
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLipid nanoparticles as the drug carrier for targeted therapy of hepatic disorders
Runxuan
Chu
 a,
Yi
Wang
*b,
Jianglong
Kong
b,
Ting
Pan
ac,
Yani
Yang
*a and
Jun
He
*a
a,
Yi
Wang
*b,
Jianglong
Kong
b,
Ting
Pan
ac,
Yani
Yang
*a and
Jun
He
*a
aNational Advanced Medical Engineering Research Center, China State Institute of Pharmaceutical Industry, 285 Gebaini Road, Shanghai 201203, P. R. China. E-mail: yyn8381@126.com; chinaynhe@163.com
bDepartment of Chemistry, Hong Kong Baptist University, Kowloon Tung, Hong Kong SAR, P. R. China. E-mail: yiwang2@hkbu.edu.hk
cDepartment of Pharmaceutics School of Pharmacy, China Pharmaceutical University, Nanjing 210009, P. R. China
First published on 24th April 2024
Abstract
The liver, a complex and vital organ in the human body, is susceptible to various diseases, including metabolic disorders, acute hepatitis, cirrhosis, and hepatocellular carcinoma. In recent decades, these diseases have significantly contributed to global morbidity and mortality. Currently, liver transplantation remains the most effective treatment for hepatic disorders. Nucleic acid therapeutics offer a selective approach to disease treatment through diverse mechanisms, enabling the regulation of relevant genes and providing a novel therapeutic avenue for hepatic disorders. It is expected that nucleic acid drugs will emerge as the third generation of pharmaceuticals, succeeding small molecule drugs and antibody drugs. Lipid nanoparticles (LNPs) represent a crucial technology in the field of drug delivery and constitute a significant advancement in gene therapies. Nucleic acids encapsulated in LNPs are shielded from the degradation of enzymes and effectively delivered to cells, where they are released and regulate specific genes. This paper provides a comprehensive review of the structure, composition, and applications of LNPs in the treatment of hepatic disorders and offers insights into prospects and challenges in the future development of LNPs.
1. Introduction
In 1965, liposomes emerged as an early version of LNPs and became a versatile nanocarrier platform that can transport both hydrophobic and hydrophilic drugs, including small molecular chemical drugs,5 antibody drugs,6 and nucleic acid drugs.7–9 Liposomes represent the first successful translation of nanodrug delivery systems from concept to clinical application.10,11 Currently, numerous liposomes have been approved and effectively utilized in clinical practice.12,13 Compared to liposomes, subsequent lipid nanocarriers, such as solid lipid nanoparticles and cationic lipid–nucleic acid complexes, exhibited more intricate structures and better physical stability.14,15 LNPs offer a valuable carrier system for treating various diseases by encapsulating and delivering nucleic acids to specific locations in the body.16,17LNP technology enables gene editing and protein replacement. Patisiran/Onpattro® was the first RNA interference therapy approved by the Food and Drug Administration (FDA) in 2018.18 After small molecule drugs and antibody therapeutics were invented, researchers proposed a third generation of treatments, i.e., “smart” nanomedicines.19,20 The design of nanomedicines aims to optimize the efficiency of biological processes while minimizing potential adverse effects through targeted drug delivery.21–23 Nanomedicine facilitated the realization of gene editing technology, which broadly encompasses the delivery of modified nucleic acids to regulate specific protein expression in cells for treating various diseases. LNPs have been increasingly utilized as versatile platforms for nucleic acid delivery due to their ability to overcome major obstacles in gene therapy, namely, degradation and limited cellular uptake of nucleic acids.24 For example, to prepare vaccines for coronavirus disease 2019 (COVID-19), scientists only needed to determine the sequence of virus-related messenger RNA (mRNA) before encapsulating the mRNA in LNPs.25,26 Compared to traditional vaccines, mRNA vaccines are more efficient and safer for patients.27,28 In addition to mRNA, LNPs can transport and deliver DNA to produce essential proteins that are deficient or dysfunctional in genetic disorders.29,30 Additionally, small interfering RNA (siRNA) and antisense oligonucleotides (ASOs) can be utilized to decrease the production of disease-causing proteins, while clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (CRISPR/Cas9) could be employed to edit or deactivate defective genes.31 LNPs based on these innovative therapeutic modalities have the potential to significantly revolutionize the treatment of rare genetic and incurable diseases in the future. This article reviews the application of LNPs in liver-targeting treatments and surveys the status of clinical trials of LNPs (Scheme 1).32–34
 | ||
| Scheme 1 LNPs were employed to facilitate gene delivery. The LNPs applications in the treatment of hepatic disorders were reviewed. Adapted with permission.1 Copyright 2023, Springer Nature. Adapted with permission.2 Copyright 2022, Springer Nature. WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. Adapted with permission.3 Copyright 2021, Springer Nature. Adapted with permission.4 Copyright 2022, Elsevier B.V. | ||
2. Overview, composition, and formation mechanism of LNPs
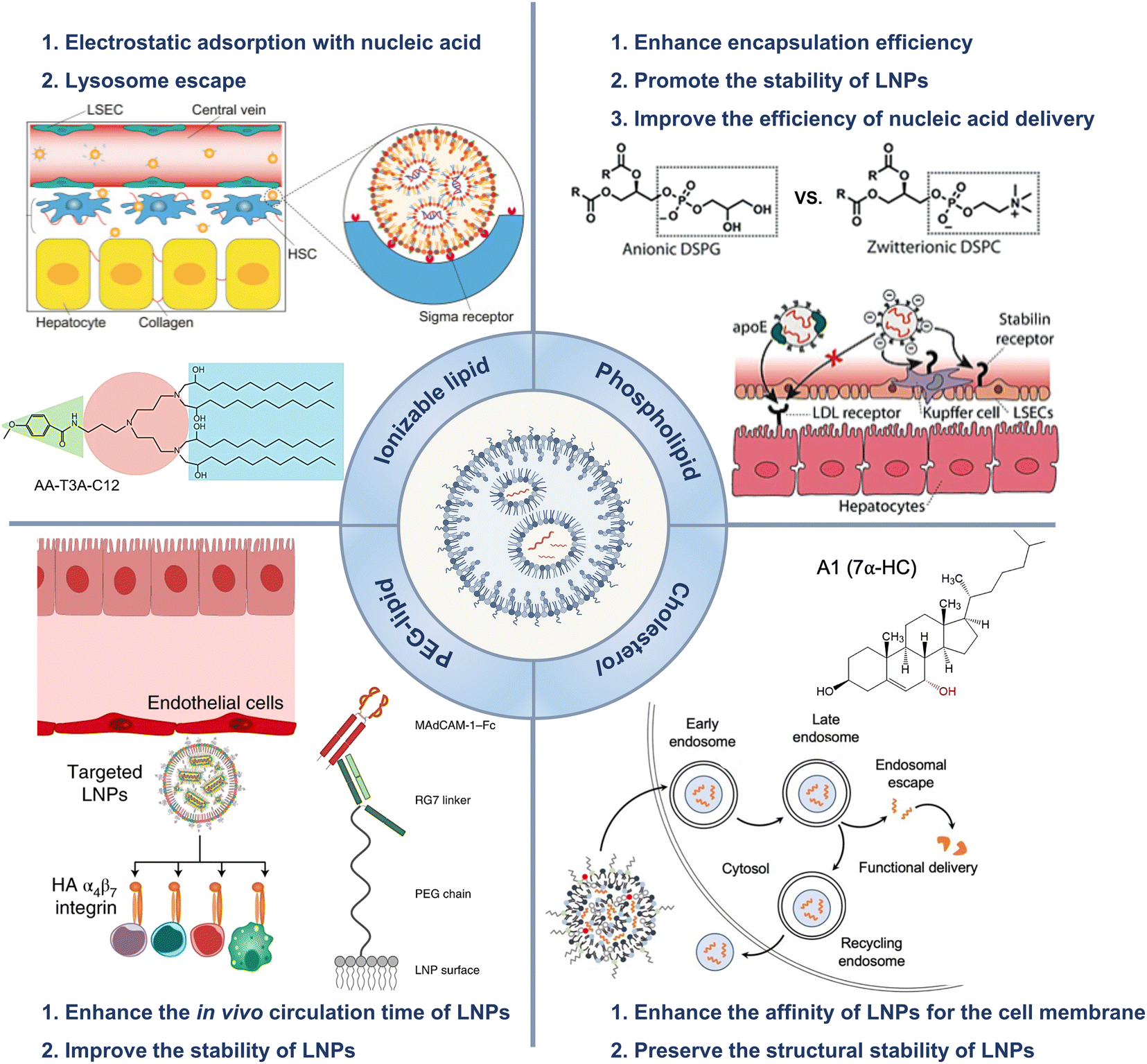
All the current FDA-approved LNP formulations, such as BNT162b2, Comirnaty® of Pfizer-BioNTech and mRNA-1273, and Spikevax® of Moderna, are based on the following kinds of lipids: (1) an ionizable lipid, (2) phospholipids, such as 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), (3) cholesterol, and (4) a polyethylene glycol (PEG)–lipid conjugate.35,36 These constituents facilitated the formation of monodisperse nanoparticles (NPs) and the efficient encapsulation of nucleic acid, while also improving the stability, increasing the cellular uptake, and promoting the endosomal escape of the nucleic acid cargo (Fig. 1(A) and (B)).16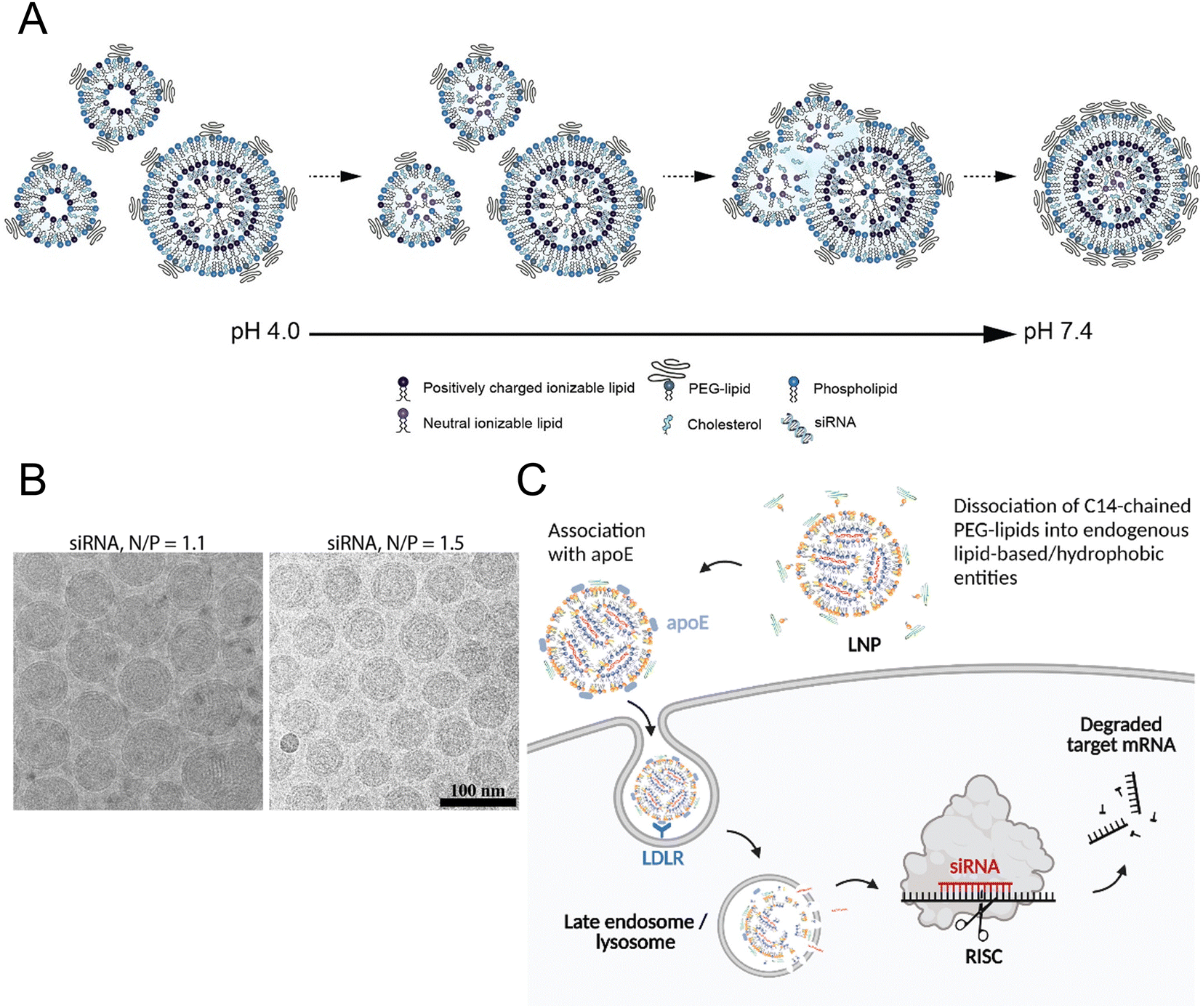 | ||
| Fig. 1 The mechanism of LNP formation. (A) The preparation of LNPs involved the formation of the compact lipid monolayer vesicles that encapsulated the siRNA under acidic conditions at pH 4.0. As the pH increased to 7.4, more cationic lipids became neutral, reducing the electrostatic repulsion and destabilizing the bilayer structures to facilitate the vesicle fusion. During the process of fusion, PEG–lipid, DSPC, and cholesterol migrated to the outer monolayer of the growing LNPs while the ionizable lipid accumulated in the interior to form an oil droplet phase at its core. Equilibrium was achieved when a sufficient concentration of PEG–lipid in the outer monolayer effectively inhibited the inter-LNP fusion. Adapted with permission.16 Copyright 2018, American Chemical Society. (B) The LNPs with the ratios of the amount of the amino lipid nitrogen to the amount of the siRNA phosphate (N/P) of 1.1 and 1.5 were observed using cryo-transmission electron microscope (cryo-TEM). Adapted with permission.16 Copyright 2018, American Chemical Society. (C) Proposed mechanism for Onpattro® uptake. Adapted with permission.37 Copyright 2022, Elsevier B.V. | ||
2.1 Ionizable lipids
The hydrophilic and negatively charged nature of nucleic acids hinder their passive diffusion across the cell membrane.38 However, upon binding to serum proteins, nucleic acids become susceptible to phagocytic cells and endogenous nuclease degradation.39 Therefore, delivery systems are necessary to protect nucleic acids from degradation and facilitate their effective uptake by target cells.40 Cationic lipids are the most appropriate formulation for addressing these issues, although their cytotoxicity remains a major concern.41,42To avoid the general safety problem of cationic lipids, scientists have proposed the use of ionizable lipids.43 This type of lipid had an apparent acid dissociation constant (pKa) below pH 7.0, which allowed the nucleic acids to be efficiently loaded under acidic conditions at pH 4.0 while maintaining a neutral surface charge density at pH 7.4.44 This strategy enabled better regulation of the circulation characteristics of LNPs within the physiological system, thereby ensuring optimal biological safety.45 The LNPs maintained electrical neutrality in the bloodstream, but upon encountering the acidic environment of endosomes, the LNPs underwent a positive charge transition that triggered membrane destabilization and subsequent release of nucleic acid cargo (Fig. 1(C)).37 The pH sensitivity of LNPs might explain their efficient cellular uptake, but this could not account for their extensive hepatic accumulation. Although these LNPs lack specific targeting ligands for hepatocytes, they can bind to apolipoprotein E (ApoE) upon entering systemic circulation, thereby promoting endocytosis by hepatocytes.46
Over the past two decades, a considerable number of ionizable lipids have been developed and evaluated by researchers to improve the transfection efficiency of LNPs (Fig. 2(A)). [3-(Dimethylamino)-2-octadec-9-enoyloxypropyl] octadec-9-enoate (DODAP) was the first reported ionizable lipid, and 4-(dimethylamino)-butanoic acid (6Z,9Z,28Z,31Z)-heptatriaconta-6,9,28,31-tetraen-19-yl-4-(dimethylamino) butanoate (DLin-MC3-DMA), an ionizable lipid with a pKa value of 6.44, was identified from a library of 56 ionizable lipids utilized in the development of Onpattro®.47,48 LNP technology was employed in the development of nucleic acid vaccines for COVID-19, wherein scientists identified two ionizable lipids with saturated tails and conical structures, namely, ALC-0315 (2-hexyl octylic acid 1,1′-[[(4-hydroxy-2,6-diisopropylphenyl)amino]adipic] acetate) and SM-102 (8-[(2-hydroxyethyl)-6-[6-oxygen(11 alkyl hexyl)amino]-1-octylnonyl octylic acid ester]) (Fig. 2(B)).49,50
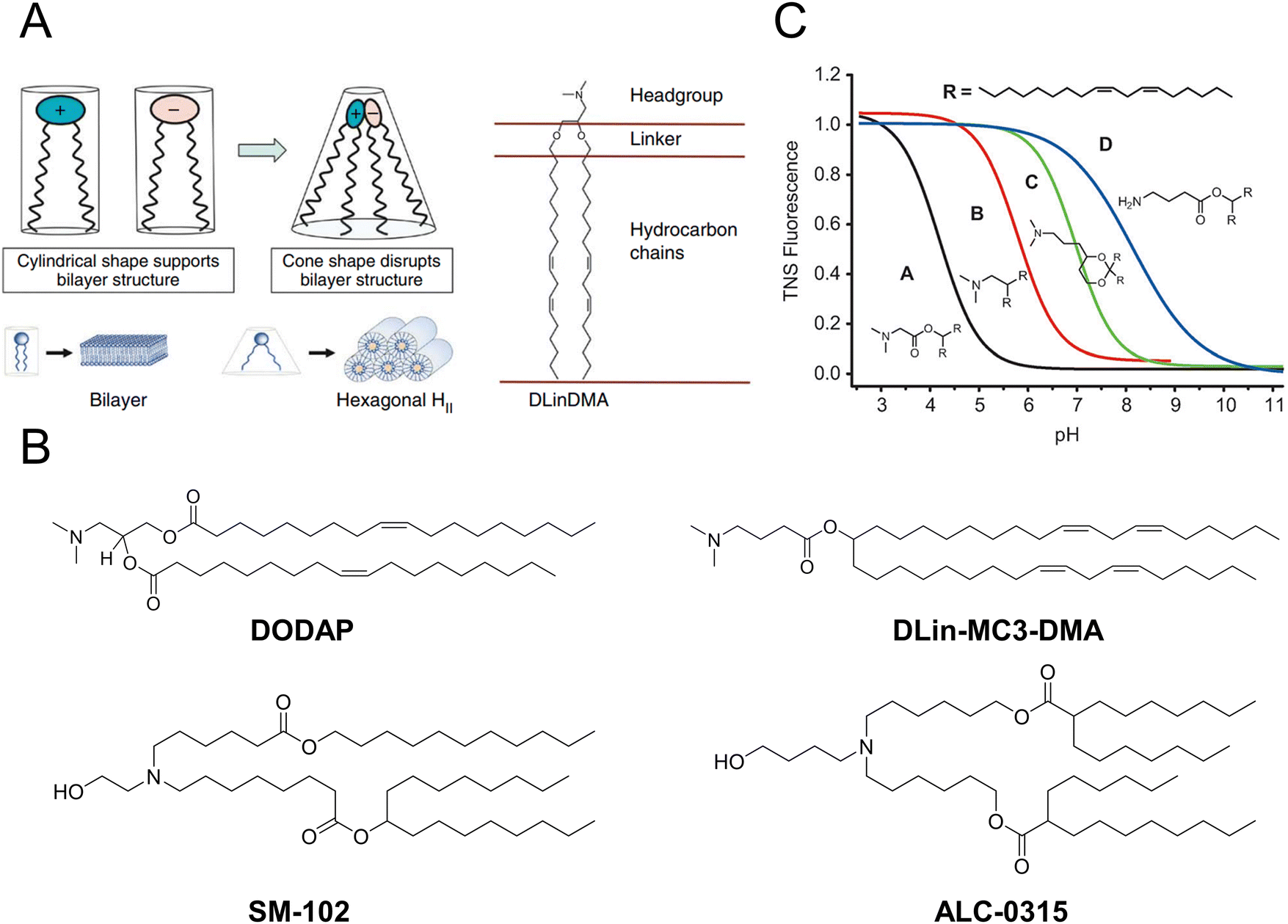 | ||
| Fig. 2 The structure and characteristics of LNPs were elucidated. (A) The ionizable lipids 1,2-dilinoleyloxy-n,n-dimethyl-3-aminopropane (DLinDMA) were composed by the headgroup, linker, and hydrocarbon chain. The ionizable lipids and anionic endosomal membrane lipids exhibited a cylindrical shape that facilitated the bilayer packing. When the ionizable and anionic lipids were mixed, the combined headgroup area was reduced compared to the sum of the individual headgroup areas due to the ion pairing. The molecular shape encouraged the nonbilayer phases, similar to the hexagonal HII phase shown here, which could cause membrane fusion or disruption because a bilayer structure could not be maintained. Adapted with permission.53 Copyright 2010, Springer Nature America. (B) The chemical structures of the mentioned ionizable lipids of DODAP, DLin-MC3-DMA, SM-102, and ALC-0315. (C) The pKa value of LNP was determined through the fluorescence titration using 2-(p-toluidine)-6-naphthalene sulfonic acid (TNS). Adapted with permission.48 Copyright 2012, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Jayaraman et al. reported that the optimal pKa range for lipids was 6.2–6.5, which plays a crucial role in facilitating efficient hepatic gene silencing in vivo for LNPs (Fig. 2(C)).48 Another study compared the gene silencing efficacy of LNPs containing different ionizable lipids, ALC-0315 and MC3, in both hepatocytes and hepatic stellate cells (HSCs). The results demonstrated that ALC-0315-based LNPs achieved potent siRNA-mediated gene silencing in both cell lines. The process for screening ionizable lipids requires extensive experimental verification and continuous optimization.51 Yu et al. reported that the premodification of cationic lipids with aminoglycosides and the utilization of LNPs could enhance the efficiency of mRNA delivery. The modified LNPs could deliver luciferase (Luc) mRNA at a dose of 0.05 mg kg−1, resulting in an average luminescence intensity of 107 (a.u.) in the liver. An engineered tdTomato mouse model was used to demonstrate the potential application of this efficient mRNA delivery system for gene editing.52
2.2 PEGylated lipids (PEG–lipids)
PEG–lipids are an essential component of LNP formulations and account for approximately 1.5% of the total molar percentage.54 Lipids contain a hydrophilic PEG polymer and a hydrophobic lipid anchor, in which the lipid domain is deeply embedded in the LNPs while the polyethylene glycol domain extended outwards.55 Moreover, PEG–lipids are commonly utilized in liposomal systems to extend the duration of circulation. These lipids function as a physical barrier that hinders the binding of plasma proteins to NPs and prevents the rapid clearance of NPs by the reticuloendothelial system (RES).56,57It was reported that PEG–lipid-containing LNPs exhibited remarkable stability in buffer at 4 °C for up to three weeks, as evidenced by the particle size, polydispersity index (PDI), zeta potential, and encapsulation efficiency measured during storage.58,59 The shelf life of the modified Moderna mRNA COVID-19 vaccine (Spikevax®) from the European Medicines Agency (EMA) is currently known to be 9 months between −25 °C and −15 °C or 30 days at 2–8 °C.60 The Pfizer-BioNTech vaccine (Comirnaty®) should be stored between −90 °C and −60 °C for up to 9 months or at 2–8 °C for up to 5 days.61,62 The transfection efficiency of LNPs was influenced by the proportion of PEG-loaded lipids incorporated in the formulation. Lokugamage et al. presented an LNP delivery system designed to efficiently deliver atomization-mediated therapeutic RNA to the lungs. The lipid composition, molar ratio, and structure of the LNPs were systematically optimized. The results revealed that the absence of PEG–lipid constituents in the LNPs had a significant impact on the stability and properties of the dispersions, thereby affecting the transfection efficiency of the LNPs. These findings confirmed the crucial role played by PEG-loaded lipids in LNP formulations.63
2.3 Role of helper lipids
Helper lipids, including cholesterols and phospholipids, were employed to enhance the stability of LNPs during storage and circulation.64LoPresti et al. compared the delivery efficiency of LNPs in which the standard helper lipid DOPE was replaced with different lipids. The findings indicated that the substitution of conventional helper lipids with anionic lipids or cationic lipids would impact the hepatic targeting of LNPs, which could lead to the redirection of LNPs towards the spleen or lung.68 In another study, scientists screened a library of 96 LNPs in a high-throughput manner and demonstrated that several formulations containing DOPE exhibited preferential accumulation in the liver. LNPs containing DSPC as a helper lipid were observed to accumulate primarily in the spleen, indicating that LNPs can be tailored for specific purposes based on the choice of helper lipids.69 Álvarez-Benedicto et al. modulated the phospholipids in LNPs to investigate the roles of helper lipids in LNP formulations. The study indicated that phospholipids containing phosphatidyl ethanolamine (PE) head groups exhibited consistent superiority in delivering mRNA both in vitro and in vivo. Zwitterionic phospholipids facilitated liver targeting, while negatively charged phospholipids redirected LNPs from the liver to the spleen.70
Patel et al. designed a library of LNPs containing hydroxycholesterols and assessed the impact of these compounds on the delivery of mRNA to T cells by exploiting endosomal trafficking mechanisms. The results suggested that replacing 25% and 50% of cholesterol with 7α-hydroxycholesterol in LNPs led to increases of 1.8-fold and 2.0-fold, respectively, in the delivery of mRNA to primary human T cells in vitro, without any additional cytotoxicity or significant impact on the stability of LNPs.4
3. Nucleic acid therapeutics
The mechanism of action for nucleic acid drugs differs from that of traditional small molecule drugs. Nucleic acid therapy utilizes RNA or DNA to regulate the expression level of target proteins after gene transcription or before protein translation, thereby employing biological mechanisms to address the chemical constraints of “undruggable” targets.73The exogenous antisense oligonucleotide (ASO) is an 18–30 nt single-stranded oligonucleotide molecule that can be a heterozygous single strand of DNA, RNA, or DNA/RNA. Upon cellular uptake, the ASO complements the target sequence and induces mRNA degradation through RNase H1 activity, thereby inhibiting protein expression.74 Alternatively, the ASO can achieve selective pre-mRNA splicing and regulate gene translation through steric effects to accomplish disease treatment.75
The therapeutic effects of small interfering RNA (siRNA), a 20–25 nucleotide double-stranded RNA molecule, are achieved through posttranscriptional gene silencing. Unlike single-stranded ASOs, siRNAs contain a ‘passenger’ (sense) strand with the same sequence as the target RNA and a complementary ‘guide’ (antisense) strand.76 Upon binding to the RNA-induced silencing complex (RISC) in the cytoplasm, siRNAs undergo passenger strand removal, thereby enabling the guide strand to function as a template for precise target recognition. Activated RISCs recognize complementary mRNAs of target genes and induce the degradation of mRNAs.77 Therefore, by designing siRNA molecules with specific sequences, researchers can accurately target and degrade specific mRNA molecules, thereby inhibiting the expression of the corresponding proteins.
mRNAs are composed of hundreds or thousands of oligonucleotide molecules and upregulate the expression of target proteins in vivo. In the cytoplasm, mRNAs function by binding to ribosomes for the translation of target proteins. These translated proteins can be intracellular, secreted by cells for uptake by other cells, or can specifically interact capable of specific interactions with targets on the cell surface. Despite being unstable both in vitro and in vivo, mRNA drugs do not require nuclear localization, thus preventing mutations caused by genomic integration.78
4. Hepatic-targeted delivery and therapy
4.1 Hepatic delivery
LNPs have been extensively studied for several decades. Like other nanomaterials, which have increased vascular permeability, nanomaterials can accumulate in regions such as tumour sites or areas with inflammation or infection.79 This phenomenon is known as the enhanced permeability and retention (EPR) effect.80 The liver, the largest abdominal organ, performs at least 500 known physiological processes, such as the synthesis of numerous biochemicals,81 the detoxification of metabolites,82 and the storage of energy.83,84 Hepatocytes comprise 80% of the liver by volume, and liver sinusoidal endothelial cells (LSECs), Kupffer cells (KCs) and HSCs accounting for only 2.8%, 2.1%, and 1.4%, respectively.85Kupffer cell (KC). KCs can remove foreign particles or red blood cells from the bloodstream through phagocytosis.86 KCs constitute only 6.5% of the liver volume, yet they represent 80–90% of the tissue macrophages within the body.87 The levels of KCs are abundant in rats aged more than 20 months,88 as well as in individuals with alcohol-related liver disease.89 KCs play a pivotal role in the pathogenesis of hepatitis, fibrosis, intrahepatic cholestasis and alcoholic liver disease.89 The activation of KCs produces proinflammatory mediators, such as prostaglandins, signaling molecules, reactive oxygen species, and others, while preventing liver inflammation.87
Sinusoidal endothelial cells (SECs). SECs are considered quiescent in terms of phagocytosis. However, upon KC impairment, the SEC acquires phagocytic competence. Comprising approximately 40% of hepatic cells, the SEC serves as a crucial barrier between the blood and hepatocytes. These intercellular spaces ranging from 50 to 200 nm and aggregate to create sieve plates that enable filtration, facilitating the diffusion of numerous substances while restricting the passage of particles 80 to 500 nm in size.90 Deposition of the extracellular matrix (ECM) results causes SECs to thicken, leading to fibrosis. Above all, the SEC plays a pivotal role in regulating haemostasis, inflammatory responses, microcirculation, and immunity.91
Hepatic stellate cells (HSCs). Eighty percent of the retinoids in the HSC house are found throughout the body. Under normal physiological conditions, HSCs are quiescent.92 The activated HSCs function as antigen-presenting cells and promote the expansion of natural killer T cell.93 HSCs stimulate the generation of nitric oxide by secreting fibronectin and vascular endothelial growth factor. Retinol is thus removed from the cell, and HSCs experience morphological alterations. Fibrosis and cirrhosis result from this process, which also causes enhanced proliferation and transdifferentiation of HSCs into fibrogenic myofibroblast-like cells that release collagen scar tissue in addition to fibrogenic and inflammatory cytokines (ECM).94
Hepatocytes. Hepatocytes constitute approximately 80% of the liver volume and play a pivotal role in the metabolism of carbohydrates, lipids, and proteins. They are also involved in the secretion of bile, clotting factors, cholesterol, and protein transporters.95 Hepatocytes are highly differentiated cells with a high capacity for replication and longevity. Hepatocytes are long-living, highly differentiated cells with a high rate of reproduction. These metabolically active cells effectively break down medications, hormones, and hazardous substances that are easily removed from the body. It is usual to refer to this process as the “first pass effect.” When there is a liver injury, hepatocellular carcinoma, chronic hepatitis, or exposure to specific toxins, there is an extensive expansion of hepatocytes and cellular damage that lasts until cirrhosis.96
Large LNPs pass through the bloodstream and are subsequently taken up by resident macrophages of the mononuclear phagocyte system (MPS). Upon entering the liver, LNPs can be recognized and internalized by KCs, thereby hindering their delivery to target cells. Once LNPs reaches the target cell, they encounter formidable barriers, including cellular internalization and endosomal escape. In addition, size and surface modification affect the route of internalization and intracellular fate. The acidic pH environment of lysosomes and the presence of cytoplasmic nucleases, particularly nucleic acids, pose significant challenges to the maintenance of cargo integrity.100,101 However, despite their relatively high delivery efficiency, only 1–4% of the phagocytosed LNPs successfully achieved lysosomal escape.102
In addition, LNPs can potentially elicit immune responses, such as inflammatory and cell-mediated immune responses. The high incidence of adverse reactions associated with LNPs could be attributed to their specific route of administration, higher drug dosages, and distinct tissue distribution pattern.103 These responses may lead to the clearance or restriction of LNPs in the body.34
Liver passive targeting. Passive targeting utilizes the properties of NPs, such as composition, particle size, and charge, to targeted deliver the drugs to specific areas within the organism. This process involves several mechanisms, such as endocytosis, fusion, adsorption, and material exchange in various tissues and cells, and can be achieved by utilizing capillary interception or leveraging the enhanced capillary permeability exhibited by the disease tissues. The liver's 100–150 nm vascular fenestration, which allows for selectivity towards either hepatocytes and HSCs or SECs and KCs on one side (>100 nm), is the primary factor influencing the passive targeting of liver cells (<100 nm). Apart from these methods, deformable nanocarriers bigger than 400 nm have the potential to extravasate into the Disse space by forceful extrusion.104
In some diseases, it is challenging to precisely deliver drugs to specific cell types in the liver through passive targeting. For example, in viral hepatitis or nonalcoholic steatohepatitis (NASH), the designated target cell is the hepatocyte;105 the KC or endothelial cell is the key target cell in acute liver inflammation;106 the HSC is the key target cell in liver fibrosis or cirrhosis;107 the bile duct epithelial cell is the key target cell in primary biliary cirrhosi;108 and the tumour cell is the key target cell in liver cance.109 Consequently, passive targeted drug delivery cannot be deemed selective targeted drug delivery.110
Liver active targeting. Active targeting involves the utilization of NPs as “missiles” to precisely deliver drugs to the desired target site, thereby enhancing therapeutic efficacy. Active targeting techniques offer the potential for targeted therapies and the mitigation of undesired side effects even within an organ.
Endogenous mechanism-mediated active targeting. Following intravenous delivery, serum proteins quickly coat nanoparticles (NPs), forming a coating known as the “protein corona”. This layer controls the biodistribution of NPs within the body and significantly alters their surface characteristics. By selectively adsorbing specific plasma proteins onto the NP surface, researchers can achieve targeted delivery to specific organs.111
ApoE is a component of the biomolecular crown that functions as an endogenous targeting ligand and is essential for the physiological clearance of lipoproteins and hepatocellular uptake.112 It is a crucial component that influences LNP-mediated nucleic acid delivery. ApoE, which is predominantly synthesized by hepatocytes and most abundant in the liver, contributes to the preferential accumulation of intravenous LNPs in the liver. LNP delivery systems target hepatocytes through ApoE, which interacts to the low-density lipoprotein receptor (LDLR) expressed on hepatocytes.113
Martin et al. investigated the relationship between the ApoE receptor and LNPs both in vitro and in vivo. They employed an exogenous ligand known as multivalent N-acetylgalactosamine (GalNAc), which has a high affinity for the hepatocyte-expressed asialoglycoprotein receptor (ASGPR). The results validated the viability of incorporating both active and passive targeting strategies for RNAi therapeutics in LNP formulations.114 In another study, small-angle neutron scattering was used by the researchers in conjunction with solvent, cholesterol, and selective lipid deuteration to clarify the composition of LNPs and the distribution of lipid components both in the presence and absence of ApoE. The binding of ApoE caused lipids to redistribute within both the shell and core regions, consequently impacting the internal structure of the LNPs and leading to mRNA release.115
Receptor-mediated active targeting. A ligand with high affinity for the receptor, such as a peptide, protein, antibody, or carbohydrate, can be attached to the surface of LNPs. LNPs can have a ligand, such as a protein, peptide, antibody, or carbohydrate, attached to their surface that has a high affinity for the receptor. LNPs can be selectively targeted by ligands to recognize specific receptors on the surface of target cells in a precise and efficient manner. This approach can specifically target relevant liver cell types associated with a disease while reducing side-effects on other liver cell types.
Kim et al. reported engineered LNPs for the specific delivery of RNA into hepatocytes and LSECs, and they examined the effects of LNP particle size and PEGylated lipid content on ApoE-mediated cellular uptake. The LNPs were modified with mannose moieties to achieve active targeting to LSECs while minimizing nonspecific cellular uptake. The selectivity and potential for actively targeting LSECs in the liver of engineered LNPs with mannose moieties surpassed those of conventional LNPs.116 In another study, researchers constructed a stable nucleic acid lipid particle delivery system loaded with mannose-modified HMGB1-siRNA (mLNP-siHMGB1) to target liver macrophages. The findings demonstrated the effective coadministration of mLNPs-siHMGB1 and DHA for the treatment of NASH.117 Zhang et al. prepared pPB peptide-modified LNPs for targeted siRNA delivery to HSCs with the aim of suppressing HMGB1 and treating liver cirrhosis. Anticipatedly, pPB-conjugated LNPs loaded with HMGB1 siRNA efficiently suppressed HSC proliferation, apoptosis, collagen deposition, and survival.118
Lipid composition-mediated active targeting. The lipid composition or structure can be customized to actively target LNPs. For example, Ni et al. reported that piperazine-containing ionizable lipids (Pi-Lipids) preferentially deliver mRNA to immune cells in vivo without targeting ligands.119 In another study, researchers encapsulated the mRNA encoding CAR in a piperazine-based LNP that targets liver macrophages. In the hepatocellular carcinoma (HCC) mice model, the LNP system significantly enhanced the phagocytic function of liver macrophages, reduced tumour burden, and prolonged survival time.120 Qiu et al. synthesized a library of lipidoids (N-series LNPs) that contained amide bonds. The in vivo results revealed that the N-series LNPs predominantly delivered mRNA to the lungs following systemic administration. Interestingly, O-series lipidoids, which incorporate ester bonds into their tails, tended to specifically deliver mRNA to the liver.111,121
In addition to the changes in ionizable cationic lipids, alterations in helper lipids impact LNP targeting. Roy et al. developed an mRNA-loaded LNP delivery system for targeted delivery to the hepatic RES. By replacing DSPC with DSPG in the Onpattro® formulation, the surface charge of LNPs were changed from neutral to negative without affecting their performance. In embryonic zebrafish and mouse models, the transformed LNPs significantly enhanced mRNA expression both globally in the liver and specifically within hepatic RES cell subpopulations.2
4.2 Treatments for hepatic disorders
Hepatic disorders have emerged as major causes of morbidity and mortality worldwide in recent decades.122 According to the global burden of disease (GBD) project, hepatic disorders, including acute hepatitis, cirrhosis, and hepatocellular carcinoma, accounted for more than 2 million deaths in 2010 and represented approximately 4% of all global fatalities.123 Despite the progress made in vaccines and antiviral medications, hepatic disorders remain a growing global burden due to several factors, including increased life expectancy, sedentary lifestyles, and overconsumption of food.124,125 It is estimated that in China, approximately one-fifth of the population suffers from hepatic disorders,126 including hepatitis B virus (HBV), hepatitis C virus (HCV), liver fibrosis,127 liver cancer, NASH,128 alcoholic hepatic disorders (ALD),129 and drug-induced liver damage. The inherent liver-targeting property of LNPs was demonstrated by the preferential accumulation of LNPs within hepatocytes.130,131 Scientists have utilized the hepatic targeting capability of LNPs and encapsulated nucleic acids in LNPs for targeted treatments of associated diseases.132Amino acid (AA) metabolism disorders. The maintenance of AA homeostasis is indispensable for all living organisms, as it profoundly influences metabolism and the pathogenesis of diseases.140 Recently, significant attention has been focused on the role of AAs as crucial signalling molecules in the regulation of energy and metabolic homeostasis.141,142 In addition to their involvement in protein metabolism, AAs function as crucial molecular regulators of glucose and lipid metabolism.143 The liver serves as the primary site for AA metabolism and harbours a diverse array of enzymes that facilitate this process, resulting in metabolic aberrations in AA that can directly impact hepatic functions.144
Individuals with fumarylacetoacetate hydrolase (FAH) mutations at birth are susceptible to the development of hepatorenal tyrosinemia type I, which can lead to renal dysfunction, liver failure, neurological impairments, and an increased risk of malignancy.145 Cheng et al. prepared dendrimer lipid nanoparticles (mDLNPs) based on 5A2-SC8 lipids to efficiently deliver FAH mRNA specifically to the liver (Fig. 3(A) and (B)).146 The mDLNPs effectively produced functional FAH protein, restored normal enzyme activity, stabilized body weight, and extended the survival of FAH−/− knockout mice. The results showed that in a mouse model, 44% of hepatocytes in the liver exhibited high levels of FAH protein. In addition, FAH−/− mice treated with FAH mDLNPs displayed similar levels of total bilirubin (TBIL), alanine aminotransferase (ALT), and aspartate aminotransferase (AST) as wild-type C57BL/6 mice and maintained a normal weight throughout the one-month treatment period (Fig. 3(C)). Another research group evaluated the effectiveness of an mRNA-loaded therapeutic approach in rescuing FAH-deficient mice. Repeated intravenous or intramuscular administration of human FAH mRNA-encapsulated LNPs resulted in the synthesis of FAH proteins in deficient mouse livers and restored normal hepatic morphology and body weight.147
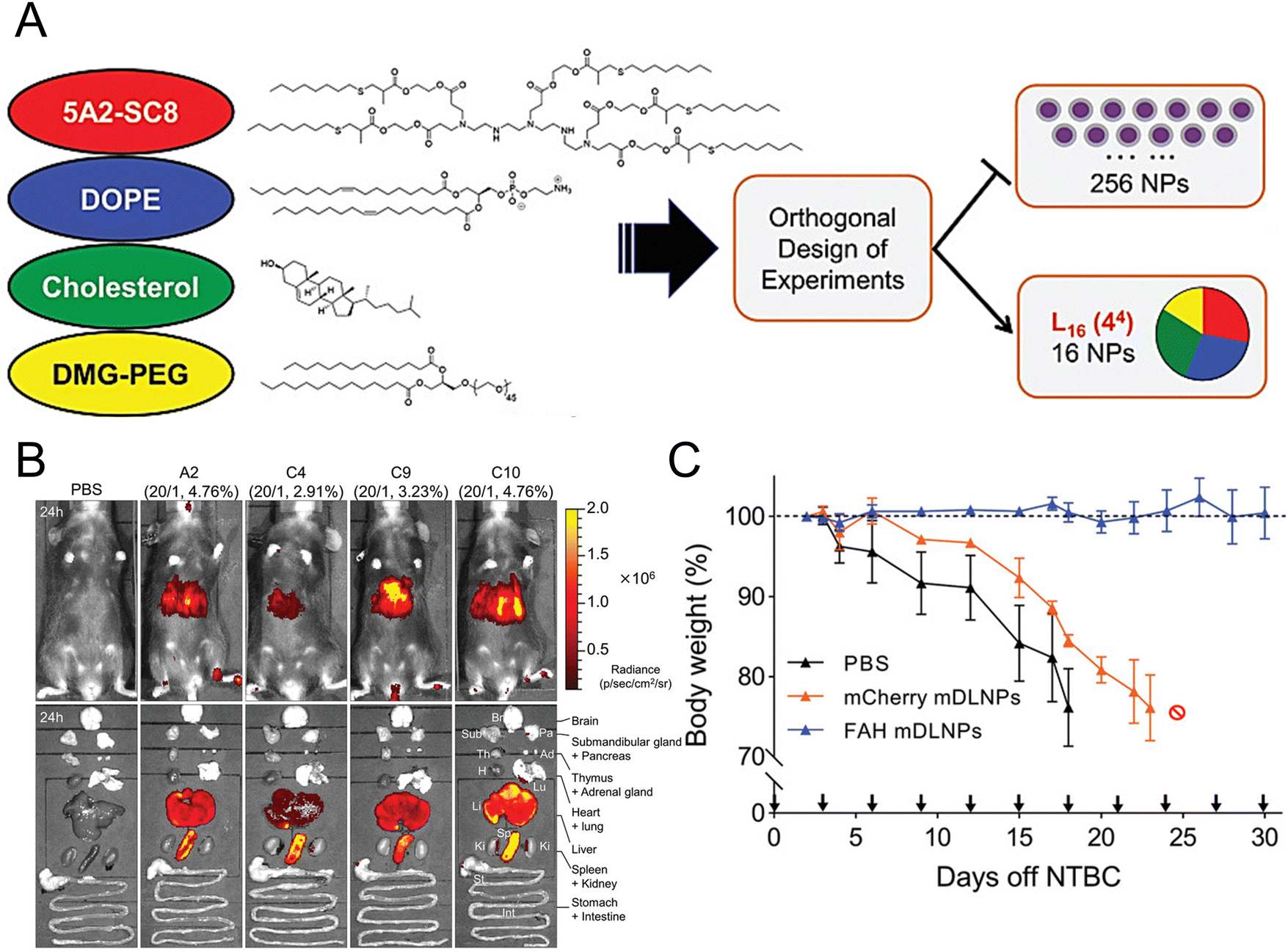 | ||
| Fig. 3 Design strategies for mDLNPs that could effectively deliver the FAH mRNA to the liver. (A) Systematic optimization of DLNPs was achieved through the design of experiment calculations. (B) Luc mRNA was loaded in the DLNP to further assess the in vivo delivery efficacy of the DLNP. (C) During a one-month therapeutic intervention study, the weights of FAH−/− mice were recorded. Adapted with permission.146 Copyright 2018, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Phenylketonuria (PKU) is an inborn metabolic condition caused by a defect in the functioning phenylalanine hydroxylase (PAH), which results in the accumulation of phenylalanine (Phe) in the blood and organs of patients. This disease can cause developmental delay, neurological deficits, and behavioural abnormalities. Cacicedo et al. used mouse PAH mRNA-loaded LNPs for the treatment of PKU mouse models.148 After repeated administrations of LNPs, the accumulation of Phe in the serum, liver, and brain rapidly decreased. Truong et al. systemically administered human arginase 1 (hARG1) mRNA-encapsulated LNPs to a murine ARG1 knockout model. The findings revealed that the level of hepatic arginase expression and activity were increased, resulting in the reinstatement of ureagenesis in the liver. Compared to the negative control group, the experimental group treated with LNPs exhibited prolonged survival and maintained a normal weight without any evidence of hepatotoxicity. Furthermore, normal levels of ammonia and arginine were detected in the plasma, while the amount of hepatic guanidinoacetic acid, a type of guanidine compound, was reduced.149
Organic acid metabolism disorders. Organic acid metabolism disorders, also known as organic acidaemia or organic aciduria, are genetic hepatic disorders caused by deficiencies in nutrient metabolism enzymes that lead to the accumulation of carboxylic acids and their metabolites.150 The primary clinical manifestations include acute acidosis, hyperammonaemia, and toxic encephalopathy in neonates and infants.151
Jiang et al. proposed an enzyme replacement strategy for the management of propionic acidaemia (PA). A rare and potentially fatal inherited metabolic disease can be caused by the absence of mitochondrial propionyl-CoA carboxylase (PCC), which consists of six alpha-PCC (PCCA) and six beta-PCC (PCCB) subunits. Scientists have used human PCCA mRNA and human PCCB mRNA-encapsulated biodegradable LNPs to successfully produce functional PCC enzymes in the liver.152 In another study, a human methylmalonyl-CoA mutase (hMUT) mRNA-loaded biodegradable LNP was developed for the treatment of methylmalonic acidemia (MMA), a metabolic disorder with limited therapeutic options. After LNPs were administered in two distinct mouse models of MMA, a significant reduction (ranging from 75% to 85%) was observed in the plasma level of methylmalonic acid, while the level of hepatic expression and activity of the hMUT protein increased. Heterozygous MMA-treated mice showed significant improvements in survival and weight gain but exhibited no signs of hepatic toxicity or inflammation.153
Glycogen storage disease (GSD). GSD is a congenital genetic metabolic disorder caused by the deficiency of various enzymes involved in glycogen metabolism, resulting in impaired glycogen storage or breakdown.154 Adequate glucose levels are essential for muscles and organs to properly function. The incidence rate of glycogen storage disorders is approximately 1 in 20
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 25000 newborns.155 The prevalent types of GSD included types I, II, III, and IV, with type I being the most common. Patients might suffer from life-threatening hypoglycaemia and long-term hepatic complications. Cao et al. developed a novel treatment for GSD 1a, a type of GSD I, by utilizing LNPs to encapsulate human G6Pase-α-encoded mRNAs. This approach exhibited excellent tolerability and efficacy upon repeated dosing in a murine model.156
000 to 25000 newborns.155 The prevalent types of GSD included types I, II, III, and IV, with type I being the most common. Patients might suffer from life-threatening hypoglycaemia and long-term hepatic complications. Cao et al. developed a novel treatment for GSD 1a, a type of GSD I, by utilizing LNPs to encapsulate human G6Pase-α-encoded mRNAs. This approach exhibited excellent tolerability and efficacy upon repeated dosing in a murine model.156
Hyperbilirubinemia. Crigler–Najjar syndrome (CN1) is a rare disorder that affects bilirubin metabolism, and results from mutations in uridine diphosphate glucuronosyl transferase 1A1 (UGTIAI).157 The disease is characterized by elevated levels of bilirubin, resulting in jaundice. In one study, Greig et al. demonstrated the therapeutic efficacy of human UGTIAI mRNA-encapsulated LNPs for the treatment of hyperbilirubinemia. The LNPs could effectively deliver mRNA to the liver and hepatocytes (the primary site of UGT1A1 activity) via receptor-mediated endocytosis. Repeated administration of the mRNA resulted in a frequency-dependent sustained increase in total bilirubin levels. Forty-two days postadministration, the serum total bilirubin levels in adult UGTIAI knockout mice returned to normal.158
Cholestasis. Wei et al. developed hABCB4 mRNA-loaded LNPs for the treatment of progressive familial intrahepatic cholestasis (PFIC3). PFIC3 is an incurable rare paediatric hepatic disorder characterized by impaired bile flow and progressive liver damage, and the only current therapeutic option is liver transplantation. The pharmacological effects of this approach were assessed both in vitro and in a BALB/c Abcb4−/− mouse model, which is susceptible to fibrosis. The results demonstrated that treatment with liver-targeted LNPs led to the re-expression of the functional hABCB4 protein and the restoration of phospholipids that were transported in cultured cells as well as in the livers of PFIC3 mice. Subsequent repeated injections of the LNPs effectively alleviated severe disease-related parameters, including inflammation, ductular reactions, and liver fibrosis, in Abcb4−/− mice.159 The above findings demonstrated the therapeutic potential of LNPs for treating inherited metabolic disorders that affect the liver.
Yang et al. developed a siRNA-loaded delivery system called cRGD-PSH-NPs, which were made of polyethyleneimine (PSH), egg phosphatidylcholine, cholesterol, DSPE-PEG2000, and survivin siRNA with DSPE-PEG2000-cRGD as the targeting ligand to enhance the specificity. The results demonstrated that compared to the unmodified NPs, the modified NPs exhibited superior gene silencing and antitumour activity in HepG2 cells. Furthermore, the modified nanoparticles demonstrated significant tumour inhibition of 74.71% in HepG2 tumour-bearing nude mice without eliciting any toxicity.163 In another study, researchers developed a novel siRNA-loaded delivery system called sTOLP, which consists of multifunctional LNPs modified with the cell-penetrating peptide oleoyl-octaarginine (OA-R8) and transferrin to deliver siRNA. The sTOLP NPs consisted of a protamine-complexed siRNA core, OA-R8, cationic and PEGylated lipids, and transferrin as a targeting ligand. Asymmetrically distributed cationic lipids could enhance the formation of ion pairs that have opposite charges to the lipids in the target membrane, thereby facilitating the endosomal escape of siRNA. Compared to other LNPs, sTOLP demonstrated a remarkable tumour inhibition efficacy of 61.7% in vivo and exhibited selective uptake by both hepatocytes and tumour cells in HepG2-bearing nude mice without inducing immunogenicity or hepatic or renal toxicity.164 Yu et al. screened LNPs incorporating lipids containing tris(2-aminoethyl)amine (TREN) and three linoleyl chains, called TRENL3, which demonstrated exceptional efficacy in siRNA transfection. Furthermore, the LNP formulation's incorporation of polyunsaturated fatty acids, such as linoleic and linolenic acid, improved the efficiency of siRNA delivery even more. These novel LNPs were found to be efficiently taken up by hepatocellular carcinoma cells in mice.165
Woitok et al. prepared an LNP-based delivery system to enhance the therapeutic potential of c-Jun N-terminal kinase 2 siRNA (siJnk2) for HCC treatment, which plays a crucial role in the inflammatory circuit underlying HCC. The LNP system contained the cationic aminolipid XL52, DSPC, cholesterol, and PEG lipid. The formulation demonstrated efficient hepatic accumulation and effectively suppressed Jnk2 expression in hepatocytes, ultimately resulting in reduced carcinogenesis in an advanced liver cancer mouse model.166 In another study, researchers developed ultrasmall LNPs (usLNPs) that delivered both sorafenib and midkine siRNA to treat hepatocellular carcinoma in mice. The usLNPs were composed of a novel fusogenic lipid, YSK05, in addition to a variety of phospholipids. A microfluidic device was utilized to optimize the particle size to 60 nm, aiming to enhance tumour penetration efficiency. The results demonstrated that the novel combination effectively eliminated HCC in mice to 85% at an unexpectedly low dose of sorafenib (2.5 mg kg−1), which could not be achieved through treatment with sorafenib alone. Furthermore, this approach eradicated 70% of sorafenib-resistant HCC in mice, indicating its potential for clinical applications in HCC treatment.167
Rybakova et al. established an in vitro-transcribed mRNA-encapsulated LNP system for the in vivo delivery of trastuzumab, a monoclonal antibody that targets HER2/ERBB2 and is commonly used for cancer treatment. Compared to the administration of trastuzumab protein, intravenous delivery of LNPs resulted in an improved pharmacokinetic profile for the produced protein and enhanced efficacy in mice bearing tumours. Following administration, the LNPs demonstrated remarkable efficacy in reducing tumour volume and enhancing survival rates in HER2-positive mouse tumour models.168 In another study, researchers have developed LNPs coencapsulated with anti-microRNA (miRNA) 27 and sorafenib, called G-S27LN. They found a distinctive cationic switchable lipid (CSL3) that was applied to encapsulate miRNA and impart pH-responsive properties to the LNPs. CSL3/DSPC/Chol/DSPE-PEG/DSPE-PEG-MAL, conjugated with the anti-Glypican 3 antibody, in a molar ratio of 40/25/20/10/5 made up the final composition of the lipids. G-S27LN demonstrated a specific affinity for HepG2 cancer cells overexpressing GPC3. In liver cancer mouse models, LNPs significantly suppressed tumour burden and reduced tumour cell proliferation without any observed toxicity.169 Lai et al. reported that interleukin-12 (IL-12) mRNA-encapsulated LNPs could effectively impede the progression of MYC oncogene-driven HCC. Compared to those in the control group, the MYC-induced HCC transgenic mice treated with the mRNA-encapsulated LNPs exhibited increased survival rates and a significant reduction in liver tumour burden, as determined by dynamic magnetic resonance imaging.170
Wang et al. demonstrated the essential role of both C-C-chemokine ligand 2 (CCL2) and C-C-chemokine ligand 5 (CCL5) in the development of liver malignancies. To counteract the immunosuppressive process, a highly potent and specific single-domain antibody called BisCCL2/5i was developed to bispecifically bind and neutralize CCL2 and CCL5. The mRNA encoding BisCCL2/5i was encapsulated in a clinically approved LNP platform containing DLin-MC3-DMA, DOPE, C14-PEG2000 and cholesterol. Combined therapy with BisCCL2/5i mRNA-loaded LNPs and a PD-1 ligand (PD-L1) inhibitor generated a robust antitumour effect and improved survival in syngeneic mouse models of primary hepatocellular carcinoma, liver metastasis of colorectal cancer, and liver metastasis of pancreatic cancer.171 In another study, researchers investigated the effectiveness of a multiplexed dendrimer LNP delivery system for precise gene editing in tumours (Fig. 4(A)). The system utilized 5A2-SC8 as the ionizable amino lipid dendrimer, along with DOPE, PEG lipid, and cholesterol. This approach utilized focal adhesion kinase (FAK) siRNA, Cas9 mRNA, and small guide RNA (sgRNA) to enhance cellular uptake and tumour penetration, resulting in an over 10-fold increase in gene-editing efficiency within tumour spheroids by suppressing FAK (Fig. 4(B)).172 The siFAK + CRISPR-PD-L1-LNPs system effectively mitigated ECM stiffness and disrupted PD-L1 expression via CRISPR/Cas gene editing, resulting in significant inhibition of tumour growth and metastasis in a liver cancer mouse model (Fig. 4(C) and (D)).
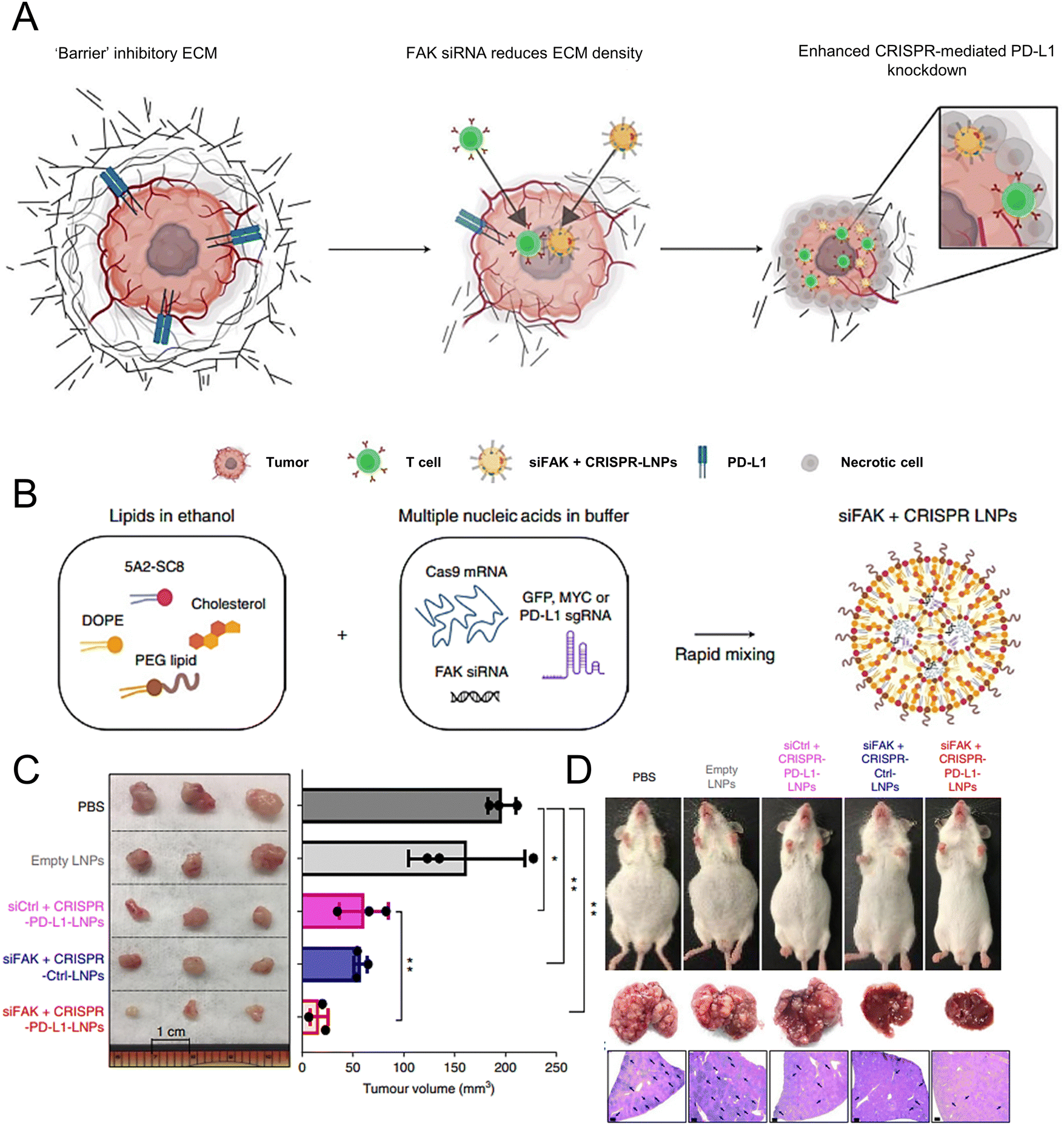 | ||
| Fig. 4 The designed LNPs (siFAK + CRISPR-LNPs) for the tumour-targeted gene editing. (A) Schematic representation of the tumour targeting mechanisms to enable the double-checkpoint blockade therapy. (B) Triple loading of the FAK siRNA, Cas9 mRNA, and sgRNA into the 5A2-SC8 LNPs was schematically illustrated. (C) The tumour size demonstrated the therapeutic effectiveness of the siFAK +CRISPR-PD-L1-LNPs in vivo. (D) Representative whole-body and liver images of the mice treated with different LNPs for 55 days. Adapted with permission.172 Copyright 2022, Springer Nature America. | ||
In one study, researchers reported cationic lipid 2-dioleyloxy-N,N-dimethyl-3-aminopropane (DODMA)-based LNPs for the delivery of the liver-specific tumour suppressor miR-122 to HCC cells. The system, called LNP-DP1, consisted of DODMA, egg phosphatidylcholine, cholesterol, and cholesterol–polyethylene glycol (Chol–PEG). These findings suggested that Chol–PEG containing LNP-DP1 showed optimal delivery efficiency. This study demonstrated that the systemic delivery of miR-122 via LNP-DP1 significantly inhibited the expression of miR-122 target genes in both normal liver and diethylnitrosamine (DEN)-induced liver tumours in miR-122 knockout mice. Furthermore, the use of LNP-DP1 containing miR-122 markedly inhibited the growth of HCC xenografts in a mouse model.173 In another study, Wang et al. incorporated oleic acid (OA) into the LNP formulation and observed a significant enhancement in the delivery efficiency of siRNA and miRNA. They formulated a series of cationic LNPs with varying helper lipids, including cholesterol, PCs, and unsaturated fatty acids. For confirmation, miR-122 was chosen and encapsulated in LNPs to demonstrate the hepatic delivery efficacy of LNPs in both tumour cell and mouse models. Compared to Lipofectamine 2000, the OA-incorporated LNPs delivered miR-122 more efficiently, resulting in a 1.8-fold increase in mature miR-122 expression and a 20% decrease in the amount of B-cell lymphoma-w, a target of miR-122. The results indicated that OA-incorporated LNPs exhibit significant potential as RNA-based therapeutic nanocarriers for targeted therapy of hepatic disorders.174
Zhang et al. utilized pPB peptide-modified LNPs to deliver high mobility group box-1 (HMGB1) siRNA and heat shock protein 47 (HSP47) siRNA, resulting in enhanced therapeutic efficacy for the treatment of liver cirrhosis. The components and molar ratios employed for the preparation of pPB-modified LNPs were as follows: DSPE-PEG-pPB:DLin-MC3-DMA:PEG-DMG:DSPC:cholesterol in a ratio of 2:40:3:10:45. The results showed that the HMGB1-siRNA-encapsulated LNPs exhibited superior therapeutic efficacy for liver cirrhosis in murine models compared to that of the HSP47-siRNA-encapsulated LNPs. LNPs can target HSCs, prevent activation and proliferation, silence HMGB1 gene expression, reduce protein release, suppress the development of fibrosis in the liver, and increase survival time in cirrhotic mouse models through the pPB peptide.180 Jia et al. also prepared stable polypeptide pPB-modified nucleic acid LNPs, pPB-SNALPs, with diameters ranging from 110 to 130 nm. The components used in the preparation included DSPE-PEG-pPB, DLin-MC3-DMA, PEG-DMG, DSPC, and cholesterol. These LNPs were specifically designed for the targeted delivery of HSP47 siRNAs to the liver for the treatment of hepatic fibrosis. Compared to unmodified SNALP, pPB-SNALPs exhibited greater liver distribution and HSC uptake in vivo, as well as greater uptake by both LX-2 cells and primary mouse HSCs in vitro. Moreover, pPB-SNARPs exhibited an augmented inhibitory effect on thioacetamide-induced hepatic fibrosis in a murine model. These findings suggested that pPB-SNALPs have great potential as drug delivery systems for gene therapy for liver fibrosis.181
Han et al. synthesized AA-lipidoids, which are lipidoids functionalized with anisamide (AA) ligands, and identified the highly potent AA-T3A-C12 lipid for targeted RNA delivery to activated fibroblasts (Fig. 5(A) and (B)). The ethanol lipid solution, comprising AA-T3A-C12, DSPC, C14-PEG, and cholesterol, was rapidly mixed with an acidic aqueous solution containing HSP47 siRNA in a microfluidic device to formulate AA-T3A-C12/siHSP47 LNPs. Compared with MC3-based LNPs, AA-T3A-C12-based LNPs showed superior RNA delivery and transfection efficiency in activated HSCs. In a mouse model of liver fibrosis induced by carbon tetrachloride (CCl4), AA-T3A-C12 achieved approximately 65% gene silencing of HSP47, resulting in a significant reduction in collagen deposition and hepatic fibrosis (Fig. 5(C) and (D)).1 The results demonstrated the potential of AA-lipidoids as a targeted antifibrotic therapy against activated fibroblasts. Lawitz et al. reported an efficient and safe LNP formulation, BMS-986263, which delivered siRNA molecules that targeted HSP47 for the treatment of advanced fibrosis. This phase 2 study was designed to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics of BMS-986263 once every week for 12 weeks in patients with advanced HCV-related hepatic fibrosis. After treatment, the plasma concentrations of BMS-986263 increased in a dose-proportional manner without any significant accumulation during weekly dosing. Patients with liver fibrosis showed significant improvement.182
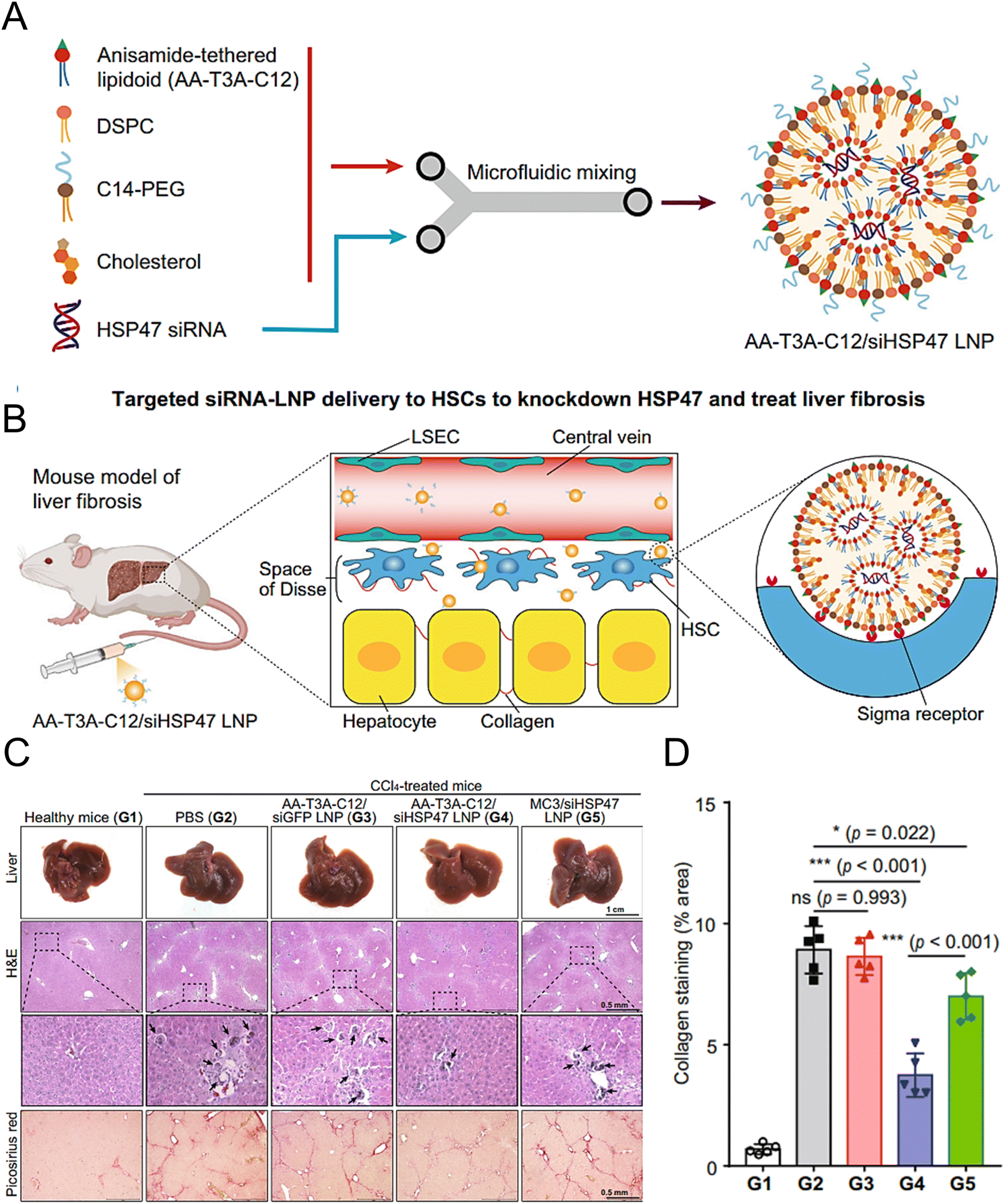 | ||
| Fig. 5 Design and validation of the AA-T3A-C12 LNP for the targeted delivery to HSCs. (A) Preparation of the ligand-tethered lipidoid nanoparticles for the targeted delivery of siRNA to the HSCs in the treatment of liver fibrosis. (B) The AA-T3A-C12/siHSP47 LNP was delivered specifically to activate HSCs for the treatment of liver fibrosis by suppressing HSP47 expression. The PEGs on the surface of the LNPs were removed during the blood circulation in vivo, exposing the multivalent anisamide ligands on the surface of the LNPs that strongly bind with the Sigma receptors overexpressed on the activated HSCs for uptake. (C) Macroscopic, histopathological, and biochemical analyses were conducted to evaluate the liver fibrosis. (D) Morphometric analysis of the picrosirius red stained areas using ImageJ software. Adapted with permission.1 Copyright 2023, Springer Nature. | ||
In one study, cationic lipid C12-200-based LNPs were used to target procollagen α1(I) siRNA to treat liver fibrosis in mice. The results indicated that the LNPs could target activated HSCs and reduce collagen deposition, thereby impeding the progression of liver fibrosis. The LNP delivery platform demonstrated minimal adverse effects, rendering it a promising therapeutic option for treating fibrotic hepatic disorders.183 Qiao et al. developed an efficient LNP delivery system that specifically targeted HSCs by encapsulating two siRNAs, collagen I (Co1lαl) to inhibit collagen synthesis and tissue inhibitor metalloproteinase-1 (TIMP-1) to promote collagen degradation. The hyperbranched lipoid-based LNPs were composed of amphiphilic cationic hyperbranched lipids (C15-PA) and the helper lipid Chol-PEG-VA. The functional LNPs exhibited remarkable delivery capability and transfection efficiency in vitro. In vivo, the dual siRNA-loaded LNPs significantly reduced collagen accumulation in fibrotic mice to levels approaching those of the control group. Repeated administration of LNPs did not cause notable toxicity or tissue inflammation.184
Yang et al. developed biodegradable LNPs loaded with human transcription factor hepatocyte nuclear factor alpha (HNF4A) mRNA and investigated the restoration of hepatic functions in various mouse models of liver fibrosis. HNF4A directly targets and activates paraoxonase 1, thereby regulating macrophages and stellate cells to reduce liver fibrosis. Repeated administration of HNF4A mRNA-encapsulated LNPs exhibited a potent therapeutic effect on four distinct murine models of liver fibrosis induced by hepatotoxins and cholestasis. Based on preclinical evidence, HNF4A mRNA therapy may be a promising strategy for reversing liver fibrosis and cirrhosis.185 In another study, Hu et al. developed an LNP system that encapsulated the relaxin gene and a miR-30a-5p mimic and selectively targeted activated HSCs in fibrotic liver tissue. Researchers previously believed that the antifibrotic peptide hormone relaxin directly reverses hepatic stellate cell activation, thereby facilitating the resolution of liver fibrosis. Upon the uptake of LNPs by activated HSCs, hepatic macrophages undergo a phenotypic transition from profibrogenic to proresolving. Combined gene therapy confirmed synergistic antifibrotic effects in murine models of liver fibrosis.186
Qiu et al. constructed a lipid-based LNP delivery platform, which consisted of 306-O12B, cholesterol, phospholipids (DSPC, DOPE, and DOPC), and DMG-PEG2000, to edit the Angptl3 genome in vivo. In the livers of wild-type C57BL/6 mice, this formulation demonstrated specific and effective Angptl3 gene knockdown, which also led to significant decreases in the levels of LDL cholesterol and triglycerides.190 Musunuru et al. used LNPs for the efficient and precise modification of disease-related genes in live cynomolgus monkeys. After a single injection of LNPs in the mouse model, the levels of proprotein convertase subtilisin/kexin type 9 (PCSK9) and LDL cholesterol simultaneously reduced by approximately 90% and 60% in vivo, respectively, and remained stable for at least 8 months.191
Suzuki et al. developed a degradable lipid L101, which was incorporated into LNPs that exhibited potent and sustained gene-silencing activity in mice. The LNPs were formulated by combining equal amounts of L101 with the internal standard lipid, following the composition ratio of L101/L021/DSPC/cholesterol/PEG-DMG as 30/30/8.5/30/1.5. The median effective dose of siRNA was approximately 0.02 mg kg−1. The L101-based LNPs underwent rapid hepatic metabolism and exhibited excellent tolerability in rats. A single intravenous administration of the PCSK9-targeting siRNA in cynomolgus monkeys resulted in greater than 90% protein silencing, with a measurable reduction in plasma LDL cholesterol levels of 50%, which persisted for two months.192 In another study, researchers examined the effectiveness of a library of biodegradable LNPs in delivering ASOs to both human cell cultures and mouse models. LNPs based on three lipids, namely, 113-O14B-3, 113-O16B-3, and 306-O12B-3, were identified as effective vectors for delivering GFP-silencing ASOs to cells expressing GFP, and helper lipids, such as cholesterol, DOPE, and PEG lipid, were added to the formulation. Compared to Lipofectamine 2000, the LNPs demonstrated superior delivery efficiency and exhibited lower cytotoxicity in vitro. Subsequently, the effectiveness of these LNPs in facilitating PCSK9 mRNA knockdown through the delivery of the PCSK9-silencing ASO to the mouse liver was evaluated.193
Tadin-Strapps et al. developed a novel mouse model that was genetically modified to carry a single copy of the human CETP transgene and a single copy of an LDL receptor mutation. Compared to normal mice, the novel model showed elevated levels of plasma LDL cholesterol even when a low-fat diet was consumed. The six ApoB siRNA sequences were encapsulated in the LNP system, which contained the cationic lipid CLinDMA, cholesterol, and PEG-DMG. LNP formulation effectively induced hepatic ApoB mRNA knockdown in a mouse model, resulting in significant reductions in the expression of serum ApoB proteins and fatty acid-related genes and sustained decreases in the levels of serum total cholesterol, triglycerides, and LDL.194
Nonalcoholic steatohepatitis (NASH). NASH is a hepatic lipid metabolism disorder caused by nonalcoholic and toxin-related factors.125 The disorder initially begins with the accumulation of fat in hepatocytes, progressing to steatohepatitis, cirrhosis, and ultimately hepatocellular carcinoma. Cholesterol was identified as an important molecule involved in this process.195
Rizvi et al. reported a highly efficient and safe LNP delivery system composed of nucleoside-modified mRNA loaded with hepatocyte growth factor (HGF) and epidermal growth factor (EGF) mRNA. The in vitro results demonstrated that LNPs could efficiently transfect all hepatocytes in the liver, including endothelial cells and KCs. Furthermore, HGF mRNA-loaded LNPs demonstrated remarkable efficacy in promoting hepatocyte proliferation. In a murine model of chronic liver injury resembling NASH, the codelivery of HGF and EGF mRNA via LNPs significantly improved hepatic steatosis and accelerated the restoration of liver functions.196 In another study, researchers employed a targeted LNP delivery system modified with the mannose receptor for the delivery of the HMGB1 siRNA mLNP-siHMGB1 and coadministered docosahexaenoic acid (DHA) for NASH treatment (Fig. 6(A)).197 mLNP-siHMGB1 was prepared using DSPE-PEG-Man, DLin-MC3-DMA, PEG-DMG, DSPC, and cholesterol. The specific targeting of macrophages via mannose receptors by mLNP-siHMGB1 effectively silenced the HMGB1 gene and reduced HMGB1 protein release in the liver (Fig. 6(B) and (C)). This process led to the modulation of liver macrophages towards an anti-inflammatory M2 phenotype, resulting in the alleviation of hepatic lobular inflammation and bullous steatosis in the NASH mouse model (Fig. 6(D)). Moreover, hepatic function in NASH model mice was restored following 8 weeks of combined treatment with mLNPs-siHMGB1 and DHA.
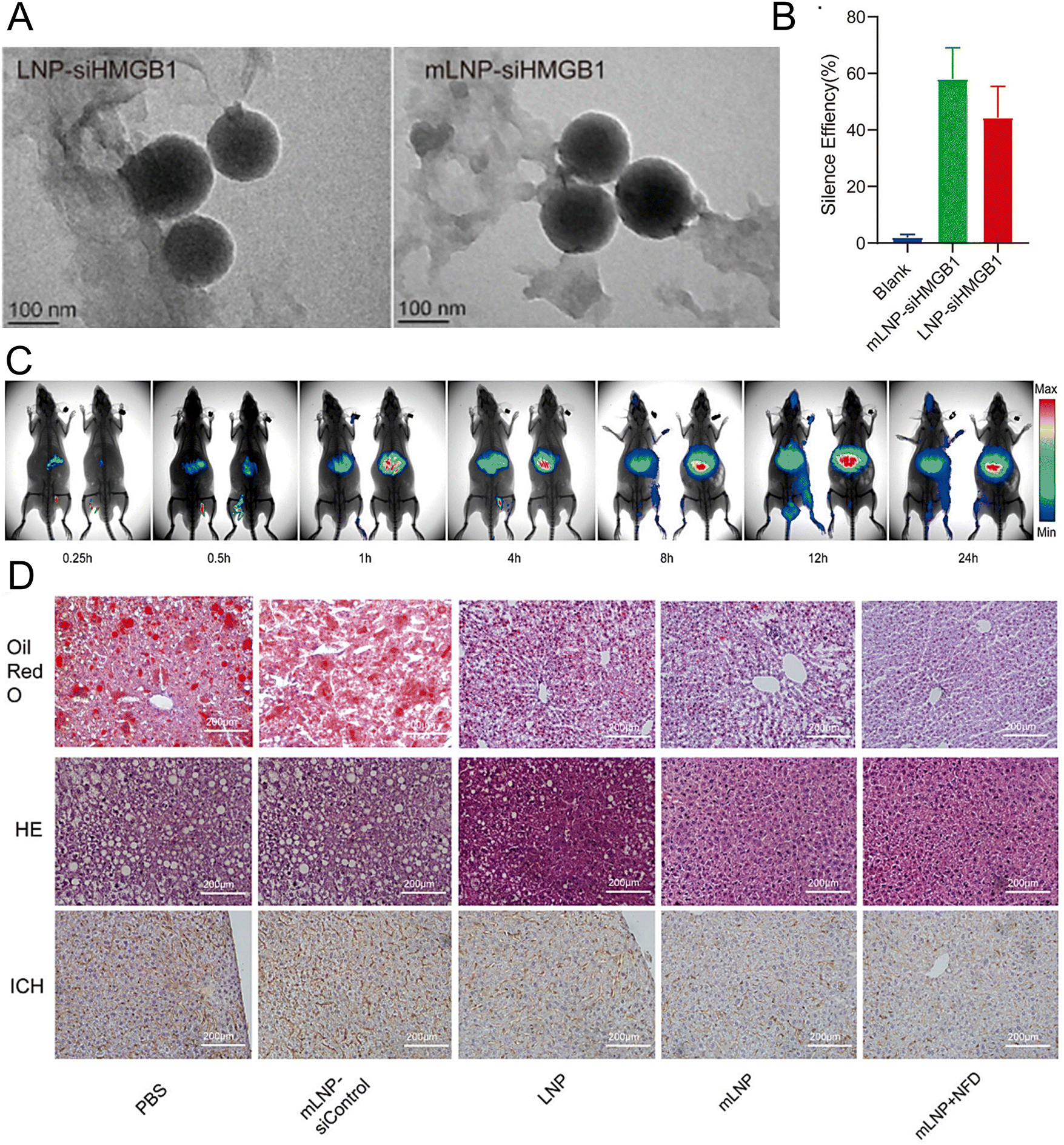 | ||
| Fig. 6 The treatment of NASH can be achieved by targeting at the mannose receptor pathway. (A) The LNP-siHMGB1 and mLNP-siHMGB1 exhibited spheroidal structures in TEM images. (B) The efficacy of LNP-siHMGB1 and mLNP-siHMGB1 in gene silencing. (C) After intravenous injection, the fluorescence intensity of the liver was significantly higher in the mLNP-siHMGB1 group (on the right) than the LNP-siHMGB1 group (on the left). (D) The hepatic steatosis and liver inflammation were effectively ameliorated following the treatment with mLNP-siHMGB1, as evidenced by the oil red O staining, HE staining, and immunohistochemical analysis of the liver tissues along with the corresponding quantitative results. Adapted with permission.197 Copyright 2022, Elsevier B.V. | ||
Homozygous familial hypercholesterolemia (HoFH). Familial hypercholesterolemia (FH) is an inherited disorder resulting from mutations in key genes involved in LDL catabolism, leading to elevated levels of LDL cholesterol. HoFH is clinically characterized by the presence of multiple xanthomas and tendon xanthomata, as well as a predisposition to the early onset of atherosclerotic cardiovascular disease.198,199
Kasiewicz et al. developed an LNP formulation delivery system loaded with adenine base editor mRNA and gRNA that targets ANGPTL3. LNPs employ GalNAc as a ligand to specifically target the liver. In the cynomolgus monkey model of HoFH, after treatment with the formulated LNPs, the levels of the ANGPTL3 protein in the whole liver and blood of the cynomolgus monkey were reduced by approximately 60% and 94%, respectively, whereas the standard LNPs yielded minimal editing. Moreover, the editing efficiency achieved by the formulated LNPs in the wild-type model was comparable to that achieved by the unmodified LNPs. Thus, a potent new tool was provided for the efficient in vivo delivery of nucleic acid.200
Acute liver injury. Acute liver injury is a condition in which the liver becomes inflamed due to factors such as drug toxicity, viral infection, excessive alcohol consumption, immune response, and radiation damage.201 Inflammation often accompanies acute liver injury, resulting in hepatocyte damage and apoptosis as well as elevated levels of transaminases in the blood that can impair normal hepatic functions.202
Kawase et al. developed biodegradable LNPs based on asymmetric ionizable lipids, which included the ionizable lipids L120, DSPC, cholesterol, and PEG-DMG. These LNPs demonstrated both high potency and excellent tolerability. These LNPs were encapsulated in siRNA targeting interferon regulatory Factor 5 (Irf5), which was subsequently assessed for its anti-inflammatory efficacy in mice. In vitro studies demonstrated that LNPs could be taken up by tissue-resident macrophages, particularly liver macrophages. The results indicated that compared to the control group, the mice injected with the Irf5 siRNA-encapsulated LNPs exhibited significantly lower levels of serum AST, ALT, and inflammatory cytokines.203 Yin et al. developed a novel delivery system that incorporated glycyrrhizic acid (GA) and polyene phosphatidylcholine (PPC) into LNPs, along with DOTAP, DSPE-PEG, and cholesterol. The modified LNPs were designed to enhance cellular uptake, promote gene silencing efficacy, increase siRNA stability, and reduce cytotoxicity. Subsequently, it was discovered that modified LNPs loaded with nuclear factor kappa-B (NF-κb) siRNA can mitigate acute liver injury, as supported by the results of histological examination, haematological analysis, and cytokine profiling. These findings suggest that LNP formulations are valuable tools for investigating macrophage function in vivo and hold promise as potential therapeutic agents for inflammatory diseases in the future.204
Hepatitis B virus (HBV). Sato et al. reported that the incorporation of GalNAc into LNPs could effectively reduce hepatotoxicity, particularly when a significant amount of LNPs was accumulated in the liver. Furthermore, the LNP system was encapsulated with YSK13-C3/chol/DSG-mPEG2k/GalNAc3-PEG2k-DSG. In a chimeric mouse model of HBV infection, the administration of LNPs resulted in a significant reduction in both the amount of HBV genomic DNA and relative antigens without indication of toxicity.205 In another study, an advanced mRNA-LNP drug encoding three anti-HBsAg antibody genes, G12-scFv, G12-scFv-Fc and G12-IgG, was developed for the treatment of HBV. The LNP formulation was prepared using SM-102, DSPC, cholesterol, and DMG-PEG2000. The results showed that the injection of a single dose of LNPs containing both G12-scFv-Fc mRNA and G12-IgG mRNA significantly reduced the serum HBsAg level in treated mice within 30 days.206
Hepatitis C virus (HCV). Dallas et al. utilized short synthetic shRNA (sshRNA)-encapsulated LNPs to silence the expression of genes associated with the HCV internal ribosome entry site. LNPs consisted of cholesterol, DPPC, PEG-C-DMA, and the cationic lipid DLinDMA at a molar ratio of 34.3
:7.1:1.4:57.1. In vivo imaging demonstrated efficient hepatic uptake and sustained retention of sshRNA in the liver, with a half-life of approximately 3 weeks for clearance after a single administration. These results suggested that sshRNA-loaded LNPs show promise as therapeutic agents for HCV.207
Haemophilia A (HA) and Haemophilia B (HB). Haemophilia is a hereditary bleeding disorder caused by an abnormal gene that encodes a coagulation factor, resulting in spontaneous or prolonged bleeding after minor injuries. Haemophilia is an X-linked recessive haemorrhagic disorder, predominantly occurs in male patients and is rare in female patients. Patients with HA and HB lacked coagulation factor VIII (FVIII, F8I22I) and coagulation factor IX (FIX, F9Mut), respectively.208
Han et al. developed and optimized LNPs for the efficient delivery of both Cas9 mRNA and a single guide RNA targeting antithrombin (AT) to the liver.209 LNPs were formulated using 246C10, DOPE, cholesterol, and PEG–ceramide (Fig. 7(A)). The endogenous regulator AT negatively modulates the generation of thrombin. From the results obtained in the HA and HB mouse models, it was observed that the bleeding-associated phenotypes were restored without liver-induced toxicity (Fig. 7(B)–(D)). Furthermore, notable immune reactions against Cas9 were observed, confirming the safety and efficacy of LNP-mediated CRISPR-Cas9 delivery as a sustainable approach for haemophilia therapy. In another study, a formulation of NP was reported for the delivery of two distinct mRNA transcripts encoding human erythropoietin (hEPO) and the factor IX (hFIX) protein. This formulation contained the cationic lipidoid C12-200, DOPE, cholesterol, and DMG-PEG2K. The dose-dependent production of proteins from each mRNA construct was observed in vivo. Subsequently, an increase in haematocrit was observed in the mouse model, indicating that exogenous mRNA-derived proteins were preserved. The protein remained at high levels (μg mL−1) and was persistently secreted one week after injection. The activity of the protein was confirmed through pharmacodynamic measurements and therapeutic interventions in an HB disease model.210 Chen et al. employed FVIII mRNA-encapsulated LNPs for the treatment of HA. The luminescent signal of the LNPs was primarily observed in the liver 4 h after intravenous injection of the IVIS, confirming the efficacy of LNP-mediated hepatic targeting. Subsequently, HA mice were administered LNPs carrying various variants of human FVIII mRNAs, resulting in rapid and sustained expression of the FVIII protein. These enduring treatment strategies hold great potential for treating patients with both HA and HB.211
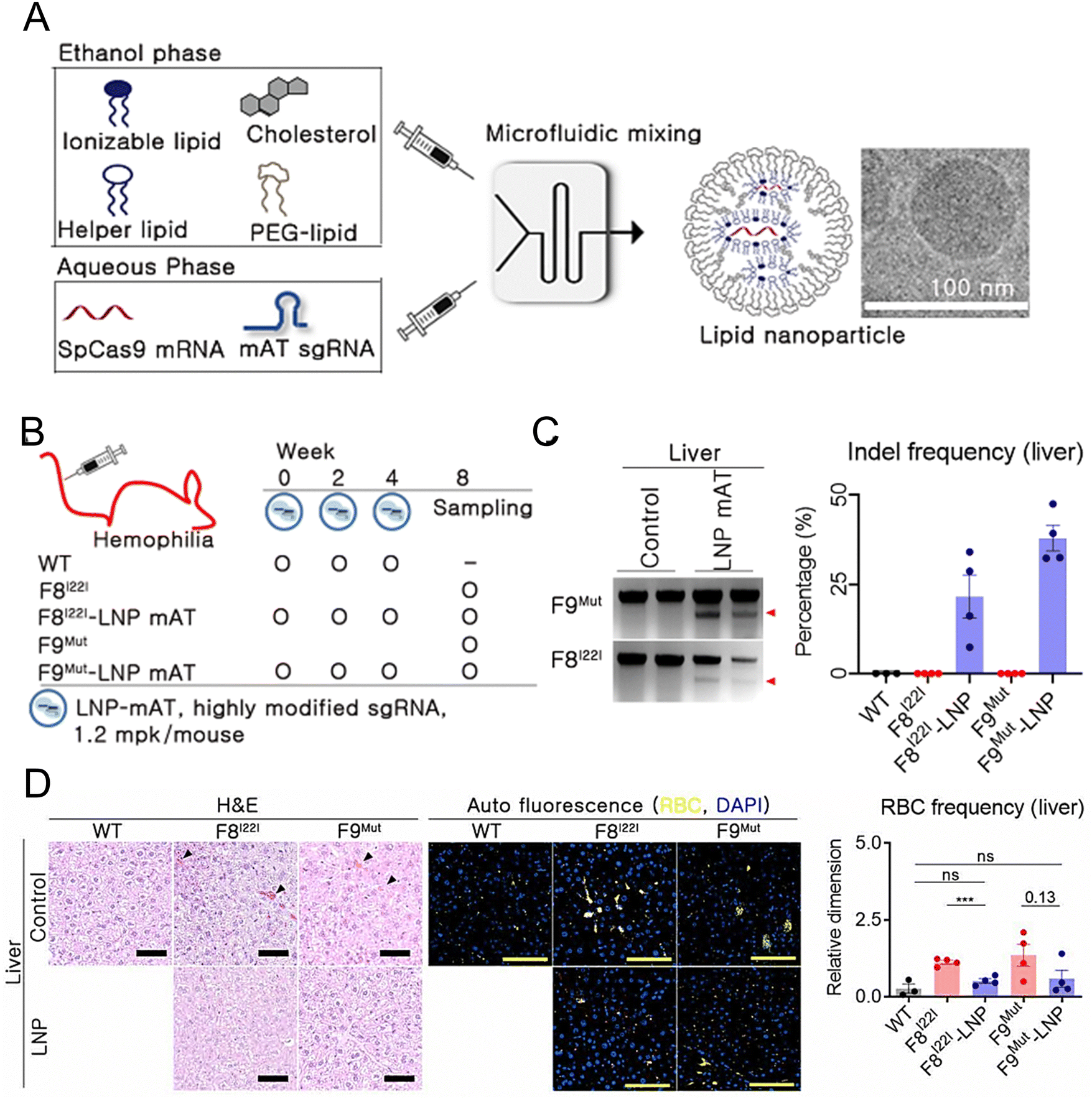 | ||
| Fig. 7 Enhanced haemophilia therapy through the LNP-mediated CRISPR-Cas9 delivery. (A) The LNPs were formulated using a microfluidic mixing system for in vivo gene delivery. (B) Simplified schematic illustrated in vivo gene targeting using the LNP-CRISPR-mAT. (C) The liver tissue of F8I22I and F9Mut exhibited average indel frequencies of 22% and 38%, respectively. (D) Red blood cells (RBC) in the interstitial liver tissue were quantified using the immunofluorescence staining to detect the autofluorescence signals at 470 and 540 nm, with the black triangles marking their location on the H&E-stained liver tissue. Adapted with permission.209 Copyright 2022, National Academy of Sciences. | ||
Transthyretin amyloidosis (ATTR). ATTR disease is a life-threatening condition characterized by the hepatic production of misfolded TTR proteins, which progressively accumulate in nerve and cardiac tissues. The inheritance pattern of ATTR amyloidosis follows an autosomal dominant pattern, and its clinical phenotype is distinguished by either amyloid polyneuropathy or cardiomyopathy. Following the onset of symptoms, patients with amyloid cardiomyopathy typically have a survival time of 2 to 6 years, while those suffering from amyloid polyneuropathy without cardiomyopathy usually have a life expectancy of 4 to 17 years.212,213
Finn et al. reported the development of an LNP-mediated delivery system for CRISPR/Cas9 editing in vivo, resulting in the knockdown of more than 97% of mouse TTR proteins. The knockdown persisted for at least 12 months. This delivery system contained the biodegradable lipid LP01, the accessory lipid PEG-DMG, Cas9 mRNA, and sgRNA. The efficacy of the LNP formulation was also shown in a rat model, in which the formulation effectively delivered the CRISPR/Cas9 components to reduce the levels of the TTR serum protein. These findings underscore the potential of this platform as an efficient genome editing tool.214 In another study, researchers demonstrated that ApoE was an important mediator of TTR silencing using Patisiran/Onpattro®, which was the first siRNA-loaded LNP approved for clinical use in patients with ATTR amyloidosis. The results showed that patisiran could efficiently reach liver cells through binding to ApoE. Although ApoE polymorphisms might affect its binding to hepatic receptors, the effectiveness of patisiran remains unaffected.215
5. The challenges of LNP technology
Despite the promising laboratory results, several challenges must be overcome before LNP technology is successfully translated into real-world applications.The homogeneity and size of the NPs are crucial parameters that need to be regulated. Researchers have shown that the size of NPs significantly influences drug pharmacokinetics by influencing the in vivo half-life,216 tissue penetration,217 and organ-specific absorption.218 In comparison to traditional preparation methods, the microfluidic methodology has been designed to synthesis LNPs with smaller particle sizes, a more homogenous distribution, increased batch-to-batch repeatability, and enhanced encapsulation effectiveness of nucleic acid medicines.219–221 From medication discovery to clinical application, scalable synthesis of LNPs with uniform particle sizes and size distributions is still a significant problem.222–224 Two approaches have been put forth to do LNP synthesis at a better throughput: channel size enlargement and parallelization.
By implementing numerous mixing channels in parallel, the parallelization technique increases the total flow rate to a substantially higher level than that of a single mixing channel. This approach significantly reduces development costs by optimizing parameters, such as flow rate ratios and lipid concentrations, using only one mixing channel. To ensure consistency and stability across all channels, the parallelization approach requires extensive advanced manufacturing and engineering methods.225–227
The channel-size enlarging method achieves greater injection volume and productivity by expanding the size of a single microfluidic channel.222,228 This method simplifies the equipment manufacturing process and increases the stability and reliability of production. It is difficult for a single mixer to synthesize LNPs with consistent properties at different flow rates. Researchers have improved upon the channel size enlarging method and developed an equidistant channel size enlarging method. After applying this improved method to inertial flow mixers with the same structure but different channel sizes, batch synthesis of LNPs with consistent performance can be achieved by controlling the flow resistance.229
The preparation of LNP formulations remains challenging due to potential issues such as the aggregation, leakage, and fusion of LNPs and susceptibility to degradation of the encapsulated mRNA and siRNA. For the stability of LNPs, while stable lipids can be incorporated into LNPs to maintain short-term stability, long-term storage and extended shelf life of the product are often achieved through freezing. The instability of mRNA-LNPs at 4 °C, which is often attributed to the inherent instability of the mRNA payload rather than the LNP system, is a challenge in this field.230,231 For example, the Moderna COVID-19 vaccine must be stored at −20 °C, whereas the Pfizer vaccine can be preserved for up to six months in an ultralow temperature freezer at −70 °C but only for five days in a refrigerator at 2–8 °C. Inadequate storage conditions might result in increased transportation expenses, whereas temperature fluctuations could compromise the quality of the product. Therefore, further investigation of the improvement in stability is necessary to eliminate the need for cold chain transport. Clinical trials testing nonfrozen and lyophilized preparations are underway. (NCT05137236, NCT05085366, NCT04816669) In one study, researchers evaluated the influence of pH, temperature, and lyophilization on LNP efficacy in HeLa cells. Under pH 7 aqueous conditions, the LNPs were more stable when stored at 2 °C for 150 days than when stored in a −20 °C freezer or at room temperature. Although aggregation and loss of efficacy were observed in LNPs subjected to freeze–thaw cycles, their stability was maintained when cryoprotectants, such as trehalose and sucrose, were used.232 The research results of Packer et al. indicate that the presence of electrophilic impurities of ionizable cationic lipid components is a reason for the instability of LNPs. The formation of lipid-mRNA adducts will reduce the activity of the mRNA-LNP product, and impurities introduced during the production process may affect the stability of the vaccine even under refrigerated conditions. Therefore, it is crucial to monitor and control the formation of lipid adducts during the research, development, and manufacturing of LNP-formulated nucleic acid products.233
6. Limitations of the present LNP technology
It has been shown that LNPs are both powerful and secure preventative vaccines as well as efficient delivery systems for therapeutic treatments. It is difficult to evaluate the nucleic acid payload distribution and capacity of LNPs, nevertheless. To analyze the payload distribution and capacity of mRNA LNPs, Li et al. devised a multicolor fluorescence spectroscopic technique that included a fluorescence coincidence analysis, a quantitative fluorescence deconvolution algorithm, and a single-molecule detection (SMD) platform. Understanding the links between structure, property, and function in future delivery systems requires knowledge of this information.2346.1 Immune stimulation
Identifying the amount of nucleic acid in LNPs enables scientists to better determine the injection dosage and the quantity of lipids used, and excessive amounts of nucleic acid can result in a range of adverse reactions. For instance, reported immune responses to COVID-19 vaccines include allergic reactions and autoimmune disorders. The CDC reported that there were roughly five occurrences of deadly anaphylaxis for every million vaccination doses given.235 Myocardial and immune thrombotic thrombocytopenia,236,237 IgA vasculitis,238 and autoimmune diseases were among the adverse effects to COVID-19 vaccinations that might occur.239,240 Modulating the immune response to LNPs could be achieved through three strategies: (1) manipulating the composition and characteristics of LNPs; (2) adding an adjuvant; and (3) adjusting the administration method.Researchers can modify the physicochemical properties of LNPs by adjusting the formulation procedure. The four lipids can be changed, additional ingredients can be added, and the composition ratio of each lipid can be changed during the LNP formation process. The characteristics of LNPs might be affected by even minute variations. The size of LNPs in the formulation, for example, is altered by varying the formulation rate or the PEG molar ratio.218 Phospholipids could be added to or substituted for charged lipids to change the charge of lipoproteins.241 Moreover, PEG lipids can be modified using certain methods.242
The inclusion of adjuvants improves the efficacy of LNPs, which stimulate innate immune responses and establish a localized immunocompetent environment at the injection site, thereby modifying the nature, strength, and efficacy of adaptive immune responses.243 For example, before Onpattro is administered, it is necessary to administer acetaminophen, glucocorticoids, and H1/H2 blockers to manage potential immune reactions during infusion.34 Apart from merely incorporating an adjuvant, scientists have also produced ionizable lipids that serve this purpose.244 Imidazole lipid (DOG-IM4), a new immunostimulatory ionizable lipid structure, was produced in one study. The components of DOG-IM4 were an imidazole-based ionizable head group, a dioleoyl lipid tail, and a polyoxyethylene linker that connected the head and tail. The outcomes showed that improving immunization was caused by the imidazole head group of lipids.245
LNPs are administered by a variety of techniques, including intramuscular (IM), intradermal (ID), subcutaneous (SC), intravenous (IV), and intranasal (IN).246–250 Researchers discovered that distinct routes of administration had synergistic effects in addition to merely comparing the routes of delivery. Mao et al. created the “prime (IM) and spike (IN)” vaccination strategy, which uses unadjuvanted IN to activate mucosal immunological memory in the respiratory system by leveraging preexisting immunity brought on by primary IM immunization. The mice with incomplete vaccination showed protection against deadly SARS-CoV-2 infection, enhanced systemic immunity, and robust resident memory B and T-cell responses in the prime and spike groups as compared to groups that received only IM or IN injections, the researchers showed.251
6.2 Targeting ability
In most instances, upon injection into the bloodstream, LNPs undergo spontaneous adsorption of ApoE onto their surface, leading to hepatic accumulation,252 but excessive liver homing of LNPs remains a drawback. Technologies that target the extrahepatic delivery of LNPs facilitate the precise administration of therapeutics to specific cells or tissues. The current options for overcoming the limitations of LNPs in liver targeting include modifying the administration route and adjusting the lipid composition.The biodistribution of LNPs can be significantly influenced by the administration route. Many studies have shown that the IM and SC routes can be utilized for delivering therapeutics to lymph nodes, which are primarily inhabited by antigen-presenting cells (APCs), T cells, and dendritic cells (DCs).217 The term “pulmonary administration” refers to the direct delivery of medications to the lungs, including inhalation and IN methods. Additionally, this approach can get around the problem of KCs’ cellular uptake of LNPs during systemic circulation.253
It is possible to influence tissue tropism after intravenous injection by merely changing the components of LNPs. Because of their differing affinities for ApoE, researchers found in one study that LNPs carrying DSPC as helper lipids concentrated in the spleen whereas LNPs containing DOPE accumulated in the liver.254 Chen et al. combined selective organ targeting (SORT) molecules with the four traditional components of LNP to accomplish extrahepatic distribution.241 The biodistribution of LNPs was impacted by interactions between the SORT molecule's charge and serum proteins. The tropism of mRNA/LNPs from the liver to the spleen and lung was changed by the addition of anionic and cationic lipids, respectively. Therefore, to create innovative delivery materials that target organs, it is essential to ascertain how particular proteins in serum interact with LNP components.
