
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly intense NIR emissive Cu4Pt2 bimetallic clusters featuring Pt(I)–Cu4–Pt(I) sandwich kernel†
Rui-Ru
Zhong‡
,
Mo
Xie‡
 ,
Cui-Zhou
Luan
,
Lin-Mei
Zhang
,
De-Bo
Hao
,
Shang-Fu
Yuan
* and
Tao
Wu
*
,
Cui-Zhou
Luan
,
Lin-Mei
Zhang
,
De-Bo
Hao
,
Shang-Fu
Yuan
* and
Tao
Wu
*
College of Chemistry and Materials Science, Guangdong Provincial Key Laboratory of Functional Supramolecular Coordination Materials and Applications, Jinan University, Guangzhou 510632, P. R. China. E-mail: sfyuan@jnu.edu.cn; wutao@jnu.edu.cn
First published on 23rd April 2024
Abstract
Metal nanoclusters (NCs) capable of near-infrared (NIR) photoluminescence (PL) are gaining increasing interest for their potential applications in bioimaging, cell labelling, and phototherapy. However, the limited quantum yield (QY) of NIR emission in metal NCs, especially those emitting beyond 800 nm, hinders their widespread applications. Herein, we present a bright NIR luminescence (PLQY up to 36.7%, ∼830 nm) bimetallic Cu4Pt2 NC, [Cu4Pt2(MeO–C6H5–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C)4(dppy)4]2+ (dppy = diphenyl-2-pyridylphosphine), with a high yield (up to 67%). Furthermore, by modifying the electronic effects of R in RCC− (R = MeO–C6H5, F–C6H5, CF3–C6H5, Nap, and Biph), we can effectively modulate phosphorescence properties, including the PLQY, emission wavelength, and excited state decay lifetime. Experimental and computational studies both demonstrate that in addition to the electron effects of substituents, ligand modification enhances luminescence intensity by suppressing non-radiation transitions through intramolecular interactions. Simultaneously, it allows the adjustment of emitting wavelengths by tuning the energy gaps and first excited triplet states through intermolecular interactions of ligand substituents. This study provides a foundation for rational design of the atomic-structures of alloy metal NCs to enhance their PLQY and tailor the PL wavelength of NIR emission.
C)4(dppy)4]2+ (dppy = diphenyl-2-pyridylphosphine), with a high yield (up to 67%). Furthermore, by modifying the electronic effects of R in RCC− (R = MeO–C6H5, F–C6H5, CF3–C6H5, Nap, and Biph), we can effectively modulate phosphorescence properties, including the PLQY, emission wavelength, and excited state decay lifetime. Experimental and computational studies both demonstrate that in addition to the electron effects of substituents, ligand modification enhances luminescence intensity by suppressing non-radiation transitions through intramolecular interactions. Simultaneously, it allows the adjustment of emitting wavelengths by tuning the energy gaps and first excited triplet states through intermolecular interactions of ligand substituents. This study provides a foundation for rational design of the atomic-structures of alloy metal NCs to enhance their PLQY and tailor the PL wavelength of NIR emission.
Introduction
Atomically precise coinage metal nanoclusters (NCs), bridging the gap between discrete metal complexes and plasmonic nanomaterials, exhibit fascinating absorption characteristics and photoluminescence (PL) properties.1–3 These luminescent metal NCs, especially those with near-infrared emission, demonstrate superior biocompatibility, photo-stability, and reduced toxicity compared to organic dyes or semiconductor quantum dots (QDs), allowing diverse applications in chemical sensing, cell labelling, and bio-imaging.3–5 The well-defined molecular and supramolecular structures of metal NCs facilitate the rationalization of their optical properties, offering benefits for the systematic regulation of luminescence performance through intra-cluster assembly and inter-cluster interactions.6 However, the PL quantum yields (PLQYs) of most metal NCs with NIR emission in the solid state are typically lower than 20%, often less than 5%.7–18 Constrained by strong non-radiative processes as governed by the energy gap law,19 the development of metal NCs with highly intense NIR emission remains a significant challenge but is of great interest for exploring their potential applications in luminescent solar concentrators20,21 and optical refrigerators.22In recent years, various approaches have been employed to achieve highly photoluminescent metal NCs.23,24 One effective method is the alloying strategy, which involves doping heteroatoms into pure metal NCs, enabling the modulation of physical and chemical properties due to the synergistic effect.25,26 For instance, Liu et al. investigated the PL intensity of M1Ag24(SR)18 (M = Ag, Au, Pd, or Pt) NCs and found the Pt1Ag24(SR)18 exhibiting dual emission at both 810 and 770 nm with the highest PLQY of 18.6% in acetonitrile.27 Additionally, a ligand-exchange-induced structural transformation strategy was designed to synthesize [PtAg28(BDT)12(TPP)4]4− (TPP = triphenylphosphine; BDT = 1,3-benzenedithiolate). Compared to Ag29 with the same atomic structure, the Pt-doped sample exhibited a ∼2.3-fold enhancement in the PL intensity and a red-shift to 720 nm.28 In this regard, the incorporation of Pt, known for its strong spin–orbit coupling, through increasing metallophilic interaction has proven to be effective in tuning NIR luminescence in homoleptic metal clusters.29–31 Despite platinum-contained gold and silver nanoclusters have been well-established, as cheaper congeners, the synthesis of copper–platinum alloy NCs has remained elusive. The main challenges are attributed to (i) the big difference of half-cell reduction potential between Cu(I)/Cu(0) (0.52 V vs. NHE) and Pt(II)/Pt(0) (1.19 V vs. NHE) and (ii) the large gap of van der Waals radius between Cu (140 pm) and Pt (175 pm). Until now, only two Pt-containing Cu NCs have been reported. Among them, [Pt2Cu34(PET)22Cl4]2− (PET = 2-phenylethanethiolate) has been isolated with a metal-exchange process, but its luminescent properties have not yet been studied.32 The PtCu18 NCs display NIR phosphorescence in their crystalline state, but with PLQYs of less than 3%.33 In this regard, exploring simpler and more efficient synthesis strategies to construct Cu–Pt bimetallic clusters, especially those with strong NIR emission, would provide valuable insights into the underlying luminescence mechanisms.
In this work, we present a novel Pt-contained Cu NCs formulated as [Cu4Pt2(MeO–PhCC)4(dppy)4]2+ (MeO-Cu4Pt2, MeO–PhCCH = 4-methoxyphenylacetylene; dppy = diphenyl-2-pyridylphosphine). Interestingly, the cluster exhibits a bright NIR luminescence with an unprecedentedly high PLQY of 36.7% centered at 830 nm in solid state. Both experimental and computational studies demonstrate that modification of the ligand substituents can improve luminescence intensity by suppressing the non-radiation transitions and modulate emitting wavelength with intra- and intermolecular interactions, which provides valuable insights into the structure–property correlation.
Experimental methods
Synthesis of [Cu4Pt2(MeO–C6H5–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) C)4(dppy)4](PF6)2 (MeO-Cu4Pt2)
C)4(dppy)4](PF6)2 (MeO-Cu4Pt2)
CH (10 μL, 0.08 mmol). A pale-yellow solution formed after stirring for 5 minutes. Then, sodium hydroborate (NaBH4, 3 mg, 0.08 mmol) in 200 μL of ethanol was slowly added. The solution rapidly changed to orange, gradually turned to brown, and eventually to dark purple. The reaction mixture was stirred for 4 hours at room temperature. Then the purple solution was centrifuged for 3 min at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm. The resulting dark purple filtrate was transferred into a thin tube, and subject to the diffusion of toluene at 5 °C to afford black purple crystals after three days (16.6 mg, yield 67% based on Pt).
