
Reinvestigation of the internal glycan rearrangement of Lewis a and blood group type H1 epitopes†
Vasilis
Kontodimas
a,
Murat
Yaman
 ac,
Kim
Greis
bde,
Maike
Lettow
bd,
Kevin
Pagel
bd and
Mateusz
Marianski
*ac
ac,
Kim
Greis
bde,
Maike
Lettow
bd,
Kevin
Pagel
bd and
Mateusz
Marianski
*ac
aDepartment of Chemistry, Hunter College, The City University of New York, New York, NY 10065, USA. E-mail: mateusz.marianski97@login.cuny.edu
bFritz-Haber-Intitut der Max Planck Gesellschaft, 14195 Berlin, Germany
cPh.D. Programs in Chemistry and Biochemistry, The Graduate Center, The City University of New York, New York, NY 10016, USA
dInstitute of Chemistry and Biochemistry, Freie Universität Berlin, 14195 Berlin, Germany
eLaboratory of Organic Chemistry, Department of Chemistry and Applied Biosciences, ETH Zürich, 8093 Zürich, Switzerland
First published on 24th April 2024
Abstract
Protonated ions of fucose-containing oligosaccharides are prone to undergo internal glycan rearrangement which results in chimeric fragments that obfuscate mass-spectrometric analysis. Lack of accessible tools that would facilitate systematic analysis of glycans in the gas phase limits our understanding of this phenomenon. In this work, we use density functional theory modeling to interpret cryogenic IR spectra of Lewis a and blood group type H1 trisaccharides and to establish whether these trisaccharides undergo the rearrangement during gas-phase analysis. Structurally unconstrained search reveals that none of the parent ions constitute a thermodynamic global minimum. In contrast, predicted collision cross sections and anharmonic IR spectra provide a good match to available experimental data which allowed us to conclude that fucose migration does not occur in these antigens. By comparing the predicted structures with those obtained for Lewis x and blood group type H2 epitopes, we demonstrate that the availability of the mobile proton and a large difference in the relative stability of the parent ions and rearrangement products constitute the prerequisites for the rearrangement reaction.
Introduction
Carbohydrates – or glycans – play a range of modulatory roles in biological systems, such as mediating intercellular communication,1 cell adhesion,2 and signaling to the extracellular matrix.3 Accordingly, changes in the glycome – the sum of all glycans on a cell surface – are associated with cancer,4 immune response,5 or acquired immunity.6 These signaling functions are facilitated by complex assembly rules and the glycans’ large structural heterogeneity;7 understanding the relationship between their structural details and derived function constitutes one of the major goals of glycomics.8Resolving the structure of an unknown glycan frequently requires a combination of several orthogonal separation and characterization techniques: liquid chromatography,9–12 mass spectrometry (MS) and multistage MS (MSn), possibly augmented by specialized fragmentation techniques,13–16 ion-mobility spectrometry (IMS),17 or molecular ion spectroscopy.18–20 After the initial separation, the information about the structure of an analyte is recorded in its fragmentation MS spectrum. Crucially for the structural assignment, this spectrum consists of bond- and ring-crossing fragments which can be used to reconstruct the analyte's composition, as well as connectivity and configuration of the glycosidic bonds.20–24 However, it has been observed that – in a process similar to peptide scrambling25 – electrospray ionization (ESI) or collision-induced dissociation (CID) can alter the glycan sequence which results in additional non-native fragments being recorded in the mass spectrum.26–33 Although the phenomenon is associated mostly with fucose-containing protonated glycans – hence it is often called fucose migration28,34–37 – similar rearrangements have been observed for rhamnose,38 mannose,39 and xylose.40 The structural motifs that trigger this rearrangement, as well as its mechanism and likelihood of occurrence, remain unknown after more than 40 years since their first observation.30,31,41
Glycans that originate from so-called Type 1 (T1) or Type 2 (T2) precursors can be particularly affected by this rearrangement. These disaccharides constitute the core structure of blood group epitopes which are common structural motifs present in N- and O-glycans and are associated with glycan modulatory42–44 and recognition45,46 functions. The main difference between these two precursors is in the type of glycosidic linkage connecting two saccharides, D-galactopyranose (Gal) and D-N-acetylglucosamine (GlcNAc). In the T1 antigen, Gal is connected to GlcNAc with the β(1 → 3) glycosidic bond, whereas in the T2 antigen the same building blocks are connected with the β(1 → 4) bond instead. These precursors are typically modified by a set of fucosyl transferases which attach a fucose moiety, a C6-deoxy L-monosaccharide, to produce Lewis or blood group type H antigens (Fig. 1A). In general, T1-derived antigens are synthesized and expressed in neutrophils, lymphocytes, and monocytes, whereas T2-derived antigens are mainly expressed in epithelial tissues, such as in the intestines, skin, and oral mucosa.47 Importantly, the functions of these glycans are communicated by the additional fucose moiety which is particularly prone to migration and can impact the reliability of the mass-spectrometric analysis.28,34,36,37
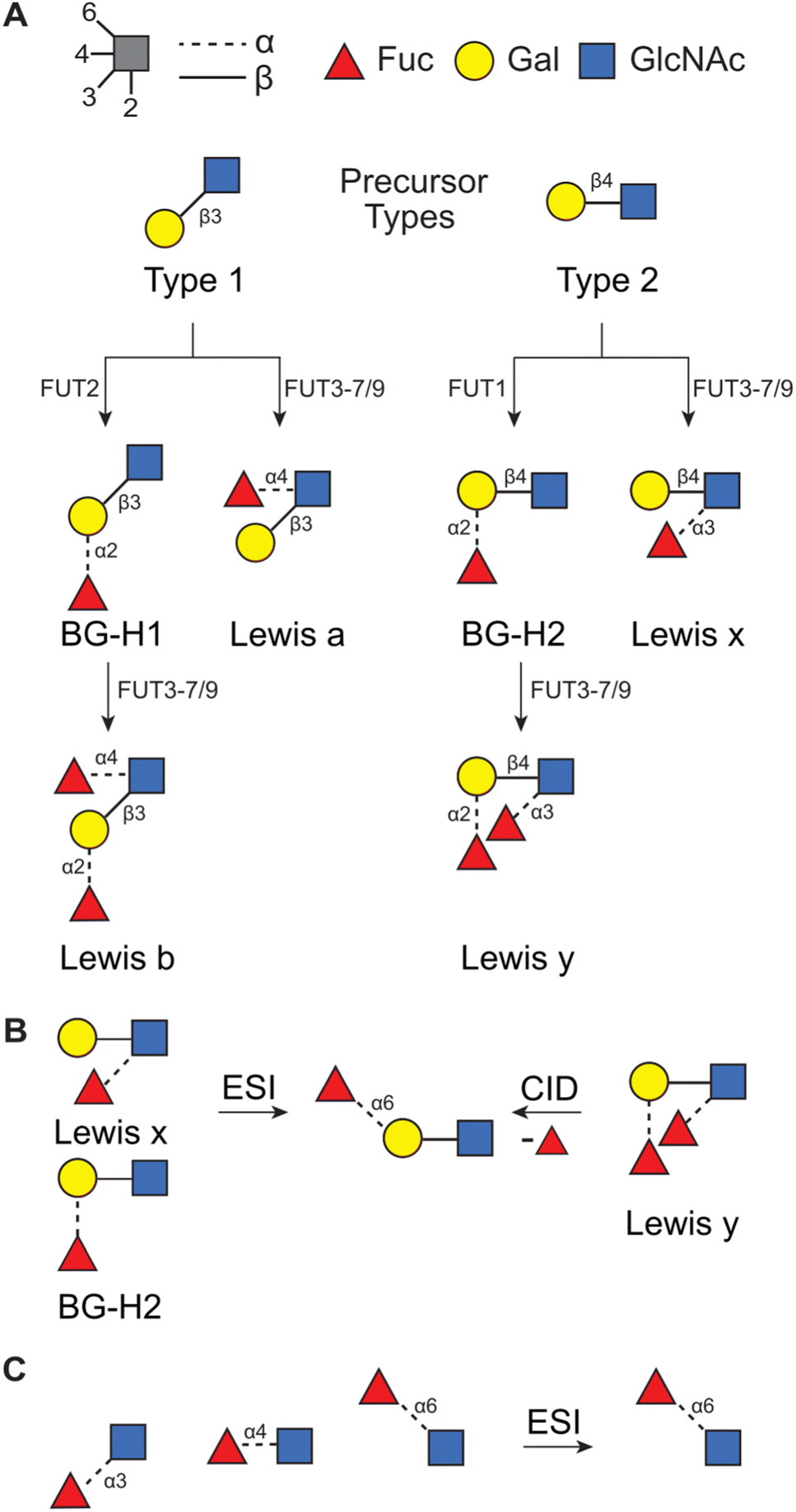 | ||
| Fig. 1 (A) The synthetic pathway for T1- and T2-derived antigens. Products of fucose migration in (B) T2-derived antigens32 and in (C) a series of Fuc → GlcNAc disaccharides.33 | ||
Recently, we have used a combination of experimental (IMS, radical-directed dissociation (RDD) MS, and cryogenic-ion IR spectroscopy), and theoretical (density functional theory (DFT) calculations) techniques to determine the fucose migration products in T2-derived Lewis x (Lex) and blood group antigen H2 (BG-H2).31,32,48 First, we have reported that the protonated [Lex + H]+ and [BG-H2 + H]+ trisaccharide ions, which differ in the point of attachment of the fucosyl group (Fig. 1B), yield identical mid-IR fingerprints. Because two structurally distinct glycans must present unique IR signatures,49 we concluded that either one or both glycans undergo a rearrangement reaction which leads to a common product.31 This reaction is promoted by a labile proton, whereas a lithium cation, a sodium cation, and an immobilized proton inhibit the rearrangement.48 Recently, we have compared the experimental collision cross sections (CCSs) and cryogenic IR spectra against the theoretical predictions simulated with DFT.32 The calculations revealed that both Lex and BG-H2 trisaccharide ions undergo rearrangement which leads to the formation of a new, non-native ion (Fig. 1B). To deduce the structure of the rearrangement product, we considered 16 possible glycosidic linkages between the fucose and the T2 core and we found that the best match between the experimental and predicted IR spectra was obtained by an isomer which has a fucose attached with the α(1 → 6) glycosidic bond to the terminal galactose. Because this isomer constitutes a global thermodynamic minimum among all regio- and stereoisomers of the fucosylated T2 glycans, we concluded that the rearrangement of the intact ion is driven by an energy gradient leading to a more stable isomer. Moreover, the rearrangement does not require CID activation and occurs in intact glycan ions, most likely during the ESI process. In a similar study, Moge et al. observed that protonated Fuc → GlcNAc disaccharides rearrange to a more stable Fucα(1 → 6) GlcNAc isomer (Fig. 1C).33
In this work, we aim to deepen our understanding of the structural motifs that drive the fucose migration by interrogating whether a similar phenomenon occurs in the T1-derived antigens. By comparing the likelihood of the migration and its tentative products in these two model systems, i.e. T1- and T2-derived antigens, we are able to trace potential reaction triggers. A different glycosidic linkage between Gal and GlcNAc in Lea and BG-H1 antigens alters the spatial arrangement and conformational flexibility of the analyte ions which affects the mobility of the proton and the relative stability of their regio- and stereoisomers. The mechanisms proposed in the literature include either a ring-opening step50 or a transfer of the fucosyl group in a single step.51 Both mechanisms require a specific spatial arrangement of the mobile proton and the fucose moiety to promote the first step of the rearrangement reaction – either protonation of the fucose ring oxygen or its glycosidic bond.32
Previously published CCSs52 and cryogenic IR spectra of the BG-H1 and Lea antigens31 were, without the context provided by the theoretical calculations, inconclusive. In the study of Mucha et al., the IR spectra of the two ions showed enough difference to conclude that the fucose rearrangement is rather unlikely.31 In contrast, Manz et al. reported identical CCSs of Lea and BG-H1 protonated ions which was explained as the fucose migration phenomenon leading to a common ion.52 Here, by comparing these experimental data against the DFT-predicted reference values, we demonstrate that the change in the glycan connectivity results in the inhibition of the internal rearrangement during the cryogenic ion IR spectroscopy measurement. This is mainly caused by impairing the mobility of the proton, which – in lowest-energy conformers of [BG-H1 + H]+ and [Lea + H]+ – does not have access to the fucose moiety. Instead of being a part of an extensive H-bond network, the proton is contained in a tight H-bond between the amide group and GlcNAc ring oxygen. The fact that not only the composition of glycans but also their connectivity defines their fragmentation properties has important implications for MS-based glycomics.
Methods
The overview of the conformational search protocol for the most stable isomers of the fucosylated T1 antigen is shown in Fig. 2. First, we have assembled 28 regio- and stereoisomers which differed with respect to the type of the glycosidic bond between the fucose moiety and T1 core, and the orientation of the hydroxy group at the anomeric carbon. The (1 → 1) glycosidic bond was excluded from the analysis. The amide group was set in a trans configuration, and the additional proton was added to its carbonyl oxygen which is the most basic protonation site and was found to be a proton carrier in the T2 antigens.32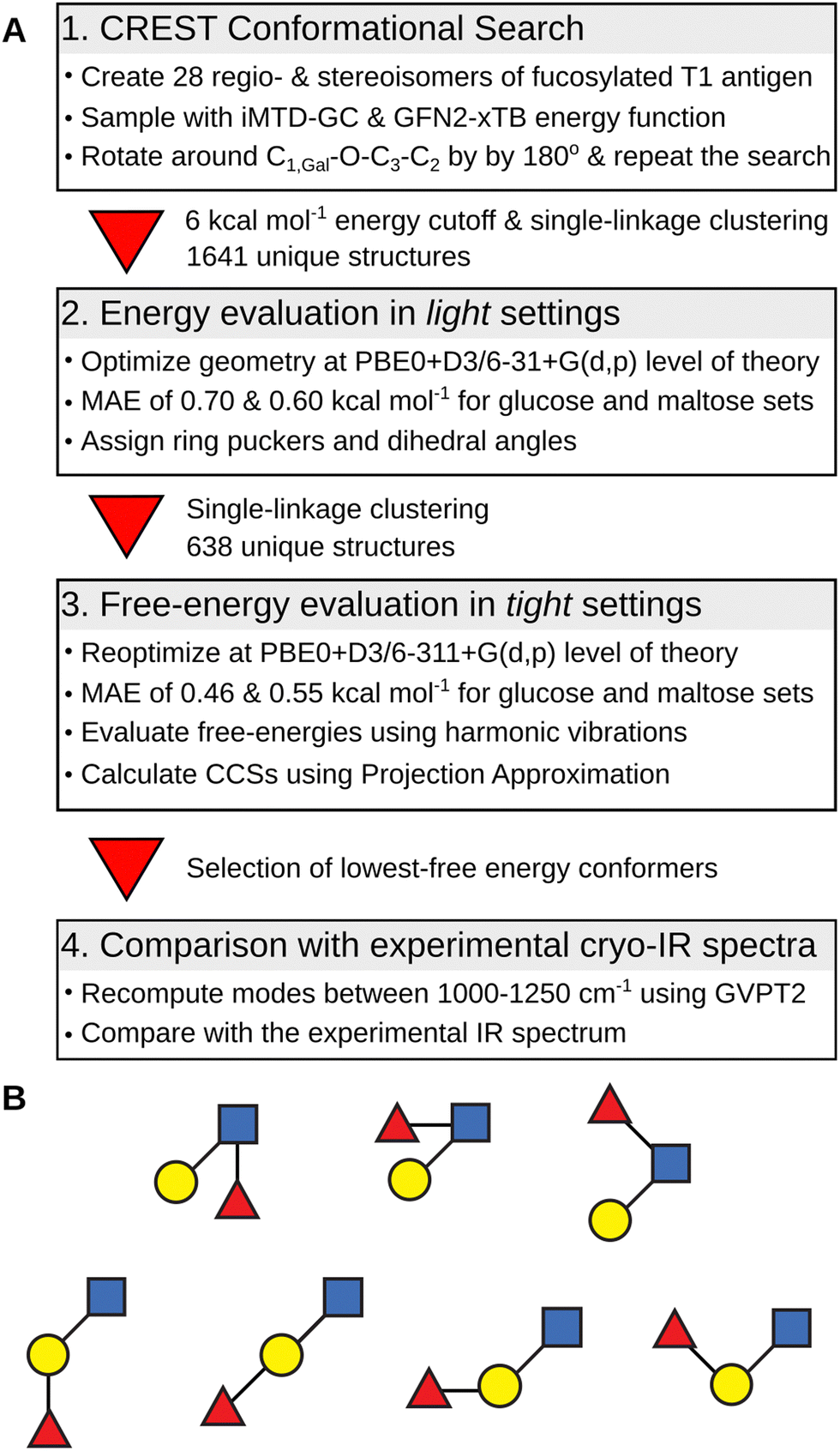 | ||
| Fig. 2 (A) The conformational search protocol used for sampling of the structural space of the fucosylated T1 antigens. (B) Investigated isomers of fucosylated T1 antigens in SNFG notation. Bond configuration is omitted for clarity. | ||
The conformational search protocol has been implemented in the CarPpy Python package.53,54 We start by performing a structural search using CREST which uses iterative metadynamics simulations and genetic operations to sample molecular conformational space.55 For the energy function, we have selected the semiempirical GNF2-xTB method, which is available in the xtb package,56 because of its excellent performance for the mono- and disaccharide benchmark set.57,58 In the previous work on Lex and BG-H2, we observed that the outcome of sampling with CREST strongly depends on the starting structure, and the rotation around the β(1 → 4) glycosidic bond between Gal and GlcNAc constitutes the highest-energy barrier.32 Thus, to improve the sampling of the conformational space of T1-derived antigens, we complemented each structural search with a second run that was initiated from a modified lowest-energy structure found in the first step. This modification included rotation around the C1,Gal−O−C3−C2 dihedral angle of the β(1 → 3) glycosidic by approximately 180 degrees. In 25% of systems, this approach resulted in an increasing number of unique conformers found by CREST (Table S1, ESI†). Next, for each pair of searches, we collected all conformers within the energy window of 6 kcal mol−1 above the lowest energy structures, clustered them using a single-linkage algorithm, merged both searches, and removed duplicates. The clustering used energy-dependent root-mean-squared deviation (RMSD) cutoffs of 1.5, 2.0, and 2.5 Å2, for structures within, respectively, 0.0 and 2.0, 2.0 and 4.0, and above 4.0 kcal mol−1 the lowest-energy structure. The energy-dependent cutoff selects more low-energy conformers that differ by few structural details and limits the number of higher-energy conformers to only representative structures. These structures were then subjected to geometry optimizations using Gaussian 1659 using the PBE0 hybrid exchange–correlation functional augmented with Grimme's D3 dispersion correction and a 6-31+G(d,p) basis set.60,61 This level of theory, which we abbreviate as light settings, shows an average 0.65 kcal mol−1 mean absolute error (MAE) when tested on a benchmark set of 205 monosaccharides and 223 disaccharides (Fig. S1 and S2, ESI†).57 The selection of structures used in the benchmark set is discussed in more detail therein. For each optimized structure we computed theoretical CCS using the projection approximation implemented in the Sigma code (PACCS),62 and analyzed glycosidic bond orientations and ring puckers.63 The functions required to assign ring puckers have been implemented in CarPpy.54 These conformational hierarchies and resulting distributions of dihedral angles and ring puckers are shown in Fig. S3–S16 (ESI†). The number of structures that have been obtained for each isomer at respective stages of the search are reported in Table S2 (ESI†).
In the next step, we increased the level of theory and derived the harmonic free-energies of most stable conformers. All structures of each isomer were clustered again using tighter criteria of 1.0, 1.5, and 2.0 Å2 RMSD cutoffs and 0.0–2.0, 2.0–5.0, and 5.0 < kcal mol−1 energy windows. Unique structures were then reoptimized in a larger 6-311+G(d,p) basis set, which we defined as tight settings, that were then used for the evaluation of harmonic frequencies. These settings provide an average MAE of 0.50 kcal mol−1 for the mono- and disaccharide benchmark set, and a more accurate free-energy estimate.64 The accuracy of the single-point relative energies of most stable conformers of these ions was further validated against the RI-MP2/CBS(3,4) level of theory (Table S3, ESI†): PBE0 + D3 yielded the smallest MAE of 0.30 kcal mol−1 among 12 tested density functionals. The resulting conformational hierarchies of [Lea + H]+, [BG-H1 + H]+, and three other non-native trisaccharide ions, Fucα(1 → 6)Galβ(1 → 3)GlcNAc ([a16 + H]+) and Galβ(1 → 3)[Fucα(1 → 6)]GlcNAc ([an16 + H]+) are shown in Fig. 3, while the conformational hierarchies of the remaining isomers are shown in Fig. S17–S20 (ESI†). A summary of the relative free energies of the most stable isomers and the differences between the experimental CCS of 144 Å2,52 and their predicted PACCSs is shown in Table 1.
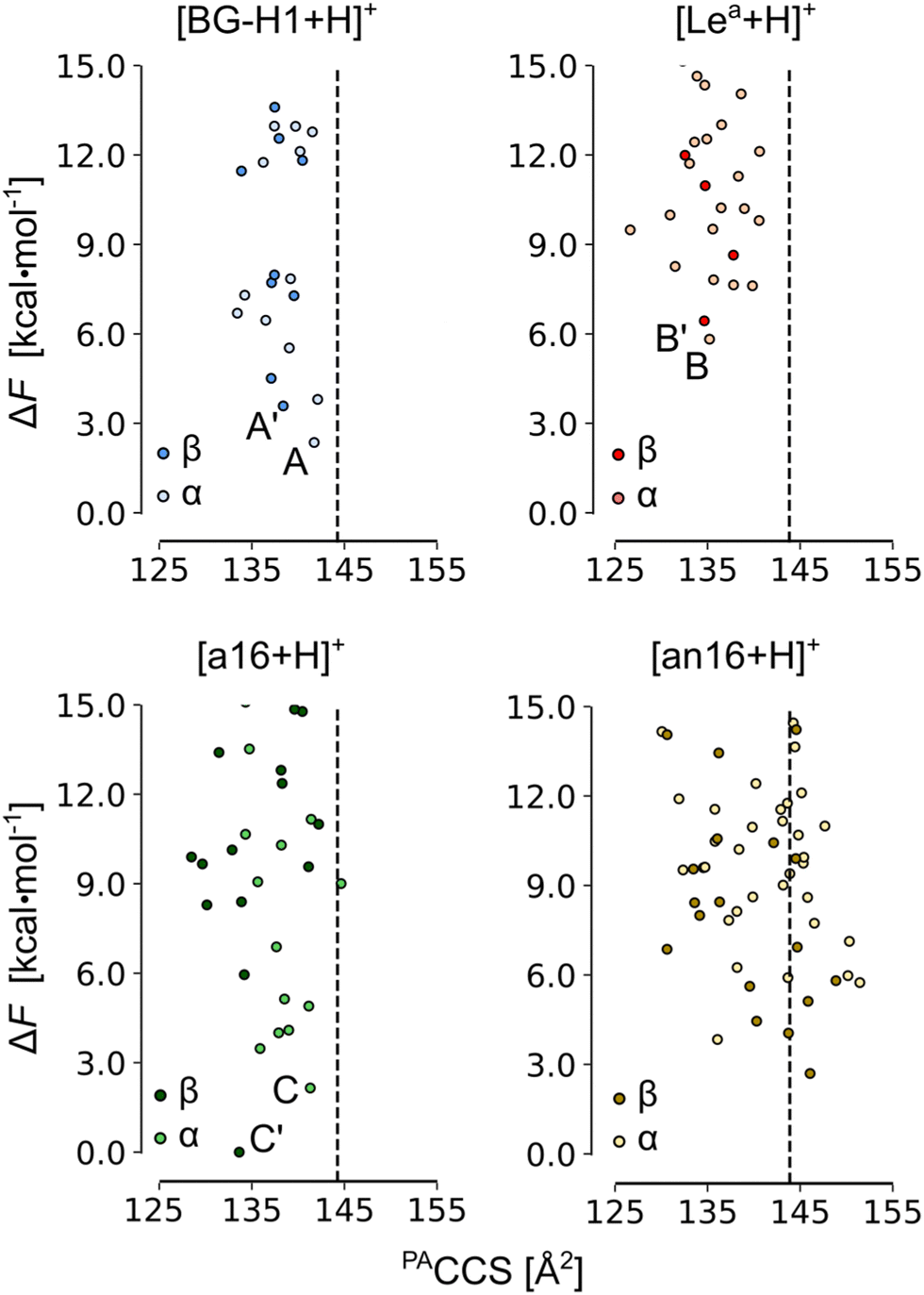 | ||
| Fig. 3 The PACCS vs. relative free energy (ΔF) of [Lea +H]+, [BG-H1 + H]+, [a16 + H]+, and [an16 + H]+. The free energies were computed at the PBE0+D3/6-311+G(d,p) level of theory. The dashed line indicates the experimental DTCCS of 144 Å2, and the free energies are plotted relative to the lowest-free energy conformer of [β-a16 + H]+. The labels are explained in Table 1. Conformers for which IR spectra and structures are discussed in the text are highlighted. | ||
| Anomer | Anomer | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| α | β | α | β | ||||||||
| Fuc | Label | ΔF | ΔC | ΔF | ΔC | Fuc | Label | ΔF | ΔC | ΔF | ΔC |
| α1 → 2N | — | 6.0 | 4 | 7.0 | 1 | β1 → 2N | — | 8.0 | 5 | 10.5 | 0 |
| α1 → 4N | Lea | 5.8 | −8 | 6.4 | −8 | β1 → 4N | — | 4.5 | −15 | 3.6 | −1 |
| α1 → 6N | an16 | 3.8 | −6 | 2.5 | 2 | β1 → 6N | bn16 | 1.3 | −10 | 0.5 | 0 |
| α1 → 2G | BG-H1 | 2.4 | 0 | 3.6 | −3 | β1 → 2G | b12 | 3.3 | −5 | 2.3 | 0 |
| α1 → 3G | — | 6.9 | 2 | 4.4 | 0 | β1 → 3G | — | 6.8 | 1 | 4.1 | 4 |
| α1 → 4G | — | 7.1 | −5 | 5.5 | −11 | β1 → 4G | b14 | 6.3 | −5 | 1.7 | −3 |
| α1 → 6G | a16 | 2.1 | −2 | 0.0 | −8 | β1 → 6G | — | 4.2 | −2 | 4.1 | −7 |
In the last step, we selected 18 of the lowest free-energy conformers of the ions shown in Fig. 3 to calculate the anharmonic vibrational spectrum using the generalized vibrational perturbation theory (GVPT2).65–67 Recently, Yang et al. examined the performance of different functionals at basis sets for vibrational spectral evaluation and reported that most of the hybrid functionals perform well for the model systems when used with anharmonic corrections.68 Harmonic modes present in the 1000–1250 cm−1 fingerprint region of the spectrum (modes 85–130) were selected to compute the anharmonic corrections. The spectra are fully labeled in Tables S4–S21 (ESI†). The anharmonic modes were then redshifted by 30 cm−1 and the amide I band, computed on the harmonic level, was scaled by 0.965 – consistent with our previous work.32,69,70 These predicted spectra were then compared with the available cryogenic IR spectra of intact protonated ions of BG-H1 and Lea to establish whether they undergo fucose migration.31
Results and discussion
First, we will discuss the effect of the position of the fucose residue on the relative free energies of the fucosylated T1 antigens. The comparison of the relative stability of the most stable conformers of each isomer reveals that ions of neither Lea nor BG-H1 constitute the thermodynamic global minimum. Instead, other isomers of fucosylated T1 antigen are thermodynamically more stable (Table 1) and the most stable isomer is decided by the orientation of the hydroxyl group at the anomeric carbon. The most stable α-anomer has the fucose moiety attached with the β(1 → 6) glycosidic bond to the GlcNAc residue ([α-bn16 + H]+), whereas the most stable β-anomer has the fucose attached to the α(1 → 6) linkage to the Gal ([β-a16 + H]+), which is the same glycosidic linkage that was found to be the rearrangement product of the Lex and BH-H2 antigens.32The relative stability of isomers can be used to propose potential rearrangement products. The free energies of the α-anomers suggest that [BG-H1 + H]+ does not undergo the rearrangement reaction, as the potential products – [α-a16 + H]+ (ΔF = −0.3 kcal mol−1) and [α-bn16 + H]+ (ΔF = −1.1 kcal mol−1) – are only marginally more stable than the parent ion. Other tentative isomers, which are less stable than the parent ions (Table 1), do not present a plausible rearrangement product. On the other hand, [Lea + H]+ is more prone to the rearrangement reaction because multiple other isomers, such as [α-an16 + H]+ (ΔF = −2.0 kcal mol−1), [α-BG-H1 + H]+ (ΔF = −3.4 kcal mol−1), [α-a16 + H]+ (ΔF = −3.7 kcal mol−1), and [α-bn16 + H]+ (ΔF = −4.5 kcal mol−1) are thermodynamically more stable. These energy differences, however, are smaller than the 8.3 kcal mol−1 difference reported for the Lex ions.
For the β-anomeric form, there exist several isomers that are more stable than either of the parent ions. The global minimum is formed by the [β-a16 + H]+ ion and is closely followed by another isomer, [β-bn16 + H]+, which is only 0.5 kcal mol−1 less stable. In effect, the differences between [β-Lea + H]+ and [β-BG-H1 + H]+ and the global minimum increase to, respectively, 6.4 and 3.6 kcal mol−1. In addition, few other ions, such as [b12 + H]+, [b14 + H]+, and [an16 + H]+, are also more stable than the BG-H1 ions. Thus, taking into account only the relative thermodynamic stability of isomers of T1-fucosylated antigens, β-anomers appear more likely to undergo the rearrangement towards one of the several stable isomers. At the same time, previous NMR studies showed that GlcNAc in solution exists dominantly in the α-anomeric form, thus the rearrangement of the less abundant β-anomers may be less impactful on the MS data.71
Next, we added the structural dimension to the analysis. Previously, we reported CCSs of a series of blood group type H and Lewis antigens52 and native protonated ions of Lea and BG-H1 have the same CCS value of 144 Å2, which was measured with drift tube IMS in helium buffer gas. Because the CCS of metal-cation adducts of the same trisaccharides yielded a difference of a few Å2, we concluded that either one or both of the protonated ions undergo the rearrangement. The comparison of CCSs predicted for the lowest free-energy conformers of BG-H1 and Lea (Table 1) agrees with this observation. The simulated CCS of [α-BG-H1 + H]+ matches precisely the experimental value, and the CCS of the β-anomer is within a range of 3 Å2. In contrast, both the lowest free-energy anomers of [Lea + H]+, as well as other higher energy conformers (Fig. 3), have a CCSs 8 Å2 smaller than the experimental CCS of 144 Å2. Thus, the native protonated ions of Lea indeed rearrange during the IMS measurements. The predicted CCSs of the most stable conformers change with respect to the orientation of the hydroxylic group at the reducing end which can affect the chemical identity of the product. For the α-anomer, the only low-energy isomer whose CCS matches the experimental value is [a16 + H]+. The other stable isomer, [bn16 + H]+, has CCS smaller by 10 Å2. Interestingly, the β-anomers behave in the opposite way: the CCS of [a16 + H]+ is 8 Å2 smaller, whereas the CCS of [bn16 + H]+ matches the experimental CCS perfectly. Another low-energy isomer of the β form, [β-an16]+, also displays CCS close to the experimental value. Therefore, the comparison of the CCSs suggests that the [BG-H1 + H]+ ion does not undergo the rearrangement, but in order to explain the CCSs measured for the less stable Lea ion one must consider the rearrangement. We speculate that the rearrangement will lead to non-native ions: [α-a16 + H]+, [β-bn16 + H]+ and [β-an16 + H]+.
To corroborate these predictions, we compared the simulated spectra of 18 lowest free-energy conformers of five most stable isomers with the previously published cryogenic IR data (Fig. 4 and Fig. S21–25, ESI†).31 These spectra were recorded using an ion trap maintained at room temperature, which corresponds to the “warm trap” in the recently published work.32 A typical IR spectrum of a glycan is composed of three main regions (Fig. 4): the 1000–1250 cm−1 region which shows highly delocalized C–O and C–C stretching modes, the 1250–1600 cm−1 region which is mostly composed of C–OH bending modes, and the 1600–1700 cm−1 region which features the amide I band vibrations (if a glycan carries an amide group). The first two regions are populated by characteristic and reproducible vibrations, which can be used to identify unknown glycans.49 Unfortunately, these bands are difficult to assign to specific structural motifs and do not provide straightforward means for the identification of a glycan composition or types of glycosidic bonds without comparing the spectra against a reference experimental or simulated spectra.18 The reported experimental spectra of Lea and BG-H1 ions are somewhat similar to each other. Most prominently, they both share the dominant band at 1085 cm−1 while the differences are rather small: Lea has a broader shoulder between 1060–1085 cm−1, but has a less intense shoulder above 1100 cm−1; Lea also lacks two bands at 1150 and 1200 cm−1. The position of the amide band, which is very sensitive to its environment and guided us in deducing chemical structures of glycosyl donors69,70 and the rearrangement product of T2 antigens,32 appears at the same wavelength for both trisaccharides. Thus, because of the minute differences between the experimental spectra of the two parent ions, we were previously unable to conclude whether Lea and BG-H1 ions undergo the rearrangement reaction.31
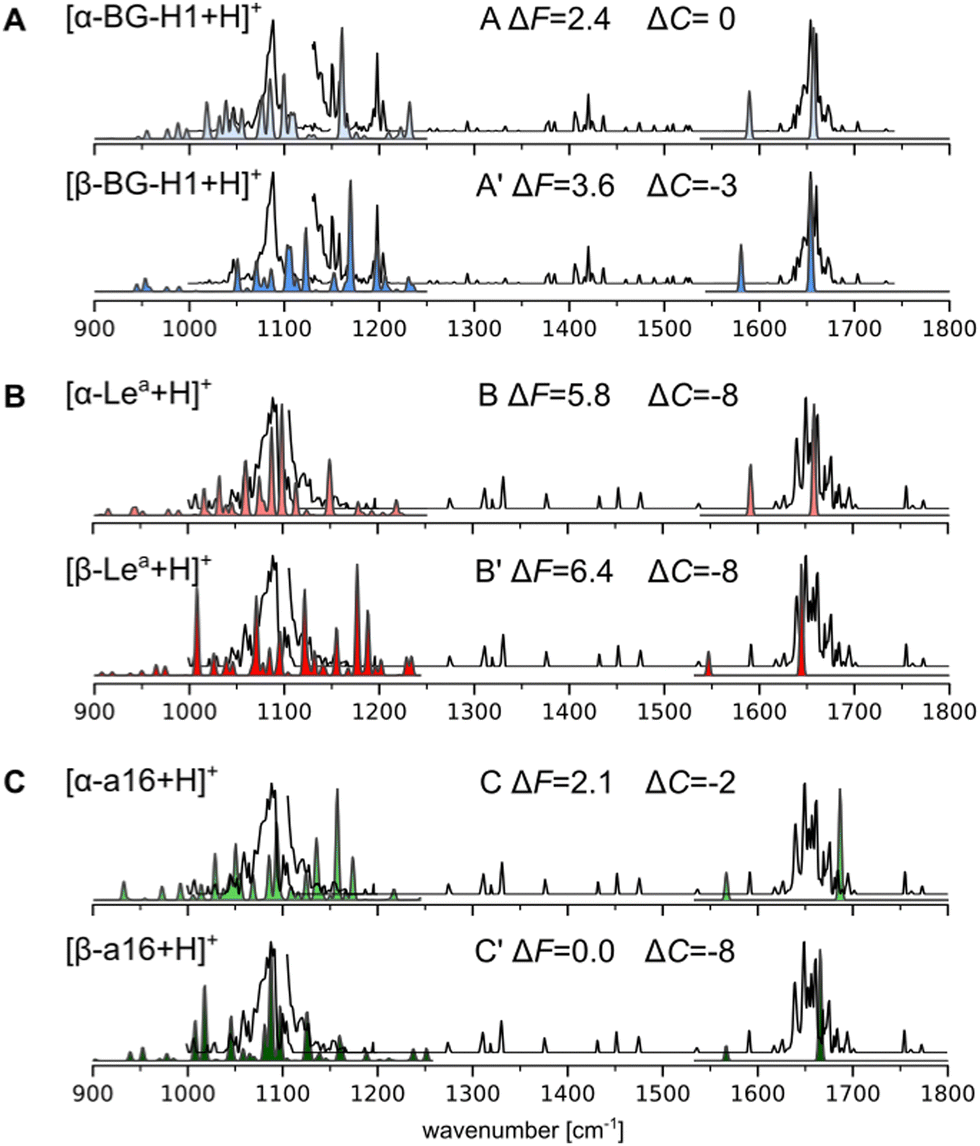 | ||
| Fig. 4 Simulated vibrational spectra of the lowest-free energy conformers of (A) [Lea + H]+, (B) [BG-H1 + H]+, and (C) [a16 + H]+ compared to the experimental IR spectrum of [BG-H1 + H]+ (A) and [Lea + H]+ (B) and (C), shown using a black line. The disconnected experimental line indicates two regions where a different laser power was used.72 The spectra between 1000 and 1250 cm−1 are computed using anharmonic corrections, while the amide I band is calculated using harmonic vibrations. ΔF and ΔC indicate respectively the relative free energies and a difference between the predicted and experimental CCSs. | ||
Herein, we used the GVPT2 method to generate reference anharmonic vibrational spectra to compare with the experimental data and reveal the chemical identity of the ions. Comparison of the spectra of the α-anomer of the protonated BG-H1 reveals several features that agree with the experimental data (Fig. 3A). First, the large band at 1090 cm−1 and its shoulder at lower wavenumbers match several bands in the simulated spectrum, while an intense simulated band at 1155 cm−1 provides a good match to one of two bands visible in the 1150–1160 cm−1 region. The β-anomer of the same conformer provides several other bands that match remaining features visible in the experimental spectrum: a second band at 1160 cm−1, and another two peak-band at 1200 cm−1. Furthermore, the amide band, predicted at 1650 cm−1 for both isomers, matches the experimental spectrum. The only feature that is not well represented in the simulated spectrum is the dominant band at 1090 cm−1. The theory, however, predicts multiple weaker bands in this region, each of which has inherent error due to the functional used in this work.68 An overlap of predicted bands might generate the intense fingerprint in the cryogenic IR. Other conformers are less stable by more than 3 kcal mol−1 and will not contribute to the spectrum (Fig. S21, ESI†). Thus, the general agreement between the simulated and experimental spectrum of BG-H1 further reinforces the interpretation that this trisaccharide does not undergo internal glycan rearrangement. This conclusion is supported not only by the overall match between the spectra, but by the three metrics we use for this assignment: thermodynamic stability, comparable CCS, and matching IR spectrum.
The simulated and experimental spectra of the Lea ion also provide a surprisingly compelling match with the experimental spectrum (Fig. 3B). The anharmonic spectrum of [α-Lea + H]+ yields several bands in the region of 1050–1100 cm−1 which well match the broad shoulder visible in the spectrum of the tentative Lea ions. In addition, the simulated spectrum does not display any other significant features in the fingerprint region, in agreement with the experiment. Even the amide band, which is predicted to be at 1655 cm−1 matches the experimental band exactly. The apparent agreement between the experimental and simulated spectra of Lea, however, is contradicted by a large difference between the predicted and measured CCS values. Interestingly, the β-anomer does not improve the match with the experimental spectrum. The fingerprint region looks very different and there are other bands at 1000 and 1200 cm−1 that do not show in the experiment. Higher energy conformers, which have larger CCSs, do not improve the IR fit with the experimental data either (Fig. S22, ESI†). Thus, based on this data, we are not able to confidently answer whether Lea undergoes the migration, and we will investigate whether other low-energy isomers would provide both a better match to the experimental IR spectrum than [Lea + H]+ ions and a comparable CCS.
In recent work, Moge et al. reported33 the rearrangement product of fucose-containing disaccharides that carried a fucose attached by an α(1 → 6) bond to GlcNAc. The rearrangement product that features a similar glycosidic linkage, [an16 + H]+, is among the most stable isomers and is more stable by 2.0 and 3.9 kcal mol−1 than, respectively, the α and β-anomers of Lea. However, while the [α-an16 + H]+ ion provides a good match to the experimental IR spectrum, the CCS is 6 Å2 smaller than the experimentally measured value (Fig. S24, ESI†). The CCS of the β-anomer, on the other hand, agrees better with the experimental value, but its IR spectrum provides an overall worse match with the experiment. Another stable glycosidic linkage is formed by fucose attached to the same oxygen but in the β-configuration. Both α- and β-anomers of [bn16 + H]+ belong to the most stable isomers of the respective anomeric form and the simulated spectra of two low-energy conformers of the α-anomer match the experimental spectra quite well (Fig. S25, ESI†). Unfortunately, these conformers are too compact, having the predicted CCSs smaller by 10 and 7 Å2 than the experimental value. The β-anomer, on the other hand, has a CCS that matches the experimental 144 Å2, but the match of the vibrational fingerprint is not satisfactory. Next, we considered α(1 → 6) with the galactose, which was the linkage observed for the Lex and BG-H2 trisaccharides (Fig. 4C and Fig. S23, ESI†). This point of attachment constitutes the second most stable isomers among α-anomers and the most stable isomer among β-anomers. In principle, the β-anomer provides a very good fit to the IR spectrum of the Lea ions; however, they once again suffer from the mismatch of the CCSs. The α-anomer, on the other hand, has a closer match with respect to CCS, but the fit of the IR fingerprint is poorer.
Finally, Sastre Toraño et al. suggested fucose attachment to the amide group of the GlcNAc.51 This suggestion was based on the comparison of arrival time distributions (ATDs) of unknown and reference glycans using IMS. Our calculations show that the fucose attached to this position is not only destabilized by at least 6 kcal mol−1 when compared to the global minimum but also the ions have too large CCSs. Nevertheless, we find that this position in the T1 antigens is more stable than in the previously reported T2 antigens, which were 10.9 kcal mol−1 less stable than the global minimum.32 It is likely that the fucose attached to the amide group might become even more stable in the larger oligosaccharides studied in that work, but they do not form the rearrangement product in our study.
In summary, none of the analyzed isomers of fucosylated T1 trisaccharides match all three metrics we use to confirm the structure of the rearrangement product: thermodynamic stability, comparable CCS, and matching IR fingerprint. In principle, the best match between the simulated and experimental IR spectrum of Lea is simply achieved by the parent ion, [α-Lea + H]+, which leads to the conclusion that fucose in the T1 antigen does not undergo migration. However, this does not explain why Manz et al. reported that native protonated ions of Lea and BG-H1 have identical CCSs. One possible explanation is that the IMS measurements on the Synapt G2-S HDMS instrument were carried under harsher conditions than those used to record the cryogenic-ion IR spectrum and these conditions could induce migration. The measured CCSs of 144 Å2 suggest that in Lea the fucose moiety could migrate to either O6 of the Gal ([a16 + H]+) or the GlcNAc ([an16 + H]+ or [bn16 + H]+), however, without the IR spectrum of the ion after migration we can only speculate about its structure. Based on this consideration, we conclude that the recorded IR spectrum indeed shows the [Lea + H]+ ions, and fucose migration does not occur.
Next, we will address the question of which structural motifs present in the Lewis and blood group type H antigens promote (or not) the rearrangement. A comparison of the lowest energy structures of α- and β-anomers of [BG-H1 + H]+ (Fig. 5A) shows that these two ions differ only by the orientation of the hydroxy group at the anomeric carbon. In both cases, the GlcNAc adopts a B1,4 ring pucker, where the protonated amide forms a tight H-bond with the ring oxygen of GlcNAc and another one between the N–H group and O6 of the galactose. This conformer is additionally stabilized by two H-bonds, one between O4H of GlcNAc and the ring oxygen of fucose, and one between O2H of Gal and O4 of the fucose moiety. In this conformation, the proton is locked in a strong H-bond with the ring oxygen and the glycosidic bond between Gal and Fuc does not have direct contact with the amide group. Furthermore, both potential acceptors of the proton that could initiate the fucose migration, the ring oxygen and the oxygen in the glycosidic bond between fucose and galactose,32 are positioned away from the protonated amide group. Thus, there most likely exists a large energy barrier associated with the proton shift that inhibits the rearrangement of the BG-H1 trisaccharides.
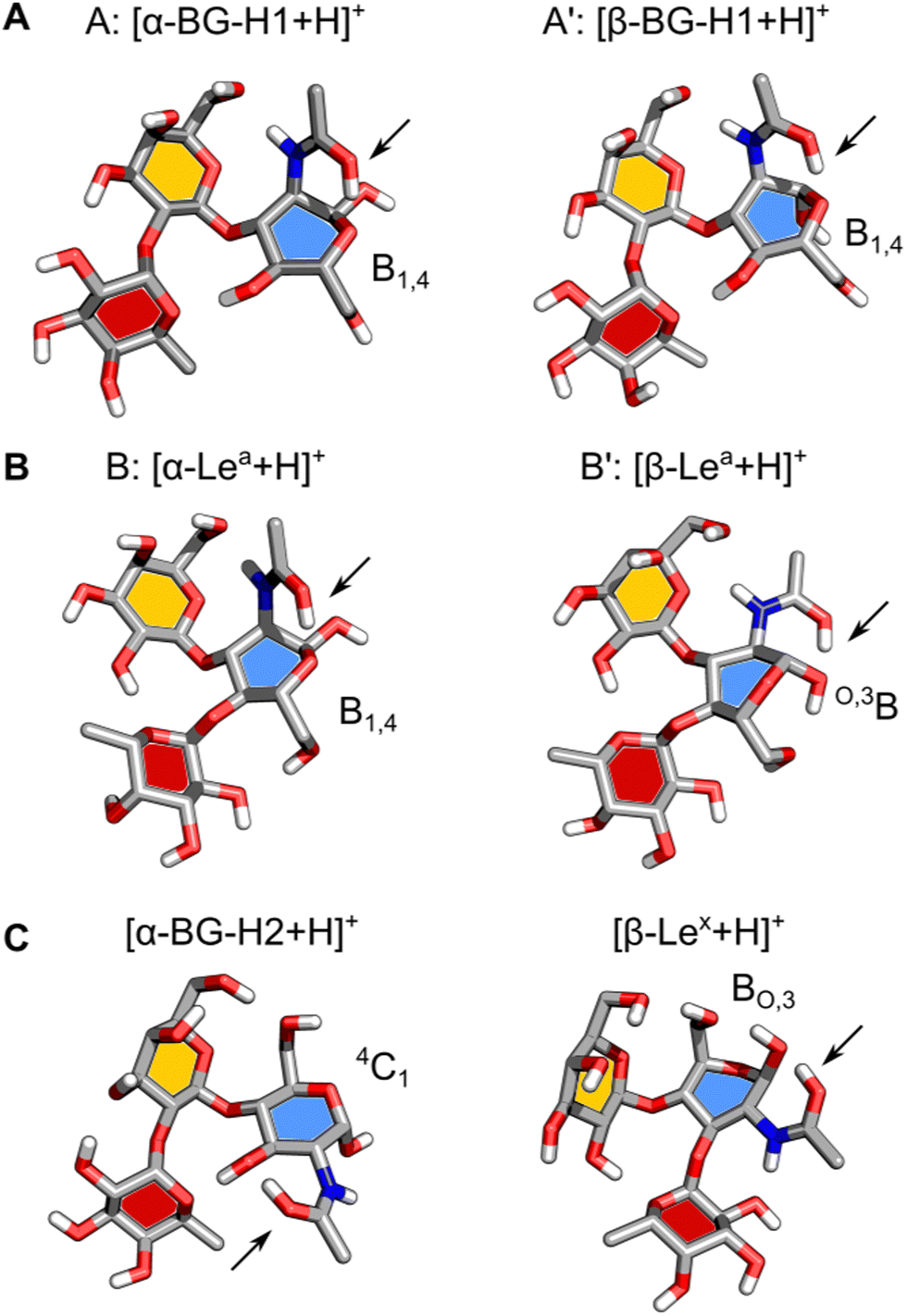 | ||
| Fig. 5 The structures of the lowest-free energy conformers of isomers of protonated ions of fucosylated T1 (A) and (B) and T2 (C) antigens. The ring pucker of the reducing-end GlcNAc is shown and the protonated amide is highlighted with an arrow. The respective monosaccharides are color-coded according to their SNFG symbols: GlcNAc – blue, Gal – yellow, and Fuc – red. | ||
The structures of α- and β-anomers of [Lea + H]+ appear somewhat similar (Fig. 5B) as well. They both share the orientation of the glycosidic bonds and stabilizing H-bonds between O2 of Fuc and O3H of Gal, and N–H of GlcNAc and O6 of Gal. In the α-anomer, however, the protonated amide group is oriented in an axial position of a B1,4 ring pucker and forms a H-bond with the ring oxygen, and another one between the N–H and O7 on Gal, as it was observed for the BG-H1 ions. The shared orientation of the amide group confirms why these two saccharides have the same amide band position in the IR spectrum. In the β-anomer of Lea, GlcNAc adopts a O,3B ring pucker which is supported by a H-bond between the amide group in an equatorial orientation and the axial hydroxy group on the anomeric carbon.
Why would Lea then undergo the migration after sufficient activation, as it happened during IMS measurements? The main difference between the structures of the BG-H1 and Lea ions is the position of the fucose glycosidic bond. In the latter, the fucose is linked by the α(1 → 4) glycosidic bond with GlcNAc which – contrary to the α(1 → 2) Gal bond in BG-H1 – can be reached by the proton acetyl group via rotation of the C2–N bond. It is possible that ion activation rotates the amide group which would be able to shift the proton towards either the glycosidic bond or the fucose ring oxygen which would trigger the rearrangement. A rotation around the Galβ(1 → 3) GlcNAc glycosidic bond, in a mechanism analogous to that postulated for the BG-H2 and Lex trisaccharides, would position O6 of the galactose close to the protonated glycosidic bond.32 After the rotation, the fucose transfer from O4 on GlcNAc to the adjacent O6 on Gal would form the [a16 + H]+ ion.
Finally, it is illustrative to compare the predicted structures of Lea and BG-H1 with Lex and BG-H2 to understand why a different arrangement of monosaccharides promotes fucose migration only in the latter pair (Fig. 5C). Different connectivity of monosaccharides changes the lowest-energy conformers of these sugars. In the case of [Lex + H]+, we observed that the Fuc α(1 → 3) GlcNAc glycosidic bond is adjacent to the protonated amide group. Such conformation requires little energy to shift the proton and initiate the rearrangement reaction. Interestingly, [BG-H2 + H]+ adopts a very similar conformation. Here, the fucose moiety is separated from the protonated amide group by the hydroxyl group at GlcNAc. This group can shuttle the proton between the amide and the fucose, but it requires more energy than in the case of Lex, which explains a larger activation barrier of BG-H2 ions for initiating the rearrangement. This agrees with the observation that the lower temperature of the ion trap during cryogenic-ion IR spectroscopy measurements was able to quench the isomerization kinetics of the BG-H2 ions.32 In Lea and BG-H1 saccharides, the proton is immobilized in a tight H-bond between the amide group and the ring oxygen of GlcNAc which is further supported by an interaction with O6 on the galactose. These interactions increase the kinetic barrier for the proton transfer and effectively quench the reaction.
Conclusions
First, we observe that not only the availability of the mobile proton, but also its proximity to the glycosidic bond of fucose is important for the likelihood of fucose migration. In our previous work,32 the lowest-energy conformers of Lex and BG-H2 featured a protonated amide in direct proximity to the fucose glycosidic bond. This conformation supplied a mobile proton able to promote internal glycan rearrangement. The examination of the lowest energy structures of the isomers of T1-fucosylated antigens revealed that the reported IR data indeed show intact ions of [Lea + H]+ and [BG-H1 + H]+. This suggests that the fucose migration does not occur for these antigens as readily as it was observed for the [Lex + H]+ and [BG-H2 + H]+ ions. The rearrangement is inhibited due to the lack of a mobile proton that would have access to the fucose moiety. This happens because the protonated amide group is locked by a strong H-bond between the amide group and the ring oxygen of GlcNAc and another one between the amide group and the O6 of Gal. The fucose glycosidic bond is also positioned away from the protonated amide which increases the energy barrier needed to activate the rearrangement. Furthermore, the free energy difference between the parent ion and the migration product appears to be another important factor driving the reaction. For T1-derived antigens, the energy difference is smaller than that observed for T2 antigens, which correlates with the decreased likelihood of the fucose migration, although sufficiently strong activation can trigger the rearrangement towards thermodynamically more stable isomers. Finally, we demonstrate that the glycan sequence, determined by the connectivity and configuration of the glycosidic bonds, affects the fragmentation properties and the likelihood of the fucose migration.Author contributions
ML, KP, and MM conceived the research. VK, MY, KG, and MM carried out the molecular modeling. All authors contributed to the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. G. thanks the Fonds National de la Recherche, Luxembourg, for funding the project GlycoCat (13549747). M. M. gratefully acknowledges the funding from the National Institute of General Medical Sciences (SC2GM135145). K. P. acknowledges generous funding from the European Research Council, ERC-2019-CoG-863934-GlycoSpec.References
- J. Li, H.-C. Hsu, J. D. Mountz and J. G. Allen, Unmasking Fucosylation: from Cell Adhesion to Immune System Regulation and Diseases, Cell Chem. Biol., 2018, 25, 499–512 CrossRef CAS PubMed.
- S. Tahata, K. Raymond, M. Quade, S. Barnes, S. Boyer, S. League, A. Kumanovics, R. Abraham, E. Jacob, P. Menon and E. Morava, Defining the mild variant of leukocyte adhesion deficiency type-II (SLC35C1-congenital disorder of glycosylation) and response to L-fucose therapy: Insights from two new families and review of the literature, Am. J. Med. Genet., Part A, 2022, 188, 2005–2018 CrossRef CAS PubMed.
- A. Varki, Biological roles of glycans, Glycobiology, 2017, 27, 3–49 CrossRef CAS PubMed.
- D. Thomas, A. K. Rathinavel and P. Radhakrishnan, Altered glycosylation in cancer: A promising target for biomarkers and therapeutics, Biochim. Biophys. Acta, Rev. Cancer, 2021, 1875, 188464 CrossRef CAS PubMed.
- C. Reily, T. J. Stewart, M. B. Renfrow and J. Novak, Glycosylation in health and disease, Nat. Rev. Nephrol., 2019, 15, 346–366 CrossRef PubMed.
- G. Stefanetti, F. Borriello, B. Richichi, I. Zanoni and L. Lay, Immunobiology of Carbohydrates: Implications for Novel Vaccine and Adjuvant Design Against Infectious Diseases, Front. Cell. Infect. Microbiol., 2022, 11 Search PubMed.
- D. B. Werz, R. Ranzinger, S. Herget, A. Adibekian, C. W. Von der Lieth and P. H. Seeberger, Exploring the structural diversity of mammalian carbohydrates (“glycospace”) by statistical databank analysis, ACS Chem. Biol., 2007, 2, 685–691 CrossRef CAS PubMed.
- S. J. North, S. Chalabi, M. Sutton-Smith, A. Dell and S. M. Haslam, in Handbook of Glycomics, ed. R. D. Cummings, J. M. Pierce, Academic Press, San Diego, 2010, pp. 263–327 Search PubMed.
- G. C. M. Vreeker and M. Wuhrer, Reversed-phase separation methods for glycan analysis, Anal. Bioanal. Chem., 2017, 409, 359–378 CrossRef CAS PubMed.
- G. R. Guile, P. M. Rudd, D. R. Wing, S. B. Prime and R. A. Dwek, A Rapid High-Resolution High-Performance Liquid Chromatographic Method for Separating Glycan Mixtures and Analyzing Oligosaccharide Profiles, Anal. Biochem., 1996, 240, 210–226 CrossRef CAS PubMed.
- K. R. Anumula High-Sensitivity and High-Resolution Methods for Glycoprotein Analysis. Anal. Biochem. 2000, 283, 17–26 Search PubMed.
- K. R. Anumula, Advances in fluorescence derivatization methods for high-performance liquid chromatographic analysis of glycoprotein carbohydrates, Anal. Biochem., 2006, 350, 1–23 CrossRef CAS PubMed.
- R. A. Zubarev, N. L. Kelleher and F. W. McLafferty, Electron Capture Dissociation of Multiply Charged Protein Cations. A Nonergodic Process, J. Am. Chem. Soc., 1998, 120, 3265–3266 CrossRef CAS.
- J. E. P. Syka, J. J. Coon, M. J. Schroeder, J. Shabanowitz and D. F. Hunt, Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 9528–9533 CrossRef CAS PubMed.
- W. R. Alley Jr., Y. Mechref and M. V. Novotny, Characterization of glycopeptides by combining collision-induced dissociation and electron-transfer dissociation mass spectrometry data, Rapid Commun. Mass Spectrom., 2009, 23, 161–170 CrossRef PubMed.
- C. Singh, C. G. Zampronio, A. J. Creese and H. J. Cooper, Higher Energy Collision Dissociation (HCD) Product Ion-Triggered Electron Transfer Dissociation (ETD) Mass Spectrometry for the Analysis of N-Linked Glycoproteins, J. Proteome Res., 2012, 11, 4517–4525 CrossRef CAS PubMed.
- J. Hofmann, H. S. Hahm, P. H. Seeberger and K. Pagel, Identification of carbohydrate anomers using ion mobility-mass spectrometry, Nature, 2015, 526, 241–244 CrossRef CAS PubMed.
- E. Mucha, A. Stuckmann, M. Marianski, W. B. Struwe, G. Meijer and K. Pagel, Indepth structural analysis of glycans in the gas phase, Chem. Sci., 2019, 10, 1272–1284 RSC.
- S. Warnke, A. Ben Faleh and T. R. Rizzo, Toward High-Throughput Cryogenic IR Fingerprinting of Mobility-Separated Glycan Isomers, ACS Meas. Sci. Au, 2021, 1, 157–164 CrossRef CAS PubMed.
- M. Grabarics, M. Lettow, C. Kirschbaum, K. Greis, C. Manz and K. Pagel, Mass Spectrometry-Based Techniques to Elucidate the Sugar Code, Chem. Rev., 2022, 122, 7840–7908 CrossRef CAS PubMed.
- L. R. Ruhaak, G. Xu, Q. Li, E. Goonatilleke and C. B. Lebrilla, Mass Spectrometry Approaches to Glycomic and Glycoproteomic Analyses, Chem. Rev., 2018, 118, 7886–7930 CrossRef CAS PubMed.
- C. Gray and S. L. Flitsch, in Coupling and Decoupling of Diverse Molecular Units in Glycosciences, ed. Witczak, Z. J., Bielski, R., Springer International Publishing, Cham, 2018, pp. 225–267 Search PubMed.
- P. Hong, H. Sun, L. Sha, Y. Pu, K. Khatri, X. Yu, Y. Tang and C. Lin, GlycoDeNovo – an Efficient Algorithm for Accurate de novo Glycan Topology Reconstruction from Tandem Mass Spectra, J. Am. Soc. Mass Spectrom., 2017, 28, 2288–2301 CrossRef CAS PubMed.
- Z. Chen, J. Wei, Y. Tang, C. Lin, C. E. Costello and P. Hong, GlycoDeNovo2: An Improved MS/MS-Based De Novo Glycan Topology Reconstruction Algorithm, J. Am. Soc. Mass Spectrom., 2022, 33, 436–445 CrossRef CAS PubMed.
- R. W. Vachet, B. M. Bishop, B. W. Erickson and G. L. Glish, Novel Peptide Dissociation: Gas-Phase Intramolecular Rearrangement of Internal Amino Acid Residues, J. Am. Chem. Soc., 1997, 119, 5481–5488 CrossRef CAS.
- D. J. Harvey, T. S. Mattu, M. R. Wormald, L. Royle, R. A. Dwek and P. M. Rudd, Internal residue loss”: Rearrangements occurring during the fragmentation of carbohydrates derivatized at the reducing terminus, Anal. Chem., 2002, 74, 734–740 CrossRef CAS PubMed.
- M. Wuhrer, M. L. M. Robijn, C. A. M. Koeleman, C. I. A. Balog, R. Geyer, A. M. Deelder and C. H. Hokke, A novel Gal(beta1-4)Gal(beta1-4)Fuc(alpha1-6)-core modification attached to the proximal N-acetylglucosamine of keyhole limpet haemocyanin (KLH) N-glycans, Biochem. J., 2004, 378, 625 CrossRef CAS PubMed.
- M. Wuhrer, C. A. M. Koeleman, C. H. Hokke and A. M. Deelder, Mass spectrometry of proton adducts of fucosylated N-glycans: fucose transfer between antennae gives rise to misleading fragments, Rapid Commun. Mass Spectrom., 2006, 20, 1747–1754 CrossRef CAS PubMed.
- N. Hashii, N. Kawasaki, S. Itoh, Y. Nakajima, A. Harazono, T. Kawanishi and T. Yamaguchi, Identification of Glycoproteins Carrying a Target Glycan-Motif by Liquid Chromatography/Multiple-Stage Mass Spectrometry: Identification of Lewis x-Conjugated Glycoproteins in Mouse Kidney, J. Proteome Res., 2009, 8, 3415–3429 CrossRef CAS PubMed.
- M. Wuhrer, A. M. Deelder and Y. E. van der Burgt, Mass spectrometric glycan rearrangements, Mass Spectrom. Rev., 2011, 30, 664–680 CrossRef CAS PubMed.
- E. Mucha, M. Lettow, M. Marianski, D. A. Thomas, W. B. Struwe, D. J. Harvey, G. Meijer, P. H. Seeberger, G. von Helden and K. Pagel, Fucose Migration in Intact Protonated Glycan Ions: A Universal Phenomenon in Mass Spectrometry, Angew. Chem., Int. Ed., 2018, 57, 7440–7443 CrossRef CAS PubMed.
- M. Lettow, K. Greis, E. Mucha, T. R. Lambeth, M. Yaman, V. Kontodimas, C. Manz, W. Hoffmann, G. Meijer, R. R. Julian, G. von Helden, M. R. Marianski and K. Pagel, Decoding the Fucose Migration Product during Mass-Spectrometric Analysis of Blood Group Epitopes, Angew. Chem., Int. Ed., 2023, e202302883 CAS.
- B. Moge, B. Schindler, O. Yeni and I. Compagnon, Fucose Migration Pathways Identified Using Infrared Spectroscopy, Angew. Chem., Int. Ed., 2023, 62, e202300538 CrossRef CAS PubMed.
- B. Ernst, D. R. Muller and W. J. Richter, False sugar sequence ions in electrospray tandem mass spectrometry of underivatized sialyl-Lewis-type oligosaccharides, Int. J. Mass Spectrom. Ion Processes, 1997, 160, 283–290 CrossRef CAS.
- X. Chen and G. C. Flynn, Analysis of N-glycans from recombinant immunoglobulin G by on-line reversed-phase high-performance liquid chromatography/mass spectrometry, Anal. Biochem., 2007, 370, 147–161 CrossRef CAS PubMed.
- A. Broberg, High-performance liquid chromatography/electrospray ionization ion-trap mass spectrometry for analysis of oligosaccharides derivatized by reductive amination and N,N-dimethylation, Carbohydr. Res., 2007, 342, 1462–1469 CrossRef CAS PubMed.
- N. Desai, D. A. Thomas, J. Lee, J. Gao and J. L. Beauchamp, Eradicating mass spectrometric glycan rearrangement by utilizing free radicals, Chem. Sci., 2016, 7, 5390–5397 RSC.
- L. P. Brull, W. Heerma, J. Thomas-Oates, J. Haverkamp, V. Kovacik and P. Kovac, Loss of internal 1 → 6 substituted monosaccharide residues from underivatized and per-O-methylated trisaccharides, J. Am. Soc. Mass Spectrom., 1997, 8, 43–49 CrossRef CAS.
- M. Wuhrer, C. A. M. Koeleman and A. M. Deelder, Hexose rearrangements upon fragmentation of N-glycopeptides and reductively aminated N-glycans, Anal. Chem., 2009, 81, 4422–4432 CrossRef CAS PubMed.
- E. S. Hecht, P. L. Loziuk and D. C. Muddiman, Xylose Migration During Tandem Mass Spectrometry of N-Linked Glycans, J. Am. Soc. Mass Spectrom., 2017, 28, 729–732 CrossRef CAS PubMed.
- M. McNeil, Elimination of internal glycosyl residues during chemical ionization-mass spectrometry of per-O-alkylated oligosaccharide-alditols, Carbohydr. Res., 1983, 123, 31–40 CrossRef CAS.
- F.-F. Li, J.-J. Liu, D.-W. Liu, B. Lin, Y.-Y. Hao, J.-P. Cong, L.-C. Zhu, S. Gao, S.-L. Zhang and M. Iwamori, Lewis Y regulates signaling molecules of the transforming growth factor β pathway in ovarian carcinoma-derived RMG-I cells, Int. J. Oncol., 2012, 40, 1196–1202 CrossRef CAS PubMed.
- M. A. Carrascal, M. Silva, J. S. Ramalho, C. Pen, M. Martins, C. Pascoal, C. Amaral, I. Serrano, M. J. Oliveira, R. Sackstein and P. A. Videira, Inhibition of fucosylation in human invasive ductal carcinoma reduces E-selectin ligand expression, cell proliferation, and ERK 1/2 and p38 MAPK, Mol. Oncol., 2018, 12, 579–593 CrossRef CAS PubMed.
- S. Hong, L. Feng, Y. Yang, H. Jiang, X. Hou, P. Guo, F. L. Marlow, P. Stanley and P. Wu, In Situ Fucosylation of the Wnt Co-receptor LRP6 Increases Its Endocytosis and Reduces Wnt/β-Catenin Signaling, Cell Chem. Biol., 2020, 27, 1140–1150 CrossRef CAS PubMed.
- O. M. T. Pearce, Cancer glycan epitopes: biosynthesis, structure and function, Glycobiology, 2018, 28, 670–696 CrossRef CAS PubMed.
- C. P. Stowell and S. R. Stowell, Biologic roles of the ABH and Lewis histo-blood group antigens Part I: infection and immunity, Vox Sang., 2019, 114, 426–442 CrossRef CAS PubMed.
- V. Ravn and E. Dabelsteen, Tissue distribution of histo-blood group antigens, Rev. Article, 2000, 108, 1–28 CAS.
- M. Lettow, E. Mucha, C. Manz, D. A. Thomas, M. Marianski, G. Meijer, G. von Helden and K. Pagel, The role of the mobile proton in fucose migration, Anal. Bioanal. Chem., 2019, 411, 4637–4645 CrossRef CAS PubMed.
- E. Mucha, A. I. Gonzalez Florez, M. Marianski, D. A. Thomas, W. Hoffmann, W. B. Struwe, H. S. Hahm, S. Gewinner, W. Schollkopf, P. H. Seeberger, G. von Helden and K. Pagel, Glycan Fingerprinting via Cold-Ion Infrared Spectroscopy, Angew. Chem., Int. Ed., 2017, 56, 11248–11251 CrossRef CAS PubMed.
- D. J. Harvey, T. S. Mattu, M. R. Wormald, L. Royle, R. A. Dwek and P. M. Rudd, “Internal Residue Loss”: Rearrangements Occurring during the Fragmentation of Carbohydrates Derivatized at the Reducing Terminus, Anal. Chem., 2002, 74, 734–740 CrossRef CAS PubMed.
- J. Sastre Torano, I. A. Gagarinov, G. M. Vos, F. Broszeit, A. D. Srivastava, M. Palmer, J. I. Langridge, O. Aizpurua-Olaizola, V. J. Somovilla and G.-J. Boons, Ion-Mobility Spectrometry Can Assign Exact Fucosyl Positions in Glycans and Prevent Misinterpretation of Mass-Spectrometry Data After Gas-Phase Rearrangement, Angew. Chem., Int. Ed., 2019, 58, 17616–17620 CrossRef CAS PubMed.
- C. Manz, M. Grabarics, F. Hoberg, M. Pugini, A. Stuckmann, W. B. Struwe and K. Pagel, Separation of isomeric glycans by ion mobility spectrometry – the impact of fluorescent labelling, Analyst, 2019, 144, 5292–5298 RSC.
- M. Marianski, CarPpy: Carbohydrate Parser in Python. https://github.com/marianski-lab/CarPpy.
- S. Rahim; M. Yaman and M. Marianski Manuscript in preparation.
- P. Pracht, F. Bohle and S. Grimme, Automated exploration of the low-energy chemical space with fast quantum chemical methods, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192 RSC.
- C. Bannwarth, S. Ehlert and S. Grimme, GFN2-xTB—An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- M. Marianski, A. Supady, T. Ingram, M. Schneider and C. Baldauf, Assessing the Accuracy of Across-the-Scale Methods for Predicting Carbohydrate Conformational Energies for the Examples of Glucose and α-Maltose, J. Chem. Theory Comput., 2016, 12, 6157–6168 CrossRef CAS PubMed.
- L. Kong and R. A. Bryce, Modeling pyranose ring pucker in carbohydrates using machine learning and semi-empirical quantum chemical methods, J. Comput. Chem., 2022, 43, 2009–2022 CrossRef CAS PubMed.
- M. J. Frisch, Gaussian 16 Revision B.01, 2016, Gaussian Inc., Wallingford CT Search PubMed.
- J. P. Perdew, M. Ernzerhof and K. Burke, Rationale for mixing exact exchange with density functional approximations, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef CAS.
- S. Grimme, J. Antony, T. Schwabe and C. Muck-Lichtenfeld, Density functional theory with dispersion corrections for supramolecular structures, aggregates, and complexes of (bio)organic molecules, Org. Biomol. Chem., 2007, 5, 741–758 RSC.
- G. von Helden, M. T. Hsu, N. Gotts and M. T. Bowers, Carbon cluster cations with up to 84 atoms: structures, formation mechanism, and reactivity, J. Phys. Chem., 1993, 97, 8182–8192 CrossRef CAS.
- A. D. Hill and P. J. Reilly, Puckering Coordinates of Monocyclic Rings by Triangular Decomposition, J. Chem. Inf. Model., 2007, 47, 1031–1035 CrossRef CAS PubMed.
- M. Bursch, J.-M. Mewes, A. Hansen and S. Grimme, Best-Practice DFT Protocols for Basic Molecular Computational Chemistry, Angew. Chem., Int. Ed., 2022, 61, e202205735 CrossRef CAS PubMed.
- V. Barone, Anharmonic vibrational properties by a fully automated second-order perturbative approach, J. Chem. Phys., 2005, 122, 014108 CrossRef PubMed.
- M. Piccardo, J. Bloino and V. Barone, Generalized vibrational perturbation theory for rotovibrational energies of linear, symmetric and asymmetric tops: Theory, approximations, and automated approaches to deal with medium-to-large molecular systems, Int. J. Quantum Chem., 2015, 115, 948–982 CrossRef CAS PubMed.
- K. B. Beć and C. W. Huck, Breakthrough Potential in Near-Infrared Spectroscopy: Spectra Simulation. A Review of Recent Developments, Front Chem., 2019, 7, 48 CrossRef PubMed.
- Q. Yang, M. Mendolicchio, V. Barone and J. Bloino, Accuracy and Reliability in the Simulation of Vibrational Spectra: A Comprehensive Benchmark of Energies and Intensities Issuing From Generalized Vibrational Perturbation Theory to Second Order (GVPT2), Front. Astron. Space Sci., 2021, 8 Search PubMed.
- E. Mucha, M. Marianski, F.-F. Xu, D. A. Thomas, G. Meijer, G. von Helden, P. H. Seeberger and K. Pagel, Unravelling the structure of glycosyl cations via cold-ion infrared spectroscopy, Nat. Commun., 2018, 9, 4174 CrossRef PubMed.
- M. Marianski, E. Mucha, K. Greis, S. Moon, A. Pardo, C. Kirschbaum, D. Thomas, G. Meijer, G. von Helden, K. Gilmore, P. Seeberger and K. Pagel, Direct Evidence for Remote Participation in Galactose Building Blocks during Glycosylations Revealed by Cryogenic Vibrational Spectroscopy, Angew. Chem., Int. Ed., 2020, 59(15), 6166–6171 CrossRef CAS PubMed.
- J. Kwon, A. Ruda, H. F. Azurmendi, J. Zarb, M. D. Battistel, L. Liao, A. Asnani, F.-I. Auzanneau, G. Widmalm and D. I. Freedberg, Glycan Stability and Flexibility: Thermodynamic and Kinetic Characterization of Nonconventional Hydrogen Bonding in Lewis Antigens, J. Am. Chem. Soc., 2023, 145, 10022–10034 CrossRef CAS PubMed.
- W. Schollkopf; S. Gewinner; H. Junkes; A. Paarmann; G. von Helden; H. P. Bluem and A. M. M. Todd The new IR and THz FEL facility at the Fritz Haber Institute in Berlin. Advances in X-ray Free-Electron Lasers Instrumentation III. 2015; 95121L.
Footnote |
| † Electronic supplementary information (ESI) available: xyz-coordinates of 18 lowest energy conformers and their vibrational band assignment, benchmark data, and structural analysis of 28 isomers of fucosylated T1 antigens. See DOI: https://doi.org/10.1039/d3cp04491b |
| This journal is © the Owner Societies 2024 |