DOI:
10.1039/D4BM00313F
(Review Article)
Biomater. Sci., 2024,
12, 3045-3067
Beyond nanoparticle-based oral drug delivery: transporter-mediated absorption and disease targeting†
Received
29th February 2024
, Accepted 16th April 2024
First published on 19th April 2024
Abstract
Various strategies at the microscale/nanoscale have been developed to improve oral absorption of therapeutics. Among them, gastrointestinal (GI)-transporter/receptor-mediated nanosized drug delivery systems (NDDSs) have drawn attention due to their many benefits, such as improved water solubility, improved chemical/physical stability, improved oral absorption, and improved targetability of their payloads. Their therapeutic potential in disease animal models (e.g., solid tumors, virus-infected lungs, metastasis, diabetes, and so on) has been investigated, and could be expanded to disease targeting after systemic/lymphatic circulation, although the detailed paths and mechanisms of endocytosis, endosomal escape, intracellular trafficking, and exocytosis through the epithelial cell lining in the GI tract are still unclear. Thus, this review summarizes and discusses potential GI transporters/receptors, their absorption and distribution, in vivo studies, and potential sequential targeting (e.g., oral absorption and disease targeting in organs/tissues).
 Hana Cho | Hana Cho is a research professor of pharmacy at The Catholic University of Korea (CUK) in the Republic of Korea. She obtained her double B.S. in Chemistry and Bioengineering and her Ph.D. in Pharmaceutics at CUK. After completing her degree, she underwent postdoctoral training at the University of Utah. Her research area is drug delivery, primarily focusing on developing effective non-viral gene delivery systems and subcellular organelle-targeted drug delivery systems. |
 Kang Moo Huh | Kang Moo Huh is a professor in the Department of Polymer Science and Engineering & Materials Science and Engineering at Chungnam National University (CNU) in the Republic of Korea. He received his Ph.D. from the School of Materials Science, Japan Advanced Institute of Science and Technology (JAIST), in 2002. He conducted his postdoctoral experiences in drug delivery at Korea Institute of Science and Technology (KIST) and Purdue University. He joined CNU as a faculty member in 2004. His current research interests include designing bioactive polymer-, nanocarrier-, and thermogel-based systems and their drug delivery and biomedical applications. |
 Han Chang Kang | Han Chang Kang is a professor of pharmacy at The Catholic University of Korea (CUK) in the Republic of Korea. He received his Ph.D. in Pharmaceutics and Pharmaceutical Chemistry at the University of Utah (UU), in 2007. After completing his degree, he worked as a post-doctoral fellow and a research assistant professor at UU. In 2011, he joined the College of Pharmacy, CUK, as a full-time instructor and now works as a professor. He is also an associate editor or editorial board member for Acta Pharmaceutica Sinica B, NanoConvergence, Journal of Pharmaceutical Investigation, and Journal of the Korean Chemical Society. His research interest is drug delivery and nanomedicine, mainly focusing on designing and developing cell/organelle/signaling molecule/transporting molecule-targeted nanoscale systems for delivering genes, proteins, and chemicals. |
1. Introduction
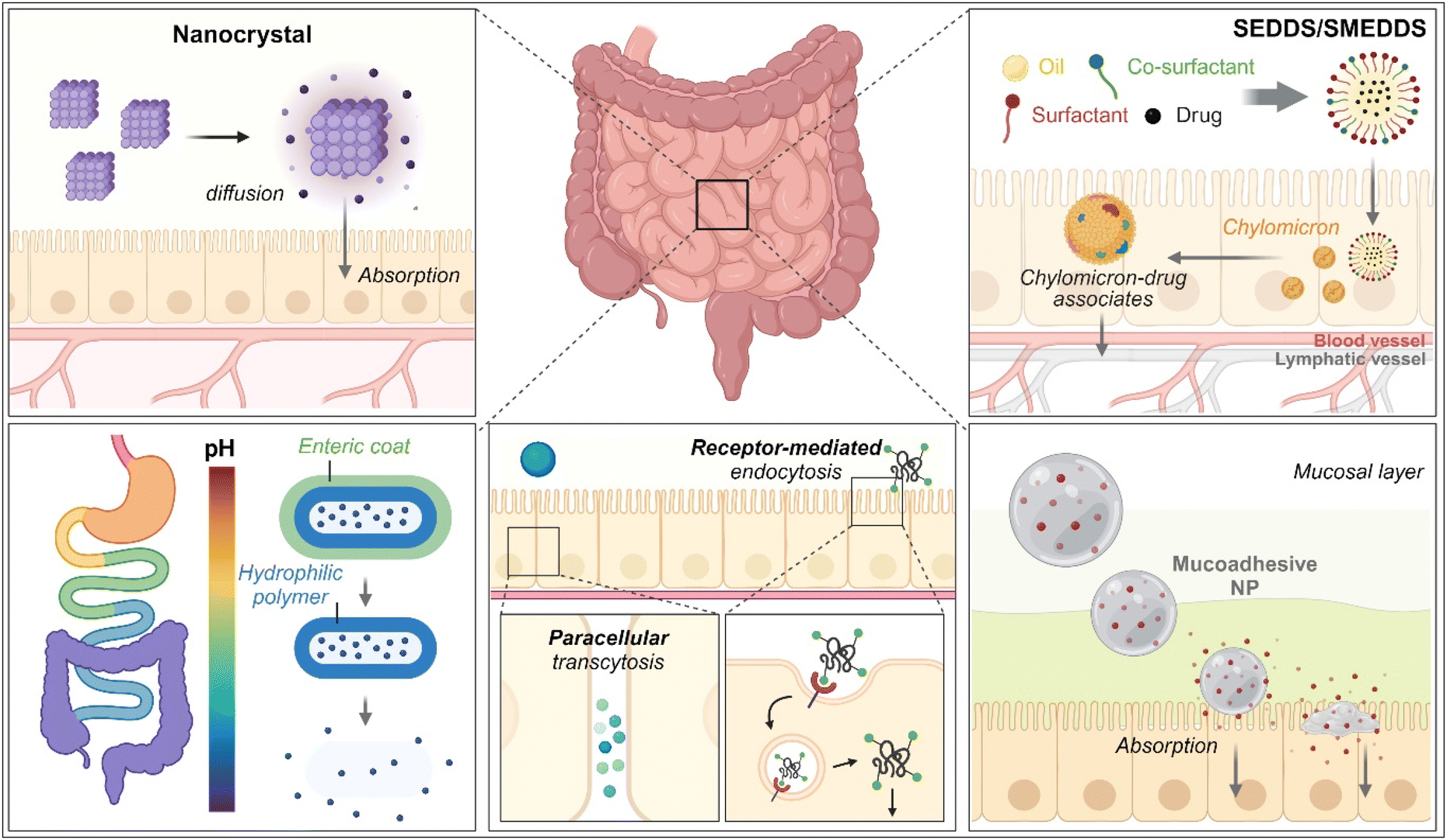
As therapeutics, low molecular weight chemical drugs (<1 kDa) and high molecular weight biological drugs (>2 kDa, e.g., peptides, proteins, siRNA, mRNA, pDNA, etc.) are administered into the body through various routes. The oral route is the most attractive because it is characterized by high patient compliance, requires no aid from medical professionals in dosing, has cost-effectiveness and mostly minor sterility constraints, and has flexible dosage design in production (e.g., solution, powder, suspension, capsule, tablet, etc.). However, the exposure of therapeutics to the chemical, biochemical, or biological environments of the gastrointestinal (GI) tract has frequently caused problems such as poor water-solubility, poor chemical/biological stability (e.g., degradation, denaturation, aggregation, hydrolysis, etc.), poor oral absorption, and so on. Thus, to solve these issues, various oral formulations and strategies such as nanocrystals, enteric-coated dosage forms (ECDFs), self-emulsifying/self-microemulsifying drug delivery systems (SEDDSs/SMEDDSs), micron-sized/nanosized drug delivery systems (MDDSs/NDDSs), and so on have been developed (Fig. 1).
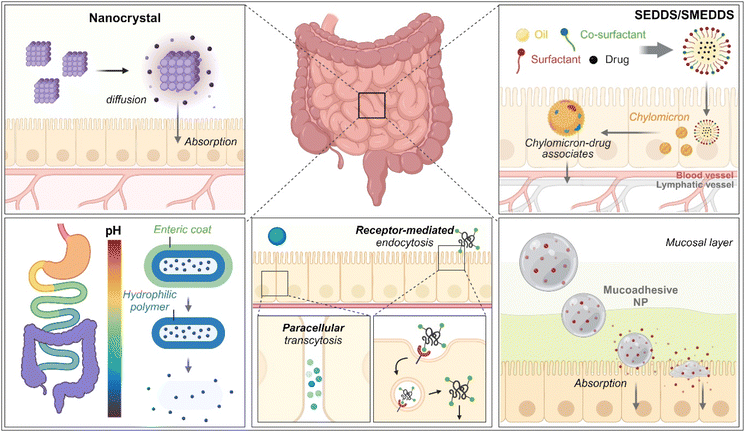 |
| | Fig. 1 Various strategies of oral drug delivery systems. | |
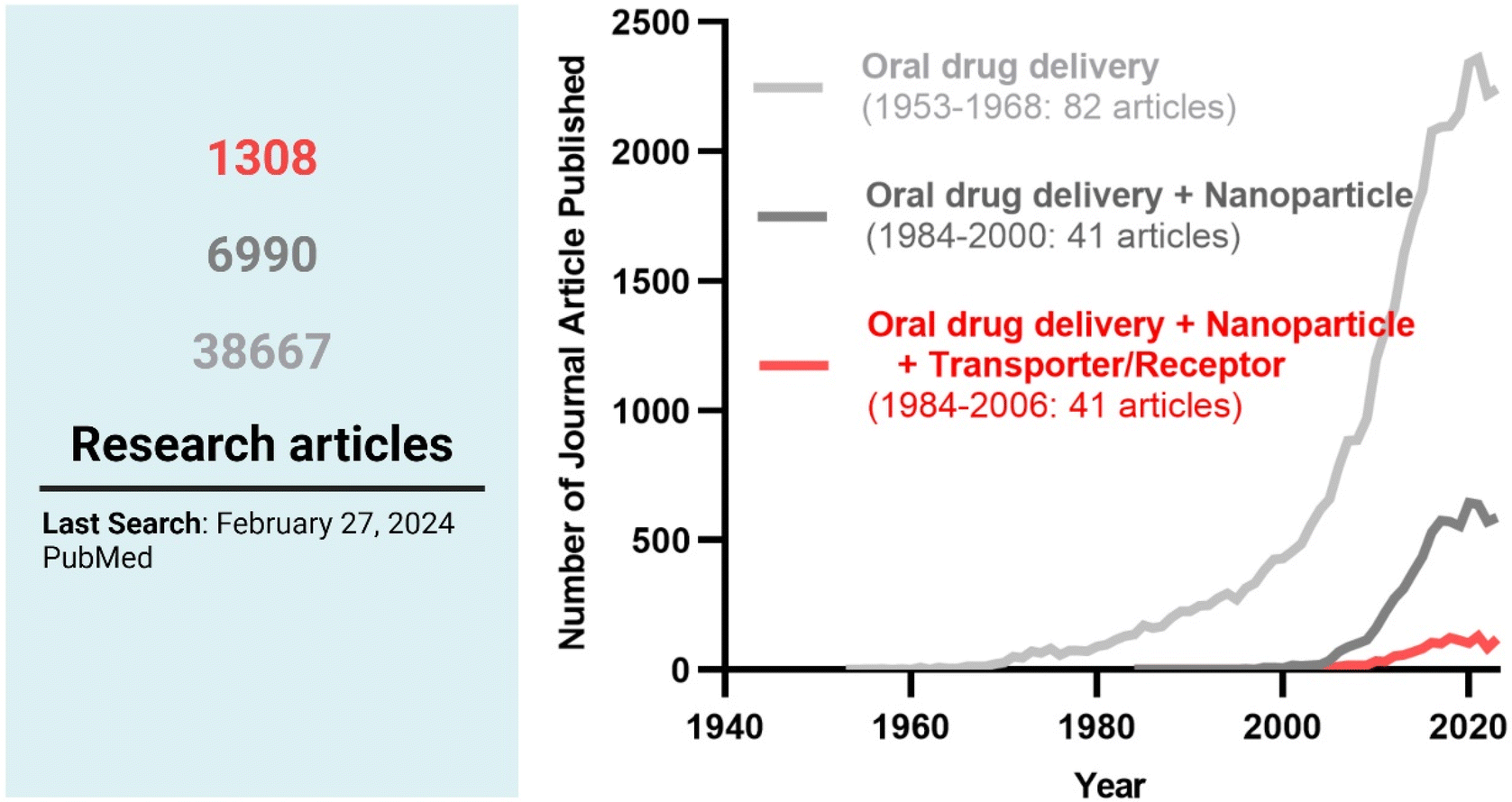
Among the strategies, nanocrystals, SEDDSs/SMEDDSs, and MDDSs/NDDSs have been used to improve the water solubility of payloads. In contrast, ECDFs and MDDSs/NDDSs could protect their payloads from the GI environments and provide targeted delivery or absorption. In addition, the intact particles of SMEDDSs and NDDSs could be absorbed through the GI tract's epithelial cell lining due to their nanoscale sizes. Depending on their intracellular trafficking and exocytosis, NDDSs could deliver their payloads into the enterocytes and their neighboring immune cells and reach various organs (e.g., lungs, liver, brain, tumors, etc.) after systemic or lymphatic circulation. In particular, introducing ligands to bind selectively with transporters/receptors expressed in both the oral and pathological sites could improve oral absorption and therapeutic outcomes while minimizing side effects. These facts have triggered the design strategies for oral drug formulations to shift from solubility and protection to targetability, resulting in starting transporter/receptor-mediated oral delivery at the nanoscale (Fig. 2). Thus, this review will introduce the potential of GI transporter/receptor-mediated oral absorption and discuss its expansion to disease targeting.
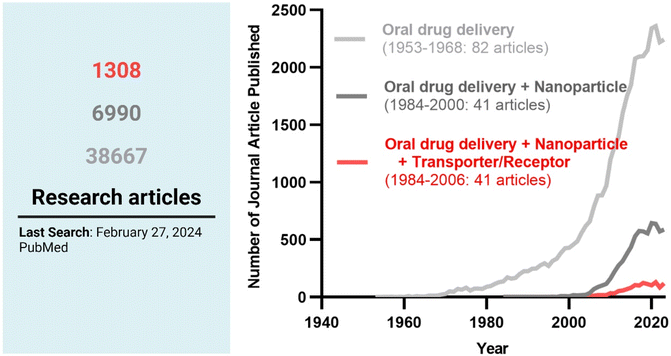 |
| | Fig. 2 The research trends in oral drug delivery, oral NDDSs, and transporter/receptor-mediated oral NDDSs. | |
2. Potential GI transporters/receptors for oral absorption
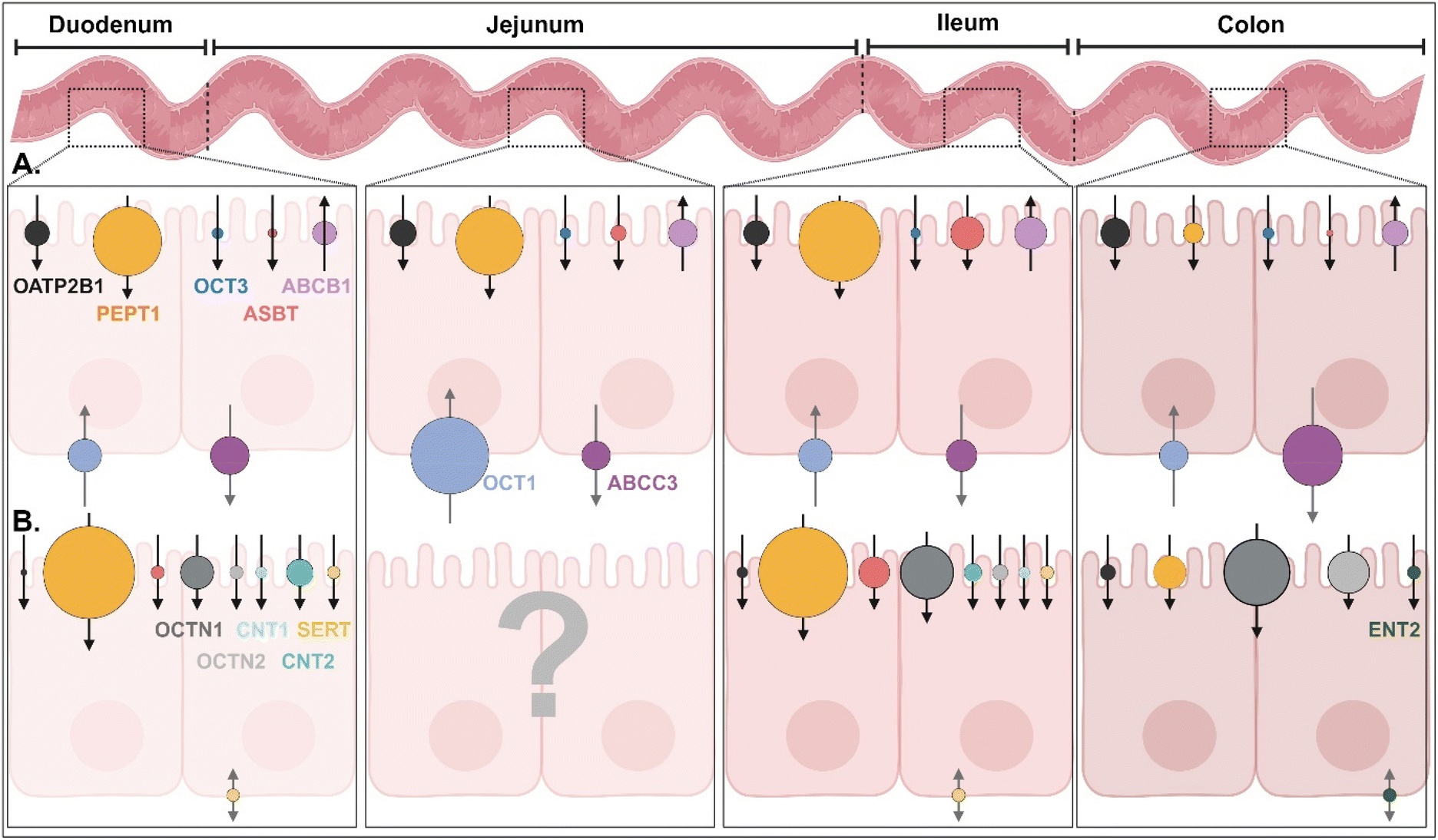
In the GI tract, various transporters and receptors are expressed (Fig. 3 and ESI Table 1†) and deliver their corresponding substrates (e.g., drugs, nutrients, etc.) (Table 1 and ESI Table 2†) through the epithelial cells (e.g., enterocytes) to their neighboring immune cells (e.g., macrophage, dendritic cells), systemic circulation, or lymphatic circulation. The expression levels and substrate-transporting activity of the GI transporters/receptors could be regulated to take up the recommended daily allowance (RDA) of specific substrates such as sugars, bile acids (BAs), vitamins, etc. In addition, the expression levels and transporting activities could be increased or decreased depending on the disease status (Table 2 and ESI Table 3†).1 Nevertheless, we are focused on the expression levels of the GI transporters/receptors rather than their transporting activity because substrate (or ligand)-decorated NDDSs-bound target GI transporters/receptors would follow endocytosis, unlike transport of substrate through a channel in the transporter.2,3 Thus, this section will discuss the potential GI transporters/receptors that their corresponding substrate-decorated NDDSs could use.
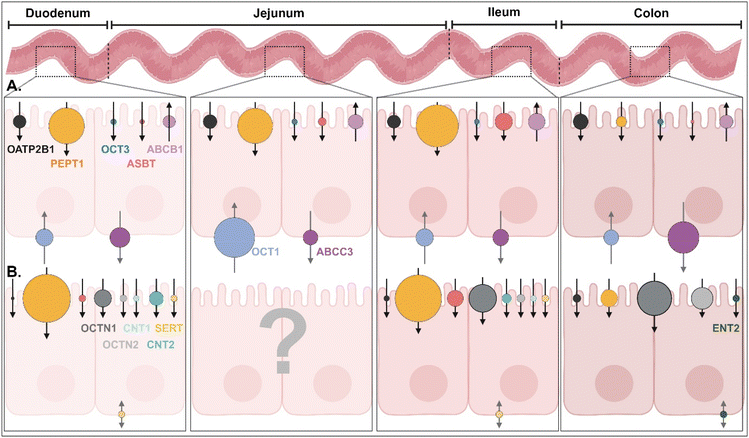 |
| | Fig. 3 (A) Quantitative protein expression levels of transporters/receptors and (B) relative mRNA expression levels of transporters/receptors in the human intestine4,5 (detailed values are shown in ESI Table 1†). OATP2B1, organic anion transporting polypeptides 2B1; PEPT1, peptide transporter 1; OCT1/3, organic cation transporter 1/3; ASBT, apical sodium-dependent bile acid transporter; ABCB1/C3, ATP-binding cassette transporter B1/C3; SERT, serotonin transporter; OCTN1/2, organic cation/carnitine transporter 1/2; CNT1/2, concentrative nucleoside transporter 1/2; ENT2, equilibrative nucleoside transporter 2. | |
Table 1 Potential substrates of various GI transporters and receptors1
| Transporter |
Slightly soluble (>1 mg mL−1) |
Very slightly soluble or practically insoluble (<1 mg mL−1) |
| ASBT |
Cholic acid sodium salt, deoxycholic acid sodium salt, glycocholic acid sodium salt, taurocholic acid sodium salt |
Benzothiazepine (and derivatives), cholic acid, deoxycholic acid, naphthol derivates |
| ATB0,+ |
Carnitine, propionyl-L-carnitine |
Acetylcarnitine |
| CNT2 |
Adenosine, cladribine, didanosine, floxuridine, formycin B, inosine, mizoribine, ribavirin, uridine, zidovudine |
Clofarabine, fluoropyrimidine, guanosine |
| ENT1 |
Adenosine, capecitabine, cladribine, cytosine, fialuridine, fludarabine, gemcitabine, ribavirin, uridine, thymidine, thymine |
Guanine, guanosine |
| GLUT2 |
Glucose |
— |
| GLUT5 |
Fructose |
— |
| MCT1 |
β-D-Hydroxybutyric acid, γ-hydroxybutyric acid, L-lactic acid, pyruvic acid, salicylates, valproic acid |
Nateglinide |
| OATP2B1 |
Aliskiren, fexofenadine |
Amiodarone, atorvastatin, bosentan, DHEAS, estrone-3-sulfate, glibenclamide, talinolol, telmisartan, L-thyroxine |
| OCTN1 |
Acetylcholine, carnitine, doxorubicin, entecavir, ergothioneine, gabapentin, imatinib, ipratropium, metformin, oxaliplatin, pregabalin, pyrilamine, verapamil |
Mitoxantrone, quinidine, tiotropium |
| OCTN2 |
Carnitine, cephaloridine, emetine, entecavir, imatinib, ipratropium, verapamil |
Etoposide, spironolactone, tiotropium |
| OSTα/β |
Cholic acid sodium salt, deoxycholic acid sodium salt, glycocholic acid sodium salt, PGE2, taurocholic acid |
Cholic acid, deoxycholic acid, DHEAS, digoxin |
| PAT1 |
Betaine, L-tryptophan |
— |
| PCFT |
— |
Folic acid, 5-methyltetrahydrofolate |
| PEPT1 |
5-Aminolevulinic acid, carnosine, cephalexin, penicillin G (benzylpenicillin), D-Phe-Ala, valacyclovir |
Cefadroxil, glibenclamide, nateglinide |
| SGLT1 |
Glucose, galactose |
— |
| SMVT1 |
Pantothenic acid |
Biotin, lipoic acid |
| SVCT1 |
L-Ascorbic acid |
— |
Table 2 The change in the expression level of the GI transporters/receptors in the status of diseases1,6–25
| Disease |
Increased expression |
Decreased expression |
| Alzheimer’s disease |
— |
MCT1 |
| Cancers |
FRα (Colorectal). PEPT1 (Colorectal) |
— |
| Cholestasis |
OSTα, OSTβ |
ASBT, OSTβ |
| Cushing syndrome |
OATP2B1, PEPT1 |
— |
| Diabetes |
ASBT, GLUT2, GLUT5, PCFT, PEPT1, SGLT1 |
— |
| Hyperthyroidism |
|
PEPT1 |
| IBD |
FRβ, ICAM-1, mannose receptor, OATP2B1, PEPT1, SVCT2, TfR |
ASBT, ATB0,+, GLUT5, MCT1, OCTN2, OSTα, OSTβ, SVCT1, SMVT |
| Infection |
FcRn |
— |
| Obesity |
ASBT, PEPT1 |
— |
2.1. GI transporters for BAs
Apical sodium-dependent bile acid transporter (ASBT), which is mainly expressed in the epithelial cells of the small intestine (particularly the distal ileum), plays a crucial role in maintaining BA homeostasis and lipid metabolism because the BAs in the intestine are transported into the blood (e.g., the portal circulation system) via the intestinal epithelial cells and their reabsorbed quantity is almost 95% (equivalent to 12–18 g/70 kg in a day).26 Also, it is known that the expression levels of ASBT decrease by about 31% and 75% in Crohn's disease and obstructive cholestasis, respectively.6,7 In contrast, the levels increase in obesity and diabetes.8,9 Thus, various BAs such as glycocholic acid (GCA), deoxycholic acid (DCA), taurocholic acid (TCA), and cholic acid (CA) have been considered as ligands to bind the ASBT selectively.
2.2. GI transporters for vitamins
Folic acid (FA; vitamin B9) or folate is an essential nutrient for various cellular processes such as DNA synthesis, repair, and methylation, and plays a vital role in cell growth and division. Its cellular uptake is mediated by either proton-coupled folate transporter (PCFT) or folate receptor (FR), and it is known that FR has a higher binding affinity with folate than PCFT (especially at pH 7.4). However, when folate exists in the GI tract, its absorption and transport are mainly carried out by PCFT rather than FR because PCFT is primarily expressed in the apical membrane of the duodenum and the proximal jejunum, whereas there are no FRs in the healthy intestine.27,28 In particular, PCFT works under weakly acidic conditions (e.g., pH 5.8–6.0) because the transporter needs protons.27 In addition, pathological conditions could increase its expression levels, or lead to up-regulate of the FR expression level. The expression level of PCFT increased by about 12-fold in diabetic rats.29 It was also reported that FRα and FRβ are highly expressed in colorectal cancer and inflamed colon cells (e.g., macrophages in inflammatory bowel disease (IBD)), respectively.10,11 Thus, folate, naringenin, and their derivatives (e.g., N5,N10-dimethyl tetrahydrofolate) have been considered to bind either the PCFT, FR, or both specifically.
Biotin (vitamin B7) and pantothenic acid (vitamin B5) play roles in fatty acid synthesis and energy production and are directly absorbed by the sodium-dependent multivitamin transporter (SMVT) expressed in the brush-border membrane of the small intestine.30–32 The expression level of SMVT was reduced in inflammatory disorders.12 However, the oral absorption of cobalamin (vitamin B12) occurs by Cubam (referred to as a multi-ligand receptor in the epithelial cells of the distal ileum) after forming the intrinsic factor (IF)–B12 complex.33 The absorbed IF–B12 complex in the ileal cells is dissociated to IF and vitamin B12, and the vitamin B12 exocytoses to the blood by multidrug resistance 1 (MDR1). Ascorbic acid (ascorbate, vitamin C) is absorbed by sodium-dependent vitamin C transporter (SVCT) 1 and is exocytosed by SVCT2 in the small intestine. SVCT1 is located at the apical membrane of the epithelial cells in the small intestine (mostly jejunum) and the colon, whereas SVCT2 is expressed in their basolateral membrane.34 Interestingly, when the extracellular concentrations of vitamin C are high, the expression levels of SVCT in the neighboring cells are increased.35 However, ulcerative colitis and Crohn's disease reduced the expression level of SVCT by about three times.13,14 Thus, various vitamins (e.g., B5, B7, B12, C) and derivatives have preferentially been considered ligands to bind the SMVT, SVCT, or Cubam.
2.3. GI transporters for saccharides
All tissues and cells have various glucose transporters (GLUTs) and sodium-dependent glucose cotransporters (sodium-glucose linked transporters; SGLTs) because glucose and sugars, transported by their concentration-dependent GLUTs and concentration-independent SGLTs, play an essential role in regulating cellular metabolism, producing energy, and maintaining blood sugar homeostasis. In particular, enterocytes have three essential sugar transporters, GLUT2, GLUT5, and SGLT1: (1) GLUT2, expressed in the basolateral membrane, transports the absorbed monosaccharides (e.g., glucose, fructose, galactose) into the blood (i.e., the portal venous system),36 (2) GLUT5, expressed in the apical membrane, specifically absorbs fructose from the GI tract to the cells,37 and (3) SGLT1 absorbs glucose and galactose into the cells.38 In particular, the expression levels of GLUT5 and SGLT1 are increased by about four times in diabetic patients compared with healthy people.15,16 Thus, sugars and their derivatives have been considered ligands that preferentially bind the GLUT, SGLT, or both.
Mannose receptor, expressed in intestinal macrophages or dendritic cells (DCs), recognizes mannose, fucose, and N-acetylglucosamine present on the surface of pathogens (e.g., bacteria, fungi, parasites) and takes them up by endocytosis for further degradation.39 IBD and colitis triggered the overexpression of mannose receptors in antigen-presenting cells (APCs).17 Thus, mannose, fucose, N-acetylglucosamine, and their derivatives have been considered ligands that bind the mannose receptor selectively.
2.4. GI transporters for cationic molecules
Carnitine is essential in fatty acid metabolism because carnitine delivers a fatty acid into the mitochondria matrix after forming a fatty acid–carnitine conjugate, e.g., acylcarnitine.40 Then, the β-oxidation of acylcarnitine produces energy. The process is essential in all tissues and cells but is more critical in skeletal or cardiac muscles that require much energy. In the GI tract, the carnitine is transported by organic cation/carnitine transporter 2 (OCTN2) under a proton gradient (i.e., H+-dependent OCTN2) because the transporter is expressed in the epithelial cells of the small intestine.41 Its carnitine transporting activity is exceptionally high in the duodenum and jejunum, resulting in about 50 times higher carnitine levels inside than outside the cells.41 In addition, the expression level of OCTN2 was reduced in Crohn's disease (about 5-fold) and colorectal cancers.18,19,42 When depending on Na+, OCTN2 can transport tetraethylammonium and ergothioneine. Thus, carnitine and its derivatives have been considered ligands that bind the OCTN2 selectively.
2.5. GI receptors for antibodies
The neonatal Fc receptor for IgG (FcRn) is mainly expressed in the epithelial cells of the proximal small intestine and the APCs of the intestinal lamina propria.43 The receptor is involved in transporting and recycling IgG (IgG1, IgG2) antibodies, improving their half-life and immune efficacy. FcRn binds tightly with the Fc of IgG in the duodenum and proximal jejunum because the receptor–Fc interaction is strong at acidic pH values (<pH 6.5), unlike neutral pH values (>pH 7.0) for its dissociation.44 In particular, dissimilar to rodents, FcRn is expressed in humans even after adulthood. Its expression in the enterocytes and DCs of the intestine was increased by TNFα (e.g., induced by inflammatory disorders such as Crohn's disease and ulcerative colitis) and viral infection (e.g., caused by gastroenteritis virus).20 Thus, Fc and Fc fragments have preferentially been considered ligands to bind the FcRn.
In healthy people, intercellular adhesion molecule 1 (ICAM-1) is not expressed in enterocytes but mainly in endothelial cells and leukocytes. It mediates cell-to-cell adhesion and interaction within the immune system during inflammation, immune response, and surveillance. However, when forming inflammatory environments (e.g., IBD), the expression of ICAM-1 increases in the epithelial cells of the small intestine to recruit immune cells.21 ICAM-1-targeted antibodies can be internalized by ICAM-1-mediated endocytosis.
2.6. GI transporters for anionic molecules
Monocarboxylate transporter 1 (MCT1) transports monocarboxylate such as lactate, pyruvate, and short-chain fatty acids (SCFA; e.g., acetate, propionate, butyrate) into cells in an H+-dependent manner.45 For example, MCT1 mainly transports butyrate and acetate in the small and large intestines (expressed in both apical and basolateral membranes of the intestinal epithelial cells) and the liver.41 In high energy-requiring tissues (e.g., skeletal muscle, heart, brain), the transporter mainly carries pyruvate and lactate,46 whereas MCT1 in cancer cells pumps lactate out.47 In particular, the expression level of MCT1 in the enterocytes of IBD patients was decreased by 60%.22 Thus, lactate, pyruvate, acetate, propionate, and butyrate have been considered as ligands that bind MCT1 preferentially.
Organic anion-transporting polypeptides (OATPs) are expressed in the epithelial cells of the small intestine and transport anionic or amphiphilic molecules of hormones, BAs, drugs, and toxins. In particular, ulcerative colitis and Crohn's disease up-regulated their expression levels by 4–10 times.19
2.7. GI transporters for peptides and amino acids
Peptide transporter 1 (PEPT1), highly expressed in the epithelial cells of the small intestine (specifically, jejunum and ileum), transports small peptides (e.g., tripeptides, L-valine, L-isoleucine, L-phenylalanine, Val-Gly, Tyr-Val), peptide analogs (e.g., Gly-Sar), or peptide-like drugs (e.g., bestatin, β-lactam antibiotics, L-DOPA-L-Phe) in the GI tract to the cells.23 In particular, targeting the transporter has been utilized for designing PEPT1-targeted prodrugs (e.g., derived from drugs not well absorbed through the GI tract) because its transporting capacity is large enough (i.e., not saturated).48 Also, the expression levels of PEPT1 are increased in the colon of IBD, diabetes, or colorectal tumor.23,24 Thus, various peptides and their derivatives have preferentially been considered ligands that bind PEPT1.
Amino acid transporter B0,+ (ATB0,+) and proton-coupled amino acid transporter 1 (PAT1) are mainly expressed in the epithelial cells of the distal ileum and the colon. The former transports neutral (e.g., leucine, isoleucine, valine, cysteine) or cationic amino acids (e.g., lysine, arginine) into cells, whereas the latter absorbs polar and nonpolar amino acids (e.g., proline, alanine, glycine) in a proton-dependent (or acid-activated) manner.49,50 In particular, IBD was found to reduce the expression level of ATB0,+ by ten times.25 Thus, amino acids and their derivatives have been considered ligands that bind ATB0,+ or PAT1.
3. Understanding mechanisms in GI transporter/receptor-mediated nanoparticle absorption and transcytosis
Using potential GI transporters/receptors, their corresponding substrate-decorated NDDSs have been investigated for improving oral absorption, and meaningful findings for their therapeutic effects have been reported. Many potential substrates and their hydrophilic derivatives listed in Table 1 could be applied, despite the few substrates that have been used. Nevertheless, to expand the design purpose of NDDSs from GI transporter/receptor-mediated enhanced oral absorption to effective disease targeting (i.e., maximizing therapeutic effects and minimizing unwanted effects in the pathological sites such as organs/tissues, cells, or organelles), understanding of the paths and mechanisms of GI transporter/receptor-targeted NDDSs is required. Most analyses in cellular entry, intracellular trafficking, and exocytosis were performed based on comparative experiments in the presence/absence of inhibitors, which affect three key oral absorption steps (Table 3). Thus, this section will describe how the designed NDDSs enter cells through GI transporters/receptors, how their intracellular trafficking occurs, and whether they are in systemic or lymphatic circulation after exocytosis.
Table 3 Inhibitors of cellular internalization, intracellular trafficking, and exocytosis mechanisms
| Mechanism |
Target mechanism |
Inhibitors |
| Cellular internalization |
Clathrin-mediated endocytosis |
Chlorpromazine |
| Caveolae-mediated endocytosis |
Nystatin |
| Genistein |
| Indomethacin |
| Filipin |
| Macropinocytosis |
5-(N-Ethyl-N-isopropyl)amiloride |
| Colchicine |
| Lipid raft-mediated endocytosis |
Methyl-β-cyclodextrin |
| Clathrin/caveolae-independent endocytosis |
Lovastatin |
| Energy-dependent endocytosis |
4 °C (low temperature) |
| Dynamin |
Dynasore |
| Actin filament |
Cytochalasin D |
| Cdc42 |
ZCL278 |
| |
| Intracellular trafficking |
Acidification of endosome |
Bafilomycin A1 |
| Endosomal sequestration |
Chloroquine |
| ER-Golgi transport |
Brefeldin |
| |
| Exocytosis |
Chylomicron |
Cycloheximide |
| OSTα/β |
17α-Ethinylestradiol |
| Golgi-mediated protein transport |
Monensin |
3.1. BA-decorated NDDSs
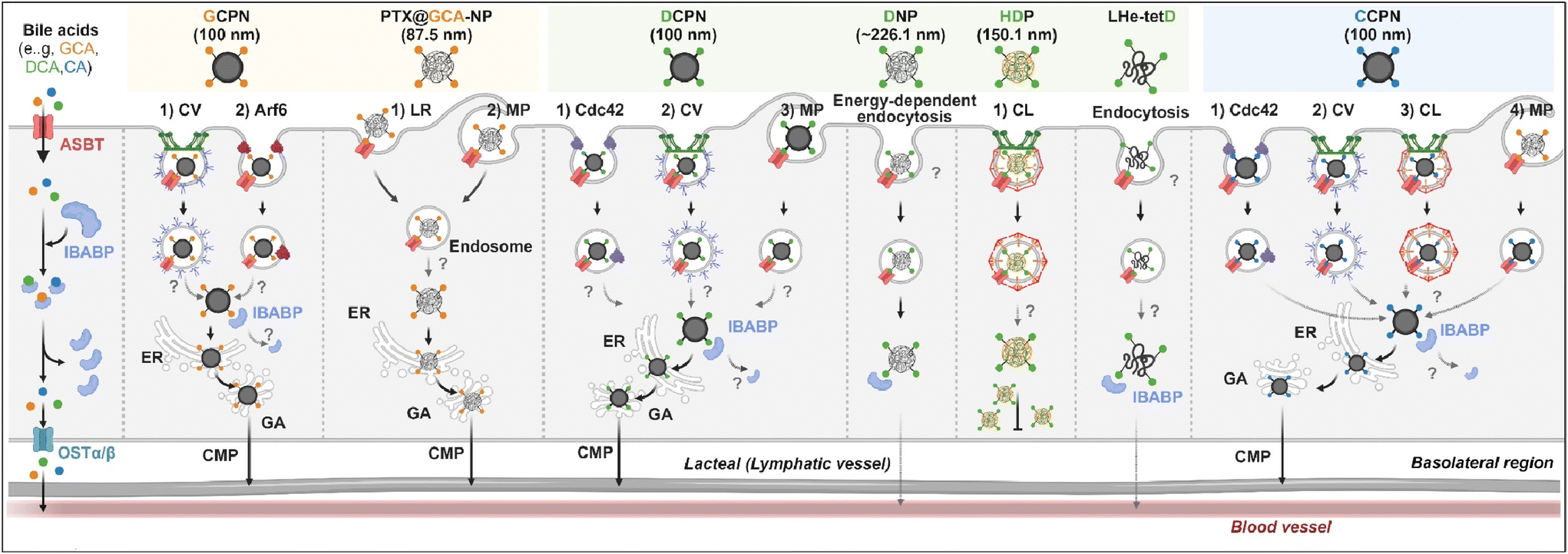
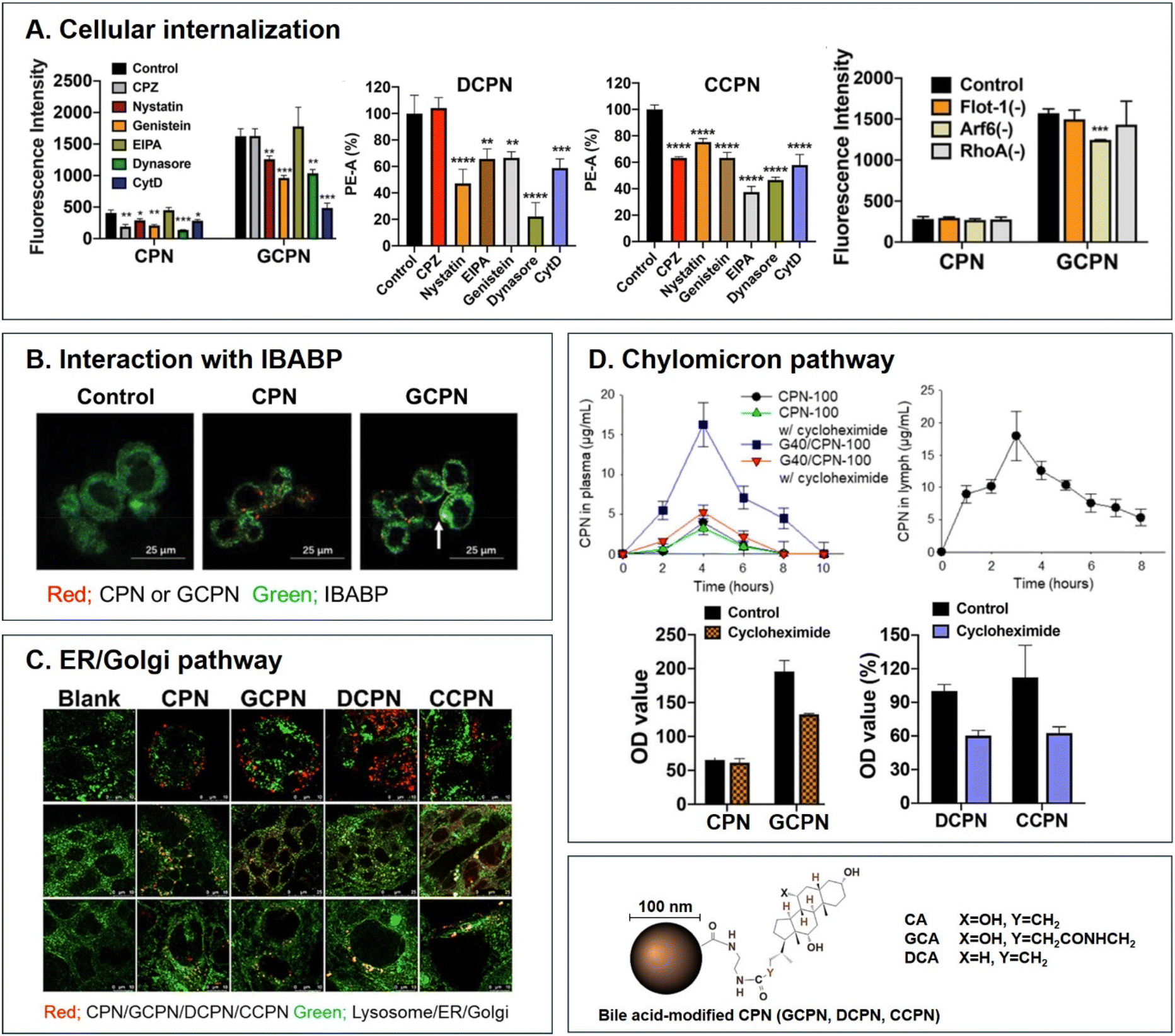
Various BAs such as GCA, DCA, TCA, and CA are ligands binding ASBT. All BA-decorated NDDSs followed endocytosis mechanisms to enter the epithelial cells in the small intestine. Still, the detailed mechanisms of their entry, intracellular trafficking, and exocytosis depend on the kind of BA and the size of its NDDS (Fig. 4). Bae et al. designed BA-decorated solid nanoparticles (NPs) (i.e., polystyrene beads) of 100 nm and applied the BA-NPs to ASBT-expressing SK-BR-3 and Caco-2 cells.3,51,52 After using various endocytosis/macropinocytosis inhibitors to understand their cellular entry, it was found that GCA-NPs (i.e., GCPN) follow caveolae-mediated or Arf-mediated endocytosis (Fig. 5). The cellular entry of DCA-NPs (i.e., DCPN) was affected by caveolae-mediated endocytosis, Cdc42-mediated endocytosis, or macropinocytosis, and CA-NPs (i.e., CCPN) underwent a combination of caveolae-mediated endocytosis, clathrin-mediated endocytosis, Cdc42-mediated endocytosis, or macropinocytosis. The BA-NP-bound ASBT complexes underwent different endocytosis mechanisms depending on the type of BA. However, studies on the colocalization of BA-NPs with the ileal bile acid-binding protein (IBABP) or Golgi/ER marker and the inhibition of transcytosis with the chylomicron pathway showed similar intracellular trafficking and exocytosis. Namely, the BA-NP-bound IBABP followed the ER/Golgi pathway and then moved from the cells to lymphatic vessels (via the chylomicron pathway). However, two steps are still unclear: (1) the endolysosomal escape of BA-NPs after cell entry and (2) the dissociation of the BA-NP-bound IBABP complex during the ER/Golgi pathway. Similarly, Huang et al. constructed PTX or quercetin-loaded liposomes (87.5 nm) and then coated the liposomes with GCA–chitosan conjugates. After the GCA-liposomes (i.e., PTX@GCA-NP)-bound ASBT complexes were taken up by enterocytes via lipid raft-mediated endocytosis or macropinocytosis, the liposomes went through the ER/Golgi pathway in Caco-2 cells. Then, they moved forward to the lymphatic vessel.53 However, other DCA-decorated NPs showed different intracellular trafficking and different exocytosis. For example, DCA-chitosan NPs (i.e., DNP)54 and low molecular weight heparin-tetrameric DCA conjugates (i.e., LHe-tetD)2 followed ASBT-mediated endocytosis, bound IBABP, and then moved to the blood. In particular, heparin-DCA/protamine complexes (i.e., HDP) bound ASBT and followed caveolae-mediated endocytosis in ASBT-expressing MDCK cells. Then, HDP was not exocytosed but retained in enterocytes.55 Although the BA is the same, findings are still debated: (1) the blood via unknown routes versus the lymphatic vessels via the ER-Golgi pathway and (2) exocytosed from the cells versus retained in the cells. Understanding of these could help the design of BA-decorated NDDSs.
