
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Vat photopolymerization using catalytic chain transfer polymerization (CCTP) derived reactive oligomers to influence mechanical properties†
Wai Hin
Lee
 ab,
Zhongyuan
Wan
b,
Ataulla
Shegiwal
a and
David
Haddleton
*b
ab,
Zhongyuan
Wan
b,
Ataulla
Shegiwal
a and
David
Haddleton
*b
aHalcyon3D Ltd, 27f Ignite House, Venture Centre, Coventry, CV4 7EZ, UK
bDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: D.M.Haddleton@warwick.ac.uk
First published on 3rd July 2024
Abstract
ω-Vinyl terminated reactive oligomers of 2-ethylhexyl methacrylate and poly(ethylene glycol)methacrylate (PEGMA) were synthesized via catalytic chain transfer polymerization (CCTP) and subsequent addition–fragmentation chain transfer (AFCT) polymerization. The reactive oligomers were then incorporated into 3D-printing resins composed of isobornyl acrylate and poly(ethylene glycol)diacrylate (PEGDA). The formulations were used in digital light processing (DLP) 3D printing, and the mechanical properties of the printed parts evaluated. The effects of concentration, molecular weight and chemistry of the reactive oligomers additives were investigated. It was found that EHMA based reactive oligomers acted as plasticizers resulting in the weakening of the printed objects, whereas PEGMA based reactive oligomers transformed the objects from stiff and brittle to ductile elastomer-like materials.
Introduction
Vat photopolymerization based 3D printing is an emerging technique to fabricate polymeric materials with high resolution and volume within a relatively short time. Compared to other techniques, vat photopolymerization has the advantages of low instrument cost and high robustness towards different materials.1–3 To tailor the properties and/or functionalize the products, a small quantity of additive can be added to the resin instead of changing the entire formulation. Ideally, a reactive additive is advantageous to avoid leaching and deterioration of properties over time.Prepolymers are a class of additives commonly employed in curing resins. In essence, polymer is dissolved in the resin as solvent which will incorporate into the objects after curing. However, this method typically required relatively high additive content (up to 50% w/w). Also, increased viscosity and polymerization-induced phase separation can be issues when a high concentration, or high molecular weight of prepolymer is applied.4–8 One solution is to use reactive low molecular weight reactive prepolymers/oligomers to maintain a low viscosity and miscibility while leaching is mitigated as a result of copolymerization, in contrast to conventional non-reactive small molecular additives.9,10
Catalytic chain transfer polymerization (CCTP) is a well-established polymerization technique to synthesise oligomers with a ω-vinyl group using certain low spin Co(II) complexes as chain transfer agents.11–13 The ω-vinyl terminated oligomers, termed as reactive oligomers, can further copolymerize with other monomers to obtain graft/block architectures and therefore are good candidates for reactive (polymerizable) components in formulations. Owing to the simple in situ generation of Co catalysts,14 extremely low catalyst loading (ppm to ppk level) and facile post-synthesis deactivation of catalyst by air, CCTP has been widely adopted commercially to synthesize useful reactive oligomers. The incorporation of CCTP-derived reactive oligomers has been applied in adhesive and emulsion industries. Mitsubishi Chemical Corp. patented the use of CCTP-derived PMMA oligomers in resin compositions to improve adhesive performance.15 It was found that a non-reactive copolymer which did not use CCTP weakened the materials, presumably due to the more random distribution of MMA moiety in the copolymer and loss of its hardening ability in the adhesive. Analogously, Rohm and Haas Co. also reported the use of methacrylic acid reactive oligomers from aqueous solution CCTP in poly(butyl acrylate) coatings, with the shear resistance significantly enhanced by the graft copolymers derived from the reactive oligomers compared to the MAA-BA copolymers.16 3M have commercialized a CCTP derived crosslinker known as an addition–fragmentation monomer (AFM) in dental resins to mitigate polymerization induced shrinkage and stress accumulation in dental resin.17
In academic research, D'Agosto has shown how PMMA reactive oligomers can be used to make PE-b-PMMA block copolymers be effectively acting as a radical chain stopper in the radical polymerisation of ethene in both solution and emulsion.18,19 When added to radical polymerisation of less reactive methacrylates they undergo very efficient RAFT to give multi block polymethacrylates again in both solution and emulsion polymerisation conditions.20 The capabilities of these reactive oligomers has also been demonstrated by application in soap-free emulsion polymerization in the synthesis of functional nanoparticles.21–23
Conversely, 3D printing mediated by controlled radical polymerization has drawn only a small amount of attention. Boyer et al. investigated 3D printing mediated by RAFT derived macroinitiators and the effects of polymer architecture on the nanoscale segregation and mechanical properties.24–26 Worrell has explored the addition of cobaloxime catalyst at ppm level in the 3D printing of dimethacrylates and achieved control in gel point and thermomechanical properties.27
Recently, we have reported the application of an addition–fragmentation monomer in 3D printing to improve shrinkage stress relaxation.28 As an extension of this work, we aimed to investigate the versatility of CCTP-derived reactive oligomers in photolithographic 3D printing. Herein, we report the synthesis of reactive oligomers of ethylhexyl methacrylate (EHMA), poly(ethylene glycol)methacrylate (PEGMA) with different molecular weights and the effects of the addition of these to 3D printing resin formulations. It was found that when the reactive oligomer was compatible with the bulk monomer, the product was plasticized by the reactive oligomer leading to a weakened product. Meanwhile, an incompatible reactive oligomer resulted in a good reinforcement effect without significant mechanical deterioration. This current work offers a facile route to attain a broad spectrum of mechanical properties of 3D printing materials with low content (≤10% w/w) of additive.
Experimental details
Materials
Ethylhexyl methacrylate (98%), poly(ethylene glycol)methyl ether methacrylate (Mw: 300 g mol−1), dimethylglyoxime (≥99%), isobornyl acrylate, poly(ethylene glycol)diacrylate (MW: 250 g mol−1), poly(ethylene glycol)diacrylate (Mw: 700 g mol−1) and alizarin were purchased from Sigma-Aldrich. Pyridine (Extra pure) and cobalt(II) acetate tetrahydrate (98%) were purchased from Fisher Scientific. Azobisisobutyronitrile (AIBN) (98%) was supplied by Wako chemicals. Phenylbis(2,4,6-trimethylbenzoyl)-phosphine oxide (BAPO) (96%) was supplied by Tokyo Chemical Industry UK Ltd. All chemicals were used as received.Instrumentation
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching band.
O stretching band.
Owing to the inherent softness of the materials the Young's modulus from the linear fit of stress strain curve at low strain region (ca. 1%) was heavily susceptible to the region selected and errors due to insufficient measurement points. Herein, a method modified from Maláč's method was adopted to calculate the specific deformation energy by integrating the stress strain curve.29,30 To mitigate the error by random selection, the point at local maximum stress, or the knee point when no local maximum was observed, was chosen as the reference point according to Kucherskii's method. The determination of knee point was as followed.31 In brief, an empirical 15th order polynomial was fitted to the entire stress strain curve to minimize the noises, and the knee point was the minimum of the first derivative of the fitted curve (Fig. S.1†).
Experimental procedures
The reaction mixture and AIBN stock solution were deoxygenated by purging with N2 for 1 h. The reaction mixture was then transferred to a 90 °C oil bath for 30 min to equilibrate the temperature and allow dissolution of the cobalt salts. AIBN solution (30 mL) was added into the reaction mixture using a N2 filled syringe to initiate the reaction. The reaction was allowed to proceed for 3 h, then terminated by allowing to cool to ambient temperature and opened to air. The conversion and degree of polymerization were determined from 1H NMR and the molecular weight distribution was acquired using SEC in chloroform.
