 0.15 cm2 V−1 s−1 were also respectively achieved for TPA-An and TBA-An-based transistors for single crystal transistors. Deriving at 2,6- and 9,10-positions of anthracene simultaneously have been extensively studied by researchers as well. (2,6-Diphenylanthracene-9,10-diyl)bis(ethyne-2,1-diyl)bis(triethylsilane) (TES-DPA) was also designed by incorporating an enlarged π-conjugation group along the long axis and bulky substitution at the short axis, demonstrating X-aggregation in the solid-state with a PLQY of up to 77.3% and a charge transport single crystal mobility of 1.47 cm2 V−1 s−1.44 2,6-Diphenyl-9,10-diphenylalkynylanthracene (DP-BPEA) was developed by introducing phenyl groups at the 2,6-positions of anthracene and benzyne groups at the 9,10-positions of anthracene.73 The molecules adopt a coplanar herringbone packing mode with an effective conjugated area of about one benzene between the parallel molecules, showing a maximum single crystal mobility of 1.37 cm2 V−1 s−1 and a fluorescence quantum yield of 32% for single crystals. Generally, derivation at the 2,6-positions of anthracene avoid the steric hindrance effect between the substituent and the central anthracene core, maintaining a good planarity of the molecule and effectively extend the π-conjugation to the greatest extent. In the case of derivation at the 9,10-positions of anthracene, the introduction of aromatic groups at the 9,10-positions of anthracene with a “carbon–carbon double/triple bond bridge” is a feasible way for π-conjugation expansion, while introducing aromatic groups at the 9,10-positions through the carbon–carbon single bond will produce large steric hindrance between the substituent group and the anthracene core, which leads to the large torsion angle between the substituent and the anthracene core, and is disadvantageous for the π-conjugation extension and charge transport.
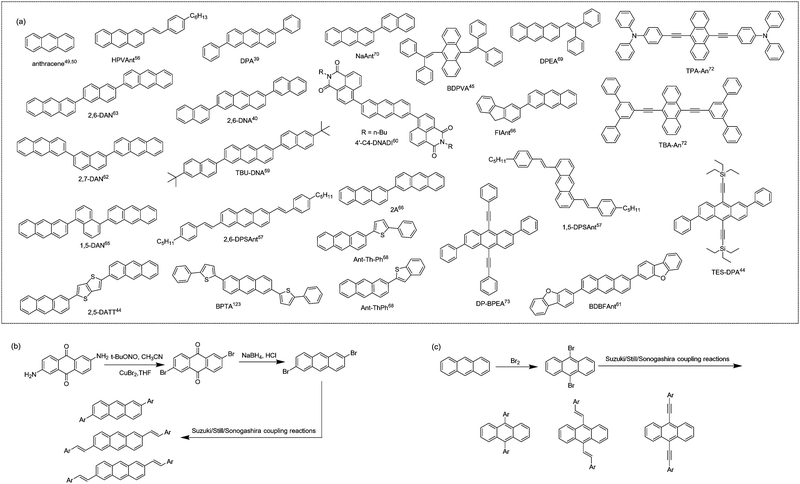 |
| | Fig. 3 (a) Chemical structures of anthracene and representative anthracene-derived organic fluorescent semiconductors. (b) General synthetic route to 2,6-substituted anthracene derivatives. (c) General synthetic route to 9,10- substituted anthracene derivatives. | |
3.2. Fluorene-derivatives
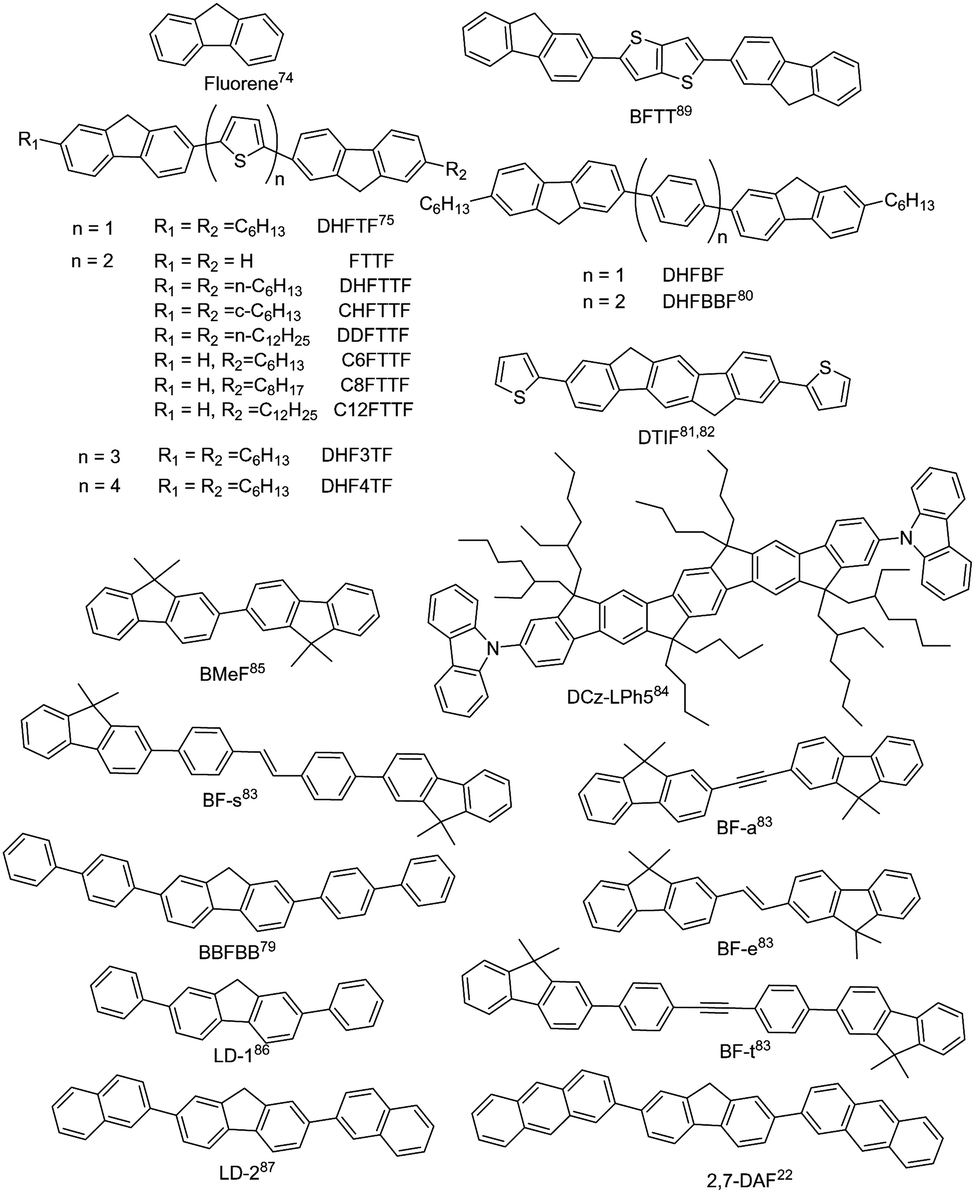
Fluorene is an excellent building block for the design of organic luminescent materials, especially for wide bandgap organic semiconductors.74 Research on fluorene-derived organic semiconductors can be dated back to long time ago. Representative small-molecule fluorene-derived organic fluorescent semiconductors are demonstrated in Fig. 4. In 2001, Bao et al. reported the electrical properties of FTTF and DH-FTTF by linking bithiophene with fluorene, showing the highest thin film mobility of 0.11 cm2 V−1 s−1 for DH-FTTF.75 Later, they developed a series of terminal alkyl substituted FTTFs, the thin film hole mobilities of which are in the range of 0.012–0.184 cm2 V−1 s−1.76–79 A series of fluorene-phenylene oligomers were also reported, showing high ionization potentials and excellent chemical stability, among which DHFBBF demonstrated the highest mobility up to 0.32 cm2 V−1 s−1 for thin film OFETs.80 Wang et al. conducted a systematic study on the synthesis and properties of 2,6-substituted indenofluorene derivatives, among which the thiophene substituted indenofluorene (DTIF) demonstrated a thin film mobility of 0.012 cm2 V−1 s−1 and a quantum efficiency of 24.5% for the film deposited at room temperature.81,82 Kazlauskas et al. designed several new bifluorene compounds (BF-a, BF-t, BF-e, and BF-s), showing a low nonradiative decay rate, high PLQY (72–82%) and short lifetime for single crystals.83 Huang et al. also reported a carbazole-end-capped ladder-type oligo-(p-phenylene) based hybrid oligomer, DCz-LPh5. The hole mobility of the DCz-LPh5 thin film was 0.001 cm2 V−1 s−1,84 determined by the space-charge limited current (SCLC) method. A PLQY value of 25% was obtained for the DCz-LPh5 film. Later in 2021, they reported a wide bandgap organic fluorescent semiconductor, 2,2′-bi(9,9′-dimethylfluorene) (BMeF),85 exhibiting crystallization-enhanced emission behavior with a high PLQY of up to 75% and a considerable charge transport mobility of 0.18 cm2 V−1 s−1 for the nanocrystal. Low threshold dual-color lasing behavior from ultraviolet (392 nm) to deep-blue (415 nm) was also demonstrated for BMeF. Dong et al. reported a series of fluorene-derived organic fluorescent semiconductors by introducing the aryl group at the 2,7-positions of fluorene.22,86,87 2,7-diphenyl-9H-fluorene (LD-1) demonstrated integrated optoelectronic properties with a relatively high mobility of 0.25 cm2 V−1 s−1 and an excellent PLQY of 60.3% for single crystals.86 2,7-Di(2-naphthyl)-9H-fluorene (LD-2) was also designed by replacing the benzene unit with a naphthalene unit at the 2,7-positions of the fluorene core, showing a higher single crystal mobility of 2.7 cm2 V−1 s−1 and a PLQY of 64%, which might be attributed to the large intermolecular transfer integrals together with a fast radiative transition rate.87 2,7-Di(2-anthryl)-9H-fluorene (2,7-DAF) was also reported, exhibiting a high single crystal carrier mobility of 2.16 cm2 V−1 s−1 and strong excimer emission with a PLQY of 47.4% for single crystals.22 Overall, in seeking for high performance organic fluorescent semiconductors, the derivation on fluorene mainly focuses on the aromatic extension at the 2,7-positions to enhance the effective conjugation area for efficient charge transport; however, the reaction activity on the 9-position should not be neglected from the perspective of material stability.88–90 For example, compared with biphenyl substituted thieno[3,2-b]thiophene (BPTT), bis(9H-fluorene-2-yl) substituted thieno[3,2-b]thiophene (BFTT) showed poor UV stability due to the keto-defect formation in fluorene units via photooxidative reactions.89
