
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn-surface polymerization reactions of dibrominated hexaphenylbenzene influenced by densely packed self-assembly†
Hiroaki
Ooe
 and
Takashi
Yokoyama
*
and
Takashi
Yokoyama
*
Faculty of Science, Yokohama City University, 22-2 Seto, Kanazawa-ku, Yokohama 236-0027, Japan. E-mail: tyoko@yokohama-cu.ac.jp
First published on 15th April 2024
Abstract
Controlled bottom-up fabrication of molecular nanostructures through on-surface reactions of tailor-made precursors is of scientific and technological interest. Recently, on-surface polymerization reactions influenced by precursor self-assembly have been reported. Thus, a fundamental understanding of the reaction process is a prerequisite for controlled formation. Herein, we report on the influence of molecular self-assembly of dibrominated hexaphenylbenzene (Br2-HPB) on the on-surface polymerization reactions on a Au(111) substrate. By using low-temperature scanning tunnelling microscopy (STM), we find that the polymerization of Br2-HPB proceeds while maintaining the long-range ordered self-assembly, despite a decrease in HPB–HPB distance due to debromination and successive covalent bonding of Br2-HPB. From the STM investigations of the polymerization process, we conclude that the polymerization of Br2-HPB is accompanied by molecular rotations to maintain the periodic array of the self-assembled structure, contrary to the conventional understanding of the polymerization of the self-assembled precursor layer.
Introduction
Covalent nanostructures have attracted increasing attention due to their potential applications in molecular electronics.1–8 For the fabrication of atomically precise nanostructures, a bottom-up synthesis employing thermally induced on-surface polymerization between halogenated precursor molecules has emerged as a powerful method.9 By using tailor-made precursor molecules, various types of nanostructures have been successfully produced, in which the molecular transformations from precursors into polymers have been directly visualized by scanning tunnelling microscopy (STM).9–14 For further development and successful formation of well-designed nanostructures, precise control of polymerization is essential. In recent years, precursor self-assembly has been advocated as a method for controlling polymerization, and some applied research have advanced.15–20 The currently well-accepted interpretation of how precursor self-assembly influences polymerization reactions is that dense packing restricts molecular diffusion and promotes polymerization between spatially adjacent reaction sites while maintaining molecular orientations.19–24 Following this interpretation, the steering of reaction sites and the template effect for polymerization products through precursor self-assembly have been demonstrated.19,22–25The on-surface polymerization influenced by precursor self-assembly has appeared in our previous study of 4-bromo-4′-(4-bromophenyl)-3′,5′,6′-triphenyl-1,1′:2′,1′′-terphenyl (Br2-HPB) molecules on Au(111).26 The Br2-HPB molecule is composed of a central benzene ring substituted by four phenyl and two bromophenyl rings, forming the six-bladed propeller shape (Fig. 1(a)). The molecular chiralities of Br2-HPB result from concerted rotations of the outer phenyl rings, which are labelled as R for clockwise (CW) and L for counterclockwise (CCW) rotation as shown in Fig. 1(b). On Au(111), the Br2-HPB molecules with as single-layer thickness are ordered into a self-assembled layer with unit cell vectors 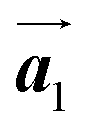 and
and 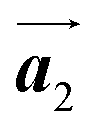 as shown in Fig. 1(c). This self-assembled layer is likely stabilized by the attractive bromophenyl–bromophenyl interactions along the
as shown in Fig. 1(c). This self-assembled layer is likely stabilized by the attractive bromophenyl–bromophenyl interactions along the 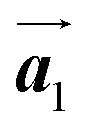 direction, as well as by the mutual interlocking of the outer phenyl rings between neighbouring molecules. Due to the linear alignment of the bromophenyl substituents of Br2-HPB, the close proximity of the bromophenyl rings would allow efficient polymerization of debrominated phenyl rings during thermal annealing. Furthermore, the R- or L-homochiral arrangements of Br2-HPB have been revealed along the linear alignment of the bromophenyl substituents in the
direction, as well as by the mutual interlocking of the outer phenyl rings between neighbouring molecules. Due to the linear alignment of the bromophenyl substituents of Br2-HPB, the close proximity of the bromophenyl rings would allow efficient polymerization of debrominated phenyl rings during thermal annealing. Furthermore, the R- or L-homochiral arrangements of Br2-HPB have been revealed along the linear alignment of the bromophenyl substituents in the 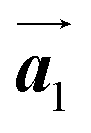 direction, and the RL-heterochiral arrangement in the
direction, and the RL-heterochiral arrangement in the  direction as indicated in Fig. 1(c).
direction as indicated in Fig. 1(c).
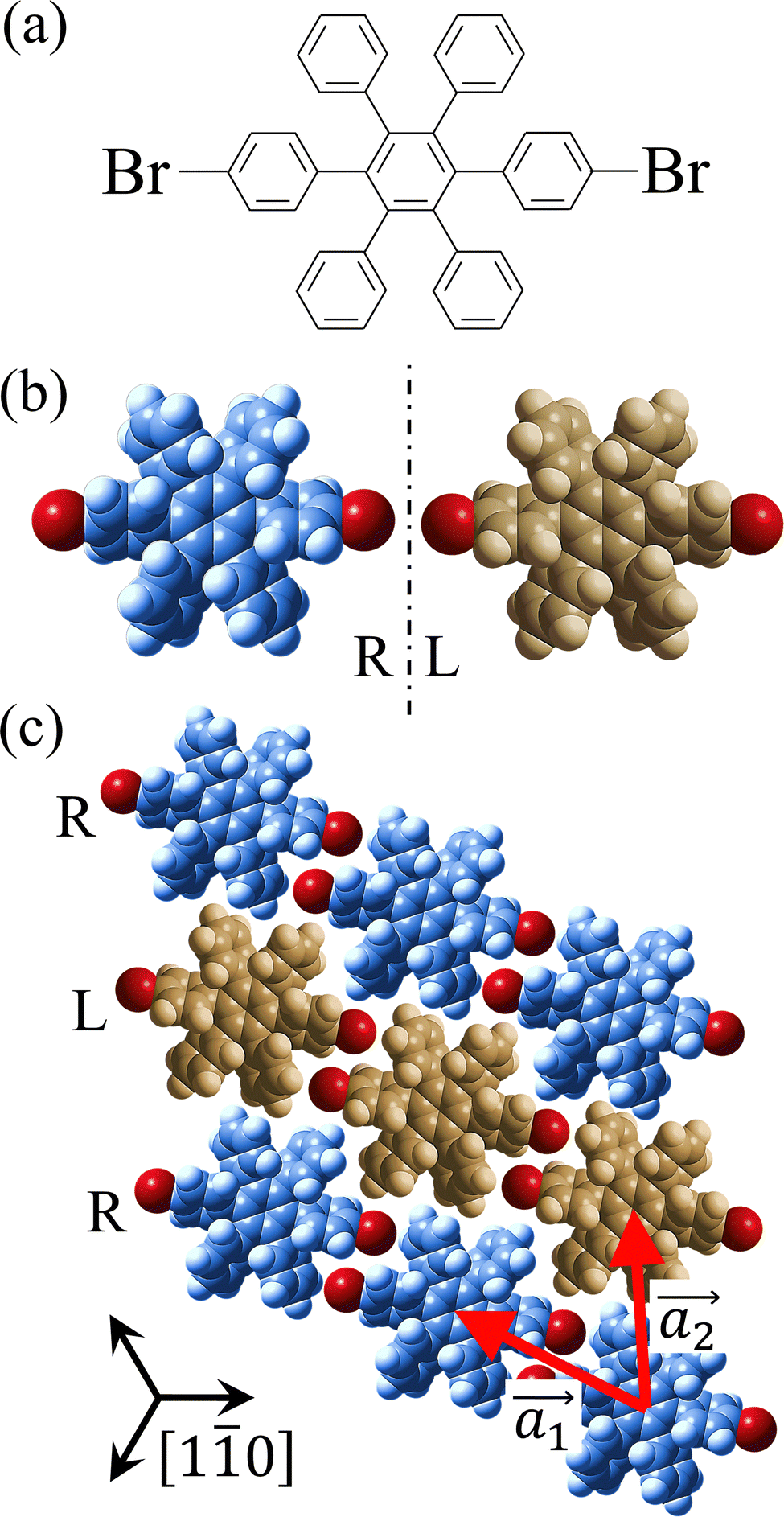 | ||
Fig. 1 (a) Chemical structure of Br2-HPB. (b) Space-filling models of the molecular chiralities. The enantiomeric R- and L-types are defined by the CW and CCW rotations of the outer phenyl rings with respect to the central benzene ring. The blue and brown coloured models represent the R- and L-types chiralities, respectively. (c) Self-assembled structure of Br2-HPB molecules described by the unit cell vectors  and and  . The lattice parameters have been obtained as . The lattice parameters have been obtained as 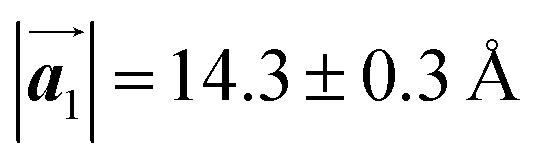 , , 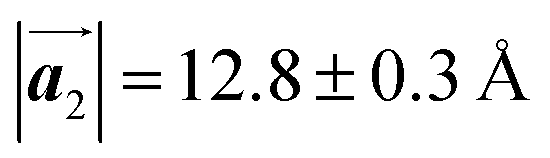 , the included angles 57.2 ± 3.1°, and the molecular orientation defined by opposite bromine atoms is rotated by about 11° from the [1 , the included angles 57.2 ± 3.1°, and the molecular orientation defined by opposite bromine atoms is rotated by about 11° from the [1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0] direction of the underlying Au(111) surface.26 The molecular chiralities tend to be ordered in R- or L-homochiral arrangements in the 0] direction of the underlying Au(111) surface.26 The molecular chiralities tend to be ordered in R- or L-homochiral arrangements in the 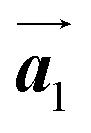 direction with a 62% probability, and in RL-heterochiral arrangement in the direction with a 62% probability, and in RL-heterochiral arrangement in the 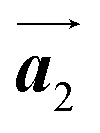 with a 81% probability. with a 81% probability. | ||
Upon thermal annealing of the self-assembled layer at 200 °C, we have reported that the debromination and sequential polymerization of the Br2-HPB precursor molecules are induced on the Au(111) surface, leading to the formation of one-dimensional (1D) hexaphenylbenzene (HPB) polymer chains.26 Surprisingly, the produced 1D HPB polymer chains were aligned to almost the same length and self-assembled into a long-range ordered structure, while both edges of the chains still remain the bromophenyl rings.
Regarding the Br2-HPB molecules on the Au(111) surface, the polymerization reactions are likely affected by the self-assembled structure, whereas how the ordering of same length HPB polymers is achieved has remained an issue. In this study, to gain deeper insights into the polymerization process from Br2-HPB precursors to HPB polymer chains, we have performed STM observations on the Br2-HPB covered Au(111) surface after short annealing at 200 °C. Herein, we present the self-assembled structure composed of both HPB polymer chains and Br2-HPB molecules, which is key to resolving the polymerization process. Based on the STM results, we discuss the detailed polymerization pathway of Br2-HPB proceeding within the self-assembled layer.
Experimental methods
All experiments were performed in an ultrahigh-vacuum (UHV) system (base pressure less than 4.0 × 10−9 Pa) with low-temperature STM. Au(111) films epitaxially grown on freshly cleaved muscovite mica were used as the substrate. The substrate surface was cleaned by repeated cycles of Ar+ sputtering and subsequent annealing at 400 °C. Then, Br2-HPB precursor molecules, purchased from Tokyo Chemical Industry, were deposited onto the clean Au(111) surface at nearly full-monolayer by sublimation from a carefully degassed quartz crucible held at 191 °C. During the molecular deposition, the substrate was maintained at room temperature. Subsequently, the sample was transferred to the low-temperature STM stage. All STM images were acquired in a constant current mode of Vs = −3.0 V and It = 20 pA at 78 K. Following confirmation of the self-assembled layer of Br2-HPB precursors by using LT-STM, the sample annealing for polymerization was carried out.Results
Fig. 2(a) shows the high-resolution STM image of the HPB polymer chains obtained after long annealing (20 hours) of the self-assembled Br2-HPB layer on the Au(111) surface at 200 °C. It is clear that the HPB polymer chains composed of 3 or 4 HPB cores are arranged in a side-by-side packing, resulting in the formation of the self-assembled straight rows of the HPB polymer chains with a single-layer thickness, as reported previously.26 The domain boundaries of the straight rows, marked by white dashed lines and black arrows in Fig. 2(a), appear dark due to the bromophenyl substituents at both edges of the HPB polymer chains, which run parallel to 0° or ±20° deviated from the [10] direction of the underlying Au(111) surface. In addition, the domain boundaries are likely stabilized by the attractive bromophenyl–bromophenyl interactions. Inside the HPB polymer chains, whereas the hexagonally arranged six lobes are expected to appear for each HPB core in the STM image, the two adjacent lobes appear to be unified between covalently bonded HPB cores.
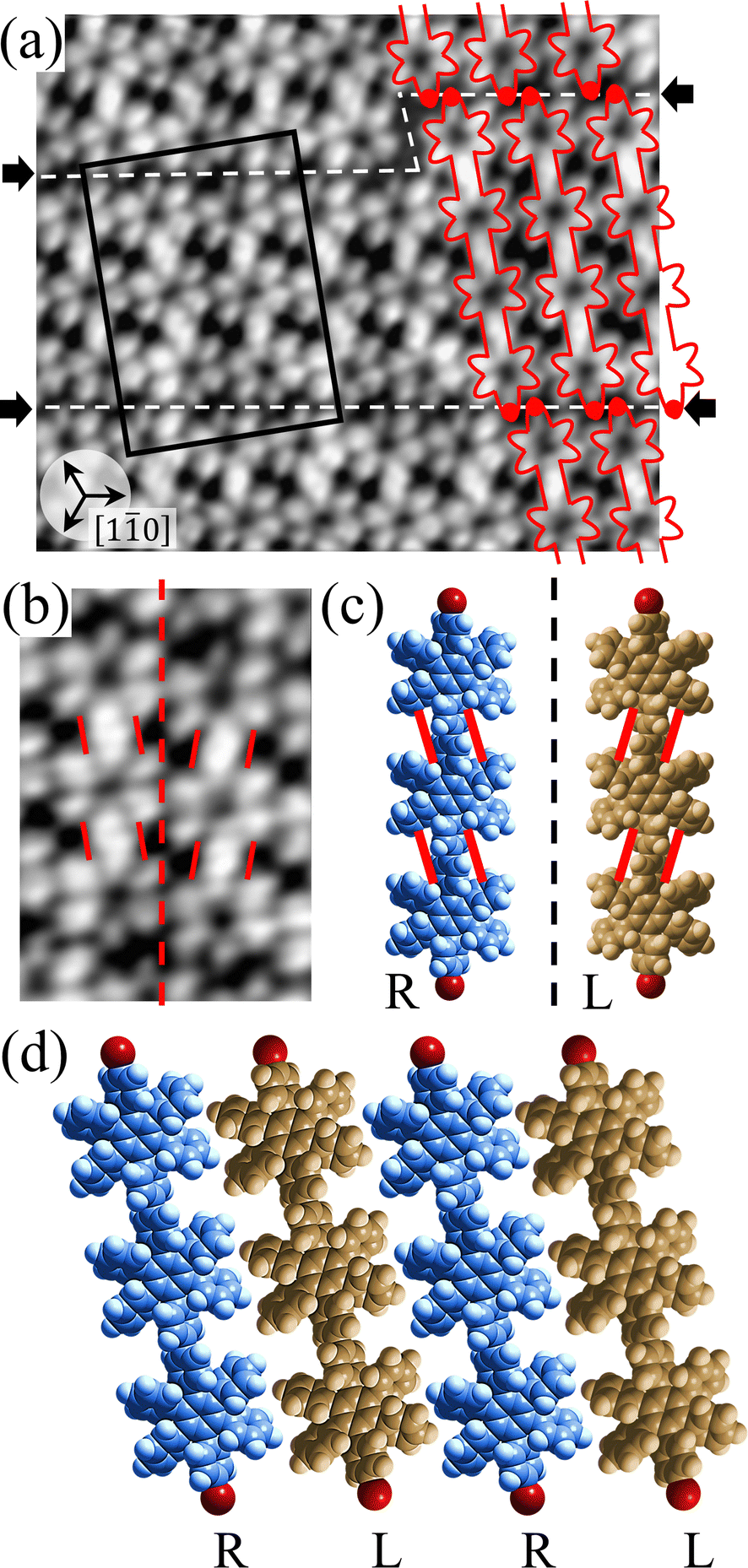 | ||
| Fig. 2 (a) STM image (10 nm × 8 nm, Vs = −3 V, and It = 20 pA) of the HPB polymer chains taken after annealing at 200 °C for 20 h, showing self-assembled straight rows of HPB polymer chains. The HPB polymer chains and bromine atoms are depicted by red outlines and red circles, respectively. The straight row is composed of the HPB polymer chains with three HPB cores on the left side, which changed to four HPB cores on the right side. White dashed lines and black arrows indicate the domain boundaries between the straight rows. Typically, the straight rows of HPB polymer chains run parallel to 0° or ±20° deviated from the [10] direction of the underlying Au(111) surface. (b) Magnified STM image (5 nm × 3 nm) of the region indicated by the black rectangle in (a) including two neighboring HPB polymer chains, in which the covalently bonded HPB cores appear to be tilted unified lobes as guided by red solid lines. The centred red dashed line is parallel to the long axes of the HPB polymer chains. (c) Space-filling models of all R- and all L-chiral HPB polymer chains. The red lines show the up-tilted sides of twisted phenyl rings between HPB cores. (d) Self-assembled structure of the straight row of HPB polymer chains exhibiting the RL-heterochiral order, in which the adjacent outer phenyl rings are in parallel configurations. | ||
For the HPB polymer chains, the chirality has not been recognized in our previous study, whereas the chiral identification of Br2-HPB molecules has been achieved by distorted appearance of the six lobes.26 In Fig. 2(a), although the lobes appear as symmetric shapes, we find that the unified lobes shared between covalently bonded HPB cores are tilted with respect to the long axis of the HPB polymer chains. The tilt directions are identical within a HPB polymer chain, and are opposite between the next chains as shown in Fig. 2(b). These two kinds of tilt directions should reflect the twisting directions of covalently bonded two phenyl rings between HPB cores as guided by red lines in Fig. 2(c). Similar twists of covalently bonded two phenyl rings have been reported for hexakis(4-iodophenyl)benzene dimer and trimer.27 From the perspective that the concerted rotations of the outer phenyl rings within each HPB core, the tilt of the unified lobes should indicate RR- or LL-homochiral junctions between polymerized HPB cores, and thus the HPB polymer chains possess either all R- or all L-chirality. We did not observe RL-heterochiral junctions within a chain, which likely result from the steric hindrance of the twisted phenyl rings. Furthermore, we find that the neighbouring HPB polymer chains exhibit RL-heterochiral arrangements with a high selectivity of about 97%, which likely result from the minimized steric hindrance between the neighbouring phenyl rings. The RL-heterochiral ordering of the homochiral rows as shown in Fig. 2(d) is similar to that for the self-assembled Br2-HPB layer as shown in Fig. 1(c).
The long-range order of the HPB polymer chains is strongly influenced by the self-assembly of Br2-HPB precursors before polymerization. In fact, we did not observe such long-range order of the HPB polymer chains after deposition on the Au(111) surface at the elevated temperature of around 200 °C, in which small domains of the HPB polymer chains were distributed randomly as shown in Fig. S1 (ESI†). According to the well-accepted interpretation of the polymerization process within the self-assembled precursor layer, the restricted molecular diffusion should lead to the preferential formation of covalent bonding between adjacent debrominated substituents while maintaining the orientations. On the other hand, the HPB–HPB distance becomes shorter through debromination and successive polymerization of Br2-HPB.
For a more detailed understanding of the polymerization process within the self-assembled Br2-HPB layer, we carried out a relatively short annealing after nearly full-monolayer deposition of Br2-HPB precursors. Fig. 3(a) shows the STM image of the sample after annealing at 200 °C for 1 hour. Compared with the periodic straight dark lines observed after 20 hours annealing, zigzag dark lines appear as indicated by red lines and red arrows in Fig. 3(a), whereas both zigzag and straight dark lines run parallel to 0° or ±20° deviated from the [10] direction of the underlying Au(111) surface. These results indicate that the polymerization reactions of Br2-HPB proceed slowly at 200 °C, and the zigzag appearance obtained after the 1 hour annealing reflects incomplete polymerization of Br2-HPB.
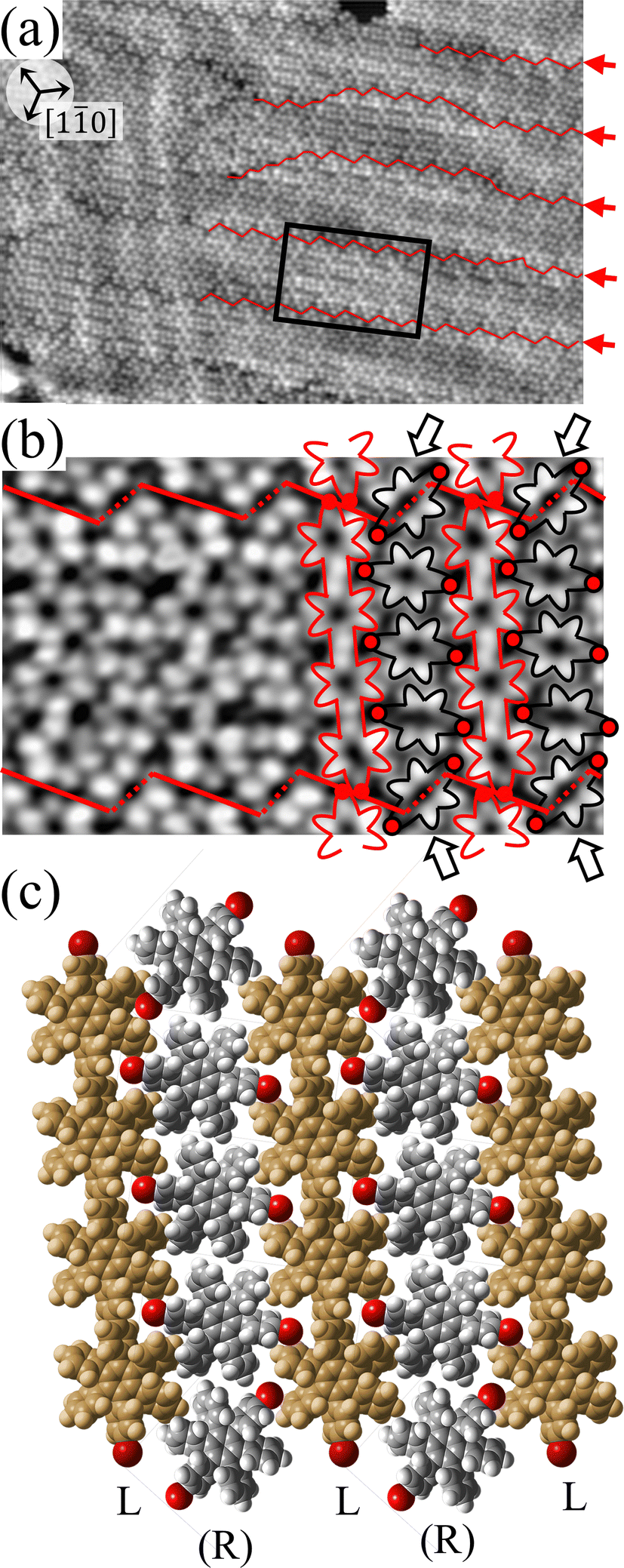 | ||
| Fig. 3 (a) STM image (40 nm × 30 nm, Vs = −3.0 V, and It = 20 pA) of Br2-HPB covered Au(111) after annealing at 200 °C for 1 h. The periodic zigzag dark lines are observed, indicated by red lines and red arrows. Typically, the zigzag dark lines run parallel to 0° or ±20° deviated from the [10] direction of the underlying Au(111) surface. (b) Enlarged STM image (10 nm × 5 nm) of the region indicated by the black rectangle in (a). The zigzag row is composed of periodically arranged HPB polymer chains and Br2-HPB molecules, depicted by red and black outlines on the right, respectively. (c) Expected self-assembled structure of the mixture phase for (b), in which the L-type HPB polymers are represented by brown, and the chiral-unidentified Br2-HPB molecules are represented by grey, respectively. For the minimized intermolecular steric hindrance, the heterochiral arrangement of homochiral rows are expected. | ||
Fig. 3(b) shows the high-resolution STM image of a zigzag row formed between two zigzag dark lines. With the help of the STM appearances of the Br2-HPB molecules and the HPB polymer chains reported previously,26 we can identify the coexistence of Br2-HPB precursors and HPB polymer chains in the zigzag row as depicted by black and red outlines, respectively. It is obvious that the HPB polymer chains, composed of the four HPB cores, are oriented in the same direction, and the Br2-HPB molecules are linearly aligned between the HPB polymer chains, forming the well-ordered mixture phase composed of both Br2-HPB precursors and HPB polymers. This result shows that approximately 50% of the Br2-HPB precursors were converted into HPB polymer chains through the 1 hour annealing at 200 °C. Furthermore, it should be noted that the bromophenyl rings appear as faint lobes, compared to the phenyl rings in the STM image obtained at the sample bias of −3.0 V,26 and thus the bromophenyl substituents still remain on the Br2-HPB molecules in the zigzag rows, as well as on both edges of the HPB polymer chains. The faint appearance of the bromophenyl rings, as indicated by red filled circles in Fig. 3(b), allows us to determine the orientations of the Br2-HPB molecules in the STM images. Fig. 3(c) shows a space-filling model of the self-assembled zigzag row, in which the Br2-HPB precursors and HPB polymer chains are alternately ordered. In the model, the bromophenyl rings of Br2-HPB molecules interlocked with neighbouring HPB polymer chains, whereas the Br2-HPB molecules located at the domain boundaries, as pointed out by arrows in Fig. 3(b), are rotated CCW by about 55°.
Between the zigzag rows, the domain boundaries are likely stabilized by the attractive bromophenyl–bromophenyl interactions between HPB polymer chains, similar to the self-assembled HPB polymer chains as shown in Fig. 2. In addition, the 55° rotation of the Br2-HPB molecules allow them to fit well into the gap between neighbouring domains. Thus, the zigzag dark lines at the domain boundaries should be associated with the dark appearance of the bromophenyl rings of the 55° rotated Br2-HPB molecules and the edges of the HPB polymer chains, as indicated by the red dashed and solid lines in Fig. 3(b), respectively. On the other hand, the zigzag dark lines disappear at the left side in Fig. 3(a), which result from the partial coexistence of the different length HPB polymer chains and the resultant partial disorder.
From the chiral identification technique for the HPB polymer chains mentioned above, we find that almost all HPB polymer chains within the zigzag rows in Fig. 3(a) have the L-chiral form. On the other hand, the chiralities of the Br2-HPB molecules located between the HPB polymer chains could not be resolved from the STM images, but the R-chiral form is expected from the suitable mutual interlocking with the HPB polymer chains. Thus, we propose the heterochiral arrangement of the homochiral rows in the mixture phase, as shown in Fig. 3(c).
According to the well-accepted interpretation of the polymerization process under the diffusion restricted conditions mentioned earlier,19–24 the sequential covalent bonding should be formed between the nearest-neighbour bromophenyl rings of Br2-HPB within the self-assembled layer in Fig. 1(c). This suggests that the HPB polymer chains grow in the 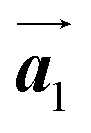 direction while maintaining the molecular orientations. However, as shown in Fig. 3(c), the bromophenyl rings of Br2-HPB in the mixture phase were not oriented parallel to the chain direction of the HPB polymers, contrary to the general expectation.
direction while maintaining the molecular orientations. However, as shown in Fig. 3(c), the bromophenyl rings of Br2-HPB in the mixture phase were not oriented parallel to the chain direction of the HPB polymers, contrary to the general expectation.
To understand HPB polymer chain formation within the self-assembled Br2-HPB layer, we carried out annealing at a slightly lower temperature of 150 °C after nearly full-monolayer deposition of Br2-HPB precursors on the Au(111) surface. Fig. 4(a) shows the STM image of the sample after annealing at 150 °C for 20 hours, in which intact Br2-HPB molecules entirely cover the Au(111) surface. As shown in Fig. 4(b), the Br2-HPB molecules retain the orientation, and the self-assembled structure of Br2-HPB is identical to that before annealing, characterized as the unit cell vectors 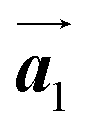 and
and  . In addition to intact Br2-HPB molecules, several polymerized HPB chains are also observed near the step edge, as indicated by red outlines in Fig. 4(b). These HPB polymer chains, composed of 2–3 HPB cores, are slightly shorter than the 3–4 cores formed after annealing at 200 °C. In the case of the 200 °C annealing, we have observed that the chain length of the HPB polymers is almost independent of the annealing time (between 1 hour and 20 hours as shown in Fig. S2, ESI†), and thus the chain length should be influenced mainly by the annealing temperature.
. In addition to intact Br2-HPB molecules, several polymerized HPB chains are also observed near the step edge, as indicated by red outlines in Fig. 4(b). These HPB polymer chains, composed of 2–3 HPB cores, are slightly shorter than the 3–4 cores formed after annealing at 200 °C. In the case of the 200 °C annealing, we have observed that the chain length of the HPB polymers is almost independent of the annealing time (between 1 hour and 20 hours as shown in Fig. S2, ESI†), and thus the chain length should be influenced mainly by the annealing temperature.
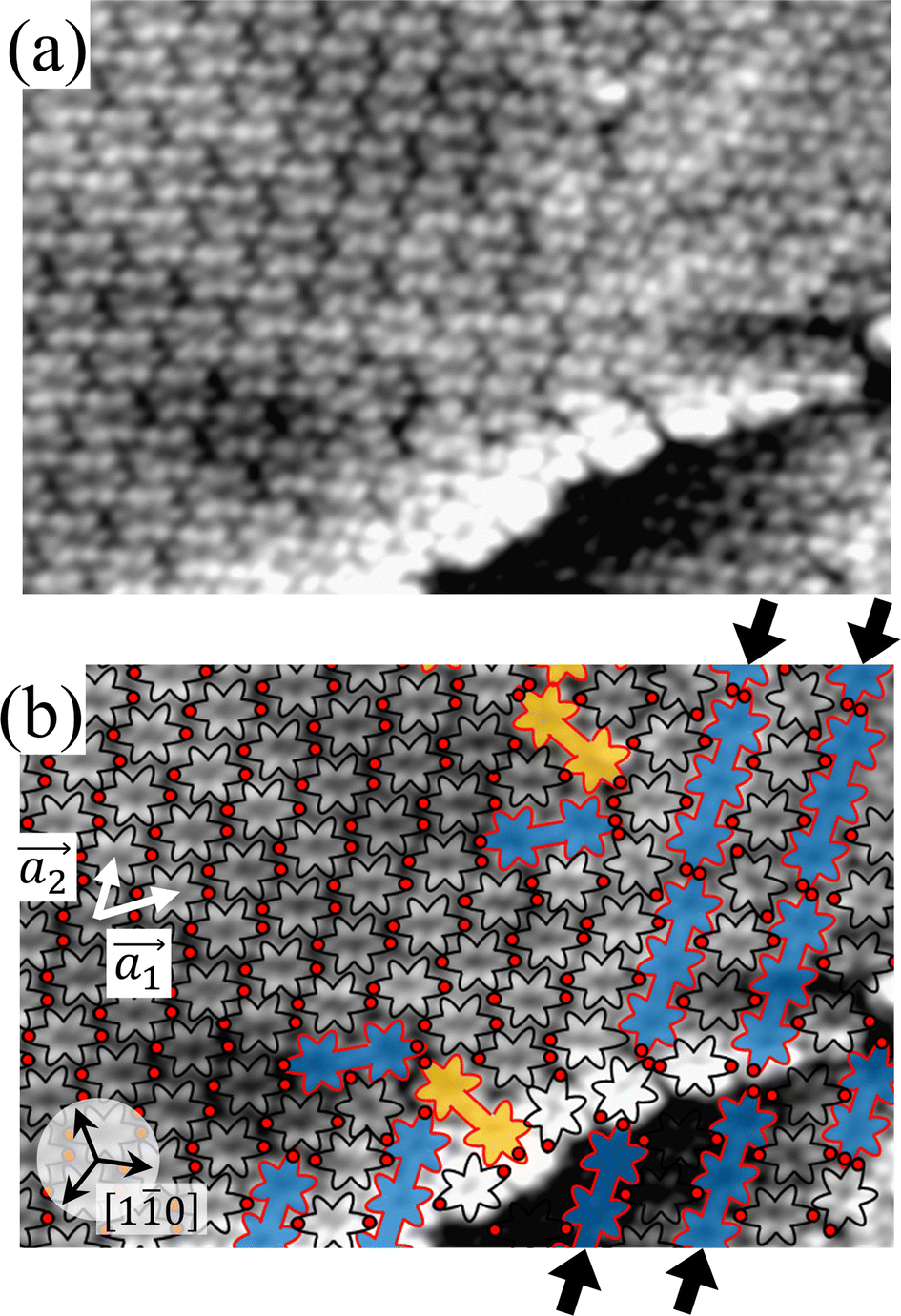 | ||
Fig. 4 (a) STM images (20 × 10 nm, Vs = −3.0 V, and It = 20 pA) of a representative area including several HPB polymer chains taken after annealing at 150 °C for 20 h. (b) STM image overlaid with molecular outlines. The Br2-HPB molecules and HPB polymer chains are depicted by black and red outlines, respectively. Blue and brown colours represent R- and L-homochiral HPB polymers, respectively. The unit cell vectors  and and  for Br2-HPB molecules remain identical to those before annealing. Black arrows indicate the preferred polymerization direction, corresponding to the for Br2-HPB molecules remain identical to those before annealing. Black arrows indicate the preferred polymerization direction, corresponding to the 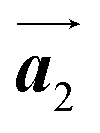 direction of the self-assembled layer of Br2-HPB precursors. direction of the self-assembled layer of Br2-HPB precursors. | ||
In Fig. 4(b), whereas three distinct orientation directions for the HPB polymer chains are observed, we find the preferential extension of HPB polymer chains in the 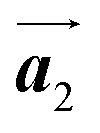 direction of the self-assembled Br2-HPB layer, contradicting our prior expectation that the polymerization proceeds in the
direction of the self-assembled Br2-HPB layer, contradicting our prior expectation that the polymerization proceeds in the 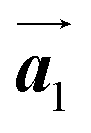 direction. In addition, a local mixture phase has been observed around the region indicated by black arrows in Fig. 4(b), in which the HPB polymer chains and Br2-HPB linear rows are alternately arranged along the [10] direction. These results indicate that the covalent bond is not formed between the nearest-neighbour bromophenyl rings along the
direction. In addition, a local mixture phase has been observed around the region indicated by black arrows in Fig. 4(b), in which the HPB polymer chains and Br2-HPB linear rows are alternately arranged along the [10] direction. These results indicate that the covalent bond is not formed between the nearest-neighbour bromophenyl rings along the 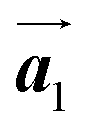 direction, but actually formed accompanied by the molecular rotations of about 60° to grow in the
direction, but actually formed accompanied by the molecular rotations of about 60° to grow in the 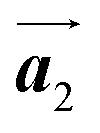 direction. Furthermore, from the chiral identification technique for the HPB polymer chains, we have revealed that the HPB polymer chains oriented in the
direction. Furthermore, from the chiral identification technique for the HPB polymer chains, we have revealed that the HPB polymer chains oriented in the 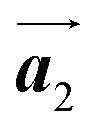 direction exhibit all-R chirality, as depicted by blue outlines in Fig. 4(b). Despite the heterochiral arrangement along the
direction exhibit all-R chirality, as depicted by blue outlines in Fig. 4(b). Despite the heterochiral arrangement along the 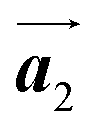 direction was observed in the self-assembled Br2-HPB layer before annealing,26 the homochiral HPB polymer chains oriented in the
direction was observed in the self-assembled Br2-HPB layer before annealing,26 the homochiral HPB polymer chains oriented in the 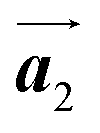 direction has been formed. This result suggests that the chiral transformations of HPB cores should be required, as well as the molecular rotations, during polymerization to form the homochiral HPB polymer chains oriented in the
direction has been formed. This result suggests that the chiral transformations of HPB cores should be required, as well as the molecular rotations, during polymerization to form the homochiral HPB polymer chains oriented in the 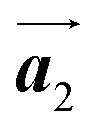 direction.
direction.
Discussion
Fig. 5(a)–(c) show space-filling models for the self-assembled structures of the Br2-HPB precursors, of the mixture, and of the HPB polymer chains, respectively. According to the well-accepted interpretation, we initially expected that the polymerization of Br2-HPB precursors was induced between the nearest-neighbour bromophenyl rings and proceeded along the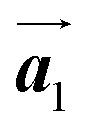 direction, but the actually produced polymer chains were aligned in the
direction, but the actually produced polymer chains were aligned in the 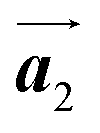 direction as shown in Fig. 5(b). This result indicates that the Br2-HPB molecules are rotated by about 60° to form the covalent bonds between debrominated substituents during the polymerization reactions, even though the dense packing in the self-assembled structure should restrict molecular diffusion. This is further supported by the fact that the remaining Br2-HPB molecules in the mixture phase in Fig. 5(b), which fit well between the HPB polymer chains, were oriented in the same direction as in the self-assembled Br2-HPB layer in Fig. 5(a).
direction as shown in Fig. 5(b). This result indicates that the Br2-HPB molecules are rotated by about 60° to form the covalent bonds between debrominated substituents during the polymerization reactions, even though the dense packing in the self-assembled structure should restrict molecular diffusion. This is further supported by the fact that the remaining Br2-HPB molecules in the mixture phase in Fig. 5(b), which fit well between the HPB polymer chains, were oriented in the same direction as in the self-assembled Br2-HPB layer in Fig. 5(a).
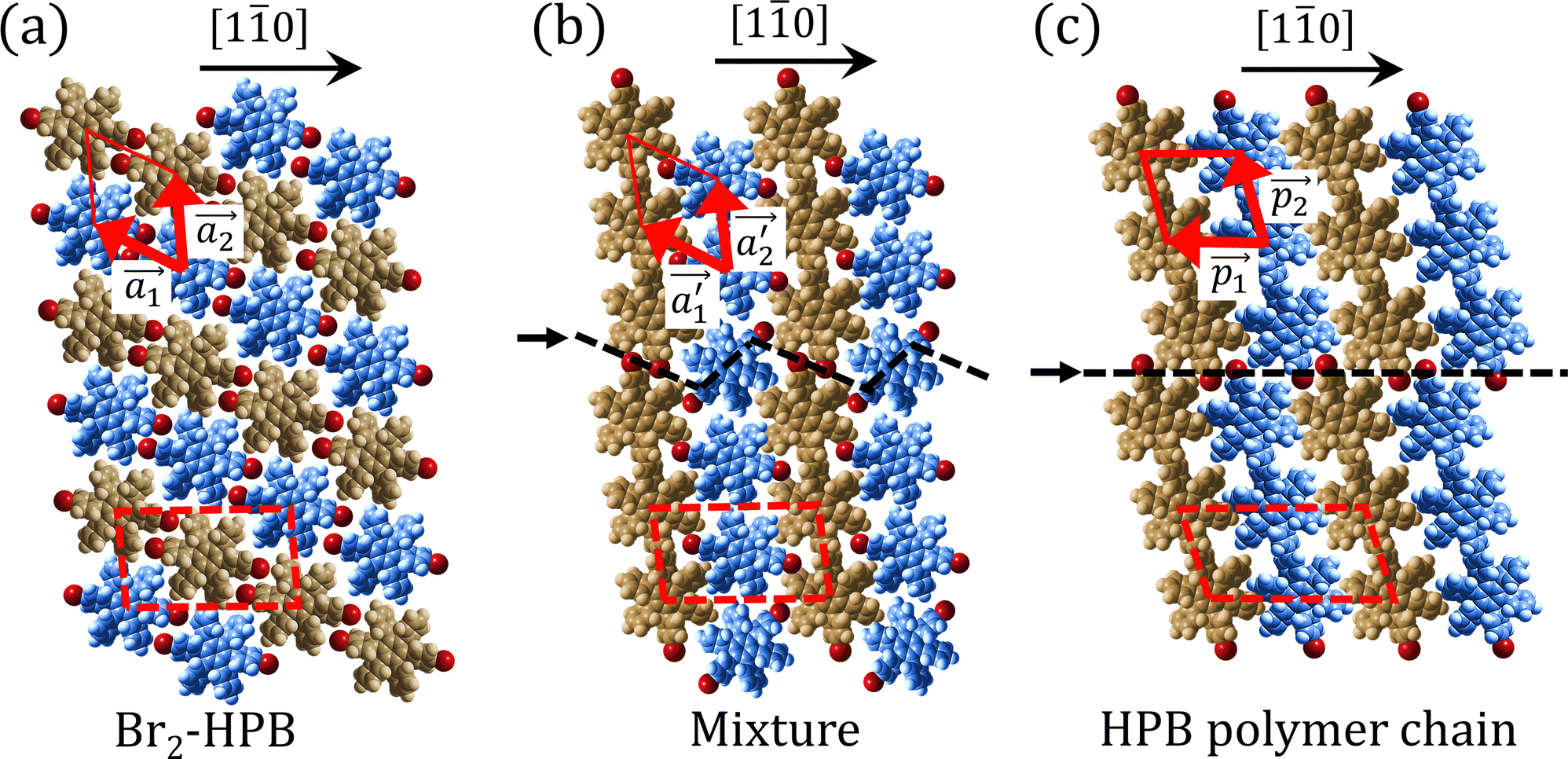 | ||
Fig. 5 The ordered self-assembled structures of (a) Br2-HPB precursors, (b) the mixture phase, and (c) HPB polymer chains. The local unit cell vectors are evaluated to be 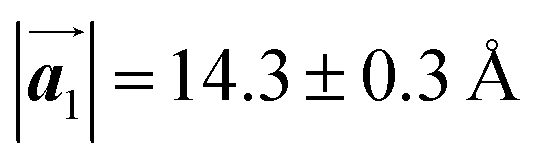 and and 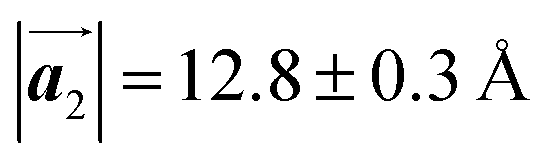 , and the included angle 57.2 ± 3.1° in (a), , and the included angle 57.2 ± 3.1° in (a), 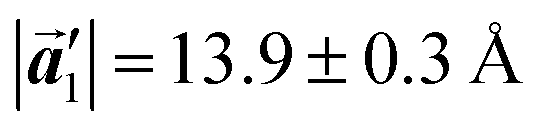 and and 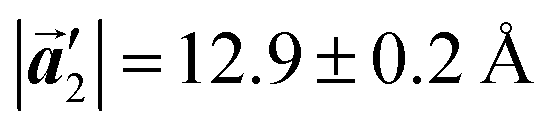 , and the included angle 54.6 ± 3.9° in (b), , and the included angle 54.6 ± 3.9° in (b), 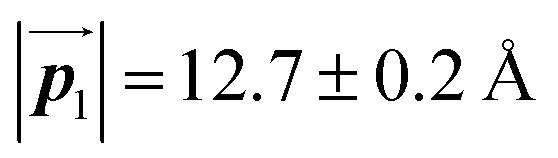 and and 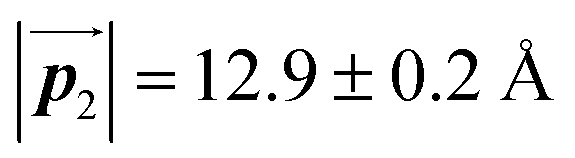 , and the included angle 75.8 ± 1.9° in (c). The blue and brown coloured models indicate the R- and L-type chiralities of HPB cores, respectively. For (b) and (c), the domain boundaries of the self-assembled structures, observed as dark lines in STM images, are indicated by black dashed lines and black arrows. The red dashed rectangles indicate the new local unit cells as centred rectangular lattices for (a) and (b), and a lattice , and the included angle 75.8 ± 1.9° in (c). The blue and brown coloured models indicate the R- and L-type chiralities of HPB cores, respectively. For (b) and (c), the domain boundaries of the self-assembled structures, observed as dark lines in STM images, are indicated by black dashed lines and black arrows. The red dashed rectangles indicate the new local unit cells as centred rectangular lattices for (a) and (b), and a lattice 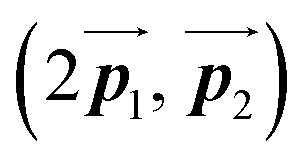 for (c). for (c). | ||
The polymerization pathway accompanied by the rotation of Br2-HPB molecules is analysed from the self-assembled structures before and after the reactions. The local unit cell vectors 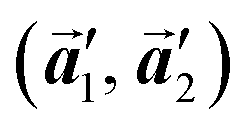 of the mixture phase, defined as in Fig. 5(b), resemble those
of the mixture phase, defined as in Fig. 5(b), resemble those 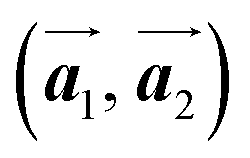 of Br2-HPB, suggesting the minimal disruption of the ordered array during polymerization. In particular, the HPB–HPB distance should be changed due to the covalent bonding through the polymerization reactions, which is estimated to be 12.9 Å by the quantum chemical calculations, corresponding to
of Br2-HPB, suggesting the minimal disruption of the ordered array during polymerization. In particular, the HPB–HPB distance should be changed due to the covalent bonding through the polymerization reactions, which is estimated to be 12.9 Å by the quantum chemical calculations, corresponding to 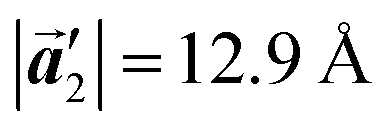 in the mixture phase. Note that this value is almost identical to the intermolecular distance of
in the mixture phase. Note that this value is almost identical to the intermolecular distance of 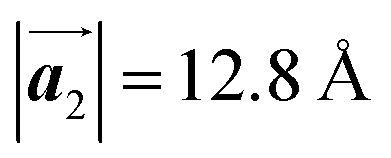 in the self-assembled structure of Br2-HPB precursors, but shorter than that of
in the self-assembled structure of Br2-HPB precursors, but shorter than that of 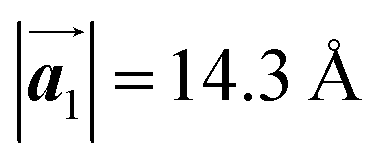 . Thus, if the polymerization reactions of Br2-HPB proceeded in the
. Thus, if the polymerization reactions of Br2-HPB proceeded in the 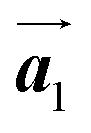 direction without large molecular rotation,
direction without large molecular rotation, 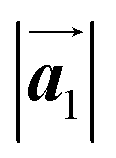 should decrease to 12.9 Å, reflecting about 13% lattice reduction along the polymer chains. This lattice mismatch might induce disorder arrangements through the polymerization. In addition to the better lattice match between
should decrease to 12.9 Å, reflecting about 13% lattice reduction along the polymer chains. This lattice mismatch might induce disorder arrangements through the polymerization. In addition to the better lattice match between 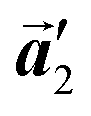 and
and 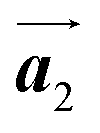 , the formation of almost the same length HPB polymer chains (3–4 HPB cores after 200 °C annealing) likely also contributes to the local order in the mixture phase. Such length-controlled polymerization should be associated with the very low reaction rate for debromination and successive covalent bonding of Br2-HPB at 200 °C due to the restricted molecular diffusion in the self-assembled layer. The slow reactions in a time range of hours allow most HPB polymer chains to reach the thermal equilibrium lengths, corresponding to 3–4 HPB cores at 200 °C. Furthermore, in the mixture phase, the same length HPB polymer chains and Br2-HPB molecules are alternately ordered, leading to the formation of the zigzag rows as shown in Fig. 3, and the domain boundary between the zigzag rows is likely stabilized by the bromophenyl–bromophenyl attractive interactions between HPB polymer chains, resulting in the two-dimensional arrangement of the zigzag rows.
, the formation of almost the same length HPB polymer chains (3–4 HPB cores after 200 °C annealing) likely also contributes to the local order in the mixture phase. Such length-controlled polymerization should be associated with the very low reaction rate for debromination and successive covalent bonding of Br2-HPB at 200 °C due to the restricted molecular diffusion in the self-assembled layer. The slow reactions in a time range of hours allow most HPB polymer chains to reach the thermal equilibrium lengths, corresponding to 3–4 HPB cores at 200 °C. Furthermore, in the mixture phase, the same length HPB polymer chains and Br2-HPB molecules are alternately ordered, leading to the formation of the zigzag rows as shown in Fig. 3, and the domain boundary between the zigzag rows is likely stabilized by the bromophenyl–bromophenyl attractive interactions between HPB polymer chains, resulting in the two-dimensional arrangement of the zigzag rows.
After the following annealing at 200 °C, the remaining Br2-HPB molecules in the mixture phase (see Fig. 5(b)) are also converted into HPB polymer chains, accompanied by the 60° rotation of Br2-HPB. As shown in Fig. 5(c), the same length HPB polymer chains pack side-by-side to form the straight rows, which is defined as the local unit cell vectors 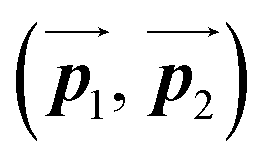 . Here, the
. Here, the 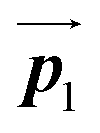 has changed in orientation from the
has changed in orientation from the 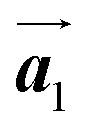 . Although the unit cell of self-assembled HPB polymer chains differs from the Br2-HPB lattice through the further polymerization, the local periodicity remains almost identical as if we took the new unit cells as the centred rectangular lattices for Fig. 5(a) and (b), and the
. Although the unit cell of self-assembled HPB polymer chains differs from the Br2-HPB lattice through the further polymerization, the local periodicity remains almost identical as if we took the new unit cells as the centred rectangular lattices for Fig. 5(a) and (b), and the 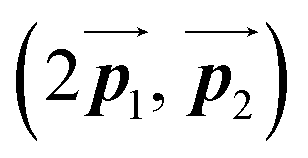 for Fig. 5(c) (see lower left in each model). The sizes of these new unit cells are 24.4 Å × 12.8 Å, 23.4 Å × 12.9 Å, and 25.0 Å × 12.9 Å, respectively. These almost identical local periodicities of the Br2-HPB precursors, of the mixture, and of the HPB polymer chains likely facilitate the conservation of the self-assembled array during polymerization. Consequently, the polymerization reactions of Br2-HPB can be completed without disruption of long-range ordering.
for Fig. 5(c) (see lower left in each model). The sizes of these new unit cells are 24.4 Å × 12.8 Å, 23.4 Å × 12.9 Å, and 25.0 Å × 12.9 Å, respectively. These almost identical local periodicities of the Br2-HPB precursors, of the mixture, and of the HPB polymer chains likely facilitate the conservation of the self-assembled array during polymerization. Consequently, the polymerization reactions of Br2-HPB can be completed without disruption of long-range ordering.
The chiral ordering of HPB cores was also transformed during the polymerization process in Fig. 5(a)–(c). Before the polymerization of Br2-HPB as shown in Fig. 5(a), due to the steric interactions of bromophenyl and phenyl rings, the R- or L-homochiral rows of Br2-HPB in the 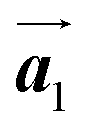 direction are formed along the linear alignment by bromophenyl–bromophenyl interactions, and the alternating ordering of the homochiral rows leads to the RL-heterochiral arrangement in the
direction are formed along the linear alignment by bromophenyl–bromophenyl interactions, and the alternating ordering of the homochiral rows leads to the RL-heterochiral arrangement in the 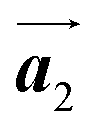 direction. On the other hand, the orientations of the chiral arrangements of HPB cores are rotated through the formation of mixture phase, as shown in Fig. 5(b). In the mixture phase, the R- or L-homochiral polymer chains are oriented in the
direction. On the other hand, the orientations of the chiral arrangements of HPB cores are rotated through the formation of mixture phase, as shown in Fig. 5(b). In the mixture phase, the R- or L-homochiral polymer chains are oriented in the 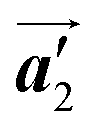 direction, whereas the homochiral rows were oriented in the
direction, whereas the homochiral rows were oriented in the 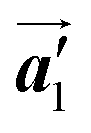 direction in the self-assembled Br2-HPB layer. This indicates that the chiral transformation occurs, simultaneously with the molecular rotations during polymerization. Furthermore, the chiral transformation is also expected for the remaining Br2-HPB molecules between the HPB polymer chains. The remaining Br2-HPB should possess the R- or L-homochiral arrangements along the
direction in the self-assembled Br2-HPB layer. This indicates that the chiral transformation occurs, simultaneously with the molecular rotations during polymerization. Furthermore, the chiral transformation is also expected for the remaining Br2-HPB molecules between the HPB polymer chains. The remaining Br2-HPB should possess the R- or L-homochiral arrangements along the 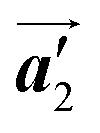 direction, which is opposite to the chirality of HPB polymer chains, minimising intermolecular steric hindrance with neighbouring HPB polymer chains as shown in Fig. 5(b). The R- or L-homochiral arrangements in the
direction, which is opposite to the chirality of HPB polymer chains, minimising intermolecular steric hindrance with neighbouring HPB polymer chains as shown in Fig. 5(b). The R- or L-homochiral arrangements in the 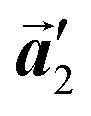 direction of Br2-HPB are different from the RL-heterochiral arrangement of Br2-HPB in the
direction of Br2-HPB are different from the RL-heterochiral arrangement of Br2-HPB in the 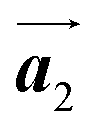 direction before the polymerization reactions, suggesting the chiral transformation of Br2-HPB during polymerization. In the [10] direction, the heterochiral arrangement in the mixture phase, composed of the R- and L-homochiral rows along the
direction before the polymerization reactions, suggesting the chiral transformation of Br2-HPB during polymerization. In the [10] direction, the heterochiral arrangement in the mixture phase, composed of the R- and L-homochiral rows along the 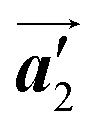 direction, remains even after the polymerization is accomplished as shown in Fig. 5(c). Thus, we successfully elucidated that the polymerization process of self-assembled Br2-HPB on Au(111) involves molecular rotation and chiral switching. In addition, the chiral ordering has been also observed across the domain boundaries in the mixture phase and the self-assembled HPB polymer chains by STM (not shown). As shown in Fig. 5(b), the R- or L-homochiral sequences of the HPB polymer chains and the Br2-HPB molecules in the mixture phase extend beyond the domain boundaries almost along the
direction, remains even after the polymerization is accomplished as shown in Fig. 5(c). Thus, we successfully elucidated that the polymerization process of self-assembled Br2-HPB on Au(111) involves molecular rotation and chiral switching. In addition, the chiral ordering has been also observed across the domain boundaries in the mixture phase and the self-assembled HPB polymer chains by STM (not shown). As shown in Fig. 5(b), the R- or L-homochiral sequences of the HPB polymer chains and the Br2-HPB molecules in the mixture phase extend beyond the domain boundaries almost along the 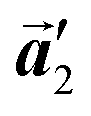 direction. In contrast, for the self-assembled HPB polymer chains in Fig. 5(c), we found the RL-heterochiral arrangement through the bromophenyl–bromophenyl contacts at the domain boundaries almost along the
direction. In contrast, for the self-assembled HPB polymer chains in Fig. 5(c), we found the RL-heterochiral arrangement through the bromophenyl–bromophenyl contacts at the domain boundaries almost along the 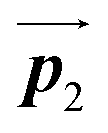 direction. These distinct chiral orderings of HPB polymer chains observed at the domain boundaries likely arise from opposite contact orientations between the bromophenyl substituents, in which the HPB polymer chains adjacent to above are positioned at the upper right in Fig. 5(b) for the mixture phase, and at the upper left in Fig. 5(c) for the self-assembled HPB polymer chains. In addition, we have observed that the orientations of the HPB polymer chains with respect to the [10] direction are slightly different in the mixture phase and in the self-assembled HPB polymer chains. Thus, the chiral ordering across the boundaries suggests the influence of contact configurations between bromophenyl substituents on the chiral coupling at domain boundary.
direction. These distinct chiral orderings of HPB polymer chains observed at the domain boundaries likely arise from opposite contact orientations between the bromophenyl substituents, in which the HPB polymer chains adjacent to above are positioned at the upper right in Fig. 5(b) for the mixture phase, and at the upper left in Fig. 5(c) for the self-assembled HPB polymer chains. In addition, we have observed that the orientations of the HPB polymer chains with respect to the [10] direction are slightly different in the mixture phase and in the self-assembled HPB polymer chains. Thus, the chiral ordering across the boundaries suggests the influence of contact configurations between bromophenyl substituents on the chiral coupling at domain boundary.
Conclusions
In conclusion, we investigated the influence of self-assembled ordering on the polymerization reactions of Br2-HPB on the Au(111) surface. While the close proximity of the bromophenyl substituents is formed by the attractive bromophenyl–bromophenyl interactions in the self-assembled layer of Br2-HPB, we found that the on-surface debromination and successive polymerization reactions of Br2-HPB proceed accompanied by about 60° rotation of the molecules. Concurrently with the polymerization accompanied by rotation of self-assembled Br2-HPB, the chirality switching of HPB cores have also occurred, forming the homochiral HPB polymer chains from the heterochiral arranged Br2-HPB precursors. It should be noted that the HPB polymer chains were formed without disruption of the ordered array even though the HPB–HPB distance was changed by polymerization. Our experimental results revealed that the intramolecular HPB–HPB periodicity (12.9 Å) of the HPB polymer chains is almost identical to the lattice size (12.8 Å) of the self-assembled Br2-HPB precursors in the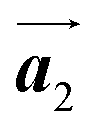 direction. The lattice conservation can be achieved by the 60° rotation of Br2-HPB during polymerization, which has been observed throughout the polymerization of Br2-HPB, such as in the mixture phase of Br2-HPB and HPB polymer chains, and in the self-assembled HPB polymer chains. Thus, we conclude that the lattice conservation of the self-assembled structures dominates the polymerization of Br2-HPB, which is accompanied by the 60° molecular rotations, although it has been believed that the polymerization reactions tend to undergo between adjacent bromophenyl rings within the densely packed self-assembled layer. In addition, we observed that the tendency of the long-range ordering of the HPB polymer chains is maintained even after the cyclodehydrogenation reactions to be transformed from HPB into hexabenzocoronene (HBC) by 400 °C annealing. As reported previously, the obtained HBC chains were aligned in almost the same direction, and local ordering was also observed.26 On the basis of our findings, on-surface reactions controlled by self-assembled ordering hold great potential for forming an ordered array of molecular nanostructures.
direction. The lattice conservation can be achieved by the 60° rotation of Br2-HPB during polymerization, which has been observed throughout the polymerization of Br2-HPB, such as in the mixture phase of Br2-HPB and HPB polymer chains, and in the self-assembled HPB polymer chains. Thus, we conclude that the lattice conservation of the self-assembled structures dominates the polymerization of Br2-HPB, which is accompanied by the 60° molecular rotations, although it has been believed that the polymerization reactions tend to undergo between adjacent bromophenyl rings within the densely packed self-assembled layer. In addition, we observed that the tendency of the long-range ordering of the HPB polymer chains is maintained even after the cyclodehydrogenation reactions to be transformed from HPB into hexabenzocoronene (HBC) by 400 °C annealing. As reported previously, the obtained HBC chains were aligned in almost the same direction, and local ordering was also observed.26 On the basis of our findings, on-surface reactions controlled by self-assembled ordering hold great potential for forming an ordered array of molecular nanostructures.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Grants-in-Aid for Scientific Research 20K05331, 20K15175, and 23K04585 from Japan Society for the Promotion of Science (JSPS).References
- S. Kubatkin, A. Danilov, M. Hjort, J. Cornil, J.-L. Brédas, N. Stuhr-Hansen, P. Hedegard and T. Bjornholm, Nature, 2003, 425, 698 CrossRef CAS PubMed.
- J. V. Barth, G. Costantini and K. Kern, Nature, 2005, 437, 671 CrossRef CAS PubMed.
- V. Barone, O. Hod and G. E. Scuseria, Nano Lett., 2006, 6, 2748 CrossRef CAS PubMed.
- M. Y. Han, B. Özyilmaz, Y. Zhang and P. Kim, Phys. Rev. Lett., 2007, 98, 206805 CrossRef PubMed.
- L. Lafferentz, F. Ample, H. Yu, S. Hecht, C. Joachim and L. Gril, Science, 2009, 323, 1193 CrossRef CAS PubMed.
- P. Ruffieux, J. Cai, N. C. Plumb, L. Patthey, D. Prezzi, A. Ferretti, E. Molinari, X. Feng, K. Müllen, C. A. Pignedoli and R. Fasel, ACS Nano, 2012, 6, 6930 CrossRef CAS PubMed.
- D. Jariwala, V. K. Sangwan, L. J. Lauhon, T. J. Marks and M. C. Hersam, Chem. Soc. Rev., 2013, 42, 2824 RSC.
- A. Maghsoumi, A. Narita, R. Dong, X. Feng, C. Castiglioni, K. Müllen and M. Tommasini, Phys. Chem. Chem. Phys., 2016, 18, 11869 RSC.
- J. Cai, P. Ruffieux, R. Jaafar, M. Bieri, T. Braun, S. Blankenburg, M. Muoth, A. P. Seitsonen, M. Saleh, X. Feng, K. Müllen and R. Fasel, Nature, 2010, 466, 470 CrossRef CAS PubMed.
- L. Lafferentz, V. Eberhardt, C. Dri, C. Africh, G. Comelli, F. Esch, S. Hecht and L. Grill, Nat. Chem., 2012, 4, 215 CrossRef CAS PubMed.
- S. Kawai, S. Saito, S. Osumi, S. Yamaguchi, A. S. Foster, P. Spijker and E. Meyer, Nat. Commun., 2015, 6, 8098 CrossRef CAS PubMed.
- L. Lafferentz, V. Eberhardt, C. Dri, C. Africh, G. Comelli, T. Dienel, L. Talirz, P. Shinde, C. A. Pignedoli, D. Passerone, T. Dumslaff, X. Feng, K. Müllen and R. Fasel, Nature, 2016, 531, 489 CrossRef PubMed.
- Q. Sun, R. Zhang, J. Qiu, R. Liu and W. Xu, Adv. Mater., 2018, 30, 1705630 CrossRef PubMed.
- M. D. Giovannantonio, Q. Chen, J. I. Urgel, P. Ruffieux, C. A. Pignedoli, K. Müllen, A. Narita and R. Fasel, J. Am. Chem. Soc., 2020, 142, 12925 CrossRef PubMed.
- H. Y. Gao, H. Wagner, D. Zhong, J.-H. Franke, A. Studer and H. Fuchs, Angew. Chem., Int. Ed., 2013, 52, 4024 CrossRef CAS PubMed.
- Q. Fan, C. Wang, Y. Han, J. Zhu, J. Kuttner, G. Hilt and J. Michael Gottfried, ACS Nano, 2014, 8, 709 CrossRef CAS PubMed.
- A. Basagni, F. Sedona, C. A. Pignedoli, M. Cattelan, L. Nicolas, M. Casarin and M. Sambi, J. Am. Chem. Soc., 2015, 137, 1802 CrossRef CAS PubMed.
- T. Wang, H. Lv, Q. Fan, L. Feng, X. Wu and J. Zhu, Angew. Chem., Int. Ed., 2017, 56, 4762 CrossRef CAS PubMed.
- J. Huang, Y. Pan, T. Wang, S. Cui, L. Feng, D. Han, W. Zhang, Z. Zeng, X. Li, P. Du, X. Wu and J. Zhu, ACS Nano, 2021, 15, 4617 CrossRef CAS PubMed.
- M. Yano, S. Yasuda, K. Fukutani and H. Asaoka, RSC Adv., 2023, 13, 14089 RSC.
- M. Ammon, T. Sander and S. Maier, J. Am. Chem. Soc., 2017, 139, 12976 CrossRef CAS PubMed.
- X. Zhou, C. Wang, Y. Zhang, F. Cheng, Y. He, Q. Shen, J. Shang, X. Shao, W. Ji, W. Chen, G. Xu and K. Wu, Angew. Chem., Int. Ed., 2017, 56, 12852 CrossRef CAS PubMed.
- Q. Fan, L. Yan, M. W. Tripp, O. Krejci, S. Dimosthenous, S. R. Kachel, M. Chen, A. S. Foster, U. Koert, P. Liljeroth and J. M. Gottfried, Science, 2021, 372, 852 CrossRef CAS PubMed.
- T. Wang, Q. Fan and J. Zhu, J. Phys. Chem. Lett., 2023, 14, 2251 CrossRef CAS PubMed.
- Q. Chen, J. R. Cramer, J. Liu, X. Jin, P. Liao, X. Shao, K. V. Gothelf and K. Wo, Angew. Chem., Int. Ed., 2017, 56, 5026 CrossRef CAS PubMed.
- H. Ooe, K. Ikeda and T. Yokoyama, J. Phys. Chem. C, 2023, 127, 7659 CrossRef CAS.
- F. Cheng, X.-J. Wu, Z. Hu, X. Lu, Z. Ding, Y. Shao, H. Xu, W. Ji, J. Wu and K. P. Loh, Nat. Commun., 2018, 9, 4871 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp00696h |
| This journal is © the Owner Societies 2024 |