The solvent and volatiles were removed by rotary evaporation at 50 °C and 150 mPa to remove most of the ethyl acetate. The product was obtained as a viscous liquid which was kept in a vacuum oven at 40 °C overnight to remove any residual volatiles. The product was stored at ambient conditions until use.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 w/w mixture of PEGDA (Mw: 250 and 700), 3 means PEGDA (Mw: 700). For example, 10E15-1 means the base resin with 10% w/w PEHMA15 reactive oligomers using PEGDA (Mw: 250) as crosslinker.
:1 w/w mixture of PEGDA (Mw: 250 and 700), 3 means PEGDA (Mw: 700). For example, 10E15-1 means the base resin with 10% w/w PEHMA15 reactive oligomers using PEGDA (Mw: 250) as crosslinker.
| Monomer | Crosslinker | Additive | |
|---|---|---|---|
| All % in w/w; all formulations contain an additional 1% w/w BAPO and 0.03% w/w alizarin as photoinitiator and photoabsorber respectively. | |||
| O-1 | 66.7% IBoA | 33.3% PEGDA 250 | — |
| O-2 | 66.7% IBoA | 16.7% PEGDA 250 | — |
| 16.7% PEGDA 700 | |||
| O-3 | 66.7% IBoA | 33.3% PEGDA 700 | — |
| 10E15-1 | 56.7% IBoA | 33.3% PEGDA 250 | 10% EHMA15 |
| 10E15-2 | 56.7% IBoA | 16.7% PEGDA 250 | 10% EHMA15 |
| 16.7% PEGDA 700 | |||
| 10E15-3 | 56.7% IBoA | 33.3% PEGDA 700 | 10% EHMA15 |
| 5E1-2 | 61.7% IBoA | 16.7% PEGDA 250 | 5% EHMA |
| 16.7% PEGDA 700 | |||
| 5E5-2 | 61.7% IBoA | 16.7% PEGDA 250 | 5% EHMA5 |
| 16.7% PEGDA 700 | |||
| 5E15-2 | 61.7% IBoA | 16.7% PEGDA 250 | 5% EHMA15 |
| 16.7% PEGDA 700 | |||
| 5P1-2 | 61.7% IBoA | 16.7% PEGDA 250 | 5% PEGMA |
| 16.7% PEGDA 700 | |||
| 5P14-2 | 61.7% IBoA | 16.7% PEGDA 250 | 5% PEGMA14 |
| 16.7% PEGDA 700 | |||
| 5P28-2 | 61.7% IBoA | 16.7% PEGDA 250 | 5% PEGMA28 |
| 16.7% PEGDA 700 | |||
| 10P1-2 | 56.7% IBoA | 16.7% PEGDA 250 | 10% PEGMA |
| 16.7% PEGDA 700 | |||
| 10P14-2 | 56.7% IBoA | 16.7% PEGDA 250 | 10% PEGMA14 |
| 16.7% PEGDA 700 | |||
| 10P28-2 | 56.7% IBoA | 16.7% PEGDA 250 | 10% PEGMA28 |
| 16.7% PEGDA 700 | |||
Results and discussion
Synthesis of reactive oligomers by CCTP and AFCT
ω-Vinyl terminated reactive oligomers of PEHMA and PEGMA were synthesized via CCTP and subsequent AFCT to attain different DP. The mechanism of CCTP and the subsequent AFCT mediated by the ω-vinyl terminated reactive oligomers were shown in Fig. S.2.†1H NMR spectra of the products were employed to determine the conversion and DP of the reactive oligomers calculated from eqn (1) and (2) for PEHMA and PEGMA respectively.
1H NMR of the PEHMA reactive oligomer synthesized via CCTP is shown in Fig. S.5.† The conversion (X) and degree of polymerization (DP) was calculated by eqn (1). X and DP of the reactive oligomer by CCTP were 97.8% and 5 respectively, and 99.5% and 15 after chain extension of EHMA via AFCT. 1H NMR (400 MHz, CDCl3) δ = 0.60–1.00 (6H, CH-CH2-CH3 of EHMA and MM), 2.41–2.71 (α-CH2-CC-COOR), 3.75–4.25 (2H, O-CH2 of EA, EHMA and MM), 5.40–5.59 (singlet, 1H, trans CCH2 of monomer and MM), 6.05–6.11 (1H, cis CCH2 of monomer), 6.13–6.26 (singlet, 1H, cis CCH2 of MM).
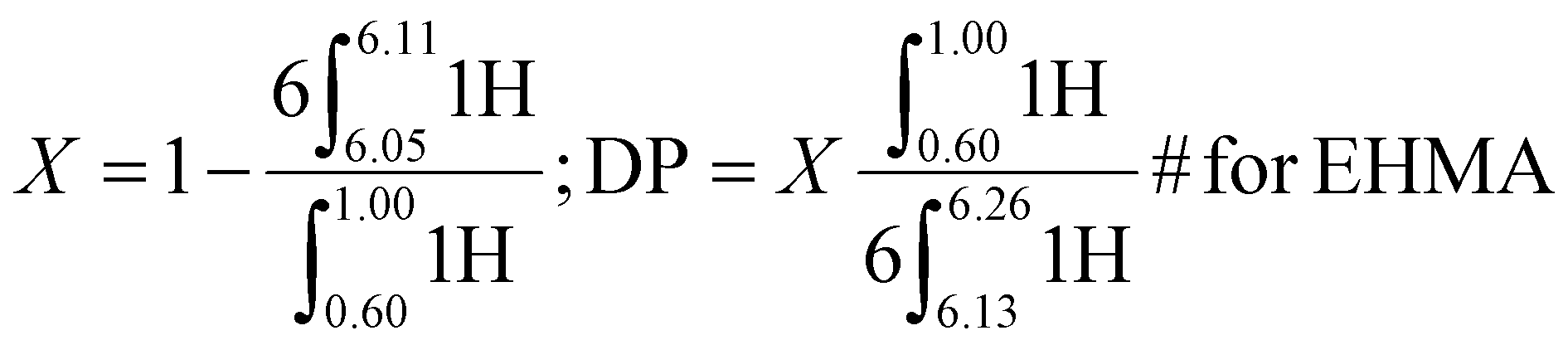 | (1) |
1H NMR of the PEGMA reactive oligomer synthesized via CCTP is shown in Fig. S.4.† The conversion (X) and degree of polymerization (DP) was calculated by eqn (2). X and DP of the reactive oligomer by CCTP were 90.0% and 14 respectively, and >99% conversion (non-detectable monomer by 1H NMR) and 28 after chain extension of PEGMA via AFCT. 1H NMR (400 MHz, CDCl3) δ = 1.87–2.03 (3H, α-CH3), 2.41–2.71 (α-CH2-CC-COOR), 3.27–3.44 (3H, CH2O-CH3), 3.45–3.59 (2H, CH2O-CH3), 3.59–3.71 (12H, –O-CH2–), 3.71–3.82 (2H, OC-OCH2-CH2O), 4.20–4.35 (2H, OC-OCH2), 5.40–5.59 (singlet, 1H, trans CCH2 of monomer and MM), 6.05–6.11 (1H, cis CCH2 of monomer), 6.13–6.26 (singlet, 1H, cis CCH2 of MM).
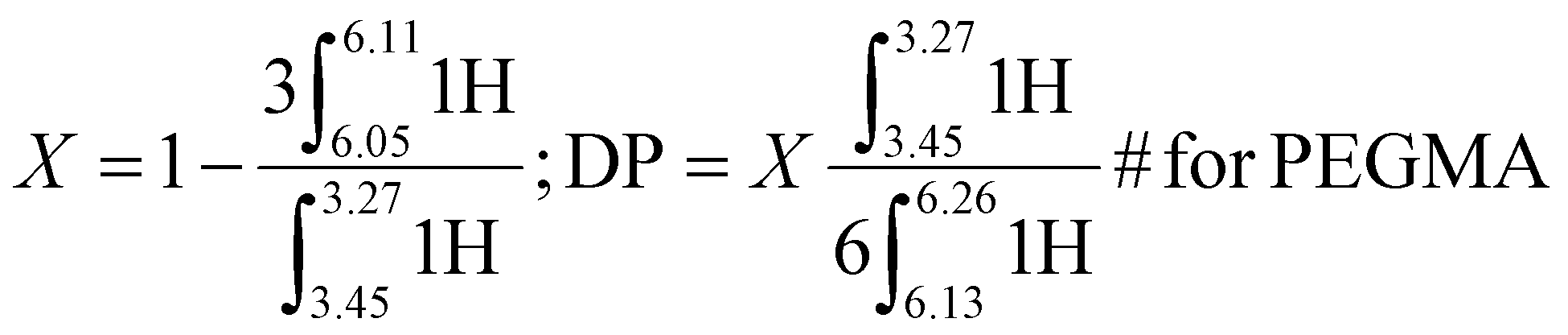 | (2) |
The molecular weight was determined by SEC in CHCl3 using calibration using PMMA standards. The molecular weight distributions of the reactive oligomers as prepared by CCTP and after chain extension by AFCT are shown in Fig. S.4.† The Mn, Mw and dispersity (Đ) are summarized in Table 2.
| M n,GPC | M w,GPC | Đ | DPNMR | X | |
|---|---|---|---|---|---|
| PEHMA5 | 1020 | 1950 | 1.91 | 5 | 97.8 |
| PEHMA15 | 2950 | 9400 | 3.18 | 15 | 99.5 |
| PEGMA14 | 4890 | 10700 |
2.19 | 14 | 90.0 |
| PEGMA28 | 9600 | 35600 |
3.70 | 28 | >99 |
Tensile testing of the 3D printed parts
3D printings are performed with isobornyl acrylate and poly(ethylene glycol)diacrylate as the base resin. This is as acrylic monomers have a fast propagation rate which shortens printing time and gives better incorporation via the formation of comb polymer.32 The specific choice of isobornyl acrylate is due to its high Tg and hydrophobicity. This facilitates storage without tackiness or softening by moisture. In addition, the resin can be handled safely in household ventilation due to the low volatility. PEGDA with different molecular weights were chosen to prevent hard and brittle materials beyond the instrumental limit for mechanical testing. All printed parts have been characterized by FT-IR spectroscopy to confirm the complete conversion by the disappearance of the band at 816 and 1636 cm−1 of the acrylicCH2 and CC from monomers (Fig. S.7†).
Tensile tests were performed to evaluate the effect of the crosslinker and additive type and its concentration. The specific tensile energy (UT), stress (σ) and strain (ε) at the damaging point (or knee point; K in subscript) and the ultimate point (U in subscript) are summarized in Table S.2† (see tensile test in Experimental section for definition and details). The significance of the knee point is that the original configuration was mechanically overcome, and further external force would lead to either complete damage (for rigid material) or internal reconfiguration (strain hardening for elastomer), whereas the ultimate point referred to the breakage of the materials.
The base resin with a crosslinker with a different spacer length demonstrated a significant effect on the tensile properties (Fig. 1). σK and UT,K were 7.28, 26.78, 45.71 MPa and 443.51, 136.62 and 70.96 MPa for O-3, O-2 and O-1 (increasing PEGDA250: PEGDA 700), and at the ultimate breakage point, σU and UT,U were 12.64, 18.87, 44.39 MPa and 796.94, 415.97 and 502.30 MPa for O-3, O-2 and O-1 respectively. These results indicated that O-1 (with PEGDA 250 only) has the strongest initial structure against deformation before the knee point, then O-2 (1:1 w/w of PEGDA 250 and PEGDA700) and O-3 (PEGDA 700 only) the least. After the knee point, O-1 was damaged rapidly upon further elongation by 1.11% (6.52% of εK). Meanwhile, O-2 and O-3 can be further elongated by 13.03% (161% of εK) and 75.34 (661% of εK) respectively.
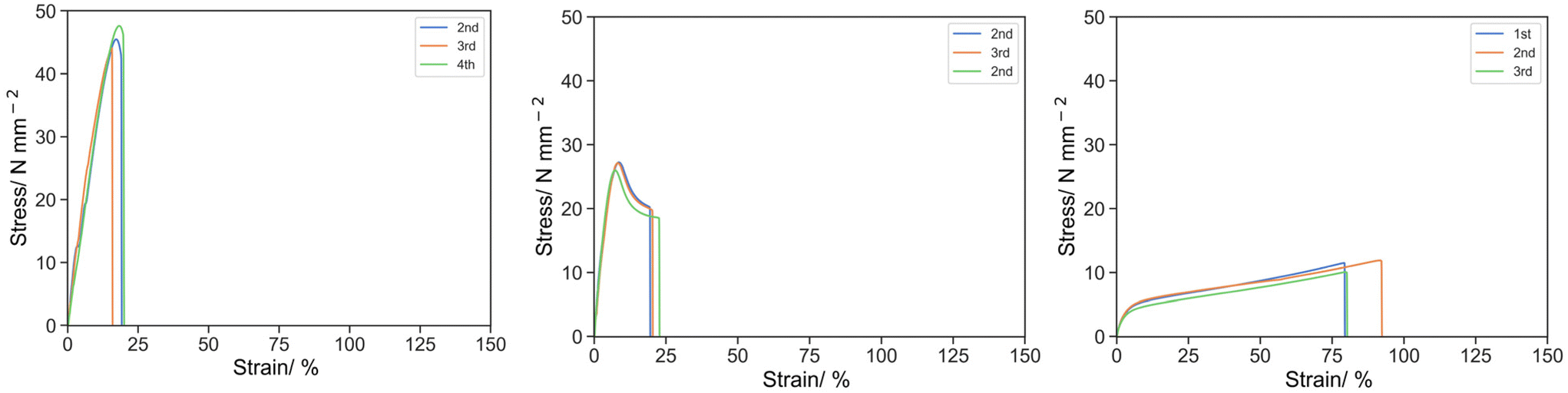 | ||
| Fig. 1 Stress strain curve by tensile test for O-1 (left), O-2 (middle) and O-3 (right) under uniaxial tension at 50 mm s−1. | ||
At the molecular level, low molecular weight PEGDA250 seems to form a compacted network with high crosslink density, reflected by the high stress and specific energy observed at the knee point. However, the short space length limits the extension of the material and therefore ultimate breakage occurs upon a slight elongation after the knee point. A less crosslinked, and consequently, a weaker network is formed by higher molecular weight PEGDA 700, at the same content by mass resulting in a smaller σK and UT,K. However, the longer spacer of PEGDA 700 unfolds under stress to allow larger elongation prior to ultimate breakage. In order to signify the effect of additives, O-2 with the moderate properties was chosen as the base resin for later studies.
ω-Vinyl terminated reactive oligomers were incorporated into the resins with a varied content, chemistry and molecular weight. 2-Ethylhexyl methacrylate and poly(ethylene glycol)methacrylate were selected here as low Tg materials with opposite hydrophilicity.
Initially, 10% w/w of IBoA was replaced by PEHMA15 reactive oligomer (Fig. 2). For 10E15-1 (PEGDA 250 only) and 10E15-2 (1:1 PEGDA 250 and PEGDA 700), weakening was observed where σK and UT,K reduced to 20.58 MPa and 77.92 MPa at 6.53% for 10E15-1, and σK and UT,K reduced to 13.44 MPa and 73.77 MPa at 8.17% for 10E15-2. In both cases, complete breakage occurred after the knee point and no strain hardening was observed. For 10E15-3 (PEGDA 700 only), the material was in fact reinforced giving a similar knee stress but improved elongation, where σK and UT,K of O-3 were 6.38 and 102.26 MPa at 22.78% respectively. Similar strain-hardening was observed after the knee point where σU and UT,U were 11.07 and 644.54 MPa at 84.16%. The weakening effect observed with the PEHMA15 reactive oligomer for 10E15-1 and 10E15-2 was attributed to low Tg of PEHMA and the polymerization kinetics of reactive oligomer with the oligomer incorporation. PEHMA has a Tg of −6 °C which softens the bulk material and reduces the specific energy. Kinetically, the rate of propagation of the reactive oligomer is slower than with the acrylic monomer resulting in a shorter kinetic primary chain length. Moreover, the reactive oligomers can also act as a chain transfer agent further reducing the kinetic chain length and thus crosslink density. The combination of these ultimately leads to weaker materials. No weakening effect was observed for 10E15-3 as the Tg of PEG spacer and the crosslink density of the material are already low in the base resin (O-3). Further introduction of low Tg component has negligible effect on the specific energy of the material.
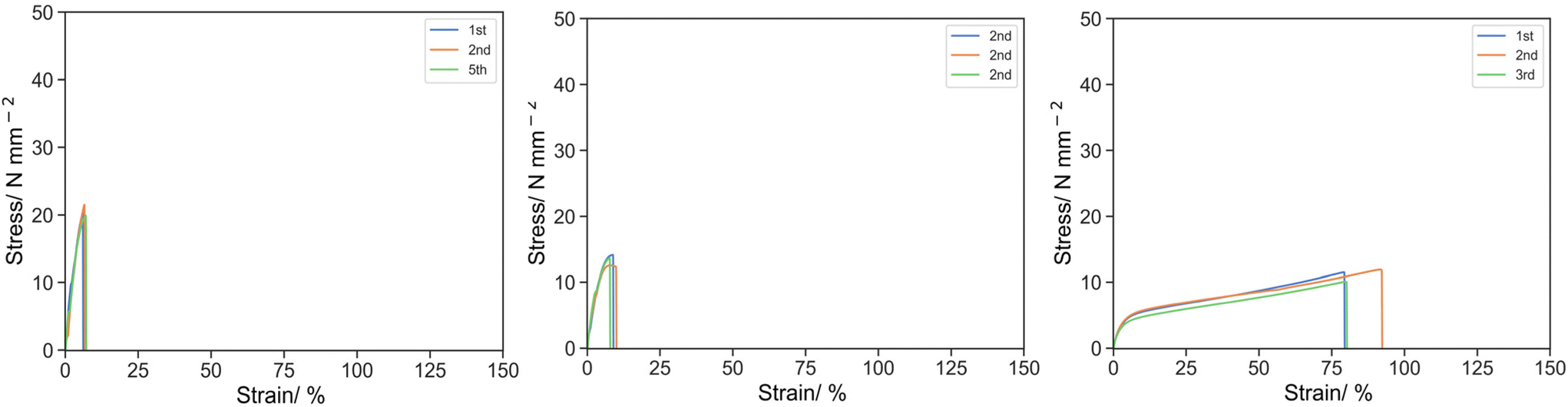 | ||
| Fig. 2 Stress strain curve for 10E15-1 (left), 10E15-2 (middle) and 10E15-3 (right) under uniaxial tension at 50 mm s−1. | ||
Subsequently, the effect of the PEHMA15 reactive oligomer concentration was investigated (Fig. 3). A clear trend in the reduction of σK and UT,K of 26.78, 19.31 and 13.44 MPa, and 136.62, 101.90, 73.77 MPa was observed with an increasing content of PEHMA15 from 0, 5 and 10% w/w, where the observed knee strain were almost identical at 8.10, 8.13 and 8.17%. At the ultimate point, the incorporation of 5% PEHMA15 slightly reduced the stress from 18.87 MPa to 15.76 MPa but improved the strain from 21.13% to 25.77%, and therefore the toughness remained similar at 415.97 and 400.31 MPa respectively, whereas complete breakage was seen at 10% PEHMA15 as previously mentioned.
 | ||
| Fig. 3 Stress strain curve for O-2 (left), 5E15-2 (middle) and 10E15-2 (right) under uniaxial tension at 50 mm s−1. | ||
It was postulated that PEHMA15 was compatible with poly(isobornyl acrylate) based resin owing to the aliphatic character acting as a plasticizer. These phenomena were similar with the mechanical properties of miscible poly(vinyl chloride)/polycaprolactone blend reported by Chiu and Min.32 The suppression and subsequently disappearance of the yielding behaviour was attributed to the reduction of Tg by PEHMA15. This enhanced the mobility of polymer segments allowing for longer extension with the compromise on lower stress. At higher reactive oligomer content (10% w/w), the crosslink density is significantly reduced due to the chain transfer activity and slower polymerization rate, and only weakening was observed. Therefore, 5% w/w additive was chosen for later studies.
The effect of the molecular weight of the reactive oligomers was demonstrated by 5E1-2, 5E5-2 and 5E15-2, using EHMA monomer, PEHMA5 and PEHMA15 reactive oligomer respectively (Fig. 4). It was observed that the molecular weight of the reactive oligomer had little but significant effect on the knee point with the stress of 23.88, 21.93, 19.31 MPa; strain of 8.44, 8.12, 8.13% and specific energy of 126.81, 112.04 and 101.90 MPa for EHMA, PEHMA5 and PEHMA15 respectively.
 | ||
| Fig. 4 Stress strain curve for 5E1-2 (left), 5E5-2 (middle) and 5E15-2 (right) under uniaxial tension at 50 mm s−1. | ||
However, at the ultimate point, 5E5-2 appears to be the weakest material with σU, εU and UT,U of 17.85 MPa, 17.92% and 302.25 MPa respectively, followed by 5E15-2 with σU, εU and UT,U of 15.76 MPa, 25.77% and 400.31 MPa. 5E1-2 was the strongest among, with σU, εU and UT,U of 19.01 MPa, 34.21% and 635.06 MPa respectively.
The variation in the mechanical properties can be rationalized by the polymerization kinetics. Moad et al. and Yamada et al. have studied the kinetics of propagation and chain transfer of ω-vinyl terminated reactive oligomers and concluded that whereas the kp drops considerably and the chain transfer activity increases from monomer to dimer, then approaches constant for further higher molecular weight.33–36 Therefore, PEHMA5 and PEHMA15 reactive oligomer induced a lower degree of crosslinking than EHMA due to slower propagation and chain transfer activity, and weaken the material. Then regarding PEHMA5 and PEHMA15, the kp and kct are similar but since PEHMA5 has a lower molecular weight, the molar concentration of reactive oligomer in 5E5-2 will be higher than 5E15-2, at the same mass fraction of reactive oligomer, and thus 5E5-2 had the slowest polymerization and degree of crosslinking and formed the weakest material.
The effect of the chemistry of the reactive oligomer was also investigated. (PEGMA)14 which has a similar Mw to PEHMA15 was synthesized via CCTP, but more polar than the aliphatic EHMA and IBoA and incorporated into the resin (Fig. 5) and has a methacrylic end group so acts as a true macromonomer (MM).
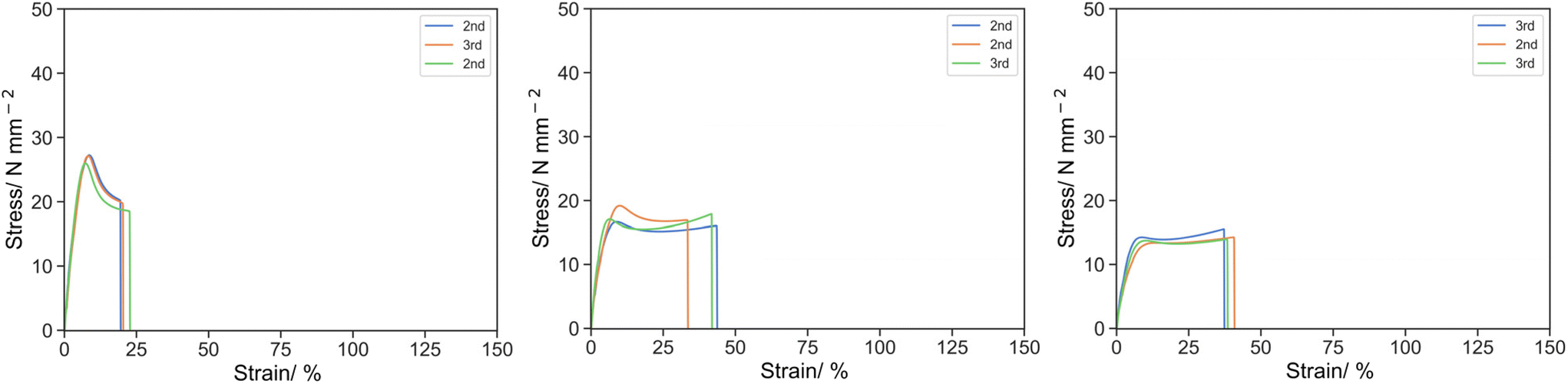 | ||
| Fig. 5 Stress strain curve for O-2 (left), 5P14-2 (middle) and 10P14-2 (right) under uniaxial tension at 50 mm s−1. | ||
The mechanical properties were distinctive to its equivalence of PEHMA15. Despite the similar suppression of the stress, the elongation of the materials was significantly improved by the inclusion of PEGMA14. σK was 26.78, 17.61 and 13.16 for O-2, 5P14-2 and 10P14-2 respectively, whereas εK was 8.10, 8.06 and 9.93, and UT,K was 136.62, 95.10 and 95.61 respectively. For the ultimate point, σU was 18.87, 16.47 and 14.06 for O-2, 5P14-2 and 10P14-2 respectively, whereas εK was 21.13, 39.61 and 38.79, and UT,K was 415.97, 609.65 and 495.59 respectively.
One of the explanations could be the phase separation of the more polar PEG moiety from aliphatic isobornyl acrylate. Reinforcement by polymerization-induced phase separation has long been studied in the case of high-impact polystyrene and PMMA/epoxy.37–40 Upon phase separation, the continuous phase remains predominantly hard component of isobornyl acrylate to contribute to the mechanical strength of the material, whereas the segregated soft component of (PEGMA300)14 enhances the elongation prior to the breakage. As the content of (PEGMA300)14 further increased, the soft phase tended to be continuous which would no longer enhance the ductility, instead a larger fraction of it also incorporated into the hard phase via copolymerization with isobornyl acrylate, led to the plasticization and softening of the material which lowered the specific energy and strength of the material.
The effects of PEGMA reactive oligomers at different molecular weights were also demonstrated (Fig. 6). At the knee point, the stress increased 7.65, 13.76 and 17.1 for 10-P1-2, 10-P14-2 and 10-P28-2, and the strain decreased 21.17, 9.93 and 9.35, which gave the specific energy of 102.26, 95.61 and 109.27 respectively. Similar trend was observed at the ultimate point, the stress was 11.23, 14.06 and 15.08, whereas the strain was 71.69, 38.79 and 30.37, and the specific energy was 615.18, 495.59 and 442.10 for 10P1-2, 10P14-2 and 10P28-2 respectively.
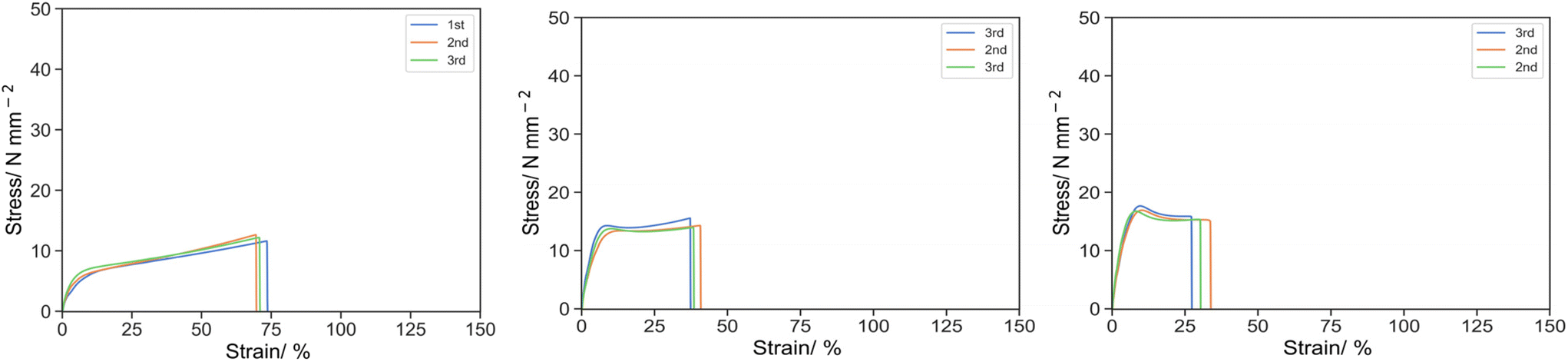 | ||
| Fig. 6 Stress strain curve for 10P1-2 (left), 10P14-2 (middle) and 10P28-2 (right) under uniaxial tension at 50 mm s−1. | ||
The rise in the knee stress and shift toward the smaller strain suggested that the structure became harder and more brittle. It was postulated that the change in mechanical properties were attributed to the polymerization-induced phase separation. In addition to the intrinsic localization in reactive oligomer, in theory, the higher the molecular weight, the more favoured the phase separation due to the higher entropy of phase separation in an incompatible polymer blend. Correlating to the mechanical properties, [monomer] demonstrated elastomeric properties with superior elongation and strain hardening before breakage. With an increase in the molecular weight of the reactive oligomer from DP = 1 to 14 then 28, the material transformed from an elastomer to a tougher and more brittle material. It was elucidated that the degree of phase separation was minuscule and a larger fraction of PEGMA was incorporated into the continuous phase and lower the Tg when PEGMA monomer was employed, resulting a ductile elastomer with the sign of superior elongation and strain hardening prior to breakage. As the molecular weight of the reactive oligomer increased, the PEGMA moiety was more favoured to phase separate from the continuous phase and resulting in a larger fraction of hard component of poly(isobornyl acrylate) remaining in the continuous phase, at the same total content of additive, and eventually led to the reduction in elongation and improvement in the mechanical strength and specific energy. Similar effects of phase separation, controlled by annealing time and temperature, on mechanical properties in polystyrene/poly(vinyl methyl ether) blend were also observed by Kim and Karasz.41
Dynamic mechanical and thermal analyses (DMTA)
DMTA was carried out to further understand the relaxation and the microstructures of the printed parts. The loss factor (tand), the ratio of loss modulus to storage modulus (G′′/G′), is a parameter related to the segmental motions, and any relaxation event raises a peak in tans. The temperature at maximum tanδ (Tmax, tanδ) and its characteristic width at 25% height (δT25%) represent the occurrence of a relaxation event and its span over a temperature range (Table S.3†). The interpretation of DMTA spectra will be based on the Tg and polymeric architecture. In brief, we herein used high Tg polymer (PIBOA, Tg: 86 °C) and low Tg reactive oligomeric additive (PEHMA or PEGMA Tg: −6 °C and −73 °C respectively), and the peak and width of tanδ indicate the Tg where segmental relaxation occurs and the degree of miscibility of the bulk polymer and the reactive oligomers. Typically, a narrow peak of tanδ suggests a homogeneously distributed bulk polymer and the reactive oligomers. As two components segregating, peak of tanδ starts broadening and eventually separate to two peaks at the Tg of the oligomers enriched and deficient domains respectively. This will give better insight to understand the origin of the tensile properties in molecular level, and the details of each formulation will be discussed as below.
The effect of the crosslinker was first demonstrated (Fig. 7). With an increasing amount of PEGDA700: PEGDA250, tanδ moving to lower temperature from 74.1 to 61.8 and 44.0 °C and the width of 25% peak height broadened from 17.2 to 18.3 and 23.9 °C for O-3, O-2 and O-1 respectively. As the fraction of PEGDA 700 increased, at the same total mass content of the crosslinker, the crosslink density reduced and thus enhanced the segmental mobility. Additionally, the extended PEG spacer length plasticizes the material to shift and broaden the peak tanδ towards lower temperature.
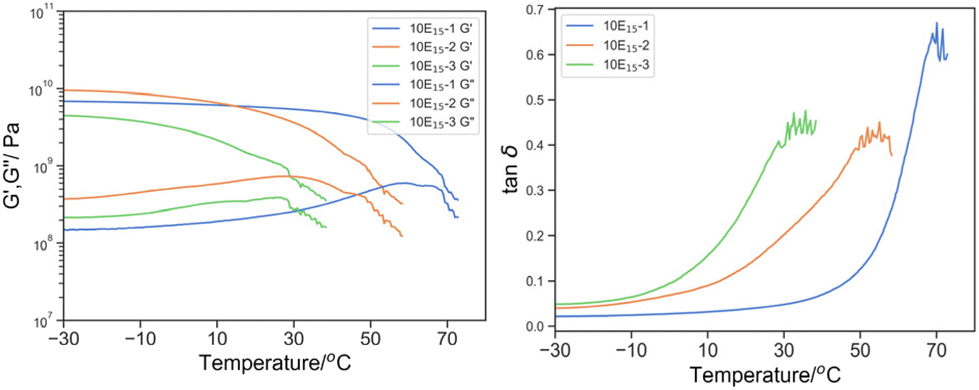 | ||
| Fig. 7 Left: G′ (solid line) and G′′ (dotted line) by DMTA of 10E15-1 (blue), 10E15-2 (green), 10E15-3 (orange); and right: its derived tanδ. | ||
The incorporation of 10% w/w PEHMA15 reactive oligomer demonstrated different effects on the materials with different crosslink density (Fig. 8). Minuscule effects were observed on 10E15-1 apart from a small reduction Tmax, tanδ (74.1 to 70.1 °C). In contrast, the 10E15-2 and 10E15-3 showed significant reduction in the Tmax, tanδ from 61.8 and 44.0 °C to 55.1 and 35.7 respectively. Moreover, significant broadening was observed in 10E15-2 from 18.3 °C to 33.4 °C for dT25%.
 | ||
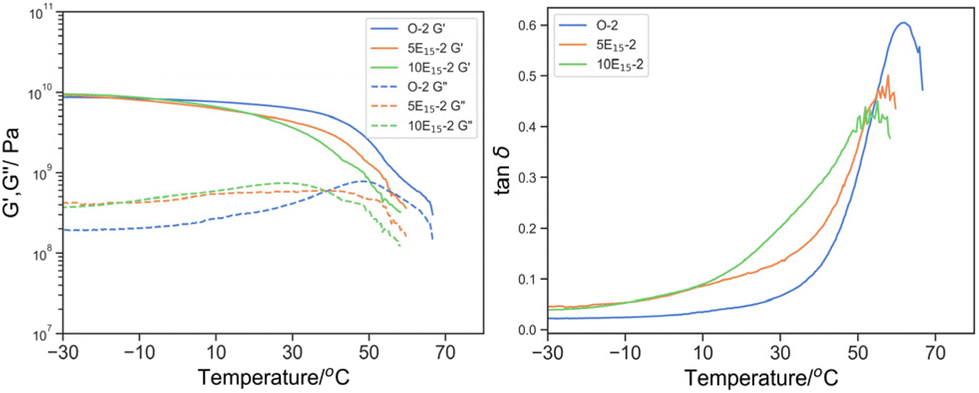
| Fig. 8 Left: G′ (solid line) and G′′ (dotted line) by DMTA of O-1 (blue), O-2 (green), O-3 (orange); and right: its derived tanδ. | ||
It was postulated that at high crosslink density, the segmental mobility was constrained by covalent bonding predominantly instead of the glass transition of polymer compositions; therefore, additional PEHMA reactive oligomer does not have a significant effect on the segmental mobility. As the crosslink density is reduced from 10E15-1 to 10E15-3, the glass transition of the bulk polymer gradually predominated the mechanical properties, and the plasticization effect by the reactive oligomer became more significant.
Considering the concentration of PEHMA15 (Fig. 9), a clear shift in the Tmax, tanδ to a lower temperature (61.8 °C for O-2, 57.8 °C for 5E15-2 and 55.1 °C for 10E15-2), peak broadening (δT25%: 17.2 °C for O-2, 22.7 °C for 5E15-2 and 33.4 °C for 10E15-2) was observed with increasing PEHMA15. This further signified the role of EHMA15 as a plasticizer in the 3D printed parts. Similar shift and broadening has been observed in DMTA of tetraglyme solvated polyurethane.42
 | ||
| Fig. 9 Left: G′ (solid line) and G′′ (dotted line) by DMTA of O-2 (blue), 5E15-2 (green), 10E15-2 (orange); and right: its derived tanδ. | ||
The effect of the molecular weight of reactive oligomer was investigated by incorporating 5% w/w of EHMA monomer, PEHMA5 and PEHMA15 reactive oligomer (Fig. 10). The molecular weight dependence of the relaxation spectra is subtle but we think significant. It was observed that Tmax, tanδ moved to high temperature from 53.8 to 54.6 and 57.8 °C was accompanied by broadening and the appearance of a secondary shoulder as the molecular weight of the reactive oligomers increased from 5E1-2, 5E5-2 to 5E15-2. While it was minimal (compared to PEGMA as discussed later), the peak broadening and eventually separation suggested the uneven distribution of plasticizing comonomer when reactive oligomers were employed, and the degree of inhomogeneity increased with the molecular weight. It was postulated that despite the chemical similarity of aliphatic isobornyl acrylate and EHMA, the reactive oligomer created localized EHMA-rich domains depleted the EHMA content in the bulk phase, resulting a higher Tmax,tanδ and peak broadening.
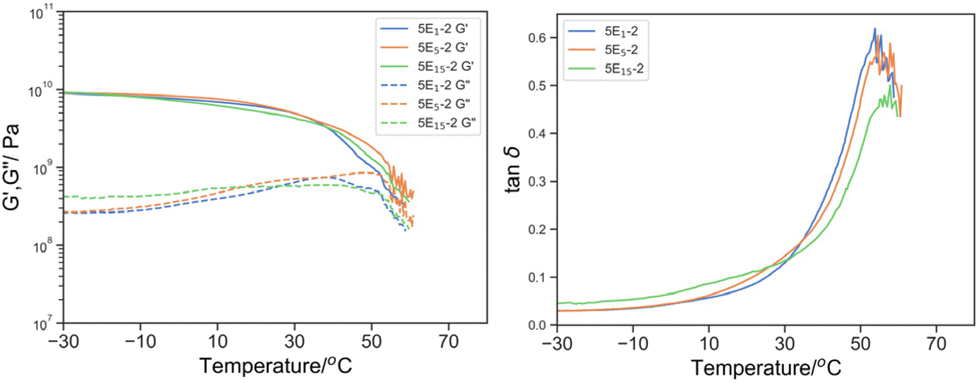 | ||
| Fig. 10 Left: G′ (solid line) and G′′ (dotted line) by DMTA of 5E1-2 (blue), 5E5-2 (green), 5E15-2 (orange); and right: its derived tanδ. | ||
The incorporation of chemically incompatible and the more polar PEGMA reactive oligomers was then studied (Fig. 11). The effects of PEGMA reactive oligomers were less noticeable than PEHMA reactive oligomer despite similar shifts in Tmax, tanδ towards lower temperature with increasing content of PEGMA14 reactive oligomer. Even at 10% w/w of reactive oligomer, only a slight secondary shoulder was observed between −10 and 30 °C, in contrast to the conspicuous broadening observed with 10% PEHMA15.
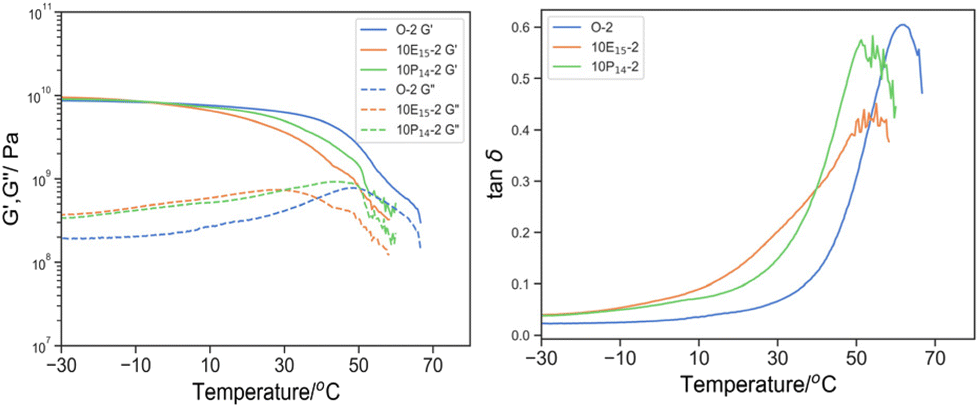 | ||
| Fig. 11 Left: G′ (solid line) and G′′ (dotted line) by DMTA of O-2 (blue), 10E15-2 (green), 10P14-2 (orange); and right: its derived tanδ. | ||
Whilst regrettably the glass transition temperature of PEGMA (−60 °C) was beyond our instrument limit, one can expect that if PEGMA reactive oligomer incorporated into the bulk material as plasticizer in the same manner as EHMA, the downshift and broadening in tanδ would be more pronounced. Conversely, Tmax, tanδ of 10P14-2 (54.1 °C) was in fact similar with 10E15-2 (55.1 °C) and δT25% was much narrower (21.7 °C vs. 33.4 °C), implying a lower content of PEGMA as plasticizer in the bulk material and the majority was phase separated to create PEGMA-rich domain whose Tg beyond the temperature range.
The concentration dependence of the PEGMA14 reactive oligomer also suggested that only a small fraction was incorporated into the bulk material (Fig. 12). The subtle reduction in Tmax, tanδ from 61.8 to 60.1 and 54.1 °C at 5% w/w and 10% w/w PEGMA14 respectively and the lack of broadening in tanδ induced by plasticization suggested that the content of PEGMA14 had insignificant effect on the bulk material. The enhanced phase separation observed with higher molecular weight reactive oligomer was more prominent with PEGMA (Fig. 13). This was demonstrated by the shift in the peak tand approaching the blank as its molecular weight increased, suggesting the content of PEGMA reactive oligomer reduced in the bulk material. The Tmax,tanδ of 10-P1-2, 10-P14-2 and 10-P28-2 was 53.6, 54.1 and 60.6 °C respectively, where the base resin (O-2) was 61.8 °C. The absence of peak broadening of tanδ, and in fact narrowing in 10-P28-2, at any molecular weight of PEGMA reactive oligomer further illustrated that the bulk material is independent to the PEGMA reactive oligomer and indicated phase separation.
 | ||
| Fig. 12 Left: G′ (solid line) and G′′ (dotted line) by DMTA of 10P1-2 (blue), 10P14-2 (green), 10P28-2 (orange); and right: its derived tanδ. | ||
 | ||
| Fig. 13
G′ (solid line) and G′′ (dotted line) by DMTA of O-2 (blue), 5P14-2 (green), 10P14-2 (orange); and right: its derived tanδ. | ||
Conclusion
In this study, PEHMA and PEGMA reactive oligomers have been synthesized via CCTP mediated by the in situ formation of cobaloxime and used to mediate subsequent addition–fragmentation chain transfer (AFCT). The reactive oligomers have been incorporated into 3D printing resins as polymerizable additives. The effects of the chemistry, molecular weight and the concentration of reactive oligomer on the mechanical properties has been studied via tensile tests and the dynamic mechanical and thermal analyses and compared to the incorporation of the original monomer. It was found that PEHMA reactive oligomers were compatible with the isobornyl acrylate based resin and acted as a plasticizer in the bulk material. This caused the weakening of the material in tensile test with the reduction in stress, specific energy and elongation. Conversely, PEGMA based additive transformed the printed parts from brittle to ductile hard materials and further to true elastomers. The origin of such transition was postulated to be the phase separation of polar PEG moiety from the aliphatic isobornyl acrylate resin. The current approach opens a facile route to access a wide range of mechanical properties of DLP 3D printed parts across using a small quantity (up to 10% w/w) of additive.Author contributions
Wai Hin Lee: writing – original draft, methodology, investigation, software, methodology, investigation, formal analysis, data curation, conceptualization. Zhongyuan Wan: methodology, investigation, data curation. Ataulla Shegiwal: writing, review & editing, visualization, supervision, resources, project administration, methodology, investigation. David Haddleton: writing, review & editing, visualization, supervision, resources, project administration, methodology, investigation, funding acquisition, conceptualization.Data availability
The data supporting this article have been included as part of the ESI† and are available from the authors on request.Conflicts of interest
DMH and AS are shareholders in Halcyon3D Ltd.Acknowledgements
Funding was received from Halcyon3D Ltd and equipment used was funded in part by EPSRC via the Polymer Characterisation Research Technology Platform (RTP) of the University of Warwick EP/V036211/1 and EP/V007688/1.References
- R. Chaudhary, P. Fabbri, E. Leoni, F. Mazzanti, R. Akbari and C. Antonini, Prog. Addit. Manuf., 2023, 8, 331–351 CrossRef.
- M. Manoj Prabhakar, A. K. Saravanan, A. Haiter Lenin, I. Jerin leno, K. Mayandi and P. Sethu Ramalingam, Mater. Today: Proc., 2021, 45, 6108–6114 Search PubMed.
- M. Pagac, J. Hajnys, Q.-P. Ma, L. Jancar, J. Jansa, P. Stefek and J. Mesicek, Polymers, 2021, 13, 598 CrossRef CAS PubMed.
- W. Li, Y. Wu, W. Liang, B. Li and S. Liu, ACS Appl. Mater. Interfaces, 2014, 6, 5726–5734 CrossRef CAS PubMed.
- W. Wang, R. Peng, Y. Gao, J. Nie and F. Sun, Macromol. Chem. Phys., 2023, 224, 2200458 CrossRef CAS.
- J. Cheng, Y. Cao, S. Jiang, Y. Gao, J. Nie and F. Sun, Ind. Eng. Chem. Res., 2015, 54, 5635–5642 CrossRef CAS.
- B. K. Kim, S. H. Kim and J. C. Song, Polymer, 1998, 39, 5949–5959 CrossRef CAS.
- Y. Wang, C. Li, X. Tuo, Y. Gong and J. Guo, J. Appl. Polym. Sci., 2021, 138, 50102 CrossRef CAS.
- W. Geurtsen, W. Spahl and G. Leyhausen, J. Biomed. Mater. Res., 1999, 44, 73–77 CrossRef CAS PubMed.
- H. Lygre, K. N. Klepp, E. Solheim and N. R. Gjerdet, Acta Odontol. Scand., 1994, 52, 150–156 CrossRef CAS PubMed.
- N. S. Enikolopyan, B. R. Smirnov, G. V. Ponomarev and I. M. Belgovskii, J. Polym. Sci., Polym. Chem. Ed., 1981, 19, 879–889 CrossRef CAS.
- A. Gridnev, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 1753–1766 CrossRef CAS.
- A. A. Gridnev and S. D. Ittel, Chem. Rev., 2001, 101, 3611–3660 CrossRef CAS PubMed.
- Du Pont, United States, 4694054A, 1987 Search PubMed.
- Mitsubishi Plastics Inc, United States, 9809731B2, 2017 Search PubMed.
- Rohm & Haas, United States, 6864309B2, 2005 Search PubMed.
- Du Pont, United States, 5028677A, 1991 Search PubMed.
- F. Baffie, G. Patias, A. Shegiwal, F. Brunel, V. Monteil, L. Verrieux, L. Perrin, D. M. Haddleton and F. D'Agosto, Angew. Chem., 2021, 133, 25560–25568 CrossRef.
- L. Sinniger, O. Boyron, P. Y. Dugas, G. Patias, D. Lester, D. M. Haddleton, V. Monteil, M. Lansalot and F. D'Agosto, Polym. Chem., 2023, 14, 4569–4579 RSC.
- N. G. Engelis, A. Anastasaki, G. Nurumbetov, N. P. Truong, V. Nikolaou, A. Shegiwal, M. R. Whittaker, T. P. Davis, D. M. Haddleton, A. Anastasaki, G. Nurumbetov, N. P. Truong, V. Nikolaou, A. Shegiwal, M. R. Whittaker, T. P. Davis and D. M. Haddleton, Nat. Chem., 2017, 9, 171–178 CrossRef CAS PubMed.
- W. H. Lee, J. R. Booth and S. A. F. Bon, Biomacromolecules, 2020, 21, 4599–4614 CrossRef CAS PubMed.
- A. Lotierzo, R. M. Schofield and S. A. F. Bon, ACS Macro Lett., 2017, 6, 1438–1443 CrossRef CAS PubMed.
- A. Lotierzo, B. W. Longbottom, W. H. Lee and S. A. F. Bon, ACS Nano, 2019, 13, 399–407 CrossRef CAS PubMed.
- V. A. Bobrin, Y. Yao, X. Shi, Y. Xiu, J. Zhang, N. Corrigan and C. Boyer, Nat. Commun., 2022, 13, 3577 CrossRef CAS PubMed.
- V. A. Bobrin, K. Lee, J. Zhang, N. Corrigan and C. Boyer, Adv. Mater., 2022, 34, 2107643 CrossRef CAS PubMed.
- X. Shi, Y. Yao, J. Zhang, N. Corrigan and C. Boyer, Small, 2023, 2305268 CrossRef PubMed.
- N. R. Bagnall, M. H. Jones, G. C. Jernigan, C. Routt, L. C. Dar and B. T. Worrell, J. Am. Chem. Soc., 2023, 145, 14202–14207 CrossRef CAS PubMed.
- Z. Wan, W. Lee, A. Shegiwal and D. Haddleton, Eur. Polym. J., 2023, 196, 112324 CrossRef CAS.
- J. Maláč, Polym. Test., 2005, 24, 790–792 CrossRef.
- J. Maláč, Polym. Test., 2006, 25, 650–655 CrossRef.
- A. M. Kucherskii, Polym. Test., 2003, 22, 503–507 CrossRef CAS.
- F.-C. Chiu and K. Min, Polym. Int., 2000, 49, 223–234 CrossRef CAS.
- C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1996, 29, 7717–7726 CrossRef CAS.
- J. Krstina, G. Moad, E. Rizzardo, C. L. Winzor, C. T. Berge and M. Fryd, Macromolecules, 1995, 28, 5381–5385 CrossRef CAS.
- T. Harada, P. B. Zetterlund and B. Yamada, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 597–607 CrossRef CAS.
- B. Yamada, S. Tagashira and S. Aoki, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 2745–2754 CrossRef CAS.
- B. J. P. Jansen, S. Rastogi, H. E. H. Meijer and P. J. Lemstra, Macromolecules, 1999, 32, 6283–6289 CrossRef CAS.
- B. J. P. Jansen, S. Rastogi, H. E. H. Meijer and P. J. Lemstra, Macromolecules, 2001, 34, 4007–4018 CrossRef CAS.
- B. J. P. Jansen, S. Rastogi, H. E. H. Meijer and P. J. Lemstra, Macromolecules, 2001, 34, 3998–4006 CrossRef CAS.
- B. J. P. Jansen, S. Rastogi, H. E. H. Meijer and P. J. Lemstra, Macromolecules, 2001, 34, 3998–4006 CrossRef CAS.
- J. H. Kim, F. E. Karasz and M. F. Malone, Polym. Eng. Sci., 1991, 31, 981–987 CrossRef CAS.
- S. T. C. Ng, C. Jolliffe, A. Goodwin, M. Forsyth and D. R. MacFarlane, Electrochim. Acta, 1998, 43, 1499–1503 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00482e |
| This journal is © The Royal Society of Chemistry 2024 |