7. Clinical trials
LNPs are a widely utilized delivery system for the safe and effective transport of nucleic acid drugs. To date, three nucleic acid drugs assisted by LNPs, namely, the ATTR siRNA drug Patisiran/Onpattro® from Alnylam, the mRNA vaccine BNT162b2 from Pfizer/BioNTech, and the mRNA vaccine mRNA-1273 from Moderna, have been approved. Compared with other nucleic acid delivery systems, LNPs exhibit superior advantages, including a high encapsulation efficiency of nucleic acids, efficient cell transfection, strong tissue penetration with low cytotoxicity and immunogenicity. The diverse biological roles of RNAs have facilitated their application in various therapeutic contexts in the liver, many of which are undergoing clinical evaluation. Tables 1 and 2 present a summary of recent clinical trials of siRNA-loaded LNPs and mRNA-loaded LNPs, respectively.| Name | Indication | Target | Administration route | Phase | Start years | NCT ID |
|---|---|---|---|---|---|---|
| ALN-TTR02 | TTR | TTR | i.v. | III | 2019 | NCT03997383 |
| BMS-986263 | NASH | HSP47 | i.v. | II | 2021 | NCT04267393 |
| Name | Indication | Target | Administration route | Phase | Start years | NCT ID |
|---|---|---|---|---|---|---|
| i.v.: intravenous injection; i.m.: intramuscular injectgion; i.v.gtt: intravenous drip. | ||||||
| mRNA-4157 | Solid tumors | Personalized neoantigens | i.m. | I | 2017 | NCT03313778 |
| NTLA-2001 | Transthyretin amyloidosis-related cardiomyopathy (ATTR-CM) | ATTR | i.v. | I | 2020 | NCT04601051 |
| mRNA-3927 | Propionic acidemia (PA) | Alpha and beta subunits of PCC | i.v. | I/II | 2021 | NCT04159103 |
| NTLA-2002 | Hereditary angioedema (HAE) | HAE | i.v. | I/II | 2021 | NCT05120830 |
| WGI-0301 | Solid tumors | Akt-1 | i.v.gtt | I | 2022 | NCT05267899 |
| ARCT-810 | Ornithine transcarbamylase (OTC) deficiency | OTC | i.v. | II | 2022 | NCT05526066 |
| OTX-2002 | HCC and other solid tumors | MYC | i.v. | I/II | 2022 | NCT05497453 |
| mRNA-3745 | GSD | GSD1a | i.v. | I | 2022 | NCT05095727 |
8. Summary and outlook
With advancements in nucleic acid drugs and cell pathogenesis genetics, targeted therapy for various human diseases at the gene level has become feasible. After 50 years of development, significant breakthroughs have been achieved in the LNP drug delivery system, which is now widely regarded as the most effective system for delivering nucleic acid drugs.In this review, we provide an overview of the potential therapeutic applications of LNPs in liver-related diseases. Although researchers have been exploring LNP technology for a decade, only one RNA-based therapeutic (Patisiran/Onpattro®) targeting hepatic disorders has been approved for commercialization in more than 30 ongoing clinical trials. This milestone marks the successful convergence of nucleic acid nanomedicine and biomedicine. LNPs present certain advantages over alternative delivery systems. However, further research is needed to enhance long-term safety, optimize immune stimulation, improve delivery volume, and target other organs.
In addition to the applications outlined in this article, LNP technology has been widely applied across various fields, including medical imaging, cosmetics, nutrition, agriculture, and nanoreactors. With the emergence of new lipid materials and advancements in methodologies, LNPs are expected to usher in a new wave of enthusiasm.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We would like to thank the members of the Novel Injectable Drug Delivery Laboratory of the National Advanced Medical Engineering Research Center for their contribution to this work. This work was supported by a grant from the Science and Technology Commission of Shanghai Municipality (23S41900200).References
- X. Han, N. Gong, L. Xue, M. M. Billingsley, R. El-Mayta, S. J. Shepherd, M.-G. Alameh, D. Weissman and M. J. Mitchell, Nat. Commun., 2023, 14, 75 CrossRef CAS PubMed.
- R. Pattipeiluhu, G. Arias-Alpizar, G. Basha, K. Y. Chan, J. Bussmann, T. H. Sharp, M. A. Moradi, N. Sommerdijk, E. N. Harris and P. R. Cullis, Adv. Mater., 2022, 34, 2201095 CrossRef CAS PubMed.
- N. Dammes, M. Goldsmith, S. Ramishetti, J. L. Dearling, N. Veiga, A. B. Packard and D. Peer, Nat. Nanotechnol., 2021, 16, 1030–1038 CrossRef CAS PubMed.
- S. K. Patel, M. M. Billingsley, C. Frazee, X. Han, K. L. Swingle, J. Qin, M.-G. Alameh, K. Wang, D. Weissman and M. J. Mitchell, J. Controlled Release, 2022, 347, 521–532 CrossRef CAS PubMed.
- Y. Li, T. Ji, M. Torre, R. Shao, Y. Zheng, D. Wang, X. Li, A. Liu, W. Zhang, X. Deng, R. Yan and D. S. Kohane, Nat. Commun., 2023, 14, 6659 CrossRef CAS PubMed.
- B. S. Pattni, V. V. Chupin and V. P. Torchilin, Chem. Rev., 2015, 115, 10938–10966 CrossRef CAS PubMed.
- K. Paunovska, D. Loughrey and J. E. Dahlman, Nat. Rev. Genet., 2022, 23, 265–280 CrossRef CAS PubMed.
- T. M. Allen and P. R. Cullis, Science, 2004, 303, 1818–1822 CrossRef CAS PubMed.
- S. Senapati, A. K. Mahanta, S. Kumar and P. Maiti, Signal Transduction Targeted Ther., 2018, 3, 7 CrossRef PubMed.
- A. Jesorka and O. Orwar, Annu. Rev. Anal. Chem., 2008, 1, 801–832 CrossRef CAS PubMed.
- N. Filipczak, J. Pan, S. S. K. Yalamarty and V. P. Torchilin, Adv. Drug Delivery Rev., 2020, 156, 4–22 CrossRef CAS PubMed.
- J. C. Kaczmarek, P. S. Kowalski and D. G. Anderson, Genome Med., 2017, 9, 60 CrossRef PubMed.
- R. van der Meel, E. Sulheim, Y. Shi, F. Kiessling, W. J. M. Mulder and T. Lammers, Nat. Nanotechnol., 2019, 14, 1007–1017 CrossRef CAS PubMed.
- H. Xing, K. Hwang and Y. Lu, Theranostics, 2016, 6, 1336–1352 CrossRef CAS PubMed.
- R. H. Fang, W. Gao and L. Zhang, Nat. Rev. Clin. Oncol., 2023, 20, 33–48 CrossRef PubMed.
- J. A. Kulkarni, M. M. Darjuan, J. E. Mercer, S. Chen, R. van der Meel, J. L. Thewalt, Y. Y. C. Tam and P. R. Cullis, ACS Nano, 2018, 12, 4787–4795 CrossRef CAS PubMed.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed.
- D. Adams, A. Gonzalez-Duarte, W. D. O’Riordan, C. C. Yang, M. Ueda, A. V. Kristen, I. Tournev, H. H. Schmidt, T. Coelho, J. L. Berk, K. P. Lin, G. Vita, S. Attarian, V. Plante-Bordeneuve, M. M. Mezei, J. M. Campistol, J. Buades, T. H. Brannagan, III, B. J. Kim, J. Oh, Y. Parman, Y. Sekijima, P. N. Hawkins, S. D. Solomon, M. Polydefkis, P. J. Dyck, P. J. Gandhi, S. Goyal, J. Chen, A. L. Strahs, S. V. Nochur, M. T. Sweetser, P. P. Garg, A. K. Vaishnaw, J. A. Gollob and O. B. Suhr, N. Engl. J. Med., 2018, 379, 11–21 CrossRef CAS PubMed.
- U. Lächelt and E. Wagner, Chem. Rev., 2015, 115, 11043–11078 CrossRef.
- Y. Wang, S. Li, X. Wang, Q. Chen, Z. He, C. Luo and J. Sun, Biomaterials, 2021, 271, 120737 CrossRef CAS PubMed.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed.
- G. Yamankurt, E. J. Berns, A. Xue, A. Lee, N. Bagheri, M. Mrksich and C. A. Mirkin, Nat. Biomed. Eng., 2019, 3, 318–327 CrossRef CAS PubMed.
- W. Jiang, C. A. von Roemeling, Y. Chen, Y. Qie, X. Liu, J. Chen and B. Y. S. Kim, Nat. Biomed. Eng., 2017, 1, 0029 CrossRef CAS.
- M. J. W. Evers, J. A. Kulkarni, R. van der Meel, P. R. Cullis, P. Vader and R. M. Schiffelers, Small Methods, 2018, 2(9), 1700375 CrossRef.
- M. Cobb, Curr. Biol., 2015, 25, R526–532 CrossRef CAS PubMed.
- N. Veiga, M. Goldsmith, Y. Granot, D. Rosenblum, N. Dammes, R. Kedmi, S. Ramishetti and D. Peer, Nat. Commun., 2018, 9, 4493 CrossRef PubMed.
- G. Zhang, T. Tang, Y. Chen, X. Huang and T. Liang, Signal Transduction Targeted Ther., 2023, 8, 365 CrossRef CAS PubMed.
- W. Ho, M. Gao, F. Li, Z. Li, X. Q. Zhang and X. Xu, Adv. Healthcare Mater., 2021, 10, e2001812 CrossRef PubMed.
- Z. He, Y. Hu, T. Nie, H. Tang, J. Zhu, K. Chen, L. Liu, K. W. Leong, Y. Chen and H.-Q. Mao, Acta Biomater., 2018, 81, 195–207 CrossRef CAS PubMed.
- S. G. Jadhav and S. F. Dowdy, Nat. Mater., 2021, 20, 575–577 CrossRef CAS PubMed.
- R. Kole, A. R. Krainer and S. Altman, Nat. Rev. Drug Discovery, 2012, 11, 125–140 CrossRef CAS PubMed.
- E. Samaridou, J. Heyes and P. Lutwyche, Adv. Drug Delivery Rev., 2020, 154, 37–63 CrossRef PubMed.
- P. R. Cullis and M. J. Hope, Mol. Ther., 2017, 25, 1467–1475 CrossRef CAS PubMed.
- C. Hald Albertsen, J. A. Kulkarni, D. Witzigmann, M. Lind, K. Petersson and J. B. Simonsen, Adv. Drug Delivery Rev., 2022, 188, 114416 CrossRef CAS PubMed.
- J. A. Kulkarni, P. R. Cullis and R. Van Der Meel, Nucleic Acid Ther., 2018, 28, 146–157 CrossRef CAS PubMed.
- J. A. Kulkarni, D. Witzigmann, S. Chen, P. R. Cullis and R. van der Meel, Acc. Chem. Res., 2019, 52, 2435–2444 CrossRef CAS PubMed.
- C. H. Albertsen, J. A. Kulkarni, D. Witzigmann, M. Lind, K. Petersson and J. B. Simonsen, Adv. Drug Delivery Rev., 2022, 188, 114416 CrossRef PubMed.
- J. D. Watson and F. H. C. Crick, Nature, 1953, 171, 737–738 CrossRef CAS PubMed.
- W. H. Hudson and E. A. Ortlund, Nat. Rev. Mol. Cell Biol., 2014, 15, 749–760 CrossRef CAS PubMed.
- I. Lostalé-Seijo and J. Montenegro, Nat. Rev. Chem., 2018, 2, 258–277 CrossRef.
- P. J. C. Lin, Y. Y. C. Tam, I. Hafez, A. Sandhu, S. Chen, M. A. Ciufolini, I. R. Nabi and P. R. Cullis, Nanomedicine, 2013, 9, 233–246 CrossRef CAS PubMed.
- G. Basha, T. I. Novobrantseva, N. Rosin, Y. Y. Tam, I. M. Hafez, M. K. Wong, T. Sugo, V. M. Ruda, J. Qin, B. Klebanov, M. Ciufolini, A. Akinc, Y. K. Tam, M. J. Hope and P. R. Cullis, Mol. Ther., 2011, 19, 2186–2200 CrossRef CAS PubMed.
- H. Lv, S. Zhang, B. Wang, S. Cui and J. Yan, J. Controlled Release, 2006, 114, 100–109 CrossRef CAS PubMed.
- Y. Sato, K. Hashiba, K. Sasaki, M. Maeki, M. Tokeshi and H. Harashima, J. Controlled Release, 2019, 295, 140–152 CrossRef CAS PubMed.
- R. L. Ball, P. Bajaj and K. A. Whitehead, Int. J. Nanomed., 2016, 12, 305–315 CrossRef PubMed.
- V. Kumar, J. Qin, Y. Jiang, R. G. Duncan, B. Brigham, S. Fishman, J. K. Nair, A. Akinc, S. A. Barros and P. V. Kasperkovitz, Mol. Ther.–Nucleic Acids, 2014, 3, e210 CrossRef PubMed.
- A. L. Bailey and P. R. Cullis, Biochemistry, 1997, 36, 1628–1634 CrossRef CAS PubMed.
- M. Jayaraman, S. M. Ansell, B. L. Mui, Y. K. Tam, J. Chen, X. Du, D. Butler, L. Eltepu, S. Matsuda and J. K. Narayanannair, Angew. Chem., Int. Ed., 2012, 124, 8657–8661 CrossRef.
- X. Han, H. Zhang, K. Butowska, K. L. Swingle, M.-G. Alameh, D. Weissman and M. J. Mitchell, Nat. Commun., 2021, 12, 7233 CrossRef CAS PubMed.
- J. Lutz, S. Lazzaro, M. Habbeddine, K. E. Schmidt, P. Baumhof, B. L. Mui, Y. K. Tam, T. D. Madden, M. J. Hope, R. Heidenreich and M. Fotin-Mleczek, npj Vaccines, 2017, 2, 29 CrossRef.
- F. Ferraresso, A. W. Strilchuk, L. J. Juang, L. G. Poole, J. P. Luyendyk and C. J. Kastrup, Mol. Pharmaceutics, 2022, 19, 2175–2182 CrossRef CAS PubMed.
- X. Yu, S. Liu, Q. Cheng, T. Wei, S. Lee, D. Zhang and D. J. Siegwart, Adv. Healthcare Mater., 2020, 9, 1901487 CrossRef CAS PubMed.
- S. C. Semple, A. Akinc, J. Chen, A. P. Sandhu, B. L. Mui, C. K. Cho, D. W. Sah, D. Stebbing, E. J. Crosley and E. Yaworski, Nat. Biotechnol., 2010, 28, 172–176 CrossRef CAS PubMed.
- X. Cheng and R. J. Lee, Adv. Drug Delivery Rev., 2016, 99, 129–137 CrossRef CAS PubMed.
- D. Bochicchio, E. Panizon, L. Monticelli and G. Rossi, Sci. Rep., 2017, 7, 6357 CrossRef CAS PubMed.
- E. Ambegia, S. Ansell, P. Cullis, J. Heyes, L. Palmer and I. MacLachlan, Biochim. Biophys. Acta, Biomembr., 2005, 1669, 155–163 CrossRef CAS PubMed.
- P. Tam, M. Monck, D. Lee, O. Ludkovski, E. Leng, K. Clow, H. Stark, P. Scherrer, R. Graham and P. Cullis, Gene Ther., 2000, 7, 1867–1874 CrossRef CAS PubMed.
- T. Suzuki, Y. Suzuki, T. Hihara, K. Kubara, K. Kondo, K. Hyodo, K. Yamazaki, T. Ishida and H. Ishihara, Int. J. Pharm., 2020, 588, 119792 CrossRef CAS PubMed.
- H. Zhang, J. Leal, M. R. Soto, H. D. Smyth and D. Ghosh, Pharmaceutics, 2020, 12, 1042 CrossRef CAS PubMed.
- S. G. Reed, M. T. Orr and C. B. Fox, Nat. Med., 2013, 19, 1597–1608 CrossRef CAS PubMed.
- M. T. M. García, Á. T. Lana, M. B. A. Agudo and M. T. R. Delgado, Enferm. Infecc. Microbiol. Clin., 2022, 40, 276 CrossRef PubMed.
- M. Tamburro, G. Ripabelli, A. D’Amico, R. De Dona, M. Iafigliola, A. Parente, N. Samprati, A. Santagata, C. Adesso, A. Natale, M. A. Di Palma, F. Cannizzaro and M. L. Sammarco, J. Community Health, 2022, 47, 814–821 CrossRef PubMed.
- M. P. Lokugamage, D. Vanover, J. Beyersdorf, M. Z. Hatit, L. Rotolo, E. S. Echeverri, H. E. Peck, H. Ni, J.-K. Yoon and Y. Kim, Nat. Biomed. Eng., 2021, 5, 1059–1068 CrossRef CAS PubMed.
- X. Cheng and R. J. Lee, Adv. Drug Delivery Rev., 2016, 99, 129–137 CrossRef CAS PubMed.
- X. Cheng and R. J. Lee, Adv. Drug Delivery Rev., 2016, 99, 129–137 CrossRef CAS PubMed.
- J. A. Kulkarni, J. L. Myhre, S. Chen, Y. Y. C. Tam, A. Danescu, J. M. Richman and P. R. Cullis, Nanomedicine, 2017, 13, 1377–1387 CrossRef CAS PubMed.
- Y. Zhu, R. Shen, I. Vuong, R. A. Reynolds, M. J. Shears, Z.-C. Yao, Y. Hu, W. J. Cho, J. Kong and S. K. Reddy, Nat. Commun., 2022, 13, 4282 CrossRef CAS PubMed.
- S. T. LoPresti, M. L. Arral, N. Chaudhary and K. A. Whitehead, J. Controlled Release, 2022, 345, 819–831 CrossRef CAS PubMed.
- R. Zhang, R. El-Mayta, T. J. Murdoch, C. C. Warzecha, M. M. Billingsley, S. J. Shepherd, N. Gong, L. Wang, J. M. Wilson and D. Lee, Biomater. Sci., 2021, 9, 1449–1463 RSC.
- E. Álvarez-Benedicto, L. Farbiak, M. M. Ramírez, X. Wang, L. T. Johnson, O. Mian, E. D. Guerrero and D. J. Siegwart, Biomater. Sci., 2022, 10, 549–559 RSC.
- T. Harayama and H. Riezman, Nat. Rev. Mol. Cell Biol., 2018, 19, 281–296 CrossRef CAS PubMed.
- M. R. Krause and S. L. Regen, Acc. Chem. Res., 2014, 47, 3512–3521 CrossRef CAS PubMed.
- X. Xie, T. Yu, X. Li, N. Zhang, L. J. Foster, C. Peng, W. Huang and G. He, Signal Transduction Targeted Ther., 2023, 8, 335 CrossRef PubMed.
- S. T. Crooke, J. L. Witztum, C. F. Bennett and B. F. Baker, Cell Metab., 2018, 27, 714–739 CrossRef CAS PubMed.
- Z. Dominski and R. Kole, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 8673–8677 CrossRef CAS PubMed.
- P. D. Zamore, T. Tuschl, P. A. Sharp and D. P. Bartel, Cell, 2000, 101, 25–33 CrossRef CAS PubMed.
- S. M. Hammond, E. Bernstein, D. Beach and G. J. Hannon, Nature, 2000, 404, 293–296 CrossRef CAS PubMed.
- S. Mitschka and C. Mayr, Nat. Rev. Mol. Cell Biol., 2022, 23, 779–796 CrossRef CAS PubMed.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS PubMed.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS PubMed.
- F. Heymann and F. Tacke, Nat. Rev. Gastroenterol. Hepatol., 2016, 13, 88–110 CrossRef CAS PubMed.
