
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetallaaromaticity involving a d0 early transition metal centre: synthesis, structure, and aromaticity of tantallapyridinazirine complexes†
Jingting
Yang‡
a,
Xin
Xu‡
b,
Zhenyang
Lin
 *b and
Zuowei
Xie
*ac
*b and
Zuowei
Xie
*ac
aDepartment of Chemistry, State Key Laboratory of Synthetic Chemistry, The Chinese University of Hong Kong, Shatin, New Territories, Hong Kong, China
bDepartment of Chemistry, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, China
cShenzhen Grubbs Institute, Department of Chemistry, Southern University of Science and Technology, Shenzhen 518055, China
First published on 14th May 2024
Abstract
Though late transition metal aromatic metallabenzenes and related heteroatom-containing analogues have been well studied, the corresponding aromatic early transition metal complexes remain elusive. Herein, we demonstrate the synthesis of aromatic, planar, and delocalised organotantallapyridinium complexes via a simple one-pot process by sequential treatment of tantalum methyl complex [η5:σ-Me2C(C5H4)(C2B10H10)]TaMe3 with alkynes and isocyanide. Single-crystal X-ray analyses, NMR spectroscopic data and DFT calculations suggest that they are aromatic tantallapyridinium complexes, a class of long-sought-after molecules. This work would shed some light on the preparation of metallaaromatics involving early transition metals.
Introduction
Aromatic chemistry has fascinated chemists for almost two centuries since Faraday isolated benzene in 1825,1 and Kekulé proposed his famous structure of benzene in 1865.2 Replacement of one CH unit in the aromatic system with an isolobal organometallic fragment delivers a class of metallaaromatic compounds, which have aroused significant synthetic and theoretical interest, for they are expected to possess both aromatic and organometallic properties,3 and extending the boundary and criteria of aromaticity.4 Represented by metallabenzene and metallabenzynes, a vast array of metallaaromatic compounds have been synthesised and characterised, varying in sizes of metallacycles, topologies of polycyclic structures (fused and spiro), types of aromaticity-contributing orbitals (π, σ), and the presence of heteroatoms or not.5 However, very narrow scope of metals is known to form isolable metallaaromatic compounds so far,5 dominated by late transition metals (Chart 1).6,7 In 2021, Xi and co-workers reported two examples of aromatic, anionic tris-spiro metalloles of V and Cr with d5 electron configuration, representing scarce examples of early transition metallaaromatic compounds.8 Still, aromatic metallacycles involving d0 metal ions remain unknown.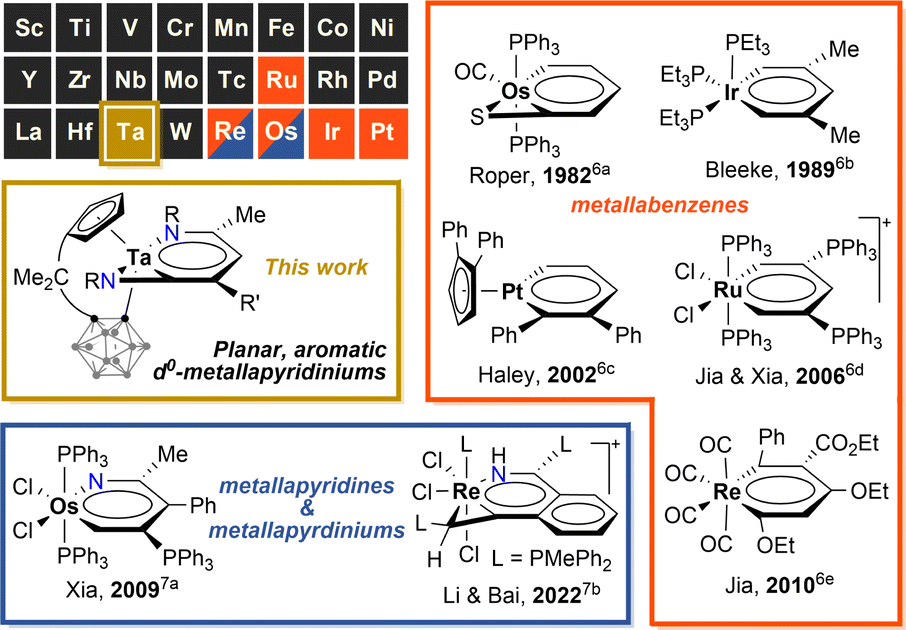 | ||
| Chart 1 Selected examples of aromatic metallabenzenes, metallapyridines and metallapyridiniums. The coloured boxes represent the elements that are known to form aromatic metallabenzenes (in orange) or metallapyridines/pyridiniums (in navy). | ||
