
Photoinduced electron transfer endows fluorogenicity in tetrazine-based near-infrared labels†
Tianruo
Shen
 a,
Xin
Li
b and
Xiaogang
Liu
*a
a,
Xin
Li
b and
Xiaogang
Liu
*a
aScience, Mathematics and Technology Cluster, Singapore University of Technology and Design, 8 Somapah Road, Singapore 487372, Singapore. E-mail: xiaogang_liu@sutd.edu.sg
bCollege of Pharmaceutical Science, Zhejiang University, 866 Yuhangtang Road, Hangzhou 310058, China
First published on 8th February 2024
Abstract
Tetrazine-functionalized fluorogenic labels are pivotal in various scientific domains, but their fluorogenicity often diminishes when the emission wavelength extends into the near-infrared (NIR) region. This investigation confirms that photoinduced electron transfer (PET) is the primary mechanism causing fluorescence quenching in such NIR labels. Furthermore, we have discovered an unprecedented photoinduced charge centralization (PCC) within the conjugated frameworks of tetrazine-integrated fluorogenic dyes. These insights into the mechanisms of fluorescence quenching pave the way for advancing tetrazine chemistry at the photophysical level. Subsequently, we introduce a rational molecular design approach for crafting tetrazine-based NIR labels that leverages the PET mechanism to significantly boost their fluorogenic response.
Introduction
Tetrazine-functionalized fluorogenic labels excel in numerous applications due to their exceptional chemo-selectivity, outstanding biocompatibility, and rapid reaction kinetics.1–3 These labels generally exhibit minimal fluorescence. However, upon undergoing click reactions like inverse electron-demand Diels–Alder (iEDDA) reactions with dienophiles or [4+1] cycloadditions with isonitriles, they produce a fluorescence turn-on effect by disrupting tetrazine fragments.4 This effect is further characterized by diverse alterations in photophysical properties, including changes in molar extinction coefficients, peak absorption/emission wavelengths, quantum yields, brightness, and fluorescence turn-on ratios, due to different types of click reactions interacting with various reactants.5,6 The most striking feature of these labels is the unique fluorescence turn-on effect. It effectively reduces background emissions, thereby enhancing the signal-to-noise ratio. As a result, tetrazine-derived labels have a broad utility in various life science fields, including bioimaging, drug delivery, and photodynamic therapy.7–11 However, although these fluorogenic labels typically exhibit high turn-on ratios in the visible region, these ratios significantly diminish when their emissions transition into the near-infrared (NIR) region. More critically, the underlying fluorescence modulation mechanism of many long-wavelength tetrazine-based probes remains elusive, hampering the rational design of NIR fluorogenic labels.Currently, the advancement of tetrazine-based fluorogenic labels predominantly concentrates on the visible region. Liu and co-workers summarized energy transfer to a dark state (ETDS) and internal conversion to a dark state (ICDS) to rationalize the fluorescence quenching mechanisms of tetrazine-based labels without and with integrated π-conjugation systems, respectively.12,13 These two mechanisms highlight the presence of an energy-stabilized dark state resulting from the n–π* transition of the tetrazine fragment (Fig. 1a left). Above the dark state, a high-hanging bright π–π* state is