000 rpm. The resulting dark purple filtrate was transferred into a thin tube, and subject to the diffusion of toluene at 5 °C to afford black purple crystals after three days (16 mg, yield 64% based on Pt).
CH (13 μL, 0.1 mmol) was dispersed in 3 mL CH2Cl2. The solution rapidly changed to orange, which gradually turned to brown, and eventually to dark purple. The reaction mixture was stirred for 6 hours at room temperature. Then the resulted solution was centrifuged for 3 min at 10000 rpm. The dark purple filtrate was transferred into a thin tube, and subject to the diffusion of toluene at 5 °C to afford black purple crystals after three days (15 mg, yield 60% based on Pt).
000 rpm. The resulting dark purple filtrate was transferred into a thin tube, and subject to the diffusion of toluene at 5 °C to afford black purple crystals after three days (16.6 mg, yield 67% based on Pt).
000 rpm. The resulting dark purple filtrate was transferred into a thin tube, and subject to the diffusion of toluene at 5 °C to afford black purple crystals after three days (16 mg, yield 64% based on Pt).
CH (13 μL, 0.1 mmol) was dispersed in 3 mL CH2Cl2. The solution rapidly changed to orange, which gradually turned to brown, and eventually to dark purple. The reaction mixture was stirred for 6 hours at room temperature. Then the resulted solution was centrifuged for 3 min at 10000 rpm. The dark purple filtrate was transferred into a thin tube, and subject to the diffusion of toluene at 5 °C to afford black purple crystals after three days (15 mg, yield 60% based on Pt).
Synthesis of [Cu4Pt2(F–C6H5–CC)4(dppy)4](PF6)2 (F-Cu4Pt2)
Using the same procedure as MeO-Cu4Pt2 cluster in method 1, whereas 4-fluorophenylacetylene (F–PhCCH, 9 μL, 0.08 mmol) was employed in place of MeO–PhCCH. The crystal yield is ca. 65% (15.8 mg, based on Pt).
Synthesis of [Cu4Pt2(CF3–C6H5–CC)4(dppy)4](PF6)2 (CF3-Cu4Pt2)
Using the same procedure as MeO-Cu4Pt2 cluster in method 1, whereas 4-(trifluoromethyl) phenylacetylene (CF3–PhCCH, 13 μL, 0.08 mmol) was employed instead of MeO–PhCCH. The crystal yield is ca. 63% (15.5 mg, based on Pt).
Synthesis of [Cu4Pt2(BiPh–CC)4(dppy)4](PF6)2 (Biph-Cu4Pt2)
Using the same procedure as MeO-Cu4Pt2 cluster in method 1, whereas 4-biphenylacetylene (BiPh–CCH, 14 mg, 0.08 mmol) was employed in place of MeO–PhCCH. The crystal yield is ca. 62% (16.1 mg, based on Pt).
Synthesis of [Cu4Pt2(Nap–CC)4(dppy)4](PF6)2 (Nap-Cu4Pt2)
Using the same procedure as MeO-Cu4Pt2 cluster in method 1, but 2-ethynyl-naphthalene (Nap–CCH, 12 mg, 0.08 mmol) was employed in place of MeO–PhCCH. The crystal yield is ca. 62% (16 mg, based on Pt).
Results and discussion
The synthesis of MeO-Cu4Pt2 cluster was carried out through a one-pot synthesis method, involving the direct reduction of Cu(CH3CN)4PF6 and K2PtCl4 in the presence of MeO–PhCCH and dppy with NaBH4 (denoted as method 1). The black-purple crystals of MeO-Cu4Pt2 were afforded by the diffusion method with a yield of ≈67% (based on Pt). Notably, NaBH4 played a pivotal role as a reducing agent in this preparation, reducing Pt(II) to monovalent Pt(I) while maintaining Cu ions in the Cu(I) state (vide infra). Alternatively, Pt(acac)2 could be employed as the platinum source instead of K2PtCl4 (method 2). This modification led to a Cu-to-Pt ratio added closer to that in the final crystals, possibly due to the chlorine ions in K2PtCl4 consuming some of the Cu atoms during the reaction. Moreover, another synthesis route for MeO-Cu4Pt2 involved utilizing a previously reported copper-hydride cluster, [Cu8H6(dppy)6](PF6),34 as the reducing agent. In this case, the platinum precursors were reduced to Pt(I) ions with the copper-hydride clusters through the internal hydrides.
The chemical composition of MeO-Cu4Pt2 was firstly determined by the electrospray ionization mass spectrometry (ESI-MS) in positive mode. As depicted in Fig. 1a, a prominent peak at 1111.11 m/z corresponds to the dicationic molecular ion [Cu4Pt2(MeO–PhCC)4(dppy)4]2+ (calcd = 1111.10). The observed isotopic pattern is in perfect agreement with the simulated one (Fig. 1a inset). A small peak with lower m/z values is identified as [Cu4Pt2(MeO–PhCC)4(dppy)3]2+, which is a fragment ion after the loss of one dppy ligand (Fig. S1†). The purity of MeO-Cu4Pt2 was further supported through nuclear magnetic resonance (NMR) and powder X-ray diffraction (PXRD). The PXRD pattern matches the simulated one generated from its single-crystal data (Fig. S2†). 1H NMR spectrum of MeO-Cu4Pt2 in CD2Cl2 exhibits broad aromatic peaks corresponding to the protons in the phenyl group of MeO–PhCC− and the phenyl and pyridyl groups of dppy while the peak at 3.7 ppm is attributed to –CH3 of MeO–PhCC− (Fig. S3†). The ratio of protons for the aromatic peaks and –CH3 is 72:12, consistent with the molecular formula of MeO-Cu4Pt2 (72:11). 31P NMR spectrum shows two peaks at 14.9 and 144.5 ppm with an intensity ratio of 2:1, which can be assigned to different coordination environments of P in dppy and PF6−, respectively (Fig. 1b). These results indicate that the cluster remains intact in the solution state. Furthermore, Fourier transform infrared spectroscopy (FT-IR) reveals the stretching vibration of CC and the P–F of PF6− at 1994 cm−1 and 841 cm−1, respectively (Fig. S4†). As displayed in Fig. S5,† the transmission electron microscope (TEM) images of MeO-Cu4Pt2 illustrate an average particle size of approximately 1 nm, confirming the highly mono-dispersed nature of the clusters and their structural integrity in solution.