Challenges in synthesising early transition metal-containing metallaaromatic systems, specifically aromatic metallabenzenes and heterometallabenzenes, are encountered from both experimental and theoretical aspects. Experimentally, the oxophilicity of d0 early transition metal complexes complicates their purification, excluding column chromatography. DFT (density functional theory) studies suggested that d0-metallabenzenes and related compounds, d0-metallapyrimidines, exhibit antiaromatic properties.9 The few isolable examples of d0-heterometallabenzenes show no convincing aromatic patterns, as evidenced by bond-length alternation and short M–N lengths (Chart 2).10 The only case of d0-metallabenzene, 1,3-dimetallabenzenes (M = Nb, Ta), presents a twisted nonplanar structure (Chart 2).11 These observations seem to point to the inaccessibility of aromatic d0-metallabenzenoids. Thus, aromatic metallabenzene and its heteroatomic analogues containing d0 early transition metals have become long-sought-after molecules.
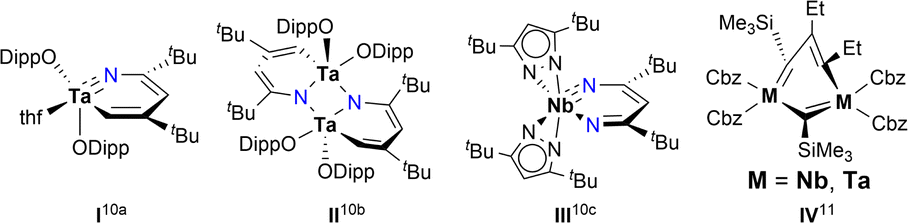 | ||
| Chart 2 Selected examples of nonaromatic d0-metallabenzenoids. | ||
During our study on organotantalum alkyne complexes bearing a multidentate bulky [Me2C(C5H4)(C2B10H10)]2− ligand, we observed sequential insertion-C–C coupling reactions with unsaturated molecules on a Ta(V) metal centre, facilely yielding a variety of d0-tantalum metallacycles.12 Based on these results, we speculate that the steric bulkiness of the σ-carboranyl ligand can effectively protect the electrophilic metal centre during multi-step insertion reactions, facilitating the formation of large metallacycles.13 To our delight, the multiple insertions of isocyanides into alkyl tantalum–alkyne complexes [η5:σ-Me2C(C5H4)(C2B10H10)]TaMe(η2-RC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH) delivered an unprecedented class of tantallapyridinazirine complexes, of which the tantallapyridinium ring manifests aromatic characters. The synthesis can be conveniently accomplished via a one-pot process from the easily prepared tantalum methyl complex [η5:σ-Me2C(C5H4)(C2B10H10)]TaMe3.14 To the best of our knowledge, it is the first example of aromatic d0-heterometallabenzene involving an early transition metal. Our findings are detailed in this article.
CH) delivered an unprecedented class of tantallapyridinazirine complexes, of which the tantallapyridinium ring manifests aromatic characters. The synthesis can be conveniently accomplished via a one-pot process from the easily prepared tantalum methyl complex [η5:σ-Me2C(C5H4)(C2B10H10)]TaMe3.14 To the best of our knowledge, it is the first example of aromatic d0-heterometallabenzene involving an early transition metal. Our findings are detailed in this article.