observed, which is induced by the light absorbance of the main fluorophore (or the entire label). The dark n–π* state dissipates the energy from the high-hanging bright π–π* state through energy transfer (EnT) or internal conversion (IC), thus leading to a weak or non-emissive precursor. Noticeably, the vertical excitation energy of the bright π–π* state gradually decreases with the bathochromic shift of the main fluorophore (or the entire label), while the energy level of the dark state induced by the tetrazine remains nearly constant. Specifically, when the bright state approaches the dark state, the potential for state crossing makes the bright state more stable after geometric relaxations (Fig. 1a middle). This suppresses the EnT/IC efficiency between the two states, consequently reducing the tetrazine-caused fluorescence quenching effect. Moreover, a NIR fluorophore always produces a lower bright state than the tetrazine's dark state during the vertical excitation (Fig. 1a right). Under this condition, the EnT/IC from the bright state to the dark state is disabled, and the fluorescence quenching is henceforward greatly inhibited. These results strongly suggest the difficulty associated with the development of tetrazine-based labels emitting in the NIR range.14–16
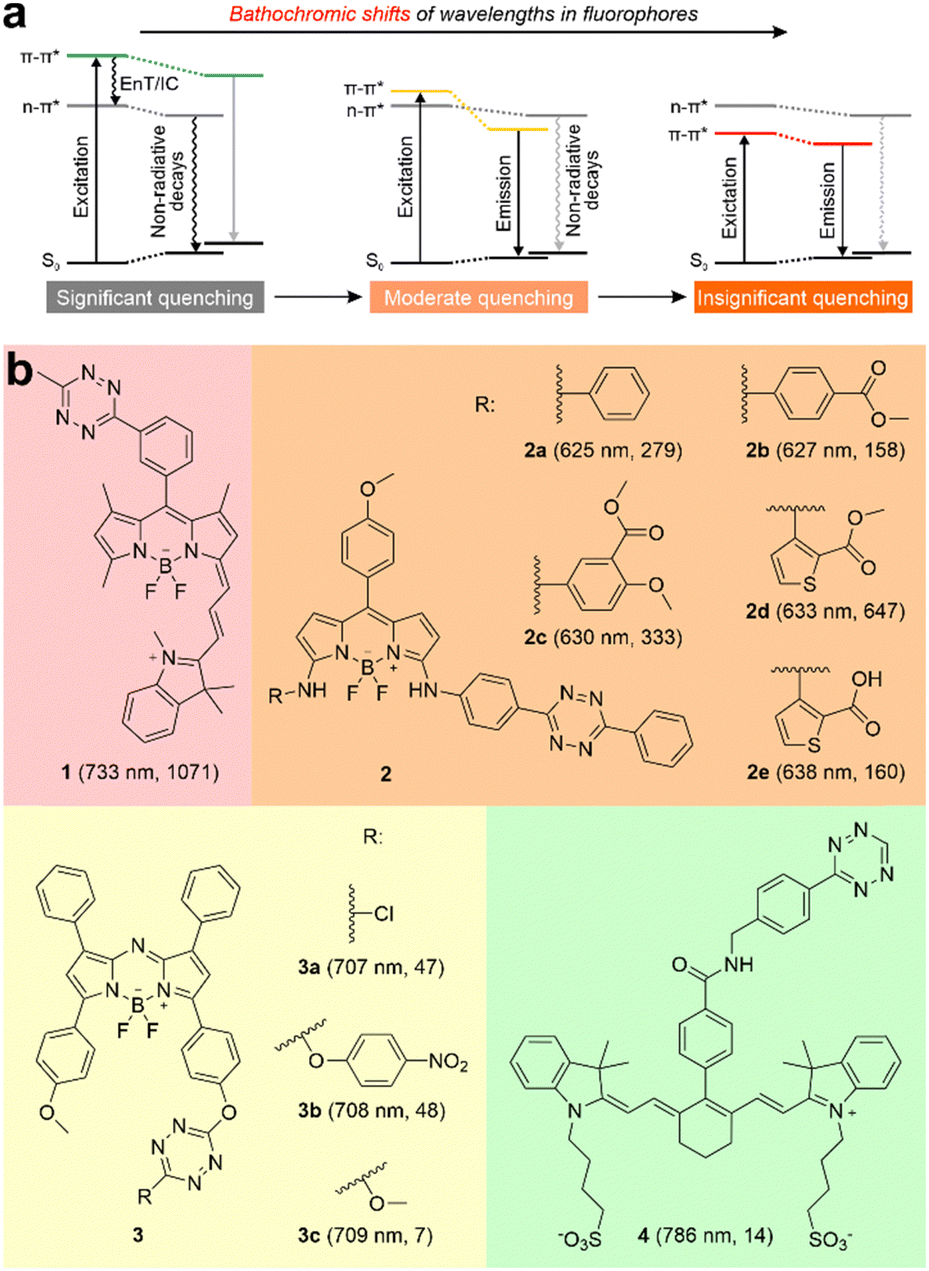 | ||
| Fig. 1 (a) Schematic illustrations of wavelength-dependent photophysical processes in tetrazine-based dyes. (b) Molecular structures of ten tetrazine-derived deep-red/NIR fluorogenic labels. The numbers in each bracket indicate the peak emission wavelength (λem) and fluorescence turn-on ratio upon iEDDA reactions, respectively. The reaction conditions are listed in Fig. S1, S2 and Table S1 (ESI†). EnT refers to energy transfer; IC denotes internal conversion. | ||
Recently, a few studies have reported several tetrazine-functionalized labels exhibiting deep-red or NIR fluorescence upon the iEDDA reactions (Fig. 1b and Fig. S1, S2, and Tables S1, S2, ESI†).17–20 Wu and O’Shea pioneered the creation of NIR fluorogenic dyes by incorporating tetrazine fragments into aza-BODIPY dyes, achieving moderate turn-on ratios of up to ∼48-fold.19 Yang and co-workers successfully produced a series of deep-red-emission BODIPY-tetrazine derivatives, achieving outstanding turn-on ratios of up to 647-fold.18 Additionally, Xu et al. developed a hybrid BODIPY-Cyanine dye peaked at 733 nm, demonstrating a remarkable turn-on ratio of 1071-fold.17 However, when tetrazine was attached to a cyanine with a longer peak emission wavelength (λem = 786 nm), the turn-on ratio was reduced to only 14-fold.20 Unfortunately, the structure–property relationship and operational mechanism of these labels are either inadequately addressed or remain subject to debate, posing challenges for the rational design of NIR-emitting tetrazine-based labels.
Herein, employing the quantum chemical calculations we showcased that photoinduced electron transfer (PET) can quench the fluorescence of red or NIR tetrazine-functionalized labels. Additionally, we uncovered a previously unrecognized variant of the photoinduced charge separation process known as photoinduced charge centralization (PCC), which specifically occurs in tetrazine-integrated fluorogenic labels with fused π-conjugations. Building upon the insights gained from the PET quenching pathway, we propose an efficient design method for the development of tetrazine-functionalized NIR labels.
Results and discussion
Rationalizing the PET quenching channel among tetrazine-derived fluorogenic labels with/toward NIR emissions
This work started by conducting detailed quantum chemical calculations to investigate the fluorescence quenching mechanism of 1 reported by Xu et al.17 The tetrazine fragment is attached to a NIR-emitting dye via a benzene ring installed at the meso-position in 1 (Fig. 1b). The computational results showed that 1 displayed distinct quenching channels from other tetrazine-based/fused fluorogenic labels in the visible region (Fig. 2a). The main fluorophore (BODIPY) in 1 emits NIR fluorescence, thus resulting a lower energy level for the bright state compared to the dark state induced by the tetrazine moiety. Specifically, during the photoexcitation of 1, the tetrazine fragment produced a locally excited (LE) state [LE (Tz), charge transfer distance (dCT) = 0.038 Å], which is a dark state with a small oscillator strength (f = 0.007) induced by the forbidden n–π* transition (Fig. 2). Below this dark state by 0.258 eV, a bright state generated by the π–π* transition in the main fluorophore (BODIPY) is observed (Fig. 2a). The large dCT (2.067 Å) suggested that this state features an intramolecular charge transfer (ICT) characteristic with a high f value (1.148, Fig. 2b).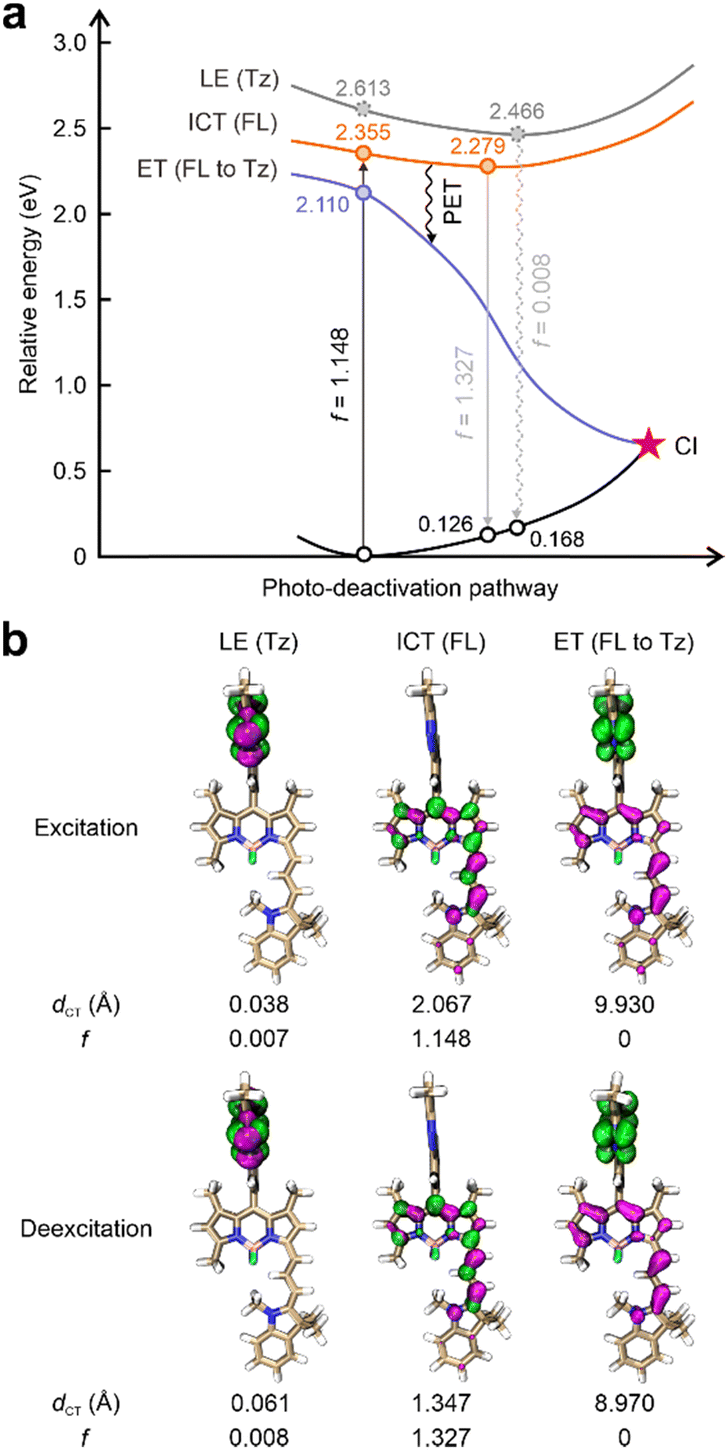 | ||
| Fig. 2 (a) Photoexcitation and deexcitation processes of 1 in dimethyl sulfoxide (DMSO). (b) Hole–electron distributions of different states with corresponding charge transfer distances (dCT) and oscillator strength (f) values of 1 in DMSO. FL: fluorophore; Tz: tetrazine; LE: locally excited; ICT: intramolecular charge transfer; ET: electron transfer; PET: photoinduced electron transfer; electron: highlighted in green; hole: highlighted in pink. | ||
This bright state [ICT (FL)] is more stable than the dark state by 0.187 eV during the deexcitation process. The traditional EnT/IC process from BODIPY to tetrazine for quenching fluorescence is therefore disabled.
Nevertheless, we have uncovered an additional pathway for fluorescence quenching in 1: PET. Beneath these two aforementioned states, another dark state with f = 0 is present. In the transition to this dark state, the electron primarily localized on the tetrazine fragment becomes effectively separated from the hole distributed across the BODIPY (dCT = 9.930 Å), showcasing a distinctive electron transfer (ET) phenomenon. In other words, BODIPY serves as the electron-donor while tetrazine acts as an electron-acceptor. As a result, the PET pathway from BODIPY to tetrazine is activated upon the geometric relaxation (Fig. 2a). The fluorescence of the BODIPY is suppressed by the subsequent conical intersection (CI) occurring between this ET state and the ground state (S0). Thus, 1 displayed almost no background emissions. After the tetrazine fragment is disintegrated via the iEDDA reaction, the dark ET state is removed. The bright π–π* state induced by the BODIPY emerges as the most stable state in the resultant (1-Pz), exhibiting notable f values in both excitation and emission processes (Fig. S2 and S3, ESI†). Consequently, the transformation from 1 to its highly emissive counterpart (1-Pz) yields a substantial increase in fluorescence intensity, as evidenced by a marked turn-on ratio.
To investigate the generalizability of this PET quenching pathway in other tetrazine-based deep-red/NIR labels, we carried out similar calculations for nine additional molecules and their respective iEDDA products (Fig. 1 and Fig. S2, S4–S14, ESI†). Among all these labels, the energy levels associated with the dark LE states resulting from the n–π* transitions in tetrazine fragments are higher than those of the bright ICT/LE states triggered by fluorophores (Fig. S4–S10, ESI†). To our delight, all the examined labels demonstrate a dark ET state, characterized by the nearly complete separation of holes (distributed on the main fluorophores) and electrons (distributed on the tetrazine segments). Hence, the subsequent transition from the bright ICT/LE state to the dark ET state takes precedence in quenching the fluorescence of each precursor. Conversely, the elimination of the dark ET state via the iEDDA reactions leads to improved fluorescence emissions in the post-reacted labels (Fig. S11–S14, ESI†).
Compared to its dominant role in deep-red and NIR fluorogenic dyes, PET plays a less significant role in quenching the fluorescence of dual-entity tetrazine-based labels with visible emissions (Fig. S15–S23, ESI†).21–24 In these visible fluorogenic dyes, the tetrazine-induced dark LE states proceed as more stable states than the bright states of the fluorophores during the photoexcitation processes. In these systems, the rate of EnT is faster than that of ET, as the ET process involves overcoming energy barriers such as exciton interactions. Therefore, the suppression of fluorescence in the tetrazine-derived labels that emit at the visible region is mainly regulated by EnT from the bright π–π* transition state to the dark n–π* transition state. A similar secondary PET pathway is also observed in other dark precursors induced by alternative quenchers (i.e., SNO and COT; Fig. S24–S26, ESI†).25,26 This is in contrast to the dominant role of PET in deep-red and NIR fluorogenic dyes, where PET plays a central role in quenching fluorescence.
Discovering a unique PCC process in the tetrazine-fused labels with integrated π-conjugations
Interestingly, we made an intriguing discovery by revealing a new form of charge transfer (CT) – charge centralization (CC) within tetrazine-fused labels featuring integrated π-conjugation (Fig. 3a, b and Fig. S27, S28, ESI†). For instance, the tetrazine fragment in 7a is incorporated as a part of the entire label, displaying excellent planarity and integrated π-conjugation. During the light excitation, the n–π* transition (dCT = 0.083 Å) of the tetrazine generated a low-lying dark state (f = 0.007). The energy of this dark state is lower than that of the bright state (f = 1.457) by 0.739 eV, which is induced by the π–π* transition (dCT = 3.196 Å) spanning throughout the entire fluorophore (including the tetrazine fragment). The fluorescence quenching can be explained by the ICDS mechanism, i.e., energy absorbed by the whole label flows to its inner tetrazine fragment via IC, resulting in label 7a being nearly non-emissive. Surprisingly, a new dark state with a low f value of 0.048 during the deexcitation is also observed in 7a (Fig. 3a). In this dark state within 7a, the hole is distributed within the entire label, while the electron concentrates on the tetrazine moiety (Fig. 3b). Both the electron and hole are part of the same π-conjugation network. This large degree of charge separation in this state resembles that during the PET process. However, the appearance of this state diverges from that of the conventional ET state, where electrons and holes are distinctly located in separate segments, isolating the PET quencher from the fluorophore. | ||
| Fig. 3 (a) Photoexcitation and deexcitation processes of 7a in water. The inset demonstrates the molecular structure of 7a. (b) Hole–electron distributions of different states with corresponding dCT and f values of 7a in water. (c) Fragment contributions (ηF) induced by tetrazine to the ET state in 1 and CC state in 7a, respectively. LE: locally excited; ICT: intramolecular charge transfer; CC: charge centralization; IC: internal conversion; PCC: photoinduced charge centralization; L: entire label; Tz: tetrazine; electron: highlighted in green; hole: highlighted in pink. | ||
To further elucidate this distinction, fragment contributions (ηF) attributed to the tetrazine moiety concerning hole, electron, and their overlap of the ET state in 1 was calculated (Fig. 3c left). The findings revealed that tetrazine has only 0.02% contribution to the hole in both excitation and deexcitation. In contrast, more than 97% of the electron is induced by tetrazine, underscoring its potency as a strong electron-acceptor. Notably, the overlap of hole–electron distributions on the tetrazine is minimal, accounting for less than 1.50% during both processes. These outcomes strongly support a conventional PET characteristic within 1, which necessitates nearly complete charge separation occurring between two independent conjugations upon photoexcitation.