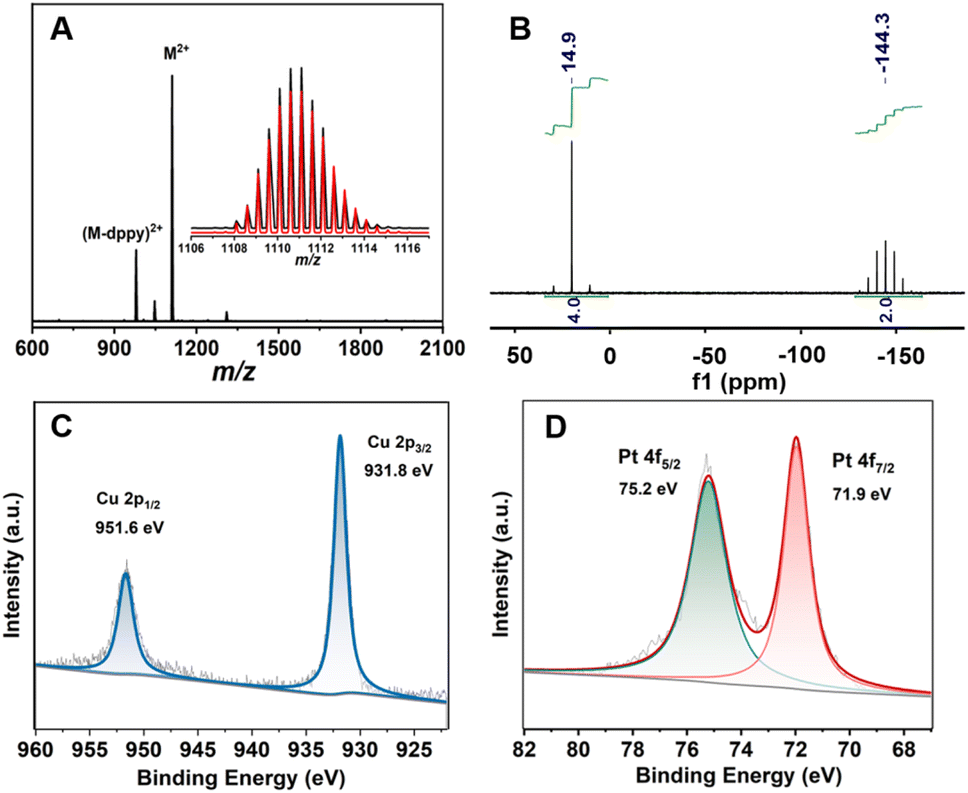 | ||
| Fig. 1 (a) ESI-MS spectra of MeO-Cu4Pt2. M = [Cu4Pt2(MeO–PhCC)4(dppy)4]. (b) 31P NMR spectrum of the MeO-Cu4Pt2. (c) Cu 2p and (d) Pt 4f XPS data for MeO-Cu4Pt2. | ||
To identify the oxidation states of constituent metal ions, X-ray photoelectron spectroscopy (XPS) was performed on MeO-Cu4Pt2. The full XPS spectra confirm the present of all expected elements (Fig. S6†). The Cu 2p3/2 peak in MeO-Cu4Pt2 is located at 931.8 eV and Cu LMM located at 569.8 eV with no shakeup satellites of Cu(II), indicating that the Cu ions exist in the “+1” valence state (Fig. 1c and S7†).35,36 In addition, the Pt 4f7/2 peak at 71.9 eV suggests an oxidation state intermediate between Pt(II) (72.4 eV) and Pt(0) (71.4 eV), supporting the present of Pt(I) in MeO-Cu4Pt2 (Fig. 1d).37–39 It should be noted that a similar Cu–Pt bimetallic cluster [Pt2Cu4{CC(3-OMe)C6H4}8]∞ has been previously reported with Pt ions in the “+2” oxidation state, while the reduced cluster with Pt(I) ions presented in this work is unprecedented. Significantly, the electron paramagnetic resonance (EPR) spectra of MeO-Cu4Pt2 display no signals indicative of unpaired spins of Pt in both crystal and solution states, implying a closed-shell electronic structure within the entire cluster (Fig. S8†).
Single-crystal X-ray diffraction analysis revealed that MeO-Cu4Pt2 crystallizes in the triclinic system with a P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group (Table S2†). It comprises two dicationic cluster [Cu4Pt2(MeO–CC)4(dppy)4]2+ and four [PF6]− counteranions in one unit cell (Fig. S9†). As shown in Fig. 2a, the octahedron-like six-metal core is surface-capped by four dppy and four alkynyl ligands. Each Pt atom in MeO-Cu4Pt2 coordinates to two terminal C atoms from different alkynyl ligands through σ bonds and two P atoms from different dppy ligands. The Cu atoms exhibit two coordination modes: Cu1 and Cu3 are coordinated to two different alkynyl ligands via π bonds, each in η2 mode, while Cu2 and Cu4 are ligated to two N atoms from different dppy ligands (Fig. 2b). Notably, the positioning of Pt atoms on the surface of the metal core in MeO-Cu4Pt2 differs from the commonly observed Pt-central-doping in reported Au, Ag, Cu, and their alloy metal NCs.28,31,32,40,41 This distinctive regional selectivity may be achieved by employing pyridylphosphine ligands, where their P and N sites selectively coordinate platinum and copper atoms based on the principle of hard and soft acid–base theory. The Cu⋯Cu distances in MeO-Cu4Pt2 range from 2.5716(12) to 2.5976(12) Å (avg. 2.5820 Å), suggesting strong Cu⋯Cu interactions.42,43 Additionally, significant Pt–Cu interactions are found in the cluster, with Pt⋯Cu distances falling within the range of 2.7219(9)–2.9163(9) Å (avg. 2.8211 Å). The average distance is shorter than the sum of the van der Waals radius of Pt and Cu (ca. 3.1 Å).29 Both the Cu⋯Cu distance (avg. 2.5820 Å) and Pt⋯Cu distance (avg. 2.8211 Å) in MeO-Cu4Pt2 are shorter than the corresponding contacts in the reported Pt(II)-based [Pt2Cu4{CC(3-OMe)C6H4}8]∞ cluster (Cu⋯Cu distance avg. 3.2922 Å and Pt⋯Cu distance avg. 3.0222 Å). These results confirm that the core structure in MeO-Cu4Pt2 is more compact.44,45 Notably, the Pt⋯Pt distance (4.3040 Å) is too long to form a metal bond. However, the sandwich structure of Pt–Cu4–Pt enables the electronic coupling between two Pt(I) atoms through the rhombus Cu4 bridge, forming a closed-shell electronic structure. Furthermore, there are two pairwise π⋯π interactions between the benzene rings of alkynyl ligands. The centroid-to-centroid distances of the aryl rings range from 3.665 to 3.779 Å (Fig. 2c). Such π⋯π interactions play an essential role in the formation and stabilization of MeO-Cu4Pt2.
space group (Table S2†). It comprises two dicationic cluster [Cu4Pt2(MeO–CC)4(dppy)4]2+ and four [PF6]− counteranions in one unit cell (Fig. S9†). As shown in Fig. 2a, the octahedron-like six-metal core is surface-capped by four dppy and four alkynyl ligands. Each Pt atom in MeO-Cu4Pt2 coordinates to two terminal C atoms from different alkynyl ligands through σ bonds and two P atoms from different dppy ligands. The Cu atoms exhibit two coordination modes: Cu1 and Cu3 are coordinated to two different alkynyl ligands via π bonds, each in η2 mode, while Cu2 and Cu4 are ligated to two N atoms from different dppy ligands (Fig. 2b). Notably, the positioning of Pt atoms on the surface of the metal core in MeO-Cu4Pt2 differs from the commonly observed Pt-central-doping in reported Au, Ag, Cu, and their alloy metal NCs.28,31,32,40,41 This distinctive regional selectivity may be achieved by employing pyridylphosphine ligands, where their P and N sites selectively coordinate platinum and copper atoms based on the principle of hard and soft acid–base theory. The Cu⋯Cu distances in MeO-Cu4Pt2 range from 2.5716(12) to 2.5976(12) Å (avg. 2.5820 Å), suggesting strong Cu⋯Cu interactions.42,43 Additionally, significant Pt–Cu interactions are found in the cluster, with Pt⋯Cu distances falling within the range of 2.7219(9)–2.9163(9) Å (avg. 2.8211 Å). The average distance is shorter than the sum of the van der Waals radius of Pt and Cu (ca. 3.1 Å).29 Both the Cu⋯Cu distance (avg. 2.5820 Å) and Pt⋯Cu distance (avg. 2.8211 Å) in MeO-Cu4Pt2 are shorter than the corresponding contacts in the reported Pt(II)-based [Pt2Cu4{CC(3-OMe)C6H4}8]∞ cluster (Cu⋯Cu distance avg. 3.2922 Å and Pt⋯Cu distance avg. 3.0222 Å). These results confirm that the core structure in MeO-Cu4Pt2 is more compact.44,45 Notably, the Pt⋯Pt distance (4.3040 Å) is too long to form a metal bond. However, the sandwich structure of Pt–Cu4–Pt enables the electronic coupling between two Pt(I) atoms through the rhombus Cu4 bridge, forming a closed-shell electronic structure. Furthermore, there are two pairwise π⋯π interactions between the benzene rings of alkynyl ligands. The centroid-to-centroid distances of the aryl rings range from 3.665 to 3.779 Å (Fig. 2c). Such π⋯π interactions play an essential role in the formation and stabilization of MeO-Cu4Pt2.