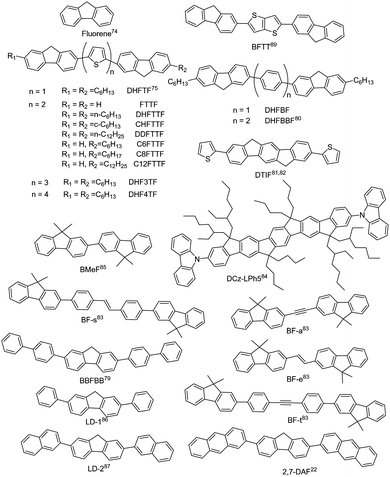 |
| | Fig. 4 Chemical structures of fluorene and representative fluorene-derived organic fluorescent semiconductors. | |
3.3. TPCOs
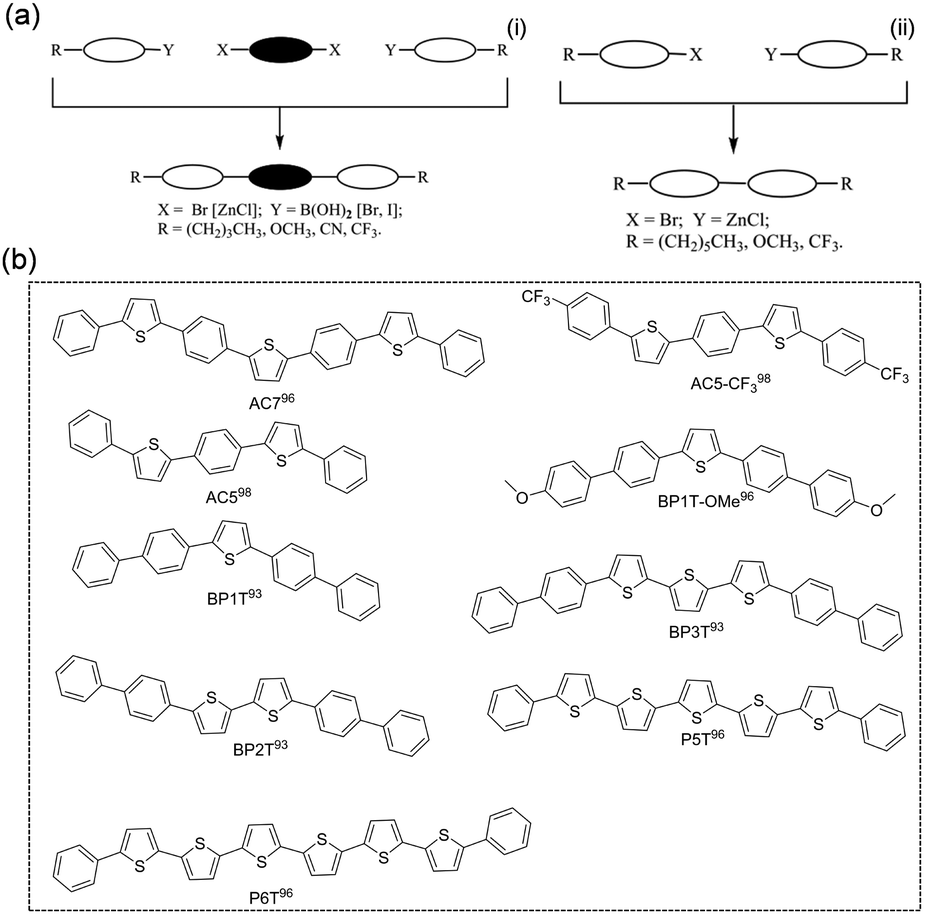
Thiophene/phenylene co-oligomers (TPCOs) are a class of organic molecules for which several numbers of thiophene rings and benzene rings are linked by a carbon–carbon single bond. TPCOs can be facilely synthesized by organosynthetic routes such as Suzuki coupling or Grignard coupling (Fig. 5a). The molecular shapes (bent, straight, and zig-zag) and the optoelectronic features (emission color, efficiency, mobility, carrier polarity, etc.) of TPCOs depend on the arrangement and numbers of thiophene rings and benzene rings or chemical modification at the molecular terminals.91,92 The fluorescence emission of the TPCO crystal covers the entire visible wavelength range from blue to red. The reported TPCOs are shown in Fig. 5b. TPCOs, including AC5, AC5-CF3, AC7, BP1T, BP2T, BP1T-OMe, BP3T, P5T, and P6T, all exhibit emissive characteristics and typical p-type charge transport characteristic.93–99 As the superstar among TPCOs, BP3T is a kind of highly luminescent material. As early as 2006, Ichikawa et al. obtained high quality BP3T crystals by an epitaxial growth method on the KCl (001) surface and found that the crystals showed a herringbone stacking mode. The PLQY obtained for BP3T at 300 K was as high as 80% and the crystals also demonstrated a self-resonant cavity laser effect.100,101 Ichikawa et al. first reported a BP3T unipolar monocrystalline transistor with a charge mobility of 0.29 cm2 V−1 s−1 with a symmetric gold electrode.102 By using asymmetric electrodes, obvious bipolar transport was obtained, and the hole and electron single crystal mobilities reached 1.64 cm2 V−1 s−1 and 0.17 cm2 V−1 s−1, respectively.103
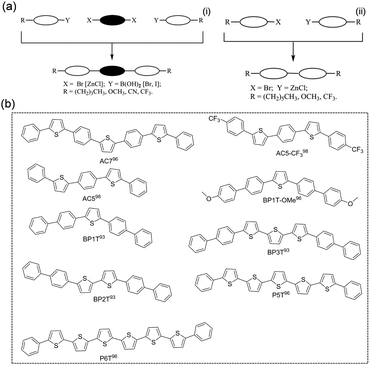 |
| | Fig. 5 (a) General synthetic pathways to terminal substituted TPCOs (left: 3-block coupling; right: 2-block coupling). Reproduced with permission.92 Copyright 2009, John Wiley and Sons. (b) Chemical structures of TPCOs-based organic fluorescent semiconductors. | |
3.4. Distyrylbenzene-derivatives
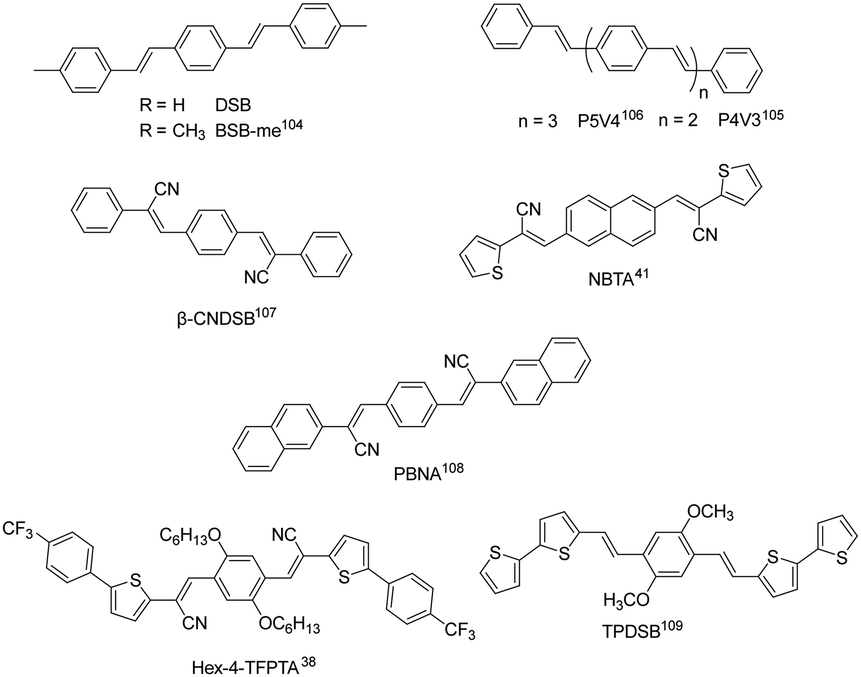
Distyrylbenzene derivatives are another excellent molecular platform to achieve high performance organic fluorescent semiconductors (Fig. 6).38,104–109 Park et al. discovered the aggregation-induced fluorescence enhancement behavior of 1-cyano-trans-1,2-bis-(4′-methylbiphenyl)-ethylene (CN-MBE) and regulated the assembly morphology, luminescence color, and amplified self-emission of such kinds of materials by modulating the molecular design and assembly conditions.110–113 Yasuda et al. reported the charge transport properties of 1,4-bis(4-methylstyryl)benzene (4MSB) and 1,4-bis(2-methylstyryl)benzene (2MSB).114 A high hole mobility of 0.13 cm2 V−1 s−1 was achieved for the 4MSB film due to the densely packed submicronsized grains with a high degree of molecular order. Adachi et al. reported BSB-Me with high PLQYs of 89% and 54%, respectively, for single crystals and thin films.104 Ma et al. reported 1,4-bis(2-cyano-2-phenylethenyl)benzene (β-CNDSB), the strong π–π interaction and intermolecular hydrogen bonding of which induce the formation of herringbone stacked two-dimensional lamellar crystals.107 β-CNDSB exhibits aggregation induced emission (AIE) behavior with the PLQY of β-CNDSB crystals reaching 70%. The ultrahigh solid-state emission might be ascribed to the planarity of the molecule and the J-aggregated mode. A cyano-substituted styrene derivative, (2Z,2′Z)-3,3′-(naphthalene-2,6-diyl)bis(2-(thiophen-3-yl)-acrylonitrile) (NBTA), via the Knoevenagel reaction was also reported.41 Three kinds of non-covalent interactions, namely, π⋯π (3.369 Å), hydrogen bond C–H⋯N (2.611 Å), and S⋯π (2.994 Å) interactions, existed in the single crystal structure. The molecules stacked in a coplanar structure with a slipped angle of 45°, showing a typical J-aggregation mode. A considerable PLQY of 37% was obtained for NBTA crystals. Hole and electron mobilities of 0.05 and 0.40 cm2 V−1 s−1 were achieved for NBTA crystal-based OLET devices. Later, they reported (2Z,2′Z)-3,3′-(1,4-phenylene)bis(2-(naphthalen-2-yl)acrylonitrile) (PBNA),108 for which cyano groups form multiple hydrogen bonds perpendicular to a linear-shape aromatic skeleton, where the energy of hydrogen bonds and π–π interactions were obviously stronger than the van der Waals forces along other directions. The stereohindrance effect originating from the naphthalene core prevents the dipole–dipole interaction, accompanied by the reduction of the nonradiative rate due to strong intermolecular interactions, which is in favor of a high emission efficiency up to 82% in crystals. The hole and electron mobilities of PBNA-based single crystal OLETs achieved 0.18 and 2.71 cm2 V−1 s−1, respectively. Park et al. developed a π-extended dicyanodistyrylbenzene molecule, Hex-4-TFPTA, the appropriate molecular stacking and highly allowed S1 → S0 transition bring about a good balance between electron transport mobility (thin film, μe = 1.0 cm2 V−1 s−1) and deep red-emissive characteristics with a thin film PLQY of 28%.38 By combining the high optical-gain of the oligomeric phenylene vinylene core–moiety with aromatic bithiophene end-groups, Fu et al. reported 1,4-dimethoxy-2,5-di[bithiophenestyryl]benzene (TPDSB). TPDSB molecules in one-dimensional microwires (1D-MWs) stack into roll π-stacks, while in two-dimensional microdisks (2D-MDs) they exhibit slip π-stacks. The two polymorphs exhibited distinct emission laser gain (1D-MWs: yellow-emissive Fabry–Perot (FP); 2D-MDs: red-emissive whispering mode (WGM) microlasers) and charge transport characteristics (1D-MWs: 1.45 × 10−4 cm2 V−1 s−1 and 2D-MDs: 1.20 × 10−2 cm2 V−1 s−1).109