Results and discussion
Synthesis and characterisation
The starting material, tantalum methyl complex [η5:σ-Me2C(C5H4)(C2B10H10)]TaMe3 (1), was conveniently synthesised via a salt metathesis method.14 The organotantalum alkynes complexes [η5:σ-Me2C(C5H4)(C2B10H10)]TaMe(η2-RCCH) [R = SiMe3 (2), tBu (3), pTol (4)] were freshly prepared via the hydrogenolysis of 1 in the presence of the corresponding alkynes.12 Treatment of in situ formed 2 with two equiv. of 2,6-dimethylphenyl isocyanide (XylNC) in toluene at room temperature afforded complex 5 in 75% isolated yield. Similarly, complexes 6 and 7 were prepared in 30% and 68% yields, respectively (Scheme 1). The lower yield of 6 could be ascribed to the steric hindrance of the t-butyl group.
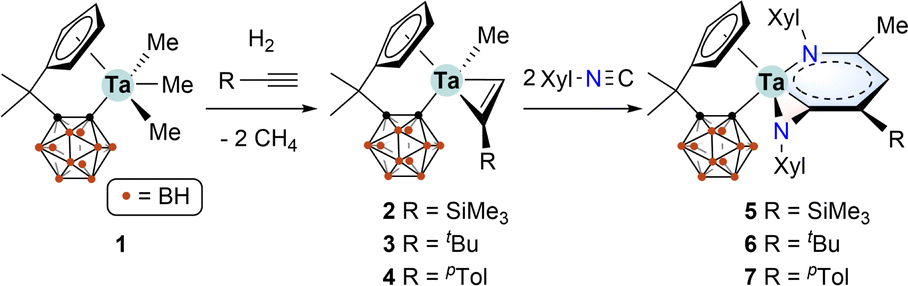 | ||
| Scheme 1 Synthesis of tantalum alkyne complexes 2–4 and tantallapyridinazirine complexes 5–7. | ||
These complexes were stable under an inert atmosphere; however, they were moisture-sensitive. They were isolated as dark brown crystals and well-characterised by single-crystal X-ray analyses and NMR spectroscopy.
Single-crystal X-ray analyses show that 5–7 share a similar core structure in which the tantalum atom is η5-bound to the cyclopentadienyl ring, σ-bound to the carboranyl cage carbon, and incorporated into a bicyclic framework consisting of a tantallapyridinium unit and a tantallaazirine moiety. The representative structure of 5 is shown in Fig. 1. The molecular structures of 6 and 7 are included in the ESI (Fig. S2 and S3†). As the key structural parameters in 5–7 are very close, we chose 5 as an example for detailed discussions.
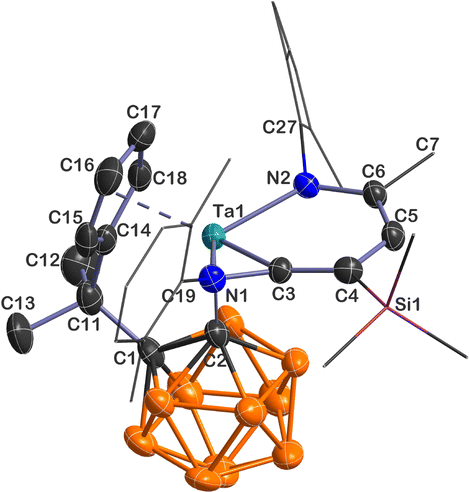 | ||
| Fig. 1 Solid-state structure of 5 drawn at the 50% probability level. For clarity, the xylyl and SiMe3 moieties are drawn in a wireframe, and the hydrogen atoms are omitted. | ||
Taking a close look at the bonding within the metallabicyclic moiety of 5, the Ta(1)–C(3) bond length (2.031(4) Å) falls at the longer end of Ta–C lengths for typical Schrock-type Ta–alkylidene complexes (1.920(6)–2.030(6) Å)15 and is shorter than a normal Ta–C(sp2) single bond (2.147(8) Å) in a tantallacyclopentadiene complex,16 suggesting some double bond character. The exocyclic Ta(1)–N(1) (1.992(3) Å) length is comparable with those found in Ta–amide complexes, and the trigonal planar geometry around N(1) is indicative of Ta(dπ) ← N(pπ) interaction.17 The Ta(1)–N(2) bond length of 2.107(3) Å is longer than the Ta(1)–N(1) one but is still within the range of Ta–N σ-bond lengths with considerable dπ ← pπ donation;18 the longer metal–nitrogen linkage is probably due to the steric repulsion between the bulky xylyl group and the carboranyl ligand. The C(3)–C(4)/C(4)–C(5)/C(5)–C(6) bond lengths (1.357(5)/1.424(6)/1.381(5) Å) are intermediate between those of typical C–C single and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds, showing some extent of bond length equalisation, and the range of the C–C lengths in 5 shows a smaller spread than those of tantallapyridine I (1.35(2)–1.45(2) Å) and tantallapyridine dimer II (1.343(6)–1.454(7) Å) (Chart 2), which share a similar TaC4N metallacycle moiety.10a,b Note that a slight extent of C–C bond length alternation is commonly observed in metallaaromatic systems, such as in a rhenabenzene (1.354(5)–1.444(5) Å).6e
C double bonds, showing some extent of bond length equalisation, and the range of the C–C lengths in 5 shows a smaller spread than those of tantallapyridine I (1.35(2)–1.45(2) Å) and tantallapyridine dimer II (1.343(6)–1.454(7) Å) (Chart 2), which share a similar TaC4N metallacycle moiety.10a,b Note that a slight extent of C–C bond length alternation is commonly observed in metallaaromatic systems, such as in a rhenabenzene (1.354(5)–1.444(5) Å).6e
The solution NMR data for characteristic metallacyclic signals in 5–7 in C6D6 are compiled in Table 1, and we chose 5 for discussion. In the 1H NMR spectrum of 5, the metallacyclic Cγ(5)–H proton was observed at 6.83 ppm as a singlet. The corresponding 6.83 and 7.05 ppm chemical shifts were recorded for 6 and 7, respectively. These exocyclic C–H resonances were downfield shifted compared with the open-chain (6.24 ppm) or cyclic (5.92–6.25 ppm) olefinic CHs for our recently reported tantalum complexes (Scheme 2a).12 Such a deshielded exocyclic proton is indicative of an aromatic environment.5 The 13C{1H} NMR spectrum of 5 displayed the signals of Cα(3), Cβ(4), Cγ(5), and Cδ(6) at 222.4, 92.7, 127.9, and 141.7 ppm, respectively. The down-field Cα chemical shift suggested a carbenoid character. The 11B{1H} NMR spectrum of 5 displayed a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:2:3 pattern ranging from −2.4 to −11.1 ppm. The solution NMR data of 6 and 7 are consistent with those of 5 (Table 1).
:3:2:3 pattern ranging from −2.4 to −11.1 ppm. The solution NMR data of 6 and 7 are consistent with those of 5 (Table 1).
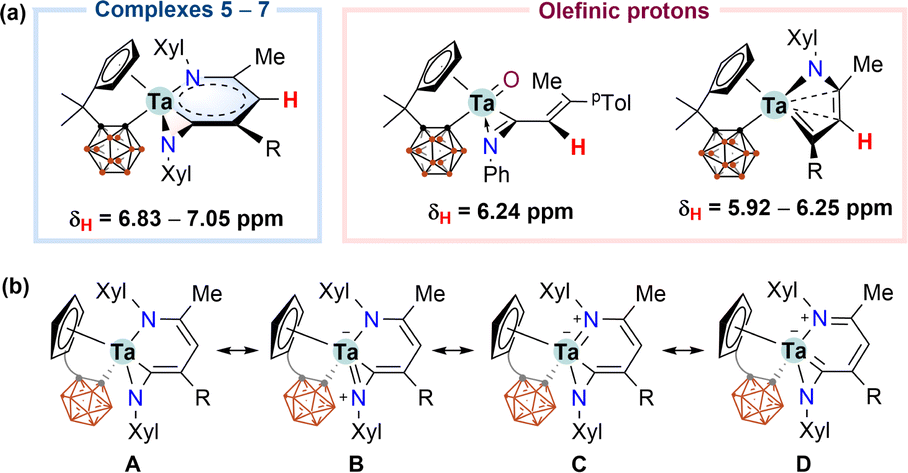 | ||
| Scheme 2 (a) 1H NMR chemical shifts for exocyclic C5H protons in 5–7 and for olefinic protons; (b) resonance structures of 5–7. | ||
The UV-vis spectra of 5, 6, and 7 (Fig. S19–S21 in the ESI†) showed the maximum absorption wavelengths λmax at 481, 480, and 491 nm, which fall in the visible light region and account for the dark colours of these complexes.