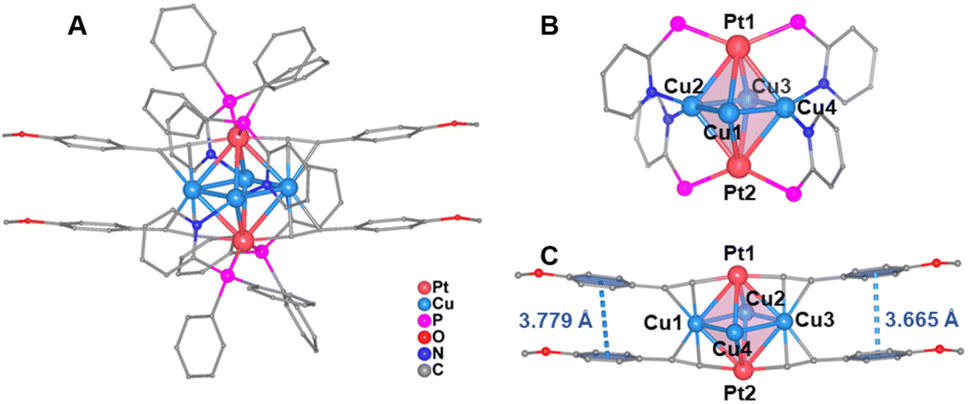 | ||
| Fig. 2 (a) The total crystal structure of MeO-Cu4Pt2. (b) Coordination motif of two dppy ligands. The benzene rings in dppy are omitted for clarity. (c) Bridging mode of four MeO–PhCC− ligands highlighting two pairs of π⋯π interactions between the benzene rings of alkynyl groups. | ||
In CH2Cl2, MeO-Cu4Pt2 presents as a purple solution. The absorption profile starts to raise from 650 nm, with four distinct absorption bands at 298, 328, 442, and 572 nm being identified (Fig. 3A). The molecular absorbance coefficient (ε) value is 3.15 × 104 M−1 cm−1 at 572 nm. Of note, copper-contained clusters with such strong absorption peaks in the visible range are rarely observed.42,46–48 We further confirm the stability of MeO-Cu4Pt2 by monitoring its optical absorption spectrum in CH2Cl2: no decomposition was observed after storage under ambient conditions for 15 hours (Fig. S10†). Interestingly, MeO-Cu4Pt2 exhibits NIR emission at 832 nm in CH2Cl2 solution upon optical excitation at 450 nm as depicted in Fig. 3A. Based on measured PLQY (2.7%) and lifetime (1.57 μs in average), the radiative decay rate constant kr and nonradiative decay rate constant knr of MeO-Cu4Pt2 (in CH2Cl2) can be calculated as 1.7 × 104 and 6.2 × 105 s−1, respectively, using the equations: τ = 1/(kr + knr); QY = kr/(kr + knr). In order to enhance the NIR emission in solution, we further explored the aggregation-induced emission enhancement (AIEE) activity of MeO-Cu4Pt2 in acetone/ethylene glycol solvent system. The degree of aggregation of metal clusters is known to be affected by the polarity of the mixed solvent, which can be manipulated by altering the volume fraction of acetone (f = volethyleneglycol/volacetone+ethyleneglycol).30,43 As illustrated in Fig. 3B, the PL intensity undergoes further enhancement with an increase in f. The highest PL intensity was attained, when f reached 90%. Transmission electron microscopy (TEM) images corroborate the gradual growth in size of cluster assembled with an increase in the fraction of the poor solvent (Fig. S11†). These results indeed support the AIEE behavior of MeO-Cu4Pt2. In addition, MeO-Cu4Pt2 was dissolved in a solution of DMSO/H2O (1:99, v/v) to assess its solubility in aqueous medium. The UV-vis absorption spectra of MeO-Cu4Pt2 in DMSO/H2O at room temperature are depicted in Fig. S12.† The QY of MeO-Cu4Pt2 in the DMSO/H2O solution was determined to be 10.1%, indicating promising prospects for biological applications.49–51
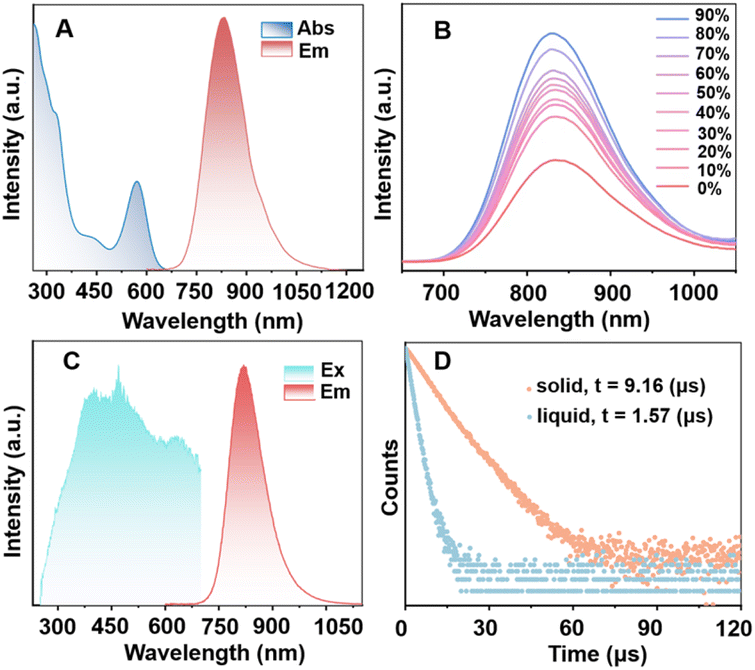 | ||
| Fig. 3 (A) The UV-vis absorption and NIR emission of MeO-Cu4Pt2 in CH2Cl2. (B) Emission spectra of MeO-Cu4Pt2 in acetone/ethylene glycol mixed solvents with different f values (cluster concentration: 1 × 10−5 mol L−1). (C) The PL excitation and emission spectra of MeO-Cu4Pt2 in the solid-state. (D) PL decay profiles for MeO-Cu4Pt2 in the solid-state and in CH2Cl2 solution. | ||
To gain further insight into the photophysical properties of MeO-Cu4Pt2, density functional theory (DFT) and time-dependent density functional theory (TD-DFT) calculations were conducted (see ESI† for detail). The simulated UV-vis absorption spectrum of MeO-Cu4Pt2 possess three major absorption peaks that is consist with the experimental spectrum (Fig. 4a). The peak I, at 572 nm, is mainly attributed to highest occupied molecular orbital (HOMO)–lowest unoccupied molecular orbital (LUMO) transition. The HOMO of MeO-Cu4Pt2 primarily occupies on the Cu4Pt2 core and alkynyl ligands, while the LUMO mainly resides on the Cu4Pt2 core (Fig. 4b and S13†). Thus, the peak I could be assigned to metal centred transition (MC). The peak II is the result of multiple electronic transitions involving HOMO → LUMO+1; HOMO → LUMO+4; HOMO−1 → LUMO; HOMO−2 → LUMO, which can be assigned as metal-to-ligand charge transfer (MLCT) characteristics. It's noteworthy that the frontier molecular orbitals are mainly distributed on Cu4Pt2 core and PhCC− ligands. Seen from Fig. 4b, the molecular orbital distribution on Cu4Pt2 core of MeO-Cu4Pt2 obey the symmetry of Oh group (Fig. S14†). The HOMO can be seen as the S-type superatomic orbital of the hexanuclear metal core while LUMO can be regarded as the P-type superatomic orbital of MeO-Cu4Pt2 (Fig. S13†).
