
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aggregation-induced emissive nanoarchitectures for luminescent solar concentrators
Elisavet
Tatsi†
 a,
Andrea
Nitti†
b,
Dario
Pasini
*b and
Gianmarco
Griffini
*a
a,
Andrea
Nitti†
b,
Dario
Pasini
*b and
Gianmarco
Griffini
*a
aDepartment of Chemistry, Materials and Chemical Engineering “Giulio Natta”, Politecnico di Milano, Piazza Leonardo da Vinci 32, 20133 Milano, Italy. E-mail: gianmarco.griffini@polimi.it
bDepartment of Chemistry and INSTM Research Unit, University of Pavia, Viale Taramelli 10, Pavia 27100, Italy. E-mail: dario.pasini@unipv.it
First published on 25th July 2024
Abstract
Aggregation-induced emission (AIE), the phenomenon by which selected luminophores undergo the enhancement of emission intensity upon aggregation, has demonstrated potential in materials and biomaterials science, and in particular in those branches for which spectral management in the solid state is of fundamental importance. Its development in the area of luminescent spectral conversion devices like luminescent solar concentrators (LSCs) is instead still in its infancy. This account aims at summarizing relevant contributions made in this field so far, with a special emphasis on the design of molecular and macromolecular architectures capable of extending their spectral breadth to the deep-red (DR) and the near-infrared (NIR) wavelengths. Because of the many prospective advantages characterizing these spectral regions in terms of photon flux density and human-eye perception, it is anticipated that further development in the design, synthesis and engineering of advanced molecular and macromolecular DR/NIR-active AIE luminophores will enable faster and easier integration of LSCs into the built environment as highly transparent, active elements for unobtrusive light-to-electricity conversion.
 Elisavet Tatsi | Elisavet Tatsi, a graduate of the University of Ioannina, obtained her Dip.Ing in Materials Science and Engineering in 2016. She completed her Ph.D. in Materials Engineering at Politecnico di Milano in 2022 under the supervision of Prof. Gianmarco Griffini, with a stint as a visiting student at the University of Melbourne under A/Prof. Wallace Wong. Her research focuses on polymer-based materials and devices for light harvesting, management, conversion, and energy storage. |
 Andrea Nitti | Andrea Nitti obtained his MSc Laurea in Chemistry (cum Laude) at the University of Bologna in 2013. He received his PhD in 2017 under supervision of Prof. Dario Pasini at the University of Pavia. During his PhD, he spent a period as a visiting student at MIT in Boston under the supervision of Prof. Timothy M. Swager. Currently he is an assistant professor at the Department of Chemistry of the University of Pavia, focusing on the sustainable organic synthesis of novel oligomeric and polymeric materials for organic electronics, and their processing using advanced manufacturing techniques. |
 Dario Pasini | Dario Pasini obtained his Ph.D. degree in Chemistry in 1997 from the University of Birmingham, U.K. (advisor: Fraser Stoddart, Nobel Prize in Chemistry 2016). After postdoctoral research at the University of California, Berkeley, U.S.A., in the group of Prof. Jean M. J. Fréchet (1997–1999), he joined the faculty of the Department of Chemistry at the University of Pavia in 2000, where he is now Full Professor of Organic Chemistry. He develops new scientific approaches in the fields of organic, supramolecular, and polymeric materials. He is a Fellow of the Royal Society of Chemistry, and the recipient of a Gutenberg Chair from the University of Strasbourg. He spent periods of research as visiting Professor with S. Matile (Geneva, CH), L. Shimizu (South Carolina, USA) and T. Swager (MIT, USA). |
 Gianmarco Griffini | Gianmarco Griffini is an Associate Professor of Science and Technology of Materials at Politecnico di Milano (Italy). He received his MSc degree in Chemical Engineering in 2005 and his Ph.D. degree in Materials Engineering from the same institution in 2012, after spending a few years in the private sector working as a process engineer. He has held visiting positions at University College London (UK), at University of California-Berkeley (USA), and at University of Castilla-La Mancha (Spain). His current research interests are focused on the development and characterization of polymer-based materials and on the study of their structure–property relationships. Areas of major interest include: materials and devices for light harvesting, management and conversion, and for energy storage; bio-derived and biodegradable polymers and composites for sustainable manufacturing; stimuli-responsive polymeric and composite materials for advanced manufacturing. In these areas, he is/has been involved as scientific coordinator in several regional, national and European projects, and as responsible for different research contracts with the private sector. |
1. Introduction
Luminescent solar concentrators (LSCs) are spectral conversion devices in which the combination of luminophores and lightguides enables the collection, re-emission and transport of solar photons, ultimately directing them towards solar cells for light-to-electricity conversion. LSC devices are able to harvest both direct and diffuse light with little angle dependence, and they are of particular interest in building integrated photovoltaic systems;1,2 however, in such applications visibly transparent LSC devices are preferable for minimal light distortion.While various strategies have been developed to improve the efficiency of LSCs, there remains significant room for improvement in different areas, including the engineering of low optical loss device architectures,3–6 the development of high optical clarity multifunctional host matrices7–10 and the design of high photoluminescence quantum yield (PLQY) emissive species.11–14 In the context of luminescent materials, aggregation-induced emission luminogens (AIEgens) expanding their spectral wavelengths into the deep red (DR) and near-infrared (NIR) ranges represent a particularly interesting approach. Indeed, while the use of luminophores with absorption and emission characteristics in the VIS range has been the most widely employed strategy to improve the performance of AIEgen-based LSCs, this has led to highly tinted devices usually characterized by a typical yellow-to-orange or red hue.6,9,15,16 In comparison, visibly transparent LSCs based on AIEgens designed to convert DR/NIR photons may offer notable advantages. These include the possibility to exploit the large photon flux available for energy use in this spectral region to boost their efficiency, while enabling seamless architectural integration with virtually no or minimal impact on the building aesthetics. Furthermore, NIR-responsive surfaces can contribute to passive cooling effects by enhancing heat dissipation, thus reducing temperature (or heat) gains.17,18 For optimized NIR absorption and emission, molecular engineering needs to be addressed through the use of different luminophores, incorporation of nanoparticles, and the design of suitable, advanced molecular or macromolecular architectures. Conventional materials give bright luminescence in dilute solutions, while the photoluminescence (PL) decreases in concentrated solutions or in the solid state due to an aggregation-caused quenching (ACQ) effect, as documented by Förster in 1954.19 Although many strategies have been reported trying to alleviate the ACQ effect, only limited achievements have been obtained since aggregation is a spontaneous process and a natural state. This phenomenon is particularly relevant for LSCs, where aggregation of luminophores is commonly found due to the high emitter concentration needed to obtain high-optical-density/high-efficiency devices.14,20 In contrast to ACQ, aggregation-induced emission (AIE) was demonstrated in 2001 by Tang et al., in which the enhancement of emission intensity upon aggregation of certain luminescent molecular species was observed.21,22 AIEgens generally consist of aromatic rotors in a twisted propeller-shaped conformation that minimizes the intermolecular π–π interactions in their aggregate state.23 Initially, a series of propeller-shaped molecules were found to possess AIE activity: hexaphenylsilole, hexaphenylbenzene,24 pentaphenylpyrrole,25 tetraphenylthiophene,26 pentaphenylphosphole oxide,27 and aryl substituted acrylates and acrylonitriles.28–30 The underlying mechanism of the AIE phenomenon is attributed to the restriction of intramolecular rotation (RIR) in the aggregated state.31–34 In the solution state, non-radiative decay pathways such as intramolecular rotations and vibrations are active, leading to low emission intensity.
AIE research has flourished in the past two decades, with an increasing number of reports in the literature, focusing on application areas as wide as optics and photonics, electronics, energy, bioimaging and therapeutics.32,35–37
In particular, recent efforts in the bio-optical field have led to the development of AIEgens with spectral response extending to the NIR-II region (1000–1700 nm) of the electromagnetic spectrum,38–40 with great potential for further exploitation in other scientific and technological areas. On the contrary, AIE research activity for LSCs has not been quite as active, with efforts mainly concentrated on AIEgens operating in the UV-visible range since their first use in lab-scale devices. In this respect, an overview of potential strategies to enable a step forward in the use of AIEgens in the field of colorless, NIR-active spectral conversion systems is still lacking.
Along these lines, this work aims to provide a detailed account on the current strengths and limitations of AIE luminophores for prospective applications in LSC devices, ultimately discussing potential pathways for their future implementation as NIR active species in visibly-transparent spectral conversion systems (Fig. 1). Specifically, this review will first give an overview of the functional nanoarchitectures based on molecular and polymeric AIE emitters currently used in LSC devices. The discussion will then be focused on the most relevant recent examples of AIEgens with absorption and emission characteristics extending to the DR/NIR spectral region, and with potential for future application in the LSC field. Finally, issues and opportunities associated with the utilization of molecular and polymeric AIEgens for DR/NIR spectral conversion in LSCs will be highlighted, presenting our vision on the design and application of tailored AIEgen architectures for such spectral conversion devices, in the hope to facilitate their development and future uptake in this field.
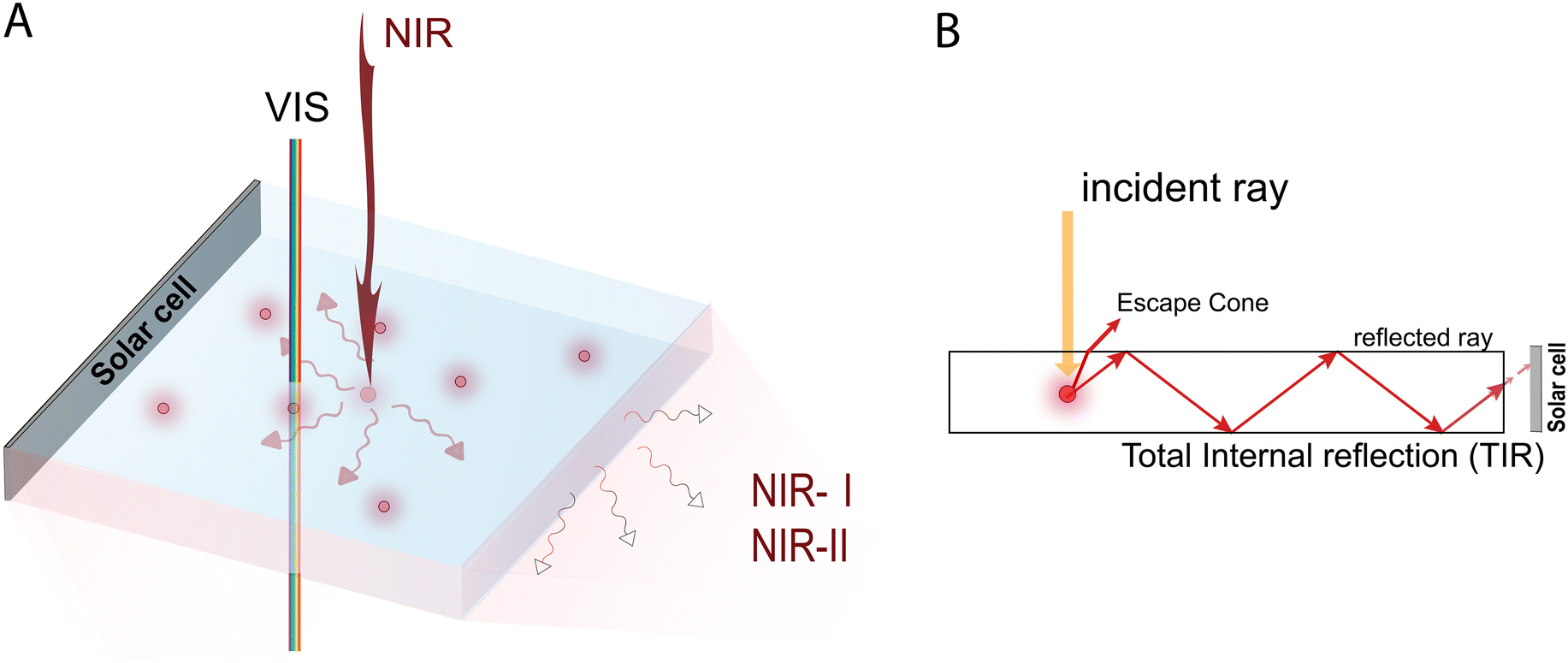 | ||
| Fig. 1 (A) Sketch of a transparent LSC, (B) working principle of LSCs. | ||
2. Discussion
2.1. AIE systems in LSCs
The use of AIEgen luminophores in the LSC literature has become increasingly popular in the past decade, and different chemical architectures have been proposed to meet the optical requirements of these devices (Fig. 2).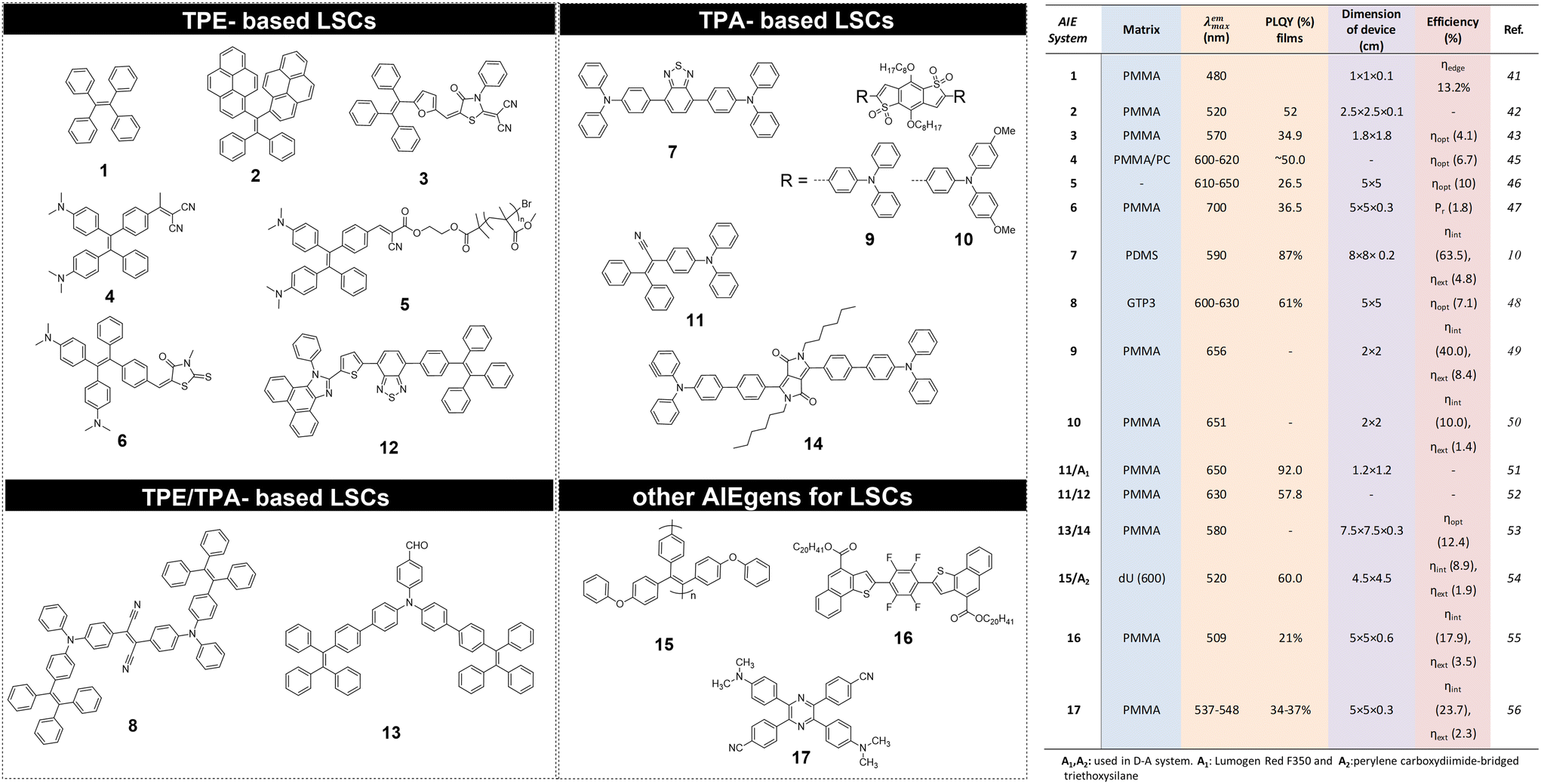 | ||
| Fig. 2 Left: Chemical structures for AIE luminophores used in LSCs; right: main photophysical parameters and performance for AIEgens used in LSCs reported in literature. Efficiency values are reported as: ηint = (no. of edge-emitted photons)/(no. of total absorbed photons); ηext = ηedge = (no. of edge-emitted photons)/(no. of total incident photons); ηopt = ISC(LSC)/(ISC(PV)G), where ISC(LSC) is the short circuit current generated by a PV cell edge-coupled to the top-illuminated LSC, ISC(PV) is the short circuit current of the same PV cell under direct illumination from the same light source, G = Atop/Aedge, with Atop = LSC top surface area, and Aedge = LSC edge surface area; Pr is the power generated by the PV cell edge-coupled to the LSC (PLSC) to the power produced by the PV cell (PPV) under direct exposure (in both cases, illumination was by an AM1.5G solar simulator under one Sun). | ||
Interestingly, most of the initial research in the field of AIE-based spectral conversion systems has centered around the utilization of AIEgens derived from tetraphenylethene (TPE, compound 1), whose AIE effect is the result of the RIR process upon aggregation. In their 2014 seminal work, Ghiggino and colleagues introduced for the first time AIEgen emitters in LSC devices using compound 1 as the core luminophore.41 Their work resulted in the fabrication of a 10 mm × 10 mm × 1 mm thin-film LSC of TPE in polymethyl methacrylate (PMMA) able to achieve an external photon efficiency ηext of 13.2% under a non-disclosed illumination source. However, it was also evidenced that the emission range of this system is not ideally suited for LSCs intended for integration with silicon or GaAs PV cells. Similarly, the absorption features poorly match the solar spectrum emission. Consequently, alternative design strategies for modifying TPE-based fluorophores have been explored to optimize AIEgen absorption and emission properties while maintaining the AIE response, despite the challenges in ensuring sufficiently high PLQY. In a subsequent study by the same research group, Banal et al. developed AIEgen luminophores based on the use of pyrene derivatives,42 whose luminescence properties are highly dependent on their regioisomeric structure: spectral shifts in their absorption and emission spectra, both in solution and in rigid polymer hosts, could be associated with the formation of weakly interacting H-aggregates, which yielded excimer-like emission with inherently large Stokes shifts (>1.0 eV, Fig. 3a).21 Through a tailored synthetic pathway, they isolated geometrically pure regioisomers of crystalline gem-substituted pyrene ethenes (compound 2). Such AIEgens, embedded in a PMMA host matrix, were used in Si–PV-coupled thin-film LSCs yielding high PLQY (52.0%) and device efficiency in line with inorganic phosphor-based LSCs of similar spectral response (ηdev = 0.44%, defined as the ratio between the electrical power produced by edge-coupled PV cells and the optical power hitting the LSC top surface). The quest to expand the emission range of TPE-based AIEgens to the red prompted Ma et al. to investigate compound 3, a D–D–π–A structure,43 which is composed of two electron-donor groups [ethene-1,1,2-triyltribenzene (D1), furan (D2)] and electron-acceptor groups [Rhodamine-CN] connected via a π-bridge.44 LSC devices incorporating 3 in PMMA exhibited absorption at 470 nm and a fluorescence peak at 570 nm, achieving an optical efficiency (ηopt) of 4.09% under simulated sunlight illumination. In a parallel effort, De Nisi et al. explored a variation of the TPE moiety, compound 4, which is based on the TPE stator and is decorated with dimethylamine and malononitrile as the electron donor (D) and the electron acceptor (A) moieties, respectively.45 The enhanced D–A interaction in this modified TPE-base AIEgen led to intense emission in the aggregate state, closely aligning with the DR portion of the solar spectrum, exhibiting VIS light absorption in the 400–550 nm wavelength range, λemmax = 600–620 nm, and a maximum PLQY of ∼50% when dispersed in PMMA. LSC systems incorporating compound 4 in polycarbonate (PC) achieved ηopt = 6.7% thanks to the better spectral matching with sunlight emission. Nonetheless, chemical compatibility issues with the PC host matrix yielded unwanted fluorescence quenching, thereby limiting device performance at high AIEgen loading. In a later study by the same research group, a TPE derivative was synthesized and employed as the initiator for preparing red-emitting compound 5via atom transfer radical polymerization (ATRP).46 This method was selected to gain finer control over the positioning of the luminophore in the macromolecular backbone, to avoid phase interferences/segregation between the host matrix (PMMA) and the luminescent species and thus maximize LSC performance. The PMMA/5 films displayed emission characteristics dependent on the fluorophore concentration, with emission peaking at 650 nm and a significant Stokes shift of 170 nm. The PLQY peaked at 26.5% for compound 5 content of 0.98 wt%. In LSC applications, compound 5 showcased a maximum ηopt of 7.05%, with a PMMA/5 film containing 1 wt% of compound 5. In a later study the same group47 reported a new derivative (compound 6) featuring dimethylamino push group and a 3-methyl-rhodanine pull moiety. The optical properties of compound 6 were systematically determined in various solvents, revealing absorption peaks at 303–307 nm, 370–374 nm, and 489–504 nm.
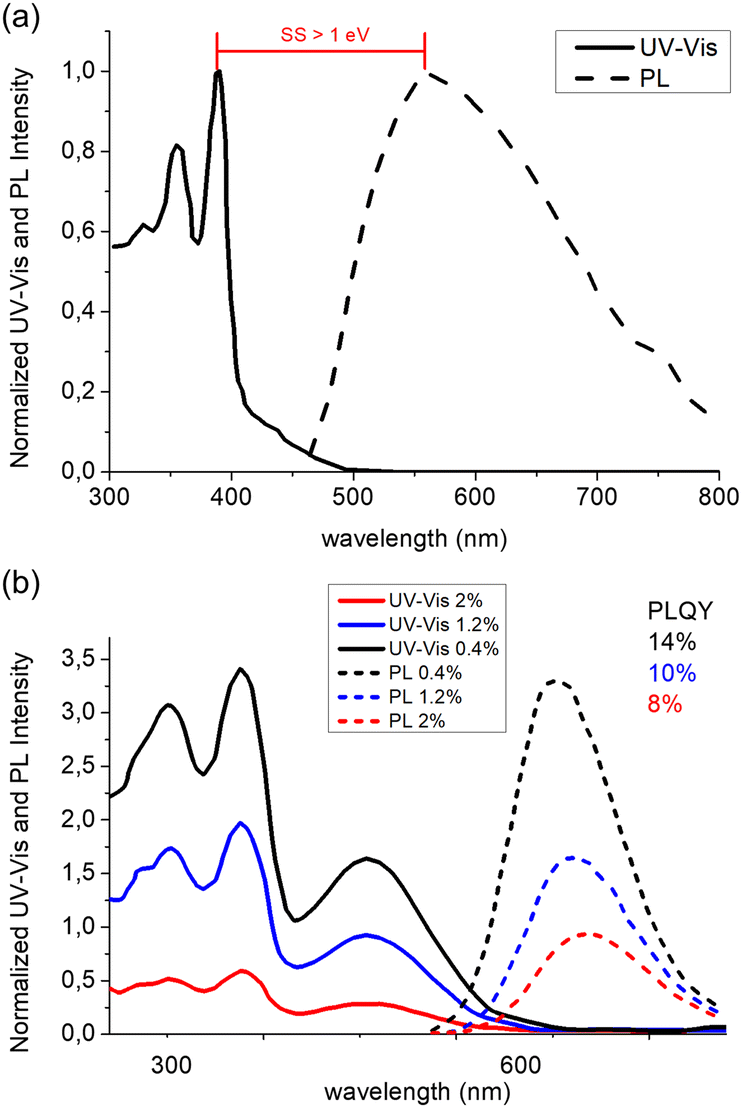 | ||
| Fig. 3 (a) UV-Vis (solid) and PL (dashed) spectra in PMMA matrix of compound 2. (b) UV-Vis and PL spectra of compound 6 in PMMA matrix at concentrations of 0.4% (black), 1.2% (blue) and 2% (red) respectively. Partially reproduced from ref. 42 and 47. Copyright 2015 and 2022 Wiley. | ||
Compound 6 exhibited an emission peak at 705 nm when aggregated in THF/water mixtures, indicative of its AIE behavior. Compound 6 also displayed crystallization-induced enhanced emission (CIEE) in the solid-state, indicating brighter fluorescence in the crystalline state compared to the amorphous state. When integrated into PMMA and poly(cyclohexyl methacrylate) (PCMA) films, compound 6 exhibited absorption bands in the 300–500 nm range, with the fluorescence band spanning the 500–800 nm range. Maximum PLQY values around 14%–16% were observed, decreasing below 10% for higher concentrations of 6 in PMMA films due to scattering following aggregate formation (Fig. 3b).
Triphenylamine (TPA)-based AIEgens have also been explored in various studies. In a recent work by Li et al., the TPA motif was exploited in combination with the benzothiadiazole moiety to obtain a near-unity-PLQY, large-Stokes shift (0.59 eV, 130 nm) AIEgen (7), offering high optical performance in polydimethylsiloxane (PDMS) LSCs.10 Compound 7 displayed absorption in the region of 360–550 nm (peaking at 461 nm) and photoluminescence in the 500–750 nm range, with a peak at 590 nm. The PDMS film exhibited high photostability, with approximately 70% of the original PLQY retained after an “accelerated aging” test equivalent to around 2.2 years of outdoor use. LSC devices were fabricated in different sizes and shapes, with ηint found to decrease slightly upon upscaling (68.5% for 3 × 3 cm2, 63.5% for 8 × 8 cm2 – planar LSC devices). Templated surface patterning was also employed to fabricate superhydrophobic LSCs, whose performance (both ηint and ηext) was however shown to be lower than for smooth devices, due to increased surface scattering. A different approach to obtain D–A AIEgen luminophores based on TPA was proposed by Geervliet et al., where the elementary electron-donating units of TPA, with one of the phenyl rings replaced by TPE, were combined with an electron-accepting fumaronitrile (FN) group (8).48,49 This D–A AIEgen was embedded in bio-based polyester matrices to obtain thin films (25 μm) exhibiting broad absorption (400–650 nm), fluorescence emission peaking at 600–630 nm, and a consistently high PLQY of 61% up to a 8 loading of 1.4 wt%. Thin-film LSC devices were obtained by embedding optimal concentrations of 8 in a random polyester copolymer of O-methylene galactarate, 1,3-propanediol and dimethyl terephthalate, yielding ηopt as high as 7.1% when edge-coupled with Si-based PV cells. More recently, authors from the same group reported the synthesis of orange/red organic emitters featuring a benzo[1,2-b:4,5-b′]dithiophene 1,1,5,5-tetraoxide central core as A unit connected to different D moieties by means of Pd-catalysed direct arylation reactions, yielding AIEgen molecules with either symmetric (D–A–D) or non-symmetric (D–A–A′) structures (9–10). The authors found that symmetric structures showed better photophysical properties for application in LSCs than their non-symmetric counterparts. In particular, 9/PMMA and 10/PMMA LSC devices exhibited ηext close to 8.4% and ηext = 0.94%.50
A recently explored strategy to enhance the efficiency of LSCs centers around the combined use of D–A pairs to enable the fluorescence resonance energy transfer (FRET) process, as will be discussed in the next section. Some of these approaches integrate AIE luminophores as either D or A species, while others resort to AIEgens for both D and A.
The first study on the FRET process in LSC applications was reported in 2004 by Banal et al., who employed the AIE donor compound 11 in conjunction with a high PLQY acceptor dye.51 AIE compound 11 is characterized by a D–π–A architecture and combines a propeller-like triphenylacrylonitrile (TPAN) group with a diarylamine, yielding high PLQY in the solid state (100% compared to 0.8% in solution of THF) and an emission centered at 550 nm. Blending 11 with the chosen acceptor (A1) in various ratios within PMMA for LSC devices yielded efficient energy transfer properties (86%). This approach kept the contribution of the acceptor absorption relatively low, minimizing reabsorption losses while maximizing light harvesting by the donor. Furthermore, 11 exhibited resistance to fluorescence quenching at high concentrations (>50 wt%), thus enabling efficient energy transfer in PMMA films. In a later study from the same group, a different FRET D–A pair (11/12) was introduced, with 12 as candidate A to replace the acceptor dye (A1) from their previous study.52 The acceptor 12 is based on TPE and phenanthro[9,10-d]imidazole with benzo-2,1,3-thiadiazole in its molecular backbone. While 12 exhibited AIE properties, it was susceptible to concentration quenching in a PMMA matrix, resulting in decreased PLQY from 93% at 9 mM to 45% at 225 mM. In contrast, 11 maintained a high PLQY of ∼98% even at a high concentration of 250 mM in PMMA.
In another approach by Mateen et al., a pair of AIE-active dyes, 13 and 14, was employed to enhance LSC efficiency under both outdoor and indoor lighting conditions, by exploiting the FRET process.53 The AIEgens systems were based on a tetraphenylethene-modified TPA derivative as the AIE donor (13), and a diketopyrrolopyrrole–TPA derivative as the AIE acceptor (14). The D compound 13 displayed high absorbance in the UV/blue range (320–420 nm) and emission from 400–600 nm, while 14 showed broad absorbance across the VIS spectrum and emission in the red/DR region (550–750 nm). The excellent spectral overlap between 14 absorbance and 13 emission enabled an efficient FRET process. For LSC device fabrication, a PMMA matrix was employed, with 13 consistently doped at 2 wt% and 14 varying from 0.25 wt% to 1.5 wt%. As a result, energy transfer efficiencies ηET varied between 58.5% and 97%. The resulting LSCs equipped with side reflectors and a back diffuser and incorporating the 13/14 AIEgen FRET pair yielded ηopt between 18% and 27.2% under AM 1.5G solar spectrum and white LED illumination, respectively.
In another study by Lyu et al., a conjugated polymer with AIE characteristics (polymer 15) was designed as the D moiety for FRET-based LSC devices, while perylene carboxydiimide-bridged triethoxysilane served as the A moiety, embedded in a ureasil waveguide. Polymer 15 displayed a high PLQY between 52–73% and a significant Stokes shift of approximately 150 nm in the solid state.54 When induced to aggregate, 15 exhibited a dramatic increase in emission intensity of up to 530%, validating its AIE response. The PLQY of 15 increased significantly, from 2% in a good solvent (THF) to 44% at 0.001 wt% in a ureasil matrix, reaching around 60% at 0.05 wt%. However, concentrations exceeding 5 × 10−3 wt% led to scattering losses due to enhanced aggregation, affecting the optical response of the LSC, with 5 × 10−3 wt% being optimal. LSCs incorporating both 15 and perylene-based acceptor (A2) in the diureasil waveguide demonstrated enhanced performance, with the highest internal photon efficiency (ηint) of 20.0% and external photon efficiency (ηext) of 5.5%, outperforming LSCs with 15 and A2 alone.
More recent investigations have introduced innovative chemical strategies to enhance the performance of LSCs through the utilization of AIEgen molecules with spectral breadth spanning from the VIS to the DR region. To this end, the introduction of fluorine atoms into a conjugated tetrafluorobenzene backbone structure led to the development of AIEgen 16, featuring a tetrafluorobenzene central core end-capped with naphthothiophenes.55 This luminophore was obtained through an eco-friendly synthetic process (low E-factor of 99) and demonstrated promising optoelectronic properties due to its large Stokes shift (≈4632 cm−1) and a two-fold increase in PLQY in the solid state compared to that in solution In pure THF, 16 displayed an absorption spectrum characterized by three peaks at 298 nm, 362 nm, and 385 nm, indicative of the π–π* transitions of the π-electron system.
With increasing water content in THF–H2O mixtures, absorption at 298 nm and 362 nm was shown to decrease, while a new peak emerged at ≈423 nm, indicating aggregate formation. A weak blue emission was found in THF and in low-water-fraction THF/water mixtures (Fig. 4), while a progressively stronger green emission at λmax = 509 nm appeared for water fractions higher than 35%. In thin-film, 16 exhibited UV–Vis absorption and emission properties akin to high-water-fraction solutions, but with a doubled PLQY compared to the solution. LSC devices were obtained by incorporating 16 into a PMMA matrix at varying concentrations (ranging from 0.5–17.5 wt%). The optimal AIEgen concentration was determined to be 12.5 wt%, delivering the highest ηext = 3.5% and maintaining a stable ηint = ∼18%. Concentrations exceeding 12.5 wt% yielded reduced emission intensity and decline in ηext, because of reabsorption, self-absorption phenomena, and limited luminophore solubility. Large-scale LSCs (10.0 × 10.0 × 0.6 cm3 and 15.0 × 15.0 × 0.6 cm3) were also successfully fabricated using the optimized 16 concentration, highlighting the scalability potential of this approach.
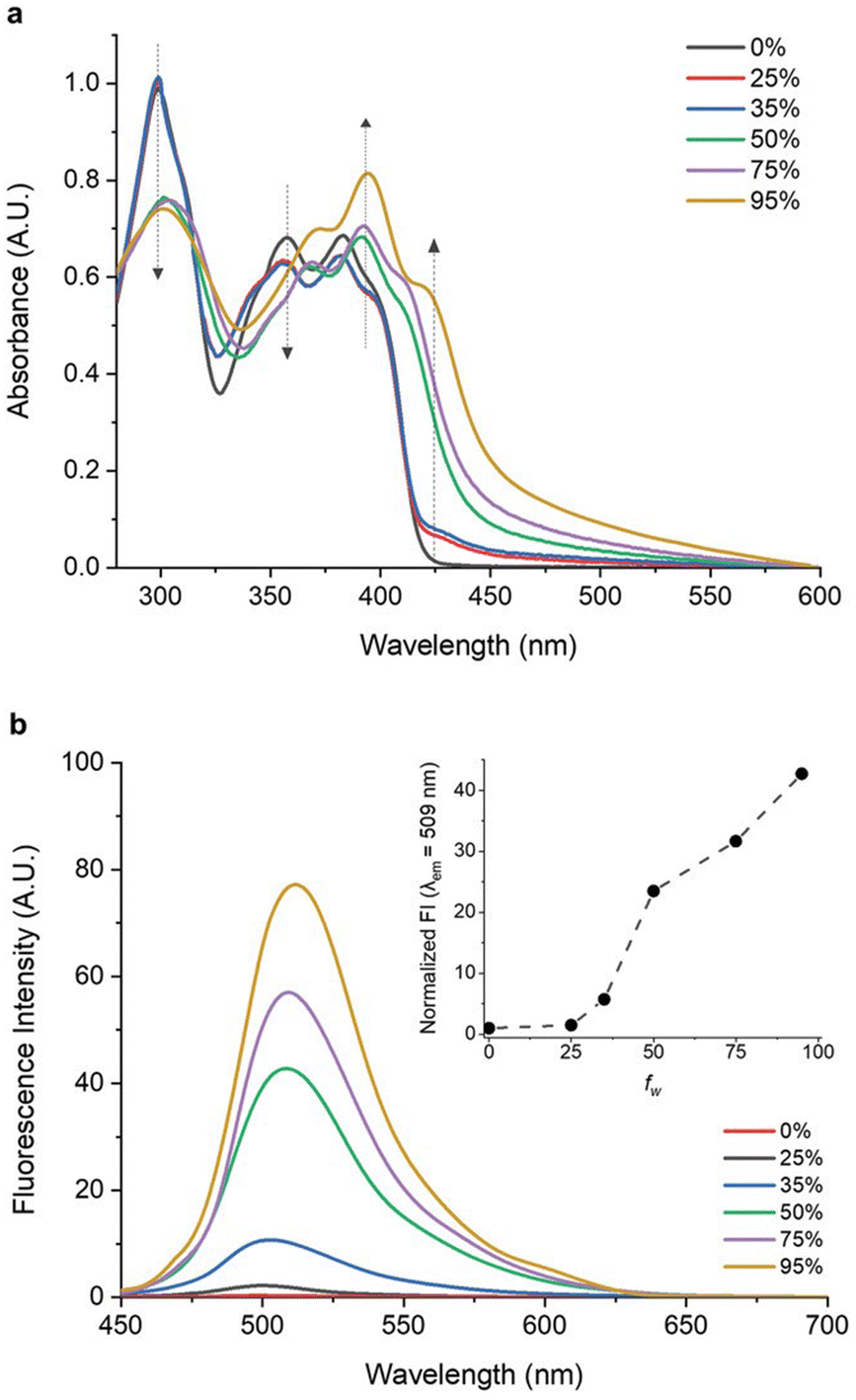 | ||
| Fig. 4 (a) UV–vis spectra of dilute solutions of 16 in THF–H2O mixtures with different volume fractions of water (fw). (b) PL spectra of 16 in THF–H2O mixtures at different fw values. The inset shows the normalized PL intensity at as fw changes. Partially reproduced with permission from ref. 55. Copyright 2021 Wiley. | ||
Another strategy proposed by Meti et al. focused on D–A type AIEgens exhibiting efficient intramolecular charge transfer (ICT) behaviour, ultimately leading to a large Stokes shift (>106 nm).56 In this work, a tetraphenylpyrazine-based AIEgen (17) was developed, which was symmetrically functionalized with dimethylamine (D) and cyano (A) groups. In the aggregate state, 17 presented restricted rotation of the phenyl rings, emitting light peaked at approximately 500 nm in DMSO, which red-shifted to 600 nm and intensified upon aggregation. For LSC device fabrication, 17 was dissolved in PMMA at varying concentrations, leading to maximum values of ηint = 23.7% and ηext = 2.33% at 0.01 wt% and 0.02 wt% AIEgen loading, respectively.
2.2. Design concepts towards DR/NIR AIEgens
As discussed in a recent review on AIEgens operating in the UV-vis spectral range11 and as previously detailed in this work, AIE-based luminophores are ideally suited for LSC applications as they enable high PLQY at high luminophore loadings, thereby maximizing the light harvesting and emission processes for optimized device performance. To further elaborate on this topic, in this section the discussion will be focused on the most recent attempts to extend the absorption and the emission of AIEgens to the DR/NIR spectral region, with the ultimate goal of extracting guidelines for expanding these concepts to the LSC field. To that end, the potential advantages of the AIE approach in LSCs using DR/NIR luminophores can be briefly summarized based on two key assets: on the one hand, luminophores converting NIR photons may take advantage of a larger photon flux vs. the UV-vis range, which can then be exploited for increased device efficiency; on the other hand, boosting non-radiative recombination through RIR mechanisms, as in the AIE paradigm, enables to tackle the unavoidably low PLQY of conventional NIR luminophores. Prototypical examples of UV-vis AIEgens already proposed in the LSC literature (blue background) and chemical platforms that could potentially be employed for DR/NIR-responsive LSCs (red background) are illustrated in Fig. 5, to demonstrate the tunability of the spectral response of AIEgens via tailored modification of their molecular structure, as discussed in further detail in the following.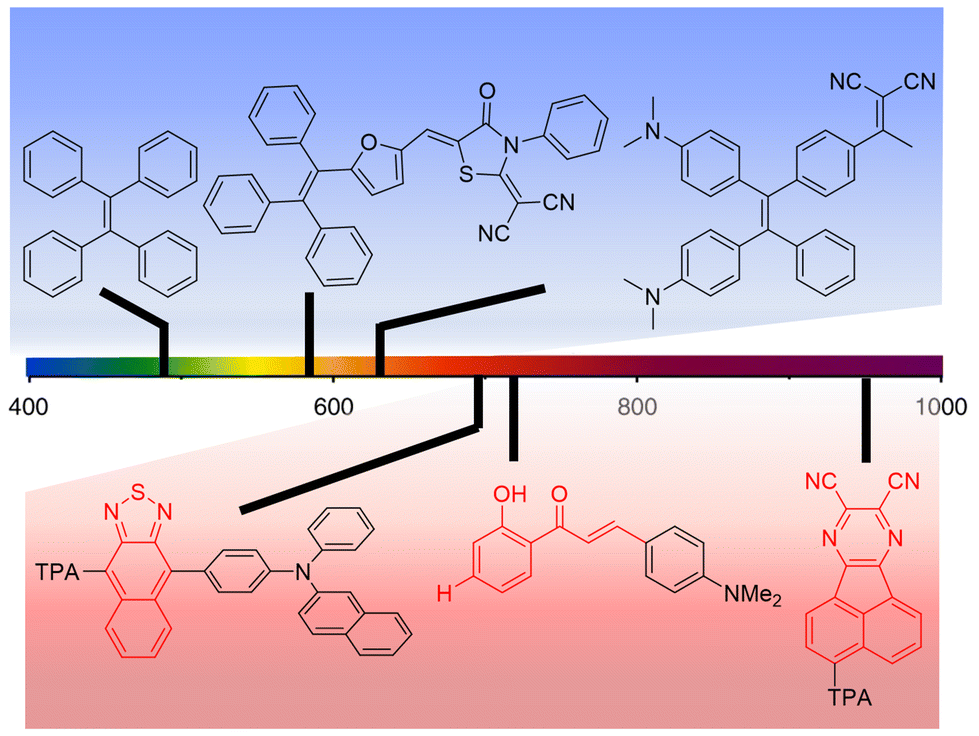 | ||
| Fig. 5 Examples of chemical structures of AIEgens with emissive properties in different spectral regions (TPA = triphenylamine). Blue background: UV-vis responsive AIEgens proposed in the LSC literature (ref. 41, 43 and 45 from left to right); red background: chemical platforms to be potentially employed for DR/NIR-responsive LSCs (ref. 57–59 from left to right). | ||
The energy separation between HOMO and LUMO levels (Eg) is the decisive factor to obtain a conventional VIS, DR or NIR organic emitter. This is generally achieved through molecular engineering (conjugation length, bond length alternation and push–pull architecture) and through the control of supramolecular interactions (molecular π–π stacking, hydrogen bonding and intermolecular charge transfer processes).60
According to the energy gap law,61–63 non-radiative decay rates (knr) (associated to allowed molecular rotational and vibrational modes) from excited state (S1 or T1) to the ground state (S0) exponentially increase with the decrease of Eg. Conventional DR/NIR emitters therefore suffer of larger knr compared to radiative decay rates (kr) as an intrinsic property linked with the HOMO–LUMO energy difference, thus yielding limited PLQY. However, increasing kr and reducing knr is crucial for obtaining high-efficiency spectral conversion devices making use of DR/NIR-emitters.
The AIE concept provides a powerful strategy to address the development of DR/NIR emitters with enhanced performance in the condensed/aggregated state. The AIE molecular design can overcome the problems associated with the energy gap law by reducing the contribution of the electron-vibration coupling component in the knr equation thanks to RIR mechanisms,64–67 allowing the realization of AIE-active DR/NIR emitters for several applications. As an example, the design and application of AIE-active NIR emitters was recently proposed for sensing selected ions and molecules.68–70 In another example, Yang and coworkers developed novel cyanine NIR-AIEgens incorporating naphthothiazolium propylsulfonate as acceptor unit and TPA as donor unit in a D–A “push–pull” molecular structure, exhibiting crystal-induced emission enhancement (CIEE) and morphology-dependent NIR emissive behavior.71 The brightly emissive crystalline powder with emission centered at 687 nm (ON state) is transformed into non-emissive amorphous state (OFF state) when grinding. The reversible inter-conversion between NIR emitted and non-fluorescent state was successfully applied in a physical pressure and temperature sensor.
Areas of intense development of NIR AIEgens are those of bioimaging and photodynamic therapy, as a result of the intrinsic deep tissue penetration ability of NIR light and thanks to its limited phototoxicity.72–74 Optical imaging in the VIS or even in the first NIR (NIR-I) region is restricted to superficial areas, since excitation and emission photons from these regions are partially attenuated by tissue scattering and adsorption.75–77 Very recently a second generation of AIE NIR-emitters based on the second NIR (NIR-II) emission region were explored. NIR-II in the range of 1000–1700 nm significantly reduces photon scattering, absorption, and self-fluorescence, allowing more effective biological tissue penetration and more precise visualization of the tissue of interest. The Tang group reported the first AIEgens for NIR-II imaging in 2020.78 They synthesized D–A “push–pull” AIEgens with twisted intramolecular charge transfer (TICT) properties using benzobisthiadiazole (BBTD) as strong electron-withdrawing unit and TPA units working as both donors and molecular rotors. In between the BBTD and TPA units, alkyl thiophene units were introduced to ensure a large distortion of the conjugated backbone. Notably, these emitters showed maximum emission at ∼1000 nm with a tail extended to 1600 nm and displayed a moderate PLQY of ∼9% as aggregates. The same group demonstrated for the first time the use of zwitterion-type NIR AIEgens as multifunctional theranostic agents.79,80 The molecular design in this case adopted 2,3,3-trimethylindolenine (ITB) as the electron-withdrawing-group (EWG) acceptor moiety, functionalized with 1,3-propane sultone, and TPA electron-donating-group (EDG) units as donor moieties. These NIR-II emitters showed extraordinary photothermal conversion efficiency for cancer treatment. For bioimaging applications, AIEgens are often made into nanoparticles (NPs) via the “nanoprecipitation method”, carried out by quick injection of the AIEgen solutions into aqueous media, and encapsulated with biocompatible surfactant agents to improve their biocompatibility and colloidal stability. The rapid precipitation process arranges luminophores into disordered or amorphous NPs in a loose-packing way, although crystallization is a more effective approach to maximize the packing of molecules and minimize intramolecular motions.81
Most of NIR AIEgens possess larger knr than kr in the aggregate state, and thus low or moderate PLQYs. The AIE concept alone is thus not sufficient to develop efficient solid-state NIR luminophores, as practically evidenced by the lack of high-performance NIR-organic light emitting diodes (OLEDs),82,83 NIR-organic photovoltaics (OPVs),84 and NIR-LSCs.
Besides the suppression of electron-vibration coupling, the reduction of electronic nonadiabatic coupling is another approach to reduce knr and overcome the energy gap law.64,85,86 Such nonadiabatic coupling elements are qualitatively proportional to the electronic transition density. Decreasing the overlap of electrons and holes in the excited states can reduce the non-radiative decay processes. Interestingly, such theoretical targets can be experimentally hit by resorting to photophysical processes entailing thermally activated delayed fluorescence (TADF), hybridized local and charge-transfer (HLCT) excited states, excited state intramolecular proton transfer (ESIPT) and FRET, as schematically represented in Fig. 6.
 | ||
| Fig. 6 Schematic representation of the TADF, HLCT, ESIPT and FRET photophysical processes. | ||
One interesting example of the use of such approaches is represented by the concept of aggregation-induced delayed fluorescence (AIDF), which combines the AIE strategy to the TADF process,87 initially observed in eosin, fullerene and porphyrin derivatives,88–90 and popularized by Adachi's and Zysman-Colman's groups, who used TADF molecules in OLED devices.91–95 TADF can harvest both singlet and triplet excitons simultaneously through efficient reverse intersystem crossing (RISC) using thermal energy. According to Boltzmann statistics, the RISC process can occur efficiently at a given temperature if the singlet–triplet energy gap (ΔEST) is small enough.96,97 Achieving small ΔEST is possible realizing a suitable separation of the HOMO and LUMO via molecular engineering. This approach, combined with the incorporation of AIE characteristics to the target luminophore, can represent a feasible methodology to develop DR/NIR emitters in the aggregate state.
The structure and metrics (emission wavelength λem and PLQY) for recent high performance AIDF DR/NIR emitters, both in solution and in neat films, are reported in Fig. 7 with the corresponding chemical structures.
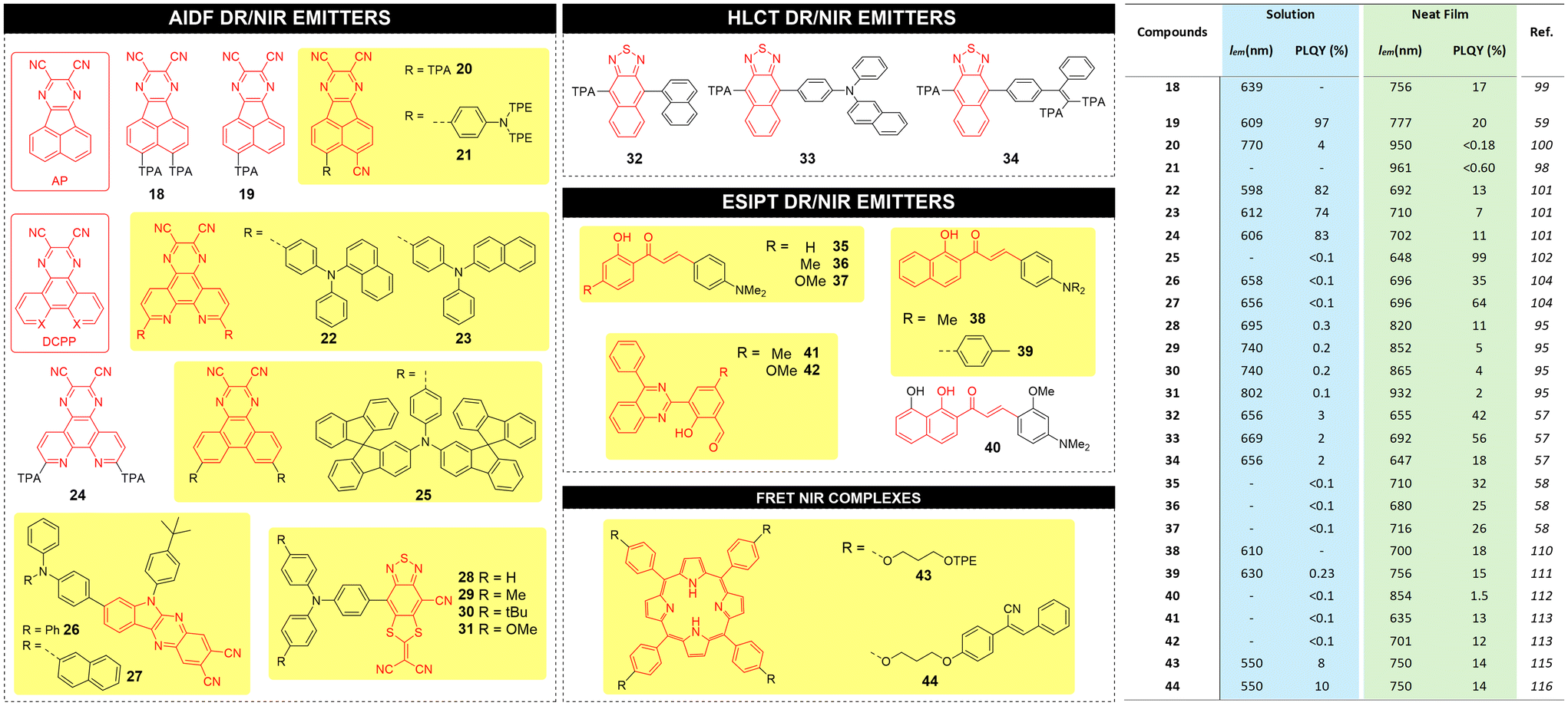 | ||
| Fig. 7 Left: chemical structures for AIDF, HLCT, ESIPT and FRET based AIE luminophores proposed in the literature; right: corresponding main photophysical parameters and performance. | ||
Bronstein and coworkers reported a NIR-AIEgen 21 based on dicyanopyrazine that is fused to the acenaphthene (AP) as the acceptor unit.98 Compared to previous AP-based materials 18–20,59,99,100 compound 21 has a TPE-based triphenylamine moiety as donor rotor unit, which confers AIE behavior and a red shift of wavelength to 961 nm in neat film. When diluted in a benzimidazole-based host matrix, luminophores 18–21 exhibit an enhancement of emission efficiency (PLQY of 63%, 98%, 9%, and 11% respectively) compared to solutions.
Tang and coworkers reported DR AIDF emitters 22–24 incorporating a pyrazino[2,3-f][1,10]phenanthroline-2,3-dicarbonitrile (DCPP) acceptor and using three different triarylamines as rotors,101 with red emission peaked at 598–620 nm in solution, and NIR emission peaked at 692–710 nm with PLQYs of 7–13% in neat films. Zhang and coworkers reported a modification in donor rotors introducing bulky spiro-fluorene moieties.102 DR emitter 25 did not show emission in dilute solution but strong emission in a host matrix with emission peaked at 648 nm and PLQY of 99%. The AIDF characteristics were confirmed by nanoaggregate formation by adding water to a solution of 25 in toluene. Ge et al. demonstrated that AIDF emitters with J-aggregate packing mode in the solid state can drastically enhance the luminescence efficiencies.103 The authors synthesized two NIR-AIDF luminophores consisting of 6-(4-(tert-butyl)phenyl)-6H-indolo[2,3-b]quinoxaline-2,3-dicarbonitrile (IQD) as planar acceptor unit and two triarylamines as donor units (compounds 26 and 27). In THF/water mixtures, PL intensities of 26 and 27 showed a swift increase when the water fraction increased to 60%. Crystal analysis revealed a face-to-face packing motif driven by C–H–π and CN–H–C interactions (J-type aggregates). A novel class of AIDF was also recently reported by the Tang group using cyano-substituted dithiafulvalene-fused benzothiadiazole (BSMCN) as the strong acceptor unit.104
AIEgens based on the HLCT excited state process provide a balance of locally excited (LE) and intramolecular charge transfer (CT) excited states, both essential to achieve NIR emission with high PLQY via efficient RISC at high-lying triplet and singlet excited states (hRISC), such as T2 to S1.105–107 LE state with a large transition moment and orbital overlap contributes to the enhancement of the PLQY, and the relatively weak CT state is beneficial for the NIR emission. By regulating the dihedral angles of the D–A segment, the character of the excited states can be modulated to form HLCT states and thus promote the hRISC process and efficient radiative decay. Recently, the Tang group reported the synthesis of three AIEgens with DR/NIR emissions implemented in OLED devices showing HLCT behaviour through a fine regulation of the excited states. The D–A architecture of such emitters (32, 33 and 34) make use of a triphenylamine-modified naphthothiadiazole derivative (TNZ) as the acceptor unit, flanked by donor rotors having different levels of steric hindrance.57 According to theoretical calculations, all emitters benefit of large energy gaps between T2 and T1, which decrease the energy dissipation by internal conversion. In addition, they exhibit a small energy difference between T2 and S1, which makes the hRISC process efficient. Compound 33 was found to possess the highest PLQY in the series in neat film form (56%), as opposed to 42% and 18% for 32 and 34, respectively.
The excited-state intramolecular proton transfer (ESIPT) process consists in a four-level photochemical process, as described in Fig. 6. ESIPT luminophores are typically found in an enol (E) form that, upon photoexcitation, promotes a reorganization of covalent bonds with consequent charge redistribution.108 This yields higher acidity of the hydrogen bond in the donor group and higher basicity of the hydrogen bond in the acceptor group, ultimately leading to fast enol (E*) to keto (K*) phototautomerization events (with kESIPT > 1012 s−1). The K* state has a short life and decays radiatively back to its electronic ground state (the original E form) with a reverse proton transfer (RPT) process.109 The change of excited state from E* to K* implies a redistribution of charge, so that ESIPT emitters have an unusually large Stokes shift when compared to traditional fluorophores, avoiding unwanted self-reabsorption and inner-filter effects that are problematic in LSC applications.
Wang and co-workers synthesized compounds 35–37 based on 2′-hydroxychalcones exhibiting ESIPT behaviour.58 Under excitation, the intramolecular hydrogen bonding induces the proton transfer from the hydroxyl group to the carbonyl group. Single crystals of compounds 35–37 exhibit NIR emission centred at 710, 680 and 716 nm, respectively, with large Stokes shifts and high PLQY (25–32%) while no emission could be recorded in diluted solutions.
ESIPT AIEgen 38 developed by the Jiang group, was found to exhibit a 20 nm red shifted λem with respect to the homologue compound 36 in its crystal form.110 A propeller-like triphenylamine donor rotor introduced into 2′-hydroxychalcone was synthesized by Fu et al. (compound 39) and was characterized by ESIPT AIE behaviour.111 Doped films of 39 exhibited emission at 596 nm with a PLQY of 10%. In contrast, the single crystal showed a strong NIR emission at 756 nm and a PLQY of 15%. Liao et al. synthesized compound 40,112 containing two adjacent intramolecular hydrogen bonds. The stepwise proton transfer process represents a true ESIPT process, with a first ultrafast proton transfer of <430 fs and the subsequent irreversible second proton transfer of ca. 1.6 ps. AIEgen 40 was used for the first realization of NIR organic single-crystal laser with emission beyond 850 nm at room temperature. Pandey et al. reported quinazoline derivatives 41–42 as ESIPT AIEgens113 with DR/NIR emission (635 and 701 nm respectively) and PLQY of 13% and 12% in the aggregate state, respectively.
Intermolecular charge transfer interactions can give rise to FRET mechanisms in the aggregate state. FRET is a photophysical process in which the excess energy of an excited species, the donor D, is transferred through space to an emitter (E) species, mediated by long-range dipole–dipole coupling (typically 1–10 nm). Using selected emitters, it is possible to push the emission into the DR/NIR region.
The FRET efficiency can be optimized by tuning the D/E spectral overlap integral, their intermolecular distance and their PLQYs.14 A sufficient spectral overlap integral requires strong overlap of the absorption spectrum of E with the emission spectrum of D. The FRET efficiency is inversely proportional to the distance between D and E, so that shortening intermolecular distance or increasing the luminophore concentration typically helps to increase the efficiency of FRET systems. Furthermore, FRET efficiency is also directly proportional to the PLQY of D and E.
The use of the FRET approach in combination with AIEgens has been reported by the Yang group, using a porphyrin emitter linked to two UV-vis AIEgens donors.114,115 Porphyrins possess excellent NIR emission in solution but strong ACQ effect in the aggregate state. Both FRET system 43–44 showed high NIR emission in both solution and solid state, with PLQY in aggregates as high as 14%. The Stokes shift of systems 43–44 was as large as 410 nm, displaying excellent fluorescence bio-imaging behavior for living cells.
3. Conclusions and outlook
In this account, we have presented an overview of the most recent research trends in the field of AIE luminophores extending their response in the DR/NIR spectral regions, with potential for application in transparent LSCs. This is a rapidly evolving multidisciplinary area that can profit of an additional development boost from the opportunities given by the building-integrated photovoltaic paradigm, where the interplay between visible transparency, aesthetics and device efficiency represents a key asset for successful exploitation.The AIE concept requires supramolecular aggregation of a high number of molecules to reveal itself. As a result of such aggregation, new supramolecular entities are formed, which naturally belong to the nanoscale domain. To that end, a detailed understanding of the changes in the chemical–physical and photophysical properties of such aggregated, nanostructured architectures is of fundamental importance when moving from the molecular to the nanoscale level, so as to ensure their effective use in light-management devices.
In this respect, there is certainly large room for improvement, both in the engineering of innovative device architectures, and in the chemistry, photophysics and nanoscale organization of the emissive molecules. In particular, relevant guidelines for designing effective molecular and macromolecular NIR-responsive AIEgens for spectral conversion systems are becoming progressively more evident. Here, we briefly summarize our vision and outlook for future development in this field:
• Molecular design strategies for NIR-compatible AIEgens: targeting a suitable energy gap represents a necessary but not a sufficient condition, since non-radiative decay rates (knr) tend to exponentially increase with decreasing energy gap. As a result, this approach needs to be accompanied by a fine tuning of the RIR mechanisms responsible for AIE behavior, through molecular engineering, intermolecular interactions and nanoscale organization, ultimately facilitating efficient light emission in the NIR region.
• Incorporation of AIEgens into polymer matrices: conventional methods of integrating luminophores into polymer matrices involve embedding them within the material in a host–guest fashion. However, direct linkage of AIEgens within the polymer matrix presents a compelling alternative. This approach allows for precise control over the spatial arrangement of AIEgens, optimizing light-harvesting efficiency. Leveraging the inherent property of AIEgens to exhibit enhanced emission intensity upon aggregation, direct incorporation into the polymer matrix may mitigate solubility issues associated with high concentrations, ultimately reducing scattering losses and enhancing device efficiency. Strategies such as block polymerization offer avenues for synthesizing tailored systems, enabling precise control over AIEgen dispersion and aggregation within the polymer matrix.
• Device engineering: current approaches for light-to-electricity conversion in LSC systems typically entail the use of edge-coupled photovoltaic devices (very often Si-based), whose spectral response is in most cases not well-matched with the emission spectrum of the luminophore. Extending the luminophore spectral breadth to the DR/NIR region requires the incorporation of lower band-gap photovoltaic systems capable of exploiting efficiently longer-wavelength photons waveguided through the LSC device. To that end, low-band-gap single junction and tandem perovskite photovoltaics and OPVs, alongside chalcogenide-based architectures, are expected to play an increasingly important role in this field as more effective and easily-tunable alternatives to Si-based devices.116–118
As emerging photovoltaic technologies are steadily progressing towards commercialization, we believe the design, development, characterization and application of DR/NIR active AIE-based luminophores in the broad area of spectral conversion devices will flourish in the next few years, with a sustained degree of innovation coming from interdisciplinary research efforts at a global scale.
Author contributions
All authors: conceptualization; D. P. and G. G.: funding acquisition; D. P. and G. G.: supervision; E. T. and A. N.: writing – original draft; D. P. and G. G.: writing – review & editing.Data availability
No new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge MUR (PRIN 2022 Prot. 2022BREBFN, acronym NIR+). A. N. acknowledge financial support from the European Union (EU) through the program” Programma Nazionale di Ripresa e Resilienza” (PNRR), Next Generation EU – “Young Researcher winner of MSCA actions or Seal Of Excellence” action (SOE_0000198). A. N. and D. P. acknowledge support from the Ministero dell'Università e della Ricerca (MUR) and the University of Pavia through the program “Dipartimenti di Eccellenza 2023–2027”.References
- K. Lee, H.-D. Um, D. Choi, J. Park, N. Kim, H. Kim and K. Seo, Cell Rep. Phys. Sci., 2020, 1, 100143 CrossRef CAS.
- C. Yang, D. Liu, M. Bates, M. C. Barr and R. R. Lunt, Joule, 2019, 3, 1803–1809 CrossRef.
- G. Panzeri, E. Tatsi, G. Griffini and L. Magagnin, ACS Appl. Energy Mater., 2020, 3, 1665–1671 CrossRef CAS.
- C. N. Eisler, L. E. Parsons, Z. Nett, C. Love, A. M. Schwartzberg and A. P. Alivisatos, Front. Photonics, 2022, 3, 932913 CrossRef.
- H. C. Bauser, C. R. Bukowsky, M. Phelan, W. Weigand, D. R. Needell, Z. C. Holman and H. A. Atwater, ACS Photonics, 2020, 7, 2122–2131 CrossRef CAS.
- K. Park, J. Yi, S.-Y. Yoon, S. M. Park, J. Kim, H.-B. Shin, S. Biswas, G. Y. Yoo, S.-H. Moon, J. Kim, M. S. Oh, A. Wedel, S. Jeong, H. Kim, S. J. Oh, H. K. Kang, H. Yang and C. J. Han, Nat. Photonics, 2024, 18, 177–185 CrossRef.
- F. Corsini, C. M. Rizzini, C. Botta, S. Turri and G. Griffini, Adv. Mater. Interfaces, 2022, 9, 2200108 CrossRef CAS.
- E. Tatsi and G. Griffini, Sol. Energy Mater. Sol. Cells, 2019, 196, 43–56 CrossRef CAS.
- E. Tatsi, G. Fortunato, B. Rigatelli, G. Lyu, S. Turri, R. C. Evans and G. Griffini, ACS Appl. Energy Mater., 2020, 3, 1152–1160 CrossRef CAS.
- X. Li, J. Qi, J. Zhu, Y. Jia, Y. Liu, Y. Li, H. Liu, G. Li and K. Wu, J. Phys. Chem. Lett., 2022, 13, 9177–9185 CrossRef CAS PubMed.
- A. Pucci, Isr. J. Chem., 2018, 58, 837–844 CrossRef CAS.
- A. Kim, A. Hosseinmardi, P. K. Annamalai, P. Kumar and R. Patel, ChemistrySelect, 2021, 6, 4948–4967 CrossRef CAS.
- S. Castelletto and A. Boretti, Nano Energy, 2023, 109, 108269 CrossRef CAS.
- B. Zhang, G. Lyu, E. A. Kelly and R. C. Evans, Adv. Sci., 2022, 9, 2201160 CrossRef PubMed.
- P. Xia, H. Sun, H. Guo, K. Zhao, C. Liang, C. Lu, Z. Wang, S. Xu and C. Wang, Adv. Funct. Mater., 2024, 2401121 CrossRef.
- P. Moraitis, R. E. I. Schropp and W. G. J. H. M. Van Sark, Opt. Mater., 2018, 84, 636–645 CrossRef CAS.
- A. Frias, M. Cardoso, A. Bastos, S. Correia, P. André, L. Carlos, V. De Zea Bermudez and R. Ferreira, Energies, 2019, 12, 451 CrossRef CAS.
- C. Yang and R. R. Lunt, Adv. Opt. Mater., 2017, 5, 1600851 CrossRef.
- Z. Qiu, X. Liu, J. W. Y. Lam and B. Z. Tang, Macromol. Rapid Commun., 2019, 40, 1800568 CrossRef PubMed.
- C. Tummeltshammer, A. Taylor, A. J. Kenyon and I. Papakonstantinou, Sol. Energy Mater. Sol. Cells, 2016, 144, 40–47 CrossRef CAS.
- F. Würthner, Angew. Chem., Int. Ed., 2020, 59, 14192–14196 CrossRef PubMed.
- J. Luo, Z. Xie, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 18, 1740–1741 RSC.
- Z. Zhao, B. He and B. Z. Tang, Chem. Sci., 2015, 6, 5347–5365 RSC.
- R. Hu, J. W. Y. Lam, Y. Liu, X. Zhang and B. Z. Tang, Chem. – Eur. J., 2013, 19, 5617–5624 CrossRef CAS PubMed.
- X. Feng, B. Tong, J. Shen, J. Shi, T. Han, L. Chen, J. Zhi, P. Lu, Y. Ma and Y. Dong, J. Phys. Chem. B, 2010, 114, 16731–16736 CrossRef CAS PubMed.
- C. T. Lai and J. L. Hong, J. Phys. Chem. B, 2010, 114, 10302–10310 CrossRef CAS PubMed.
- A. Fukazawa, Y. Ichihashi and S. Yamaguchi, New J. Chem., 2010, 34, 1537–1540 RSC.
- A. Nitti, F. Villafiorita-Monteleone, A. Pacini, C. Botta, T. Virgili, A. Forni, E. Cariati, M. Boiocchi and D. Pasini, Faraday Discuss., 2017, 196, 143–161 RSC.
- C. Botta, S. Benedini, L. Carlucci, A. Forni, D. Marinotto, A. Nitti, D. Pasini, S. Righetto and E. Cariati, J. Mater. Chem. C, 2016, 4, 2979–2989 RSC.
- A. Nitti, C. Botta, A. Forni, E. Cariati, E. Lucenti and D. Pasini, CrystEngComm, 2020, 22, 7782–7785 RSC.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- Y. Chen, J. W. Y. Lam, R. T. K. Kwok, B. Liu and B. Z. Tang, Mater. Horiz., 2019, 6, 428–433 RSC.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- H. Wang, Q. Li, P. Alam, H. Bai, V. Bhalla, M. R. Bryce, M. Cao, C. Chen, S. Chen, X. Chen, Y. Chen, Z. Chen, D. Dang, D. Ding, S. Ding, Y. Duo, M. Gao, W. He, X. He, X. Hong, Y. Hong, J.-J. Hu, R. Hu, X. Huang, T. D. James, X. Jiang, G. Konishi, R. T. K. Kwok, J. W. Y. Lam, C. Li, H. Li, K. Li, N. Li, W.-J. Li, Y. Li, X.-J. Liang, Y. Liang, B. Liu, G. Liu, X. Liu, X. Lou, X.-Y. Lou, L. Luo, P. R. McGonigal, Z.-W. Mao, G. Niu, T. C. Owyong, A. Pucci, J. Qian, A. Qin, Z. Qiu, A. L. Rogach, B. Situ, K. Tanaka, Y. Tang, B. Wang, D. Wang, J. Wang, W. Wang, W.-X. Wang, W.-J. Wang, X. Wang, Y.-F. Wang, S. Wu, Y. Wu, Y. Xiong, R. Xu, C. Yan, S. Yan, H.-B. Yang, L.-L. Yang, M. Yang, Y.-W. Yang, J. Yoon, S.-Q. Zang, J. Zhang, P. Zhang, T. Zhang, X. Zhang, X. Zhang, N. Zhao, Z. Zhao, J. Zheng, L. Zheng, Z. Zheng, M.-Q. Zhu, W.-H. Zhu, H. Zou and B. Z. Tang, ACS Nano, 2023, 17, 14347–14405 CrossRef CAS PubMed.
- Q. Xia, Y. Zhang, Y. Li, Y. Li, Y. Li, Z. Feng, X. Fan, J. Qian and H. Lin, Aggregate, 2022, 3, e152 CrossRef CAS.
- G. R. Suman, M. Pandey and A. S. J. Chakravarthy, Mater. Chem. Front., 2021, 5, 1541–1584 RSC.
- J. Li, Z. Feng, X. Yu, D. Wu, T. Wu and J. Qian, Coord. Chem. Rev., 2022, 472, 214792 CrossRef CAS.
- S. Song, Y. Wang, Y. Zhao, W. Huang, F. Zhang, S. Zhu, Q. Wu, S. Fu, B. Z. Tang and D. Wang, Matter, 2022, 5, 2847–2863 CrossRef CAS.
- Y. Li, Z. Cai, S. Liu, H. Zhang, S. T. H. Wong, J. W. Y. Lam, R. T. K. Kwok, J. Qian and B. Z. Tang, Nat. Commun., 2020, 11, 1255–1255 CrossRef CAS PubMed.
- J. L. Banal, J. M. White, K. P. Ghiggino and W. W. H. Wong, Sci. Rep., 2014, 4, 4635 CrossRef PubMed.
- J. L. Banal, J. M. White, T. W. Lam, A. W. Blakers, K. P. Ghiggino and W. W. H. Wong, Adv. Energy Mater., 2015, 5, 1500818 CrossRef.
- W. Ma, W. Li, M. Cao, R. Liu, X. Zhao and X. Gong, Org. Electron., 2019, 73, 226–230 CrossRef CAS.
- Z. Sun, S. Shi, P. Guan and B. Liu, Spectrochim. Acta, Part A, 2022, 272, 120946 CrossRef CAS PubMed.
- F. D. Nisi, R. Francischello, A. Battisti, A. Panniello, E. Fanizza, M. Striccoli, X. Gu, N. L. C. Leung, B. Z. Tang and A. Pucci, Mater. Chem. Front., 2017, 1, 1406–1412 RSC.
- R. Mori, G. Iasilli, M. Lessi, A. B. Muñoz-García, M. Pavone, F. Bellina and A. Pucci, Polym. Chem., 2018, 9, 1168–1177 RSC.
- C. Micheletti, Q. Wang, F. Ventura, M. Turelli, I. Ciofini, C. Adamo and A. Pucci, Aggregate, 2022, 3, e188 CrossRef CAS.
- T. A. Geervliet, I. Gavrila, G. Iasilli, F. Picchioni and A. Pucci, Chem. – Asian J., 2019, 14, 877–883 CrossRef CAS PubMed.
- K. Li, W. Qin, D. Ding, N. Tomczak, J. Geng, R. Liu, J. Liu, X. Zhang, H. Liu, B. Liu and B. Z. Tang, Sci. Rep., 2013, 3, 1150 CrossRef PubMed.
- M. Bartolini, C. Micheletti, A. Picchi, C. Coppola, A. Sinicropi, M. Di Donato, P. Foggi, A. Mordini, G. Reginato, A. Pucci, L. Zani and M. Calamante, ACS Appl. Energy Mater., 2023, 6, 4862–4880 CrossRef CAS PubMed.
- J. L. Banal, K. P. Ghiggino and W. W. H. Wong, Phys. Chem. Chem. Phys., 2014, 16, 25358–25363 RSC.
- B. Zhang, J. L. Banal, D. J. Jones, B. Z. Tang, K. P. Ghiggino and W. W. H. Wong, Mater. Chem. Front., 2018, 2, 615–619 RSC.
- F. Mateen, T. G. Hwang, L. F. Boesel, W. J. Choi, J. P. Kim, X. Gong, J. M. Park and S. K. Hong, Int. J. Energy Res., 2021, 45, 17971–17981 CrossRef CAS.
- G. Lyu, J. Kendall, I. Meazzini, E. Preis, S. Bayseç, U. Scherf, S. Clément and R. C. Evans, ACS Appl. Polym. Mater., 2019, 1, 3039–3047 CrossRef CAS PubMed.
- F. Corsini, A. Nitti, E. Tatsi, G. Mattioli, C. Botta, D. Pasini and G. Griffini, Adv. Opt. Mater., 2021, 9, 2100182 CrossRef CAS.
- P. Meti, F. Mateen, D. Y. Hwang, Y. E. Lee, S. K. Hong and Y. D. Gong, Dyes Pigm., 2022, 202, 110221 CrossRef CAS.
- Q. Wan, W. Dai, Y. Xie, Q. Ke, C. Zhao, B. Zhang, Z. Zeng, Z. Wang and B. Z. Tang, Chem. Eng. J., 2023, 451, 138529 CrossRef CAS.
- X. Cheng, K. Wang, S. Huang, H. Zhang, H. Zhang and Y. Wang, Angew. Chem., Int. Ed., 2015, 54, 8369–8373 CrossRef CAS PubMed.
- J. Xue, Q. Liang, R. Wang, J. Hou, W. Li, Q. Peng, Z. Shuai and J. Qiao, Adv. Mater., 2019, 31, 1808242–1808242 CrossRef PubMed.
- M.-P. Zhuo, X.-D. Wang and L.-S. Liao, Small Sci., 2022, 2, 2200029 CrossRef CAS.
- Y. R. Poh, S. Pannir-Sivajothi and J. Yuen-Zhou, J. Phys. Chem. C, 2023, 127, 5491–5501 CrossRef CAS.
- W. Siebrand, J. Chem. Phys., 2004, 46, 440–447 CrossRef.
- R. Englman and J. Jortner, Mol. Phys., 1970, 18, 145–164 CrossRef CAS.
- Y.-C. Wei, S. F. Wang, Y. Hu, L.-S. Liao, D.-G. Chen, K.-H. Chang, C.-W. Wang, S.-H. Liu, W.-H. Chan, J.-L. Liao, W.-Y. Hung, T.-H. Wang, P.-T. Chen, H.-F. Hsu, Y. Chi and P.-T. Chou, Nat. Photonics, 2020, 14, 570–577 CrossRef CAS.
- W.-C. Chen, P.-T. Chou and Y.-C. Cheng, J. Phys. Chem. C, 2019, 123, 10225–10236 CrossRef CAS.
- Q. Wu, C. Deng, Q. Peng, Y. Niu and Z. Shuai, J. Comput. Chem., 2012, 33, 1862–1869 CrossRef CAS PubMed.
- C. Deng, Y. Niu, Q. Peng, A. Qin, Z. Shuai and B. Z. Tang, J. Chem. Phys., 2011, 135, 014304 CrossRef PubMed.
- J. Jiang, H. Sun, Y. Hu, G. Lu, J. Cui and J. Hao, Chem. Commun., 2021, 57, 7685–7688 RSC.
- R. Li, L. Yan, Z. Wang and Z. Qi, J. Mol. Struct., 2017, 1136, 1–6 CrossRef CAS.
- X. Jin, L. Dong, X. Di, H. Huang, J. Liu, X. Sun, X. Zhang and H. Zhu, RSC Adv., 2015, 5, 87306–87310 RSC.
- W. Yang, C. Liu, Q. Gao, J. Du, P. Shen, Y. Liu and C. Yang, Opt. Mater., 2017, 66, 623–629 CrossRef CAS.
- Y. Cai, Z. Wei, C. Song, C. Tang, W. Han and X. Dong, Chem. Soc. Rev., 2019, 48, 22–37 RSC.
- J. Li and K. Pu, Chem. Soc. Rev., 2019, 48, 38–71 RSC.
- S. Wang, K. Zhou, X. Lyu, H. Li, Z. Qiu, Z. Zhao and B. Z. Tang, Chem. Biomed. Imaging, 2023, 1, 509–521 CrossRef CAS.
- P. Wang, Y. Fan, L. Lu, L. Liu, L. Fan, M. Zhao, Y. Xie, C. Xu and F. Zhang, Nat. Commun., 2018, 9, 2898 CrossRef PubMed.
- Z. Ma, F. Wang, W. Wang, Y. Zhong and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2021446118 CrossRef CAS PubMed.
- G. Deng, S. Li, Z. Sun, W. Li, L. Zhou, J. Zhang, P. Gong and L. Cai, Theranostics, 2018, 8, 4116–4128 CrossRef CAS PubMed.
- Y. Li, Z. Cai, S. Liu, H. Zhang, S. T. H. Wong, J. W. Y. Lam, R. T. K. Kwok, J. Qian and B. Z. Tang, Nat. Commun., 2020, 11, 1255 CrossRef CAS PubMed.
- W. Zhu, M. Kang, Q. Wu, Z. Zhang, Y. Wu, C. Li, K. Li, L. Wang, D. Wang and B. Z. Tang, Adv. Funct. Mater., 2021, 31, 2007026 CrossRef CAS.
- Y. Lin, M. Yi, X. Guan, E. Chen, L. Yang, S. Li, Y. Li and L. Zhang, J. Nanobiotechnol., 2023, 21, 49 CrossRef CAS PubMed.
- Z. Zheng, D. Li, Z. Liu, H.-Q. Peng, H. H. Y. Sung, R. T. K. Kwok, I. D. Williams, J. W. Y. Lam, J. Qian and B. Z. Tang, Adv. Mater., 2019, 31, 1904799 CrossRef CAS PubMed.
- W. Xiong, C. Zhang, Y. Fang, M. Peng and W. Sun, Polymers, 2023, 15, 98 CrossRef CAS PubMed.
- L. Tu, Y. Xie, Z. Li and B. Tang, SmartMat, 2021, 2, 326–346 CrossRef CAS.
- Y. Zhao, L. Zhang, Y. Liu, Z. Deng, R. Zhang, S. Zhang, W. He, Z. Qiu, Z. Zhao and B. Z. Tang, Langmuir, 2022, 38, 8881–8889 CrossRef PubMed.
- J. Xue, J. Xu, J. Ren, Q. Liang, Q. Ou, R. Wang, Z. Shuai and J. Qiao, Sci. China: Chem., 2021, 64, 1786–1795 CrossRef CAS.
- C. Erker and T. Basché, J. Am. Chem. Soc., 2022, 144, 14053–14056 CrossRef CAS PubMed.
- J. Guo, Z. Zhao and B. Z. Tang, Adv. Opt. Mater., 2018, 6, 1800264 CrossRef.
- C. A. Parker and C. G. Hatchard, Trans. Faraday Soc., 1961, 57, 1894–1904 RSC.
- M. N. Berberan-Santos and J. M. M. Garcia, J. Am. Chem. Soc., 1996, 118, 9391–9394 CrossRef CAS.
- A. Endo, M. Ogasawara, A. Takahashi, D. Yokoyama, Y. Kato and C. Adachi, Adv. Mater., 2009, 21, 4802–4806 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- S. Tang, J. M. dos Santos, J. Ràfols-Ribé, J. Wang, E. Zysman-Colman and L. Edman, Adv. Funct. Mater., 2023, 33, 2306170 CrossRef CAS.
- G. Hong, C. Si, A. K. Gupta, C. Bizzarri, M. Nieger, I. D. W. Samuel, E. Zysman-Colman and S. Bräse, J. Mater. Chem. C, 2022, 10, 4757–4766 RSC.
- X. Chen, S. Bagnich, R. Pollice, B. Li, Y. Zhu, R. Saxena, Y. Yin, W. Zhu, A. Aspuru-Guzik, E. Zysman-Colman, A. Köhler and Y. Wang, Adv. Opt. Mater., 2024, 12, 2301784 CrossRef CAS.
- G. Preda, A. Aricò, C. Botta, D. Ravelli, D. Merli, S. Mattiello, L. Beverina and D. Pasini, Org. Lett., 2023, 25, 6490–6494 CrossRef CAS PubMed.
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707–3714 CrossRef CAS PubMed.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed.
- D. G. Congrave, B. H. Drummond, Q. Gu, S. Montanaro, H. Francis, V. Riesgo-González, W. Zeng, C. S. B. Matthews, S. Dowland, I. A. Wright, C. P. Grey, R. H. Friend and H. Bronstein, J. Mater. Chem. C, 2022, 10, 4831–4836 RSC.
- Y. Yuan, Y. Hu, Y.-X. Zhang, J.-D. Lin, Y.-K. Wang, Z.-Q. Jiang, L.-S. Liao and S.-T. Lee, Adv. Funct. Mater., 2017, 27, 1700986 CrossRef.
- D. G. Congrave, B. H. Drummond, P. J. Conaghan, H. Francis, S. T. E. Jones, C. P. Grey, N. C. Greenham, D. Credgington and H. Bronstein, J. Am. Chem. Soc., 2019, 141, 18390–18394 CrossRef CAS PubMed.
- Z. Cai, X. Wu, H. Liu, J. Guo, D. Yang, D. Ma, Z. Zhao and B. Z. Tang, Angew. Chem., Int. Ed., 2021, 60, 23635–23640 CrossRef CAS PubMed.
- H. Wang, J.-X. Chen, L. Zhou, X. Zhang, J. Yu, K. Wang and X.-H. Zhang, Mater. Horiz., 2023, 10, 2997–3004 RSC.
- M. Zhao, M. Li, W. Li, S. Du, Z. Chen, M. Luo, Y. Qiu, X. Lu, S. Yang, Z. Wang, J. Zhang, S.-J. Su and Z. Ge, Angew. Chem., Int. Ed., 2022, 61, e202210687 CrossRef CAS PubMed.
- Y. Yu, H. Xing, D. Liu, M. Zhao, H. H.-Y. Sung, I. D. Williams, J. W. Y. Lam, G. Xie, Z. Zhao and B. Z. Tang, Angew. Chem., Int. Ed., 2022, 61, e202204279 CrossRef CAS PubMed.
- S. Wang, H. Li, Z. Song, H. Jiang, X. Zhang, C.-S. Tsang, Q. Liu, L. Y. S. Lee, D. Ma and W.-Y. Wong, J. Mater. Chem. C, 2023, 11, 8196–8203 RSC.
- H. Zhang, J. Xue, C. Li, S. Zhang, B. Yang, Y. Liu and Y. Wang, Adv. Funct. Mater., 2021, 31, 2100704 CrossRef CAS.
- A. Cesaretti, T. Bianconi, M. Coccimiglio, N. Montegiove, Y. Rout, P. L. Gentili, R. Misra and B. Carlotti, J. Phys. Chem. C, 2022, 126, 10429–10440 CrossRef CAS.
- A. Weller, Naturwissenschaften, 1955, 42, 175–176 CrossRef CAS.
- A. C. Sedgwick, L. Wu, H.-H. Han, S. D. Bull, X.-P. He, T. D. James, J. L. Sessler, B. Z. Tang, H. Tian and J. Yoon, Chem. Soc. Rev., 2018, 47, 8842–8880 RSC.
- J.-J. Wu, H. Gao, R. Lai, M.-P. Zhuo, J. Feng, X.-D. Wang, Y. Wu, L.-S. Liao and L. Jiang, Matter, 2020, 2, 1233–1243 CrossRef.
- X. Wang, Q. Liao, H. Li, S. Bai, Y. Wu, X. Lu, H. Hu, Q. Shi and H. Fu, J. Am. Chem. Soc., 2015, 137, 9289–9295 CrossRef CAS PubMed.
- J.-J. Wu, M.-P. Zhuo, R. Lai, S.-N. Zou, C.-C. Yan, Y. Yuan, S.-Y. Yang, G.-Q. Wei, X.-D. Wang and L.-S. Liao, Angew. Chem., Int. Ed., 2021, 60, 9114–9119 CrossRef CAS PubMed.
- B. K. Dwivedi, V. D. Singh, R. P. Paitandi and D. S. Pandey, ChemPhysChem, 2018, 19, 2672–2682 CrossRef CAS PubMed.
- Q. Yu, S. Chen, C. Han, H. Guo and F. Yang, J. Lumin., 2020, 220, 117017 CrossRef CAS.
- C. Han, S. Jiang, J. Qiu, H. Guo and F. Yang, New J. Chem., 2019, 43, 3317–3322 RSC.
- S. Hadke, M. Huang, C. Chen, Y. F. Tay, S. Chen, J. Tang and L. Wong, Chem. Rev., 2022, 122, 10170–10265 CrossRef CAS PubMed.
- D.-H. Lim, J.-W. Ha, H. Choi, S. C. Yoon, B. R. Lee and S.-J. Ko, Nanoscale Adv., 2021, 3, 4306–4320 RSC.
- Y. Bai, K. Lang, C. Zhao, Q. Guo, R. Zeng, J. Wang, T. Hayat, A. Alsaedi and Z. Tan, Sol. RRL, 2020, 4, 1900280 CrossRef CAS.
Footnote |
| † Equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |