
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular basis for non-invasive diagnostics of cardiac amyloids using bone tracers
Emily
Lewkowicz
,
Shobini
Jayaraman
and
Olga
Gursky
 *
*
Department of Pharmacology, Physiology & Biophysics, Boston University Chobanian and Avedisian School of Medicine, Boston, MA, USA. E-mail: gursky@bu.edu
First published on 22nd July 2024
Abstract
Amyloid diseases including Alzheimer's, Parkinson's and over 30 others are incurable life-threatening disorders caused by abnormal protein deposition as fibrils in various organs. Cardiac amyloidosis is particularly challenging to diagnose and treat. Identification of the fibril-forming protein, which in the heart is usually amyloid transthyretin (ATTR) or amyloid immunoglobulin light chain (AL), is paramount to treatment. A transformative non-invasive diagnostic modality is imaging using technetium-labeled pyrophosphate or diphosphonate bone tracers, 99mTc-PYP/DPD/HMDP. For unknown reasons, these tracers show preferential uptake by ATTR deposits. The tracer-binding moiety is unknown and potentially involves amyloid fibrils and/or amyloid-associated calcific deposits. We propose that, like in the bone, the tracers chelate to surface-bound Ca2+ in amyloid. In high-affinity protein sites, Ca2+ is coordinated by pairs of acidic residues. To identify such residues on amyloids, we harnessed atomic structures of patient-derived cardiac amyloids determined using cryogenic electron microscopy since 2019. These structures help explain why most but not all ATTR deposits uptake 99mTc-PYP/DPD/HMDP radiotracers, while in AL the opposite is true. Moreover, fibril structures help explain greater microcalcification observed in ATTR vs. AL deposits. These findings may aid the diagnostics and therapeutic targeting of cardiac amyloidosis and are relevant to other amyloids.
1. Introduction
Amyloidoses are underdiagnosed life-threatening diseases wherein normally soluble proteins or peptides deposit as insoluble fibrils in vital organs. Amyloid fibril formation involves self-assembly of many protein copies into an insoluble polymer. Over 40 fibril-forming proteins have been identified to-date that deposit as pathologic amyloids in humans.1,2 Some fibril-forming proteins such as Tau deposit in more than one amyloid disease.3 Amyloid deposition can damage cells and organs, ultimately leading to organ failure. In neurodegenerative disorders such as Alzheimer's, Parkinson's or prion diseases, amyloid fibrils deposit in the brain. In other systemic or localized amyloidoses, the afflicted organs often involve the kidneys, liver and heart.1 Amyloid deposition in the heart is lethal if untreated, and treatment of cardiac amyloidosis is particularly challenging.1,4,5 Although amyloid diseases are currently incurable, disease-modifying therapies have emerged that prolong patient life and improve its quality.6 The therapeutic outcomes depend critically on the accurate and timely diagnostics, which is a major challenge.1,4,5 This mini-review is focused on the preferred diagnostic method for cardiac amyloidosis and its molecular underpinnings.2. Recent advances in the diagnostics and treatment of cardiac amyloidosis
Cardiac amyloidosis is a group of diseases that often present as cardiomyopathy. Once considered a rare fatal disease, cardiac amyloidosis afflicts a significant fraction of patients with heart failure (up to 20% by some estimates), particularly aging men.4–6 Although the numbers keep rising as the population ages and the diagnostic methods improve, the disease can be actively managed thanks to major pharmacological advances.4–6 Cardiac amyloidosis results from the extracellular deposition of a plasma protein, which is typically amyloid transthyretin (ATTR), amyloid immunoglobulin light chains (AL) or, occasionally, amyloid apolipoprotein A-I (AApoAI).4 Co-deposition of different fibril-forming proteins perhaps due to heterogeneous cross-seeding is an emerging challenge in the field.8,9 Current pharmacotherapies for ATTR include small-molecule stabilizers of the native protein conformation (e.g. tafamidis); oligonucleotide-based gene-silencing approaches; newly developed CRISPR-Cas9-based gene editing tools; and antibody-mediated amyloid removal.6 Current pharmacotherapies for AL include chemotherapy targeting the aberrant plasma cell clone to block the generation of the amyloidogenic light chain, along with supportive therapies; new therapies are being developed, such as antibody-mediated fibril removal.7 In a subset of patients, organ transplantation (liver for ATTR or AApoAI, bone marrow for AL) can be used to block the aberrant protein secretion.4 These protein-targeting approaches necessitate timely and accurate identification of the fibril-forming protein, or amyloid typing, which is paramount for treatment.The gold standard for amyloid typing has been organ biopsy followed by immunohistochemical or mass spectrometry analysis to identify the fibrillar protein and associated “amyloid signature” proteins. The biopsy results are more accurate for the target vs. surrogate organ, e.g. heart vs. fat. However, endomyocardial biopsy is a risky procedure not suited for many patients. Since 2016, the field has been transformed by the application of noninvasive methods for diagnosis and differentiation of cardiac ATTR vs. AL, which became the preferred modality in many countries.5,10,11 The methods involve scintigraphy using technetium-labeled bone tracers pyrophosphate (99mTc-PYP), hydroxymethylene diphosphonate (99mTc-HMDP) or 3,3-diphosphono-1,2-propanodicarboxylate (99mTc-DPD), followed by 3D nuclear imaging to differentiate tracer binding to amyloid deposits vs. Ca2+ in blood (Fig. 1A and B).5 This diagnostic modality is currently used for ∼70% of patients with cardiac amyloidosis, yet gray areas remain. Amyloid diagnostics using 99mTc scintigraphy is prone to false negatives, false positives require additional testing, the method does not assess the overall amyloid load and is not suited as a readout for treatment, and the tracer binding moiety is unknown.5,10,12 Some of these issues and their potential molecular underpinnings are addressed below.