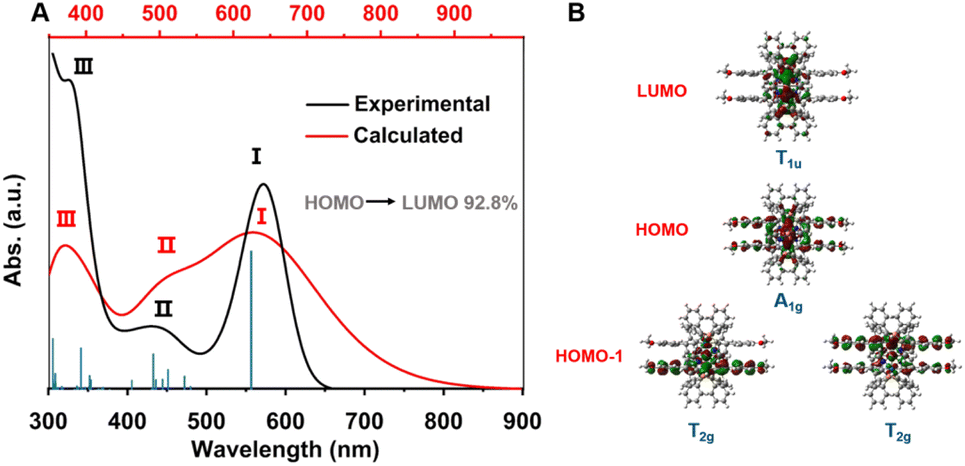 | ||
| Fig. 4 (a) Experimental optical absorption spectrum (black trace) of MeO-Cu4Pt2 in comparison to the calculated spectrum (red trace). Blue bars show the individual transitions (delta-function-like peaks showing the relative oscillator strengths). (b) Frontier orbitals of MeO-Cu4Pt2. | ||
The natural population analysis was performed to reveal the atomic charges of the coordination atoms, as shown in Fig. S15.† The Pt atoms are positively charged by +0.089 and +0.099 |e|, whereas the four Cu atoms two in one group are positively charged by +0.028 |e| and +0.054 |e|, respectively. As expected, all P and N atoms of dppy ligands are positive charge while all C atoms of alkyne moieties have negatively charged. After coordination, the total charge of metal core is calculated to be 1.622. These results confirm the assignment of all the metal atoms are with +1 charge.
Significantly, MeO-Cu4Pt2 in the solid state exhibits an intense NIR emission at room temperature with an emission peak at 824 nm upon excitation (λex) of 450 nm (Fig. 3C). The PL decay curve of MeO-Cu4Pt2 crystals demonstrates a lifetime of 9.16 μs in the microsecond range, indicative of phosphorescent emission originating from an excited triplet state (Fig. 3D).52 The PLQY of MeO-Cu4Pt2 in the solid-state was determined to be 36.7%. Remarkably, a recent breakthrough by Wang et al. involved copper substitution to form an Au16Cu6(tBuPhCC)18 cluster, achieving near-unity PLQY in deaerated solution at room temperature with an emission maximum at 720 nm.53 Compared with pure copper, copper alloy clusters, and even other coinage metals like gold and silver clusters, MeO-Cu4Pt2 exhibits the strongest luminescence in the NIR range beyond 800 nm in the solid state (Fig. S16 and Table S1†).7–18,54–56 Based on the measured PLQY (36.7%) and lifetime (9.16 μs), the kr and knr of MeO-Cu4Pt2 in the crystal state are determined to be 4.0 × 104 and 6.9 × 104 s−1, respectively. In comparison to MeO-Cu4Pt2 in the solution state, the largely reduced nonradiative transitions in the crystal significantly enhance its luminescence. This heightened NIR emission efficiency is probably attributed to the diminished nonradiative transitions (knr) facilitated by intramolecular π⋯π interactions and intermolecular C–H⋯O interactions with adjacent clusters (Fig. S17†).
Motivated by the highly luminescent nature of MeO-Cu4Pt2, we aim to systematically modulate the PL properties by altering the electronic effects of the substituents of alkynyl ligands RCC− (R = F–C6H5 for 4-fluorophenylacetylene, CF3–C6H5 for 4-(trifluoromethyl) phenylacetylene, Nap for 2-ethynyl-naphthalene, Biph for 4-biphenylacetylene).29 According to the synthetic method 1 of MeO-Cu4Pt2, F-Cu4Pt2, CF3-Cu4Pt2, Biph-Cu4Pt2, and Nap-Cu4Pt2 were successfully prepared. The chemical formulae of these Cu4Pt2 were characterized by the ESI-MS with m/z of 1086.55 for F-Cu4Pt2, 1186.55 for CF3-Cu4Pt2, 1203.14 for Biph-Cu4Pt2, and 1151.10 for Nap-Cu4Pt2, respectively, which corresponds to the molecular formulae of [Cu4Pt2(F–C6H5–CC)4(dppy)4]2+, [Cu4Pt2(CF3–C6H5–CC)4(dppy)4]2+, [Cu4Pt2(Nap–CC)4(dppy)4]2+, and [Cu4Pt2(BiPh–CC)4(dppy)4]2+ (Fig. S18 and S19†). Furthermore, FT-IR (Fig. S20†) PXRD (Fig. S21–S24), and NMR spectrum (Fig. S25–S28†) were also performed to further verify the chemical formula and purity of the above Cu4Pt2 clusters. Single-crystal X-ray diffraction analysis revealed that F-/CF3-/Biph-Cu4Pt2 are all crystallized in the monoclinic space group P21/C, expect for Nap-Cu4Pt2 which is crystallized in C2/c. These results show that F-/CF3-/Biph-/Nap-Cu4Pt2 have the same structure as MeO-Cu4Pt2 except for the different alkynyl ligands (Fig. S29 and Tables S3–S6†). Multiple C–H⋯π and π⋯π interactions stabilizing the crystals are depicted in Fig. S30–S33.†
The UV-vis absorption spectra of five R-Cu4Pt2 clusters measured in CH2Cl2 are presented in Fig. 5a. The lowest-energy peaks of the Cu4Pt2 clusters and their absorption coefficient (ε) are manifested in Table S7.† Notably, there is a gradual red-shift of the first absorption peaks as follows: Biph-Cu4Pt2 ≈ CF3-Cu4Pt2 (555 nm) < F-Cu4Pt2 (559 nm) < Nap-Cu4Pt2 (570 nm) < MeO-Cu4Pt2 (572 nm). Considering the identical core structure of Cu4Pt2, the shift of the first peaks for the five clusters in CH2Cl2 is mainly due to the electron effects of the alkyne ligands. Alkynyl ligands are typical σ–π coordination ligands, which donating electrons from σ orbitals and accept electrons via unoccupied π* orbitals. Therefore, the electron-donating effect of the σ orbital and the electron-withdrawing effect of the π* antibonding orbital of alkyne ligands jointly determine the electron effects to Cu4Pt2 core which is the luminescent centre. It should be noted that this is a case different from the ligand effects observed in metal NCs featuring phosphine and thiolate ligands.57,58 Generally, higher level σ orbital will raise the HOMO level of whole cluster and the lower level π* orbital will reduce the LUMO level of cluster. Both effects result in a smaller energy gap of metal NCs, leading to a redshift in absorptions and emissions. While the coordination between occupied orbitals should be the major effect in electronic structure. After comprehensive consideration, we valued the electron-donating capability of R substituents in the five alkyne ligands by frontier orbital analysis of alkynyl ligands (Fig. S34†).