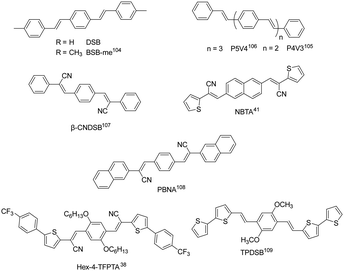 |
| | Fig. 6 Chemical structures of representative distyrylbenzene-derived organic fluorescent semiconductors. | |
3.5. Others
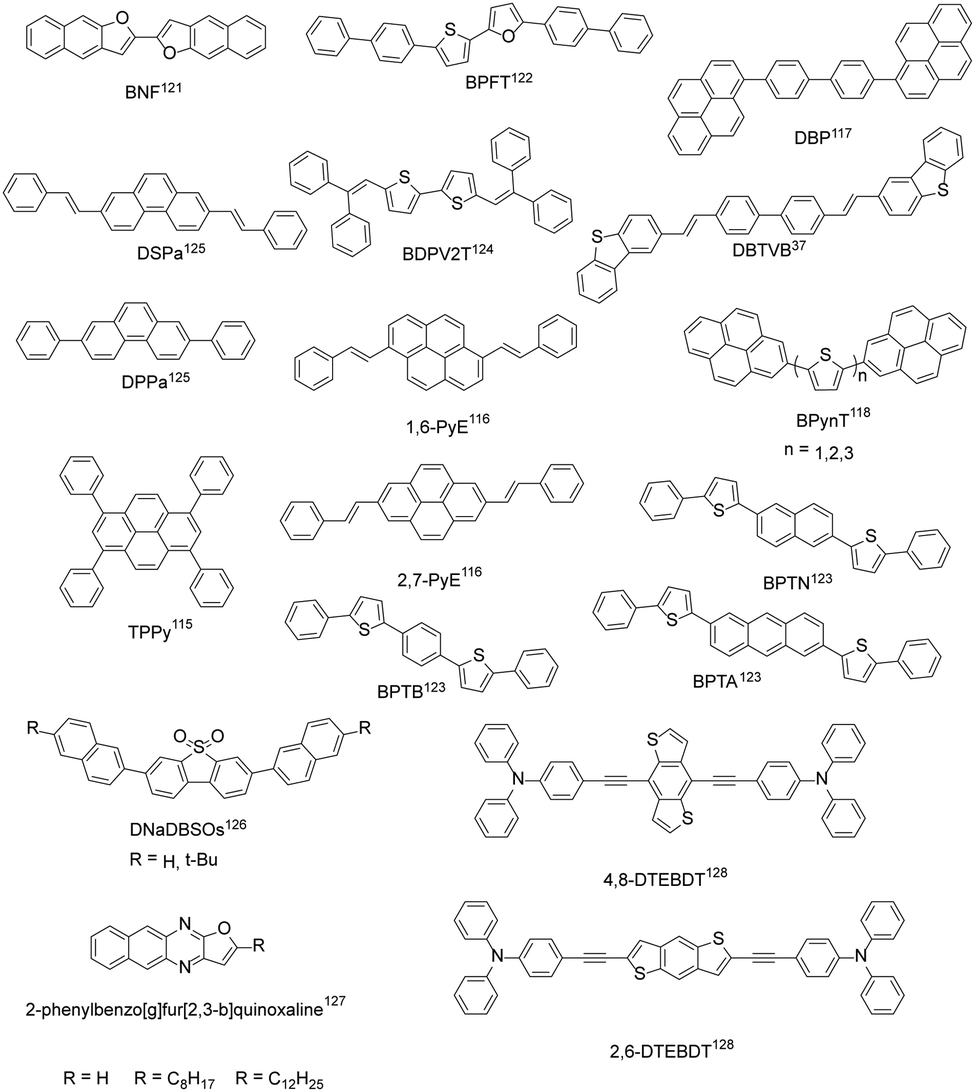
In addition to the above four common types of organic fluorescent semiconductors, there are also other kinds of molecular structures that achieve both efficient charge transport and solid-state emission (Fig. 7). Bisri et al. synthesized TPPy with a PLQY of up to 68% for single crystals and TPPy-based single crystal light-emitting devices with asymmetric electrodes show a hole mobility as high as 0.34 cm2 V−1 s−1 and an electron mobility of 7.7 × 10−2 cm2 V−1 s−1.115 Zhang et al. reported two pyrene derivatives (1,6-PyE and 2,7-PyE) by incorporating β-styrene units into the pyrene core to modulate the intermolecular interactions. The maximum hole mobility of 2,7-PyE films reaches 1.66 cm2 V−1 s−1, which is higher than that of 1,6-PyE, with PLQYs of 28.8% and 27.4%, respectively, for 1,6-PyE and 2,7-PyE crystals.116 Hong et al. synthesized the thermally stable compound DBP with a thin film mobility of 0.21 cm2 V−1 s−1 and pure blue emission with main peak at 470 nm.117 Jin et al. reported 2-positional pyrene end-capped oligothiophene cooligomers, BPynT (n = 1, 2, 3), among which BPy2T demonstrated a highest single crystal field-effect mobility of 3.3 cm2 V−1 s−1 and a PLQY value of 32% with green emission in the crystalline state.118 Due to the small atomic radius and larger electronegativity of oxygen than sulfur, increased intermolecular molecular orbital overlap and superior charge transport are expected for furan-based organic semiconductors.119 Materials containing furan groups also show great potential as high performance organic fluorescent semiconductors, especially for their unique optoelectronic features: a wide energy gap originated from the weaker aromaticity of furan rings than thiophene rings, an intense photoluminescence characteristic, and mechanofluorochromism.119,120 Takimiya et al. designed BNF with μh = 0.1 cm2 V−1 s−1 and μe = 4 × 10−2 cm2 V−1 s−1 for single crystal light-emitting transistors, PLQY = 72% (single crystals).121 BPFT showed bipolar characteristics with a hole mobility of up to 0.27 cm2 V−1 s−1 and an electron mobility of 1.3 × 10−3 cm2 V−1 s−1 for single crystals.122 Due to the unique flat and curved structures, the PLQY of BPFT single crystals was as high as 51%. Adachi et al. developed BPTA, BPTN and BPTB with anthracene, naphthalene and phenyl groups as the conjugated core, respectively. Among them, BPTA has a higher mobility (single crystals, μh = 0.14 cm2 V−1 s−1, μe = 0.19 cm2 V−1 s−1), but the weaker emission with a PLQY of 31% for crystals. The hole and electron mobility of BPTB and BPTN is an order of magnitude lower than that of BPTA, which show much stronger emission with PLQYs reaching 87% and 56%, respectively, for BPTB and BPTN single crystals.123 Tian et al. designed BDPV2T with a linear π-conjugated bithiophene backbone, exhibiting high carrier mobility (single crystal, μmax = 1.0 cm2 V−1 s−1) and solid-state emission. The PLQY of BDPV2T crystals is as high as 30%, which is higher than that of solution (11%), indicating its aggregation enhanced emission properties.124 Hu et al. synthesized phenanthrene derivatives (DPPa) with a single crystal mobility of up to 1.6 cm2 V−1 s−1 and a single crystal PLQY of up to 37.13%.125 The results showed that phenanthrene derivatives can be comparable with their anthracene isomers. Fu et al. designed 4,4′-bis(2-dibenzothiophenyl-vinyl)-biphenyl (DBTVB) and prepared its herringbone packed ultra-thin single crystal microplate, which achieved balanced mobilities of μh = 3.55 ± 0.5 and μe = 2.37 ± 0.5 cm2 V−1 s−1, a high solid-state emission efficiency of 85%, and striking low-threshold laser characteristics.37 Theoretical and experimental studies revealed that strong electron coupling and small reorganization energy ensure efficient charge transport, while exciton vibration effects and negligible π–π orbital overlaps lead to the strong emission of H-aggregates. Dibenzothiophene sulfone-based blue-emissive organic semiconductors (DNaDBSOs) were also reported, which demonstrated superior solid-state PLQYs of 46–67% (powder) and ambipolar-transporting properties.126 Ma et al. developed a series of 2-phenylbenzo[g]furo[2,3-b]quinoxaline derivatives and revealed that benzyl substituted benzo[g]furo[2,3-b]quinoxaline demonstrated the best integrated optoelectronic properties due to the better crystallinity and continuity of the film.127 By replacing the anthracene core with benzo[1,2-b:4,5-b′]dithiophene, 4,8-DTEBDT and 2,6-DTEBDT were reported, exhibiting single crystal charge carrier mobilities of up to 0.25 and 0.06 cm2 V−1 s−1, together with single crystal PLQYs of 51% and 45%, respectively.128
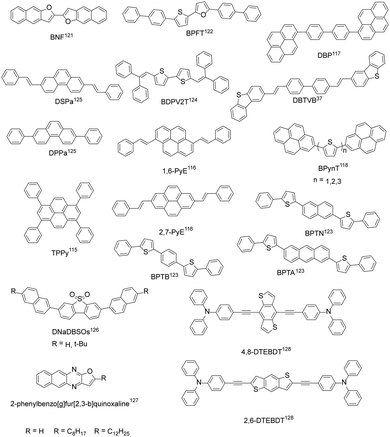 |
| | Fig. 7 Chemical structures of other organic fluorescent semiconductors reported. | |
4. Optoelectronic applications
Organic fluorescent semiconductors, as a unique class of organic materials, have found their special application scenarios in organic optoelectronics. Up to now, high performance organic fluorescent semiconductors have achieved successful application progress in devices such as OLETs, OLEDs, and organic lasers, which we will briefly discuss in the following.
4.1. OLETs
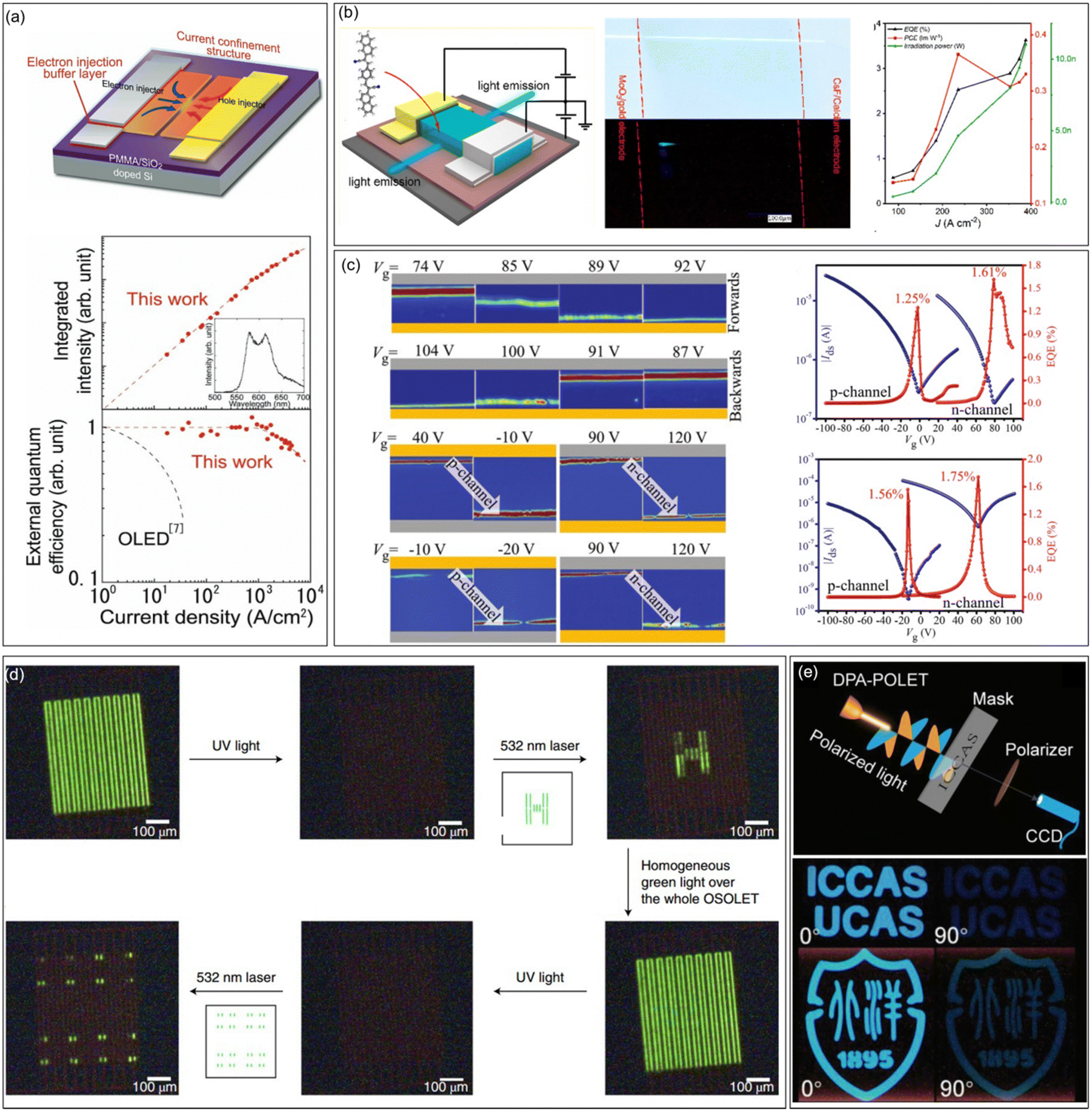
An OLET is a kind of integrated optoelectronic device that combines the light-emitting characteristics of OLEDs and the switching function of OFETs. It is the smallest component that may be integrated with the OLED and OFET. The achievement of high-performance OLETs is of great significance to reduce the size of organic circuits and the final realization of electrically pumped organic lasers. High mobility emissive organic semiconductors are the cornerstone for high performance OLETs, especially for single component OLETs. An ideal OLET requires an active layer with high mobility, strong luminescence, and balanced carrier transport. Since the first OLET with tetracene thin films as the active layer reported in 2003, OLETs have witnessed significant achievements in terms of device efficiency, current density, luminance, emission colour diversity, and so on.29 Adachi et al. reported the ambipolar single crystal OLET based on BSB-Me with almost equal electron and hole mobilities (∼0.005 cm2 V−1 s−1), and the external quantum efficiency (EQE) was roughly estimated to be 0.2%.104 Takenobu et al. constructed high performance OLETs based on the BP3T single crystal. By the insertion of CaF as an electron-injection buffer layer and introduction of a current-confinement architecture, the EQE reached ∼1% and the maximum current density is enhanced to 33 kA cm−2 (Fig. 8a).129 Ma et al. reported a series of OLETs with oligostyrenes as the active layer. For instance, the OLET device based on PBNA exhibited brilliant blue emission, reaching an EQE of 3.63%, which is one of the highest values for single component OLETs at that time (Fig. 8b).108 Hu et al. conducted comprehensive studies on OLETs with anthracene derivatives as active layers. For example, OLETs based on 2,6-DPA and 2,6-DNA achieved good ambipolar charge transport with spatially controlled light emission across the channel (Fig. 8c). High EQE values (2,6-DPA: 1.61% and 2,6-DNA: 1.75%), brightness (2,6-DPA: 1210 cd m−2 and 2,6-DNA: 3180 cd m−2) and current density (2,6-DPA: 1.3 kA cm−2 and 2,6-DNA: 8.4 kA cm−2) were also achieved.130 Furthermore, by adjusting the dopant ratio, a large color gamut as high as 59% National Television System Committee standard together with a high current density of 326.4 kA cm−2 were achieved for the OLETs based on molecular doped organic active layers, indicating great potential in displays and related optoelectronic applications.131 Except for the enhancements of the overall performances of OLETs, functionalized OLETs based on organic fluorescent semiconductors were also investigated, which might bring more possibilities for the future application of OLETs. For example, Samorì et al. constructed optically switchable OLETs by blending emissive semiconductors (F8, F8T2, and MDMO-PPV) with photochromic diarylethenes (DAEs), achieving reversible modulation of charge transport and electroluminescence of three primary colours in the same device (Fig. 8d).132 Taking advantage of the anisotropy of organic semiconductor single crystals, polarized OLETs based on 2,6-DPA were successfully constructed, demonstrating a degree of polarization comparable to completely linearly polarized light (Fig. 8e).133
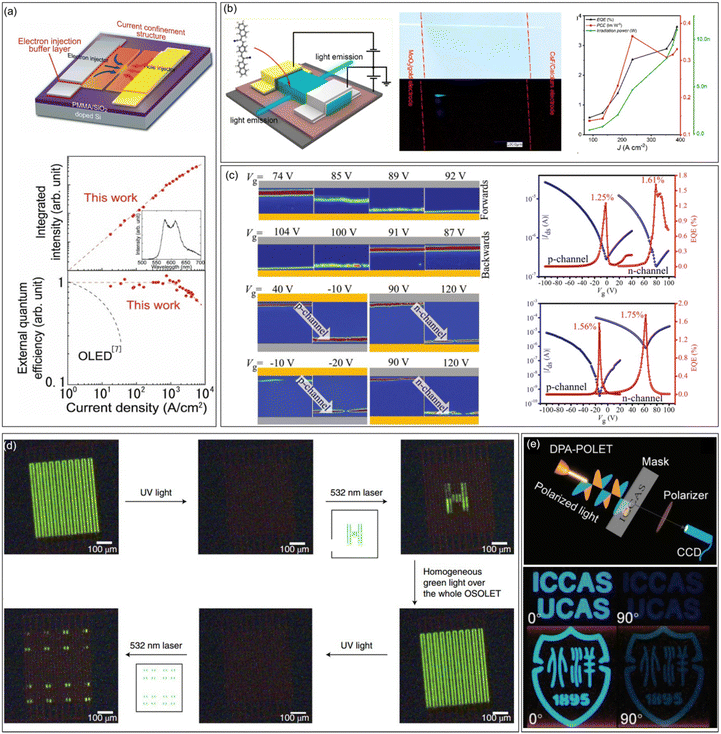 |
| | Fig. 8 Representative applications in OLETs. (a) Design principle for an improved organic single-crystal light emitting transistor with extremely high current density and current-density dependence of the integrated intensity of the luminescence spectra (top) and external quantum efficiency (bottom) in the ambipolar BP3T-OLETs. Reproduced with permission.129 Copyright 2012, John Wiley and Sons. (b) Device structure of PBNA-based single crystal OLETs, photographs of the green crystal under natural light (upper) and electrical driving (lower) in OLETs, and the EQE, PCE, and irradiation power changes as a function of current density. Reproduced with permission.108 Copyright 2021, American Chemical Society. (c) A series of color-coded images for 2,6-DPA and 2,6-DNA-based OLETs extracted from light emission captured by CCD operating under forward and backward, and EQE versus Vg for 2,6-DPA-OLET and 2,6-DNA-OLET within both p- and n-channel regions. Reproduced with permission.130 Copyright 2019, John Wiley and Sons. (d) Emitting pattern created and erased within single OLETs. Reproduced with permission.132 Copyright 2019, Springer Nature. (e) Schematic of the imaging system with DPA-OPLET as the light source and the images of imaging by DPA-OPLET with polarization angles of 0° (left pictures) and 90° (right pictures). Reproduced with permission.133 Copyright 2023, John Wiley and Sons. | |
4.2. OLEDs
Since Tang et al. reported the first heterojunction-structured OLED in 1987 and greatly lowered the operating voltage of OLEDs, OLEDs have shown great application prospects in the field of organic displays and lighting due to their advantages of flexibility, lightness and self-emission, etc.134–136 During the past few decades, organic light-emitting diodes have witnessed great progress in terms of material structure diversity, luminous mechanism, device performance, and device stability. Generally, OLEDs are sandwich-like structures composed of an anode, an organic emissive functional layer, and a metal cathode. In practical applications, considering device structures (top emission/bottom emission) and different features of the organic emissive layer, functional layers, such as electron/hole injection/transporting layer, electron/hole blocking layer, etc., are usually introduced to improve the optoelectronic performance of the device. Organic fluorescent semiconductors, serving as a unique class of luminescent materials, also demonstrate their application potential in OLEDs as the active emissive layer, especially in the fabrication of single crystal OLEDs with high mobility or high current density, which we will discuss several representative examples below.
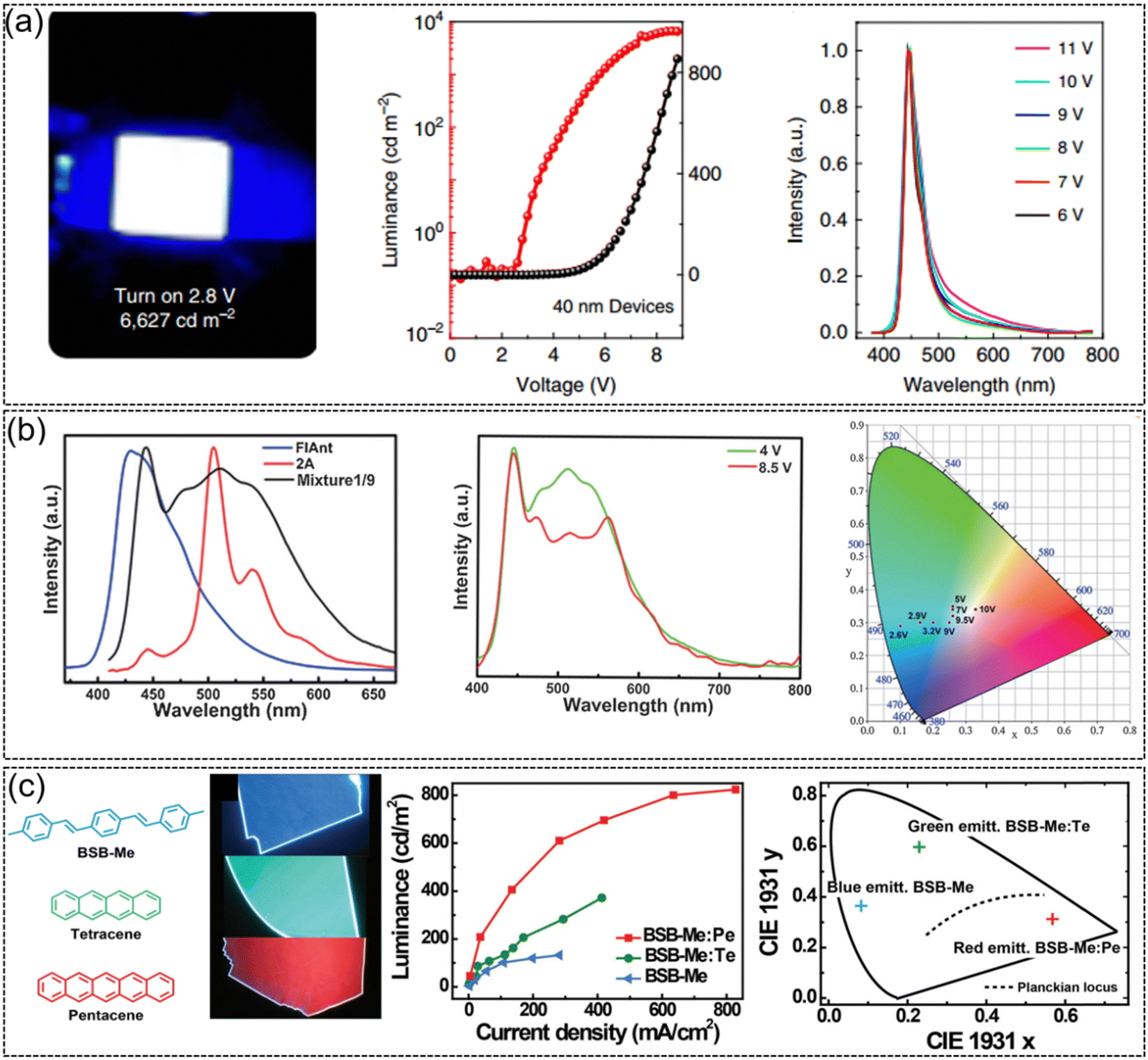
2,6-DPA, which not only exhibits ultrahigh carrier mobility, but also demonstrates strong solid-state emission with a PLQY of 41.2% for single crystals. As a luminescent molecule, 2,6-DPA-based OLEDs emit pure blue emission with a brightness of up to 6627 cd m−2 and a turn on voltage of 2.8 V (Fig. 9a). The 2,6-DPA-based OLED arrays could be successfully driven by 2,6-DPA-based transistors, suggesting the great application potential of 2,6-DPA in organic active displays.39 High mobility white emissive organic semiconductors were also achieved by doping a small amount of FlAnt in 2A (Fig. 9b).66 Because of the similar molecular and crystal stacking structures of FlAnt and 2A, a small amount of FlAnt doping in 2A (μ = 2.98 cm2 V−1 s−1) did not significantly reduce the mobility of thin film devices (μ = 1.56 cm2 V−1 s−1) but efficiently modulated the emission color. OLEDs based on mixtures (FlAnt: 2A = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9) exhibited a low turn on voltage of 3 V, a high brightness of 2560 cd m−2 and an EQE of 0.16% at a drive voltage of 10 V. The CIE coordinates move from blue-green emission of 2A (0.1, 0.29) to pure white emission (0.33, 0.34) for the mixtures upon driving voltage modulation.
:9) exhibited a low turn on voltage of 3 V, a high brightness of 2560 cd m−2 and an EQE of 0.16% at a drive voltage of 10 V. The CIE coordinates move from blue-green emission of 2A (0.1, 0.29) to pure white emission (0.33, 0.34) for the mixtures upon driving voltage modulation.
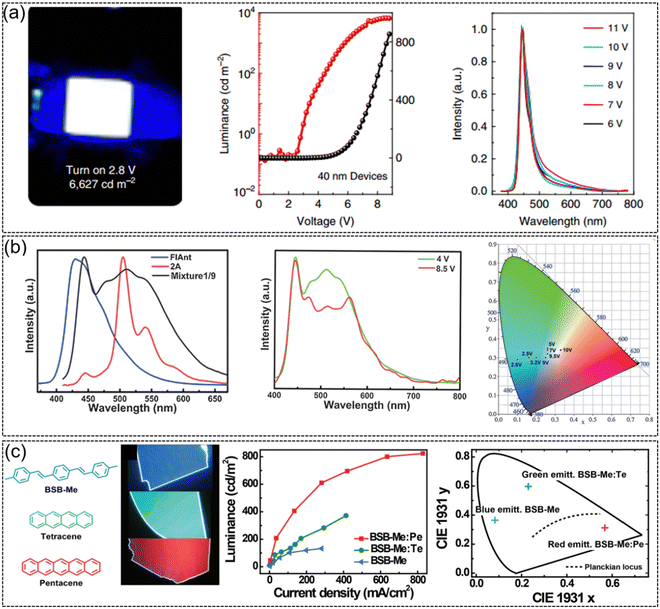 |
| | Fig. 9 Representative applications in OLEDs. (a) Strong blue emission from OLEDs based on DPA (2 mm × 2 mm), current density–voltage-luminance characteristics of the OLEDs and EL spectra operated at different voltages. Reproduced with permission.39 Copyright 2015, Springer Nature. (b) EL spectra in OLEDs based on FlAnt, 2A, and mixture1/9, EL spectra of mixture1/9 at 4 and 8.5 V and CIE coordinates at different driving voltages in OLED devices based on mixture1/9. Reproduced with permission.66 Copyright 2016, John Wiley and Sons. (c) Chemical structures and top-view photographs of BSB-Me, BSB-Me:Te, and BSB-Me:Pe organic crystals under UV-light irradiation, EL performance of BSB-Me, BSB-Me:Te, and BSB-Me:Pe single-crystal-based OLEDs and corresponding 1931 CIE coordinate diagrams of blue, green, and red light emission with CIE coordinates of (0.56, 0.31), (0.23, 0.59), and (0.08, 0.36), respectively. Reproduced with permission.137 Copyright 2017, John Wiley and Sons. | |
Due to the advantages of fewer grain boundaries, long-range highly ordered molecular stacking, and low defect density, organic semiconductor single crystals generally present better optoelectronic properties (mobility, emission efficiency, exciton diffusion length, etc.) than the polycrystalline or amorphous counterpart. The research on single crystal OLEDs can be dated back to 1963, when Pope et al. prepared OLEDs using the bulk anthracene single crystal and two silver paste electrodes and achieved electroluminescence at an extremely high voltage of 400 V and low 100 μA cm−2 was achieved.138 Later, Hotta et al. constructed low driven voltage single crystal OLEDs based on the needle-like BP2T single crystal.139 Compared with traditional OLED devices, OLED devices based on the organic semiconductor single crystal are expected to effectively improve the carrier mobility and the current density. Furthermore, owing to the intrinsic anisotropy of the single crystal, high-efficiency polarized emission might be achieved from single crystal OLEDs.140 Therefore, except for the conventional OLED with the thin film organic active layer, high performance organic fluorescent semiconductors were also intensively applied in single crystal OLEDs. Using pure blue emissive single crystal BSB-Me as the host molecule and tetraphene (Te) and pentaphene (Pe) as the guest molecules, high performance single crystal OLEDs were achieved (Fig. 9c).137 Through dipole–dipole interactions, effective energy transfer occurred from BSB-Me as the donor to Te or Pe as the acceptor, and three primary electroluminescence emissions were realized. The maximum brightness and current efficiency of BSB-Me:Pe single crystal OLEDs achieve 820 cd m−2 and 0.9 cd A−1, respectively, which is one of the highest performances for organic single crystal OLEDs at that time. An ambipolar organic single crystal OLED was successfully developed by growing mixed crystals of n-type material BTPB (μe = 1.83 cm2 V−1 s−1) and p-type material BSB-Me (μh = 0.12 cm2 V−1 s−1).141 By adjusting the mixing ratio of BSB-Me and BTPB, the mobility of holes and electrons could be controlled to be well balanced. The maximum brightness of the mixed ambipolar crystal OLED is greatly increased from 132 cd m−2 of the unipolar crystal to 1116 cd m−2. The efficiency of the ambipolar single crystal OLED also exhibits three times enhancement compared with that of the unipolar single crystal OLEDs. In a mixture ratio of 1:1:10 (pentaphene:BTPB:BSB-Me) doped single crystal OLEDs, after optimization, the maximum brightness, current efficiency and EQE achieved 5467 cd m−2, 2.82 cd A−1 and 1.64%, which are among the best performances for single crystal OLEDs at that time. For single crystal OLEDs, it is not only the optical and electrical properties of the material that affect the comprehensive performance of the device, the crystallization characteristics, or whether it is possible to prepare large-area high-quality organic semiconductor single crystals for device fabrication, is also one of the important factors that determine device behavior. Besides, up to now, most high-performance single crystal OLEDs are based on organic fluorescent lamellar single crystal semiconductors, after solving the problems such as how to obtain high-quality (lamellar) single crystals, how to avoid crystal damage during device construction, and how to achieve good contact and efficient charge injection between the electrodes and the single crystal, etc., we believe the possibility of the realization of single crystal OLEDs based on phosphorescence and (MR-)thermally activated delayed fluorescence (TADF) materials.135,142,143
4.3. Organic lasers
Except for the applications in OLETs, OLEDs and organic fluorescent semiconductors with lasing characteristics are also candidates for the study of electrically pumped organic lasers (EPOLs), which show great promise in smart display technologies and related optoelectronic circuits.144 Up to now, organic semiconductor-based electrically pumped organic lasers (EPOLs) were still in the infancy stage, and the reported studies of organic semiconductors lasers were based on a light pumped mode, among which we will summarize several representative exemplifications below.
Hotta et al. reported the narrowed emission spectra from the AC5 crystal with its full width at half maximum down to ∼6 nm, and the maximum net optical gain coefficient is as high as 75 cm−1, suggesting its potential lasing characteristics.145 Because of a drain current increase in the ambipolar regime, spectral narrowing was also observed for the BP3T single crystal OLET (Fig. 10a).103 It should also be noted that similar lasing characteristics were also observed in other TPCOs, such as BP2T-OMe,145 BP1T-CN,146etc. The edge emission of the BDPV2T crystals is brighter than their main body, indicating excellent optical waveguide emission in the solid-state, which is a prerequisite for generating photoamplification and initiating laser light. The amplified spontaneous emission (ASE) properties of the BDPV2T crystals were characterized using a pulsed laser of 355 nm as an excitation source, exhibiting a threshold of 80 μJ cm−2, an optical amplification threshold as low as 8 kW cm−2, and an optical net gain as high as 70 cm−1, which is attributed to the high solid-state PLQY and good crystal quality.124 Clear whispering-gallery-mode (WGM) lasing characteristics can be observed from the hexagonal DBTVB microcrystals upon 400 nm femtosecond laser excitation (Fig. 10b).37 With the increase of the laser pump density (P), the top of the 0-1 jump appears as a set of sharp peaks near 469 nm, showing an ultralow threshold at Pth = 5.95 μJ cm−2. The photoluminescence decay time of the DBTVB crystals shortens to less than 20 ps as the pump intensity increases from P = 0.7 Pth to P = 1.3 Pth, indicating the presence of an excited emission process. The single crystal of LD-1 exhibits lasing thresholds of 71 and 53 μJ cm−2 at emission peaks of 390 nm and 410 nm, and high quality factors (Q) of ∼3100 and ∼2700 (Fig. 10c).86 LD-2 was also reported for low-threshold lasers with a higher single crystal charge transport mobility (2.7 cm2 V−1 s−1), low-threshold lasing properties of 9.43 μJ cm−2 and 9.93 μJ cm−2 at emission peaks of 420 nm and 443 nm, and high Q values of 2131 and 1684.87
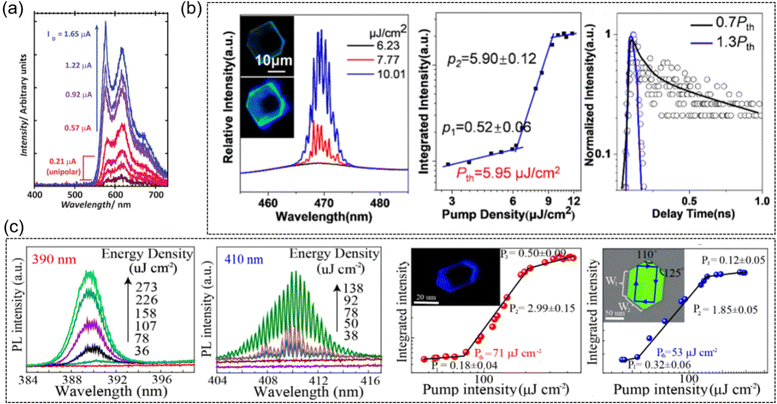 |
| | Fig. 10 Representative applications in lasers. (a) Current-dependent spectral evolution, measured in real time. Reproduced with permission.103 Copyright 2009, John Wiley and Sons. (b) High-resolution PL spectra of a hexagonal microplate at different pump densities of 400 nm femtosecond excitation lasers, integrated intensity of the peak at 469 nm as a function of pump density and corresponding PL decay at 469 nm below and above the lasing threshold. Reproduced with permission.37 Copyright 2022, American Chemical Society. (c) High-resolution lasing spectra of the LD-1 crystal at 390 and 410 nm at different pump densities, and the corresponding integrated area of the peak (410 nm, 390 nm) as a function of the pump density. Reproduced with permission.86 Copyright 2020, American Chemical Society. | |
5. Conclusions
In this review, the recent advances in small molecule organic fluorescent semiconductors are briefly summarized from the aspects of the aggregation structures, the structures and properties of the materials, and the related optoelectronic applications. As a special class of organic functional materials, organic fluorescent semiconductors are the material basis for organic integrated optoelectronic device and are crucial for the final realization of EPOLs. Though impressive progress has been achieved in recent years, there still remain several challenges below:
(i) From the perspective of electrical properties, although various material structures with considerably high optoelectronic properties have been developed, the design and synthesis of n-type and “truly ambipolar”, especially air stable n-type and “truly ambipolar” without the usage of active metal electrodes, organic fluorescent semiconductors are needed, which are of great significance for the final application of organic fluorescent semiconductor-based optoelectronic devices.
(ii) From the perspective of optical properties, red-emissive organic semiconductors are highly demanded for the construction of full color displays. Among the reported single component organic fluorescent semiconductors, the main emission peaks range from ∼400 nm to ∼680 nm, with the emission colors changing from blue-violet to orange-red; however, few molecules demonstrate pure red emission, yet red color is one of the three basic colors for full color displays. Therefore, high performance red-emissive organic semiconductors are highly desired.
Author contributions
Lingxu Zhao: literature investigation and writing the original draft. Jie Li: conceiving and supervising this study. Liqiang Li: giving valuable suggestions. Wenping Hu: giving valuable suggestions.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by National Natural Science Foundation of China (21905199 and 52121002), the Tianjin Natural Science Foundation (23JCYBJC01790), and the State Key Laboratory of Luminescent Materials and Devices, South China University of Technology (2024-skllmd-25).
Notes and references
- H. Shirakawa, E. J. Louis, A. G. MacDiarmid, C. K. Chiang and A. J. Heeger, J. Chem. Soc., Chem. Commun., 1977, 578–580 RSC.
- C. L. Wang, H. L. Dong, W. P. Hu, Y. Q. Liu and D. B. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- J. E. Anthony, A. Facchetti, M. Heeney, S. R. Marder and X. W. Zhan, Adv. Mater., 2010, 22, 3876–3892 CrossRef CAS PubMed.
- J. G. Mei, Y. Diao, A. L. Appleton, L. Fang and Z. N. Bao, J. Am. Chem. Soc., 2013, 135, 6724–6746 CrossRef CAS PubMed.
- G. Saito and Y. Yoshida, Bull. Chem. Soc. Jpn., 2007, 80, 1–137 CrossRef CAS.
- X. M. Xu, Y. F. Yao, B. W. Shan, X. Gu, D. Q. Liu, J. Y. Liu, J. B. Xu, N. Zhao, W. P. Hu and Q. Miao, Adv. Mater., 2016, 28, 5276–5283 CrossRef CAS PubMed.
- H. J. Chen, Y. L. Guo, G. Yu, Y. Zhao, J. Zhang, D. Gao, H. T. Liu and Y. Q. Liu, Adv. Mater., 2012, 24, 4618–4622 CrossRef CAS PubMed.
- Y. W. Jiang, Z. T. Zhang, Y. X. Wang, D. L. Li, C. T. Coen, E. Hwaun, G. Chen, H. C. Wu, D. L. Zhong, S. M. Niu, W. C. Wang, A. Saberi, J. C. Lai, Y. L. Wu, Y. Wang, A. A. Trotsyuk, K. Y. Loh, C. C. Shih, W. H. Xu, K. Liang, K. L. Zhang, Y. H. Bai, G. Gurusankar, W. P. Hu, W. Jia, Z. Cheng, R. H. Dauskardt, G. C. Gurtner, J. B. H. Tok, K. Deisseroth, I. Soltesz and Z. N. Bao, Science, 2022, 375, 1411–1417 CrossRef CAS PubMed.
- X. H. Ding, C. X. Wei, L. Z. Wang, J. Yang, W. X. Huang, Y. Z. Chang, C. J. Ou, J. Y. Lin and W. Huang, SmartMat, 2023, e1213 CrossRef.
- Y. Diao, L. Shaw, Z. Bao and S. C. B. Mannsfeld, Energy Environ. Sci., 2014, 7, 2145–2159 RSC.
- C. Wei, L. Li, Y. Zheng, L. Wang, J. Ma, M. Xu, J. Lin, L. Xie, P. Naumov, X. Ding, Q. Feng and W. Huang, Chem. Soc. Rev., 2024, 53, 3687–3713 RSC.
- G. H. Gelinck, H. E. A. Huitema, E. Van Veenendaal, E. Cantatore, L. Schrijnemakers, J. Van der Putten, T. C. T. Geuns, M. Beenhakkers, J. B. Giesbers, B. H. Huisman, E. J. Meijer, E. M. Benito, F. J. Touwslager, A. W. Marsman, B. J. E. Van Rens and D. M. De Leeuw, Nat. Mater., 2004, 3, 106–110 CrossRef CAS PubMed.
- P. A. Ersman, R. Lassnig, J. Strandberg, D. Y. Tu, V. Keshmiri, R. Forchheimer, S. Fabiano, G. Gustafsson and M. Berggren, Nat. Commun., 2019, 10, 5053 CrossRef PubMed.
- J. H. Oh, H. W. Lee, S. Mannsfeld, R. M. Stoltenberg, E. Jung, Y. W. Jin, J. M. Kim, J. B. Yoo and Z. N. Bao, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6065–6070 CrossRef CAS PubMed.
- Y. A. Huang, X. Gong, Y. C. Meng, Z. W. Wang, X. S. Chen, J. Li, D. Y. Ji, Z. M. Wei, L. Q. Li and W. P. Hu, Nat. Commun., 2021, 12, 21 CrossRef CAS PubMed.
- R. Shi, S. Jiao, Q. Yue, G. Gu, K. Zhang and Y. Zhao, Exploration, 2022, 2, 20220066 CrossRef CAS PubMed.
- H. Y. Guo, H. C. Dai and C. L. Wang, ChemPlusChem, 2023, 88, e202300026 CrossRef CAS PubMed.
- C. Melzer and H. von Seggern, Nat. Mater., 2010, 9, 470–472 CrossRef CAS PubMed.
- M. Muccini, Nat. Mater., 2006, 5, 605–613 CrossRef CAS PubMed.
- M. A. McCarthy, B. Liu, E. P. Donoghue, I. Kravchenko, D. Y. Kim, F. So and A. G. Rinzler, Science, 2011, 332, 570–573 CrossRef CAS PubMed.
- C. C. Zhang, P. L. Chen and W. P. Hu, Small, 2016, 12, 1252–1294 CrossRef CAS PubMed.
- Z. Y. Xie, D. Liu, Z. N. Zhao, C. Gao, P. Wang, C. X. Jiang, X. F. Liu, X. T. Zhang, Z. J. Ren, S. K. Yan, W. P. Hu and H. L. Dong, Angew. Chem., Int. Ed., 2024, 63, e202319380 CrossRef CAS PubMed.
- A. Hepp, H. Heil, W. Weise, M. Ahles, R. Schmechel and H. von Seggern, Phys. Rev. Lett., 2003, 91, 157406 CrossRef PubMed.
- V. Podzorov, E. Menard, A. Borissov, V. Kiryukhin, J. A. Rogers and M. E. Gershenson, Phys. Rev. Lett., 2004, 93, 086602 CrossRef CAS PubMed.
- V. C. Sundar, J. Zaumseil, V. Podzorov, E. Menard, R. L. Willett, T. Someya, M. E. Gershenson and J. A. Rogers, Science, 2004, 303, 1644–1646 CrossRef CAS PubMed.
- D. R. Maulding and B. G. J. Roberts, J. Org. Chem., 1969, 34, 1734–1736 CrossRef CAS.
- J. E. Anthony, J. S. Brooks, D. L. Eaton and S. R. Parkin, J. Am. Chem. Soc., 2001, 123, 9482–9483 CrossRef CAS PubMed.
- X. T. Zhang, H. L. Dong and W. P. Hu, Adv. Mater., 2018, 30, 1801048 CrossRef PubMed.
- Z. S. Qin, H. K. Gao, H. L. Dong and W. P. Hu, Adv. Mater., 2021, 33, 2007149 CrossRef CAS PubMed.
- Y. W. Liu, Y. L. Guo and Y. Q. Liu, Small Struct., 2021, 2, 2000083 CrossRef CAS.
- Z. T. Liu, G. X. Zhang and D. Q. Zhang, Chem. – Eur. J., 2016, 22, 462–471 CrossRef CAS PubMed.
- P. P. Yu, Y. G. Zhen, H. L. Dong and W. P. Hu, Chem, 2019, 5, 2814–2853 CAS.
- C. L. Wang, H. L. Dong, L. Jiang and W. P. Hu, Chem. Soc. Rev., 2018, 47, 422–500 RSC.
- J. L. Brédas, J. P. Calbert, D. A. da Silva and J. Cornil, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5804–5809 CrossRef PubMed.
- S. Q. Ma, S. J. Du, G. C. Pan, S. T. Dai, B. Xu and W. J. Tian, Aggregate, 2021, 2, e96 CrossRef CAS.
- F. C. Spano, Acc. Chem. Res., 2010, 43, 429–439 CrossRef CAS PubMed.
- F. Yin, J. B. De, M. H. Liu, H. Huang, H. Geng, J. N. Yao, Q. Liao and H. B. Fu, Nano Lett., 2022, 22, 5803–5809 CrossRef CAS PubMed.
- S. Oh, J. H. Kim, S. K. Park, C. H. Ryoo and S. Y. Park, Adv. Opt. Mater., 2019, 7, 1901274 CrossRef CAS.
- J. Liu, H. T. Zhang, H. L. Dong, L. Q. Meng, L. F. Jiang, L. Jiang, Y. Wang, J. S. Yu, Y. M. Sun, W. P. Hu and A. J. Heeger, Nat. Commun., 2015, 6, 10032 CrossRef CAS PubMed.
- J. Li, K. Zhou, J. Liu, Y. G. Zhen, L. Liu, J. D. Zhang, H. L. Dong, X. T. Zhang, L. Jiang and W. P. Hu, J. Am. Chem. Soc., 2017, 139, 17261–17264 CrossRef CAS PubMed.
- Y. J. Wan, J. Deng, W. L. Wu, J. D. Zhou, Q. Niu, H. Y. Li, H. K. Yu, C. Gu and Y. G. Ma, ACS Appl. Mater. Interfaces, 2020, 12, 43976–43983 CrossRef CAS PubMed.
- Z. Xie, B. Yang, L. Liu and Y. Ma, J. Mol. Eng. Mater., 2013, 01, 1340002 CrossRef.
- Z. Q. Xie, B. Yang, F. Li, G. Cheng, L. L. Liu, G. D. Yang, H. Xu, L. Ye, M. Hanif, S. Y. Liu, D. G. Ma and Y. G. Ma, J. Am. Chem. Soc., 2005, 127, 14152–14153 CrossRef CAS PubMed.