In comparison, during the vertical excitation phase of 7a, tetrazine exhibits a distribution of 10.11% to holes and 54.04% to electrons (Fig. 3c right and Table S3, ESI†), respectively. The overlap percentage between hole and electron reaches 23.38%. Following geometric relaxation, charges predominantly migrated to the tetrazine fragment (98.36%). Concurrently, this tetrazine fragment also contributed to the hole, indicated by a value of 10.38%. Although this contribution is relatively small, it results in a non-negligible hole–electron overlap (31.95%). This quasi-dark state is hence denominated as a charge centralization (CC) state. Moreover, similar dark states have also been identified in other tetrazine-integrated fluorogenic labels (Fig. S27–S29 and Table S3, ESI†). These findings stand in contrast to the PET characteristics exhibited by 1.
Therefore, we introduced the term “photoinduced charge centralization” (PCC) to describe this unique pathway, which rationalizes the secondary fluorescence quenching channel in tetrazine-integrated labels possessing fused π-conjugated systems. This stands in contrast to conventional PET, where the quencher is separated from the π-conjugated system of the fluorophore, resulting in inter-conjugation PET.
Utilizing the PET pathway to develop tetrazine-based NIR labels with improved performance by comprehending the structure–property relationships
The PET quenching pathway inspired us to explore the factors that can influence the PET efficiency in the tetrazine-based NIR labels. The efficient PET requires a small energy difference between the lowest unoccupied molecular orbitals (LUMO) of electron-donor and electron-acceptor (ΔEL), given that the tetrazine acts as an electron acceptor (Fig. 4a). The efficiency of PET quenching is reflected by the fluorescence turn-on ratio upon iEDDA reactions. Notably, labels in Sets 1 and 2 demonstrated strong fluorescence turn-on effects (158–1071-fold) upon click reactions (Fig. 1b). In contrast, the fluorescence enhancement ratios of post-reacted labels in Sets 3 and 4 were relatively weaker (7–48-fold). | ||
| Fig. 4 Illustrations of (a) activated and (b) deactivated PET processes utilizing the frontier molecular orbitals of tetrazine-based NIR labels. (c) Molecular structures of 8a–d and (d) corresponding electronic energies of HOMOs and LUMOs of main fluorophores in DMSO ((d) the dashed line indicates the average LUMO energy of tetrazine). | ||
Our analyses of the frontier orbitals unveiled labels within Sets 1 and 2 demonstrated small ΔEL (< 0.21 eV), indicating the LUMO energy of the main fluorophore closely approached to that of tetrazine (Fig. S30–S32, ESI†).27 Besides, the calculated distances (represented by dCT) between the electron-donor (main fluorophore) and the electron-acceptor (tetrazine) is < 10 Å, ensuring efficient PET (Fig. 2b and Fig. S4–S8, ESI†).28,29 The fluorescence of 1 and 2a–e thereby experienced substantial quenching of their fluorescence due to the rapid PET, resulting in remarkable fluorescence turn-on ratios upon iEDDA reactions (Fig. 1b).
Set 3 molecules contrastingly demonstrated large LUMO gaps, averaging at 0.552 eV (Fig. 4b and Fig. S33, ESI†). In addition, 4 displayed a notable dCT (12.945 Å; Fig. S10 and S34, ESI†). As a consequence, these four labels exhibited reduced fluorescence turn-on ratios after reacting with dienophiles (Fig. 1b).
The NIR fluorophores with bathochromically shifted peak absorption/emission wavelengths (λabs/λem) often correlate with the reduced electronic gaps between the highest occupied molecular orbital (HOMO) and LUMO (ΔEH–L).30 For enhancing PET efficiency in a tetrazine-based NIR label, it is crucial that the LUMO energy of the main fluorophore remains relatively close to that of the tetrazine. Therefore, an ideal decrease in ΔEH–L should predominantly result from an elevation of the HOMO energy level (Fig. 4a). To this end, we have affixed tetrazine fragments to various rhodamine derivatives to investigate the differences in ΔEL and ΔEH–L among these labels (Fig. 4c). Incorporating heteroatoms into the rhodamine framework elicits a redshift in λabs/λem,31–33 evidencing through the decreased ΔEH–L (Fig. 4d). It is worth noting that the ΔEH–L decrease in 8b and 8d comparing to 8a is mainly attributed to the substantially decreased LUMO energies, while their HOMO energies remain nearly unchanged (∼5.931 eV). For instance, the ΔEH–L of 8d is 0.576 eV lower than that of 8a. Yet, the sharp drop in LUMO energy increases the ΔEL to 0.431 eV in 8d, which is inconducive to efficient PET (Fig. S35, ESI†). In contrast to 8a, the LUMO energy of 8c decreased by 0.333 eV, accompanied by a noteworthy increment in the HOMO energy (0.153 eV; Fig. S35a and S35c, ESI†). This strongly implies that the introduction of a silicon atom (Si) into the rhodamine skeleton can retain a small ΔEH–L in the main fluorophore while ensuring that the PET occurs from Si-rhodamine to tetrazine (ΔEL = 0.214 eV and Fig. S35c, ESI†).