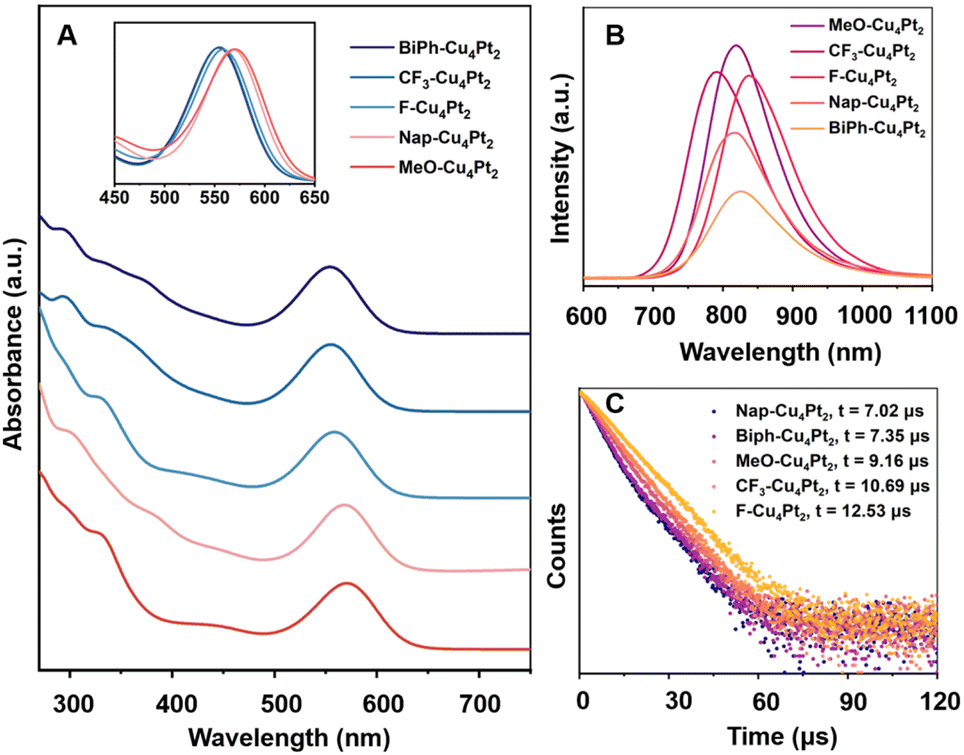 | ||
| Fig. 5 (a) The UV-vis absorption of five Cu4Pt2 in CH2Cl2. Insets: amplified graph of their normalized first absorption peaks. (b) PL emission and (c) PL decay profiles of five Cu4Pt2 crystals at ambient temperature. | ||
UV–vis diffuse-reflectance spectra (UV–vis DRS) of all R-Cu4Pt2 clusters were recorded, revealing several broad absorption peaks tailing to 800 nm (Fig. S35†). The HOMO–LUMO gaps, derived from UV–vis DRS, are shown in Fig. S36.† Among these, F-Cu4Pt2 exhibits the smallest energy gap (1.78 eV), while CF3-Cu4Pt2 has the largest gap (1.88 eV) (Table 1). Notably, in the solid-state, all the five Cu4Pt2 clusters emit bright NIR luminescence at ambient temperature (Fig. 5b). The PL decay curve of all Cu4Pt2 crystals show the microsecond time regime with single exponential function for fitting (Fig. 5c and S37†). The PL emission peaks exhibits a red-shift in the order of CF3-Cu4Pt2 (791 nm) → Nap-Cu4Pt2 (818 nm) → MeO-Cu4Pt2 (824 nm), corresponding to the increasing electron-donating capability in the order CF3 → Nap → MeO, with a decrease in the HOMO–LUMO gaps. The unexpected red shift for F-Cu4Pt2 probably attributed to the additional intermolecular F⋯π interactions, associated with the smallest optical energy gap (Fig. S30†). Similarly, the increased intra- and intermolecular π⋯π interaction of Biph-Cu4Pt2 likely contributes to its red shift in emission (Fig. S32†). The subtle changes observed in the energy gap and first excited triplet state result in a change to the transition dipole moment and thereby influencing the kr values in accordance with the Fermi's golden rule.
| Cluster | Gap (eV) | λ em (nm) | QY (%) | τ (μs) | π⋯π distance (Å) | Dihedral angle (°) | K r (104 s−1) | K nr (104 s−1) |
|---|---|---|---|---|---|---|---|---|
| a Gap, optical gap of the crystal samples; τav, average lifetime; kr, radiative decay rate; knr, nonradiative decay rate; PLQY = kr/(knr + kr); τ = 1/(knr + kr). | ||||||||
| MeO-Cu4Pt2 | 1.79 | 824 | 36.7 | 9.16 | 3.665/3.779 | 2.561/4.523 | 4.0 | 6.9 |
| CF3-Cu4Pt2 | 1.88 | 791 | 32.8 | 10.69 | 3.828/3.909 | 5.577/6.972 | 3.1 | 6.3 |
| F-Cu4Pt2 | 1.78 | 840 | 31.8 | 12.53 | 3.750/3.812 | 6.817/12.388 | 2.5 | 5.4 |
| Nap-Cu4Pt2 | 1.80 | 818 | 23.0 | 7.02 | 3.963/3.963 | 13.829/13.829 | 3.3 | 11.0 |
| Biph-Cu4Pt2 | 1.86 | 825 | 13.9 | 7.35 | 3.714/3.870 | 6.855/11.788 | 1.9 | 11.7 |
With respect to the PL intensity, the observed PLQY of Cu4Pt2 clusters follow the order of Biph-Cu4Pt2 (13%) → Nap-Cu4Pt2 (23%) → F-Cu4Pt2 (31%) → CF3-Cu4Pt2 (32%) → MeO-Cu4Pt2 (36%), corresponding to the intensity of intramolecular interactions. Typically, restricting the vibrational and rotational motions of molecules suppresses their non-radiative recombination pathways, thereby effectively enhancing the luminescence. In the present study, the intramolecular π⋯π distance and the dihedral angle between the benzene rings of alkynyl ligands serve as the crucial indicators for assessing molecular structural rigidity (Fig. 6 and Table 1). For the brightly luminous MeO-Cu4Pt2, it has the shortest π⋯π interaction distance and the smallest dihedral angle of benzene rings. In contrast, for the weekly luminous Biph-Cu4Pt2, its aromatic rings are connected by a single C–C bond, resulting in severe vibration that increases the non-radiation transitional process. The calculated rate constants Kr and Knr of the five Cu4Pt2 clusters support the above results (Table 1).