According to the structural and spectroscopic data, the overall bonding pictures of the metallabicyclic moiety in 5–7 can be described as a resonance hybrid of forms A, B, C, and D. (Scheme 2b). Structure A stands for the chelated vinyl amide form of the metallacyclic system without M–N dπ–pπ interaction, while structures B and C demonstrate the M–N dπ–pπ interaction that leads to trigonal planar geometry for both nitrogen atoms. Structures C and D account for the delocalisation over the tantallapyridinium ring, in which structure D demonstrates the carbene character of Cα(3), consistent with the downfield resonance observed from 13C NMR spectra (Table 1).
Computational studies
Concerning the co-planar structures of 5–7 with considerable bond length equalisation, we wondered whether the tantallapyridinium and tantallaazirine units are aromatic. To address this issue, we employed density functional theory (DFT) using the ωB97XD functional (see the ESI† for computational details) to calculate the nucleus-independent chemical shift (NICS) values for 5, which are normally used as a measure of aromaticity.19 NICS(0)/NICS(1)/NICS(1)zz values of −12.3/−13.9/−22.2 and −4.3/−7.8/−31.9 ppm were obtained for the tantallapyridinium (b) and tantallaazirine (a) rings, respectively (Scheme 3a). While the negative NICS values of the 3-membered tantallaazirine ring are attributable to the influence of anisotropy around the metal,4c the large negative NICS(1)zz value of the 6-membered tantallapyridinium ring points to the metallaaromatic character.19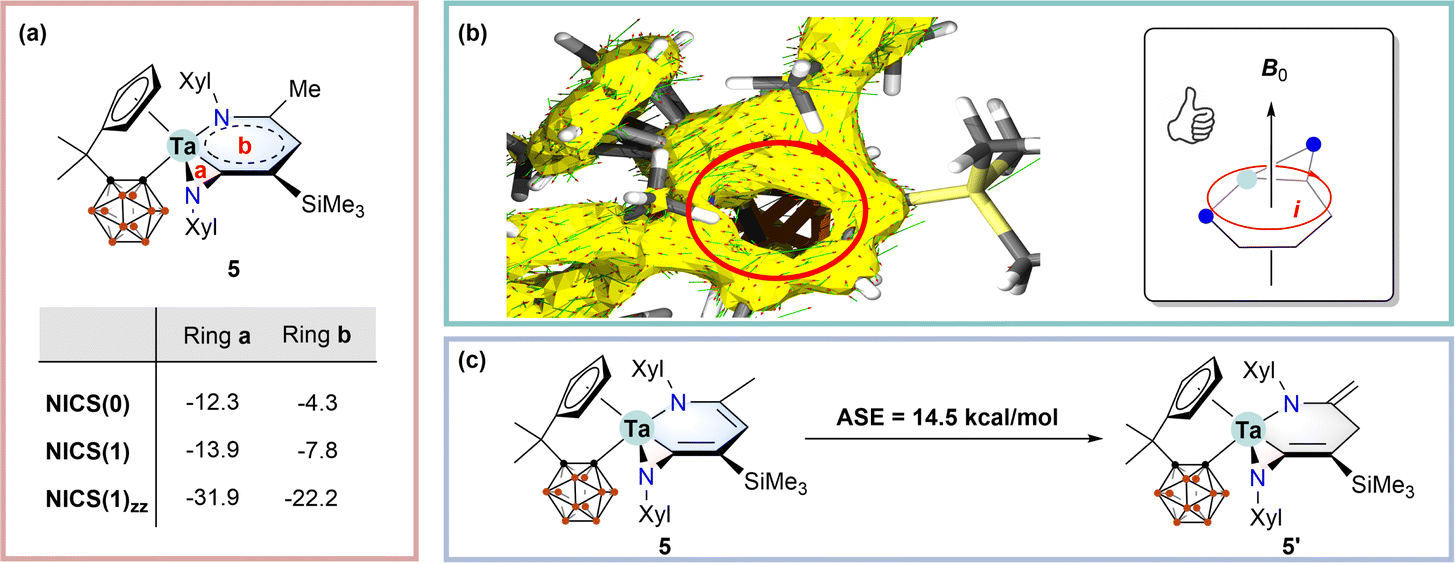 | ||
| Scheme 3 (a) Calculated nucleus-independent chemical shifts (NICSs, in ppm) for the tantallaazirine and tantallapyridinium rings in 5; (b) calculated ACID for 5 with a magnetic field pointing upward the ring plane. The yellow surface is the ACID isosurface plotted in 0.05 isovalue, and the small red-head green arrows are current density vectors. The subgraph on the right side illustrates the left-hand relationship between the external magnetic field and the induced ring current; (c) calculation of the aromatic stabilisation energy (ASE) for 5. | ||
The aromaticity of the tantallapyridinium ring in 5 is further supported by anisotropy of the induced current density (ACID) analyses. ACID is a powerful computational descriptor to visualise the delocalisation of electrons within a molecule as well as induced currents in an external magnetic field20 and has been widely used for evaluation of metallaaromaticity.4c,5g As shown in Scheme 3b, the connectivity