Expanding on our insights into the structure–property relationships, we crafted a NIR fluorogenic probe, designated as 8e, to serve as a model for our design approach (Fig. 5a).34 Relative to compound 8c, the tetrazine moiety in 8e is positioned in closer proximity to the primary fluorophore, resulting in a reduced ET distance from 7.157 Å to 5.692 Å (Fig. 5b and c). This shortened distance enhances the propensity for PET. Moreover, the higher HOMO energy level of the fluorophore in 8e, exceeding that of 8c by 0.178 eV, leads to a lower ΔEH–L of 4.048 eV (Fig. 5d and Fig. S35c, ESI†). Additionally, a decrease in ΔEL to 0.079 eV further facilitates PET activation while maintaining a narrow HOMO–LUMO gap, as evidenced in Fig. 5d. Collectively, these structural adjustments suggest that our design strategy is effective for developing high-performance tetrazine-functionalized labels with red-shifted wavelengths and enhanced fluorescence turn-on ratios posting click reactions. It is important to note that alongside peak absorption/emission wavelengths and fluorescence enhancement ratios, other photophysical parameters like quantum yields and brightness are also crucial for optimizing the fluorogenicity and practical utilities of tetrazine-based NIR labels. These factors should be considered during molecular design.
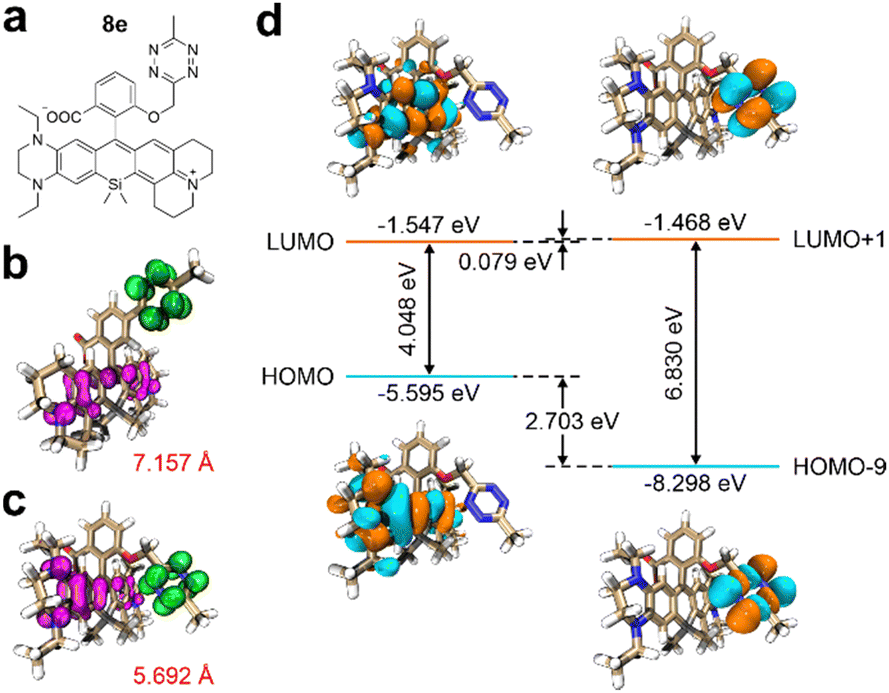 | ||
| Fig. 5 (a) Molecular structure of 8e. Optimized geometries and hole–electron distributions with the corresponding dCT of the ET states in (b) 8c and (c) 8e during the vertical excitation in DMSO. (d) Calculated electronic energies of different frontier orbitals in 8e during the vertical excitation in DMSO. Electron: highlighted in green; hole: highlighted in pink. | ||
Conclusions
In conclusion, our research underscores the pivotal role of the photoinduced electron transfer (PET) mechanism in the fluorescence attenuation of tetrazine-functionalized labels within the deep-red-to-NIR spectrum. We observed this quenching pathway even in labels that emit visible fluorescence, although PET plays a secondary role in their fluorogenicity. Importantly, our study unveils a distinct type of charge transfer, which we have termed photoinduced charge centralization (PCC), in labels where tetrazine is fused within the conjugation framework. We have also identified two critical factors for optimizing PET in these labels: a minimal electron transfer distance and a reduced ΔEL. These factors are essential for achieving marked fluorescence enhancement posting click reactions. To demonstrate these guiding principles, we have systematically engineered a tetrazine-based Si-rhodamine fluorogenic label. Our insights lay a foundational framework for advancing the design of tetrazine-based NIR labels with superior fluorogenic capabilities.Author contributions
T. Shen: conceptualization, methodology, software, investigation, formal analysis, visualization, writing – original draft, and writing – reviewing & editing; X. Li: project administration, funding acquisition, and writing – reviewing & editing; X. Liu: resources, supervision, project administration, funding acquisition, writing – reviewing & editing, and data curation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful for the support from the Ministry of Education, Singapore (No. MOET2EP10120-0007) and the Singapore University of Technology and Design (SUTD) under the SUTD-ZJU IDEA grant [SUTD-ZJU (VP) 201905]. X. Li is supported by the National Program for Support of Top-Notch Young Professionals (2021). T. Shen acknowledges the scholarship support from the Doctor of Philosophy program at SUTD. The authors are grateful for the computational resources from the National Supercomputing Centre (Singapore).Notes and references
- N. K. Devaraj, The future of bioorthogonal chemistry, ACS Cent. Sci., 2018, 4, 952–959 CrossRef CAS PubMed.
- T. Cañeque, S. Müller and R. Rodriguez, Visualizing biologically active small molecules in cells using click chemistry, Nat. Rev. Chem., 2018, 2, 202–215 CrossRef.
- S. K. Choi, J. Kim and E. Kim, Overview of syntheses and molecular-design strategies for tetrazine-based fluorogenic probes, Molecules, 2021, 26, 1868 CrossRef CAS PubMed.
- H. Sun, Q. Xue, C. Zhang, H. Wu and P. Feng, Derivatization based on tetrazine scaffolds: synthesis of tetrazine derivatives and their biomedical applications, Org. Chem. Front., 2022, 9, 481–498 RSC.
- X. Chen and Y.-W. Wu, Selective chemical labeling of proteins, Org. Biomol. Chem., 2016, 14, 5417–5439 RSC.
- Y. Deng, T. Shen, X. Yu, J. Li, P. Zou, Q. Gong, Y. Zheng, H. Sun, X. Liu and H. Wu, Tetrazine-isonitrile bioorthogonal fluorogenic reactions enable multiplex labeling and wash-free bioimaging of live cells, Angew. Chem., Int. Ed., 2024, e202319853, DOI:10.1002/anie.202319853.
- N. K. Devaraj and R. Weissleder, Biomedical applications of tetrazine cycloadditions, Acc. Chem. Res., 2011, 44, 816–827 CrossRef CAS PubMed.
- Y. Dong, Y. Tu, K. Wang, C. Xu, Y. Yuan and J. Wang, A general strategy for macrotheranostic prodrug activation: synergy between the acidic tumor microenvironment and bioorthogonal chemistry, Angew. Chem., Int. Ed., 2020, 59, 7168–7172 CrossRef CAS PubMed.
- X. Jiang, M. Li, Y. Wang, C. Wang, Y. Wang, T. Shen, L. Shen, X. Liu, Y. Wang and X. Li, 1,2,4,5-Tetrazine-tethered probes for fluorogenically imaging superoxide in live cells with ultrahigh specificity, Nat. Commun., 2023, 14, 1401 CrossRef CAS PubMed.
- E. Albitz, K. Németh, G. Knorr and P. Kele, Fluorogenic evaluation of bioorthogonally applicable tetrazine Cy3-probes for in vivo labeling schemes, Org. Biomol. Chem., 2023, 21, 7358–7366 RSC.
- Y. Wang, C. Zhang, H. Wu and P. Feng, Activation and delivery of tetrazine-responsive bioorthogonal prodrugs, Molecules, 2020, 25, 5640 CrossRef CAS PubMed.
- W. Chi, L. Huang, C. Wang, D. Tan, Z. Xu and X. Liu, A unified fluorescence quenching mechanism of tetrazine-based fluorogenic dyes: energy transfer to a dark state, Mater. Chem. Front., 2021, 5, 7012–7021 RSC.
- T. Shen, W. Zhang, P. Yadav, X. W. Sun and X. Liu, The fluorescence quenching mechanism of tetrazine-functionalized fluorogenic labels with integrated π-conjugations: internal conversion to a dark state, Mater. Chem. Front., 2023, 7, 1082–1092 RSC.
- W. Mao, W. Chi, X. He, C. Wang, X. Wang, H. Yang, X. Liu and H. Wu, Overcoming spectral dependence: a general strategy for developing far-red and near-infrared ultra-fluorogenic tetrazine bioorthogonal probes, Angew. Chem., Int. Ed., 2022, 61, e202117386 CrossRef CAS PubMed.
- L. Chen, F. Li, M. Nandi, L. Huang, Z. Chen, J. Wei, W. Chi, X. Liu and J. Yang, Towards tetrazine-based near-infrared fluorogenic dyes: is there a wavelength limit?, Dyes Pigm., 2020, 177, 108313 CrossRef CAS.
- X. Zhang, J. Gao, Y. Tang, J. Yu, S. S. Liew, C. Qiao, Y. Cao, G. Liu, H. Fan, Y. Xia, J. Tian, K. Pu and Z. Wang, Bioorthogonally activatable cyanine dye with torsion-induced disaggregation for in vivo tumor imaging, Nat. Commun., 2022, 13, 3513 CrossRef CAS PubMed.
- Z. He, T. Ishizuka, Y. Hishikawa and Y. Xu, Click chemistry for fluorescence imaging via combination of a BODIPY-based 'turn-on' probe and a norbornene glucosamine, Chem. Commun., 2022, 58, 12479–12482 RSC.
- L. Chen, F. Li, Y. Li, J. Yang, Y. Li and B. He, Red-emitting fluorogenic BODIPY-tetrazine probes for biological imaging, Chem. Commun., 2022, 58, 298–301 RSC.