- J. Liu, L. Q. Meng, W. G. Zhu, C. C. Zhang, H. T. Zhang, Y. F. Yao, Z. R. Wang, P. He, X. T. Zhang, Y. Wang, Y. G. Zhen, H. L. Dong, Y. P. Yi and W. P. Hu, J. Mater. Chem. C, 2015, 3, 3068–3071 RSC.
- J. B. Zhang, B. Xu, J. L. Chen, S. Q. Ma, Y. J. Dong, L. J. Wang, B. Li, L. Ye and W. J. Tian, Adv. Mater., 2014, 26, 739–745 CrossRef.
- L. Yu, Y. X. Hu, J. Li, Z. W. Wang, H. Q. Zhang, Y. A. Huang, Y. P. Lou, Y. J. Sun, X. Y. Lu, H. P. Liu, Y. S. Zheng, S. G. Wang, X. S. Chen, D. Y. Ji, L. Q. Li and W. P. Hu, J. Mater. Chem. C, 2022, 10, 8874–8880 RSC.
- Q. Sun, T. Jiang, Q. Ou, Q. Peng and Z. G. Shuai, Adv. Opt. Mater., 2023, 11, 2202621 CrossRef CAS.
- M. H. Liu, Y. L. Wei, Q. Ou, P. Y. Yu, G. Wang, Y. A. Duan, H. Geng, Q. Peng, Z. G. Shuai and Y. Liao, J. Phys. Chem. Lett., 2021, 12, 938–946 CrossRef CAS PubMed.
- M. C. Um, J. Jang, J. Kang, J. P. Hong, D. Y. Yoon, S. H. Lee, J. J. Kim and J. I. Hong, J. Mater. Chem., 2008, 18, 2234–2239 RSC.
- L. Jiang, W. P. Hu, Z. M. Wei, W. Xu and H. Meng, Adv. Mater., 2009, 21, 3649–3653 CrossRef CAS.
- R. Katoh, K. Suzuki, A. Furube, M. Kotani and K. Tokumaru, J. Phys. Chem. C, 2009, 113, 2961–2965 CrossRef CAS.
- H. Klauk, U. Zschieschang, R. T. Weitz, H. Meng, T. Sun, G. Nunes, D. E. Keys, C. R. Fincher and Z. Xiang, Adv. Mater., 2007, 19, 3882–3887 CrossRef CAS.
- H. Meng, F. P. Sun, M. B. Goldfinger, F. Gao, D. J. Londono, W. J. Marshal, G. S. Blackman, K. D. Dobbs and D. E. Keys, J. Am. Chem. Soc., 2006, 128, 9304–9305 CrossRef CAS PubMed.
- C. L. Wang, Y. L. Liu, Z. Y. Ji, E. J. Wang, R. J. Li, H. Jiang, Q. X. Tang, H. X. Li and W. P. Hu, Chem. Mater., 2009, 21, 2840–2845 CrossRef CAS.
- C. L. Wang, Y. L. Liu, Z. M. Wei, H. X. Li, W. Xu and W. P. Hu, Appl. Phys. Lett., 2010, 96, 143302 CrossRef.
- A. Dadvand, A. G. Moiseev, K. Sawabe, W. H. Sun, B. Djukic, I. Chung, T. Takenobu, F. Rosei and D. F. Perepichka, Angew. Chem., Int. Ed., 2012, 51, 3837–3841 CrossRef CAS PubMed.
- A. Dadvand, W. H. Sun, A. G. Moiseev, F. Bélanger-Gariépy, F. Rosei, H. Meng and D. F. Perepichka, J. Mater. Chem. C, 2013, 1, 2817–2825 RSC.
- J. Liu, H. L. Dong, Z. R. Wang, D. Y. Ji, C. L. Cheng, H. Geng, H. T. Zhang, Y. G. Zhen, L. Jiang, H. B. Fu, Z. S. Bo, W. Chen, Z. G. Shuai and W. P. Hu, Chem. Commun., 2015, 51, 11777–11779 RSC.
- J. Li, Z. S. Qin, Y. J. Sun, Y. G. Zhen, J. Liu, Y. Zou, C. L. Li, X. Y. Lu, L. Jiang, X. T. Zhang, D. Y. Ji, L. Q. Li, H. L. Dong and W. P. Hu, Angew. Chem., Int. Ed., 2022, 61, e202206825 CrossRef CAS PubMed.
- Y. Zhang, C. Gao, P. Wang, Y. Liu, Z. Liu, W. Xie, H. Xu, Y. Dang, D. Liu, Z. Ren, S. Yan, Z. Wang, W. Hu and H. Dong, Angew. Chem., Int. Ed., 2023, 62, e202217653 CrossRef CAS PubMed.
- Y. Zhao, L. J. Yan, I. Murtaza, X. Liang, H. Meng and W. Huang, Org. Electron., 2017, 43, 105–111 CrossRef CAS.
- L. Zheng, Z. Qin, Z. Liu, J. Li, Y. Hu, Y. Sun, J. Li, X. Zhang, K. Zhang, H. Dong, L. Li and W. Hu, ACS Appl. Mater. Interfaces, 2024, 16, 36688–36695 CrossRef CAS PubMed.
- L. Zheng, J. Li, K. Zhou, X. Yu, X. Zhang, H. Dong and W. Hu, Nano Res., 2020, 13, 1976–1981 CrossRef CAS.
- X. X. Yu, L. Zheng, J. F. Li, P. P. Yu, Z. Y. Liu, C. G. Li, Y. Zou, X. T. Zhang and W. P. Hu, Org. Electron., 2020, 87, 105941 CrossRef CAS.
- F. Li, L. Zheng, X. X. Yu, S. Y. Li, S. Liu, H. N. Wu, Y. J. Sun, L. J. Sun and X. T. Zhang, Mater. Chem. Front., 2021, 5, 5124–5129 RSC.
- M. Y. Chen, Y. Zhao, L. J. Yan, S. Yang, Y. N. Zhu, I. Murtaza, G. F. He, H. Meng and W. Huang, Angew. Chem., Int. Ed., 2017, 56, 722–727 CrossRef CAS PubMed.
- X. X. Yu, L. Zheng, J. F. Li, L. Wang, J. L. Han, H. Y. Chen, X. T. Zhang and W. P. Hu, Sci. China: Chem., 2019, 62, 251–255 CrossRef CAS.
- J. F. Li, L. Zheng, L. J. Sun, C. G. Li, X. T. Zhang, S. S. Cheng and W. P. Hu, J. Mater. Chem. C, 2018, 6, 13257–13260 RSC.
- D. Liu, J. Li, J. Liu, X. Q. Lu, M. X. Hu, Y. Li, Z. B. Shu, Z. J. Ni, S. Ding, L. Jiang, Y. G. Zhen, X. T. Zhang, H. L. Dong and W. P. Hu, J. Mater. Chem. C, 2018, 6, 3856–3860 RSC.
- J. Li, J. Liu, Y. Zhen, L. Meng, Y. Wang, H. Dong and W. Hu, J. Mater. Chem. C, 2015, 3, 10695–10698 RSC.
- S. Z. Bisri, T. Takenobu, K. Sawabe, S. Tsuda, Y. Yomogidao, T. Yamao, S. Hotta, C. Adachi and Y. Iwasa, Adv. Mater., 2011, 23, 2753–2758 CrossRef CAS PubMed.
- J. W. Tao, D. Liu, J. B. Jing, H. L. Dong, L. J. Liu, B. Xu and W. J. Tian, Adv. Mater., 2021, 33, 2105466 CrossRef CAS PubMed.
- J. Liu, J. Y. Liu, Z. C. Zhang, C. H. Xu, Q. Y. Li, K. Zhou, H. L. Dong, X. T. Zhang and W. Hu, J. Mater. Chem. C, 2017, 5, 2519–2523 RSC.
- M. N. Yu, C. J. Ou, B. Liu, D. Q. Lin, Y. Y. Liu, W. Xue, Z. Q. Lin, J. Y. Lin, Y. Qian, S. S. Wang, H. T. Cao, L. Y. Bian, L. H. Xie and W. Huang, Chin. J. Polym. Sci., 2017, 35, 155–170 CrossRef CAS.
- H. Meng, Z. N. Bao, A. J. Lovinger, B. C. Wang and A. M. Mujsce, J. Am. Chem. Soc., 2001, 123, 9214–9215 CrossRef CAS PubMed.
- H. Meng, J. Zheng, A. J. Lovinger, B. C. Wang, P. G. Van Patten and Z. N. Bao, Chem. Mater., 2003, 15, 1778–1787 CrossRef CAS.
- J. Locklin, D. W. Li, S. C. B. Mannsfeld, E. J. Borkent, H. Meng, R. Advincula and Z. Bao, Chem. Mater., 2005, 17, 3366–3374 CrossRef CAS.
- M. L. Tang, M. E. Roberts, J. J. Locklin, M. M. Ling, H. Meng and Z. N. Bao, Chem. Mater., 2006, 18, 6250–6257 CrossRef CAS.
- T. J. Shin, H. Yang, M. M. Ling, J. Locklin, L. Yang, B. Lee, M. E. Roberts, A. B. Mallik and Z. Bao, Chem. Mater., 2007, 19, 5882–5889 CrossRef CAS.
- J. Locklin, M. M. Ling, A. Sung, M. E. Roberts and Z. N. Bao, Adv. Mater., 2006, 18, 2989–2992 CrossRef CAS.
- T. Hadizad, J. Zhang, Z. Y. Wang, T. C. Gorjanc and C. Py, Org. Lett., 2005, 7, 795–797 CrossRef CAS PubMed.
- C. Py, T. C. Gorjanc, T. Hadizad, J. Zhang and Z. Y. Wang, J. Vac. Sci. Technol., A, 2006, 24, 654–656 CrossRef CAS.
- P. Baronas, G. Kreiza, P. Adomenas, O. Adomeniene, K. Kaziauskas, J. C. Ribierre, C. Adachi and S. Jursenas, ACS Appl. Mater. Interfaces, 2018, 10, 2768–2775 CrossRef CAS PubMed.
- Q. Wei, Y. Li, J. G. Liu, Q. Y. Fang, J. W. Li, X. H. Yan, L. H. Xie, Y. Qian, R. D. Xia and W. Huang, Adv. Opt. Mater., 2017, 5, 1601003 CrossRef.
- Y. X. Li, W. Liu, M. N. Yu, X. M. Dong, C. J. Ou, M. Eginligil, R. Q. Li, X. W. Zhang, Y. J. Nie, L. H. Xie, C. X. Xu, J. Q. Liu and W. Huang, J. Mater. Chem. C, 2021, 9, 3171–3176 RSC.
- D. Liu, J. B. De, H. K. Gao, S. Q. Ma, Q. Ou, S. Li, Z. S. Qin, H. L. Dong, Q. Liao, B. Xu, Q. Peng, Z. G. Shuai, W. J. Tian, H. B. Fu, X. T. Zhang, Y. G. Zhen and W. P. Hu, J. Am. Chem. Soc., 2020, 142, 6332–6339 CrossRef CAS PubMed.
- D. Liu, X. X. Wu, C. Gao, C. G. Li, Y. S. Zheng, Y. Li, Z. Y. Xie, D. Y. Ji, X. F. Liu, X. T. Zhang, L. Q. Li, Q. Peng, W. P. Hu and H. L. Dong, Angew. Chem., Int. Ed., 2022, 61, e202200791 CrossRef CAS PubMed.