of the ACID isosurface suggests considerable electron delocalisation over the metallabicyclic moiety in 5. When a magnetic field pointing perpendicular upwards to the tantallapyridinium plane is placed, the induced current vectors, plotted as red-head green arrows on the ACID surface, display a clockwise, diatropic ring circulation (Scheme 3b). Such a diatropic-induced current is evidence of aromaticity and is consistent with the NMR deshielding for exocyclic protons and the NICS calculation.
Aromatic stabilisation energy (ASE), calculated as the reaction energy of isodesmic reactions, is another useful criterion for aromaticity, since an aromatic molecule is usually more stable than its other isomers.21 We proposed a hypothetical isodesmic reaction for 5 using the “methyl–methylene” isomerisation method recommended by Schleyer,21 to estimate the ASE of 5 (Scheme 3c). An aromatic stabilisation energy (ASE) of 14.5 kcal mol−1 was obtained by considering the relative stability of 5 with respect to the isomeric structure (5′) (Scheme 3c). The calculated ASE value is comparable to those reported for rhenabenzenes.22
To further understand the mechanistic details for the formation of 5–7, we calculated the energy profile for the reaction of 2 with two equiv. of XylNC leading to the formation of 5 (Scheme 4). Coordination of the first molecule of XylNC to 2 is the initial event of the reaction and is slightly exergonic with a small barrier, as expected. The coordination is followed by the insertion of the coordinated XylNC to the Ta–Me bond to give η2-iminoacyl η2-alkyne complex E.12 These two steps are very facile and exergonic by 28.9 kcal mol−1. The C–C bond formation contributes significantly to the stability of E. Since E is formally a 16 e− species, further coordination of the second equiv. of XylNC to E is possible, but at a stability penalty to give E′. Clearly, the further coordination promotes reductive coupling of the two η2 ligands, leading to the formation of the 5-membered metallacyclic complex F. Again, the C–C bond formation contributes significantly to the high stability of F. From F, a very facile insertion of the second coordinated XylNC into Ta–C gives G with an exo NXyl unit. Coordination of the exo NXyl unit to the metal centre followed by a very facile structural rearrangement gives the final product 5. The calculation results indicate that the formation of 5 is very exergonic by 71.5 kcal mol−1, and the reductive coupling of the two η2 ligands is rate-determining with an overall barrier of 25.1 kcal mol−1.