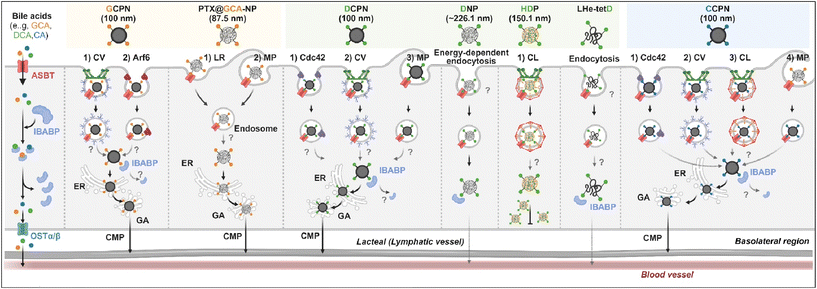 |
| | Fig. 4 Detailed mechanisms of the entry, intracellular trafficking, and exocytosis of some BA-decorated NDDSs (GCPN, GCA conjugated carboxylated polystyrene NP;1,52 PTX@GCA-NP, GCA–chitosan coated PTX@Liposome;53 DCPN, GCA conjugated carboxylated polystyrene NP;51 DNP, DCA conjugated chitosan NP;54 HDP, heparin-DCA/protamine complex;56 LHe-tetD, low molecular weight heparin-tetrameric DCA conjugate;2 CCPN, CA conjugated carboxylated polystyrene NP51). CV, caveolae-mediated endocytosis; LR, lipid-raft-mediated endocytosis; MP, macropinocytosis; CL, clathrin-mediated endocytosis; CMP, chylomicron pathway; ER, endoplasmic reticulum; GA, Golgi apparatus; IBABP, ileal bile acid binding protein; OSTα/β, organic solute transporter α/β. | |
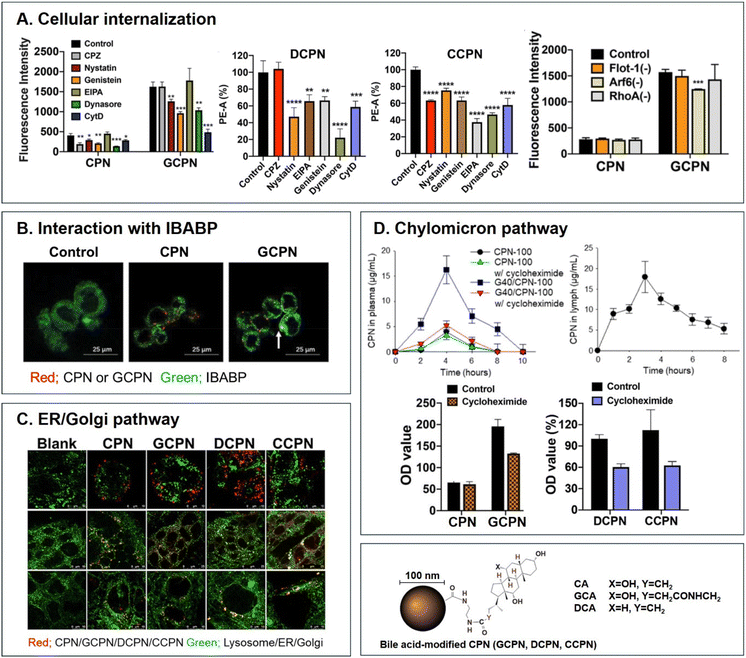 |
| | Fig. 5 Analyses of (A) cellular internalization, (B) interaction with IBABP, (C) ER/Golgi pathway, and (D) chylomicron pathway of BA-decorated polystyrene nanoparticles (i.e., GCPN, DCPN, and CCPN) (reproduced from ref. 1 with permission from American Chemical Society, copyright 2018; reproduced from ref. 51 with permission from Elsevier, copyright 2022; Reproduced from ref. 52 under the terms of the Creative Commons CC-BY License). | |
3.2. Vitamin-decorated NDDSs
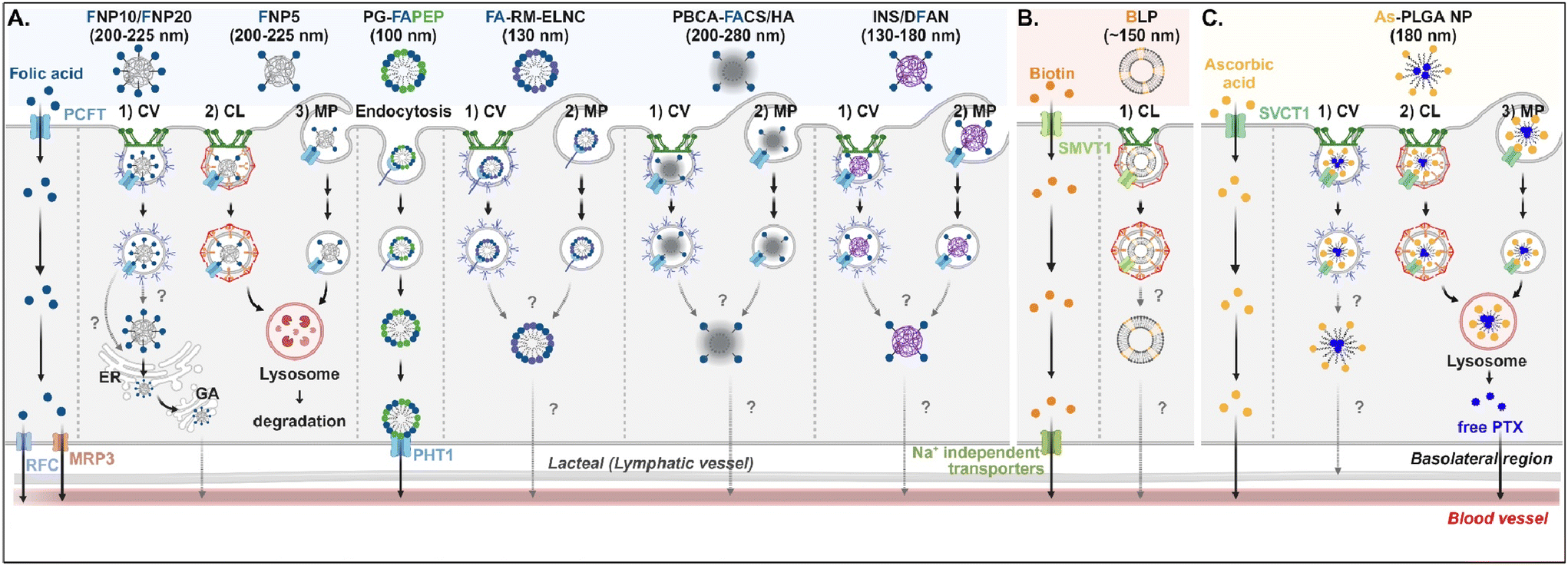
Understanding of the cellular entry of folate (vitamin B9)-mediated NDDSs via PCFT or FR is not deep, and their subsequent processes in intracellular trafficking and exocytosis have been rarely investigated (Fig. 6A). Nevertheless, Gan et al. tried to understand the absorption and transcytosis in high or low PCFT-expression cells using a ternary nanocomplex (NC) of FA-grafted chitosan, γ-poly(glutamic acid) (γ-PGA), and insulin.29 In low PCFT-expression cells (i.e., Caco-2 treated with PCFT inhibitor), after binding the FA-NCs (i.e., FNP) to PCFT, the FA-NC-bound PCFT complexes were accumulated in the lysosomes via clathrin-mediated endocytosis. Then, they were degraded, regardless of the folate contents in the NCs. Similarly, when low FA-NCs (≤5%; i.e., FNP5) were applied to high PCFT-expression cells (i.e., Caco-2), the NC-bound PCFT complexes followed clathrin-mediated endocytosis or macropinocytosis and then were sequestrated and degraded in the lysosomes. However, the FA-NCs with a higher FA content (10–20%; i.e., FNP10, FNP20) took the ER-Golgi pathway via caveolae-mediated endocytosis and followed transcytosis to the blood. In addition, when applying FA-decorated NPs (e.g., 130 nm FA-RM-ELNC,56 200–280 nm PBCA-FACS/HA,57 and 130–180 nm INS/DFAN58) having different sizes to enterocytes, their entry mechanisms were caveolae-dependent endocytosis or macropinocytosis in Caco-2 cells regardless of whether the NPs were lipid or polymer-based, and their intracellular trafficking and transcytosis mechanisms were not reported. Interestingly, Gan et al. designed dual GI transporter-targeted NPs (i.e., PG-FAPEP) composed of PCFT-targeted FA and proton-coupled oligopeptide transporter (PHT1)-targeted tripeptide because PCFT and PHT1 are localized at the apical or basolateral membrane of enterocytes, respectively (in the original article, FR was expressed instead of PCFT. However, the review expresses PCFT instead of FR because PCFT could exist rather than FR).59 After PG-FAPEP bound to PCFT, the PG-FAPEP-PCFT complexes underwent endocytosis and then released PG-FAPEP into the cytosol via tripeptide-mediated endosomal escape in Caco-2 cells. Finally, the released PG-FAPEP formed a complex with PHT1 to move further into the blood. Overall, most FA-decorated NDDSs could undergo caveolae-mediated endocytosis or macropinocytosis. However, the uptake depends on pH because an acidic pH gradient only allows cellular entry.60
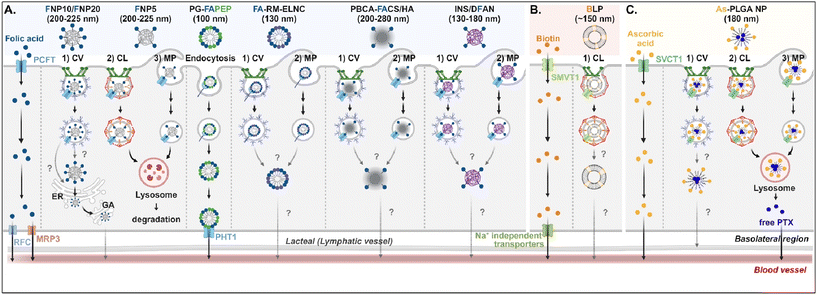 |
| | Fig. 6 Detailed mechanisms of the entry, intracellular trafficking, and exocytosis of (A) some FA-, (B) some biotin-, or (C) some ascorbic acid-decorated NDDSs (FNP, FA-grafted chitosan NP;29 PG-FAPEP, FA and tripeptide dual-modified PLGA NP;60 FA-RM-ELNC, FA-modified exenatide reverse micelle lipid nanocapsules;57 PBCA-FACS/HA, FA-conjugated chitosan and hyaluronic acid-coated poly(n-butylcyanoacrylate) NP;58 INS/DFAN, FA-modified pectin NP;59 BLP, biotinylated liposome;61 As-PLGA-NP, ascorbate-conjugated PLGA NP62). CV, caveolae-mediated endocytosis; MP, macropinocytosis; CL, clathrin-mediated endocytosis; ER, endoplasmic reticulum; GA, Golgi apparatus; RFC, reduced folate carrier; MRP3, multidrug resistance protein 3; PHT1, peptide/histidine transporter 1. | |
Biotin (vitamin B7) or ascorbic acid (vitamin C)-decorated NDDSs have been designed to target SMVT1 and SVCT1, respectively. However, their mechanisms for cellular internalization, trafficking, and exocytosis are rarely studied. Nevertheless, Wu et al. constructed 150 nm-sized biotin-decorated liposomes (BLP) made of biotin-1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE), soybean phosphatidylcholine (SPC), and cholesterol.63 The designed BLP targeted bound to biotin receptor (e.g., SMVT1), and the formed BLP–SMVT1 complex was internalized into cells via clathrin-dependent endocytosis in Caco-2 cells (Fig. 6B). Then, the blood circulation of BLP was expected, although there was no mechanism study on its endosomal release and exocytosis. In addition, Chen et al. mixed palmitoyl ascorbate (As) with poly(lactide-co-glycolide) (PLGA) and prepared As-PLGA NPs.61 The NPs first bound to SVCT1, and the formed NP-SVCT1 complexes followed three pathways, namely caveolae-mediated endocytosis, clathrin-mediated endocytosis, or macropinocytosis in Caco-2 cells (Fig. 6C). When the NPs followed caveolae-mediated endocytosis, their lymphatic circulation was expected, although there was no experimental evidence. However, when the NPs underwent clathrin-mediated endocytosis or macropinocytosis, the NPs were sequestrated in the lysosomes and were degraded, releasing their payloads, free paclitaxel (PTX). Then, the released PTX passively penetrated the basolateral membrane of the intestinal epithelial cells and moved toward the blood circulation.
3.3. Saccharide-decorated NDDSs
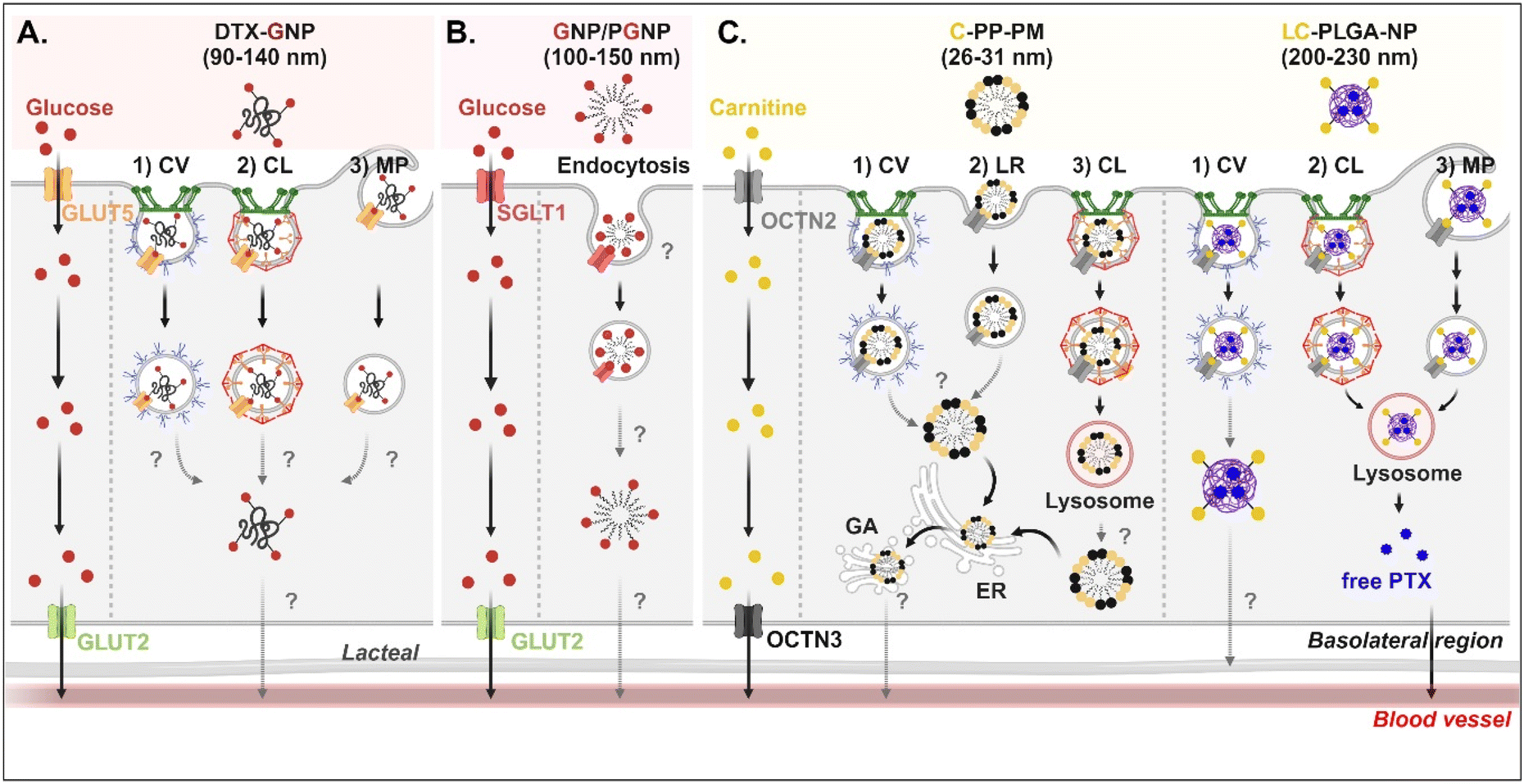
Studies on the absorption and transcytosis of saccharide-decorated NDDSs are lacking using GLUTs and SGLTs, despite these being essential transporters for the daily glucose uptake of about 250 g/70 kg adults. However, Guo et al. designed NPs (i.e., DTX-GNP) made of glucose-conjugated zein and showed their interaction with GLUTs.62 The NP-bound GLUTs were internalized by caveolae-mediated endocytosis, clathrin-dependent endocytosis, or macropinocytosis in Caco-2 cells. However, it was not explained which subtype of GLUTs were bound and which endosomal escape mechanisms, intracellular trafficking, and exocytosis pathways were involved (Fig. 7A). In addition, Zhang et al. designed two glucose-decorated NPs (i.e., a glucosylated NP (GNP) of glucose-PEG-PLGA and a glucosylated cationic NP (PGNP) of glucose-PEG-PLGA and DOTAP) to target the overexpressed SGLT in the epithelial cells of the proximal small intestine.64 After binding PGNP with SGLT, the PGNP–SGLT complex followed an SGLT1-mediated endocytic pathway (proved by endosomal markers such as Rab5 and Rab11) in Caco-2 cells (Fig. 7B). However, there is no study on their endosomal escape, intracellular trafficking, or exocytosis.
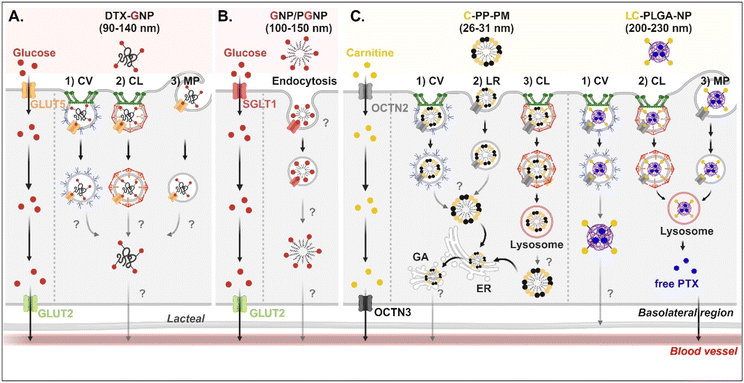 |
| | Fig. 7 Detailed mechanisms of the entry, intracellular trafficking, and exocytosis of (A) some glucose-(GLUT5-mediated), (B) some glucose-(SGLT1-mediated), or (C) some carnitine-decorated NDDSs (DTX-GNP, glucose-modified zein NP loaded with docetaxel;62 GNP, glucose-PEG-PLGA;64 PGNP, a mixed NP of glucose-PEG-PLGA and DOTAP;64 C-PP-PM, carnitine-conjugated polymeric micelles (PEOz-PLA);65 LC-PLGA-NP, L-carnitine-conjugated PLGA NP66). CV, caveolae-mediated endocytosis; LR, lipid-raft-mediated endocytosis; MP, macropinocytosis; CL, clathrin-mediated endocytosis; ER, endoplasmic reticulum; GA, Golgi apparatus. | |
3.4. Cationic molecule-decorated NDDSs
For OCTN2-targeted oral absorption, many studies selected carnitine as a cationic molecule, and carnitine-decorated NDDSs have been developed. Their cellular entry mechanisms depended on the type and size of NP. For example, Zhou et al. prepared 26–31 nm-sized carnitine-decorated poly(lactic acid) (PLA) polymeric micelles (PMs) (i.e., C-PP-PM) composed of methoxy poly(ethylene glycol)-PLA (mPEG-PLA) and carnitine-PEG-PLA.65 After C-PP-PM selectively bound with OCTN2, the PM–OCTN2 complexes followed two different endocytic pathways: (1) clathrin-mediated endocytosis and (2) caveolae- or lipid raft-mediated endocytosis in Caco-2 cells (Fig. 7C). When the PM–OCTN2 complexes followed clathrin-mediated endocytosis, the complexes passed through late endosomes and lysosomes. It was expected that C-PP-PM would be dissociated from OCTN2 and escape from lysosomes, although no precise mechanism is described. Other routes are that the PM–OCTN2 complexes would follow caveolae- or lipid raft-mediated endocytosis and then be released into the cytosol. Regardless of endocytic pathways, C-PP-PM in the cytosol took the ER-Golgi pathway and circulated in the blood. As another example, Sun et al. designed 200–230 nm-sized carnitine-decorated NPs (i.e., LC-PLGA-NP), and the NPs were prepared by mixing stearoyl-L-carnitine with PLGA.66 When the contents of stearoyl-L-carnitine were 10, 20, or 40% in LC-PLGA-NP, 10% carnitine-NPs showed the highest cellular uptake, and sodium helped to improve their uptake in Caco-2 cells. Following caveolae-mediated endocytosis, LC-PLGA-NP moved toward the lymphatic vessel (Fig. 7C). However, when the NPs followed clathrin-mediated endocytosis or macropinocytosis, the NPs accumulated in the lysosomes and then degraded to release their payloads (i.e., free PTX). The released PTX was transported to the blood through the basolateral membrane of enterocytes. Overall, their entry mechanisms affected exocytosis, resulting in determining either the blood or lymphatic pathway.
3.5. Peptide/amino acid-decorated NDDSs
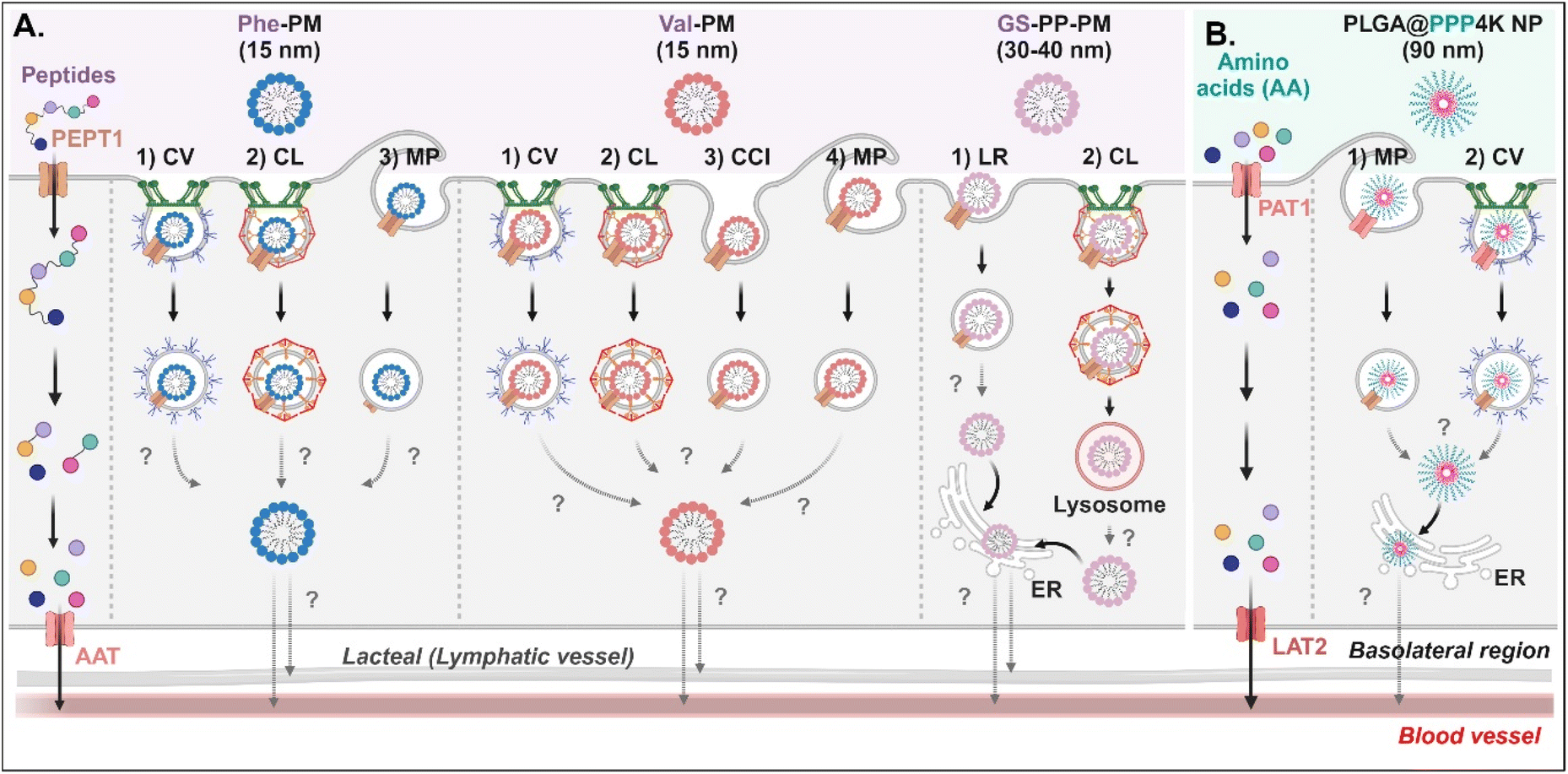
Various short peptides (e.g., dipeptides and tripeptides), amino acids, and their derivatives-decorated NDDSs enter enterocytes using PEPT1 or PAT1. Some studies have been conducted on their intracellular mechanisms, but there is limited understanding of their intracellular trafficking and exocytosis. Tu et al. synthesized Val-α-tocopherol-PEG (TSPG) or Phe-TPGS by conjugating Val or Phe to TSPG, and made their 15 nm-sized PMs (e.g., Val-PM or Phe-PM).67 After Val-PM bound to PEPT1, the formed Val-PM-PEPT1 complexes were internalized into the cells via a combination of clathrin-mediated endocytosis, caveolae-mediated endocytosis, macropinocytosis, or clathrin-/caveolae-independent endocytosis in Caco-2 cells (Fig. 8A). Similarly, Phe-PM followed a combination of clathrin-mediated endocytosis, caveolae-mediated endocytosis, or macropinocytosis. However, their endosomal escape and exocytosis were not analyzed after endocytosing Val-PM or Phe-PM. Nevertheless, in the presence of lymphatic transport inhibitors (i.e., cycloheximide), their blood concentrations (e.g., AUC) decreased. The results indicate that the exocytosis of Val-PM and Phe-PM would take the lymphatic pathway. In addition, Liu et al. prepared 30–40 nm-sized Gly-Sar-conjugated PEG-PLA NPs (i.e., GS-PP-PM) to target PEPT1.68 GS-PP-PM-bound PEPT1 was internalized into the cells through clathrin-mediated or lipid-raft-mediated endocytosis in Caco-2 cells (Fig. 8A). Their escape from the endosomes or lysosomes was unknown, but GS-PP-PM was migrated to the basolateral membrane via the ER.
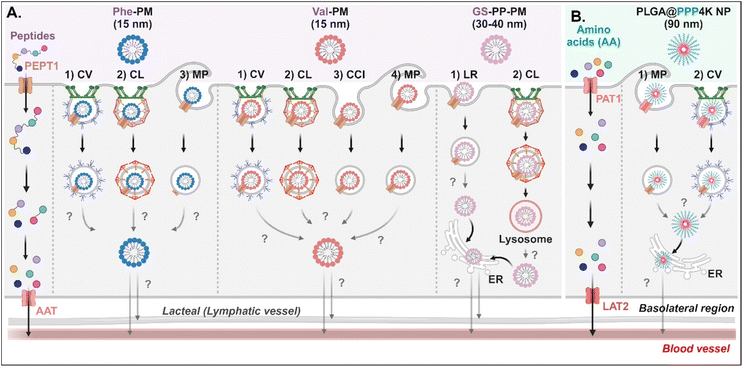 |
| | Fig. 8 Detailed mechanisms of the entry, intracellular trafficking, and exocytosis of (A) some AA (Phe or Val)- or (B) some zwitterion-decorated NDDSs (Phe-PM, Phe-conjugated PM;67 Val-PM, Val-conjugated PM;67 GS-PP-PM, Gly-Sar-conjugated PM;68 PLGA@PPP4K, PLGA/CB-PPO-PCB NP69). CV, caveolae-mediated endocytosis; LR, lipid-raft-mediated endocytosis; MP, macropinocytosis; CL, clathrin-mediated endocytosis; CCI, clathrin/caveolae-independent endocytosis; ER, endoplasmic reticulum; GA, Golgi apparatus; AAT, amino acid transporter; LAT2, L-type amino acid transporter 2. | |
For targeting PAT1, Jin et al. chose betaine as the PAT1 ligand.69 They constructed 90 nm-sized betaine-decorated NPs (i.e., PLGA-PPP2K, PLGA-PPP3K, PLGA-PPP4K) with different molecular weights of poly(propylene oxide) (PPO), which was composed of poly(carboxybetaine) (PCB)-PPO-PCB and PLGA. When entering enterocytes, the NP-bound PAT1 complexes underwent macropinocytosis or clathrin-mediated endocytosis in Caco-2 cells (Fig. 8B). Then, PLGA-PPP was released to the cytosol (despite an unknown mechanism of endosomal escape) and moved into the blood through the ER.
3.6. Antibody-decorated NDDSs
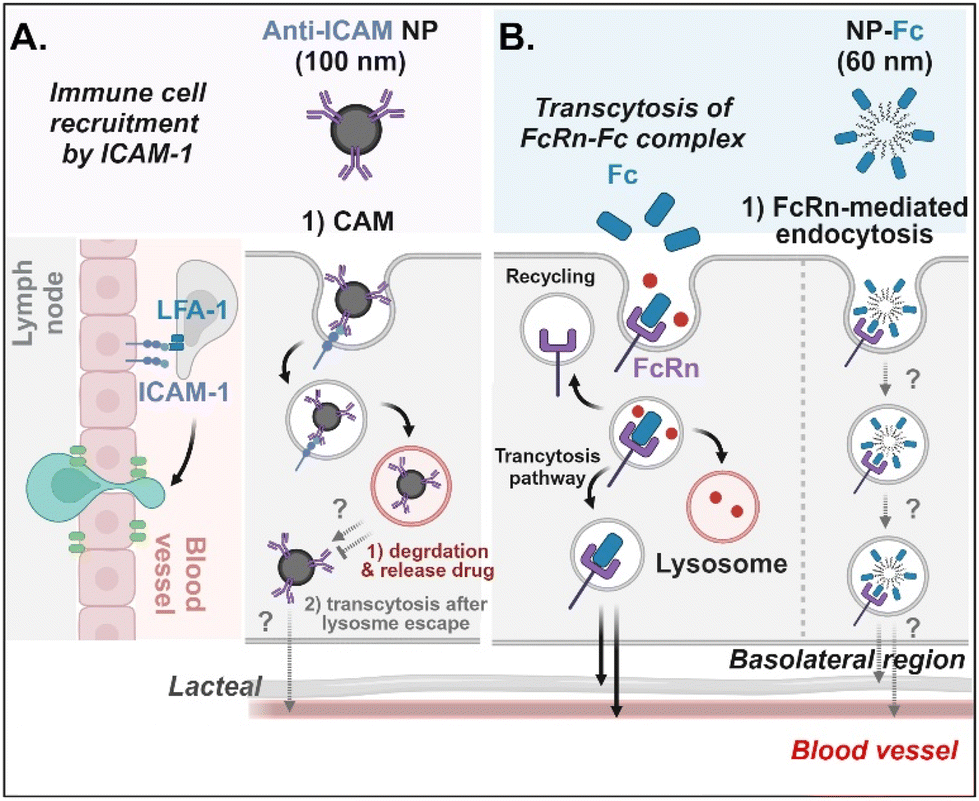
Antibodies- or antibody fragments-decorated NDDSs are mainly considered to be internalized through antibody/receptor-mediated endocytosis, and their cellular entry and exocytosis mechanisms have yet to be studied in detail. Muro et al. designed anti-ICAM-coated polystyrene beads (i.e., anti-ICAM NP) to confirm ICAM-1-mediated cellular internalization in ICAM-1-overexpressed enterocytes.70 Anti-ICAM NP-bound ICAM-1 was internalized via CAM (cell adhesion molecule)-mediated endocytosis. Interestingly, the intracellular trafficking via the lysosomes depended on the cell preparation. When Caco-2 cells were cultured on coverslips (i.e., apical and basolateral sides cannot be distinguished.), anti-ICAM NP-ICAM-1 complexes were localized to the lysosomes. However, when Caco-2 cells were cultured as a monolayer (i.e., the apical side can be distinguished from the basolateral side), anti-ICAM NP-ICAM-1 complexes moved to the basolateral region after escaping from the lysosomes (followed by an unknown mechanism) (Fig. 9A). In addition, Farokhzad et al. designed FcRn overexpressed enterocyte-targeted Fc-decorated NDDSs using Fc-conjugated PLA-PEG-Mal NP (i.e., NP-Fc).71 Although their detailed cellular entry and transcytosis mechanisms were unclear, NP-Fc-bound FcRn was internalized through FcRn-mediated endocytosis (Fig. 9B). Through comparative studies using wild-type mice and FcRn knockout mice, NP-Fc transport into the blood was confirmed after its absorption in the small intestine, although the detailed mechanisms of its intracellular trafficking, endosomal escape, and exocytosis were unknown.
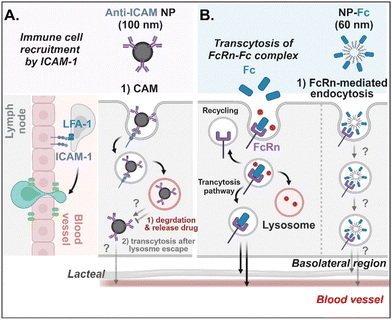 |
| | Fig. 9 Detailed mechanisms of the entry, intracellular trafficking, and exocytosis of (A) some anti-ICAM- or (B) some Fc-decorated NDDSs (anti-ICAM, ICAM-1 antibody-conjugated polystyrene beads;70 NP-Fc, Fc-conjugated PLA-PEG NP71). CAM, cell adhesion molecule-mediated endocytosis; LFA-1, lymphocyte function-associated antigen-1. | |
4.
In vivo therapeutic potential and challenges of GI transporter-mediated NDDSs
For the process from the entry to the exit of the GI tract, the detailed mechanisms and paths of substrate-decorated NDDSs still need to be clarified. Nevertheless, various NDDSs have been investigated to target GI transporters/receptors for enhanced oral absorption and therapeutic effects of their payloads. Using the NDDSs, their therapeutics have been delivered from the surroundings of the GI tract (e.g., small intestine, colon, Peyer's patch) to the blood or lymphatic circulation and further organs/tissues (e.g., liver, brain, lung, tumor, etc.). They could be applied to treat solid tumors (e.g., breast, colon, lung, colorectal liver metastasis), diabetes, virus-infected lung diseases, and so on. The size and the type of therapeutics are not limited because small chemicals, proteins/peptides, genes (e.g., siRNA), and magnetic/metallic nanoparticles have been used, and their size scale is from 0.1 nm to tens of nm. Thus, this section introduces some potential examples of GI transporter/receptor-targeted NDDSs. More examples can be found in Table 4.
Table 4 Some potential examples of GI transporter/receptor-targeted NDDSs
| Transporter/receptor |
Substrate |
Carrier (size, zeta) |
Disease |
Therapeutic outcome/oral bioavailability (oBA) |
Drug |
Ref. |
| ATV, atorvastatin; H6P, heat shock protein 65; OVX, ovariectomized rodent; pGLP-1, GLP-1 plasmid; rhPTH, human recombinant teriparatide; SNL, solid lipid nanoparticle. |
| ASBT |
GCA |
Liposome (150 nm/−40 mV) |
CT26-tumor |
Anti-tumor vaccine effect (DC maturation ↑, Th1/Th2 immunity ↑) |
OVA/poly(I:C) |
72
|
| ASBT |
GCA |
SLN (120 nm/−32 mV) |
B16F10, 4T1, CT26-tumor |
Anti-tumor effect |
DTX |
73
|
| ASBT |
GCA |
Liposome (120 nm/−32 mV) |
T2DM |
Anti-diabetic effect (blood glucose level ↓) oBA: 19.5% |
Exendin-4 |
74
|
| ASBT |
TCA |
Liposome (194 nm/negatively charged) |
T1DM |
Anti-diabetic effect (blood glucose level ↓) oBA: 34% |
Insulin |
75
|
| ASBT |
TCA |
Polyplexes (194 nm/negatively charged) |
T2DM |
Anti-diabetic effect (blood glucose level ↓) |
pGLP-1 |
76
|
| ASBT |
TCA |
Polyplexes (55 nm/6 mV) |
T2DM |
Anti-diabetic effect (blood glucose level ↓) |
pGLP-1 |
77
|
| ASBT |
TCA |
Polyplexes (230 nm/−5 mV) |
OVX |
Bone regeneration ↑ oBA: 30% |
rhPTH |
78
|
| ASBT |
DCA |
Emulsion (6–45 nm/−10–+3 mV) |
4T1-tumor |
Anti-tumor effect oBA: 20–35% |
ATV |
79
|
| ASBT |
DCA |
Polyplexes (7.64 nm/−0.36 mV) |
Osteoporosis |
Bone formation ↑ |
rhPTH |
80
|
| Cubam |
VB12 |
Dextran NP (150–280 nm) |
T1DM |
Anti-diabetic effect (blood glucose level ↓) |
Insulin |
81
|
| FcRn |
Albumin |
PLGA NP (150 nm/−8 mV) |
T1DM |
Anti-diabetic effect (blood glucose level ↓) |
Insulin |
82
|
| FcRn |
IgG1-Fc |
Polyplexes (78 nm/10 mV) |
T2DM |
Anti-diabetic effect (blood glucose level ↓) |
pGLP-1 |
83
|
| GLUT |
Glucose |
Zein NP (87.9–136.4 nm/−22–−18.5 mV) |
4T1-tumor |
Anti-tumor effect |
DTX |
62
|
| LAT1 |
Anti-CD98 |
Polyplexes (147–261 nm/7.9–17.3 mV) |
IBD |
Anti-inflammation effect |
CD98 siRNA |
84
|
| LAT1 |
Anti-CD98 |
PLGA NP (270 nm/−24 mV) |
Colon cancer |
Anti-tumor effect |
CD98 siRNA, Camptothecin |
85
|
| Mannose receptor |
Mannose |
LNP (284 nm/12.8 mV) |
IBD |
Anti-inflammation effect |
Budesonide |
86
|
| Mannose receptor |
Mannose |
Chitosan NP (322.5 nm/34.9 mV) |
Auto-immune disease |
Immune tolerance↑ |
H6P |
87
|
| MCT1 |
Butyrate |
PLGA NP (80–90 nm/−16.67–6.12 mV) |
T1DM |
Anti-diabetic effect (blood glucose level ↓) oBA: 9.28% |
Insulin |
88
|
| PCFT |
FA |
Pectin NP (130–270 nm/−26–−17 mV) |
T1DM |
Anti-diabetic effect (blood glucose level ↓) |
Insulin |
58
|
| PEPT1 |
L-Val-Val |
PLGA NP (165 nm/−4–−2 mV) |
— |
4.3-Fold higher oBA than DTX |
DTX |
89
|
| PEPT1 |
L-Val |
PLGA NP (213 nm/−14.4 mV) |
T1DM |
Anti-diabetic effect (blood glucose level ↓) |
Insulin |
90
|
| SMVT |
Biotin |
Liposome (∼150 nm) |
T2DM |
Anti-diabetic effect (blood glucose level ↓) oBA: 8.23, 12.09% |
Insulin |
63
|
4.1. Anti-tumor potential of BA-decorated NDDSs
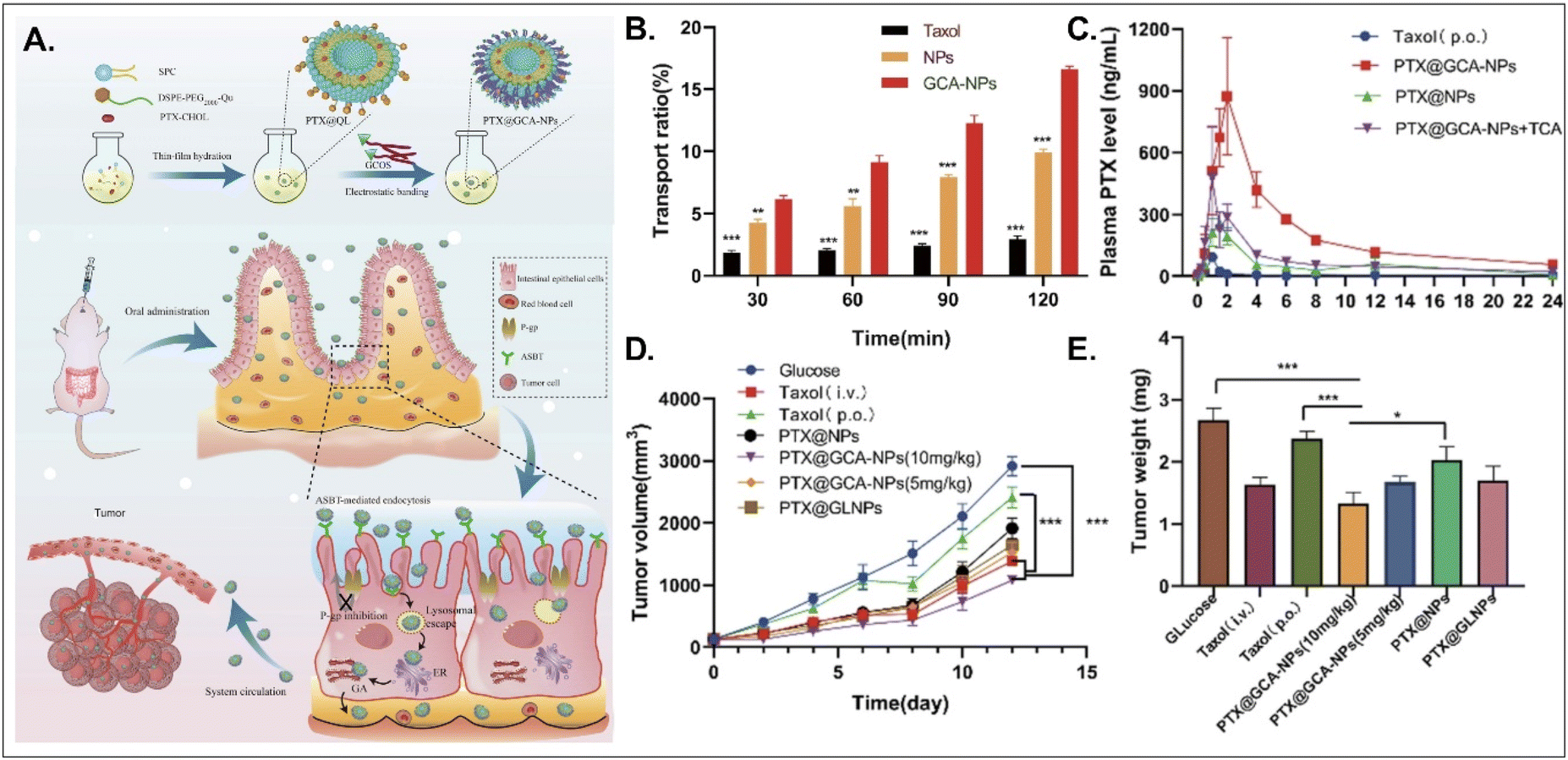
PTX, a hydrophobic anti-tumor drug that stabilizes microtubules and results in apoptosis, is absorbed into enterocytes when administered orally. Still, its oral bioavailability (oBA) is less than 1% because P-glycoprotein (Pgp) pumps it out to the GI tract.53 Thus, Liu et al. designed BA-decorated liposomes to increase the oBA of PTX (Fig. 10).53 The designed liposomes composed of DSPE-PEG-Que, PTX-cholesterol, and sphingosylphosphoricoline (SPC) were prepared and then coated with GCA–chitosan, forming PTX@GCA-NPs. GCA and Que were selected to target ASBT-mediated absorption and inhibit a drug efflux function of Pgp, respectively, in the epithelial cells of the small intestine. PTX@GCA-NPs delivered more PTX to enterocytes and further resulted in 19-fold and 4-fold higher AUC (i.e., the concentration of PTX in the blood) than two controls of Taxol® and PTX@NPs (coated with GCA-free chitosan), respectively. Also, based on the results of the reduced PTX levels in the blood when free TCA and PTX@GCA-NP were co-administered, both ASBT-mediated enterocyte entry and systemic delivery of PTX@GCA-NP were confirmed. In particular, when orally administered to an LL2 (lung cancer cell line)-xenograft mouse model, PTX@GCA-NPs showed a higher anti-tumor effect than Taxol® and PTX@NPs, whereas PTX@GLNP (excluding Que) showed a lower anti-tumor effect than PTX@GCA-NPs. These findings mean that GCA improves oral absorption of PTX, and Que blocks Pgp-mediated PTX efflux, resulting in a synergy of anti-tumor effect.
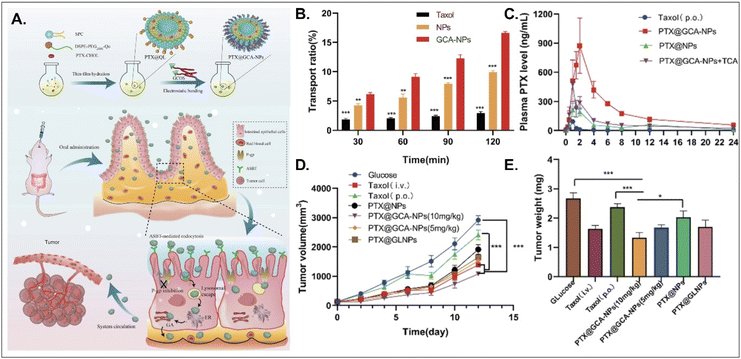 |
| | Fig. 10 Anti-tumor potential of PTX@GCA-NPs: (A) the preparation and design concept, (B) the transport ratio in the monolayer of Caco-2 cells, (C) the time-dependent plasma drug concentration, (D) the time-dependent tumor growth, and (E) the weight of the explanted tumor (reproduced from ref. 53 under the terms of the Creative Commons CC-BY 4.0 License). | |
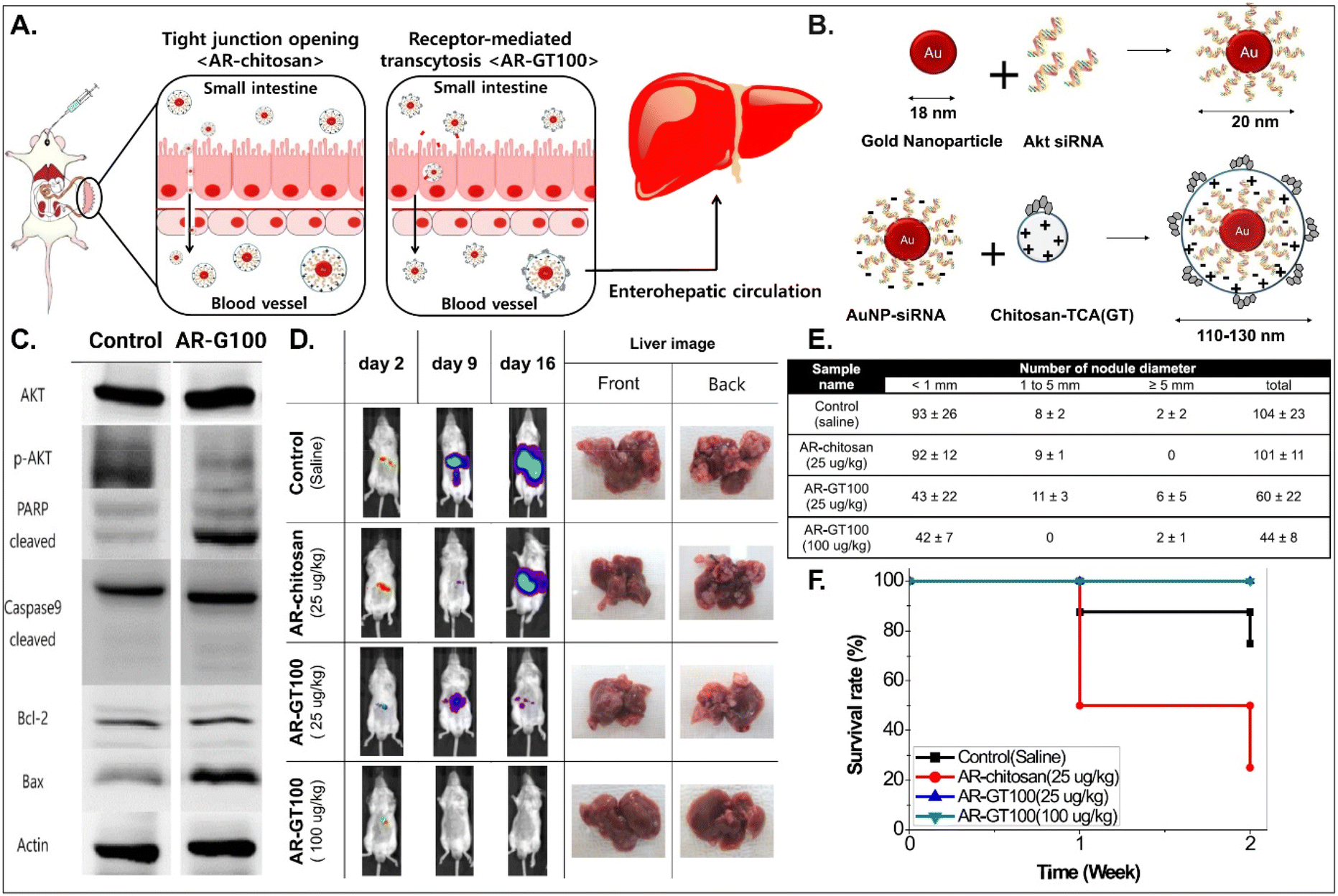
It is known that BAs are mainly reabsorbed in the small intestine and reach the liver through enterohepatic circulation. This would open up a potential for BA-decorated NDDSs to target liver diseases after ASBT-mediated oral absorption. Lee et al. planned to treat colorectal liver metastasis (CLM) involved in the AKT/PI3K pathway and designed AKT-silencing NDDSs (Fig. 11).91 Thiolated Akt siRNA was chemically linked to gold nanoparticles (AuNPs), and then the formed 20 nm-sized Akt siRNA-AuNPs (i.e., AR) were coated with TCA-conjugated chitosan (i.e., chitosan-TCA, GT), preparing 110–130 nm-sized AR-GT100. The ASBT-targeted Akt-silencing NPs (i.e., AR-GT100) showed good colloidal stability at pH 2 and 5, mimicking the GI tract. When applying AR-GT100 to Caco-2 and HepG2 cells, AR-GT100 had higher cellular uptake than AT-chitosan (i.e., a TCA-free control). When orally administered, AR-GT100 protected Akt siRNA in the GI tract, was absorbed well in the ileum, and then distributed to the liver. In a CLM mouse model using CT26 cells, the orally administered AR-GT100 decreased by 45% compared with a CLM control. In particular, AR-GT100 reduced the number and the size of metastatic tumor nodules in the liver by 43–58% more than AR-chitosan, improving mouse survival rates. Overall, the study found that the selective accumulation of AR-GT100 in the ileum and liver may be mediated by ASBT and NTCP (sodium/taurocholate cotransporting polypeptide), respectively. The findings mean that BA-decorated NDDSs could target and treat liver disorders after oral absorption.
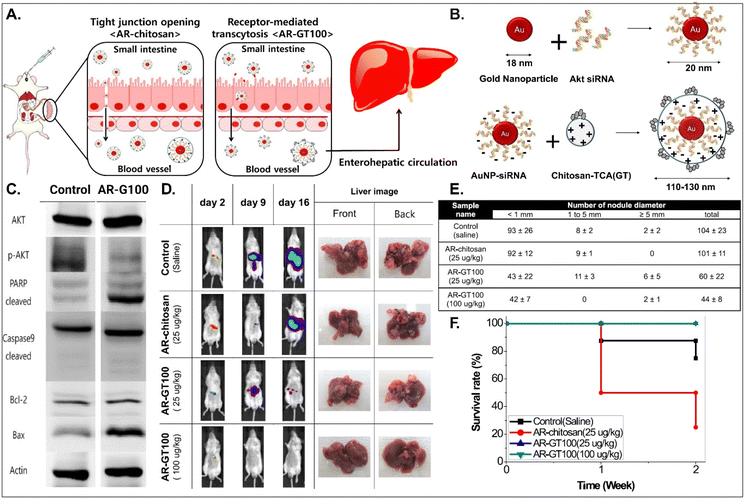 |
| | Fig. 11 Anti-tumor potential of Akt siRNA-AuNPs: (A) the design concept, (B) the preparation, (C) the immunoblot analysis involved in PI3K/AKT signaling and apoptotic pathway, (D) the progression in CLM, (E) the number of cancer nodules in the liver, and (F) the survival rate in a CLM cancer mouse model (reproduced from ref. 91 with permission from American Chemical Society, copyright 2017). | |
4.2. Anti-diabetic potential of BA-decorated NDDSs
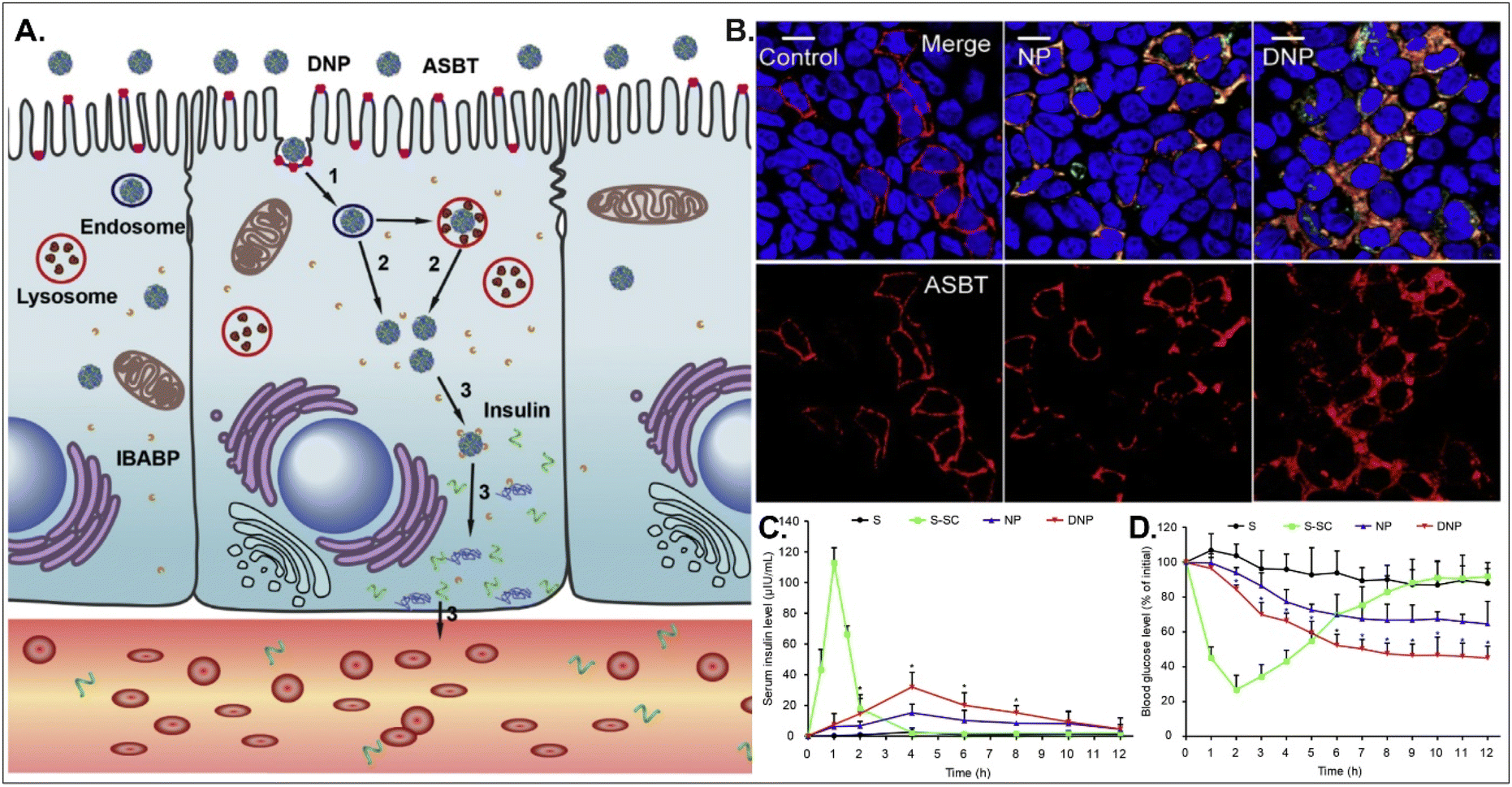
Oral absorption of peptides/proteins is limited due to their enzymatic/chemical degradation and instability in the GI tract. Therefore, many efforts to improve oBA have been made, and they have focused on overcoming obstacles such as mucous penetration, cellular entry, endosome escape, and transcytosis. For example, Fan et al. designed DCA-chitosan-based insulin NPs (i.e., DNPs), which were electrostatically made by complexing with DCA-chitosan, insulin, and γ-PGA (Fig. 12).54 Chitosan was expected to help their mucus penetration, endosomal escape, and insulin protection. Also, DCA could follow ASBT-mediated endocytosis in enterocytes. The decomposed insulin was much reduced after DNPs entered enterocytes via ASBT and escaped to the cytosol without their lysosomal sequestration. When orally administered to type 1 diabetic rats, DNPs provided about 12 times higher bioavailability of insulin than free insulin. In addition, DNPs regulated blood glucose levels well compared with free insulin. Although the mechanism of transcytosis of free insulin from enterocytes to the blood is unclear, the findings indicate that the payloads, such as peptides/proteins, could be effectively delivered to the blood via ASBT-mediated absorption.
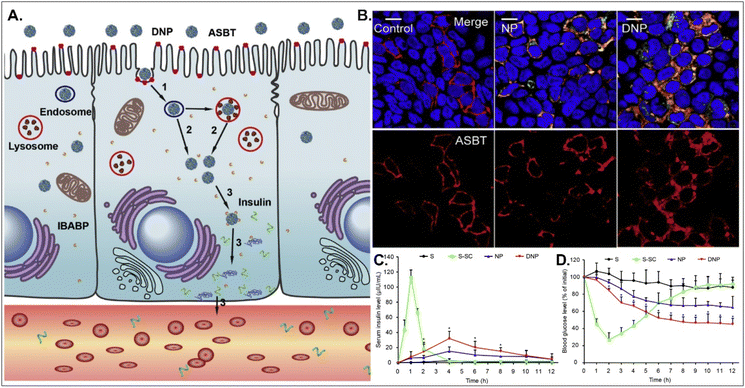 |
| | Fig. 12 Anti-diabetic potential of DNPs: (A) the expected transcytosis mechanisms, (B) the distribution of ASBT (red) and FITC-Ins (green) in Caco-2 cells, and (C) the serum insulin level, and (D) the plasma glucose level in a diabetic rat model (reproduced from ref. 54 with permission from Elsevier, copyright 2018). | |
4.3. Anti-tumor potential of vitamin-decorated NDDSs
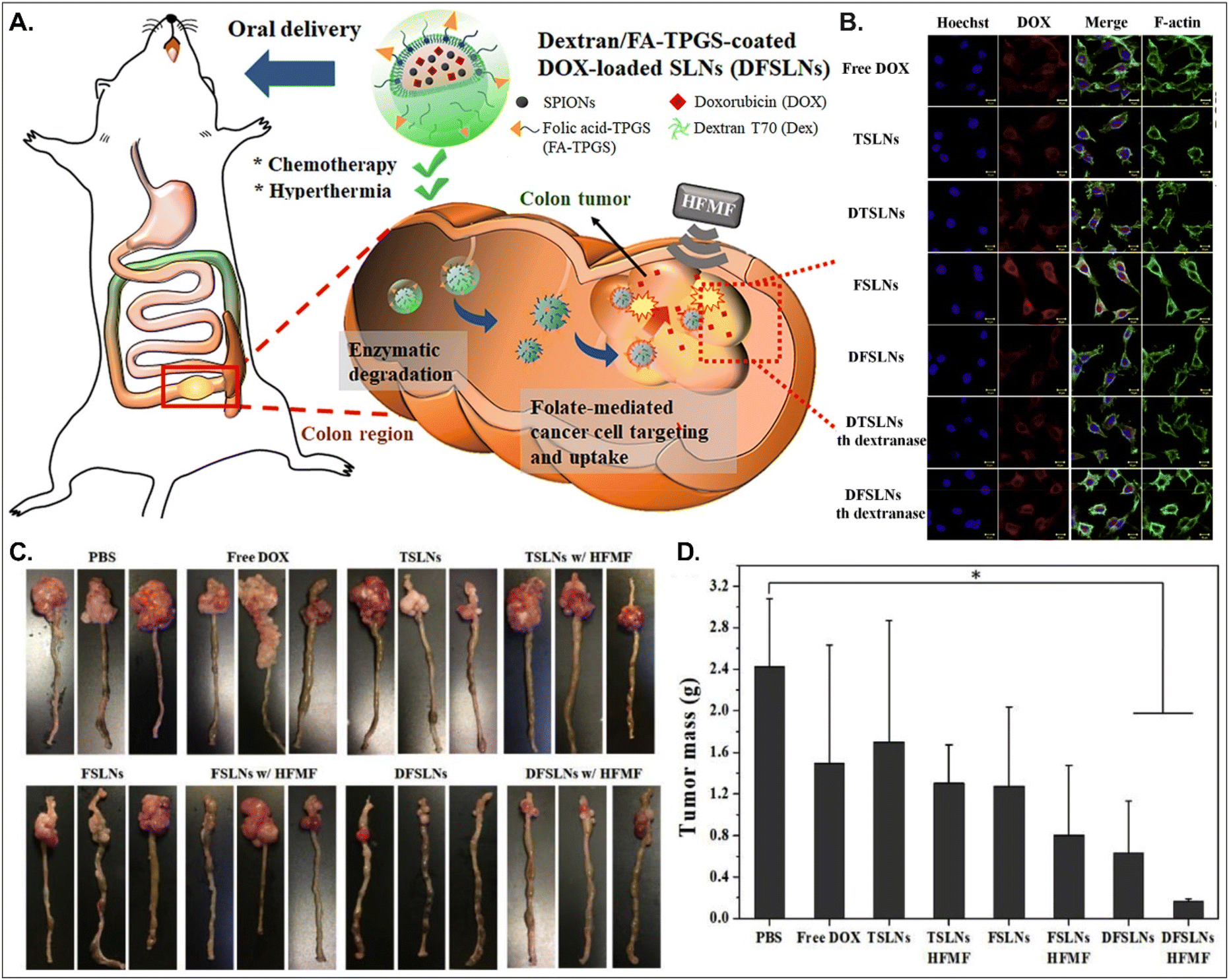
Both PCFT and FR absorb FA. PCFT exists in the duodenum and proximal jejunum, where the pH in the intestinal tract is low because PCFT transports FA in a proton-dependent manner. On the other hand, FR is expressed in the distal ileum and colon, where the pH is neutral, and shows exceptionally high expression in colorectal cancer. Therefore, to target colorectal cancer using FA-decorated NDDSs, the NDDSs should avoid PCFT-mediated absorption in the proximal intestine, but make more drugs reach the colon. Shen et al. selected a solid lipid nanoparticle (SLN) composed of trilaurin, oleate, and FA-conjugated TPGS. They loaded doxorubicin (Dox) and superparamagnetic iron oxide nanoparticles (SPIONs) into the SLN (Fig. 13).92 Then, the formed Dox-SPION@SLN was coated with dextran-conjugated octadecanol, finally forming DFSLN. In the proximal intestine, DFSLN was not interacted with and absorbed because dextran shielded FA at the surface of SLN, and FA did not use PCFT-mediated cellular internalization. DFSLN traveled to reach the distal intestine and colon, where dextranase is present. In these sites, the dextran coating on the DFSLN was dissolved and removed, and FA was exposed to the surface of SLN, resulting in FR-targeted Dox and SPIONs delivery to FR-overexpressed colon cancers. In particular, when DFSLN was applied to an orthotopic colon cancer mouse model, it showed 3-fold better tumor growth inhibition than PBS. With a high-frequency magnetic field (HFMF), DFSLN reduced tumor mass more effectively than without HFMF. Overall, the findings indicate that the shielding–deshielding strategy of GI transporter/receptor-targeted NDDSs could allow selective targeting to both transporter/receptor and organ in the GI tract for effective therapeutic outcomes.
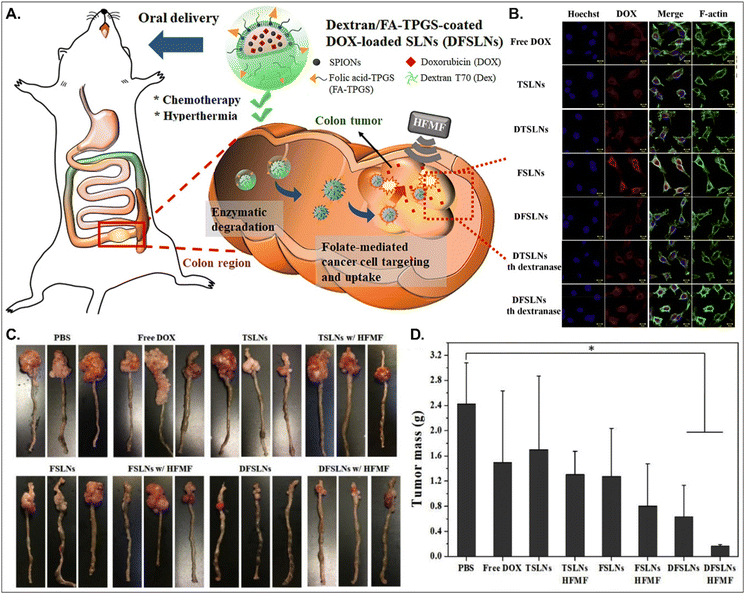 |
| | Fig. 13 Anti-tumor potential of DFSLN: (A) the design concept, (B) the cellular uptake in CT26 cells, (C) photographs of the tumor-bearing colon, and (D) the mass of the explanted tumor (reproduced from ref. 92 with permission from Elsevier, copyright 2019). | |
4.4. Anti-diabetic potential of vitamin-decorated NDDSs
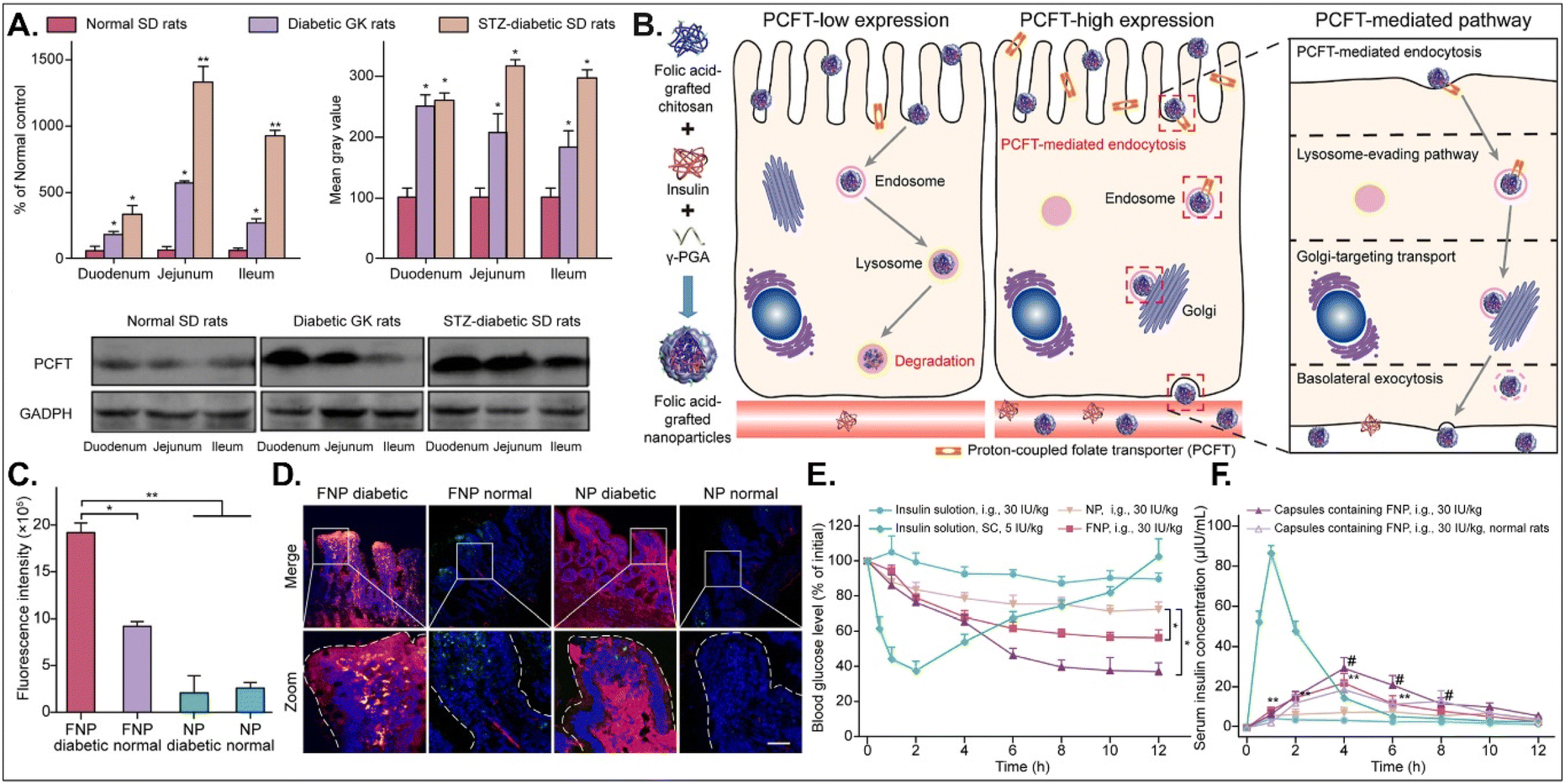
The disease status and substrate requirement strongly influence the expression levels of GI transporters/receptors. Their inhibition or activation can be targeted to treat diseases. Gan et al. found 12-fold overexpressed PCFT in diabetic rats compared with non-diabetic rats and designed FA-chitosan conjugate-based insulin NDDS (i.e., FNP) for PCFT-targeted oral absorption (Fig. 14).29 In low PCFT-expressed Caco-2 cells, FNP followed clathrin-mediated endocytosis and accumulated in the lysosomes for further insulin degradation. However, when applied to high PCFT-expressed Caco-2 cells, FNPs bound PCFT, and the FNP-PCFT complex followed caveolae-mediated endocytosis for the enterocyte entry. Then, FNP avoided lysosomal accumulation and exocytosed into the blood via the ER/Golgi pathway. When orally administered to rats, enterocytes rarely absorbed free insulin. However, orally dosed FNP accumulated in the enterocytes of diabetic rats, and the absorbed FNP decreased in the presence of free FA. In diabetic rats, FNP, unlike free insulin, reduced the blood glucose concentration by 40% after 8 hours and showed about 14% in the oBA of insulin. The findings indicate that FA-decorated NDDSs could effectively deliver insulin to the blood via PCFT-mediated oral absorption in diabetic environments.
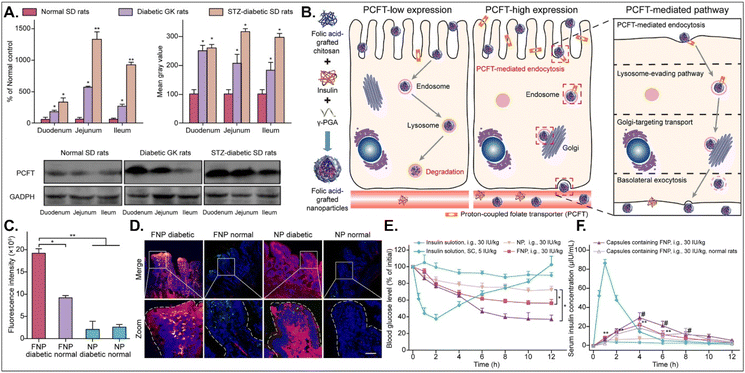 |
| | Fig. 14 Anti-diabetic potential of FNP: (A) the mRNA and protein expression levels of PCFT in non-diabetic or diabetic rats, (B) the expected transcytosis mechanisms, (C) the fluorescence intensity in two-photon images, (D) the colocalization of FNP (green) with PCFT (red), and (E) the blood glucose level and (F) the serum insulin level in diabetic rats (reproduced from ref. 29 under the terms of the Creative Commons CC-BY-ND 4.0 License). | |
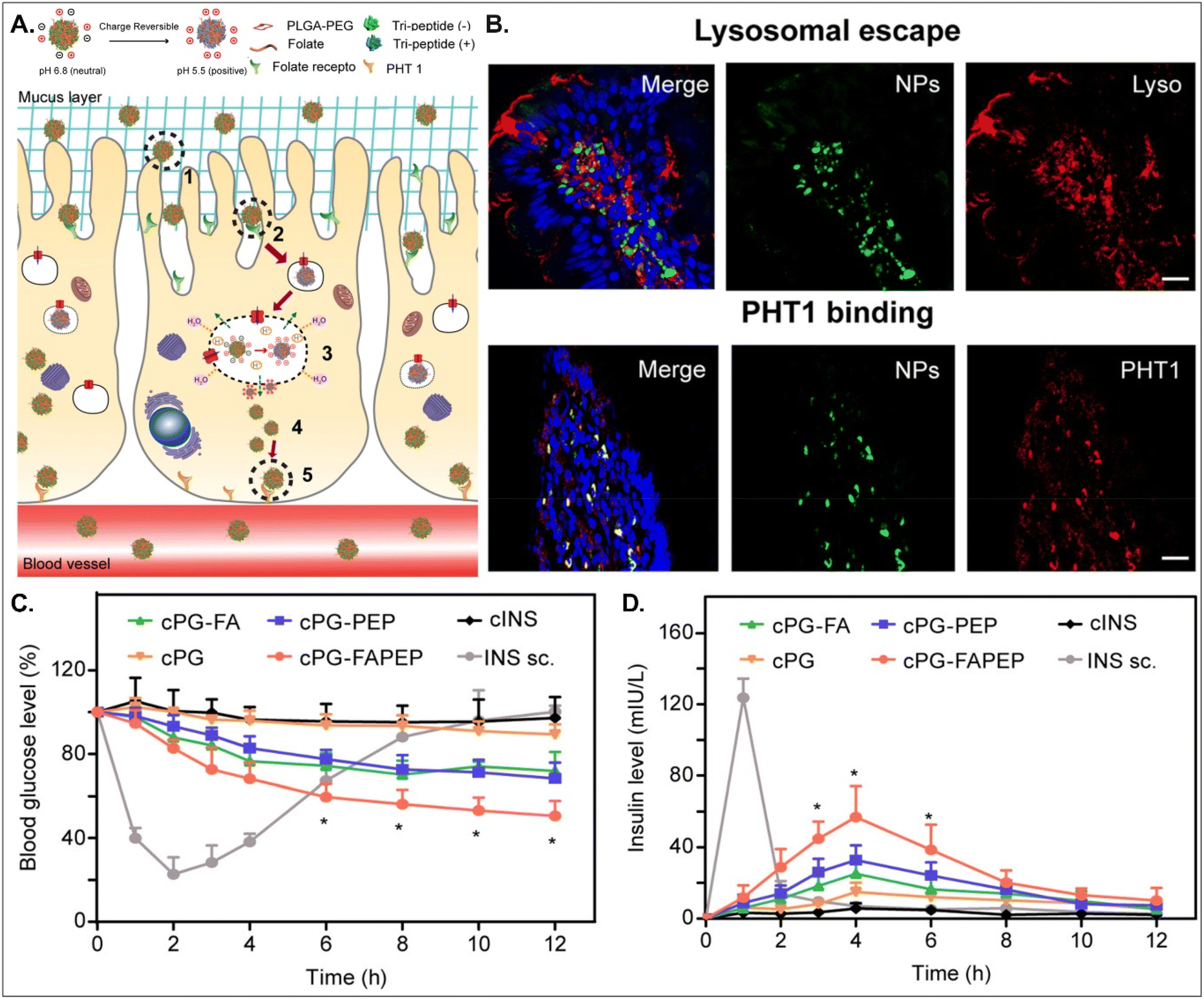
Depending on the drug, it can act within enterocytes or move to the blood after oral absorption. If its mode of action does not occur in enterocytes, its intracellular trafficking and exocytosis to the basolateral region are also necessary after absorption into enterocytes. Therefore, Gan et al. designed dual GI transporter-targeted NDDSs (i.e., PG-FAPEP), which have FA and tripeptide (i.e., Asp-Phe-Gly) for targeting FR in the apical membrane and PHT1 in the basolateral membrane of enterocytes (Fig. 15).59 The charges of the tripeptide exposed on the surface of PG-FAPEP depended on pH: neutral charges at pH 6.8 (i.e., intestinal pH) and positive charges at pH 5.5. In the intestine, the neutrally charged PG-FAPEP penetrated the mucus layer and was internalized into enterocytes via FA-FR-mediated endocytosis. In the endosomes, the tripeptide exposed on the surface of PG-FAPEP was changed to positive charges, enabling proton buffering and endosomal escape. In the cytosol, the tripeptide of the PG-FAPEP bound to PHT1, and the formed PG-FAPEP-PHT1 complex was exocytosed via a PHT1-mediated pathway. In particular, orally administered PG-FAPEP reduced blood glucose concentration to 45% after 6 hours in diabetic rats, and its oBA was about 14.3%. Overall, sequential targeting to GI transporters/receptors at the apical or the basolateral membrane of enterocytes could effectively produce enhanced cellular internalization and enhanced exocytosis of drugs, resulting in their adequate blood circulation. In addition, although the study explained FR targeting, the possibility of PCFT targeting rather than FR targeting could be more realistic due to their known expression levels in the intestine.
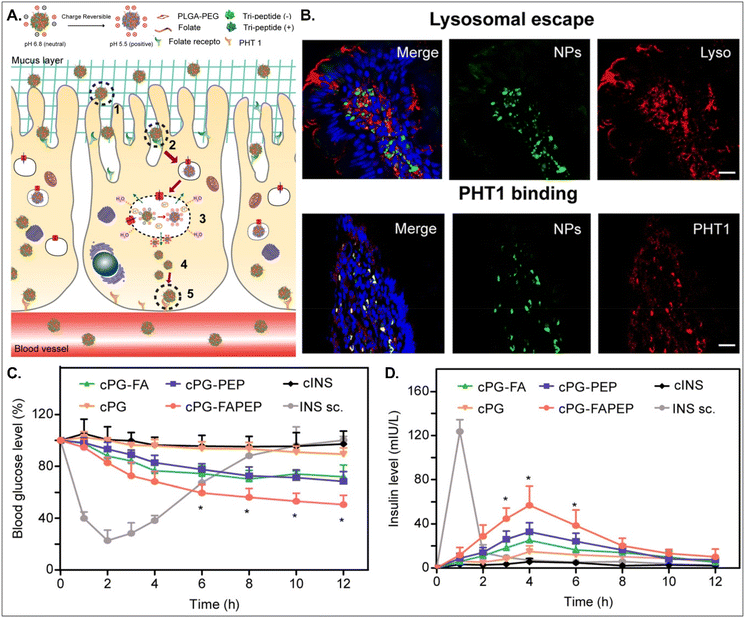 |
| | Fig. 15 Anti-diabetic potential of PG-FAPEP: (A) the design concept, (B) the colocalization of PG-FAPEP with lysosomes or PHT1 in the intestinal villi, and (C) the blood glucose level and (D) the serum insulin level in diabetic rats (reproduced from ref. 59 with permission from Elsevier, copyright 2021). | |
4.5. Anti-infective potential of saccharide-decorated NDDSs
When taking antibiotics orally, they travel down from the stomach to small intestine, and large intestine. They kill microbes in the GI tract, resulting in gut dysbiosis in the large intestine. However, when antibiotics are required to reach the lung through the blood after oral absorption, they should be absorbed in the small intestine before reaching the large intestine. Zhang et al. designed glucosylated NPs (i.e., GNP; glucose-PEG-PLGA) and glucosylated cationic NPs (PGNP; glucose-PEG-PLGA and DOTAP) to use the overexpressed SGLT in the proximal small intestine, and the designed NPs encapsulated antibiotics, ampicillin (Amp) to treat lung infection after SGLT-mediated oral absorption.64 PGNP showed much higher oral absorption and accumulation in the proximal small intestine than GNP but did not reach the large intestine. In a Streptococcus pneumoniae-mediated lung infection model, GNP-Amp and PGNP-Amp, orally administered, delivered a 5-fold higher antibacterial efficacy than free Amp. Also, two glucosylated NPs reduced the mRNA level of pro-inflammatory cytokines (i.e., IL-6, IL-1β, TNF-α) by 2-fold compared with free Amp. Pulmonary pathological and histological analyses demonstrated that two glucosylated NPs reduced inflammation and pathology. The treatment of PGNP-Amp in a lung infection model made the status of the intact alveoli close to that of non-infected, normal lungs. In particular, the designed NPs did not damage the intestinal microbial environment. The findings indicate that glucose-decorated NDDSs could undergo SGLT-mediated oral absorption and then circulate in the blood, reaching the lungs. The results show that oral administration of saccharide-decorated NDDSs could treat lung disorders.
4.6. Anti-tumor potential of antibody-decorated NDDSs
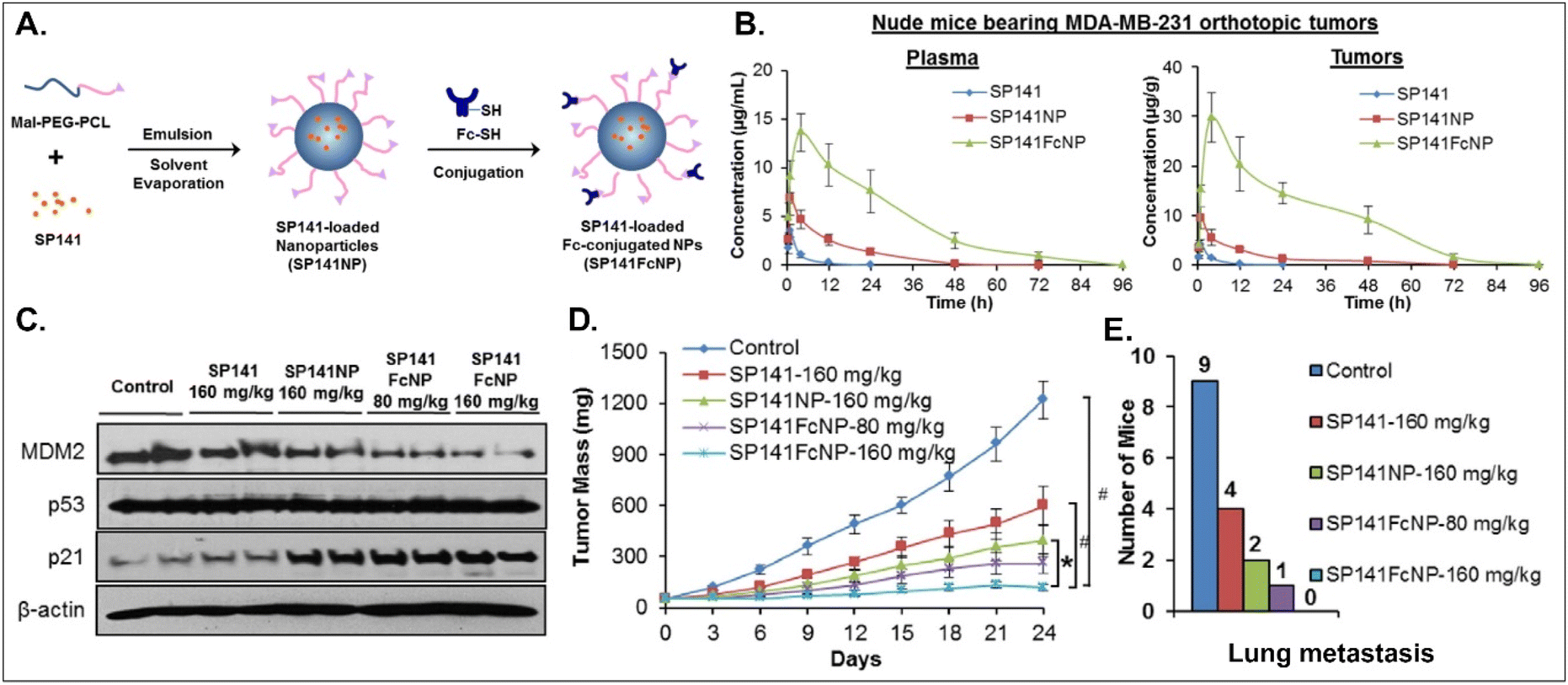
Various oral NDDSs have been developed to improve the oBA of poorly water-soluble and unstable drugs in the GI tract. To orally deliver SP141, a newly discovered inhibitor of MDM2, Qin et al. designed FcRn-targeted NPs (i.e., SP141FcNP), which were constructed by a two-step process: (1) the preparation of SP141-loaded Mal-PEG-PCL NPs (SP141NP) via the emulsion-solvent evaporation method and (2) the conjugation of Fc on the surface of SP141NP (Fig. 16).93 The designed SP141FcNP was expected to target the first FcRn on enterocytes for oral absorption and then target the second FcRn on breast cancer cells for treating solid tumors through systemic circulation. In MCF7 and MDA-MB-231 cells, the NPs effectively killed tumor cells via apoptosis and reduced the expression of MDM2. When orally administered in an MDA-MB-231 orthotopic tumor model, SP141FcNP made 28.5-fold higher and 56.5-fold higher concentrations of SP141 than free SP141 in the plasma and tumor, respectively. In particular, SP141FcNP was absorbed by FcRn in the small intestine and then was highly accumulated in the MDA-MB-231 tumor after blood circulation. Then, SP141FcNPs reduced the expression of MDM2 in the MDA-MB-231 tumor, inhibited its tumor growth by 90% compared with the control, and decreased lung metastasis. The findings indicate that antibody-decorated NDDSs could take FcRn-mediated oral absorption and circulate in the blood, reaching FcRn-expressing solid tumors.
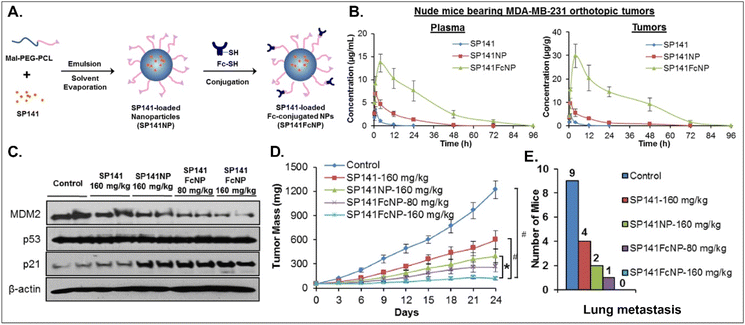 |
| | Fig. 16 Anti-tumor potential of SP141FcNP: (A) the design concept, (B) the time-dependent plasma or tumor level, (C) the expression of MDM2, (D) the time-dependent tumor mass, and (E) the numbers of mice with lung metastasis to lungs in an MDA-MB-231 orthotopic tumor-bearing mice model (reproduced from ref. 93 with permission from Elsevier, copyright 2016). | |
4.7. Some general issues of GI transporter/receptor-mediated NDDSs for clinical applications
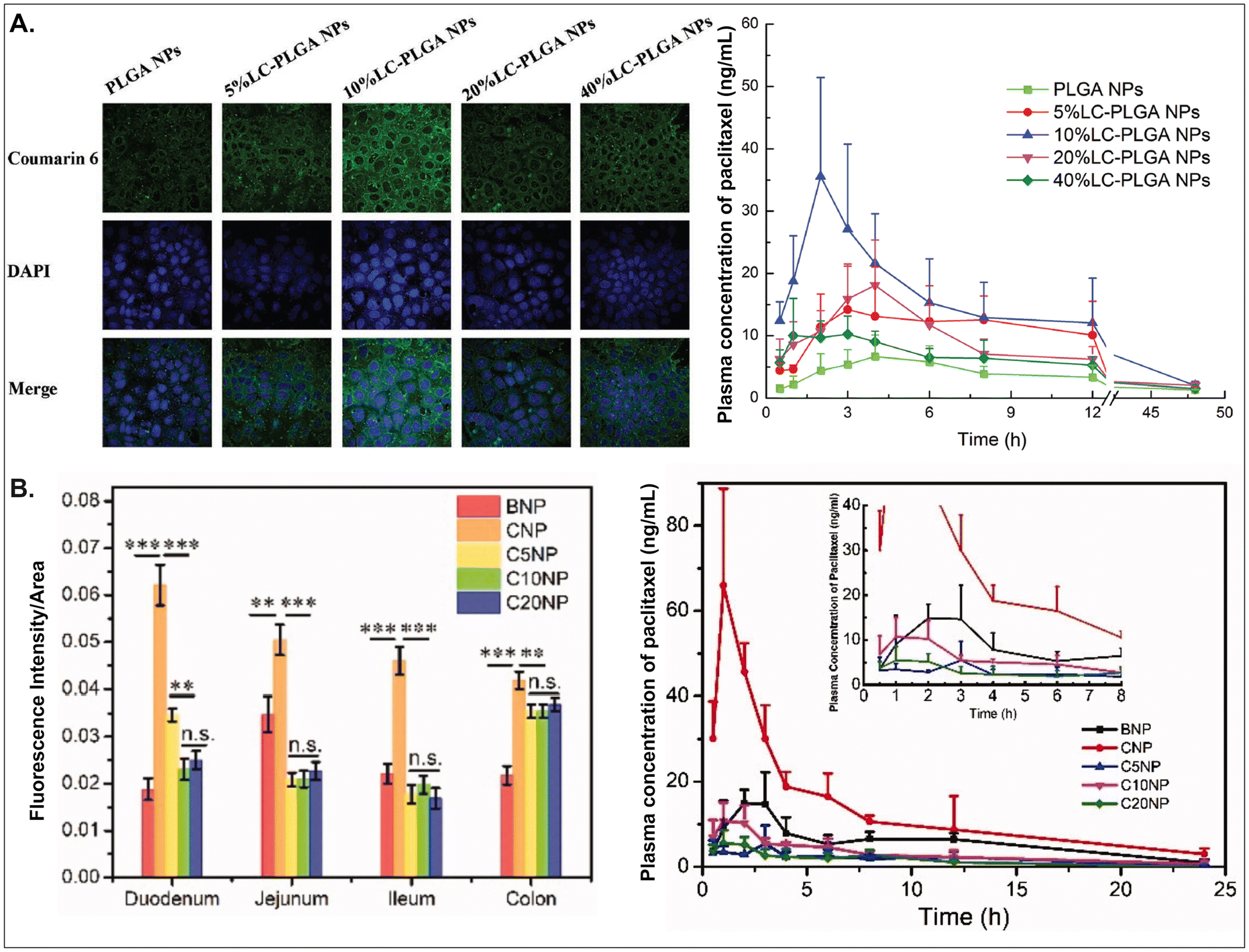
As aforementioned, various GI transporter/receptor-mediated NDDSs have been investigated and applied to animal disease models, and their therapeutic outcomes showed great potential to move further clinical applications. Nevertheless, they have some general issues relevant to colloidal stability in the GI tract and binding affinity to the GI transporters/receptors of interest. First, when NDDSs are orally administered, they move from the mouth to the stomach and then small and large intestines. During the journey in the GI tract, they may lose their colloidal stability under acidic (e.g., pH 1–5 in a fasted or fed stomach94) or enzymatic (e.g., pepsin, lipase, etc. in the stomach;95 di/amino peptidase, lactase, etc. in the small intestine;96 azoreductase, dextranase, etc. in the large intestine97) conditions before their absorption in small and large intestines because the conditions can cause their aggregation, disintegration, or degradation. Thus, some studies have monitored particle sizes in GI tract-mimicking fluids to evaluate whether nanosized delivery systems keep their colloidal stability.3,74 Nevertheless, enteric-coated capsules could be used if carrier particles are unstable in the stomach or their changes are not evaluated.29,59 Second, when substrates (or ligands) are decorated on the NP surface, their binding affinities to their corresponding transporters (or receptors) can be strongly affected by their exposed side and density/number after linking on the NP surface, and the type and length of linker molecules between substrates and NPs. For example, chemically linking substrates on the NP surface can result in partial or full masking of their binding side by the NP itself or by their neighboring substrates decorated on the NP surface. Unlike a binding between free substrates and their counterparts (e.g., transporters), steric hindrance by neighboring substrates or nanoparticular components on the surface of the same NP could reduce the binding affinity between the designed substrate and the target transporter. Nevertheless, steric hindrance-mediated influence has been frequently overlooked when designing NDDSs. In reality, when synthesizing carnitine-decorated NDDSs, the linking side of carnitine should be the hydroxyl group rather than the carboxylic acid because the latter is essential for binding to OCTN.41 Also, although no study measures a binding affinity, Sun et al. reported that carnitine's optimal contents/densities on carnitine-decorated NDDSs were about 10% for cellular uptake and oBA when preparing NDDSs by mixing carnitine-conjugated PLGA (i.e., PLGA-LC) and PLGA at various ratios (i.e., 5, 10, 20, 40%) of PLGA-LC. Increasing carnitine's contents by up to 10% increased the cellular uptake and oBA of carnitine-decorated NDDSs, and then higher carnitine contents (e.g., 20–40%) reduced the levels (Fig. 17A).66 The results indicate that too many substrates on the NP surface can inhibit or hinder the targeted binding between substrate and transporter. In addition, Chen et al. introduced different PEG lengths (i.e., 0, 500, 1000, 2000 Da) between PLGA and carnitine, making CNP, C5NP, C10NP, and C20NP, respectively. Interestingly, CNP showed the highest intestinal absorption and oBA compared with other carnitine-decorated NDDSs with lengthy PEG linkers (Fig. 17B).98 The authors inferred that an increasing flexibility (by lengthy linkers) of the cationic substrate on the NP surface could result in strong interaction with the negatively charged mucus layer instead of interaction between carnitine and OCTN. Thus, when designing oral NDDSs, the above two critical factors should be investigated in depth, further resulting in the maximized binding affinity between substrate and target transporter and the highest oBA (i.e., highest therapeutic outcomes).
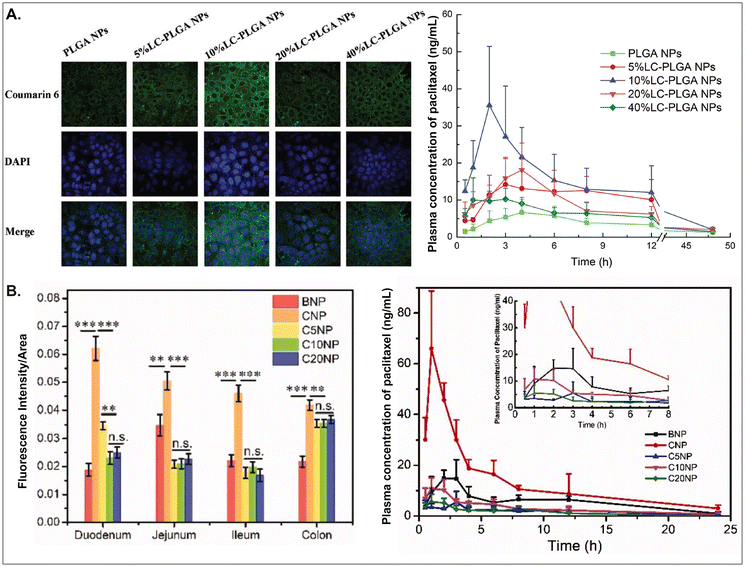 |
| | Fig. 17 (A) Cellular uptake in Caco-2 cells and pharmacokinetics profile of paclitaxel in LC-PLGA NP-administered rats, and (B) fluorescence intensity and pharmacokinetics profile of the drug in carnitine-NP-administrated rats (for A, reproduced from ref. 66 with permission from Wiley-VCH, copyright 2017; for B, reproduced from ref. 98 under the terms of the Creative Commons CC-BY 4.0 License). | |
5. Future directions beyond GI transporter/receptor-mediated NDDSs: potential for disease targeting
5.1. Potential for disease targeting dependent on the expression of GI transporters/receptors
The expression levels of potential GI transporters/receptors are not fixed, but dependent on the status of diseases (Table 2 and ESI Table 3†). For example, colorectal cancer increased the expression levels of PEPT1 and FR. IBD increased the expression levels of FRβ, ICAM-1, OATP, PEPT1, and transferrin receptors, but decreased those of ASBT, ATB0,+, MCT1, OCTN2, OSTα, OSTβ, SVCT, and SVMT. Also, various GI transporters/receptors were increased: ASBT, GLUT5, PCFT, PEPT1, and SGLT1 in diabetes; ASBT and PEPT in obesity; FcRn in infection. The change in the expression level of GI transporters/receptors could improve or limit their use for oral absorption of substrate-decorated NDDSs. If BA-decorated NDDSs are developed to target ASBT, the NDDSs could make better therapeutic outcomes in diabetes and obesity but have less efficacy in IBD and obstructive cholestasis. Targeting PEPT1 will be a good choice in treating diabetes, IBD, and obesity. However, targeting ASBT, MCT1, OCTN2, SVCT, and SVMT will be the wrong choice to treat IBD. Thus, understanding the relationship between the expression level of GI transporters/receptors and diseases will be critical for a high therapeutic effect of substrate-decorated NDDSs.
5.2. Potential for disease targeting dependent on the destination of GI transporter/receptor-mediated NDDSs
When substrate-decorated NDDSs follow GI transporter/receptor-mediated oral absorption, their cellular entry, endosomal/endolysosomal escape, intracellular trafficking, exocytosis, and blood/lymphatic circulation depend strongly on their sizes, surface characteristics (e.g., zeta-potential, charge, hydrophilicity/hydrophobicity), materials (e.g., polymers, lipids, metals, ceramics), and architectures (e.g., gels, micelles, vesicles, solid/porous particles, oil droplets). Many efforts have been made to understand and investigate GI transporter/receptor-mediated paths and mechanisms from the entry to the exit of the NDDSs through the epithelial cell lining of the intestine. However, the relevant information is still lacking. Nevertheless, if the NDDSs are well-regulated to follow the designed paths for moving to the destination of interest, they could be localized in the intestine or its surroundings, circulated in either the blood or lymphatic vessel, and distributed to organs/tissues. First, for local delivery in the intestine or its surroundings, the NDDSs might contain blockers/inhibitors of exit pathways and be localized in the epithelial cells of the small intestine and the colon and various immune cells (e.g., macrophages, DCs) in the Peyer's patches. Small intestinal bacterial overgrowth, Crohn's disease, ulcerative colitis, GI infections, and colon cancer could be targeted. Second, if substrate-decorated NDDSs release their payloads into the blood after moving to the blood, their applications would be extensive, including cancers, inflammatory disorders, infective disorders, autoimmune diseases, metabolic diseases, etc. However, their first-pass metabolism in the liver could lower their therapeutic outcomes. Third, if GI transporter/receptor-targeted NDDSs take a lymphatic pathway after an exocytosis route (e.g., a chylomicron pathway), the NDDSs could improve the bioavailability of their payloads because they bypass hepatic first-pass metabolism (or directly enter systemic circulation). In particular, the intestinal lymphatic vessel-targeted NDDSs enable the treatment of various immune diseases, including IBD, by effectively activating or suppressing various immune cells because the intestine is the largest immune organ, where 70–80% of the immune cells in the body exist. In situ genetic engineering of immune cells (e.g., T cells, macrophages, DCs, etc.) could also be possible. Fourth, suppose the designed NDDSs are distributed to the diseased organs/tissues after entering the systemic circulation directly or via the lymphatic circulation. In that case, the endothelial cells (e.g., blood–brain barrier) neighboring the target organs/tissues should express transporters/receptors, or the blood capillary neighboring the target organs/tissues (e.g., liver, spleen, bone marrow, solid tumor tissues, inflamed tissues, etc.) should be fenestrated or sinusoidal. The NDDSs could treat various disorders in the brain, liver, spleen, solid tumors, and inflamed tissues. Overall, the design strategies regulating endolysosomal escape, intracellular trafficking, or exocytosis should be investigated for a customized destination of substrate-decorated NDDSs and their encapsulated therapeutics.
5.3. Potential for disease targeting dependent on dual transporter/receptor targeting of substrate-decorated NDDSs to both intestinal barrier and disease organs
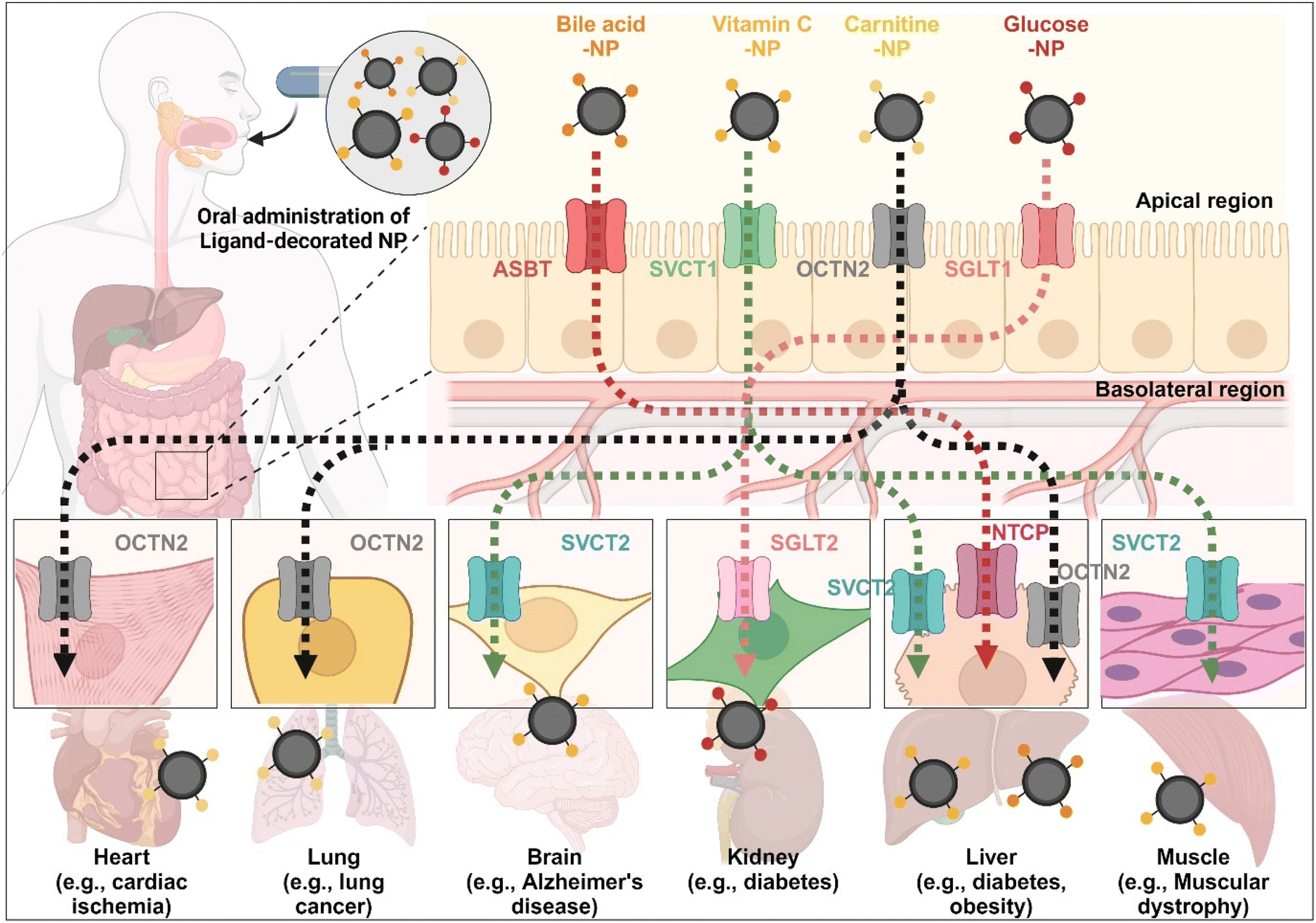
The enterocyte contains various transporters/receptors that can absorb vital nutrients and energy sources from the GI tract. The substances are transported to enterocytes via specific transporters/receptors and then distributed through the blood to the required organs. This fact indicates that the organs also have substance-corresponding transporters/receptors. Thus, substrate-decorated NDDSs can target GI transporters/receptors on enterocytes for oral absorption and then target disease-related organs expressing the same or substrate-transportable transporters/receptors for selective delivery of their payloads (Fig. 18). For example, BAs are first absorbed by enterocytes through ASBT, transported into the blood, and then reabsorbed into the liver through NTCP,91 present in the basolateral membrane of hepatocytes. Namely, BA-decorated NDDSs can deliver therapeutics for liver-related diseases such as hepatocarcinoma, cirrhotic cholangitis, and metabolic liver diseases. In particular, the NDDSs may effectively target metabolic liver diseases associated with diabetes or obesity due to increased intestinal ASBT expression in diabetic or obese patients. Second, ascorbic acid (vitamin C) is absorbed and diffused into enterocytes through SVCT1. It makes equilibrium with its oxidized form, dehydroascorbic acid (DHA). DHA is transported to the blood through GLUT.99 The brain, muscles, adrenal glands, and liver absorb vitamin C via SVCT2, making the tens of mM compared with the tens of μM in the blood. Thus, vitamin C-decorated NDDSs can transport drugs to various organs via SVCT-mediated oral absorption and organ targeting, and can be applied to treat diseases in the brain, liver, and so on. Third, utilizing OCTN2, expressed not only in enterocytes but also in the basolateral membranes of neurons, lung airways, heart, liver, and muscles, may allow carnitine-decorated NDDSs to treat disorders in the brain, lungs, heart, liver, and so on. In particular, the increased expression of OCTN2 in glioblastoma could be a good factor when selecting a target disease of OCTN2-mediated NDDSs. Fourth, glucose is absorbed into enterocytes by SGLT1 and then transported to the blood by GLUT2. Also, the kidneys reabsorb glucose by SGLT2 or GLUT1 and express it in the renal basolateral membrane.100 The kidneys of diabetic patients frequently malfunction, requiring medication to restore their function. With the increased expression of SGLT1 in the intestine of diabetic patients, glucose-decorated NDDSs could be utilized to selectively deliver therapeutics to the kidneys for treating diabetes-mediated kidney diseases. Substrate-decorated NDDSs could make sequentially dual-targeting drug delivery systems for enhanced oral absorption and disease targeting.
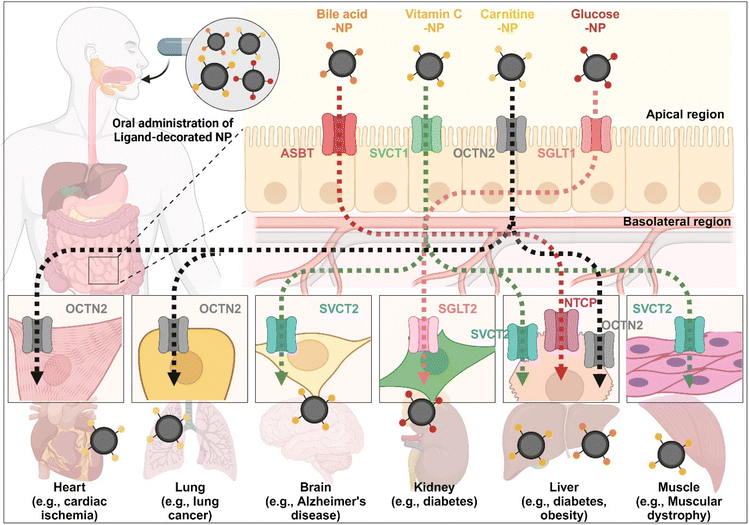 |
| | Fig. 18 Possible examples of dual-targeted substrate-decorated NDDSs for oral absorption and organ/disease targeting. | |
6. Conclusion
Various oral drug delivery systems have been investigated to overcome poor water-solubility, chemical/biological stability, oral absorption, or targetability. Among them, GI transporter/receptor-targeted NDDSs could be an essential strategy to solve all the issues mentioned earlier. The enhanced or overexpressed GI transporters/receptors could improve the payloads’ therapeutic outcomes in the NDDSs. In particular, the transcytosis-modulating design strategies of the NDDSs could tune the destinations of their payloads, treating broad-range disorders in the GI tract, the systemic/lymphatic vessel, or various organs/tissues. Thus, GI transporter/receptor-targeted NDDSs have solid potential to expand from oral absorption to disease or organ targeting.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This study was supported by the National Research Foundation of Korea (NRF), which is funded by the Korean government (for MSIT, NRF-2022R1A2C2006026 and NRF-2022M3E5F1017414; for MOE, NRF-2022R1I1A1A01064322). The cartoons in the graphic abstract, Fig. 1, 3, 4, 6, 7, 8, 9, and 18 were created with BioRender.com.
References
- M. Drozdzik, I. Czekawy, S. Oswald and A. Drozdzik, Pharmacol. Rep., 2020, 72, 1173–1194 CrossRef PubMed.
- T. A. Al-Hilal, S. W. Chung, F. Alam, J. Park, K. E. Lee, H. Jeon, K. Kim, I. C. Kwon, I.-S. Kim and S. Y. Kim, Sci. Rep., 2014, 4, 4163 CrossRef PubMed.
- K. S. Kim, K. Suzuki, H. Cho, Y. S. Youn and Y. H. Bae, ACS Nano, 2018, 12, 8893–8900 CrossRef CAS PubMed.
- M. Drozdzik and S. Oswald, Curr. Med. Chem., 2016, 23, 4468–4489 CrossRef CAS PubMed.
- Y. Meier, J. J. Eloranta, J. Darimont, M. G. Ismair, C. Hiller, M. Fried, G. A. Kullak-Ublick and S. R. Vavricka, Drug Metab. Dispos., 2007, 35, 590–594 CrossRef CAS PubMed.
- D. Jung, A. Fantin, U. Scheurer, M. Fried and G. Kullak-Ublick, Gut, 2004, 53, 78–84 CrossRef CAS PubMed.
- P. Hruz, C. Zimmermann, H. Gutmann, L. Degen, U. Beuers, L. Terracciano, J. Drewe and C. Beglinger, Gut, 2006, 55, 395–402 CrossRef CAS PubMed.
- F. Annaba, K. Ma, P. Kumar, A. K. Dudeja, R. D. Kineman, B. L. Shneider, S. Saksena, R. K. Gill and W. A. Alrefai, Am. J. Physiol.: Gastrointest. Liver Physiol., 2010, 299, G898–G906 CrossRef CAS PubMed.
- S. Sundaram, B. Palaniappan, N. Nepal, S. Chaffins, U. Sundaram and S. Arthur, Cells, 2019, 8, 1197 CrossRef CAS PubMed.
- N. Patel, L. Ghali, I. Roitt, L. P. Munoz and R. Bayford, Nanoscale Adv., 2021, 3, 5373–5386 RSC.
- S. H. Lee, J. G. Song and H.-K. Han, Acta Pharm. Sin. B, 2022, 12, 4249–4261 CrossRef CAS PubMed.
- J. Skupsky, S. Sabui, M. Hwang, M. Nakasaki, M. D. Cahalan and H. M. Said, Cell. Mol. Gastroenterol. Hepatol., 2020, 9, 557–567 CrossRef PubMed.
- S. Pérez-Torras, I. Iglesias, M. Llopis, J. J. Lozano, M. Antolín, F. Guarner and M. Pastor-Anglada, J. Crohn's Colitis, 2016, 10, 850–859 CrossRef PubMed.
- V. S. Subramanian, S. Sabui, G. A. Subramenium, J. S. Marchant and H. M. Said, Am. J. Physiol.: Gastrointest. Liver Physiol., 2018, 315, G241–G248 CrossRef CAS PubMed.
- Y. Fujita, H. Kojima, H. Hidaka, M. Fujimiya, A. Kashiwagi and R. Kikkawa, Diabetologia, 1998, 41, 1459–1466 CrossRef CAS PubMed.
- J. Dyer, I. Wood, A. Palejwala, A. Ellis and S. Shirazi-Beechey, Am. J. Physiol.: Gastrointest. Liver Physiol., 2002, 282, G241–G248 CrossRef CAS PubMed.
- P. Liu, C. Gao, H. Chen, C. T. Vong, X. Wu, X. Tang, S. Wang and Y. Wang, Acta Pharm. Sin. B, 2021, 11, 2798–2818 CrossRef CAS PubMed.
- M. Fujiya, Y. Inaba, M. W. Musch, S. Hu, Y. Kohgo and E. B. Chang, Inflamm. Bowel Dis., 2011, 17, 907–916 CrossRef PubMed.
- K. A. Wojtal, J. J. Eloranta, P. Hruz, H. Gutmann, J. Drewe, A. Staumann, C. Beglinger, M. Fried, G. A. Kullak-Ublick and S. R. Vavricka, Drug Metab. Dispos., 2009, 37, 1871–1877 CrossRef CAS PubMed.
- M. Pyzik, L. K. Kozicky, A. K. Gandhi and R. S. Blumberg, Nat. Rev. Immunol., 2023, 23, 1–18 CrossRef PubMed.
- T. M. Bui, H. L. Wiesolek and R. Sumagin, J. Leucocyte Biol., 2020, 108, 787–799 CrossRef CAS PubMed.
- R. Thibault, P. De Coppet, K. Daly, A. Bourreille, M. Cuff, C. Bonnet, J. F. Mosnier, J. P. Galmiche, S. Shirazi-Beechey and J. P. Segain, Gastroenterology, 2007, 133, 1916–1927 CrossRef CAS PubMed.
- C.-Y. Wang, S. Liu, X.-N. Xie and Z.-R. Tan, Drug Des., Dev. Ther., 2017, 2017, 3511–3517 CrossRef PubMed.
- S. A. Ingersoll, S. Ayyadurai, M. A. Charania, H. Laroui, Y. Yan and D. Merlin, Am. J. Physiol.: Gastrointest. Liver Physiol., 2012, 302, G484–G492 CrossRef CAS PubMed.
- G. D'Argenio, M. Calvani, A. Casamassimi, O. Petillo, S. Margarucci, M. Rienzo, I. Peluso, R. Calvani, A. Ciccodicola and N. Caporaso, FASEB J., 2006, 20, 2544–2546 CrossRef PubMed.
- A. F. Hofmann, Arch. Intern. Med., 1999, 159, 2647–2658 CrossRef CAS PubMed.
- R. Zhao, S. H. Min, Y. Wang, E. Campanella, P. S. Low and I. D. Goldman, J. Biol. Chem., 2009, 284, 4267–4274 CrossRef CAS PubMed.
- M. Kim, S. Pyo, C. H. Kang, C. O. Lee, H. K. Lee, S. U. Choi and C. H. Park, PLoS One, 2018, 13, e0198347 CrossRef PubMed.
- J Li, Y Zhang, A Wang, Y Qiu, W Fan, M Hovgaard, Y Li, R Wang, X Li and Y Gan, Acta Pharm. Sin. B, 2022, 12, 1460–1472 CrossRef CAS PubMed.
- M. Quick and L. Shi, Vitam. Horm., 2015, 98, 63–100 CrossRef CAS PubMed.
- R. Leonardi and S. Jackowski, EcoSal Plus, 2007, 2 DOI:10.1128/ecosalplus.3.6.3.4.
- C. Sirithanakorn and J. E. Cronan, FEMS Microbiol. Rev., 2021, 45, fuab003 CrossRef CAS PubMed.
- C. B. F. Andersen, M. Madsen, T. Storm, S. K. Moestrup and G. R. Andersen, Nature, 2010, 464, 445–448 CrossRef PubMed.
- J. C. Boyer, C. E. Campbell, W. J. Sigurdson and S.-M. Kuo, Biochem. Biophys. Res. Commun., 2005, 334, 150–156 CrossRef CAS PubMed.
- V. S. Subramanian, P. Srinivasan, A. J. Wildman, J. S. Marchant and H. M. Said, Am. J. Physiol.: Gastrointest. Liver Physiol., 2017, 312, G340–G347 CrossRef PubMed.
- C. I. Cheeseman, Gastroenterology, 1993, 105, 1050–1056 CrossRef CAS PubMed.
- V. Douard and R. P. Ferraris, Am. J. Physiol.: Endocrinol. Metab., 2008, 295, E227–E237 CrossRef CAS PubMed.
- P. Song, A. Onishi, H. Koepsell and V. Vallon, Expert Opin. Ther. Targets, 2016, 20, 1109–1125 CrossRef CAS PubMed.
- A. K. Azad, M. V. Rajaram and L. S. Schlesinger, J. Cytol. Mol. Biol., 2014, 1, 1000003 Search PubMed.
- N. Longo, M. Frigeni and M. Pasquali, Biochim. Biophys. Acta, Mol. Cell Res., 2016, 1863, 2422–2435 CrossRef CAS PubMed.
- G. Wang, L. Zhao, Y. Sun, D. Zhao, M. Sun, Z. He and Y. Wang, Asian J. Pharm. Sci., 2020, 15, 158–172 CrossRef PubMed.
- L. Kou, Q. Yao, S. Sivaprakasam, Q. Luo, Y. Sun, Q. Fu, Z. He, J. Sun and V. Ganapathy, Drug Delivery, 2017, 24, 1338–1349 CrossRef CAS PubMed.
- M. Pyzik, T. Rath, W. I. Lencer, K. Baker and R. S. Blumberg, J. Immunol., 2015, 194, 4595–4603 CrossRef CAS PubMed.
- D. C. Roopenian and S. Akilesh, Nat. Rev. Immunol., 2007, 7, 715–725 CrossRef CAS PubMed.
- A. Borthakur, S. Saksena, R. K. Gill, W. A. Alrefai, K. Ramaswamy and P. K. Dudeja, J. Cell. Biochem., 2008, 103, 1452–1463 CrossRef CAS PubMed.
- N. Vijay and M. E. Morris, Curr. Pharm. Des., 2014, 20, 1487–1498 CrossRef CAS PubMed.
- X. Sun, M. Wang, M. Wang, L. Yao, X. Li, H. Dong, M. Li, T. Sun, X. Liu and Y. Liu, Front. Cell Dev. Biol., 2020, 8, 651 CrossRef PubMed.
- E. J. Perkins and T. Abraham, Drug Metab. Dispos., 2007, 35, 1903–1909 CrossRef CAS PubMed.
- J. L. Sloan and S. Mager, J. Biol. Chem., 1999, 274, 23740–23745 CrossRef CAS PubMed.
- L. Metzner, K. Neubert and M. Brandsch, Amino Acids, 2006, 31, 111–117 CrossRef CAS PubMed.
- F. Deng and Y. H. Bae, Nanomedicine, 2023, 48, 102629 CrossRef CAS PubMed.
- F. Deng, K. S. Kim, J. Moon and Y. H. Bae, Adv. Sci., 2022, 9, 2201414 CrossRef CAS PubMed.
- W. Liu, Y. Han, X. Xin, L. Chen, Y. Liu, C. Liu, X. Zhang, M. Jin, J. Jin and Z. Gao, J. Nanobiotechnol., 2022, 20, 1–23 CrossRef PubMed.
- W. Fan, D. Xia, Q. Zhu, X. Li, S. He, C. Zhu, S. Guo, L. Hovgaard, M. Yang and Y. Gan, Biomaterials, 2018, 151, 13–23 CrossRef CAS PubMed.
- J. Park, J. U. Choi, K. Kim and Y. Byun, Biomaterials, 2017, 147, 145–154 CrossRef CAS PubMed.
- J. He, R. Ding, Y. Tao, Z. Zhao, R. Yuan, H. Zhang, A. Wang, K. Sun, Y. Li and Y. Shi, Drug Delivery, 2023, 30, 2181744 CrossRef PubMed.
- H. Cheng, S. Guo, Z. Cui, X. Zhang, Y. Huo, J. Guan and S. Mao, Int. J. Pharm., 2021, 596, 120297 CrossRef CAS PubMed.
- F. Zhang, X. Pei, X. Peng, D. Gou, X. Fan, X. Zheng, C. Song, Y. Zhou and S. Cui, Biomater. Adv., 2022, 135, 212746 CrossRef CAS PubMed.
- Z. Xi, E. Ahmad, W. Zhang, J. Li, A. Wang, Faridoon, N. Wang, C. Zhu, W. Huang, L. Xu, M. Yu and Y. Gan, J. Controlled Release, 2022, 342, 1–13 CrossRef CAS PubMed.
- Y. Yang, Y. Yin, J. Zhang, T. Zuo, X. Liang, J. Li and Q. Shen, Pharmaceutics, 2018, 10, 146 CrossRef CAS PubMed.
- Q. Luo, M. Jiang, L. Kou, L. Zhang, G. Li, Q. Yao, L. Shang and Y. Chen, Artif. Cells, Nanomed., Biotechnol., 2018, 46, 198–208 CrossRef CAS PubMed.
- Y. Xing, X. Li, W. Cui, M. Xue, Y. Quan and X. Guo, Pharmaceutics, 2022, 14, 1361 CrossRef CAS PubMed.
- X. Zhang, J. Qi, Y. Lu, W. He, X. Li and W. Wu, Nanomedicine, 2014, 10, 167–176 CrossRef CAS PubMed.
- G. Zhang, Q. Wang, W. Tao, W. Jiang, E. Elinav, Y. Wang and S. Zhu, Nat. Biomed. Eng., 2022, 6, 867–881 CrossRef CAS PubMed.
- C. He, Y. Jin, Y. Deng, Y. Zou, S. Han, C. Zhou, Y. Zhou and Y. Liu, ACS Biomater. Sci. Eng., 2020, 6, 2146–2158 CrossRef CAS PubMed.
- L. Kou, Q. Yao, M. Sun, C. Wu, J. Wang, Q. Luo, G. Wang, Y. Du, Q. Fu, J. Wang, Z. He, V. Ganapathy and J. Sun, Adv. Healthcare Mater., 2017, 6, 1700165 CrossRef PubMed.
- J. Wang, L. Wang, Y. Li, X. Wang and P. Tu, Int. J. Nanomed., 2018, 13, 7997–8012 CrossRef CAS PubMed.
- Y. Jin, Q. Liu, C. Zhou, X. Hu, L. Wang, S. Han, Y. Zhou and Y. Liu, Nanoscale, 2019, 11, 21433–21448 RSC.
- K. Liu, Y. Chen, Z. Yang and J. Jin, Int. J. Biol. Macromol., 2023, 236, 123870 CrossRef CAS PubMed.
- R. Ghaffarian, T. Bhowmick and S. Muro, J. Controlled Release, 2012, 163, 25–33 CrossRef CAS PubMed.
- E. M. Pridgen, F. Alexis, T. T. Kuo, E. Levy-Nissenbaum, R. Karnik, R. S. Blumberg, R. Langer and O. C. Farokhzad, Sci. Transl. Med., 2013, 5, 213ra167 Search PubMed.
- K. S. Kim, S. Lee, K. Na and Y. H. Bae, Adv. Healthcare Mater., 2022, 11, e2200909 CrossRef PubMed.
- K. S. Kim, Y. S. Youn and Y. H. Bae, J. Controlled Release, 2019, 311–312, 85–95 CrossRef CAS PubMed.
- K. Suzuki, K. S. Kim and Y. H. Bae, J. Controlled Release, 2019, 294, 259–267 CrossRef CAS PubMed.
- K. S. Kim, D. S. Kwag, H. S. Hwang, E. S. Lee and Y. H. Bae, Mol. Pharm., 2018, 15, 4756–4763 CrossRef CAS PubMed.
- M. Nurunnabi, S. A. Lee, V. Revuri, Y. H. Hwang, S. H. Kang, M. Lee, S. Cho, K. J. Cho, Y. Byun, Y. H. Bae, D. Y. Lee and Y. K. Lee, J. Controlled Release, 2017, 268, 305–313 CrossRef CAS PubMed.
- S. M. S. Shahriar, J. M. An, M. N. Hasan, S. S. Surwase, Y. C. Kim, D. Y. Lee, S. Cho and Y. K. Lee, Nano Lett., 2021, 21, 4666–4675 CrossRef CAS PubMed.
- J. M. An, S. M. S. Shahriar, Y. H. Hwang, S. R. Hwang, D. Y. Lee, S. Cho and Y. K. Lee, ACS Appl. Mater. Interfaces, 2021, 13, 23314–23327 CrossRef CAS PubMed.
- L. Subedi, P. Pandey, B. Khadka, J. H. Shim, S. S. Cho, S. Kweon, Y. Byun, K. T. Kim and J. W. Park, Drug Delivery, 2022, 29, 3397–3413 CrossRef CAS PubMed.
- L. Subedi, P. Pandey, S. H. Kang, K. T. Kim, S. S. Cho, K. Y. Chang, Y. Byun, J. H. Shim and J. W. Park, J. Controlled Release, 2022, 349, 502–519 CrossRef CAS PubMed.
- K. B. Chalasani, G. J. Russell-Jones, A. K. Jain, P. V. Diwan and S. K. Jain, J. Controlled Release, 2007, 122, 141–150 CrossRef CAS PubMed.
- C. Azevedo, J. Nilsen, A. Grevys, R. Nunes, J. T. Andersen and B. Sarmento, J. Controlled Release, 2020, 327, 161–173 CrossRef CAS PubMed.
- M. N. Hasan, Y. H. Hwang, J. M. An, S. M. S. Shahriar, S. Cho and Y. K. Lee, ACS Appl. Mater. Interfaces, 2020, 12, 38925–38935 CrossRef CAS PubMed.
- H. T. Nguyen, G. Dalmasso, L. Torkvist, J. Halfvarson, Y. Yan, H. Laroui, D. Shmerling, T. Tallone, M. D'Amato, S. V. Sitaraman and D. Merlin, J. Clin. Invest., 2011, 121, 1733–1747 CrossRef CAS PubMed.
- B. Xiao, E. Viennois, Q. Chen, L. Wang, M. K. Han, Y. Zhang, Z. Zhang, Y. Kang, Y. Wan and D. Merlin, ACS Nano, 2018, 12, 5253–5265 CrossRef CAS PubMed.
- G. K. Sinhmar, N. N. Shah, S. U. Rawal, N. V. Chokshi, H. N. Khatri, B. M. Patel and M. M. Patel, Artif. Cells, Nanomed., Biotechnol., 2018, 46, 565–578 CrossRef CAS PubMed.
- Y. Chen, J. Wu, J. Wang, W. Zhang, B. Xu, X. Xu and L. Zong, Diabetologia, 2018, 61, 1384–1396 CrossRef CAS PubMed.
- L. Wu, M. Liu, W. Shan, X. Zhu, L. Li, Z. Zhang and Y. Huang, J. Controlled Release, 2017, 262, 273–283 CrossRef CAS PubMed.
- Y. Du, C. Tian, M. Wang, D. Huang, W. Wei, Y. Liu, L. Li, B. Sun, L. Kou, Q. Kan, K. Liu, C. Luo, J. Sun and Z. He, Drug Delivery, 2018, 25, 1403–1413 CrossRef CAS PubMed.
- A. Jain and S. K. Jain, Acta Diabetol., 2015, 52, 663–676 CrossRef CAS PubMed.
- S. H. Kang, V. Revuri, S. J. Lee, S. Cho, I. K. Park, K. J. Cho, W. K. Bae and Y. K. Lee, ACS Nano, 2017, 11, 10417–10429 CrossRef CAS PubMed.
- M. Y. Shen, T. I. Liu, T. W. Yu, R. Kv, W. H. Chiang, Y. C. Tsai, H. H. Chen, S. C. Lin and H. C. Chiu, Biomaterials, 2019, 197, 86–100 CrossRef CAS PubMed.
- J. J. Qin, W. Wang, S. Sarkar and R. Zhang, J. Controlled Release, 2016, 237, 101–114 CrossRef CAS PubMed.
- H. D. Lemos, L. D. Prado and H. V. A. Rocha, Braz. J. Pharm. Sci., 2022, 58, e19759 CrossRef.
- M. Wickham, R. Faulks, J. Mann and G. Mandalari, Dissolution Technol., 2012, 19, 15–22 CrossRef CAS.
- M. S. Hedemann, S. Højsgaard and B. B. Jensen, J. Anim. Physiol. Anim. Nutr., 2003, 87, 32–41 CrossRef CAS PubMed.
- V. Sinha and R. Kumria, Pharm. Res., 2001, 18, 557–564 CrossRef CAS PubMed.
- L. Kou, R. Sun, S. Xiao, X. Cui, J. Sun, V. Ganapathy, Q. Yao and R. Chen, Drug Delivery, 2020, 27, 170–179 CrossRef CAS PubMed.
- S. Sivadas, A. K. Mohanty, S. Rajesh, S. K. Muthuvel and H. R. Vasanthi, J. Biomol. Struct. Dyn., 2023, 41, 15124–15136 CrossRef CAS PubMed.
- H. Koepsell, Pflugers Arch., 2020, 472, 1207–1248 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Kang Moo
Huh‡
a,
Kang Moo
Huh‡