
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuercetin-loaded nanoarchaeosomes for breast cancer therapy: a ROS mediated cell death mechanism†
Subastri
Ariraman
,
Abirami
Seetharaman
,
Kaviya Vijayalakshmi
Babunagappan
and
Swathi
Sudhakar
 *
*
Department of Applied Mechanics and Biomedical Engineering, Indian Institute of Technology Madras, Chennai, India. E-mail: swathi.s@iitm.ac.in
First published on 5th August 2024
Abstract
Breast cancer is the deadliest disease among women due to the lack of novel targeted therapies. Flavonoid compounds like quercetin exhibit excellent anticancer activity. However, quercetin's high hydrophobicity, poor solubility, and low bioavailability limit its therapeutic efficacy. Several systems, like polymeric, metallic, and liposomal nanomaterials, have been designed to enhance quercetin's therapeutic efficacy. However, these nanocarrier systems lack colloidal stability and biocompatibility and have poor drug-loading efficiency. Herein, we have synthesized thermostable quercetin-loaded nanoarchaeosomes (NAQ) for effective breast cancer treatment. The synthesized NAQ have a size of 53.5 ± 1 nm. The drug loading efficiency was 99 ± 0.2%, and sustained drug release kinetics was observed. Furthermore, to determine the in vitro anticancer potential of NAQ, we have used the breast cancer cell line MCF-7 as a model. The NAQ induced significant cell death in breast cancer cells by generating reactive oxygen species (ROS), resulting in an IC50 value of 2.5 μM. The observed IC50 value was thirty-five-fold less than the IC50 value of quercetin alone treated MCF-7 cells. The flow cytometry and cell cycle analyses confirmed that NAQ induced necroptosis with cell cycle arrest at the G0/G1 phase. The NAs and NAQ exhibit excellent biocompatibility in normal fibroblast cells NIH 3T3. Our findings suggest that NAQ have good biocompatibility, stability, and drug loading and release kinetics and can act as an effective alternative anticancer nanotherapeutic agent against breast cancer by minimizing the side effects.
1. Introduction
Chemotherapy is one of the most common treatments for breast cancer.1 However, it has severe limitations due to adverse side effects like neutropenia, alopecia, and hypersensitivity reactions. So, researchers are urged to develop novel alternative strategies to control breast cancer. Quercetin, the most common flavonoid, possesses many biological activities such as anticancer, anti-inflammatory, and antioxidant activities.2 Furthermore, several studies have proven that quercetin exhibited an antitumor effect in breast cancer.3 However, the quercetin drug molecules have severe limitations because of their low bioavailability, instability under physiological conditions, and poor solubility in aqueous solutions like human serum.4,5 Therefore, higher quercetin concentrations and frequent drug administration are required for cancer treatment, which enhances the treatment cost and adverse effects like nephrotoxicity.6 Therefore, effective quercetin delivery at the target site is a challenging process. Subsequently, a drug delivery system with polymeric, metallic, and organic nanomaterials has recently been used to overcome these effects.7 Specifically, liposomes have been used to improve quercetin delivery8,9 due to their biocompatible nature compared to other nanomaterials. However, liposomes lack colloidal stability, which leads to uncontrolled drug release at the nontarget site and decreases the therapeutic effect.10,11 Hence, in our present study, we have synthesized a highly stable novel drug delivery system, nanoarchaeosomes (NAs), for effective quercetin delivery for cancer therapy using highly thermostable archaeal lipids. Archaeal lipids consist of highly branched isoprenoid side links and ether bonds linked to the glycerol-1-phosphate backbone. Because of the ether bonds, they have better stability at extreme pH and high temperatures (40 °C to 100 °C).12,13 Furthermore, archaeal lipids exhibit excellent bioavailability and controlled drug release mechanisms at the target sites.14 Based on these significant features, we have synthesized NAs constituted of archaeal lipids to develop an effective quercetin delivery system. The quercetin-loaded NAs (NAQ) have been explored by in vitro anti-breast cancer studies on the MCF-7 breast cancer cell line. The synthesized NAs revealed high biocompatibility in normal cells. NAQ showed high drug loading efficiency and prolonged drug release over 24 hours. Furthermore, NAQ exhibited in vitro cytotoxicity against the MCF-7 breast cancer cell line with an IC50 value of 2.5 μM. FACS results also proved that NAQ induced necrosis and late apoptosis with cell cycle arrest at the G0/G1 phase. This study sheds light on utilizing nanoarchaeosomes as novel drug delivery vehicles, and further investigation in this area can be aimed at realizing their full potential for cancer therapeutic applications.2. Experimental section
2.1. Materials
Quercetin, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), 1-stearoyl-2-oleoyl-sn-glycero-3-phosphocholine (SOPC), archaeal lipids, propidium iodide and ethidium bromide (EtBr) were purchased from Sigma-Aldrich. Acridine orange (AO) was obtained from Nalgen. Phalloidin and 2′-7′ dichlorodihydrofluorescein diacetate (DCFDA) were acquired from Invitrogen and Abcam. Annexin-V APC was obtained from Biolegend. Hoechst was procured from Thermo Fisher.2.2. Preparation of nanoarchaeosomes (NAs)
NAs were synthesized as described previously by Kaviya et al..15 Briefly, SOPC and archaeal lipids were dissolved in a ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 in chloroform solvent to reach a final concentration of 1 mg ml−1 and then subjected to vacuum drying. Then, the dried lipid was reconstituted with Milli-Q water and vortexed for 5 minutes. Next, the prepared sample was sonicated at 45 °C for 30 minutes using an ultrasonic bath sonicator (B.R. Biochem Life Sciences). Then, the sample mixture was centrifuged (Eppendorf Centrifuge 5415C) at 13000 rpm for 30 minutes.
:20 in chloroform solvent to reach a final concentration of 1 mg ml−1 and then subjected to vacuum drying. Then, the dried lipid was reconstituted with Milli-Q water and vortexed for 5 minutes. Next, the prepared sample was sonicated at 45 °C for 30 minutes using an ultrasonic bath sonicator (B.R. Biochem Life Sciences). Then, the sample mixture was centrifuged (Eppendorf Centrifuge 5415C) at 13000 rpm for 30 minutes.
2.3. Dynamic light scattering (DLS) and zeta potential
The size and surface charge of synthesized NAs were evaluated using a DLS (Horiba Scientific DLS) and zeta analyzer (Horiba Scientific Zeta Analyzer). NAs were diluted in a 1:100 ratio. This dispersed suspension was taken for analysis, and the hydrodynamic diameter and surface charge of NAs were measured.
2.4. SEM, TEM, EDX, AFM, and Raman spectroscopy analyses
The morphological examination and elemental analysis of the NAs were performed using a scanning electron microscope (S-4800, Hitachi) and transmission electron microscope (JEOL JEM 3010, filament-LaB6). For SEM analysis, NAs were drop-cast onto a pre-cleaned glass slide, and the sample was allowed to dry at room temperature, followed by gold sputter coating at 2.5 kV and 20 mA at a rate of 10 nm per minute (Polaran SC7640 gold sputter). Then, morphological characterization was performed using a scanning electron microscope operated at the voltage of 1–5 kV. Following this, 10 μL of NAs was spotted on the TEM copper grids, dried at room temperature overnight, and the image was acquired using a transmission electron microscope. Furthermore, elemental analysis of NAs was performed using the energy dispersive X-ray (EDX) method. Subsequently, atomic force microscopy (Bruker, OTESPA-R3) measurements were carried out for NAs. In this study, NA suspension was placed on a glass slide (1 × 2 cm) and air-dried overnight to form a thin film over the glass slide. The dried film was scanned using an atomic force microscope (AFM) in tapping mode. Finally, images were analyzed using Nano analyzer 3.0 software. Furthermore, confocal Raman spectroscopy (WITec alpha 300R) was performed for the physical characterization of NAs. For this study, the NA suspension was drop-cast onto a pre-cleaned glass slide, and the sample was allowed to air dry for 3 hours at room temperature. Then, the Raman spectrum was collected in the range of 100 cm−1 to 3500 cm−1.2.5. Drug loading efficiency
NAQ were prepared by adding equal amounts of NAs (1 mg ml−1) and quercetin (Q) (100 μM ml−1) kept at 300 rpm for 12 hours in a ThermoMixer (Eppendorf). After incubation, NAQ were subjected to the freeze–thaw method as described in a previous study.12 Briefly, the sample was centrifuged at 13000 rpm for 30 minutes. Then, the supernatant was collected and quantified using a UV-visible spectrophotometer (Nanodrop one, Thermo Scientific) by measuring its absorbance at 375 nm. Furthermore, high-performance liquid chromatography (HPLC) (Shimadzu LC-2050C) analysis was also performed to accurately determine the free drug concentration. The HPLC analysis was carried out by injecting 25 μL of the sample in the C18 column at a 1 ml minute−1 flow rate and a column temperature of 37 °C. Methanol and water (HPLC grade) in the ratio of 60:40 (v/v) were used as a mobile phase, and a detection wavelength of 370 nm was used with a total run time of 16 minutes per injection. Data were obtained and processed using LC solution software. The standard curves were plotted using different concentrations of free quercetin (20, 40, 60, 80, and 100 μM) under the same chromatographic conditions as mentioned above. Free drug concentration was quantified from the analysis, and the drug loading efficiency (DLE%) was determined using the following formula.After drug loading, NAQ were characterized using SEM, TEM, zeta potential, AFM, and Raman spectroscopy, as described above.

2.6. Fourier transform infrared (FTIR) spectroscopy
To confirm the entrapment of quercetin on NAs, Fourier transform infrared (FTIR) spectroscopy was performed (Nicolet iS5 FT-IR). The sample pellets were prepared using KBr in a ratio of 1:100 (% w/w) using a pressure mold (0.5–1 Pa). The FTIR spectra were collected in the transmission wavelength ranges of 500–4000 cm−1.
2.7. In vitro drug release at different pH values
The drug release kinetics of NAQ was determined by the dialysis membrane method.14 In brief, the NAQ filled dialysis membrane (12–14 kDa, Himedia) was submerged in a small glass bottle containing 1× phosphate buffered saline (PBS) at different pH values (7.4 and 4.5) with continuous stirring at 150 rpm for 24 hours. Finally, samples were collected at different time intervals of 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12 and 24 hours and the samples were analyzed using the high-performance liquid chromatography (Shimadzu LC-2050C) technique.2.8. In vitro cell culture studies
000 units of penicillin and 10 mg of streptomycin in 0.9% NaCl) (Himedia) in a humidified atmosphere of 5% CO2 at 37 °C.
Furthermore, morphological changes of cells were examined under an inverted phase contrast microscope (Magnus Invi).

2.9. Stability of NAQ
To evaluate the stability of NAQ, we have used different physiological media such as Dulbecco's Modified Eagle's Medium (DMEM), 10% FBS (fetal bovine serum), 50% FBS, 1× PBS (phosphate buffered saline) pH 7.4, and 1× PBS pH 4 with an incubation period of 72 hours. Furthermore, the stability of solutions was assessed by using a DLS (Horiba Scientific DLS) and zeta analyzer (Horiba Scientific zeta analyzer).2.10. Hemolysis assay
To assess the blood compatibility of nanoformulation, the hemolytic assay was performed using different concentrations of NAs and NAQ using the method reported by Preedia et al.17 with some modifications. For this study, goat blood was freshly collected from a slaughter shop near the Indian Institute of Technology Madras (IITM), Chennai. The blood sample was centrifuged at 2500 rpm for 5 min, and RBC was separated from plasma and washed with 1× PBS two times. The purified RBCs were suspended in 1× PBS in a ratio of 1:9. Different concentrations of NAs (0.01 and 0.02 mg mL−1) and NAQ (10, 50, and 100 μM) were added to 0.2 ml of diluted RBC (1 × 106/mL) and made up to 1 ml with 1× PBS. Distilled water and 1× PBS were used as a positive and negative control, respectively. Samples were mixed gently and incubated at 37 °C for 4 h. After incubation, the mixture was centrifuged at 3000 rpm for 3 min, 100 μl of the supernatant was collected in 96 well plates, and absorbance was read at 570 nm using a microplate reader (Biotek H1m synergy). The hemolysis percent of RBCs was calculated using the following formula,(S.A. – sample absorbance; NCA – negative control absorbance; and PCA – positive control absorbance).

2.11. Cytoskeleton analysis by phalloidin–Hoechst staining
To understand the effect of NAQ on F-actin cytoskeleton derangement, MCF-7 and NIH 3T3 cells were seeded onto the 6-well plate at a density of 2 × 104 cells per mL and incubated for 24 hours. After incubation, both the cells were treated with NAs (0.05 mg) and NAQ (2.5 μM) (selected based on the IC50 concentration) and kept overnight. After the treatment, the cells were fixed with 4% paraformaldehyde (PFA) and 0.5% Triton-X for 15 minutes. After fixation, the cells were washed with 1× PBS buffer. Following this, the cells were stained with 50 μl of phalloidin and incubated for 1 hour. Then 10 μl of Hoechst (10 μg ml−1) nuclei counter stain was added into the cells and images were taken under a fluorescence microscope (Nikon Inverted Ti U Eclipse), and a merged image profile of phalloidin and Hoechst-stained cells was analyzed using ImageJ software.2.12. Determination of the intracellular ROS level
Next, intracellular ROS (Reactive Oxygen Species) levels were determined using the DCFDA staining procedure.18 Oxidation of DCFDA through ROS converts it to 2′–7′- dichlorofluorescein (DCF), which emits green fluorescence at 530 nm. Briefly, MCF-7 cells were treated with NAQ (2.5 μM) and then kept for 24 hours at 37 °C followed by incubation with 5 μL of DCFDA (20 μM) for 30 minutes in the dark. After incubation, the cells were washed with 1× PBS twice and imaged under a fluorescence microscope (Nikon Inverted Ti U Eclipse) by capturing emission at 530 nm with a magnification of 20×. Furthermore, fluorescence intensity was evaluated using ImageJ software.2.13. Cell morphological evaluation by the acridine orange (AO)/ethidium bromide (EtBr) dual staining method
The AO/EtBr dual staining method was performed based on the protocol of Gohel et al..19 Using this method, the cells undergoing apoptosis are differentiated from the viable cells through the morphological variation of apoptotic nuclei. AO and EtBr are DNA intercalating dyes. AO will be taken up by both viable and non-viable cells and stains double-stranded and single-stranded DNA, emitting green fluorescence at an excitation wavelength of 490 nm. EtBr is taken up by non-viable cells and emits red fluorescence. Furthermore, AO/EtBr-stained cells were categorized as viable (green fluorescence), early apoptotic (bright green fluorescence with condensed chromatin), late apoptotic (orange/yellow fluorescence) and non-viable cells (red fluorescence).20 Briefly, MCF-7 and NIH 3T3 cells were seeded in 6 well plates at a density of 2 × 104 cells per mL and treated with NAs alone (0.05) and NAQ (2.5 μM) (selected based on the IC50 concentration) and incubated for 24 hours. After incubation, the existing medium was aspirated, and the cells were gently washed with 1× PBS (pH 7.4) and equal volumes of fluorescent dyes AO/EtBr (100 μg mL−1) were added into the cells and incubated for 30 min at 37 °C. Subsequently, the stained cells were washed with 1× PBS and immediately the cells were viewed under a fluorescence microscope (Nikon Inverted Ti U Eclipse) at 20× magnification. Furthermore, fluorescence intensity was quantified using ImageJ software.2.14. Fluorescence-activated cell sorting (FACS) analysis
The FACS analysis of NAQ in MCF-7 was carried out using flow cytometry according to the manufacturer's instructions. Apoptosis is the mechanism of programmed cell death, which is a common mechanism for the elimination of unwanted cells from the immune system. During apoptosis, phosphatidylserine will be released through membrane blebbing (loss of membrane phospholipid asymmetry) from the inner plasma membrane to the outer membrane leaflet.21 So, phosphatidylserine expression at the cell surface plays a vital role in recognizing and removing apoptotic cells. Thus, apoptotic cells will be quantified by flow cytometry using the binding of allophycocyanin-labeled Annexin to phosphatidylserines. For this FACS analysis, MCF-7 cells were seeded in 6 well plates at a density of 1 × 105 cells per well and incubated for 24 hours, and the incubated cells were treated with 2.5 μM NAQ for 24 hours. Then, non-adhered and adhered cells were collected through centrifugation at 1500 rpm for 5 min. Then, the collected cells were washed with cold 1× PBS twice and stained with 2 μl of Annexin V-APC (allophycocyanin 4 μg ml−1) and 2 μl of propidium iodide (PI) (1 mg mL−1) in 1× binding buffer (10 mM HEPES, 140 mM sodium chloride and 2.5 mM calcium chloride) for 15 min at room temperature in the dark. Initially, unstained control samples and single control samples (Annexin and propidium iodide-stained cells) were analyzed for fluorochrome compensation. Subsequently, apoptotic and necrotic cells were quantified by flow cytometry (Beckman Coulter), and the results were analyzed using CytExpert software version 2.4.2.15. Cell cycle analysis
The diverse phases of the cell cycle were analyzed using a Beckman Coulter flow cytometer equipped with CytExpert software version 2.4. In this analysis, MCF-7 breast cancer cells were treated with NAQ (2.5 μM) and incubated for 24 hours. Subsequently, the cells were harvested through centrifugation and washed with ice-cold 1× PBS. Then, the cells were fixed with 70% cold ethanol at −20 °C for at least 2 hours. Then, the cells were rinsed with 1× PBS by centrifuging at 1500 rpm for 5 min. Finally, the cells were stained with propidium iodide (12 μg ml−1) and ribonuclease A (RNAase A) (10 μg ml−1) in 0.1% Triton X buffer at 4 °C for 15 minutes before the experiment using flow cytometry.2.16. Statistical analysis
All data are expressed as mean ± S.D. SPSS software was used for the statistical valuation of data. Statistical analysis was executed with one-way ANOVA. P < 0.05 was considered statistically significant.3. Results and discussion
3.1. Preparation and characterization of NAs
NAs were prepared according to the protocol mentioned in the Experimental section. Furthermore, size and charge characterization studies were carried out using DLS, SEM, TEM, and zeta potential analyses. From the DLS analysis, we observed that the size of the NAs was around 53.5 ± 0.9 nm (Fig. 1A). The SEM and TEM images of NAs showed a spherical shape with a size of 50 ± 2 nm (Fig. 1B and C), which was in agreement with DLS results. The zeta potential was then analyzed to determine the net surface charge of the NAs. The surface charge of the NAs showed −55 ± 1 mV (Fig. S1, ESI†). A negative zeta value indicates the NAs can exhibit high colloidal stability and less aggregation due to increased electrostatic repulsion between the colloids. Several studies stated that nanomaterials with extreme surface charges (zeta potential ranges higher than 30 mV or less than −30 mV) are highly stable and highly dispersed in the aqueous solution.22–26 Previous studies reported that nanomaterials with a size less than 100 nm have a higher cellular uptake rate through endocytosis, receptor-mediated endocytosis, and passive diffusion.27,28 Therefore, we speculate that NAs with a size less than 100 nm accumulate at target sites with increased half-life in the blood.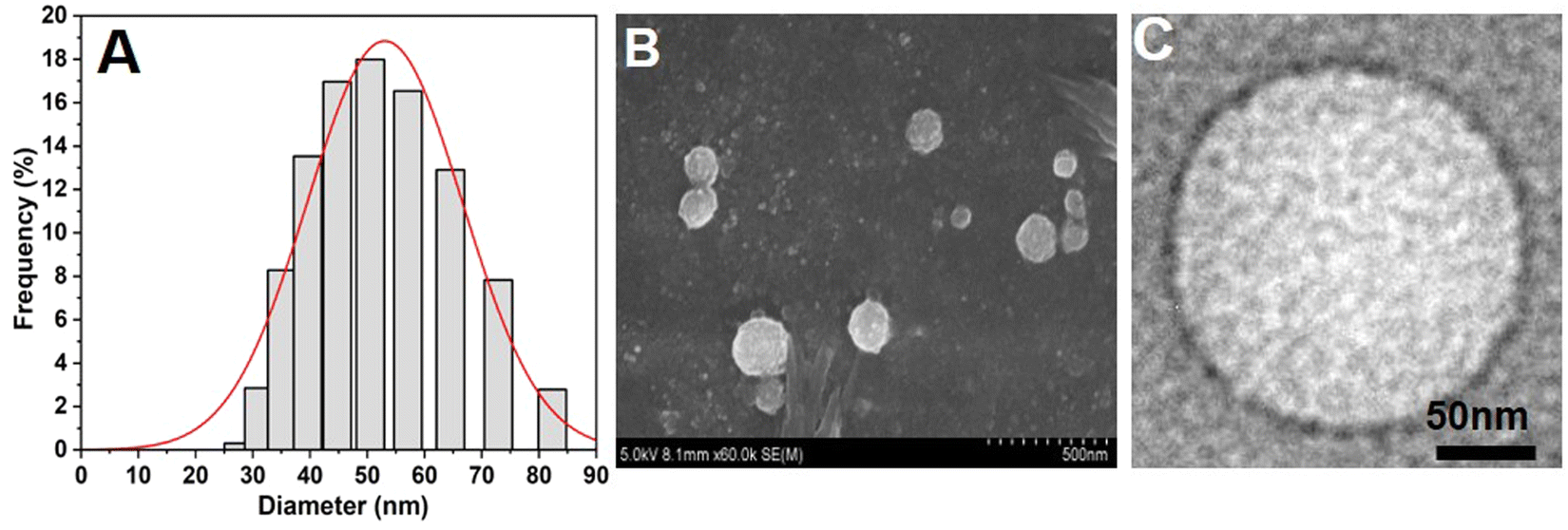 | ||
| Fig. 1 Physicochemical characterization of nanoarchaeosomes (NAs). (A) Average hydrodynamic size of NAs measured by the DLS method. (B) SEM image of NAs. (C) TEM image of NAs. | ||
3.2. Drug encapsulation efficiency
After the size and stability characterization, quercetin was loaded into NAs using the freeze–thaw method as described previously. The quercetin loading efficiency of NAs was found to be 99 ± 0.2%, which was confirmed by the HPLC technique. Furthermore, TEM and SEM (Fig. 2A and B) were performed for NAQ. There were no significant changes in the size and shape of NAQ compared to NAs. The TEM and SEM images of NAQ showed a smooth spherical shape with a size of 50 ± 3 nm (Fig. 2B). Next, the zeta potentials of NAs, quercetin, and NAQ were measured and found to be −55 ± 1 mV, +4.66 mV, and −42.1 mV (Fig. S1, ESI†). We observed that there was a significant reduction in the surface charge of NAQ compared to that of NAs, confirming that quercetin was effectively loaded into the NAs. The high drug encapsulation efficiency was achieved by the freeze–thaw method, where the ice crystals formed during freezing may induce temporary pores within the NAs, which may lead to passive diffusion and encapsulation of quercetin inside NAs. Furthermore, favorable interaction with both the acyl group and the head group of the phospholipid and hydrophobic–hydrophobic interaction between quercetin and NAs might contribute to higher encapsulation efficiency.29 Next, the three-dimensional surface contour of NAQ (100 μM) was analyzed by AFM (Fig. S2, ESI†). The results implied that the shape of the NAs (1 mg ml−1) (Fig. S2A and B, ESI†) and NAQ (Fig. S2C and D, ESI†) was spherical, with height distributions of around 86.2 nm and 56.8 nm, respectively. These results indicate that NAs are highly stable after being loaded with quercetin. The NAQ and NA diameter size may be higher than the size recorded using DLS. This could be attributed to flattening of vesicles and their interaction with the substrate surface.30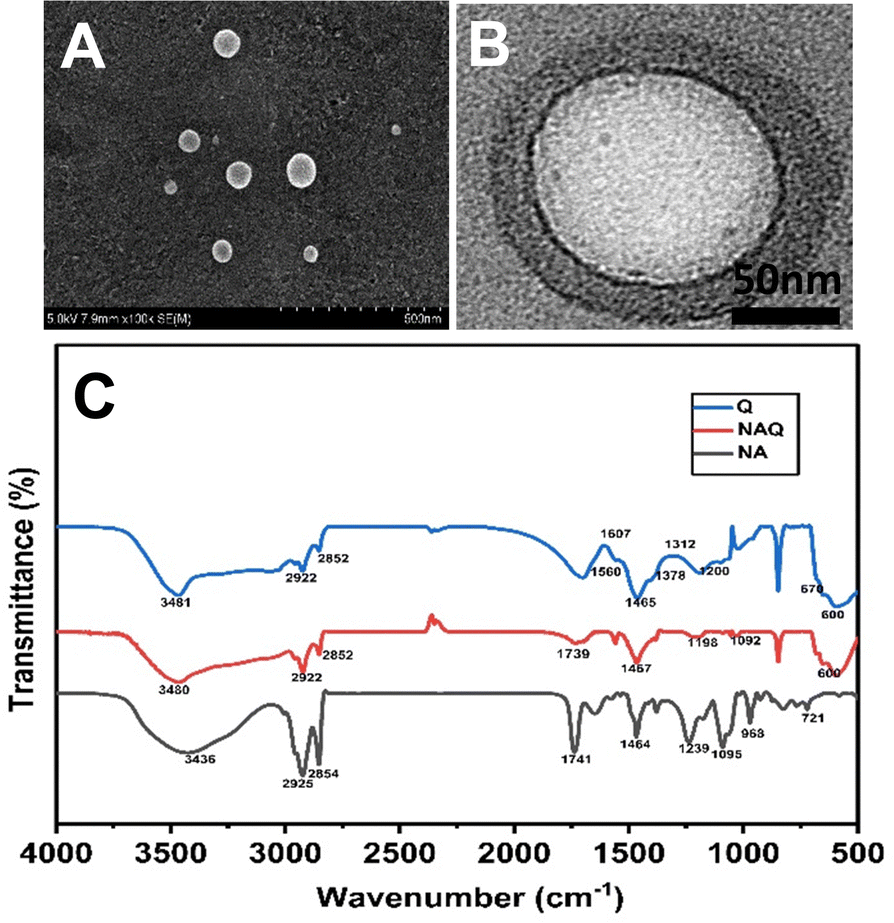 | ||
| Fig. 2 (A) TEM image of NAs. (B) SEM image of NAs. (C) FTIR spectra of NAs, quercetin loaded nanoarchaeosomes (NAQ) and quercetin alone at 25 °C. | ||
In addition, Raman spectroscopic analysis was performed to evaluate further quercetin-loaded NAs. In this study, the representative Raman spectra of NAs and NAQ are presented in Fig. S3 (ESI†). The characteristic peaks of NAs were observed at 567 cm−1, 1098 cm−1, and 2897 cm−1 (Fig. S3A, ESI†). The absorption at 1098 cm−1 is attributed to C–C stretching and that at 2894 cm−1 corresponds to anti-symmetric CH2 stretching vibration.
Next, the characteristic peak of NAQ was analyzed (Fig. S3B, ESI†). NAQ showed OH phenolic bending at 1454 cm−1, which represents the lipids and flavonoid interaction,31 confirming the quercetin loading into NAs.
3.3. Fourier transform infrared spectral analysis of NAQ
Next, the FTIR analysis was performed to investigate the molecular interactions between quercetin and NAs (Fig. 2C). The FTIR spectrum of NAs showed peaks at 2925 cm−1 corresponding to C–H stretch, 1741 cm−1 corresponding to the ester bond linkage, 1464 cm−1 corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching and 1239 cm−1 corresponding to PO2 antisymmetric stretching. The FTIR spectrum of NAs was well correlated with our previous report.15 The FTIR spectrum of quercetin showed characteristic peaks at wavelengths of 3481 cm−1 corresponding to OH stretching, 1378 cm−1 corresponding to OH bending of phenol, 1607 cm−1 and 1560 cm−1 corresponding to CC aromatic ring stretching, 1312 cm−1 corresponding to C–H aromatic hydrocarbon bending and 1200 cm−1 corresponding to CO stretching in phenol.32 Next, the analysis of the FTIR spectrum of NAQ exhibited peaks of both NAs and quercetin, confirming the quercetin loading in NAQ, with peaks at 2922 cm−1 corresponding to C–H stretch, 1741 cm−1 corresponding to the ester bond linkage, and 1239 cm−1 indicating PO2 anti-symmetric stretching. A few pure quercetin bands disappeared in the spectrum of NAQ, which specified that quercetin could be loaded well in the lipid bilayers due to hydrophobic interactions or hydrogen bonds.33 Furthermore, EDX analysis was performed to confirm quercetin's entrapment in NAs and the results are illustrated in Table S1 and in ESI† (Fig. S4).
O stretching and 1239 cm−1 corresponding to PO2 antisymmetric stretching. The FTIR spectrum of NAs was well correlated with our previous report.15 The FTIR spectrum of quercetin showed characteristic peaks at wavelengths of 3481 cm−1 corresponding to OH stretching, 1378 cm−1 corresponding to OH bending of phenol, 1607 cm−1 and 1560 cm−1 corresponding to CC aromatic ring stretching, 1312 cm−1 corresponding to C–H aromatic hydrocarbon bending and 1200 cm−1 corresponding to CO stretching in phenol.32 Next, the analysis of the FTIR spectrum of NAQ exhibited peaks of both NAs and quercetin, confirming the quercetin loading in NAQ, with peaks at 2922 cm−1 corresponding to C–H stretch, 1741 cm−1 corresponding to the ester bond linkage, and 1239 cm−1 indicating PO2 anti-symmetric stretching. A few pure quercetin bands disappeared in the spectrum of NAQ, which specified that quercetin could be loaded well in the lipid bilayers due to hydrophobic interactions or hydrogen bonds.33 Furthermore, EDX analysis was performed to confirm quercetin's entrapment in NAs and the results are illustrated in Table S1 and in ESI† (Fig. S4).
The weight percentage of carbon (C) and oxygen (O) in NAs were found to be 87.74% and 12.26%, respectively. The atomic percentages of C and O in NAs were found to be 90.51% and 9.49%. The weight percentages of C, O, and Na in quercetin were found to be 11.54%, 55.47%, and 32.99%, respectively, and atomic percentages of C, O and Na in quercetin were 16.39%, 59.14%, and 24.47%, respectively. Similarly, the weight percentages of C, O, and Na in NAQ were 12.91%, 47.58%, and 39.51%, respectively, and atomic percentages of C, O, and Na in NAQ were 18.63%, 51.57%, and 29.80%, respectively. NAQ showed higher weight and atomic percentages (47 to 52%) of O element and reduced weight and atomic percentages (12 to 18%) of C element compared to NAs. We speculate that the shift in weight% and atomic% of elements in NAQ may be due to the presence of quercetin.
3.4. Drug release study
Next, the drug release study was performed at pH 7.4 and pH 4.5, representing physiological and tumor conditions. From the drug release profile (Fig. 3), we observed that the quercetin release was 20 ± 2.3, 35 ± 3.5, 50 ± 4.2, 70 ± 3.36, 90 ± 3.12, 100 ± 3.8, 100 ± 3.33, 100 ± 2.8, and 100 ± 3.65% at 0.5, 1, 1.5, 2, 4, 6, 8, 12, and 24 h at acidic pH. At neutral pH, the drug release of 12 ± 1.13, 22 ± 2.24, 34 ± 3.23, 48 ± 3.83, 52 ± 4.12, 68 ± 5.5, 78 ± 5.83, 92 ± 4.53, and 100 ± 4.8% was observed at 0.5, 1, 1.5, 2, 4, 6, 8, 12, and 24 h. It was noted that 100% of the drugs were released within 6 hours at acidic pH while at neutral pH, it took more than 18 hours to achieve 100% quercetin release from NAs.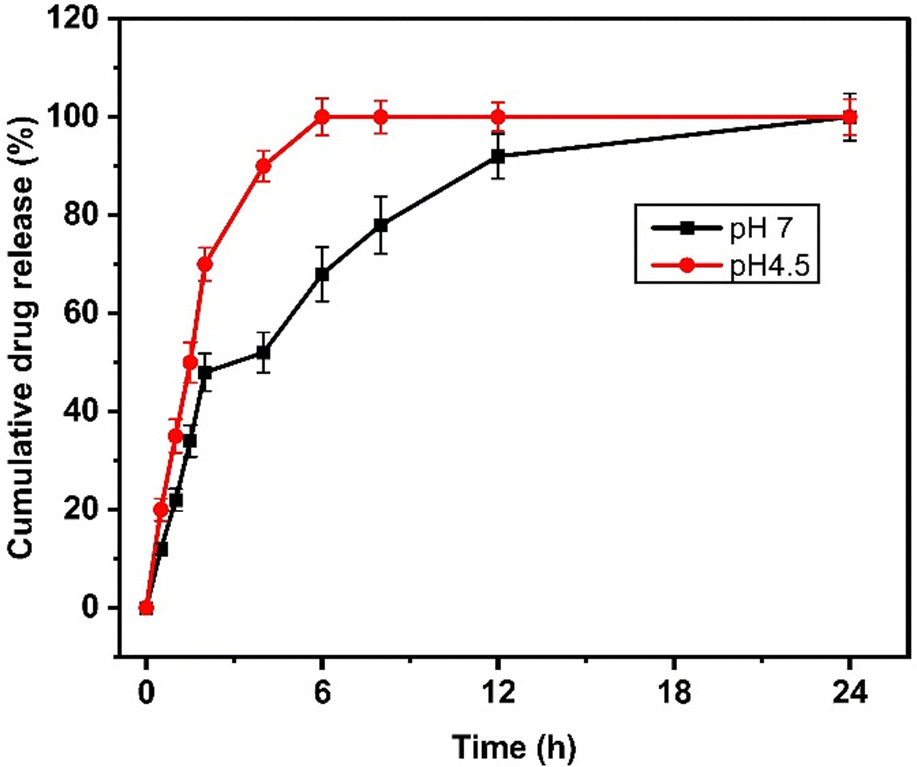 | ||
| Fig. 3 Drug release kinetics of NAQ under neutral (pH 7.4, black) and acidic conditions (pH 4.5, red). The data represent mean ± S.D. | ||
The drug release rate was higher at an acidic pH of 4.5, and this fast release of quercetin at pH 4.5 may be due to the protonation of the hydroxyl group of quercetin inducing repulsive forces, leading to increased drug release at acidic pH.34 This result also correlates well with the previous report published by Ferreira et al.,35 where the β-cyclodextrin delivery systems show significant quercetin release at an acidic pH.
3.5. Biocompatibility of NAs in NIH 3T3 cells
To understand the effects of NAs and NAQ on normal cells, the biocompatibility assay was performed using different concentrations of NAs (0.01, 0.025, and 0.05 mg) and NAQ (2.5 μM) on normal fibroblast NIH 3T3 cells. The cell viability assay results clearly showed that the NIH 3T3 cells treated with different concentrations of NAs did not show any toxicity, nuclear damage, or cell death (Fig. 4A). From the microscopic image (Fig. 4B(i) and (ii)), it was observed that the NA-treated cells showed (Fig. 4B(ii)) intact cellular morphology similar to control cells (Fig. 4B(i)). Next, AO/EtBr staining was performed, and the results are shown in Fig. 4C(i) and (ii). The NA-treated cells (Fig. 4C(ii)) emit green fluorescence, representing the viability of cells. The treated cells showed no apoptotic cell death and retained their intact nuclear structure like untreated control cells (Fig. 4C(i)). Furthermore, the effect of NAs on actin cytoskeleton organization of NIH 3T3 was studied, and the results are shown in Fig. S7 (ESI†). Fig. S7A (ESI†) presents control cells, and Fig. S7B (ESI†) presents NA-treated cells, where (i) implies phalloidin stained cells, (ii) represents Hoechst-stained cells and (iii) represents merged cells. From the figure, control cells showed cellular morphology with normal actin cytoskeleton organization. Furthermore, NA treated NIH 3T3 cells showed no noticeable actin cytoskeleton disorganization. So, we confirm that the drug delivery system NAs could not have any adverse effects on normal cells. | ||
| Fig. 4 Cell toxicity effect of NAs on normal fibroblast NIH 3T3 cells at 24 hours. (A) The MTT assay showing the cell viability of NA-treated NIH 3T3 cells at various concentrations of NAs (0.01, 0.025, and 0.05 mg). The values are shown as mean ± S.D (ns represents not significant). (B) Optical microscopic images of (i) untreated cells and (ii) NA (0.05 mg) treated cells at 10× magnification. (C) Fluorescence microscopic images of (i) untreated cells, (ii) NA (0.05 mg) treated cells using the AO/EtBr staining method at 20× magnification. (D) Graph presenting the toxic effect of NAQ (2.5 μM) on NIH 3T3 cells by the MTT method. (E). Microscopic images of (i) untreated cells and (ii) NAQ (2.5 μM) treated cells at 10× magnification. (F) Fluorescence microscopic images of (i) untreated cells and (ii) NAQ (2.5 μM) treated cells (green color fluorescence indicates normal healthy cells without any nuclear damage). The values are shown as mean ± S.D. (ns represents no significant difference between control and treated cells). | ||
Then, we tested the cell viability of NAQ (2.5 μM) treated NIH 3T3 cells. No significant decrease in viability was noticed between control and NAQ-treated cells (Fig. 4D). Fig. 4E(i) and (ii) shows the microscopic images, and Fig. 4F(i) and (ii) presents AO/EtBr staining of control and NAQ-treated NIH 3T3 cells. The microscopic and AO/EtBr assay results with green fluorescence intensity confirmed that NAQ (2.5 μM) did not cause significant cellular changes in NIH 3T3 cells and they were similar to untreated cells. The results confirmed that NAQ were biocompatible and didn’t affect normal cells. We have also confirmed that NAQ exhibited excellent stability in different physiological media and usage of the medium didn’t induce any aggregation in NAQ (Fig. S5, ESI†). Next, we investigated the hemolytic effect of NAs and NAQ at 37 °C for 4 h. In this study, NAs and NAQ did not show any hemolytic effect (Fig. S6, ESI†). The results obtained from our study indicate that NA and NAQ formulations are compatible with blood, and they may be used for intravenous administration.
3.6. In vitro anticancer effect of NAQ on the breast cancer cell MCF-7
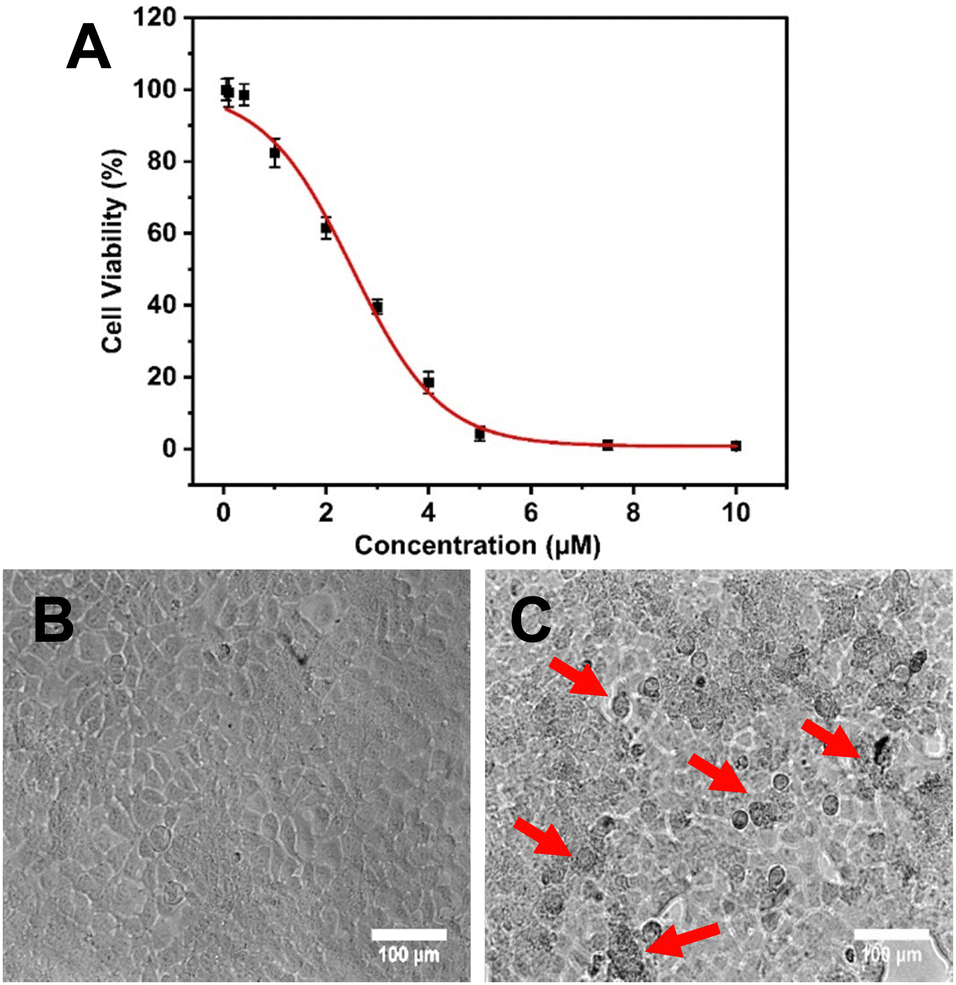 | ||
| Fig. 5 Cell cytotoxic effect of NAQ at different concentrations (0.05, 0.1, 0.4, 1, 2, 3, 4, 5, 7.5 and 10 μM) on MCF-7 at 24 hours by MTT assay. (A) IC50 determination of NAQ. (B) Optical images of untreated (control) MCF-7 cells. (C) NAQ (2.5 μM) treated MCF-7 cells at 20× magnification (red arrows indicate morphological changes and cell death). Data were expressed as mean ± S.D. | ||
Hence, it is found that NAs improved the therapeutic efficacy of quercetin by enhancing its delivery. As previously discussed, NAs alone didn’t exhibit any cytotoxicity on normal fibroblast cell lines. However, to prove that the cytotoxic effect on MCF-7 cells was only exhibited by NAQ, the cytotoxic effect of NAs at different concentrations of 0.01, 0.025, and 0.05 mg was assessed on MCF-7 cells. The results showed that NA-treated MCF-7 cells exhibited cell viability similar to untreated cells (Fig. S9A, ESI†), and no morphological changes in the cellular structure were observed (Fig. S9B, ESI†). The morphology of the untreated cells and NAQ-treated cells at IC50 concentration after 24 hours is depicted in Fig. 5. The untreated control cells showed healthy normal cell structures with intact nuclear morphology (Fig. 5B). The NAQ (2.5 μM) treated cells showed distinctive attributes of cell death (indicated by red arrows), including loss of cell membrane integrity, cell shrinkage, chromatin condensation and cell detachment as compared to untreated cells (Fig. 5C). The optical microscopic images of the MCF-7 cells treated with NAQ of different concentrations are presented in Fig S10 (ESI†). With an increase in the NAQ concentrations, the MCF-7 cells exhibited morphological changes with cell death. The enhanced cytotoxicity effect of NAQ on MCF-7 cells may be related to the NA's lipophilic properties, which endorses quercetin's intracellular uptake. Furthermore, NAs markedly increased the quercetin solubility and stability in MCF-7. Next, a further study was carried out at IC50 concentration of NAQ to elucidate possible modes of action in breast cancer treatment.
 | ||
| Fig. 6 Effect of NAQ on cytoskeleton disruption of MCF-7 cells. (A) The upper panel shows untreated control cells, and (B) the lower panel shows NAQ (2.5 μM) treated cells at 20× magnification. (i) The red channel specifies the cytoskeleton stained with phalloidin, and (ii) the blue channel indicates the nuclei stained with Hoechst. (iii) A merged image of stained cells is shown (white arrows indicate cytoskeleton damage). | ||
 | ||
| Fig. 7 The effect of NAQ on ROS production of MCF-7 cells observed using a fluorescence microscope using DCFDA assay at 20× magnification. (A) Untreated cells. (B) NAQ (2.5 μM) treated cells, (C) Graph showing the fluorescence intensity of intracellular ROS. The values indicate mean ± standard deviation of three independent tests. * represents the level of significance difference at P < 0.05 as compared to the control. | ||
 | ||
| Fig. 8 Acridine orange (AO) and ethidium bromide (EtBr) staining of MCF-7 cells after 24 h of treatment with NAQ: (A) untreated cells. (B) NAQ (2.5 μM) treated cells (yellow arrows indicate dead cells with the uptake of EtBr); (i) AO stained, (ii) EtBr stained, and (iii) merged image of cells stained with AO/EtBr. (C) Graph showing the fluorescence intensity of AO and EtBr. * denotes the significance level at P < 0.05 as compared to the control. | ||
In contrast, NAQ (2.5 μM) did not cause any toxicity in normal NIH 3T3 fibroblast cells (Fig. 4F), which may be due to lower basal intracellular ROS levels and a complete antioxidant capacity in normal cells.43 So, the normal cells are less vulnerable to the ROS stress induced by quercetin.
 | ||
| Fig. 9 FACS analysis of control and NAQ-treated MCF-7 cells represented as a dot plot in quadrants (Q1, Q2, Q3, and Q4). The left bottom quadrant (Q1) shows the percentage of live cells; the other three quadrants, Q2, Q3, and Q4, represent early apoptosis, necrosis, and late apoptosis: (A) untreated MCF-7 cells. (B) NAQ-treated MCF-7 cells. | ||
Moreover, activating necrosis in tumor cells is a potential approach to evade the failure of cancer treatments due to apoptosis resistance. Hence, NAQ may provide strategies for developing effective therapeutic agents against apoptosis-resistant cancer.
 | ||
| Fig. 10 Cell cycle profiles of (A) control (untreated) and (B) NAQ (2.5 μM) treated breast cancer cells MCF-7 through flow cytometry with propidium iodide staining. (C) Graph showing the percentages of cell cycle distribution between control and treated cells. * represents the significance level at P < 0.05 compared to control. | ||
Additionally, the percentage of cells in the S phase was significantly lower for the cells treated with NAQ than for the control. This finding concords with the previous report47 on arresting the cell at the G0/G1 phase with considerable cellular apoptosis in breast cancer cells. Our findings agreed with those of Ranganathan et al.,48 who demonstrated that quercetin-treated MCF-7 cells diminish cell proliferation through G0/G1 phase arrest.
4. Conclusions
Our study emphasizes the significance of nanoarchaeosome nanoparticles as a promising nanocarrier for cancer treatment and aids in overcoming the stumbling blocks of quercetin in cancer treatment. We designed novel and colloidally stable NAs to deliver the bioflavonoid quercetin for anticancer therapy. The NAs improved quercetin's loading and release kinetics. Additionally, NAQ showed excellent in vitro anticancer activity in MCF-7 cells with an IC50 value of 2.5 μM, which is thirty-five-fold less than the IC50 value of free quercetin. NAQ-induced apoptosis and necroptosis-related cell death through ROS production and by efficiently arresting the cells at G0/G1 phases. Therefore, our findings suggest that NAQ can be a potent anti-breast cancer agent. Nevertheless, further investigations must be carried out to evaluate the efficacy of NAQ in the in vivo breast cancer model.Author contributions
Swathi Sudhakar designed the project. Subastri Ariraman, Abirami Seetharaman, Kaviya Vijayalakshmi Babunagappan, and Swathi Sudhakar conducted the experiments and analyzed the results. Subastri Ariraman and Swathi Sudhakar wrote the manuscript. Swathi Sudhakar finalized the manuscript.Data availability
The authors declare that the work data and materials are available in the present manuscript and also the data supporting this article have been included in the ESI.†Conflicts of interest
The authors declare that there is no conflict of interest.Acknowledgements
We are grateful for the financial support from IIT Madras through the Institutions of Eminence (IoE) Scheme (Grant No. SP22231226CPETWOCSMHOC, SP22231226CPET WOHCTHOC, IP23242000AMIITM009000 and RF22230395AMNFIG009000) from the Ministry of Education, Government of India. The authors also thank Prof. Pijyush Gosh for providing the FTIR facility and Prof. Saumendra Kumar Bajpai for the fluorescence microscope facility. The authors thank Cancer Institute (WIA) Adyar, Chennai, for the FACS facility. The first author thanks Mr Sameer Basha for his immense support during drug-release studies.References
- L. Wilkinson and T. Gathani, Br. J. Radiol., 2022, 95, 20211033 CrossRef PubMed.
- J. Zhao, J. Liu, T. Wei, X. Ma, Q. Cheng, S. Huo, C. Zhang, Y. Zhang, X. Duan and X.-J. Liang, Nanoscale, 2016, 8, 5126–5138 RSC.
- M. Ezzati, B. Yousefi, K. Velaei and A. Safa, Life Sci., 2020, 248, 117463 CrossRef CAS PubMed.
- K. Srinivas, J. W. King, L. R. Howard and J. Monrad, J. Food Eng., 2010, 100, 208–218 CrossRef CAS.
- E. U. Graefe, J. Wittig, S. Mueller, A. K. Riethling, B. Uehleke, B. Drewelow, H. Pforte, G. Jacobasch, H. Derendorf and M. Veit, J. Clin. Pharmacol., 2001, 41, 492–499 CrossRef CAS PubMed.
- S. Andres, S. Pevny, R. Ziegenhagen, N. Bakhiya, B. Schäfer, K. I. Hirsch-Ernst and A. Lampen, Mol. Nutr. Food Res., 2018, 62, 1700447 CrossRef PubMed.
- J. K. Patra, G. Das, L. F. Fraceto, E. V. R. Campos, M. del, P. Rodriguez-Torres, L. S. Acosta-Torres, L. A. Diaz-Torres, R. Grillo, M. K. Swamy, S. Sharma, S. Habtemariam and H.-S. Shin, J. Nanobiotechnol., 2018, 16, 71 CrossRef PubMed.
- G. Xu, H. Shi, L. Ren, H. Gou, D. Gong, X. Gao and N. Huang, Int. J. Nanomed., 2015, 10, 2051–2063 CAS.
- D. Lombardo and M. A. Kiselev, Pharmaceutics, 2022, 14, 543 CrossRef CAS PubMed.
- L. Sercombe, T. Veerati, F. Moheimani, S. Y. Wu, A. K. Sood and S. Hua, Front. Pharmacol., 2015, 6, 286 Search PubMed.
- T. O. B. Olusanya, R. R. Haj Ahmad, D. M. Ibegbu, J. R. Smith and A. A. Elkordy, Molecules, 2018, 23, 907 CrossRef PubMed.
- G. B. Patel and G. D. Sprott, Crit. Rev. Biotechnol., 1999, 19, 317–357 CrossRef CAS PubMed.
- M. J. Altube, L. I. Caimi, C. Huck-Iriart, M. J. Morilla and E. L. Romero, Pharmaceutics, 2021, 13, 1331 CrossRef CAS PubMed.
- L. Krishnan and G. D. Sprott, J. Drug Targeting., 2003, 11, 515–524 CrossRef CAS PubMed.
- K. V. Babunagappan, A. Seetharaman, S. Ariraman, P. B. Santhosh, J. Genova, N. P. Ulrih and S. Sudhakar, Nanoscale Adv., 2024, 6, 2026–2037 RSC.
- J. C. Marcarini, M. S. F. Tsuboy, R. C. Luiz, L. R. Ribeiro, C. B. Hoffmann-Campo and M. S. Mantovani, Exp. Toxicol. Pathol., 2011, 63, 459–465 CrossRef CAS PubMed.
- E. Preedia Babu, A. Subastri, A. Suyavaran, P. Lokeshwara Rao, M. Suresh Kumar, K. Jeevaratnam and C. Thirunavukkarasu, RSC Adv., 2015, 5, 62067–62077 RSC.
- A. Subastri, A. Suyavaran, E. Preedia Babu, S. Nithyananthan, R. Barathidasan and C. Thirunavukkarasu, J. Cell. Physiol., 2018, 233, 1775–1790 CrossRef PubMed.
- A. Gohel, M.-B. McCarthy and G. Gronowicz, Endocrinology, 1999, 140, 5339–5347 CrossRef CAS PubMed.
- A. Subastri, V. Arun, P. Sharma, E. Preedia Babu, A. Suyavaran, S. Nithyananthan, G. M. Alshammari, B. Aristatile, V. Dharuman and C. Thirunavukkarasu, Chem. – Biol. Interact., 2018, 295, 73–83 CrossRef CAS PubMed.
- K. Velu, S. Panda, G. Kumar, D. Kumar, L. Shrestha, K. Ariga, K. Vasanth, S. Chinnathambi, S. Dhas and K. U. Suganya, Appl. Surf. Sci., 2019, 487, 211–217 CrossRef.
- D. Savaghebi, M. Barzegar and M. R. Mozafari, Food Sci. Nutr., 2020, 8, 299–310 CrossRef CAS PubMed.
- V. Gupta and P. Trivedi, in Lipid Nanocarriers for Drug Targeting, ed. A. M. Grumezescu, William Andrew Publishing, 2018, pp. 563–627 Search PubMed.
- M. Gumustas, C. T. Sengel-Turk, A. Gumustas, S. A. Ozkan and B. Uslu, in Multifunctional Systems for Combined Delivery, Biosensing and Diagnostics, ed. A. M. Grumezescu, Elsevier, 2017, pp. 67–108 Search PubMed.
- P. Das and M. K. Das, in Nanocosmeceuticals, ed. M. K. Das, Academic Press, 2022, pp. 95–138 Search PubMed.
- E. Joseph and G. Singhvi, in Nanomaterials for Drug Delivery and Therapy, ed. A. M. Grumezescu, William Andrew Publishing, 2019, pp. 91–116 Search PubMed.
- S. Behzadi, V. Serpooshan, W. Tao, M. A. Hamaly, M. Y. Alkawareek, E. C. Dreaden, D. Brown, A. M. Alkilany, O. C. Farokhzad and M. Mahmoudi, Chem. Soc. Rev., 2017, 46, 4218–4244 RSC.
- F. Villanueva-Flores, A. Castro-Lugo, O. T. Ramírez and L. A. Palomares, Nanotechnology, 2020, 31, 132002 CrossRef PubMed.
- J. C. Stevens Barrón, C. Chapa González, E. Álvarez Parrilla and L. A. la Rosa, Biomolecules, 2023, 13, 1158 CrossRef PubMed.
- B. Ruozi, G. Tosi, E. Leo and M. A. Vandelli, Talanta, 2007, 73, 12–22 CrossRef CAS PubMed.
- M. Huang, E. Su, F. Zheng and C. Tan, Food Funct., 2017, 8, 3198–3208 RSC.
- M. Catauro, F. Papale, F. Bollino, S. Piccolella, S. Marciano, P. Nocera and S. Pacifico, Sci. Technol. Adv. Mater., 2015, 16, 35001 CrossRef PubMed.
- D. Liu, H. Hu, Z. Lin, D. Chen, Y. Zhu, S. Hou and X. Shi, J. Photochem. Photobiol., B., 2013, 127, 8–17 CrossRef CAS PubMed.
- A. Samadi, M. Pourmadadi, F. Yazdian, H. Rashedi, M. Navaei-Nigjeh and T. Eufrasio-da-Silva, Int. J. Biol. Macromol., 2021, 182, 11–25 CrossRef CAS PubMed.
- M. Ferreira, D. Gomes, M. Neto, L. A. Passarinha, D. Costa and Â. Sousa, Pharmaceutics, 2023, 15, 936 CrossRef CAS PubMed.
- A. Karimian, M. Majidinia, A. Moliani, F. Alemi, Z. Asemi, B. Yousefi and A. F. Naghibi, Pathol. – Res. Pract., 2022, 240, 154143 CrossRef CAS PubMed.
- A. Sharma, V. Panwar, J. Thomas, V. Chopra, H. S. Roy and D. Ghosh, Colloids Surf., B, 2021, 200, 111572 CrossRef CAS PubMed.
- M. Kaur, D. Prasher, A. Sharma, D. Ghosh and R. Sharma, Environ. Sci. Pollut. Res. Int, 2023, 30, 38869–38885 CrossRef CAS PubMed.
- K. Burridge and K. Wennerberg, Cell, 2004, 116, 167–179 CrossRef CAS PubMed.
- B. Deepa, H. V. Babaji, J. V. Hosmani, A. W. H. Alamir, S. Mushtaq, A. T. Raj and S. Patil, Appl. Sci., 2019, 9, 5147 CrossRef CAS.
- S. S. Kamble, J. Choudhari, R. Nimma, T. V. S. Kumar, K. K. Patil, S. V. Hese, B. S. Dawane and R. N. Gacche, Cancer Rep., 2022, 5, e1600 CrossRef CAS PubMed.
- P. Biswas, D. Dey, P. K. Biswas, T. I. Rahaman, S. Saha, A. Parvez, D. A. Khan, N. J. Lily, K. Saha, M. Sohel, M. M. Hasan, S. Al Azad, S. Bibi, M. N. Hasan, M. Rahmatullah, J. Chun, M. A. Rahman and B. Kim, Int. J. Mol. Sci., 2022, 23, 11746 CrossRef CAS PubMed.
- L. Gibellini, M. Pinti, M. Nasi, S. De Biasi, E. Roat, L. Bertoncelli and A. Cossarizza, Cancers, 2010, 2, 1288–1311 CrossRef CAS PubMed.
- T. Geetha, V. Malhotra, K. Chopra and I. P. Kaur, Indian J. Exp. Biol., 2005, 43, 61–67 CAS.
- A. Kocyigit, I. Koyuncu, M. Dikilitas, F. Bahadori and B. Turkkan, Asian Pac. J. Trop. Biomed., 2016, 6, 872–880 CrossRef CAS.
- M.-B. Meng, H.-H. Wang, Y.-L. Cui, Z.-Q. Wu, Y.-Y. Shi, N. G. Zaorsky, L. Deng, Z.-Y. Yuan, Y. Lu and P. Wang, Oncotarget, 2016, 7, 57391–57413 CrossRef PubMed.
- X.-H. Deng, H.-Y. Song, Y.-F. Zhou, G.-Y. Yuan and F.-J. Zheng, Exp. Ther. Med., 2013, 6, 1155–1158 CrossRef CAS PubMed.
- S. Ranganathan, D. Halagowder and N. D. Sivasithambaram, PLoS One, 2015, 10, 1–21 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00258j |
| This journal is © The Royal Society of Chemistry 2024 |