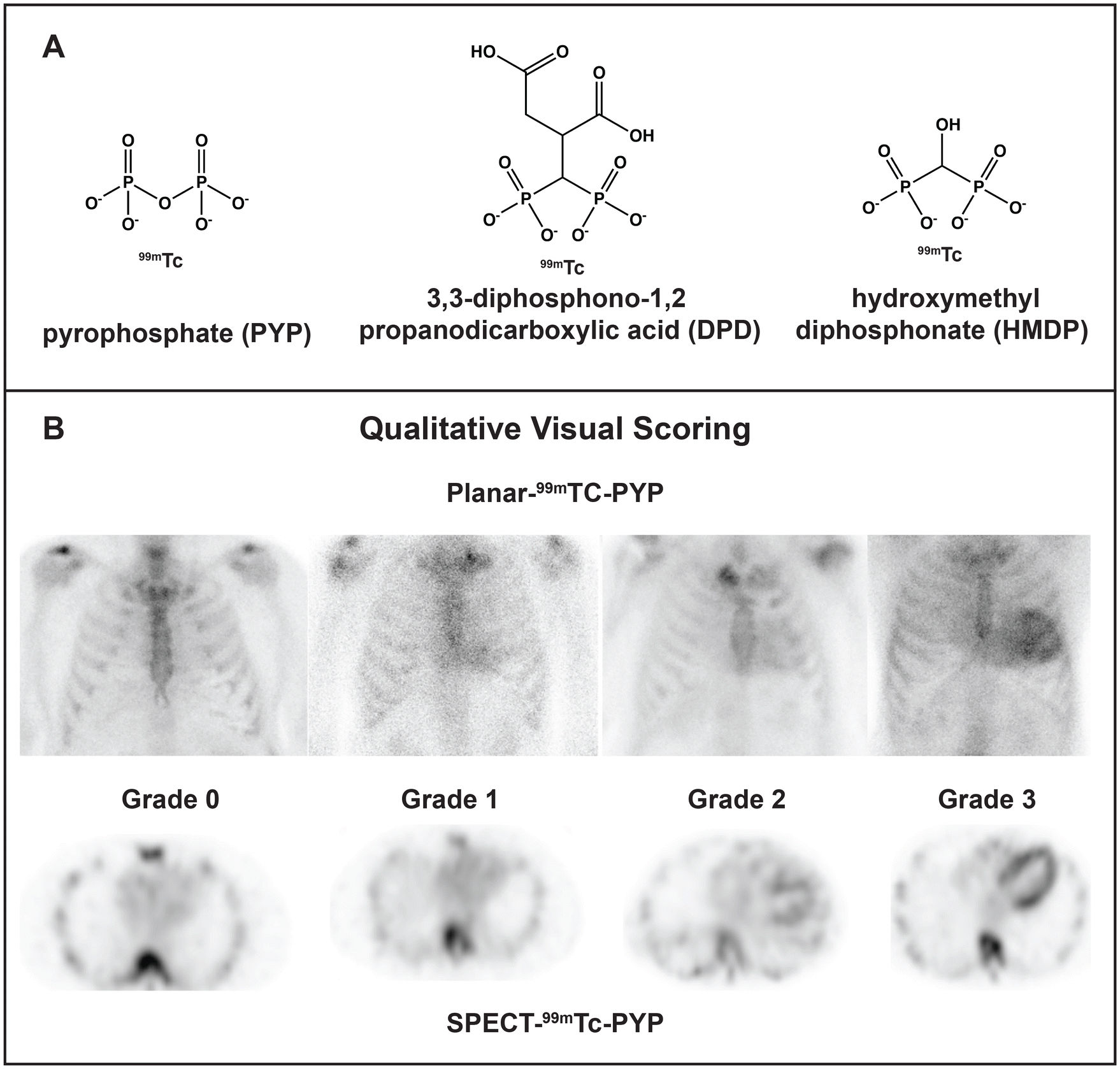 | ||
| Fig. 1 Technetium-labeled bone tracers used for diagnostic imaging of cardiac amyloid. (A) 99mTc-labeled radiotracer molecules. 99mTc-PYP is preferentially used in the USA, while 99mTc-DPD and 99mTc-HMDP are widely used in Europe.5 (B) Radiographs using 99mTc-PYP scintigraphy and single-photon emission computed tomography (SPECT) to visualize and quantify the tracer uptake in the heart. Numbers indicate the degree of uptake, from grade 0 (no uptake) to grade 3 (high uptake); grades 2 and 3 are often observed in untreated ATTR patients. Reproduced from Dorbala et al.11 with permission from Elsevier, copyright 2020. | ||
3. Key challenges and unanswered questions in amyloid typing
For unknown reasons, the 99mTc-PYP/DPD/HMDP radiotracers preferentially (although not only and not always) colocalize with ATTR vs. AL or AApoAI deposits.5,10 This differential uptake enables one to distinguish between ATTR and other types of cardiac amyloidosis. Although misinterpretation of the images remains the major source of false positives and false negatives, there are other sources of error. In fact, some AL patients show substantial radiotracer uptake, leading to false positives in ATTR diagnostics. Still, if the uptake is high (grade 2 or 3 on the Perugini scale, Fig. 1B) and AL amyloidosis has been excluded by other methods, ATTR can be diagnosed with high certainty. Furthermore, false negatives in 99mTc scintigraphy have also been reported, and are thought to stem from the low amyloid load at early disease stages, differences in TTR expression, and/or tracer uptake by most but not all ATTR deposits.5,10 Notably, the reasons for the fiber-specific tracer uptake are unknown.Furthermore, although the 99mTc scintigraphy provides a useful diagnostic tool, recent reports show that it is not a good readout for the changes in cardiac function in response to treatment. ATTR patients treated with tafamidis (a small-molecule stabilizer of the native transthyretin tetramer) or with an RNAi-based gene silencing therapy show decreased radiotracer uptake, yet their cardiac function does not improve.12 We and others hypothesize that the tracer is taken up only by the surfaces of newly formed amyloids, while in mature amyloids tracer binding sites are blocked by other molecules deposited on amyloid surface. In fact, amyloid deposits usually localize in the areas of excessive collagen deposition, or fibrosis, suggesting amyloid sequestration by the extracellular matrix molecules such as collagen fibrils. Intimate amyloid-collagen interactions have been implicated in the collagen-induced inhibition of amyloid phagocytosis and clearance in vivo and in vitro.13 Moreover, amyloid-collagen superstructures have been observed in patient-derived AL fibrils using a combination of cryo-EM and cryo-electron tomography.14 We posit that intimate interactions of amyloids with collagens and perhaps other extracellular matrix components interfere with the tracer uptake and influence the biological properties of amyloids and associated calcification. Such interactions extend to other protein amyloids15 and are actively explored by us and others.
Perhaps the major gap in our understanding of 99mTc scintigraphy of amyloids is that the radiotracer-binding moiety is unknown. This moiety potentially involves amyloid fibrils and/or calcific deposits associated with cardiac amyloidosis.16 Calcification in collagen-rich regions, which for unknown reasons is greater in cardiac ATTR vs. AL patients,16,17 is probably not the only mechanism explaining preferential tracer uptake by ATTR deposits.5,17 Identifying the radiotracer-binding moiety is necessary to elucidate the molecular basis of 99mTc scintigraphy, understand its limitations, and potentially identify new therapeutic targets.
4. Calcium binding and fibril-specific radiotracer uptake by ATTR amyloids
In bone and calcific deposits, PYP and diphosphonate radiotracers chelate to Ca2+ ions.18 Our central premise is that, in addition to calcific deposits, protein amyloids uptake PYP/DPD/HMPD radiotracers via arrays of surface-bound Ca2+ ions. Metal ion binding to amyloid has been predicted3,19 and observed experimentally20 (Fig. 2A). Moreover, several lines of experimental evidence suggest that phosphates in 99mTc-PYP/DPD/HMPD radiotracers can bind Ca2+ in amyloid.21 Current evidence includes: in vitro binding of 99mTc-DPD to synthetic amyloid fibrils;22 destabilization and aggregation of native transthyretin upon calcium binding in vitro;23 animal model studies showing that the anti-ATTR antibody enhances rapid disappearance of the amyloid-associated 99mTc-PYP signal in an ATTR rat model;21 deposition of calcium phosphate immediately adjacent to cardiac amyloid fibrils of ATTR (but not AL) observed in some clinical human studies.17,24 Taken together, these findings suggest radiotracer binding to amyloid surface. Below we propose structural underpinnings for such binding and its fibril specificity.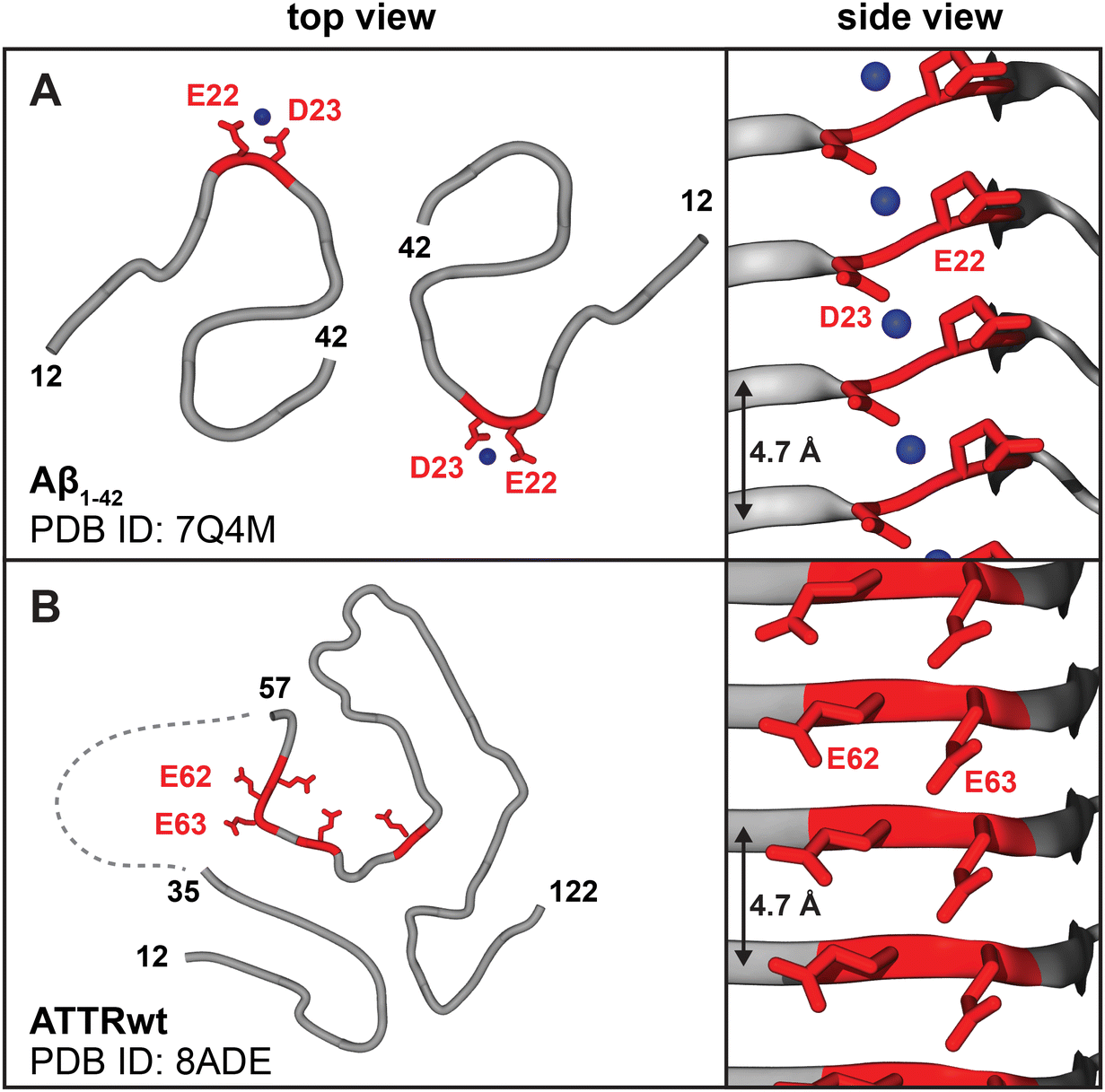 | ||
| Fig. 2 Amyloids display periodic ligand binding sites on the fibril surface, including acidic residue pairs that can bind calcium. (A) Cryo-EM structures of patient-derived Aβ1–42 type-I and type-II fibers show periodic arrays of metal ions bound by acidic residue pairs, Glu22-Asp23;20 type-II fibril structure is shown. (B) Cryo-EM structures of ATTR type-A fibrils feature surface arrays of acidic pairs, e.g. Glu62-Glu63 in the major “closed-gate” polymorph. Linker residues 35–56 are cleaved or disordered (dashed line),29 providing access to the Glu62-Glu63 site that probably binds Ca2+. | ||
ATTR features two fibril types: the predominant type-A fibrils contain both full-length and fragmented TTR, while type-B fibrils contain only full-length TTR. Wild-type TTR (in ATTRwt) and most ATTR variants (in ATTRv, which typically feature single-point substitutions) form type-A fibrils; few ATTRv (Phe64Leu and Tyr114Cys) feature type-B fibrils; Val30Met forms either type-A or type-B fibrils in different patients. For unknown reasons, only type-A fibrils uptake radiotracers.5 Amyloid fibril structures suggest an explanation.
5. Amyloid fibril structures suggest surface arrays of Ca2+ binding sites
Calcium binding sites in proteins are highly variable in metal ion coordination, which can involve 4–8 oxygens from acidic and polar groups including water molecules, and range in affinity from millimolar to nanomolar.25 High-affinity Ca2+ binding sites involve two acidic residues that balance the charge; in contiguous sites, these residues are close in the primary structure, which minimizes the entropic penalty of binding and increases the affinity. Notably, some but not all amyloids feature exposed contiguous pairs of acidic residues (Fig. 2 and 3). Such residues form periodic arrays along the fibril surface (Fig. 2), which is unique to amyloid assemblies and stems from their shared structural features.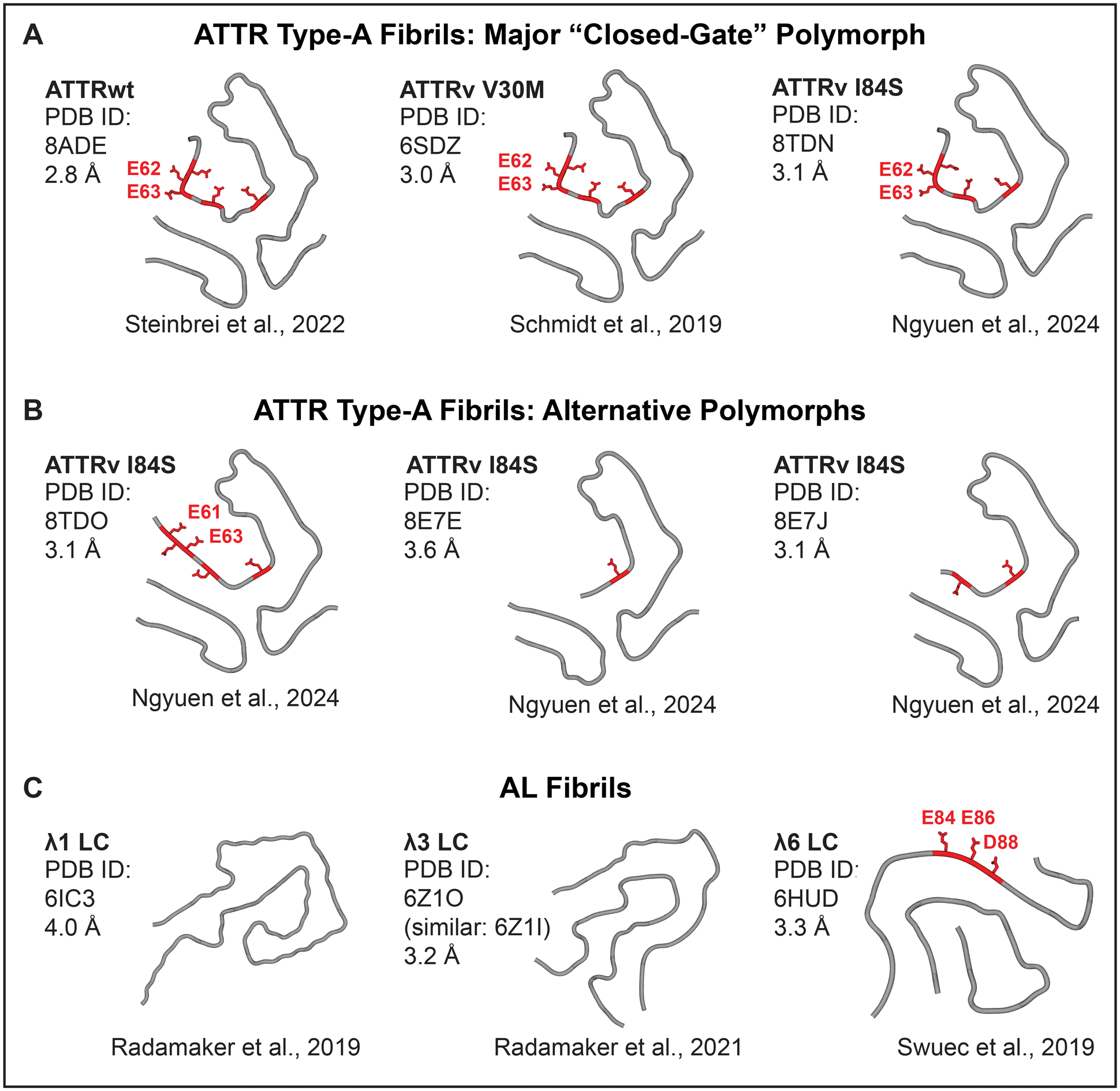 | ||
| Fig. 3 Cryo-EM structures of cardiac patient-derived ATTR and AL fibrils suggest calcium-mediated radiotracer binding sites. ATTR type-A fibrils have similar folds with alternative “gate” conformations leading to fibril polymorphism. (A) The major “closed-gate” polymorph. (B) Alternative polymorphs. All patients featured the “closed-gate” polymorph; some had additional polymorphs.31 Acidic-rich segment Glu61-Glu62-Glu63-Phe64-Val65-Glu66 (red). (C) Cardiac AL fibrils are highly polymorphic. Only one structure features an exposed acidic-rich segment, Glu80-Asp81-Glu82-Ala83-Glu84-Asp84 (red). | ||
All known structures of patient-derived amyloid fibrils contain flattened protein molecules stacked in parallel in-register β-sheets connected by intermolecular hydrogen bonds, wherein identical residues spaced at 4.7–4.8 Å form periodic arrays (Fig. 2A and B).26 In amorphous calcium phosphate (ACP), which is the precursor of the hydroxyapatite mineral in the bone and calcific deposits, the shortest Ca–Ca spacing is also 4.7 Å. This stereochemical complementarity between amyloid and ACP suggests that arrays of acidic residues (Glu or Asp) on the amyloid surface can bind ACP and Ca2+, facilitating both hydroxyapatite formation27 and Ca2+-mediated bone tracer uptake. If so, why do type-A ATTR fibrils preferentially uptake bone tracers?
The answers are suggested by the atomic structures of patient-derived type-A ATTR fibrils, a number of which have been determined by cryo-EM since 2019.28–31 All ATTRwt and ATTRv fibrils, including Val20Ile, Gly47Glu, Val30Met, Ile84Ser, and Val122Ile, adopt a similar amyloid fold.31 This similarity reflects common primary structures that typically differ by just one amino acid substitution in ATTRv vs. ATTRwt. N-terminal (Pro11-Lys35) and C-terminal (Gly57-Thr123) residue fragments comprise fibril cores; variable “gate” segment (Gly57-Gly67) generates structural polymorphism (Fig. 3B). All patients feature a “closed-gate” polymorph (Fig. 2B and 3A) containing exposed Glu62-Glu63 pair from the sole acidic-rich segment, Glu61-Glu62-Glu63-Phe64-Val65-Glu66. This pair forms a periodic ionic array along the fibril side; similarly, parenchymal type-I and type-II fibrils of Alzheimer's Aβ1–42 peptide feature Glu22-Asp23 arrays that bind metal ions observed by cryoEM (Fig. 2A). We postulate that in ATTR type-A fibrils, exposed acidic pairs from Glu61-Glu66 segment bind Ca2+ ions that form surface arrays and mediate both calcification and bone tracer uptake. Conformational variability, structural disordering and/or partial truncation of the “gate” segment reported in some patient-specific polymorphs (Fig. 3B) potentially contribute to variable patient-specific tracer uptake observed by 99mTc scintigraphy.
This hypothetical mechanism is consistent with lack of tracer uptake by ATTR type-B fibrils, whose atomic structures are currently unknown. We speculate that in full-length transthyretin comprising type-B fibrils, residues 35–56 linking the “gate” to the N-terminal segment may obstruct Glu62-Glu63 and/or change the amyloid conformation to block the radiotracer binding. Patient-derived type-B fibrils structures, which are currently under investigation, will test this idea.
Unlike ATTR, antibody light chain variable domains that comprise AL deposits have highly variable amino acid sequences, with each AL patient featuring a unique primary structure of the fibril-forming protein. This translates into structural variability of AL amyloid folds, several of which have been determined since 2019 (Fig. 3C).32–34 Of the four available structures of cardiac AL fibrils, only one features exposed acidic pairs from the sole acidic-rich segment, Glu80-Asp81-Glu82-Ala83-Glu84-Asp84. We propose that: (i) Ca2+ and radiotracer binding by such exposed acidic pairs potentially produces false positives in ATTR scintigraphy; (ii) variable (typically diminished) exposure of the acidic-rich segment Glu80-Asp84 in AL fibrils probably contributes to variable (generally diminished) tracer uptake and microcalcification detected in AL vs. ATTR patients.10,16,17
The sole currently available AL patient-derived fibril structure that features an exposed acidic-rich segment belongs to λ6 subfamily of the λ isotype, which is responsible for approximately 1/4 and 3/4 of all AL cases, respectively.7 Amino acid sequence in this segment is highly conserved in the λ isotype, but in κ isotype (which accounts for nearly 1/4 of AL cases) this sequence is less acidic and less conserved.35 Due to high sequence and structural variability of the light chain variable domains, which form the fibril core in AL deposits, more structural studies of patient-derived AL fibrils are needed to definitively link exposed acidic-rich segments with false positives in the ATTR diagnostics using 99mTc scintigraphy.
6. Summary, open question and future directions
In conclusion, atomic structures of patient-derived cardiac amyloids support the notion that fibril-specific uptake of 99mTc-labeled bone tracers involves calcium-mediated pyrophosphate or diphosphonate binding to amyloids, as well as to amyloid-associated calcific deposits. We postulate that stereochemical complementarity between the organic (acidic residue pairs on amyloid surface) and inorganic phases (Ca2+/ACP and 99mTc-PYP/DPD/HMDP) promotes both calcification and bone tracer uptake by type-A ATTR fibrils and by a subset of AL fibrils featuring acidic residue pairs on their surface (Fig. 3). This new concept helps explain why both calcification and tracer uptake are generally increased in ATTR vs. AL patients with cardiac amyloidosis.Several questions remain open. First, relative amounts of PYP/DPD/HMDP tracers that bind to amyloid vs. amyloid-associated calcific deposits in vivo remain to be determined. Second, it remains unclear why 99mTc-labeled methyl diphosphonate bone tracer (MDP) shows low sensitivity for cardiac amyloidosis. Greater rotational flexibility of PYP and DPD/HMDP might be a factor contributing to their increased binding to amyloid as compared to MDP. Third, the proposed concept applies to bone tracers that chelate to Ca2+, such as PYP and diphosphonates; the other type of currently used 18F-labeled positron emission tomography bone tracer NaF binds through a different mechanism.36 Fourth, tracer uptake in vivo is probably influenced by other factors, including calcium-binding extracellular matrix components such as collagens14,27 and heparan sulfate, a ubiquitous amyloid co-factor.3 These and other auxiliary molecules are found in amyloid deposits and, we propose, can directly bind to amyloids and influence their formation, structure, interactions with other ligands, and biological properties. Future studies of amyloid complexes with these factors, Ca2+, and radiotracers will test these concepts, and will also explore potential binding of Ca2+ and radiotracers to pre-fibrillar amyloid oligomers. Finally, cryo-EM studies of other patient-derived amyloids, including type-B ATTR fibrils and additional AL fibril polymorphs, will shed more light on the structural and ligand binding properties of these remarkable protein polymers.
Author contributions
EL – Visualization, methodology, writing – review and editing. SJ – Investigation, writing – review and editing, funding acquisition. OG – Conceptualization, methodology, writing – original draft, review and editing, funding acquisition.Ethical approval
This research does not involve any human or animal studies (exemption 4 by the Institutional Review Board). The research was conducted in full compliance with institutional ethics rules (IRB approval protocol # 20-2249).Abbreviations
| Aβ | Alzheimer's amyloid-beta peptide |
| ACP | Amorphous calcium phosphate |
| AApoAI | Amyloid apolipoprotein A-I |
| AL | Amyloid light chain |
| ATTR | Amyloid transthyretin |
| Cryo-EM | Electron cryo-microscopy |
| DPD | 3,3-Diphosphono-1,2-propanodicarboxylate |
| HMDP | Hydroxymethylene diphosphonate |
| PYP | Pyrophosphate |
Data availability
All original data for the minireview “Molecular basis for non-invasive diagnostics of cardiac amyloids using bone tracers” has been previously published. Atomic structures of amyloids are available in the protein data bank (pdb); access numbers (PDB ID) are included in the manuscript. The published paper will be available via PubMed and upon request from authors.Conflicts of interest
None of the authors has any conflicts of interests to declare.Acknowledgements
This research was supported by NIH grants R01 GM067260 and R01 GM135158. We thank Drs Navneet Narula and Jagat Narula who stimulated our interest in cardiac amyloid calcification.References
- J. N. Buxbaum, A. Dispenzieri, D. S. Eisenberg, M. Fändrich, G. Merlini, M. J. M. Saraiva, Y. Sekijima and P. Westermark, Amyloid nomenclature 2022: update, novel proteins, and recommendations by the International Society of Amyloidosis (ISA) nomenclature committee, Amyloid, 2022, 29, 213–219 CrossRef PubMed.
- N. Leung and S. H. Nasr, Update on classification, etiology, and typing of renal Amyloidosis: A review, Am. J. Kidney Dis., 2024 DOI:10.1053/j.ajkd.2024.01.530.
- E. Lewkowicz, S. Jayaraman and O. Gursky, Protein amyloid cofactors: Charged side-chain arrays meet their match?, Trends Biochem. Sci., 2021, 46, 626–629 CrossRef CAS PubMed.
- J. M. Griffin, H. Rosenblum and M. S. Maurer, Pathophysiology and therapeutic approaches to cardiac amyloidosis, Circ. Res., 2021, 128, 1554–1575 CrossRef CAS PubMed.
- R. Saro, D. Pavan, A. Porcari, G. Sinagra and M. Mojoli, Lights and shadows of clinical applications of cardiac scintigraphy with bone tracers in suspected amyloidosis, J. Clin. Med., 2023, 12, 7605 CrossRef CAS PubMed.
- A. Porcari, G. Sinagra, J. D. Gillmore, M. Fontana and P. N. Hawkins, Breakthrough advances enhancing care in ATTR amyloid cardiomyopathy, Eur. J. Intern. Med., 2024, 123, 29–36 CrossRef CAS PubMed.
- V. Sanchorawala, Systemic light chain amyloidosis, N. Engl. J. Med., 2024, 390, 2295–2307 CrossRef PubMed.
- K. Konstantoulea, N. Louros, F. Rousseau and J. Schymkowitz, Heterotypic interactions in amyloid function and disease, FEBS J., 2022, 289, 2025–2046 CrossRef CAS PubMed.
- U. Thelander , Presented in part at the XIX International Symposium on Amyloidosis, Rochester MN, USA, May 2024.
- J. D. Gillmore, M. S. Maurer, R. H. Falk, G. Merlini, T. Damy, A. Dispenzieri, A. D. Wechalekar, J. L. Berk, C. C. Quarta, M. Grogan, H. J. Lachmann, S. Bokhari, A. Castano, S. Dorbala, G. B. Johnson, A. W. J. M. Glaudemans, T. Rezk, M. Fontana, G. Palladini, P. Milani, P. L. Guidalotti, K. Flatman, T. Lane, F. W. Vonberg, C. J. Whelan, J. C. Moon, F. L. Ruberg, E. J. Miller, D. F. Hutt, B. P. Hazenberg, C. Rapezzi and P. N. Hawkins, Nonbiopsy diagnosis of cardiac transthyretin amyloidosis, Circulation, 2016, 133, 2404–2412 CrossRef CAS PubMed.
- S. Dorbala, S. Cuddy and R. H. Falk, How to image cardiac amyloidosis, JACC Cardiovasc. Imaging, 2020, 13, 1368–1383 CrossRef PubMed.
- R. Rettl, R. Calabretta, F. Duca, C. Binder, C. Kronberger, R. Willixhofer, M. Poledniczek, C. Donà, C. Nitsche, D. Beitzke, C. Loewe, M. Auer-Grumbach, D. Bonderman, S. Kastl, C. Hengstenberg, R. Badr Eslam, J. Kastner, J. Bergler-Klein, M. Hacker and A. Kammerlander, Reduction in 99m Tc-DPD myocardial uptake with therapy of ATTR cardiomyopathy, Amyloid, 2024, 31, 42–51 CrossRef CAS PubMed.
- J. W. Jackson, J. S. Foster, E. B. Martin, S. Macy, C. Wooliver, M. Balachandran, T. Richey, R. E. Heidel, A. D. Williams, S. J. Kennel and J. S. Wall, Collagen inhibits phagocytosis of amyloid in vitro and in vivo and may act as a ‘don't eat me’ signal, Amyloid, 2023, 30, 249–260 CrossRef CAS PubMed.
- S. Ricagno, T. Schulte, A. Chaves-Sanjuan, V. Speranzini, K. Sicking, G. Mazzini, P. Rognoni, S. Caminito, P. Milani, C. Marabelli, A. Corbelli, L. Diomede, F. Fiordaliso, L. Anastasia, C. Pappone, G. Merlini, M. Bolognesi, M. Nuvolone, R. Fernandez-Busnadiego and G. Palladini, Helical superstructures between amyloid and collagen VI in heart-derived fibrils from a patient with Light Chain Amyloidosis, Res. Sq., 2023 DOI:10.21203/rs.3.rs-3625869/v1 , preprint.
- C. L. Hoop, J. Zhu, S. Bhattacharya, C. A. Tobita, S. E. Radford and J. Baum, Collagen I weakly interacts with the β-sheets of β2-microglobulin and enhances conformational exchange to induce amyloid fformation, J. Am. Chem. Soc., 2020, 142, 1321–1331 CrossRef CAS PubMed.
- U. Thelander, G. T. Westermark, G. Antoni, S. Estrada, A. Zancanaro, E. Ihse and P. Westermark, Cardiac microcalcifications in transthyretin (ATTR) amyloidosis, Int. J. Cardiol., 2022, 352, 84–91 CrossRef PubMed.
- A. Mori, Y. Saito, K. Nakamura, T. Iida, S. Akagi, M. Yoshida, M. Taniyama, T. Miyoshi and H. Ito, Microcalcification and 99mTc-Pyrophosphate uptake without increased bone metabolism in cardiac tissue from patients with transthyretin cardiac amyloidosis, Int. J. Mol. Sci., 2023, 24, 1921 CrossRef CAS PubMed.
- G. P. Keeling, F. Baark, O. L. Katsamenis, J. Xue, P. J. Blower, S. Bertazzo and R. T. M. de Rosales, 68Ga-bisphosphonates for the imaging of extraosseous calcification by positron emission tomography, Sci. Rep., 2023, 13, 14611 CrossRef CAS PubMed.
- L. R. Martín, L. R. Santiago, I. V. Korendovych, M. Sodupe and J.-D. Maréchal, in Methods in Enzymology, ed. I. V. Korendovych, Academic Press, 2024, vol. 697, pp. 211–245 Search PubMed.
- Y. Yang, D. Arseni, W. Zhang, M. Huang, S. Lövestam, M. Schweighauser, A. Kotecha, A. G. Murzin, S. Y. Peak-Chew, J. Macdonald, I. Lavenir, H. J. Garringer, E. Gelpi, K. L. Newell, G. G. Kovacs, R. Vidal, B. Ghetti, B. Ryskeldi-Falcon, S. H. W. Scheres and M. Goedert, Cryo-EM structures of amyloid-β 42 filaments from human brains, Science, 2022, 375, 167–172 CrossRef CAS PubMed.
- J. George, M. Rappaport, S. Shimoni, S. Goland, I. Voldarsky, Y. Fabricant, O. Edri, V. Cuciuc, S. Lifshitz, S. Tshori and M. Fassler, A novel monoclonal antibody targeting aggregated transthyretin facilitates its removal and functional recovery in an experimental model, Eur. Heart J., 2020, 41, 1260–1270 CrossRef CAS PubMed.
- F. E. Buroni, M. G. Persico, L. Lodola, M. Concardi and C. Aprile, In vitro study: binding of 99m Tc-DPD to synthetic amyloid fibrils, Curr. Issues Pharm. Med. Sci., 2015, 28, 231–235 CrossRef CAS.
- C. Cantarutti, M. C. Mimmi, G. Verona, W. Mandaliti, G. W. Taylor, P. P. Mangione, S. Giorgetti, V. Bellotti and A. Corazza, Calcium binds to transthyretin with low affinity, Biomolecules, 2022, 12, 1066 CrossRef CAS PubMed.
- M. A. Stats and J. R. Stone, Varying levels of small microcalcifications and macrophages in ATTR and AL cardiac amyloidosis: implications for utilizing nuclear medicine studies to subtype amyloidosis, Cardiovasc. Pathol., 2016, 25, 413–417 CrossRef CAS PubMed.
- X. Wang, M. Kirberger, F. Qiu, G. Chen and J. J. Yang, Towards predicting Ca2+ –binding sites with different coordination numbers in proteins with atomic resolution, Proteins, 2009, 75, 787–798 CrossRef CAS PubMed.
- P. C. Ke, R. Zhou, L. C. Serpell, R. Riek, T. P. J. Knowles, H. A. Lashuel, E. Gazit, I. W. Hamley, T. P. Davis, M. Fändrich, D. E. Otzen, M. R. Chapman, C. M. Dobson, D. S. Eisenberg and R. Mezzenga, Half a century of amyloids: Past, present and future, Chem. Soc. Rev., 2020, 49, 5473–5509 RSC.
- S. Jayaraman, N. Narula, J. Narula and O. Gursky, Amyloid and collagen templates in aortic valve calcification, Trends Mol. Med., 2024 Search PubMed , S1471-4914(24)00115-1.
- M. Schmidt, S. Wiese, V. Adak, J. Engler, S. Agarwal, G. Fritz, P. Westermark, M. Zacharias and M. Fändrich, Cryo-EM structure of a transthyretin-derived amyloid fibril from a patient with hereditary ATTR amyloidosis, Nat. Commun., 2019, 10, 5008 CrossRef PubMed.
- M. Steinebrei, J. Gottwald, J. Baur, C. Röcken, U. Hegenbart, S. Schönland and M. Schmidt, Cryo-EM structure of an ATTRwt amyloid fibril from systemic non-hereditary transthyretin amyloidosis, Nat. Commun., 2022, 13, 6398 CrossRef CAS PubMed.
- I. Iakovleva, M. Hall, M. Oelker, L. Sandblad, I. Anan and A. E. Sauer-Eriksson, Structural basis for transthyretin amyloid formation in vitreous body of the eye, Nat. Commun., 2021, 12, 7141 CrossRef CAS PubMed.
- B. A. Nguyen, V. Singh, S. Afrin, A. Yakubovska, L. Wang, Y. Ahmed, R. Pedretti, M. d. C. Fernandez-Ramirez, P. Singh, M. Pękała, L. O. Cabrera Hernandez, S. Kumar, A. Lemoff, R. Gonzalez-Prieto, M. R. Sawaya, D. S. Eisenberg, M. D. Benson and L. Saelices, Structural polymorphism of amyloid fibrils in ATTR amyloidosis revealed by cryo-electron microscopy, Nat. Commun., 2024, 15, 581 CrossRef CAS PubMed.
- P. Swuec, F. Lavatelli, M. Tasaki, C. Paissoni, P. Rognoni, M. Maritan, F. Brambilla, P. Milani, P. Mauri, C. Camilloni, G. Palladini, G. Merlini, S. Ricagno and M. Bolognesi, Cryo-EM structure of cardiac amyloid fibrils from an immunoglobulin light chain AL amyloidosis patient, Nat. Commun., 2019, 10, 1269 CrossRef PubMed.
- L. Radamaker, Y.-H. Lin, K. Annamalai, S. Huhn, U. Hegenbart, S. O. Schönland, G. Fritz, M. Schmidt and M. Fändrich, Cryo-EM structure of a light chain-derived amyloid fibril from a patient with systemic AL amyloidosis, Nat. Commun., 2019, 10, 1103 CrossRef PubMed.
- L. Radamaker, S. Karimi-Farsijani, G. Andreotti, J. Baur, M. Neumann, S. Schreiner, N. Berghaus, R. Motika, C. Haupt, P. Walther, V. Schmidt, S. Huhn, U. Hegenbart, S. O. Schönland, S. Wiese, C. Read, M. Schmidt and M. Fändrich, Role of mutations and post-translational modifications in systemic AL amyloidosis studied by cryo-EM, Nat. Commun., 2021, 12, 6434 CrossRef CAS PubMed.
- R. van der Kant, N. Louros, J. Schymkowitz and F. Rousseau, Thermodynamic analysis of amyloid fibril structures reveals a common framework for stability in amyloid polymorphs, Structure, 2022, 30, 1178–1189 CrossRef CAS PubMed.
- L. X. Zhang, P. Martineau, V. Finnerty, G. Giraldeau, M.-C. Parent, F. Harel and M. Pelletier-Galarneau, Comparison of 18F-sodium fluoride positron emission tomography imaging and 99mTc-pyrophosphate in cardiac amyloidosis, J. Nucl. Cardiol., 2022, 29, 1132–1140 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2024 |