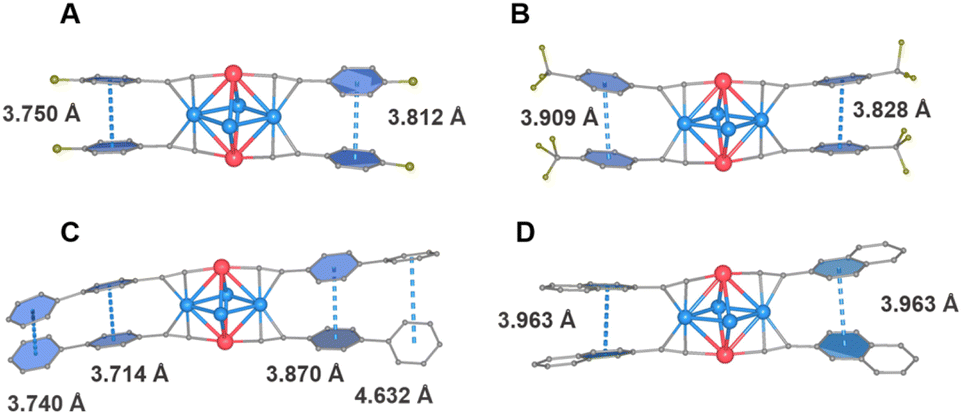 | ||
| Fig. 6 The π⋯π intramolecular interactions of (a) F-Cu4Pt2, (b) CF3-Cu4Pt2, (c) Biph-Cu4Pt2, and (d) Nap-Cu4Pt2. | ||
Conclusions
In summary, we have successfully synthesized a series of NIR emissive Cu–Pt bimetallic NCs with high yield via a one-pot method. Compared to previous work, the present study reveals three significant features: (1) a remarkable high yield (67%) of Cu–Pt bimetallic NCs achieved through a one-pot synthesis method, in contrast to the post-doping strategy commonly used in reported Pt- and Pd-doped copper clusters; (2) the presence of Pt on the surface of the metal core, diverging from the widely observed Pt-doping in the central region of metal NCs; (3) adjustment of the PL properties, such as luminescence intensity and wavelength, in the Cu4Pt2 clusters were accomplished through ligand modification-induced electronic effects and intra- and intermolecular interactions. This work not only presents a straightforward and effective approach for synthesizing alloy NCs but also provides insights into the structure–optical properties relationship, particularly in terms of NIR emission.Data availability
Data are available on request to the authors.Author contributions
S.-F. Y. and T. W. conceived the research, designed the research approach, and supervised the study; R.-R. Z. designed and performed the experiments and conducted the data analysis with the help of C.-Z. L., L.-M. Z. and D.-B. H.; M. X. carried out the DFT calculations; R.-R. Z., M. X., S.-F. Y. and T. W. wrote the manuscript with input from all the authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by National Natural Science Foundation of China (22071165, 92261205, 22201103, and U22A20432). We are grateful to Dr Zong-Jie Guan (Hunan University) for his helpful discussions regarding the DFT calculations.Notes and references
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- Y. Tao, M. Li, J. Ren and X. Qu, Chem. Soc. Rev., 2015, 44, 8636–8663 RSC.
- N. Goswami, K. Zheng and J. Xie, Nanoscale, 2014, 6, 13328–13347 RSC.
- X.-R. Song, N. Goswami, H.-H. Yang and J. Xie, Analyst, 2016, 141, 3126–3140 RSC.
- X. Kang and M. Zhu, Coord. Chem. Rev., 2019, 394, 1–38 CrossRef CAS.
- X. Wan, W. W. Xu, S. Yuan, Y. Gao, X. Zeng and Q. Wang, Angew. Chem., Int. Ed., 2015, 54, 9683–9686 CrossRef CAS PubMed.
- M.-M. Zhang, X.-Y. Dong, Z.-Y. Wang, X.-M. Luo, J.-H. Huang, S.-Q. Zang and T. C. W. Mak, J. Am. Chem. Soc., 2021, 143, 6048–6053 CrossRef CAS PubMed.
- C. Cerretani, H. Kanazawa, T. Vosch and J. Kondo, Angew. Chem., Int. Ed., 2019, 58, 17153–17157 CrossRef CAS PubMed.
- Z. Wang, R. Senanayake, C. M. Aikens, W.-M. Chen, C.-H. Tung and D. Sun, Nanoscale, 2016, 8, 18905–18911 RSC.
- K.-G. Liu, F. Bigdeli, H.-J. Li, J.-Z. Li, X.-W. Yan, M.-L. Hu and A. Morsali, Inorg. Chem., 2020, 59, 6684–6688 CrossRef CAS.
- M. Hembury, N. Beztsinna, H. Asadi, J. B. Van Den Dikkenberg, J. D. Meeldijk, W. E. Hennink and T. Vermonden, Biomacromolecules, 2018, 19, 2841–2848 CrossRef CAS PubMed.
- S. Zhu, X. Wang, S. Li, L. Liu and L. Li, ACS Appl. Mater. Interfaces, 2020, 12, 11063–11071 CrossRef CAS PubMed.
- V. Rück, C. Cerretani, V. A. Neacşu, M. B. Liisberg and T. Vosch, Phys. Chem. Chem. Phys., 2021, 23, 13483–13489 RSC.
- H. Hu, P. Huang, O. J. Weiss, X. Yan, X. Yue, M. G. Zhang, Y. Tang, L. Nie, Y. Ma, G. Niu, K. Wu and X. Chen, Biomaterials, 2014, 35, 9868–9876 CrossRef CAS PubMed.
- Y. Chen, D. M. Montana, H. Wei, J. M. Cordero, M. Schneider, X. L. Guével, O. Chen, O. T. Bruns and M. G. Bawendi, Nano Lett., 2017, 17, 6330–6334 CrossRef CAS PubMed.
- N. Uehara, N. Sonoda and C. Haneishi, Colloids Surf., A, 2018, 538, 14–22 CrossRef CAS.
- W.-D. Si, C. Zhang, M. Zhou, W.-D. Tian, Z. Wang, Q. Hu, K.-P. Song, L. Feng, X.-Q. Huang, Z.-Y. Gao, C.-H. Tung and D. Sun, Sci. Adv., 2023, 9, eadg3587 CrossRef CAS PubMed.
- Y.-C. Wei, S. F. Wang, Y. Hu, L.-S. Liao, D.-G. Chen, K.-H. Chang, C.-W. Wang, S.-H. Liu, W.-H. Chan, J.-L. Liao, W.-Y. Hung, T.-H. Wang, P.-T. Chen, H.-F. Hsu, Y. Chi and P.-T. Chou, Nat. Photonics, 2020, 14, 570–577 CrossRef CAS.
- F. Meinardi, A. Colombo, K. A. Velizhanin, R. Simonutti, M. Lorenzon, L. Beverina, R. Viswanatha, V. I. Klimov and S. Brovelli, Nat. Photonics, 2014, 8, 392–399 CrossRef CAS.
- F. Meinardi, F. Bruni and S. Brovelli, Nat. Rev. Mater., 2017, 2, 17072 CrossRef CAS.
- T. Sadi, I. Radevici and J. Oksanen, Nat. Photonics, 2020, 14, 205–214 CrossRef CAS.
- J. Ge, X. Chen, J. Yang and Y. Wang, Analyst, 2021, 146, 803–815 RSC.
- X. Kang and M. Zhu, Chem. Soc. Rev., 2019, 48, 2422–2457 RSC.
- E. Khatun and T. Pradeep, ACS Omega, 2021, 6, 1–16 CrossRef CAS PubMed.