- M. W. Robinson, C. Harmon and C. O’Farrelly, Cell. Mol. Immunol., 2016, 13, 267–276 CrossRef CAS PubMed.
- E. Trefts, M. Gannon and D. H. Wasserman, Curr. Biol., 2017, 27, R1147–r1151 CrossRef CAS PubMed.
- P. Kubes and C. Jenne, Annu. Rev. Immunol., 2018, 36, 247–277 CrossRef CAS PubMed.
- R. Böttger, G. Pauli, P.-H. Chao, N. A. Fayez, L. Hohenwarter and S.-D. Li, Adv. Drug Delivery Rev., 2020, 154, 79–101 CrossRef PubMed.
- P. Thomas, in Encyclopedia of Cancer, ed. M. Schwab, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp.1963–1965 Search PubMed.
- M. Bilzer, F. Roggel and A. L. Gerbes, Liver Int., 2006, 26, 1175–1186 CrossRef CAS PubMed.
- S. N. Hilmer, V. C. Cogger and D. G. Le Couteur, J. Gerontol., Ser. A, 2007, 62, 973–978 CrossRef PubMed.
- G. Kolios, V. Valatas and E. Kouroumalis, World J. Gastroenterol., 2006, 12, 7413–7420 CrossRef CAS PubMed.
- H. Ruf and B. J. Gould, Eur. Biophys. J., 1999, 28, 1–11 CrossRef CAS PubMed.
- D. Benten, A. Follenzi, K. K. Bhargava, V. Kumaran, C. J. Palestro and S. Gupta, Hepatology, 2005, 42, 140–148 CrossRef CAS PubMed.
- A. Geerts, Semin. Liver Dis., 2001, 21, 311–335 CrossRef CAS PubMed.
- F. Winau, G. Hegasy, R. Weiskirchen, S. Weber, C. Cassan, P. A. Sieling, R. L. Modlin, R. S. Liblau, A. M. Gressner and S. H. Kaufmann, Immunity, 2007, 26, 117–129 CrossRef CAS PubMed.
- S. L. Friedman, Physiol. Rev., 2008, 88, 125–172 CrossRef CAS PubMed.
- S. R. Wang, G. Renaud, J. Infante, D. Catala and R. Infante, In Vitro Cell. Dev. Biol., 1985, 21, 526–530 CrossRef CAS PubMed.
- S. Donthamsetty, V. S. Bhave, C. S. Kliment, W. C. Bowen, W. M. Mars, A. W. Bell, R. E. Stewart, A. Orr, C. Wu and G. K. Michalopoulos, Hepatology, 2011, 53, 587–595 CrossRef CAS PubMed.
- S. Novakowski, K. Jiang, G. Prakash and C. Kastrup, Sci. Rep., 2019, 9, 552 CrossRef CAS PubMed.
- T. Kanazawa, K. Morisaki, S. Suzuki and Y. Takashima, Mol. Pharmaceutics, 2014, 11, 1471–1478 CrossRef CAS PubMed.
- J. K. Lam, W. Liang and H. K. Chan, Adv. Drug Delivery Rev., 2012, 64, 1–15 CrossRef CAS PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- D. Lechardeur, K. J. Sohn, M. Haardt, P. B. Joshi, M. Monck, R. W. Graham, B. Beatty, J. Squire, H. O’Brodovich and G. L. Lukacs, Gene Ther., 1999, 6, 482–497 CrossRef CAS PubMed.
- S. Liu, Q. Cheng, T. Wei, X. Yu, L. T. Johnson, L. Farbiak and D. J. Siegwart, Nat. Mater., 2021, 20, 701–710 CrossRef CAS PubMed.
- J. Szebeni, G. Storm, J. Y. Ljubimova, M. Castells, E. J. Phillips, K. Turjeman, Y. Barenholz, D. J. A. Crommelin and M. A. Dobrovolskaia, Nat. Nanotechnol., 2022, 17, 337–346 CrossRef CAS PubMed.
- E. L. Romero, M. J. Morilla, J. Regts, G. A. Koning and G. L. Scherphof, FEBS Lett., 1999, 448, 193–196 CrossRef CAS PubMed.
- A. Wieckowska, A. J. McCullough and A. E. Feldstein, Hepatology, 2007, 46, 582–589 CrossRef CAS PubMed.
- V. Thakur, M. R. McMullen, M. T. Pritchard and L. E. Nagy, J. Gastroenterol. Hepatol., 2007, 22(Suppl 1), S53–S56 CAS.
- S. L. Friedman and M. B. Bansal, Hepatology, 2006, 43, S82–88 CrossRef CAS PubMed.
- H. Robertson, J. A. Kirby, W. W. Yip, D. E. Jones and A. D. Burt, Hepatology, 2007, 45, 977–981 CrossRef CAS PubMed.
- I. Fabregat, C. Roncero and M. Fernández, Liver Int., 2007, 27, 155–162 CrossRef CAS PubMed.
- S. Taurin, H. Nehoff and K. Greish, J. Controlled Release, 2012, 164, 265–275 CrossRef CAS PubMed.
- M. Qiu, Y. Tang, J. Chen, R. Muriph, Z. Ye, C. Huang, J. Evans, E. P. Henske and Q. Xu, Proc. Natl. Acad. Sci. U. S. A., 2022, 119(8), e2116271119 CrossRef CAS PubMed.
- M. Kockx, M. Traini and L. Kritharides, J. Mol. Med., 2018, 96, 361–371 CrossRef CAS PubMed.
- K. Paunovska, A. J. Da Silva Sanchez, M. P. Lokugamage, D. Loughrey, E. S. Echeverri, A. Cristian, M. Z. C. Hatit, P. J. Santangelo, K. Zhao and J. E. Dahlman, Nano Lett., 2022, 22, 10025–10033 CrossRef CAS PubMed.
- A. Akinc, W. Querbes, S. De, J. Qin, M. Frank-Kamenetsky, K. N. Jayaprakash, M. Jayaraman, K. G. Rajeev, W. L. Cantley and J. R. Dorkin, Mol. Ther., 2010, 18, 1357–1364 CrossRef CAS PubMed.
- F. Sebastiani, M. Yanez Arteta, M. Lerche, L. Porcar, C. Lang, R. A. Bragg, C. S. Elmore, V. R. Krishnamurthy, R. A. Russell, T. Darwish, H. Pichler, S. Waldie, M. Moulin, M. Haertlein, V. T. Forsyth, L. Lindfors and M. Cárdenas, ACS Nano, 2021, 15, 6709–6722 CrossRef CAS PubMed.
- M. Kim, M. Jeong, S. Hur, Y. Cho, J. Park, H. Jung, Y. Seo, H. Woo, K. Nam and K. Lee, Sci. Adv., 2021, 7, eabf4398 CrossRef CAS PubMed.
- J. E. Zhou, L. Sun, L. Liu, Y. Jia, Y. Han, J. Shao, J. Wang, Y. Wang, L. Yu and Z. Yan, J. Controlled Release, 2022, 343, 175–186 CrossRef CAS PubMed.
- J. Zhang, H. Shen, J. Xu, L. Liu, J. Tan, M. Li, N. Xu, S. Luo, J. Wang, F. Yang, J. Tang, Q. Li, Y. Wang, L. Yu and Z. Yan, ACS Nano, 2020, 14, 6305–6322 CrossRef CAS PubMed.
- H. Ni, M. Z. C. Hatit, K. Zhao, D. Loughrey, M. P. Lokugamage, H. E. Peck, A. D. Cid, A. Muralidharan, Y. Kim, P. J. Santangelo and J. E. Dahlman, Nat. Commun., 2022, 13, 4766 CrossRef CAS PubMed.
- Z. Yang, Y. Liu, K. Zhao, W. Jing, L. Gao, X. Dong, Y. Wang, M. Han, C. Shi, C. Tang, P. Sun, R. Zhang, Z. Fu, J. Zhang, D. Zhu, C. Chen and X. Jiang, J. Controlled Release, 2023, 360, 718–733 CrossRef CAS PubMed.
- M. Qiu, Z. Glass, J. Chen, M. Haas, X. Jin, X. Zhao, X. Rui, Z. Ye, Y. Li, F. Zhang and Q. Xu, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(10), e2020401118 CrossRef CAS PubMed.
- M. C. S. Wong, J. L. W. Huang, J. George, J. Huang, C. Leung, M. Eslam, H. L. Y. Chan and S. C. Ng, Nat. Rev. Gastroenterol. Hepatol., 2019, 16, 57–73 CrossRef PubMed.
- J. D. Stanaway, A. D. Flaxman, M. Naghavi, C. Fitzmaurice, T. Vos, I. Abubakar, L. J. Abu-Raddad, R. Assadi, N. Bhala, B. Cowie, M. H. Forouzanfour, J. Groeger, K. M. Hanafiah, K. H. Jacobsen, S. L. James, J. MacLachlan, R. Malekzadeh, N. K. Martin, A. A. Mokdad, A. H. Mokdad, C. J. L. Murray, D. Plass, S. Rana, D. B. Rein, J. H. Richardus, J. Sanabria, M. Saylan, S. Shahraz, S. So, V. V. Vlassov, E. Weiderpass, S. T. Wiersma, M. Younis, C. Yu, M. El Sayed Zaki and G. S. Cooke, Lancet, 2016, 388, 1081–1088 CrossRef PubMed.
- S. K. Asrani, H. Devarbhavi, J. Eaton and P. S. Kamath, J. Hepatol., 2019, 70, 151–171 CrossRef PubMed.
- Z. Younossi, Q. M. Anstee, M. Marietti, T. Hardy, L. Henry, M. Eslam, J. George and E. Bugianesi, Nat. Rev. Gastroenterol. Hepatol., 2018, 15, 11–20 CrossRef PubMed.
- M. A. Aldersley and J. G. O’Grady, Drugs, 1995, 49, 83–102 CrossRef CAS PubMed.
- V. Hernandez-Gea and S. L. Friedman, Annu. Rev. Phytopathol., 2011, 6, 425–456 CrossRef CAS PubMed.
- Z. M. Younossi, J. Hepatol., 2019, 70, 531–544 CrossRef PubMed.
- J. S. Bajaj, Nat. Rev. Gastroenterol. Hepatol., 2019, 16, 235–246 CrossRef PubMed.
- R. Böttger, G. Pauli, P.-H. Chao, N. Al Fayez, L. Hohenwarter and S.-D. Li, Adv. Drug Delivery Rev., 2020, 154–155, 79–101 CrossRef PubMed.
- D. Witzigmann, J. A. Kulkarni, J. Leung, S. Chen, P. R. Cullis and R. van der Meel, Adv. Drug Delivery Rev., 2020, 159, 344–363 CrossRef CAS PubMed.
- J. Hu, Y. Xu, J. Hao, S. Wang, C. Li and S. Meng, Protein Cell, 2012, 3, 364–371 CrossRef CAS PubMed.
- N. Gonzaludo, J. W. Belmont, V. G. Gainullin and R. J. Taft, Genet. Med., 2019, 21, 1781–1789 CrossRef PubMed.
- J. Menon, M. Vij, D. Sachan, A. Rammohan, N. Shanmugam, I. Kaliamoorthy and M. Rela, World J. Transplant., 2021, 11, 161 CrossRef PubMed.
- R. Hegarty, N. Hadzic, P. Gissen and A. Dhawan, Eur. J. Pediatr., 2015, 174, 1387–1392 CrossRef CAS PubMed.
- C. R. Ferreira, D. Cassiman and N. Blau, Mol. Genet. Metab., 2019, 127, 117–121 CrossRef CAS PubMed.
- J. S. Kim, K. M. Kim, S. H. Oh, H. J. Kim, J. M. Cho, H.-W. Yoo, J.-M. Namgoong, D. Y. Kim, K.-H. Kim and S. Hwang, Pediatr. Gastroenterol. Hepatol. Nutr., 2015, 18, 48–54 CrossRef PubMed.
- A. Zarrinpar and R. W. Busuttil, Nat. Rev. Gastroenterol. Hepatol., 2013, 10, 434–440 CrossRef CAS PubMed.
- Y. Granot and D. Peer, Semin. Immunol., 2017, 34, 68–77 CrossRef CAS PubMed.
- B. Carroll, V. I. Korolchuk and S. Sarkar, Amino Acids, 2015, 47, 2065–2088 CrossRef CAS PubMed.
- J. Jung, H. Zeng and T. Horng, Nat. Cell Biol., 2019, 21, 85–93 CrossRef CAS PubMed.
- P. Strzyz, Nat. Rev. Mol. Cell Biol., 2015, 16, 703 Search PubMed.
- Z. Dai, Z. Wu, W. Zhu and G. Wu, in Recent Advances in Animal Nutrition and Metabolism, ed. G. Wu, Springer International Publishing, Cham, 2022, pp.127–143 Search PubMed.
- D. Gyamfi and K. O. Danquah, in Molecular Aspects of Alcohol and Nutrition, ed. V. B. Patel, Academic Press, San Diego, 2016, pp.3–15 Search PubMed.
- A. Dursun, R. K. Özgül, S. Sivri, A. Tokatlı, A. Güzel, L. Mesci, M. Kılıç, D. Aliefendioglu, F. Özçay, M. Gündüz and T. Coşkun, JIMD Reports - Case and Research Reports, 2011/1: Case and Research Reports, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp.17–21 Search PubMed.
- Q. Cheng, T. Wei, Y. Jia, L. Farbiak, K. Zhou, S. Zhang, Y. Wei, H. Zhu and D. J. Siegwart, Adv. Mater., 2018, 30, 1805308 CrossRef PubMed.
- M. L. Cacicedo, C. Weinl-Tenbruck, D. Frank, S. Wirsching, B. K. Straub, J. Hauke, J. G. Okun, N. Horscroft, J. B. Hennermann and F. Zepp, Mol. Ther.–Methods Clin. Dev., 2022, 26, 294–308 CrossRef CAS PubMed.
- M. L. Cacicedo, C. Weinl-Tenbruck, D. Frank, M. J. Limeres, S. Wirsching, K. Hilbert, M. A. Pasha Famian, N. Horscroft, J. B. Hennermann and F. Zepp, Front. Bioeng. Biotechnol., 2022, 10, 993298 CrossRef PubMed.
- B. Truong, G. Allegri, X.-B. Liu, K. E. Burke, X. Zhu, S. D. Cederbaum, J. Häberle, P. G. Martini and G. S. Lipshutz, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 21150–21159 CrossRef CAS PubMed.
- K. Tanaka, in Biology of Brain Dysfunction, ed. G. E. Gaull, Springer, US, Boston, MA, 1975, vol. 3, pp.145–214 Search PubMed.
- M. R. Seashore, in Rosenberg's Molecular and Genetic Basis of Neurological and Psychiatric Disease, ed. R. N. Rosenberg and J. M. Pascual, Academic Press, Boston, 5th edn, 2015, pp.601–606 Search PubMed.
- L. Jiang, J.-S. Park, L. Yin, R. Laureano, E. Jacquinet, J. Yang, S. Liang, A. Frassetto, J. Zhuo and X. Yan, Nat. Commun., 2020, 11, 5339 CrossRef CAS PubMed.
- D. An, A. Frassetto, E. Jacquinet, M. Eybye, J. Milano, C. DeAntonis, V. Nguyen, R. Laureano, J. Milton and S. Sabnis, EBioMedicine, 2019, 45, 519–528 CrossRef PubMed.
- P. S. Kishnani, J. Goldstein, S. L. Austin, P. Arn, B. Bachrach, D. S. Bali, W. K. Chung, A. El-Gharbawy, L. M. Brown, S. Kahler, S. Pendyal, K. M. Ross, L. Tsilianidis, D. A. Weinstein, M. S. Watson and ACMG Work Group on Diagnosis and Management of Glycogen Storage Diseases Type VI and IX, Genet. Med., 2019, 21, 772–789 CrossRef PubMed.
- W. B. Hannah, T. G. J. Derks, M. L. Drumm, S. C. Grünert, P. S. Kishnani and J. Vissing, Nat. Rev. Dis. Primers, 2023, 9, 46 CrossRef PubMed.
- J. Cao, M. Choi, E. Guadagnin, M. Soty, M. Silva, V. Verzieux, E. Weisser, A. Markel, J. Zhuo, S. Liang, L. Yin, A. Frassetto, A. R. Graham, K. Burke, T. Ketova, C. Mihai, Z. Zalinger, B. Levy, G. Besin, M. Wolfrom, B. Tran, C. Tunkey, E. Owen, J. Sarkis, A. Dousis, V. Presnyak, C. Pepin, W. Zheng, L. Ci, M. Hard, E. Miracco, L. Rice, V. Nguyen, M. Zimmer, U. Rajarajacholan, P. F. Finn, G. Mithieux, F. Rajas, P. G. V. Martini and P. H. Giangrande, Nat. Commun., 2021, 12, 3090 CrossRef CAS PubMed.
- S. Fagiuoli, E. Daina, L. D’Antiga, M. Colledan and G. Remuzzi, J. Hepatol., 2013, 59, 595–612 CrossRef PubMed.
- J. A. Greig, J. K. Chorazeczewski, V. Chowdhary, M. K. Smith, M. Jennis, J. C. Tarrant, E. L. Buza, K. Coughlan, P. G. Martini and J. M. Wilson, Mol. Ther.–Methods Clin. Dev., 2023, 29, 32–39 CrossRef CAS PubMed.
- G. Wei, J. Cao, P. Huang, P. An, D. Badlani, K. A. Vaid, S. Zhao, D. Q. Wang, J. Zhuo and L. Yin, J. Hepatol., 2021, 74, 1416–1428 CrossRef CAS PubMed.
- E. C. Lo, A. N. Rucker and M. P. Federle, Semin. Radiat. Oncol., 2018, 28, 267–276 CrossRef PubMed.
- P. A. Ott, F. S. Hodi, H. L. Kaufman, J. M. Wigginton and J. D. Wolchok, J. Immunotherap. Cancer., 2017, 5, 16 CrossRef PubMed.
- B. Sangro, P. Sarobe, S. Hervás-Stubbs and I. Melero, Nat. Rev. Gastroenterol. Hepatol., 2021, 18, 525–543 CrossRef PubMed.
- S. Yang, D. Wang, X. Zhang, Y. Sun and B. Zheng, Drug Delivery, 2021, 28, 995–1006 CrossRef CAS PubMed.
- L. Yuhuan, Y. Kongtong, B. Ye, Q. Yuhang, S. Yating, L. Yujing, X. Jing and T. Lesheng, ACS Appl. Mater. Interfaces, 2016, 8, 26613–26621 CrossRef PubMed.
- B. Yu, S.-H. Hsu, C. Zhou, X. Wang, M. C. Terp, Y. Wu, L. Teng, Y. Mao, F. Wang and W. Xue, Biomaterials, 2012, 33, 5924–5934 CrossRef CAS PubMed.
- M. M. Woitok, M. E. Zoubek, D. Doleschel, M. Bartneck, M. R. Mohamed, F. Kießling, W. Lederle, C. Trautwein and F. J. Cubero, Cell Death Dis., 2020, 11, 343 CrossRef CAS PubMed.
- M. A. Younis, I. A. Khalil, Y. H. Elewa, Y. Kon and H. Harashima, J. Controlled Release, 2021, 331, 335–349 CrossRef CAS PubMed.
- Y. Rybakova, P. S. Kowalski, Y. Huang, J. T. Gonzalez, M. W. Heartlein, F. DeRosa, D. Delcassian and D. G. Anderson, Mol. Ther., 2019, 27, 1415–1423 CrossRef CAS PubMed.
- Z. Wang, K. Zhao, Y. Zhang, X. Duan and Y. Zhao, Pharm. Res., 2019, 36, 1–13 CrossRef PubMed.
- I. Lai, S. Swaminathan, V. Baylot, A. Mosley, R. Dhanasekaran, M. Gabay and D. W. Felsher, J. Immunotherap. Cancer., 2018, 6, 1–11 Search PubMed.
- Y. Wang, K. Tiruthani, S. Li, M. Hu, G. Zhong, Y. Tang, S. Roy, L. Zhang, J. Tan and C. Liao, Adv. Mater., 2021, 33, 2007603 CrossRef CAS PubMed.
- D. Zhang, G. Wang, X. Yu, T. Wei, L. Farbiak, L. T. Johnson, A. M. Taylor, J. Xu, Y. Hong and H. Zhu, Nat. Nanotechnol., 2022, 17, 777–787 CrossRef CAS PubMed.
- S.-h Hsu, B. Yu, X. Wang, Y. Lu, C. R. Schmidt, R. J. Lee, L. J. Lee, S. T. Jacob and K. Ghoshal, Nanomedicine, 2013, 9, 1169–1180 CrossRef CAS PubMed.
- X. Wang, B. Yu, W. Ren, X. Mo, C. Zhou, H. He, H. Jia, L. Wang, S. T. Jacob and R. J. Lee, J. Controlled Release, 2013, 172, 690–698 CrossRef CAS PubMed.
- Y.-S. Lim and W. R. Kim, Clin. Liver Dis., 2008, 12, 733–746 CrossRef PubMed.
- T. A. Wynn and T. R. Ramalingam, Nat. Med., 2012, 18, 1028–1040 CrossRef CAS PubMed.
- C. Bonnans, J. Chou and Z. Werb, Nat. Rev. Mol. Cell Biol., 2014, 15, 786–801 CrossRef CAS PubMed.
- T. Tsuchida and S. L. Friedman, Nat. Rev. Gastroenterol. Hepatol., 2017, 14, 397–411 CrossRef CAS PubMed.
- T. Kisseleva and D. Brenner, Nat. Rev. Gastroenterol. Hepatol., 2021, 18, 151–166 CrossRef PubMed.
- J. Zhang, H. Shen, J. Xu, L. Liu, J. Tan, M. Li, N. Xu, S. Luo, J. Wang and F. Yang, ACS Nano, 2020, 14, 6305–6322 CrossRef CAS PubMed.
- Z. Jia, Y. Gong, Y. Pi, X. Liu, L. Gao, L. Kang, J. Wang, F. Yang, J. Tang and W. Lu, Mol. Pharmaceutics, 2018, 15, 53–62 CrossRef CAS PubMed.
- E. J. Lawitz, D. E. Shevell, G. S. Tirucherai, S. Du, W. Chen, U. Kavita, A. Coste, F. Poordad, M. Karsdal and M. Nielsen, Hepatology, 2022, 75, 912–923 CrossRef CAS PubMed.
- C. Jiménez Calvente, A. Sehgal, Y. Popov, Y. O. Kim, V. Zevallos, U. Sahin, M. Diken and D. Schuppan, Hepatology, 2015, 62, 1285–1297 CrossRef PubMed.
- J.-B. Qiao, Q.-Q. Fan, C.-L. Zhang, J. Lee, J. Byun, L. Xing, X.-D. Gao, Y.-K. Oh and H.-L. Jiang, J. Controlled Release, 2020, 321, 629–640 CrossRef CAS PubMed.
- T. Yang, M. Poenisch, R. Khanal, Q. Hu, Z. Dai, R. Li, G. Song, Q. Yuan, Q. Yao and X. Shen, J. Hepatol., 2021, 75, 1420–1433 CrossRef CAS PubMed.
- M. Hu, Y. Wang, Z. Liu, Z. Yu, K. Guan, M. Liu, M. Wang, J. Tan and L. Huang, Nat. Nanotechnol., 2021, 16, 466–477 CrossRef CAS PubMed.
- S. Kathiresan, O. Melander, C. Guiducci, A. Surti, N. P. Burtt, M. J. Rieder, G. M. Cooper, C. Roos, B. F. Voight, A. S. Havulinna, B. Wahlstrand, T. Hedner, D. Corella, E. S. Tai, J. M. Ordovas, G. Berglund, E. Vartiainen, P. Jousilahti, B. Hedblad, M.-R. Taskinen, C. Newton-Cheh, V. Salomaa, L. Peltonen, L. Groop, D. M. Altshuler and M. Orho-Melander, Nat. Genet., 2008, 40, 189–197 CrossRef CAS PubMed.
- Y.-Y. Zhang, Z.-Y. Fu, J. Wei, W. Qi, G. Baituola, J. Luo, Y.-J. Meng, S.-Y. Guo, H. Yin and S.-Y. Jiang, Science, 2018, 360, 1087–1092 CrossRef CAS PubMed.
- B.-B. Chu, Y.-C. Liao, W. Qi, C. Xie, X. Du, J. Wang, H. Yang, H.-H. Miao, B.-L. Li and B.-L. Song, Cell, 2015, 161, 291–306 CrossRef CAS PubMed.
- M. Qiu, Z. Glass, J. Chen, M. Haas, X. Jin, X. Zhao, X. Rui, Z. Ye, Y. Li and F. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2020401118 CrossRef CAS PubMed.
- K. Musunuru, A. C. Chadwick, T. Mizoguchi, S. P. Garcia, J. E. DeNizio, C. W. Reiss, K. Wang, S. Iyer, C. Dutta and V. Clendaniel, Nature, 2021, 593, 429–434 CrossRef CAS PubMed.
- Y. Suzuki, K. Hyodo, T. Suzuki, Y. Tanaka, H. Kikuchi and H. Ishihara, Int. J. Pharm., 2017, 519, 34–43 CrossRef CAS PubMed.
- L. Yang, F. Ma, F. Liu, J. Chen, X. Zhao and Q. Xu, Mol. Ther.–Nucleic Acids, 2020, 19, 1357–1367 CrossRef CAS PubMed.
- M. Tadin-Strapps, L. B. Peterson, A.-M. Cumiskey, R. L. Rosa, V. H. Mendoza, J. Castro-Perez, O. Puig, L. Zhang, W. R. Strapps, S. Yendluri, L. Andrews, V. Pickering, J. Rice, L. Luo, Z. Chen, S. Tep, B. Ason, E. P. Somers, A. B. Sachs, S. R. Bartz, J. Tian, J. Chin, B. K. Hubbard, K. K. Wong and L. J. Mitnaul, J. Lipid Res., 2011, 52, 1084–1097 CrossRef CAS PubMed.
- D. Q. Huang, A. G. Singal, Y. Kono, D. J. H. Tan, H. B. El-Serag and R. Loomba, Cell Metab., 2022, 34, 969–977 CrossRef CAS PubMed.
- F. Rizvi, E. Everton, A. R. Smith, H. Liu, E. Osota, M. Beattie, Y. Tam, N. Pardi, D. Weissman and V. Gouon-Evans, Nat. Commun., 2021, 12, 613 CrossRef CAS PubMed.
- J.-E. Zhou, L. Sun, L. Liu, Y. Jia, Y. Han, J. Shao, J. Wang, Y. Wang, L. Yu and Z. Yan, J. Controlled Release, 2022, 343, 175–186 CrossRef CAS PubMed.
- A. K. Soutar and R. P. Naoumova, Nat. Clin. Pract. Cardiovasc. Med., 2007, 4, 214–225 CrossRef CAS PubMed.
- G. Migliara, V. Baccolini, A. Rosso, E. D’Andrea, A. Massimi, P. Villari and C. De Vito, Front Public Health, 2017, 5, 252 CrossRef PubMed.
- L. N. Kasiewicz, S. Biswas, A. Beach, H. Ren, C. Dutta, A. M. Mazzola, E. Rohde, A. C. Chadwick, C. Cheng, K. Musunuru, S. Kathiresan, P. Malyala, K. G. Rajeev and A. M. Bellinger, bioRxiv, 2021.
- M. R. McGill and H. Jaeschke, in Studies on Hepatic Disorders, ed. E. Albano and M. Parola, Springer International Publishing, Cham, 2015, pp.199–214 Search PubMed.
- V. Arroyo, R. Moreau, P. S. Kamath, R. Jalan, P. Ginès, F. Nevens, J. Fernández, U. To, G. García-Tsao and B. Schnabl, Nat. Rev. Dis. Primers, 2016, 2, 16041 CrossRef PubMed.
- W. Kawase, D. Kurotaki, Y. Suzuki, H. Ishihara, T. Ban, G. R. Sato, J. Ichikawa, H. Yanai, T. Taniguchi, K. Tsukahara and T. Tamura, Mol. Ther.–Nucleic Acids, 2021, 25, 708–715 CrossRef CAS PubMed.
- Q. Yin, X. Song, P. Yang, W. Yang, X. Li, X. Wang and S. Wang, Nanomedicine, 2023, 48, 102649 CrossRef CAS PubMed.
- Y. Sato, H. Matsui, N. Yamamoto, R. Sato, T. Munakata, M. Kohara and H. Harashima, J. Controlled Release, 2017, 266, 216–225 CrossRef CAS PubMed.
- B. Chen, Y. Chen, J. Li, C. Wang, W. Song, Y. Wen, J. Lin, Y. Wu and T. Ying, mBio, 2022, 13, e0161222 CrossRef PubMed.
- N. M. Belliveau, J. Huft, P. J. C. Lin, S. Chen, A. K. K. Leung, T. J. Leaver, A. W. Wild, J. B. Lee, R. J. Taylor, Y. K. Tam, C. L. Hansen and P. R. Cullis, Mol. Ther.–Nucleic Acids, 2012, 1, e37 CrossRef PubMed.
- K. Batt, S. Xing, M. Kuharic, M. Bullano, J. Caicedo, S. Chakladar, R. Markan and S. Farahbakhshian, Haemophilia, 2023, 29, 809–818 CrossRef PubMed.
- J. P. Han, M. Kim, B. S. Choi, J. H. Lee, G. S. Lee, M. Jeong, Y. Lee, E.-A. Kim, H.-K. Oh and N. Go, Sci. Adv., 2022, 8, eabj6901 CrossRef CAS PubMed.
- F. DeRosa, B. Guild, S. Karve, L. Smith, K. Love, J. R. Dorkin, K. J. Kauffman, J. Zhang, B. Yahalom, D. G. Anderson and M. W. Heartlein, Gene Ther., 2016, 23, 699–707 CrossRef CAS PubMed.
- C.-Y. Chen, D. M. Tran, A. Cavedon, X. Cai, R. Rajendran, M. J. Lyle, P. G. V. Martini and C. H. Miao, Mol. Ther.–Nucleic Acids, 2020, 20, 534–544 CrossRef CAS PubMed.
- Y. Sekijima, J. Neurol., Neurosurg. Psychiatry, 2015, 86, 1036–1043 CrossRef PubMed.
- M. S. Maurer, J. H. Schwartz, B. Gundapaneni, P. M. Elliott, G. Merlini, M. Waddington-Cruz, A. V. Kristen, M. Grogan, R. Witteles, T. Damy, B. M. Drachman, S. J. Shah, M. Hanna, D. P. Judge, A. I. Barsdorf, P. Huber, T. A. Patterson, S. Riley, J. Schumacher, M. Stewart, M. B. Sultan and C. Rapezzi, N. Engl. J. Med., 2018, 379, 1007–1016 CrossRef CAS PubMed.
- J. D. Finn, A. R. Smith, M. C. Patel, L. Shaw, M. R. Youniss, J. van Heteren, T. Dirstine, C. Ciullo, R. Lescarbeau, J. Seitzer, R. R. Shah, A. Shah, D. Ling, J. Growe, M. Pink, E. Rohde, K. M. Wood, W. E. Salomon, W. F. Harrington, C. Dombrowski, W. R. Strapps, Y. Chang and D. V. Morrissey, Cell Rep., 2018, 22, 2227–2235 CrossRef CAS PubMed.
- C. Niemietz, O. Nadzemova, A. Zibert and H. H. J. Schmidt, Amyloid, 2020, 27, 45–51 CrossRef CAS PubMed.
- Y. Wei, L. Quan, C. Zhou and Q. Zhan, Nanomedicine, 2018, 13, 1495–1512 CrossRef CAS PubMed.
- T. Nakamura, M. Kawai, Y. Sato, M. Maeki, M. Tokeshi and H. Harashima, Mol. Pharmaceutics, 2020, 17, 944–953 CrossRef CAS PubMed.
- S. Chen, Y. Y. C. Tam, P. J. C. Lin, M. M. H. Sung, Y. K. Tam and P. R. Cullis, J. Controlled Release, 2016, 235, 236–244 CrossRef CAS PubMed.
- P. H. Huang, S. Zhao, H. Bachman, N. Nama, Z. Li, C. Chen, S. Yang, M. Wu, S. P. Zhang and T. J. Huang, Adv. Sci., 2019, 6, 1900913 CrossRef CAS PubMed.
- N. Kimura, M. Maeki, Y. Sato, Y. Note, A. Ishida, H. Tani, H. Harashima and M. Tokeshi, ACS Omega, 2018, 3, 5044–5051 CrossRef CAS PubMed.
- C. C. L. Cheung and W. T. Al-Jamal, Int. J. Pharm., 2019, 566, 687–696 CrossRef CAS PubMed.
- J. Y. Han, J. N. La Fiandra and D. L. DeVoe, Nat. Commun., 2022, 13, 6997 CrossRef CAS PubMed.
- J. M. Lim, A. Swami, L. M. Gilson, S. Chopra, S. Choi, J. Wu, R. Langer, R. Karnik and O. C. Farokhzad, ACS Nano, 2014, 8, 6056–6065 CrossRef CAS PubMed.
- S. J. Shepherd, C. C. Warzecha, S. Yadavali, R. El-Mayta, M. G. Alameh, L. Wang, D. Weissman, J. M. Wilson, D. Issadore and M. J. Mitchell, Nano Lett., 2021, 21, 5671–5680 CrossRef CAS PubMed.
- N. M. Belliveau, J. Huft, P. J. Lin, S. Chen, A. K. Leung, T. J. Leaver, A. W. Wild, J. B. Lee, R. J. Taylor, Y. K. Tam, C. L. Hansen and P. R. Cullis, Mol. Ther.–Nucleic Acids, 2012, 1, e37 CrossRef PubMed.
- D. Desai, Y. A. Guerrero, V. Balachandran, A. Morton, L. Lyon, B. Larkin and D. E. Solomon, Nanomedicine, 2021, 35, 102402 CrossRef CAS PubMed.
- M. J. Toth, T. Kim and Y. Kim, Lab Chip, 2017, 17, 2805–2813 RSC.
- K. S. Elvira, X. Casadevall i Solvas, R. C. Wootton and A. J. deMello, Nat. Chem., 2013, 5, 905–915 CrossRef CAS PubMed.
- Z. Ma, H. Tong, S. Lin, L. Zhou, D. Sun, B. Li, C. Tian and J. Chu, Nano Res., 2024, 17, 2899–2907 CrossRef CAS.
- E. Kon, U. Elia and D. Peer, Curr. Opin. Biotechnol, 2022, 73, 329–336 CrossRef CAS PubMed.
- D. J. A. Crommelin, T. J. Anchordoquy, D. B. Volkin, W. Jiskoot and E. Mastrobattista, J. Pharm. Sci., 2021, 110, 997–1001 CrossRef CAS PubMed.
- R. L. Ball, P. Bajaj and K. A. Whitehead, Int. J. Nanomed., 2017, 12, 305–315 CrossRef CAS PubMed.
- M. Packer, D. Gyawali, R. Yerabolu, J. Schariter and P. White, Nat. Commun., 2021, 12, 6777 CrossRef CAS PubMed.
- S. Li, Y. Hu, A. Li, J. Lin, K. Hsieh, Z. Schneiderman, P. Zhang, Y. Zhu, C. Qiu, E. Kokkoli, T. H. Wang and H. Q. Mao, Nat. Commun., 2022, 13, 5561 CrossRef CAS PubMed.
- K. A. Risma, K. M. Edwards, D. S. Hummell, F. F. Little, A. E. Norton, A. Stallings, R. A. Wood and J. D. Milner, J. Allergy Clin. Immunol., 2021, 147, 2075–2082 CrossRef CAS PubMed.
- B. Bozkurt, I. Kamat and P. J. Hotez, Circulation, 2021, 144, 471–484 CrossRef CAS PubMed.
- D. V. Parums, Med. Sci. Monit., 2021, 27, e932899 CAS.
- E. Rista, A. Strakosha, K. Saliaj, F. Ymeri and M. Ikonomi, Cureus, 2023, 15, e33938 Search PubMed.
- A. Flemming, Nat. Rev. Immunol., 2021, 21, 72 CrossRef CAS PubMed.
- Y. Chen, Z. Xu, P. Wang, X. M. Li, Z. W. Shuai, D. Q. Ye and H. F. Pan, Immunology, 2022, 165, 386–401 CrossRef CAS PubMed.
- Q. Cheng, T. Wei, L. Farbiak, L. T. Johnson, S. A. Dilliard and D. J. Siegwart, Nat. Nanotechnol., 2020, 15, 313–320 CrossRef CAS PubMed.
- T. Suzuki, Y. Suzuki, T. Hihara, K. Kubara, K. Kondo, K. Hyodo, K. Yamazaki, T. Ishida and H. Ishihara, Int. J. Pharm., 2020, 588, 119792 CrossRef CAS PubMed.
- J. K. Tom, T. J. Albin, S. Manna, B. A. Moser, R. C. Steinhardt and A. P. Esser-Kahn, Trends Biotechnol., 2019, 37, 373–388 CrossRef CAS PubMed.
- L. Miao, L. Li, Y. Huang, D. Delcassian, J. Chahal, J. Han, Y. Shi, K. Sadtler, W. Gao, J. Lin, J. C. Doloff, R. Langer and D. G. Anderson, Nat. Biotechnol., 2019, 37, 1174–1185 CrossRef CAS PubMed.
- M. Ripoll, M. C. Bernard, C. Vaure, E. Bazin, S. Commandeur, V. Perkov, K. Lemdani, M. C. Nicolaï, P. Bonifassi, A. Kichler, B. Frisch and J. Haensler, Biomaterials, 2022, 286, 121570 CrossRef CAS PubMed.
- V. Francia, R. M. Schiffelers, P. R. Cullis and D. Witzigmann, Bioconjugate Chem., 2020, 31, 2046–2059 CrossRef CAS PubMed.
- K. Van der Jeught, S. De Koker, L. Bialkowski, C. Heirman, P. Tjok Joe, F. Perche, S. Maenhout, S. Bevers, K. Broos, K. Deswarte, V. Malard, H. Hammad, P. Baril, T. Benvegnu, P. A. Jaffrès, S. A. A. Kooijmans, R. Schiffelers, S. Lienenklaus, P. Midoux, C. Pichon, K. Breckpot and K. Thielemans, ACS Nano, 2018, 12, 9815–9829 CrossRef CAS PubMed.
- G. Anderluzzi, G. Lou, S. Woods, S. T. Schmidt, S. Gallorini, M. Brazzoli, R. Johnson, C. W. Roberts, D. T. O’Hagan, B. C. Baudner and Y. Perrie, J. Controlled Release, 2022, 342, 388–399 CrossRef CAS PubMed.
- J. L. Schnyder, H. M. Garcia Garrido, C. A. De Pijper, J. G. Daams, C. Stijnis, A. Goorhuis and M. P. Grobusch, Travel Med. Infect. Dis., 2021, 41, 102007 CrossRef CAS PubMed.
- L. Van Hoecke, K. Roose, M. Ballegeer, Z. Zhong, N. N. Sanders, S. De Koker, X. Saelens and S. Van Lint, Mol. Ther.–Nucleic Acids, 2020, 22, 373–381 CrossRef CAS PubMed.
- T. Mao, B. Israelow, M. A. Peña-Hernández, A. Suberi, L. Zhou, S. Luyten, M. Reschke, H. Dong, R. J. Homer, W. M. Saltzman and A. Iwasaki, Science, 2022, 378, eabo2523 CrossRef CAS PubMed.
- A. Akinc, W. Querbes, S. De, J. Qin, M. Frank-Kamenetsky, K. N. Jayaprakash, M. Jayaraman, K. G. Rajeev, W. L. Cantley, J. R. Dorkin, J. S. Butler, L. Qin, T. Racie, A. Sprague, E. Fava, A. Zeigerer, M. J. Hope, M. Zerial, D. W. Sah, K. Fitzgerald, M. A. Tracy, M. Manoharan, V. Koteliansky, A. Fougerolles and M. A. Maier, Mol. Ther., 2010, 18, 1357–1364 CrossRef CAS PubMed.
- E. Sadauskas, H. Wallin, M. Stoltenberg, U. Vogel, P. Doering, A. Larsen and G. Danscher, Part. Fibre Toxicol., 2007, 4, 10 CrossRef PubMed.
- R. Zhang, R. El-Mayta, T. J. Murdoch, C. C. Warzecha, M. M. Billingsley, S. J. Shepherd, N. Gong, L. Wang, J. M. Wilson, D. Lee and M. J. Mitchell, Biomater. Sci., 2021, 9, 1449–1463 RSC.
| This journal is © The Royal Society of Chemistry 2024 |