- D. Wu and D. F. O'Shea, Fluorogenic NIR-probes based on 1,2,4,5-tetrazine substituted BF2-azadipyrromethenes, Chem. Commun., 2017, 53, 10804–10807 RSC.
- R. Swann, S. Slikboer, A. Genady, L. R. Silva, N. Janzen, A. Faraday, J. F. Valliant and S. Sadeghi, Tetrazine-derived near-infrared dye for targeted photoacoustic imaging of bone, J. Med. Chem., 2023, 66, 6025–6036 CrossRef CAS PubMed.
- M. E. Graziotto, L. D. Adair, A. Kaur, P. Verite, S. R. Ball, M. Sunde, D. Jacquemin and E. J. New, Versatile naphthalimide tetrazines for fluorogenic bioorthogonal labelling, RSC Chem. Biol., 2021, 2, 1491–1498 RSC.
- L. G. Meimetis, J. C. Carlson, R. J. Giedt, R. H. Kohler and R. Weissleder, Ultrafluorogenic coumarin-tetrazine probes for real-time biological imaging, Angew. Chem., Int. Ed., 2014, 53, 7531–7534 CrossRef CAS PubMed.
- S. J. Siegl, J. Galeta, R. Dzijak, A. Vazquez, M. Del Rio-Villanueva, M. Dracinsky and M. Vrabel, An extended approach for the development of fluorogenic trans-cyclooctene-tetrazine cycloadditions, ChemBioChem, 2019, 20, 886–890 CrossRef CAS PubMed.
- S. J. Siegl, J. Galeta, R. Dzijak, M. Dracinsky and M. Vrabel, Bioorthogonal fluorescence turn-on labeling based on bicyclononyne-tetrazine cycloaddition reactions that form pyridazine products, ChemPlusChem, 2019, 84, 493–497 CrossRef CAS PubMed.
- R.-Y. Guo, Y.-T. Zhang, S. Chanmungkalakul, H.-R. Guo, Y. Hu, J. Li, X. Liu, Y. Zang and X. Li, Bioinspired design of reversible fluorescent probes for tracking nitric oxide dynamics in live cells, CCS Chem., 2021, 3, 116–128 CrossRef CAS.
- S. Chanmungkalakul, S. A. A. Abedi, F. J. Hernández, J. Xu and X. Liu, The dark side of cyclooctatetraene (COT): photophysics in the singlet states of “self-healing” dyes, Chin. Chem. Lett., 2023, 109227, DOI:10.1016/j.cclet.2023.109227.
- W. Chi, J. Chen, W. Liu, C. Wang, Q. Qi, Q. Qiao, T. M. Tan, K. Xiong, X. Liu, K. Kang, Y. T. Chang, Z. Xu and X. Liu, A general descriptor deltae enables the quantitative development of luminescent materials based on photoinduced electron transfer, J. Am. Chem. Soc., 2020, 142, 6777–6785 CrossRef CAS PubMed.
- P. Werther, K. Yserentant, F. Braun, K. Grussmayer, V. Navikas, M. Yu, Z. Zhang, M. J. Ziegler, C. Mayer, A. J. Gralak, M. Busch, W. Chi, F. Rominger, A. Radenovic, X. Liu, E. A. Lemke, T. Buckup, D. P. Herten and R. Wombacher, Bio-orthogonal red and far-red fluorogenic probes for wash-free live-cell and super-resolution microscopy, ACS Cent. Sci., 2021, 7, 1561–1571 CrossRef CAS PubMed.
- A. P. de Silva, T. S. Moody and G. D. Wright, Fluorescent PET (photoinduced electron transfer) sensors as potent analytical tools, Analyst, 2009, 134, 2385–2393 RSC.
- T. Shen, Y. Gao, C. Wang, Z. Xu and X. Liu, Methine-quinoidal fragment induces significant bathochromic shifts in organic dyes, J. Phys. Chem. B, 2021, 125, 1447–1452 CrossRef CAS PubMed.
- F. Deng and Z. Xu, Heteroatom-substituted rhodamine dyes: structure and spectroscopic properties, Chin. Chem. Lett., 2019, 30, 1667–1681 CrossRef CAS.
- T. B. Ren, W. Xu, W. Zhang, X. X. Zhang, Z. Y. Wang, Z. Xiang, L. Yuan and X. B. Zhang, A general method to increase Stokes shift by introducing alternating vibronic structures, J. Am. Chem. Soc., 2018, 140, 7716–7722 CrossRef CAS PubMed.
- W. Chi, D. Tan, Q. Qiao, Z. Xu and X. Liu, Spontaneously blinking rhodamine dyes for single-molecule localization microscopy, Angew. Chem., Int. Ed., 2023, 62, e202306061 CrossRef CAS PubMed.
- X. Wu, D. Tan, Q. Qiao, W. Yin, Z. Xu and X. Liu, Molecular origins of the multi-donor strategy in inducing bathochromic shifts and enlarging Stokes shifts of fluorescent proteins, Phys. Chem. Chem. Phys., 2022, 24, 15937–15944 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details and additional results. See DOI: https://doi.org/10.1039/d3qm01217d |
| This journal is © the Partner Organisations 2024 |