- B. Kobin, S. Behren, B. Braun-Cula and S. Hecht, J. Phys. Chem. A, 2016, 120, 5474–5480 CrossRef CAS PubMed.
- Y. Y. Noh, R. Azumi, M. Goto, B. J. Jung, E. H. Lim, H. K. Shim, Y. Yoshida, K. Yase and D. Y. Kim, Chem. Mater., 2005, 17, 3861–3870 CrossRef CAS.
- L. L. Liu, S. Tang, M. R. Liu, Z. Q. Xie, W. Zhang, P. Lu, M. Hanif and Y. G. Ma, J. Phys. Chem. B, 2006, 110, 13734–13740 CrossRef CAS PubMed.
- S. Hotta and T. Yamao, J. Mater. Chem., 2011, 21, 1295–1304 RSC.
- T. Katagiri, S. Ota, T. Ohira, T. Yamao and S. Hotta, J. Heterocycl. Chem., 2007, 44, 853–862 CrossRef CAS.
- H. Yanagi, Y. Araki, T. Ohara, S. Hotta, M. Ichikawa and Y. Taniguchi, Adv. Funct. Mater., 2003, 13, 767–773 CrossRef CAS.
- Y. Wang, R. Kumashiro, Z. F. Li, R. Nouchi and K. Tanigaki, Appl. Phys. Lett., 2009, 95, 103306 CrossRef.
- K. Yamane, H. Yanagi, A. Sawamoto and S. Hotta, Appl. Phys. Lett., 2007, 90, 162108 CrossRef.
- S. Hotta, T. Yamao, S. Z. Bisri, T. Takenobu and Y. Iwasa, J. Mater. Chem. C, 2014, 2, 965–980 RSC.
- T. Katagiri, Y. Shimizu, K. Terasaki, T. Yamao and S. Hotta, Org. Electron., 2011, 12, 8–14 CrossRef CAS.
- T. Yamao, Y. Shimizu, H. Kuriki, T. Katagiri and S. Hotta, Jpn. J. Appl. Phys., 2010, 49, 01AB01 CrossRef.
- T. Yamao, H. Kuriki, T. Miki and S. Hotta, J. Nanosci. Nanotechnol., 2009, 9, 165–168 CrossRef CAS PubMed.
- S. Kanazawa, M. Ichikawa, T. Koyama and Y. Taniguchi, ChemPhysChem, 2006, 7, 1881–1884 CrossRef CAS PubMed.
- S. Hotta, M. Goto, R. Azumi, M. Inoue, M. Ichikawa and Y. Taniguchi, Chem. Mater., 2004, 16, 237–241 CrossRef CAS.
- K. Nakamura, M. Ichikawa, R. Fushiki, T. Kamikawa, M. Inoue, T. Koyama and Y. Taniguchi, Jpn. J. Appl. Phys., 2005, 44, L1367–L1369 CrossRef CAS.
- S. Z. Bisri, T. Takenobu, Y. Yomogida, H. Shimotani, T. Yamao, S. Hotta and Y. Iwasa, Adv. Funct. Mater., 2009, 19, 1728–1735 CrossRef CAS.
- H. Nakanotani, R. Kabe, M. Yahiro, T. Takenobu, Y. Iwasa and C. Adachi, Appl. Phys. Express, 2008, 1, 091801 CrossRef.
- H. Nakanotani, M. Saito, H. Nakamura and C. Adachi, Appl. Phys. Lett., 2009, 95, 033308 CrossRef.
- H. Nakanotani, M. Saito, H. Nakamura and C. Adachi, Appl. Phys. Lett., 2009, 95, 103307 CrossRef.
- J. Deng, Y. X. Xu, L. Q. Liu, C. F. Feng, J. Tang, Y. Gao, Y. Wang, B. Yang, P. Lu, W. S. Yang and Y. G. Ma, Chem. Commun., 2016, 52, 2370–2373 RSC.
- L. Q. Liu, C. Cai, Z. J. Zhang, S. T. Zhang, J. Deng, B. Yang, C. Gu and Y. G. Ma, ACS Mater. Lett., 2021, 3, 428–432 CrossRef CAS.
- Q. Liao, Z. Wang, Q. G. Gao, Z. Y. Zhang, J. H. Ren, J. B. De, X. S. Zhang, Z. Z. Xu and H. B. Fu, J. Mater. Chem. C, 2018, 6, 7994–8002 RSC.
- B. K. An, D. S. Lee, J. S. Lee, Y. S. Park, H. S. Song and S. Y. Park, J. Am. Chem. Soc., 2004, 126, 10232–10233 CrossRef CAS PubMed.
- B. K. An, S. K. Kwon, S. D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410–14415 CrossRef CAS PubMed.
- R. Kabe, H. Nakanotani, T. Sakanoue, M. Yahiro and C. Adachi, Adv. Mater., 2009, 21, 4034–4038 CrossRef CAS.
- H. Nakanotani and C. Adachi, Adv. Opt. Mater., 2013, 1, 422–427 CrossRef.
- T. Yasuda, M. Saito, H. Nakamura and T. Tsutsui, Jpn. J. Appl. Phys., 2006, 45, L313–L315 CrossRef CAS.
- S. Z. Bisri, T. Takahashi, T. Takenobu, M. Yahir, C. Adachi and Y. Iwasa, Jpn. J. Appl. Phys., 2007, 46, L596–L598 CrossRef CAS.
- H. Ju, K. Wang, J. Zhang, H. Geng, Z. Liu, G. Zhang, Y. Zhao and D. Zhang, Chem. Mater., 2017, 29, 3580–3588 CrossRef CAS.
- J. Kwon, J.-P. Hong, S. Lee and J.-I. Hong, New J. Chem., 2013, 37, 2881–2887 RSC.
- K. Oniwa, H. Kikuchi, H. Shimotani, S. Ikeda, N. Asao, Y. Yamamoto, K. Tanigaki and T. Jin, Chem. Commun., 2016, 52, 4800–4803 RSC.
- H. Tsuji and E. Nakamura, Acc. Chem. Res., 2017, 50, 396–406 CrossRef CAS PubMed.
- H. Tsuji, G. M. O. Favier, C. Mitsui, S. Lee, D. Hashizume and E. Nakamura, Chem. Lett., 2011, 40, 576–578 CrossRef CAS.
- K. Niimi, H. Mori, E. Miyazaki, I. Osaka, H. Kakizoe, K. Takimiya and C. Adachi, Chem. Commun., 2012, 48, 5892–5894 RSC.
- K. Oniwa, T. Kanagasekaran, T. Jin, M. Akhtaruzzaman, Y. Yamamoto, H. Tamura, I. Hamada, H. Shimotani, N. Asao, S. Ikeda and K. Tanigaki, J. Mater. Chem. C, 2013, 1, 4163–4170 RSC.
- T. Komori, H. Nakanotani, T. Yasuda and C. Adachi, J. Mater. Chem. C, 2014, 2, 4918–4921 RSC.
- S. Q. Ma, K. Zhou, M. X. Hu, Q. Y. Li, Y. J. Liu, H. T. Zhang, J. B. Jing, H. L. Dong, B. Xu, W. P. Hu and W. J. Tian, Adv. Funct. Mater., 2018, 28, 1802454 CrossRef.
- J. Li, C. Li, L. Sun, X. Zhang, S. Cheng and W. Hu, Sci. China: Chem., 2019, 62, 916–920 CrossRef CAS.
- Q. B. Li, Y. H. Zhang, J. F. Lin, Y. Zou, P. Wang, Z. S. Qin, Y. S. Wang, Y. Li, Y. Zhang, C. Gao, Y. P. Zang, W. P. Hu and H. L. Dong, Angew. Chem., Int. Ed., 2023, 62, e202308146 CrossRef CAS PubMed.
- G. Wang, J. Li, Y. X. Li, D. D. Wang, J. J. Zhang, Y. Wu, Y. G. Zhen, Q. X. Tang, H. Ma, W. P. Hu, Z. X. Wu and A. K. Y. Jen, J. Mater. Chem. C, 2019, 7, 9690–9697 RSC.
- J. W. Tao, Z. Y. Liu, D. Liu, H. L. Dong, J. B. Jing, J. X. Song, L. J. Liu, B. Xu and W. J. Tian, Aggregate, 2023, 4, e269 CrossRef CAS.
- K. Sawabe, M. Imakawa, M. Nakano, T. Yamao, S. Hotta, Y. Iwasa and T. Takenobu, Adv. Mater., 2012, 24, 6141–6146 CrossRef CAS PubMed.
- Z. S. Qin, H. K. Gao, J. Y. Liu, K. Zhou, J. Li, Y. Y. Dang, L. Huang, H. X. Deng, X. T. Zhang, H. L. Dong and W. P. Hu, Adv. Mater., 2019, 31, 1903175 CrossRef PubMed.
- Z. S. Qin, C. Gao, H. K. Gao, T. Y. Wang, H. L. Dong and W. P. Hu, Sci. Adv., 2022, 8, eabp8775 CrossRef CAS PubMed.
- L. L. Hou, X. Y. Zhang, G. F. Cotella, G. Carnicella, M. Herder, B. M. Schmidt, M. Pätzel, S. Hecht, F. Cacialli and P. Samorì, Nat. Nanotechnol., 2019, 14, 347–353 CrossRef CAS PubMed.
- Z. S. Qin, T. Y. Wang, H. K. Gao, Y. Li, H. L. Dong and W. P. Hu, Adv. Mater., 2023, 35, 2301955 CrossRef CAS.
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- B. Geffroy, P. Le Roy and C. Prat, Polym. Int., 2006, 55, 572–582 CrossRef CAS.
- R. Ding, J. Feng, F. X. Dong, W. Zhou, Y. Liu, X. L. Zhang, X. P. Wang, H. H. Fang, B. Xu, X. B. Li, H. Y. Wang, S. Hotta and H. B. Sun, Adv. Funct. Mater., 2017, 27, 1604659 CrossRef.
- M. Pope, H. P. Kallmann and P. Magnante, J. Chem. Phys., 1963, 38, 2042–2043 CrossRef CAS.
- H. Yanagi, T. Morikawa and S. Hotta, Appl. Phys. Lett., 2002, 81, 1512–1514 CrossRef CAS.
- M.-H. An, R. Ding, X.-L. Zhang, S.-N. Chen, Y.-N. Wang, G.-D. Ye, Q.-C. Zhu, N.-K. Chen, Y. Liu, J. Feng and H.-B. Sun, Optica, 2022, 9, 121–129 CrossRef CAS.
- M. H. An, R. Ding, Q. C. Zhu, G. D. Ye, H. Wang, M. X. Du, S. N. Chen, Y. Liu, M. L. Xu, T. Xu, W. Wang, J. Feng and H. B. Sun, Adv. Funct. Mater., 2020, 30, 2002422 CrossRef CAS.
- Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685–690 CrossRef CAS PubMed.
- X. Li, S. Fu, Y. Xie and Z. Li, Rep. Prog. Phys., 2023, 86, 096501 CrossRef PubMed.
- K. Wang and Y. S. Zhao, Chem, 2021, 7, 3221–3231 CAS.
- T. Yamao, K. Yamamoto and S. Hotta, J. Nanosci. Nanotechnol., 2009, 9, 2582–2585 CrossRef CAS PubMed.
- K. Yamashita, T. Nakahata, T. Hayakawa, Y. Sakurai, T. Yamao, H. Yanagi and S. Hotta, Appl. Phys. Lett., 2014, 104, 253301 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab,
Liqiang
Li
*ab,
Liqiang
Li