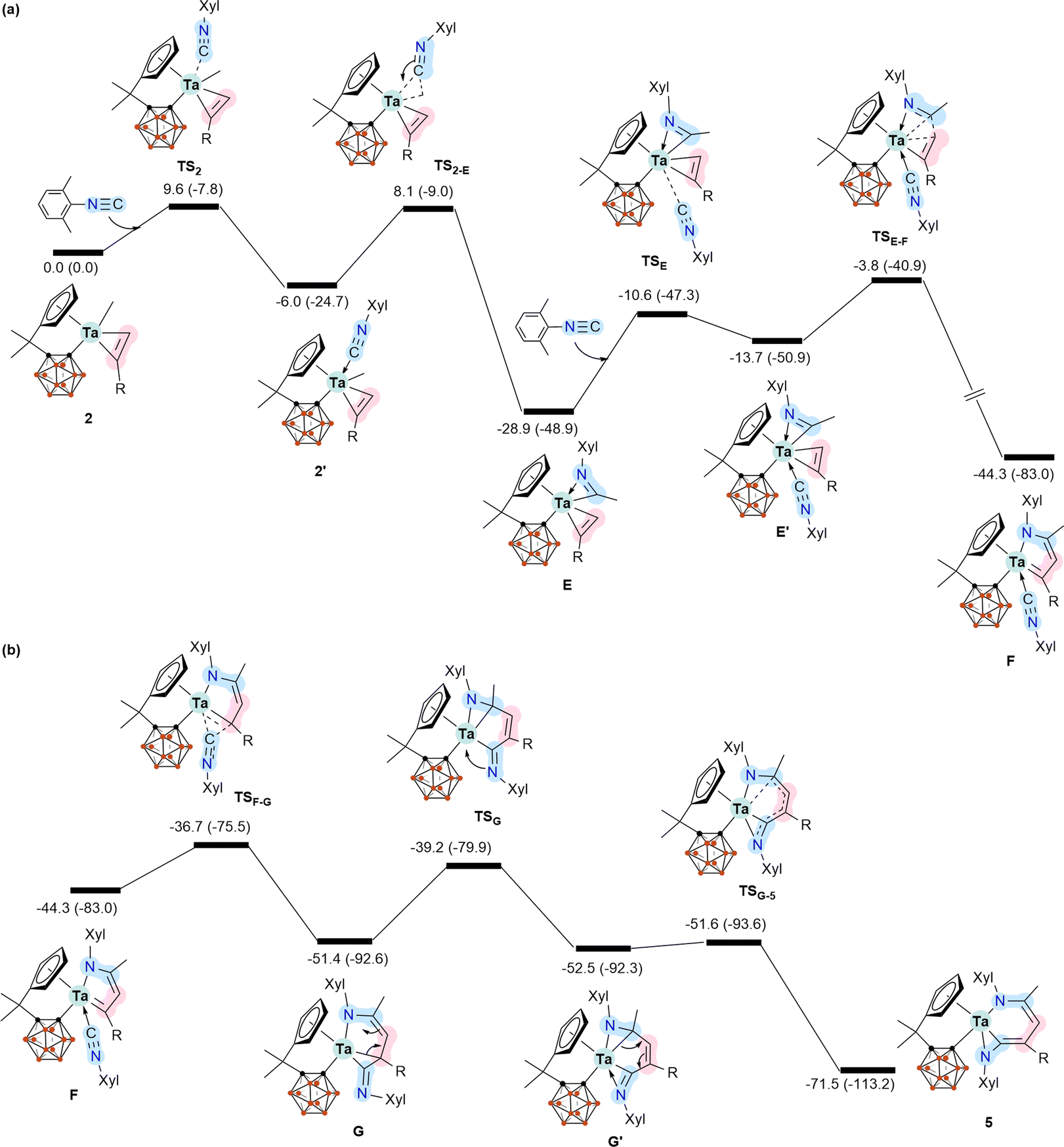 | ||
| Scheme 4 Energy profile calculated for the reaction of 2 with two molecules of XylNC leading to the formation of 5. For clarity, the energy profile is separated into two parts (a and b). The relative free energies and electronic energies (in parentheses) are given in kcal mol−1. | ||
Conclusions
We have successfully synthesised three tantallapyridinazirine complexes (5–7) bearing a linked cyclopentadienyl–carboranyl ligand via treating tantalum alkyne complexes with two equiv. of isocyanide. The aromaticity of the resultant tantallapyridinium ring is evidenced by the planar structure, equalised bond lengths, and the deshielded exocyclic proton signal in 1H NMR. Such aromaticity has also been supported by DFT calculations. Complexes 5–7 represent the first examples of metallaaromatics involving d0 early transition metal ions. This work also offers a new venue for the synthesis of such kinds of complexes.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
Z. L. and Z. X. directed and conceived this project. J. Y. conducted the experiments. X. X. performed the theoretical work. All authors discussed the results and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the Research Grants Council of HKSAR (Project No. SRFS2021-4S05 to Z. X. and 16302222 to Z. L.) and the Southern University of Science and Technology (to Z. X.).Notes and references
- M. Faraday, Philos. Trans. R. Soc. London, 1825, 115, 440 CrossRef
.
-
(a) F. A. Kekulé, Bull. Soc. Chim. Fr., 1865, 3, 98 Search PubMed
- D. L. Thorn and R. Hoffmann, Nouv. J. Chim., 1979, 3, 39 CAS
-
(a) H. S. Rzepa, Chem. Rev., 2005, 105, 3697 CrossRef CAS
- Selected reviews for metallaaromatic compounds:
(a) J. R. Bleeke, Acc. Chem. Res., 2007, 40, 1035 CrossRef CAS PubMed
- Selected articles for aromatic metallabenzenes:
(a) G. P. Elliott, W. R. Roper and J. M. Waters, J. Chem. Soc. Chem. Commun., 1982, 811 RSC
- Selected articles for aromatic metallapyridines or metallapyridiniums:
(a) B. Liu, H. Wang, H. Xie, B. Zeng, J. Chen, J. Tao, T. B. Wen, Z. Cao and H. Xia, Angew. Chem., Int. Ed., 2009, 48, 5430 CrossRef CAS PubMed
- Z. Huang, Y. Zhang, W.-X. Zhang, J. Wei, S. Ye and Z. Xi, Nat. Commun., 2021, 12, 1319 CrossRef CAS PubMed
-
(a) D. B. Lawson and R. L. DeKock, J. Phys. Chem. A, 1999, 103, 1627 CrossRef CAS
-
(a) K. J. Weller, I. Filippov, P. M. Briggs and D. E. Wigley, Organometallics, 1998, 17, 322 CrossRef CAS
- R. D. Profilet, P. E. Fanwick and I. P. Rothwell, Angew. Chem., Int. Ed., 1992, 31, 1261 CrossRef
- J. Yang and Z. Xie, Organometallics, 2023, 42, 1347 CrossRef CAS
-
(a) Z. Xie, Acc. Chem. Res., 2003, 36, 1 CrossRef CAS PubMed
- H. Tsurugi, Z. Qiu, K. Yamamoto, R. Arteaga-Muller, K. Mashima and Z. Xie, Organometallics, 2011, 30, 5960 CrossRef CAS
-
(a) M. R. Churchill, F. J. Hollander and R. R. Schrock, J. Am. Chem. Soc., 1978, 100, 647 CrossRef CAS
- J. R. Strickler, P. A. Wexler and D. E. Wigley, Organometallics, 1988, 7, 2067 CrossRef CAS
- A. S. Batsanov, A. V. Churakov, J. A. K. Howard, A. K. Hughes, A. L. Johnson, A. J. Kingsley, I. S. Neretin and K. Wade, J. Chem. Soc., Dalton Trans., 1999, 3867 RSC
-
(a) N. C. Tomson, J. Arnold and R. G. Bergman, Dalton Trans., 2011, 40, 7718 RSC
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Chem. Rev., 2005, 105, 3842 CrossRef CAS PubMed
- D. Geuenich, K. Hess, F. Köhler and R. Herges, Chem. Rev., 2005, 105, 3758 CrossRef CAS PubMed
- P. v. R. Schleyer and F. Pühlhofer, Org. Lett., 2002, 4, 2873 CrossRef CAS PubMed
- R. Lin, K.-H. Lee, K. C. Poon, H. H. Y. Sung, I. D. Williams, Z. Lin and G. Jia, Chem.–Eur. J., 2014, 20, 14885 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, complete characterisation data, computational details, and NMR spectra. CCDC 2288668–2288670 for 5–7, respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02629b |
| ‡ These authors contributed equally to this article. |
| This journal is © The Royal Society of Chemistry 2024 |