- S. Hossain, Y. Niihori, L. V. Nair, B. Kumar, W. Kurashige and Y. Negishi, Acc. Chem. Res., 2018, 51, 3114–3124 CrossRef CAS PubMed.
- X. Liu, J. Yuan, C. Yao, J. Chen, L. Li, X. Bao, J. Yang and Z. Wu, J. Phys. Chem. C, 2017, 121, 13848–13853 CrossRef CAS.
- M. S. Bootharaju, S. M. Kozlov, Z. Cao, M. Harb, M. R. Parida, M. N. Hedhili, O. F. Mohammed, O. M. Bakr, L. Cavallo and J.-M. Basset, Nanoscale, 2017, 9, 9529–9536 RSC.
- Z.-Y. Wang, J.-Y. Wang, L.-Y. Zhang, M. Yang, X. Zhang and Z.-N. Chen, J. Mater. Chem. C, 2020, 8, 5174–5182 RSC.
- Y.-P. Li, X.-X. Fan, Y. Wu, X.-C. Zeng, J.-Y. Wang, Q.-H. Wei and Z.-N. Chen, J. Mater. Chem. C, 2017, 5, 3072–3078 RSC.
- X. Kang, M. Zhou, S. Wang, S. Jin, G. Sun, M. Zhu and R. Jin, Chem. Sci., 2017, 8, 2581–2587 RSC.
- S. Lee, M. S. Bootharaju, G. Deng, S. Malola, H. Häkkinen, N. Zheng and T. Hyeon, J. Am. Chem. Soc., 2021, 143, 12100–12107 CrossRef CAS PubMed.
- J.-J. Fang, Z. Liu, Z.-Y. Wang, Y.-P. Xie and X. Lu, Angew. Chem., Int. Ed., 2024, e202401206 Search PubMed.
- S.-F. Yuan, H.-W. Luyang, Z. Lei, X.-K. Wan, J.-J. Li and Q.-M. Wang, Chem. Commun., 2021, 57, 4315–4318 RSC.
- J.-H. Huang, L.-Y. Liu, Z.-Y. Wang, S.-Q. Zang and T. C. W. Mak, ACS Nano, 2022, 16, 18789–18794 CrossRef CAS PubMed.
- M. Qu, F. Zhang, D. Wang, H. Li, J. Hou and X. Zhang, Angew. Chem., Int. Ed., 2020, 59, 6507–6512 CrossRef CAS PubMed.
- H. Yin, S. Zhao, K. Zhao, A. Muqsit, H. Tang, L. Chang, H. Zhao, Y. Gao and Z. Tang, Nat. Commun., 2015, 6, 6430 CrossRef CAS PubMed.
- Q. Liu, Z. Yan, J. Gao, E. Wang and G. Sun, ACS Appl. Mater. Interfaces, 2020, 12, 24683–24692 CrossRef CAS PubMed.
- M. S. Bootharaju, S. M. Kozlov, Z. Cao, M. Harb, N. Maity, A. Shkurenko, M. R. Parida, M. N. Hedhili, M. Eddaoudi, O. F. Mohammed, O. M. Bakr, L. Cavallo and J.-M. Basset, J. Am. Chem. Soc., 2017, 139, 1053–1056 CrossRef CAS PubMed.
- K. K. Chakrahari, R. P. B. Silalahi, T. Chiu, X. Wang, N. Azrou, S. Kahlal, Y. Liu, M. Chiang, J. Saillard and C. W. Liu, Angew. Chem., Int. Ed., 2019, 58, 4943–4947 CrossRef CAS PubMed.
- H. Shen and T. Mizuta, Chem.–Asian J., 2017, 12, 2904–2907 CrossRef CAS.
- S. Nematulloev, R. Huang, J. Yin, A. Shkurenko, C. Dong, A. Ghosh, B. Alamer, R. Naphade, M. N. Hedhili, P. Maity, M. Eddaoudi, O. F. Mohammed and O. M. Bakr, Small, 2021, 17, 2006839 CrossRef CAS PubMed.
- M. Zhang, X. Dong, Z. Wang, H. Li, S. Li, X. Zhao and S. Zang, Angew. Chem., Int. Ed., 2020, 59, 10052–10058 CrossRef CAS PubMed.
- J. P. H. Charmant, J. Forniés, J. Gómez, E. Lalinde, R. I. Merino, M. T. Moreno and A. G. Orpen, Organometallics, 1999, 18, 3353–3358 CrossRef CAS.
- Y. Hua, J. Huang, Z. Shao, X. Luo, Z. Wang, J. Liu, X. Zhao, X. Chen and S. Zang, Adv. Mater., 2022, 34, 2203734 CrossRef CAS PubMed.
- M. Olaru, E. Rychagova, S. Ketkov, Y. Shynkarenko, S. Yakunin, M. V. Kovalenko, A. Yablonskiy, B. Andreev, F. Kleemiss, J. Beckmann and M. Vogt, J. Am. Chem. Soc., 2020, 142, 373–381 CrossRef CAS PubMed.
- A. W. Cook, Z. R. Jones, G. Wu, S. L. Scott and T. W. Hayton, J. Am. Chem. Soc., 2018, 140, 394–400 CrossRef CAS PubMed.
- S. Lee, M. S. Bootharaju, G. Deng, S. Malola, W. Baek, H. Häkkinen, N. Zheng and T. Hyeon, J. Am. Chem. Soc., 2020, 142, 13974–13981 CrossRef CAS PubMed.
- M. Chen, Z. Lei, W. Feng, C. Li, Q.-M. Wang and F. Li, Biomaterials, 2013, 34, 4284–4295 CrossRef CAS PubMed.
- W. Zhong, K. Liang, W. Liu and L. Shang, Chem. Sci., 2023, 14, 8823–8830 RSC.
- W. Zhong, X. Yan, S. Qu and L. Shang, Aggregate, 2023, 4, e245 CrossRef CAS.
- Z. Wu, J. Liu, Y. Gao, H. Liu, T. Li, H. Zou, Z. Wang, K. Zhang, Y. Wang, H. Zhang and B. Yang, J. Am. Chem. Soc., 2015, 137, 12906–12913 CrossRef CAS PubMed.
- W.-Q. Shi, L. Zeng, R.-L. He, X.-S. Han, Z.-J. Guan, M. Zhou and Q.-M. Wang, Science, 2024, 383, 326–330 CrossRef CAS PubMed.
- W.-D. Tian, W.-D. Si, S. Havenridge, C. Zhang, Z. Wang, C. M. Aikens, C.-H. Tung and D. Sun, Sci. Bull., 2024, 69(2), 40–48 CrossRef CAS PubMed.
- W.-D. Si, C. Zhang, M. Zhou, Z. Wang, L. Feng, C.-H. Tung and D. Sun, Sci. Adv., 2024, 10, eadm6928 CrossRef CAS PubMed.
- T. Jia, Z.-J. Guan, C. Zhang, X.-Z. Zhu, Y.-X. Chen, Q. Zhang, Y. Yang and D. Sun, J. Am. Chem. Soc., 2023, 145, 10355–10363 CrossRef CAS PubMed.
- X.-K. Wan, Q. Tang, S.-F. Yuan, D. Jiang and Q.-M. Wang, J. Am. Chem. Soc., 2015, 137, 652–655 CrossRef CAS PubMed.
- N. Kobayashi, Y. Kamei, Y. Shichibu and K. Konishi, J. Am. Chem. Soc., 2013, 135, 16078–16081 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Characterization details, X-ray crystallographic data. CCDC 2330685–2330689. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc01022a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |