
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Free drug and ROS-responsive nanoparticle delivery of synergistic doxorubicin and olaparib combinations to triple negative breast cancer models†
Robert J.
Cavanagh
*a,
Patrícia F.
Monteiro
ad,
Cara
Moloney
 ab,
Alessandra
Travanut
a,
Fatemeh
Mehradnia
a,
Vincenzo
Taresco
c,
Ruman
Rahman
b,
Stewart G.
Martin
b,
Anna M.
Grabowska
b,
Marianne B.
Ashford
d and
Cameron
Alexander
*a
ab,
Alessandra
Travanut
a,
Fatemeh
Mehradnia
a,
Vincenzo
Taresco
c,
Ruman
Rahman
b,
Stewart G.
Martin
b,
Anna M.
Grabowska
b,
Marianne B.
Ashford
d and
Cameron
Alexander
*a
aSchool of Pharmacy, University of Nottingham, NG7 2RD, UK. E-mail: robert.cavanagh1@nottingham.ac.uk; cameron.alexander@nottingham.ac.uk
bSchool of Medicine, BioDiscovery Institute, University of Nottingham, NG7 2RD, UK
cSchool of Chemistry, University of Nottingham, NG7 2RD, UK
dAdvanced Drug Delivery, Pharmaceutical Sciences, R&D, AstraZeneca, Macclesfield, UK
First published on 15th February 2024
Abstract
Combinations of the topoisomerase II inhibitor doxorubicin and the poly (ADP-ribose) polymerase inhibitor olaparib offer potential drug–drug synergy for the treatment of triple negative breast cancers (TNBC). In this study we performed in vitro screening of combinations of these drugs, administered directly or encapsulated within polymer nanoparticles, in both 2D and in 3D spheroid models of breast cancer. A variety of assays were used to evaluate drug potency, and calculations of combination index (CI) values indicated that synergistic effects of drug combinations occurred in a molar-ratio dependent manner. It is suggested that the mechanisms of synergy were related to enhancement of DNA damage as shown by the level of double-strand DNA breaks, and mechanisms of antagonism associated with mitochondrial mediated cell survival, as indicated by reactive oxygen species (ROS) generation. Enhanced drug delivery and potency was observed with nanoparticle formulations, with a greater extent of doxorubicin localised to cell nuclei as evidenced by microscopy, and higher cytotoxicity at the same time points compared to free drugs. Together, the work presented identifies specific combinations of doxorubicin and olaparib which were most effective in a panel of TNBC cell lines, explores the mechanisms by which these combined agents might act, and shows that formulation of these drug combinations into polymeric nanoparticles at specific ratios conserves synergistic action and enhanced potency in vitro compared to the free drugs.
Introduction
Breast cancer is a continuing global health concern, and despite advances in surgery and radiotherapy, there are still requirements for treatment when these options fail.1 Triple negative breast cancer (TNBC) is a subtype of this disease, in which the expression of estrogen (ER) and progesterone receptors (PR) is reduced, and accounts for 15–20% of all breast cancer cases.2,3 In addition, the human epidermal growth factor receptor 2 (HER 2) is not overexpressed as in other breast cancers, making TNBC particularly difficult to treat,4–6 with poor prognoses and low overall survival.7 The 5 years survival of TNBC patients is approximately 70%, compared to 90% in other breast cancer subtypes, and decreases to approximately 10% following disease metastasis.8,9 Chemotherapy is long-established for use in treating breast cancers, but many current drugs have undesirable side-effects, including systemic toxicity and limited efficacy, particularly in TNBC. The therapeutic window of some of these drugs can be improved by the use of delivery vehicles, and liposomal formulations of the topoisomerase II inhibitor, doxorubicin, have been used in patients for over 25 years with clear benefits in terms of quality of life.10Despite these successes, the heterogeneity of many cancers means that single agent chemotherapy is not always effective, and combination therapies are increasingly being used.11 The combination of doxorubicin with the poly (ADP-ribose) polymerase (PARP) inhibitor, olaparib, has shown promising results in preclinical and early stage clinical studies, and recently olaparib has been evaluated in platinum-resistant ovarian cancer in combination with liposomal doxorubicin.12 However, the differing pharmacokinetics of a small molecule free drug, compared to a liposomal carrier, introduces inherent complications in terms of dosing. In addition, the differences in cell entry mechanisms for drugs and nanoparticles13 mean that overexpressed efflux pumps in certain cancer cell types might alter the relative intracellular concentrations of the drugs.14
As a consequence, there are some important potential advantages of encapsulating or conjugating combinations of drug molecules in a single carrier. The concept of co-delivery of drugs by polymer carriers and liposomes has been well-established in vitro15,16 and recently has been extended to efficacy in humans.17 Furthermore, the variety of mechanisms by which some anti-cancer drugs act mean that there can be advantages of co-delivery beyond avoidance of efflux pumps in resistant cell lines.18 These can include directing drugs to specific intracellular targets to induce apoptotic pathways or sensitisation of the cell to other cytotoxic processes such as radiation.19–22 There is accordingly a clear rationale for incorporating combinations of drugs in carriers for many cancers, and for breast cancer the specific combination of doxorubicin and olaparib, in the same nanoparticle. In previous studies it has been shown that TNBC cells can exhibit altered redox environments, with upregulation of intracellular glutathione.23–25 However, it is also the case that reactive oxygen species (ROS) can be present in higher concentrations in TNBC compared to other breast cancer subtypes26 and is believed to play a pro-tumorigenic role by maintaining the oncogenic signalling required for proliferation and survival.27,28 Moreover, increased ROS generation may be induced therapeutically following radiotherapy, or via the action of chemotherapy drugs.29,30 Polymer therapeutic and nanoparticle delivery systems have been designed to exploit abnormal redox environments in cancer cells by both reduction-sensitive31–34 and oxidation-sensitive chemistries.35,36 In this work we have evaluated combinations of doxorubicin and olaparib, delivered via nanoparticle carriers containing an oxidatively-cleavable thioketal group cross-linking the micellar-like cores, and demonstrate that the formulations retain the advantages of specific drug![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :drug ratios in synergistic cytotoxicity in TNBC cell lines.
:drug ratios in synergistic cytotoxicity in TNBC cell lines.
Results
We set out initially to assess any synergistic or antagonistic effects of combinations of doxorubicin and olaparib, and then to encapsulate them in nanoparticulate carriers at ratios shown to be most active against a series of TNBC cell lines.Effects of individual drugs on breast cancer cells
The potencies of doxorubicin (DOX) and olaparib (OLA) were studied in three breast cancer cells (BCC) lines, the luminal subtype line MCF-7 and the triple negative MDA-MB-231 and MDA-MB-468 breast cancer lines. Cells were cultured in standard 2D models and as 3D spheroid models. Measured drug potencies (Table 1) showed that in both 2D and spheroid culture models TNBC lines were relatively less sensitive to DOX than the luminal breast cancer line (MCF-7), with higher DOX IC50 values calculated in the latter. In general, OLA was determined to be approximately two orders of magnitude less cytotoxic than the DNA damaging agent DOX in the BCC lines tested. Comparison of IC50 values between cell cultures models, revealed that 3D spheroids were more resistant to DOX and OLA treatment than their 2D cultured counterparts, in accordance with prior literature;37 DOX demonstrated approximately 6-fold, 18-fold and 29-fold increased resistance in MDA-MB-231, MDA-MB-468 and MCF-7 cells respectively, and OLA 2-fold, 8-fold and 5-fold increased resistance in MDA-MB-231, MDA-MB-468 and MCF-7 cells respectively, when comparing 2D to 3D spheroid culture.| Cell line | 2D cultured cells | 3D cultured cells | ||
|---|---|---|---|---|
| Doxorubicin | Olaparib | Doxorubicin | Olaparib | |
| MDA-MB-231 | 0.97 ± 0.15 μM | 101 ± 13 μM | 5.5 ± 1.0 μM | 217 ± 28 μM |
| MDA-MB-468 | 0.23 ± 0.14 μM | 28 ± 5.0 μM | 4.1 ± 0.7 μM | 213 ± 18 μm |
| MCF-7 | 0.09 ± 0.01 μM | 74 ± 2.1 μM | 2.6 ± 0.8 μM | 343 ± 46 μM |
Identification of synergistic DOX and OLA combinations
Following the assessment of single drug potency, the effects of DOX and OLA combinations were evaluated in the three cell lines over a 48-hour period. Drug combinations were investigated at 5 different molar ratios, and the combination index (CI) values were calculated for each ratio. In accordance with the median-effect algorithm established by Chou and Talalay,38,39 CI values categorise and quantify drug–drug combinations at specific ratios as antagonistic (CI > 1.1), additive (CI = 0.9–1.1) or synergistic (CI < 0.9).The CI results are presented in Fig. 1 as a heat map for each DOX:OLA ratio and demonstrate that drug–drug synergy occurred in a molar-ratio dependent manner. In the TNBC lines (MDA-MB-231 and MDA-MB-468), synergistic efficacy was observed at molar ratios wherein DOX was the dominant drug (100:1 and 10:1) or when applied at equal amounts with OLA (1:1). The highest synergy, as quantified by CI value, was observed in MDA-MB-231 cells at these ratios. Interestingly, only an additive effect was elicited at these molar ratios in the luminal MCF-7 cell line. At higher OLA ratios, antagonist effects were induced, or, in the case of MDA-MB-468 at 1:10 (DOX:OLA), additive effects. It can be noted that stronger antagonistic effects, as denoted by increasing CI values, were induced at the highest OLA ratio 1:100.
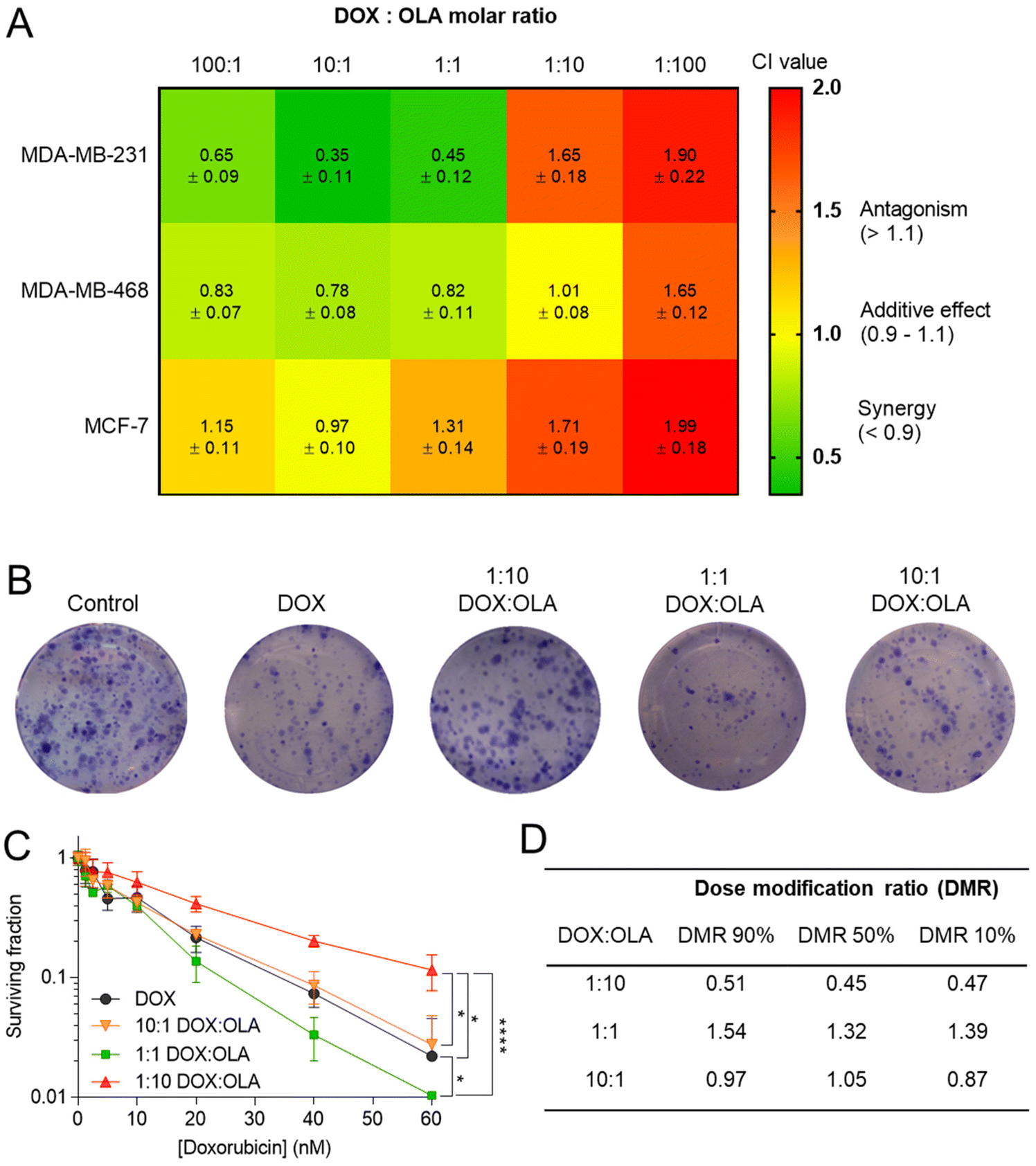 | ||
| Fig. 1 (A) Combination index (CI) values (mean ± S.D) for DOX and OLA at different molar ratios using Tou and Chalay method for quantification of synergy.38,39 Data generated from PrestoBlue metabolic assays following drug combination treatment. (B) Clonogenic survival assay in MDA-MB-231 cells, (C) subsequent dose–response plotting and (D) table of calculated dose modification ratio. Statistical significance tested using two-way ANOVA and Tukey's multiple comparisons tests (*, p < 0.05; ****, p < 0.0001). | ||
These initial observations were made using a metabolic activity-based assay (PrestoBlue™), and thus to derive further mechanistic detail, clonogenic survival assays were undertaken in MDA-MB-231 cells, whereby the capability of a single cell to form a colony was assessed following single or combination drug therapy. Levels of cytotoxicity in cells treated with DOX were approximately three orders of magnitude greater than in cells treated with OLA, with significant inhibition of colony formation observed at concentrations ≥5.0 nM and ≥2.5 μM for DOX and OLA, respectively (Fig. 1 and ESI Fig. 2†). Molar ratios 10:1, 1:1 and 1:10 (DOX:OLA) at DOX concentrations in a range of 0.6–60 nM were then applied and the effects on colony formation measured. Significantly decreased colony survival was observed at molar ratios of 1:1 and significantly increased survival noted at 1:10 when compared to DOX alone treatment. Dose modification ratios (DMR) of DOX:OLA combinations compared to DOX alone indicate an antagonistic effect with 1:10 combination (DMR < 1), synergistic effects with 1:1 (DMR > 1), and additive effects with 10:1 (DMR ≈ 1).
In order to study DOX:OLA combinations in a more physiologically relevant model, 3D spheroids of MCF-7, MDA-MB-231 and MDA-MB-468 were generated. Displayed in Fig. 2 as a heat map, the CI values of DOX:OLA combinations indicated similar trends to those observed in 2D cultured cells. Namely, in MDA-MB-231 and MDA-MB-468 TNBC spheroids, synergy was observed in molar ratios higher in DOX (100:1 and 10:1) or equal drug ratios (1:1), and antagonism induced at higher OLA ratios (1:10 and 1:100). Again, as with 2D models, MCF-7 spheroids only demonstrated additive effects (at 100:1, 10:1 and 1:1) or antagonistic effects (1:10 and 1:100). These observations made in spheroids via ATP level measurement (CellTiter Glo 3D assay), and subsequent IC50 and CI value calculations, were supported by morphological and spheroid volume analysis performed via microscopy (Fig. 2).
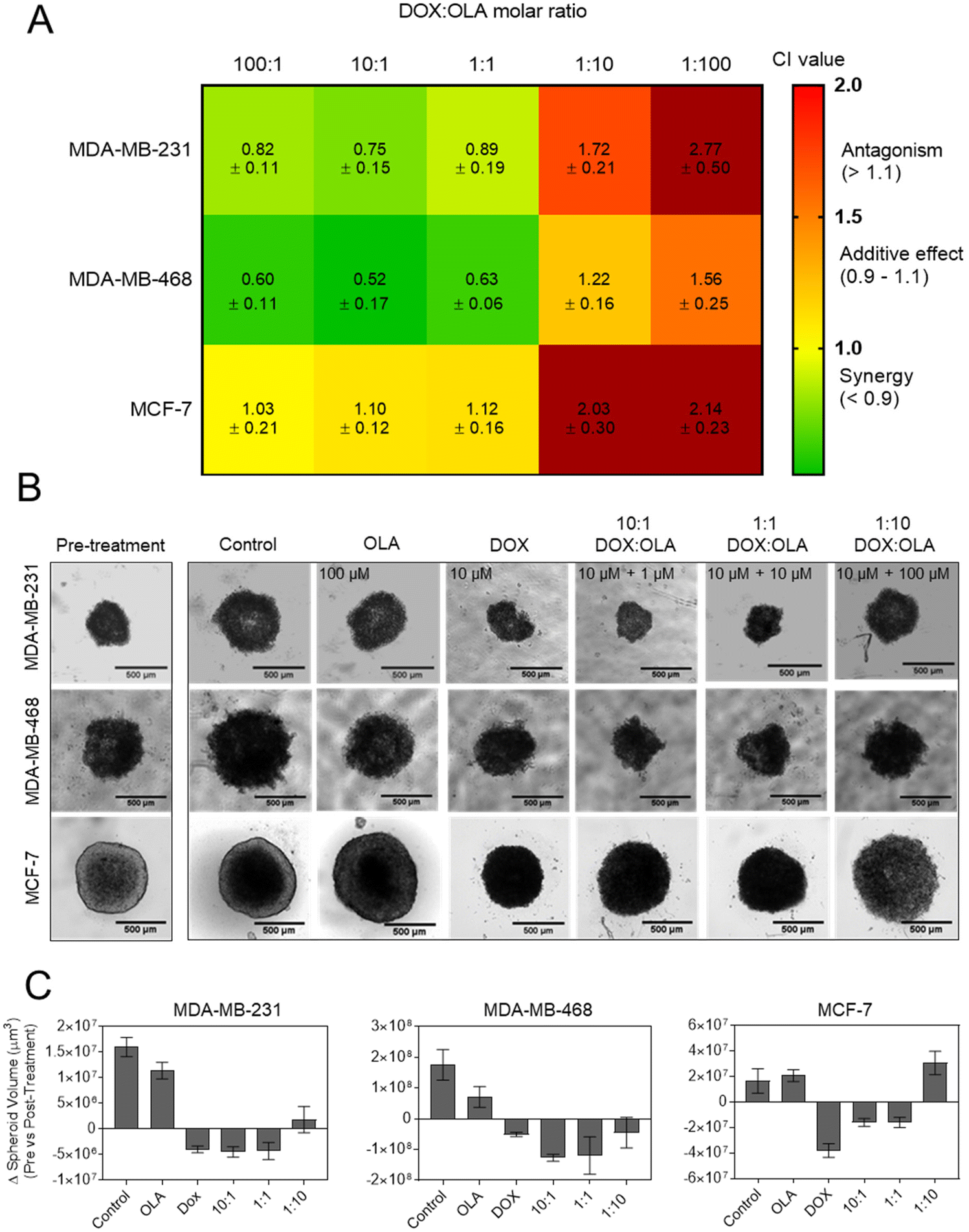 | ||
| Fig. 2 Synergism of DOX:OLA combinations in 3D BCCs spheroid models. (A) CI values generated following treatment and assaying with CellTiter Glo 3D probing cellular ATP levels. (B) Micrographs of spheroid morphologies pre- and post-treatment (72 h exposure) and (C) subsequent spheroid volume analysis. Scale bar is 500 μm and volume change presented as mean ± S.D. | ||
Apoptosis assessment with caspase activation
Following measurements of drug potency via metabolic activities and cell survival, the potential of DOX, OLA and DOX:OLA combinations to induce apoptosis was studied in MDA-MB-231 cells via the measurement of caspase-3/7 activation (Fig. 3). DOX applied at a concentration of 10 nM induced significant caspase-3/7 activation, relative to untreated control. OLA was shown to require concentrations ≥10 μM to induce significant caspase-3/7 activation (ESI Fig. 3†). When solutions of 10 nM DOX were supplemented with either 1 or 10 nM OLA, to represent combination molar ratios of 10:1 and 1:1 (DOX:OLA) respectively, enhanced caspase activation was observed relative to the single drug controls (Fig. 3A and B). However, application of 10 nM DOX in combination with 100 nM OLA (representing 1:10 DOX:OLA ratio) resulted in the abolishment of caspase activation, reducing levels to those comparable to the vehicle control (Fig. 3C). Indeed, the 1:10 (DOX:OLA) combination was observed to required drug concentrations 100-fold higher (1 μM DOX + 10 μM OLA) in order to induce levels of caspase activation that were significantly higher than single DOX treatment (ESI Fig. 3†). Together, the caspase-3/7 activation assays supported the noted molar ratio-dependency of synergy or antagonism with DOX:OLA combinations observed in 2D and 3D cytotoxicity experiments (Fig. 1 and 2).
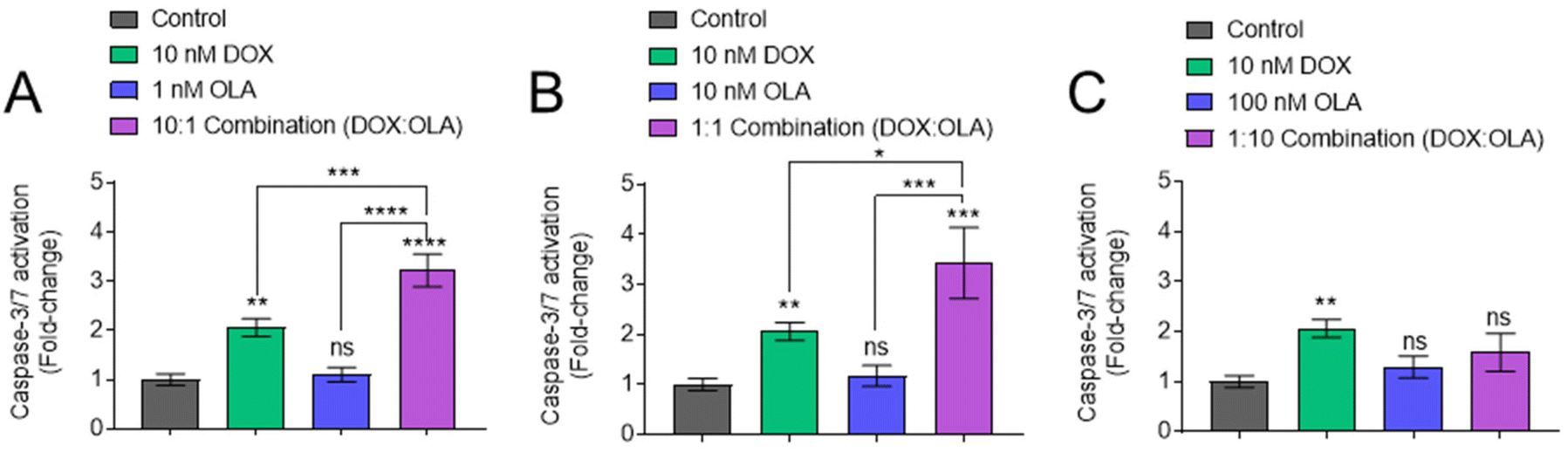 | ||
| Fig. 3 Detection of caspase-3/7 activation. MDA-MB-231 cells cultured in 2D manner and incubated with single, combination or the vehicle control for 24 hours. Levels of caspase-3/7 activation were then probed using CellEvent caspase-3/7 detection reagent. Variations in dosing of OLA compared to DOX are shown in (A), 1 nm OLA, (B) 10 nM OLA, and (C) 100 nm OLA. Values normalised to vehicle control. Statistical significance tested using one-way ANOVA and Dunnett's multiple comparisons tests (*, p < 0.05; **, p < 0.005; ***, p < 0.0005; ****, p < 0.0001). Data presented as mean ± S.D. | ||
Investigation of mechanisms for synergistic drug combination efficacy
To investigate the mechanism of synergy of the DOX and OLA combinations, the level of double stranded DNA breaks (DSBs) was assessed in MDA-MB-231 and MCF-7 cells using the γH2AX assay (Fig. 4). The data demonstrated that 1 μM DOX induced significantly increased levels of γH2AX foci, indicating increased DSBs, compared to the untreated controls for both cell lines. Treatment with OLA generated significant increases in γH2AX foci at concentrations of 1 and 10 μM but not for 0.1 μM OLA in both MDA-MB-231 and MCF-7 cells. In MCF-7 cells, DOX and OLA combinations, tested at molar ratios of 10:1, 1:1 and 1:10, did not elicit any significant increases in γH2AX foci relative to DOX mono-treatment (Fig. 4). Indeed, only comparable γH2AX foci numbers were seen at 10:1 (foci, 38 ± 8) and 1:1 DOX:OLA (foci, 39 ± 13) combinations compared to DOX (foci, 43 ± 15), and a non-significant (P = 0.31), decrease in foci at the 1:10 combination (foci, 32 ± 5). Together with previously calculated CI values (Fig. 1 and 2), the γH2AX assay thus indicated that only additive or antagonist effects were induced by DOX and OLA combinations in MCF-7 luminal breast cancer cells. In MDA-MB-231 cells, significant increases in γH2AX foci were observed with DOX and OLA combinations compared to treatment with DOX alone (foci, 43 ± 7). Notably, a significant increase with 10:1 (foci, 63 ± 11; P = 0.055) and 1:1 (foci, 89 ± 11; P = 0.004) DOX:OLA combinations, combinations that were previously determined synergistic via CI calculations (Fig. 1 and 2). The 1:10 DOX:OLA combination, in support with antagonistic CI values, induced significantly decreased γH2AX foci (foci, 23 ± 3; P = 0.011) relative to the DOX mono-treatment control.
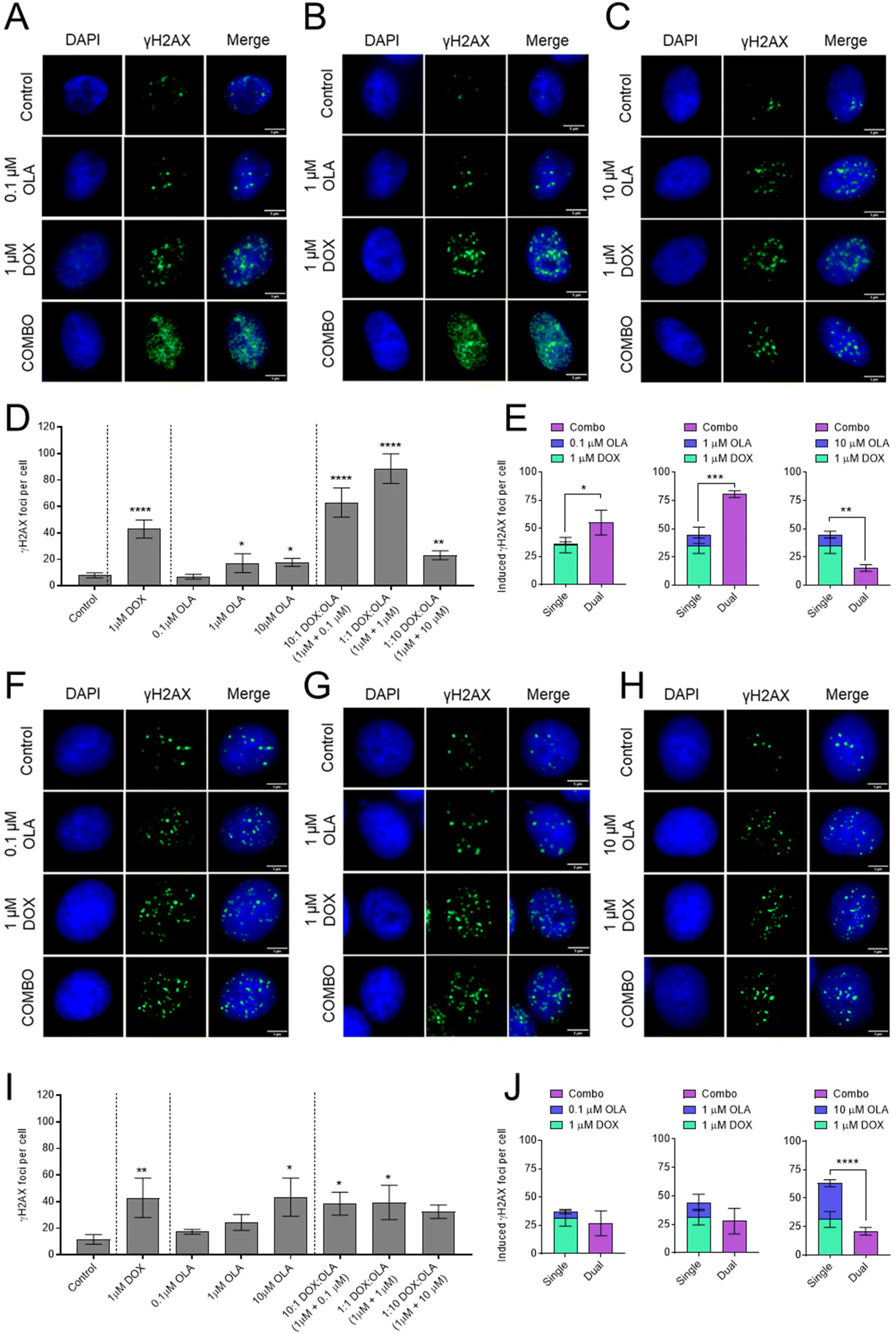 | ||
| Fig. 4 Immunofluorescence microscopy of γH2AX foci as a marker of dsDNA breaks in (A–E) MDA-MB-231 and (F–J) MCF-7cells. Cells exposed to single dosed DOX (1 μM), OLA (0.1–10 μM) and DOX:OLA combinations for 24 hours. DOX concentration was kept constant at 1 μM and OLA concentration ranged from (A and F) 0.1 μM, (B and G) 1 μM, and (C and H) 10 μM as either single drug or in combination with DOX, to represent DOX:OLA combinations of 10:1, 1:1 and 1:10, respectively. Scale bar, 5 μm. (D and I) Foci quantification was performed using ImageJ and data presented in bar charts (mean ± S.D). (E and J) Induced foci per cell data presented for response additivity analysis. Statistical significance tested using one-way ANOVA and Dunnett's multiple comparisons tests to compare (D and I) treatments to control and, (E and J) summation of single treatments to combination treatments (*, p < 0.05; **, p < 0.005; ***, p < 0.0005; ****, p < 0.0001). | ||
Investigation of redox species induction via combination drug treatments
To explore the potential mitochondrial or redox mechanisms of DOX and OLA combinations, drug induced ROS generation was studied in MDA-MB-231 TNBC cells (Fig. 5). DOX has been previously found to increase intracellular ROS generation via redox cycling and the Fenton reaction.40 In MDA-MB-231 cells it can be observed that significant increases in ROS, as determined by the CM-H2DCFA assay, were induced, in an approximately linear time-dependent manner, within the first 60 minutes of exposure to 1 μM DOX (Fig. 5A). On the other hand, OLA was observed to induce a transient ROS spike evident at 15 minutes of exposure which subsided to basal levels by 30 minutes. The ROS spike appeared to be concentration dependent, with the largest magnitude observed to approx. 2-Fold basal levels with 10 μM OLA, 1.5-fold with 1 μM OLA, and a slight, but significant increase to 1.2-fold with 0.1 μM OLA (Fig. 5A). Following the OLA-induced ROS burst at 15 minutes, no further significant increase in ROS was observed with OLA within a 60-minute time frame. For drug combination treatments, intracellular ROS profiles demonstrated aspects of both DOX and OLA in an exposure time-dependent manner. Notably, for 1 μM DOX with 1 μM OLA or 10 μM OLA (1:1 and 1:10 molar ratios, respectively), the transient ROS burst at 15 minutes was experienced in a comparable manner to OLA alone treatment. ROS levels subsequently returned to basal levels at 30 minutes, as observed with OLA treatment, followed by an increase in ROS levels observed at 60 minutes. This latter ROS increase was likely resulting from the effects of DOX, which in DOX-only treatments increased ROS at this time point. In DOX:OLA combinations, this DOX-associated ROS generation at 60 minutes appeared significantly lower in DOX combinations with 1 μM OLA or 10 μM OLA than in DOX alone treatment. Together, the data thus indicated that OLA might suppress DOX-mediated ROS generation when applied at these concentrations.
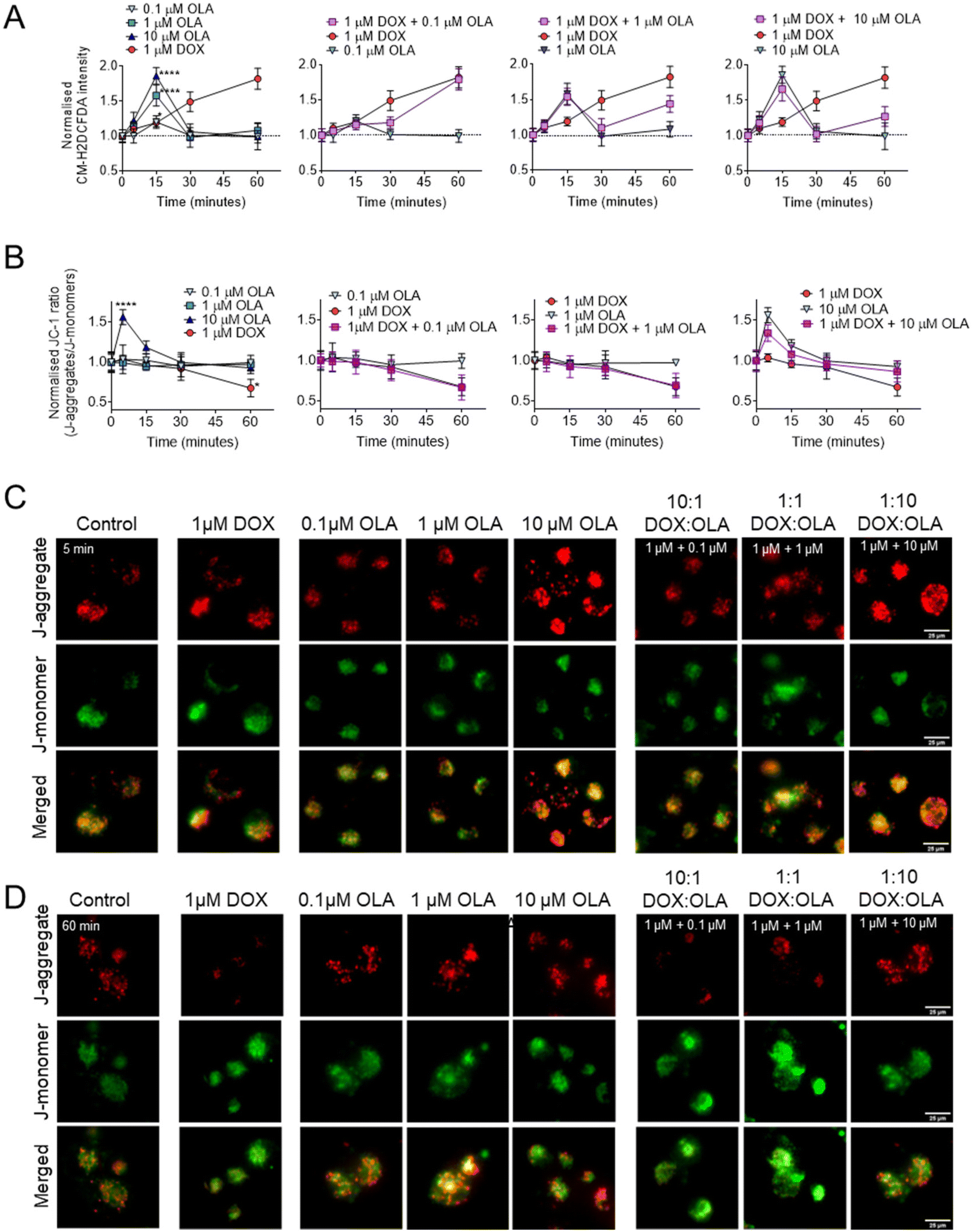 | ||
| Fig. 5 Mitochondrial effects of single drugs and drug combinations in MDA-MB-231 cells. (A) Intracellular ROS generation assessed with CM-H2DCFA. (B) Mitochondrial membrane potential measured using JC-1 assay and accompanying JC-1 images captured at (C) 5 minutes and (D) 60 minutes of exposure. Data presented as mean ± S.D. Scale bar is 25 μm. | ||
To investigate further, mitochondrial membrane potentials (ΔΨm) were studied using a JC-1 assay (Fig. 5B and C). When applied as mono-treatment, 1 μM DOX was observed to induce significant ΔΨm depolarisation at 60 minutes of drug exposure. In addition to DOX-mediated DNA damage, overproduction of ROS is known to have detrimental effects on mitochondrial processes such as the electron transport chain and mitochondrial membrane permeability.40 Thus, the DOX-mediated ΔΨm depolarisation was likely a consequence of the observed DOX-associated ROS generation (Fig. 5A). Application of 0.1 μM and 1 μM OLA was not observed to induce any alterations in ΔΨm. However, 10 μM OLA was noted to elicit ΔΨm hyperpolarisation in a transient manner, peaking at 5 minutes, with a decreased but still significant hyperpolarisation at 10 minutes and a return to baseline levels at 15 minutes of exposure. Interestingly, mitochondrial hyperpolarisation has been reported before with PARP inhibition and associated with conferring mitochondrial protection and cell survival.41 In the current work, ΔΨm hyperpolarisation appeared to precede the OLA-mediated ROS burst, suggesting both events may be associated, with ΔΨm hyperpolarisation potentially driving increased mitochondrial ROS generation, an event that has been reported elsewhere, albeit under different conditions.42
In combinations of DOX and OLA, no significant differences were observed from the DOX alone treatment. A gradual ΔΨm depolarisation was found when either 0.1 μM or 1 μM OLA were applied alongside 1 μM DOX (Fig. 5B). However, application of 10 μM OLA with 1 μM DOX, resulted in ΔΨm hyperpolarisation at 5 minutes. This effect was similar, albeit reduced, relative to treatment with OLA alone, and was followed by an apparent ΔΨm depolarisation at 60 minutes. This suggested that 10 μM OLA may have conferred some protection against the ΔΨm depolarisation mediated by DOX at 60 minutes, an effect likely mediated by the reduced ROS generation noted with this combination at 60 minutes. Taken together, these data suggested that antagonism, observed in combinations where DOX < OLA (i.e. 1:10, 1:100), may have resulted from OLA-mediated mitochondrial protection. This phenomenon we associate with ΔΨm hyperpolarisation, a consequent ROS spike, and ultimately a suppression of DOX-mediated ROS generation.
Nanoparticle formulation and uptake in BCCs
It has been suggested that some combination therapies may be enhanced by co-delivery of the therapeutic agents by a single carrier, so that variations in the individual pharmacokinetic profiles of the drugs can be avoided and simultaneous intracellular dosing achieved by the delivery agent. Thus, to investigate any synergies of drug combinations delivered intracellularly, we chose to prepare polymeric nanoparticles as delivery vehicles for the drug combinations. For the architectures and chemistries of the carriers, we selected micellar-like nanoparticles, employing a poly(ethyleneglycol) component to provide colloidal stability and reduce protein adsorption, and a mixed poly(D,L-lactide-co-2-azidocaprolactone) block with the pendant azide groups installed (Fig. 6) to enable facile core-cross-linking via click chemistries.43 The synthetic strategy was adapted from that which we used for reduction-responsive polymers for docetaxel delivery in TNBC,44 but in this case we prepared an oxidatively-sensitive dithioketal45 between the cross-link points to replace the disulfide linker we employed previously. The oxidation-cleavable cross-linker (DTK, Fig. 6) was chosen with a view to potential subsequent application in radiation-treated cells, as first-line interventions for breast cancer usually involve radiotherapy.30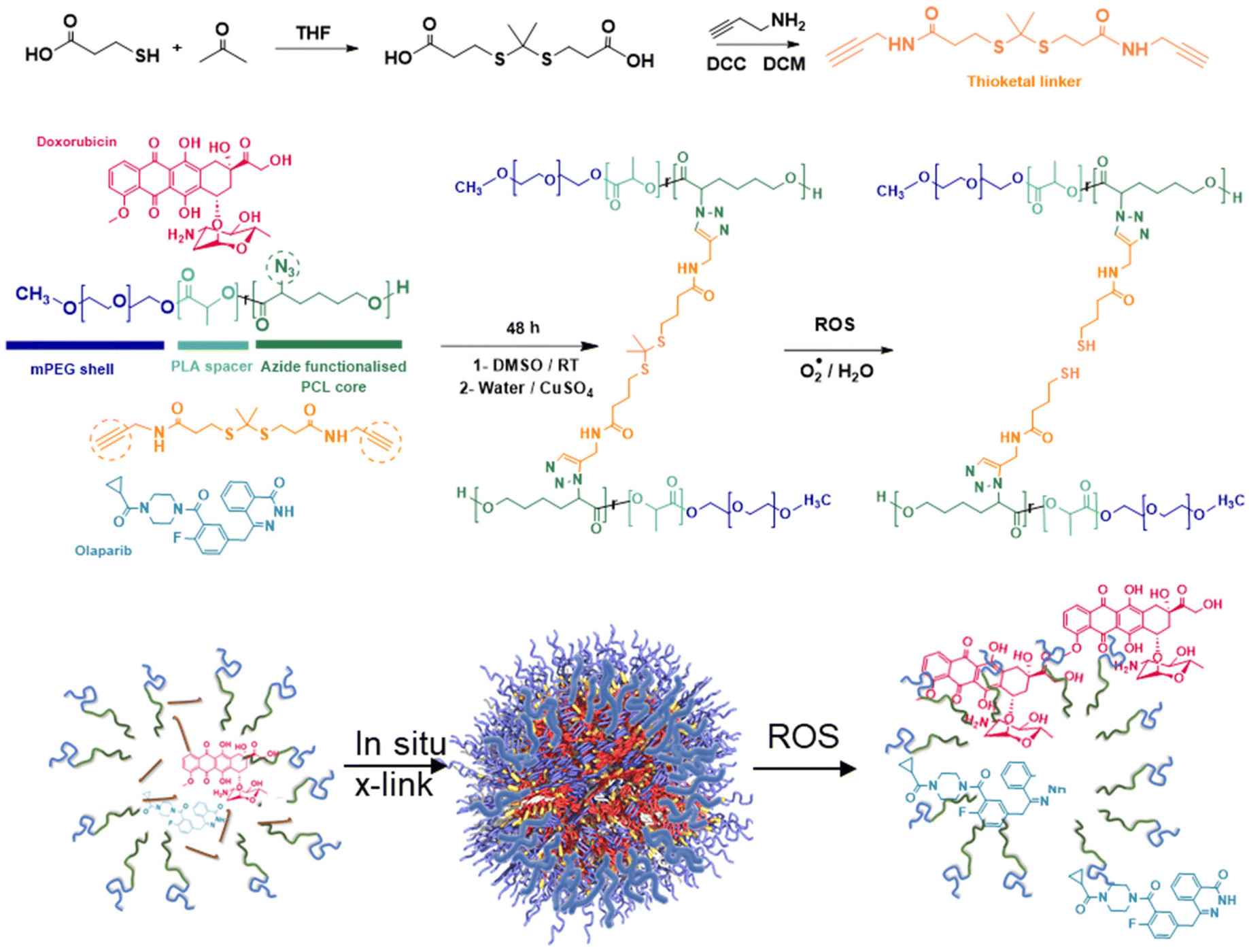 | ||
| Fig. 6 Synthetic scheme for the preparation of ROS-responsive nanoparticles. A bis(alkynyl) thioketal cross-linker is reacted with mPEG-poly(lactide-co-poly(azido-caprolactone)) in the presence of DOX and OLA to form drug-loaded nanoparticles designed to release payload in the presence of reactive oxygen species (ROS). | ||
In addition, to enable intracellular tracking of the NPs via microscopy, Cy5-tagged NPs were prepared via reaction of ∼1% of the azide groups on the polymeric back bone with alkyne-functional Cy5 fluorophore. ‘Core’ cross-linking of the polymers was carried out in a similar way, in this case by reacting the azide-groups with 3,3′-(propane-2,2-diylbis(sulfanediyl))bis(N-(prop-2-yn-1-yl)propanamide) pre-mixed with/without DOX and OLA in DMSO, with a solution of ascorbic acid followed by copper sulphate in water under a nitrogen atmosphere. Extensive extraction with EDTA solutions was used to remove copper ions which might otherwise be toxic to the cells. The resultant cross-linked polymer nanoparticles were sufficiently colloidally stable that they could be freeze-dried, and when resuspended in water, exhibited diameters of 120–130 nm (Table 2). By varying amounts of drugs in the ‘feed’ solutions in DMSO, it was possible to encapsulate combinations of DOX:OLA at various ratios. However due to the variability associated with encapsulating two drugs with distinct physiochemical properties, formulating the exact ratios used in initial studies (10:1, 1:1, 1:10) was technically challenging. The exact determined DOX:OLA ratios in the prepared NPs were 13:1 (as the closest ratio to 10:1), 1:1.2 (≈1:1) and 1:15 (as the closest to the desired 1:10). Previous in vitro CI synergy assessments (Fig. 1 and 2) demonstrated comparable synergistic effects at 100:1 vs. 10:1 and comparable antagonistic effects at 1:10 vs. 1:100, thus a degree of variation from the exact desired ratios was deemed acceptable. For simplicity these are hereby referred to as their approximate, rounded ratios, however it should be noted that all in vitro studies use for control comparison unformulated counterparts at their ‘exact’ ratio (13:1, 1:1.2 and 1:15).
| Formulation | Drug combination | Particle size, nm | Polydispersity index |
|---|---|---|---|
| mPEG-b-PLA-PCL-DTK 1 | — | 129 | 0.05 |
| mPEG-b-PLA-PCL-DTK 2 | DOX:OLA 13:1 (≈10:1) |
124 | 0.27 |
| mPEG-b-PLA-PCL-DTK 3 | DOX:OLA 1:1.2 (≈1:1) |
119 | 0.06 |
| mPEG-b-PLA-PCL-DTK 4 | DOX:OLA 1:15 (≈1:10) |
111 | 0.10 |
In the absence of oxidative conditions, the polymer NPs were stable for up to 24 h under the conditions used for cell culture, and did not markedly alter in size or polydispersity when stored at 4 °C (Fig. S4†), but incubation of the polymer NPs with hydrogen peroxide solutions resulted in the rapid loss of discrete particles as evidenced by poor correlation functions in DLS, indicating particle breakdown via the oxidising species (Fig. S5†). We chose a hydrogen peroxide solution at much higher concentrations (50 μM) than the reactive oxygen species (ROS) present in normal tissue, but which we reasoned might occur transiently in localised areas of high oxidative stress in cancer cells. Indeed, measurement of ROS levels in breast cancer cell lines using the general oxidative stress indicator probe CM-H2DCFDA indicated raised levels of oxidative species in the cancer cell lines (Fig. S6†). Incubation of cross-linked and non cross-linked polymer NPs, and those prepared with a non-degradable cross-linker,44 indicated no loss of metabolic activity in MCF-7, MDA-MB-231 and MDA-MB-468 cells, and no evidence of membrane disruption in the absence of encapsulated drugs (Fig. S7†). These experiments suggested that any effects observed in TNBC cells treated with polymer NPs containing drugs would not be due to activities of the carriers, but only the drugs.
The internalisation of NPs was studied in breast cancer cells (Fig. 7). For these initial studies we prepared non-drug loaded Cy5 labelled NPs (Cy5-NPs) by mixing a labelled mPEG-PLA-PCL polymer, in which a proportion of the azide groups were reacted with Cy5 alkyne, with the mPEG-PLA-PCL-azide prior to cross-linking with the dithioketal. Particle sizing for these NPs was problematic due to absorbance by the Cy5 label close to the laser wavelength of the light scattering instrumentation, but we reasoned that the sizes and charges would be representative of the ‘parent’ cross-linked polymers as the preparation methods were largely the same.46 Fluorescence microscopy images demonstrated the uptake and intracellular accumulation of the Cy5-NPs in MCF-7, MDA-MB-231 and MDA-MB-468 cells (Fig. 7A). Image analysis and Cy5-NPs signal quantification revealed that NP internalisation was time-dependent and comparable between cell lines at 0.5 h (Fig. 7B). However, it was found that at 1 h and 2 h, NPs uptake was approximately 2-fold higher in MCF-7 relative to MDA-MB-231 and MDA-MB-468 lines, with the latter two TNBC lines exhibiting similar levels of internalised NPs. Furthermore, the use of Lysotracker dyes enabled the extent of co-localisation of the NPs with active lysosomal compartments to be measured at the 2 h timepoint (Fig. 7C). The Pearson correlation coefficient (PCC) values for NP-Cy5 and Lysotracker labels in MDA-MB-468 and MDA-MB-231 cells at 2 h were 0.58 ± 0.07 and 0.64 ± 0.10, respectively, indicating that a proportion of the NPs were external to lysosomal compartments by this time.
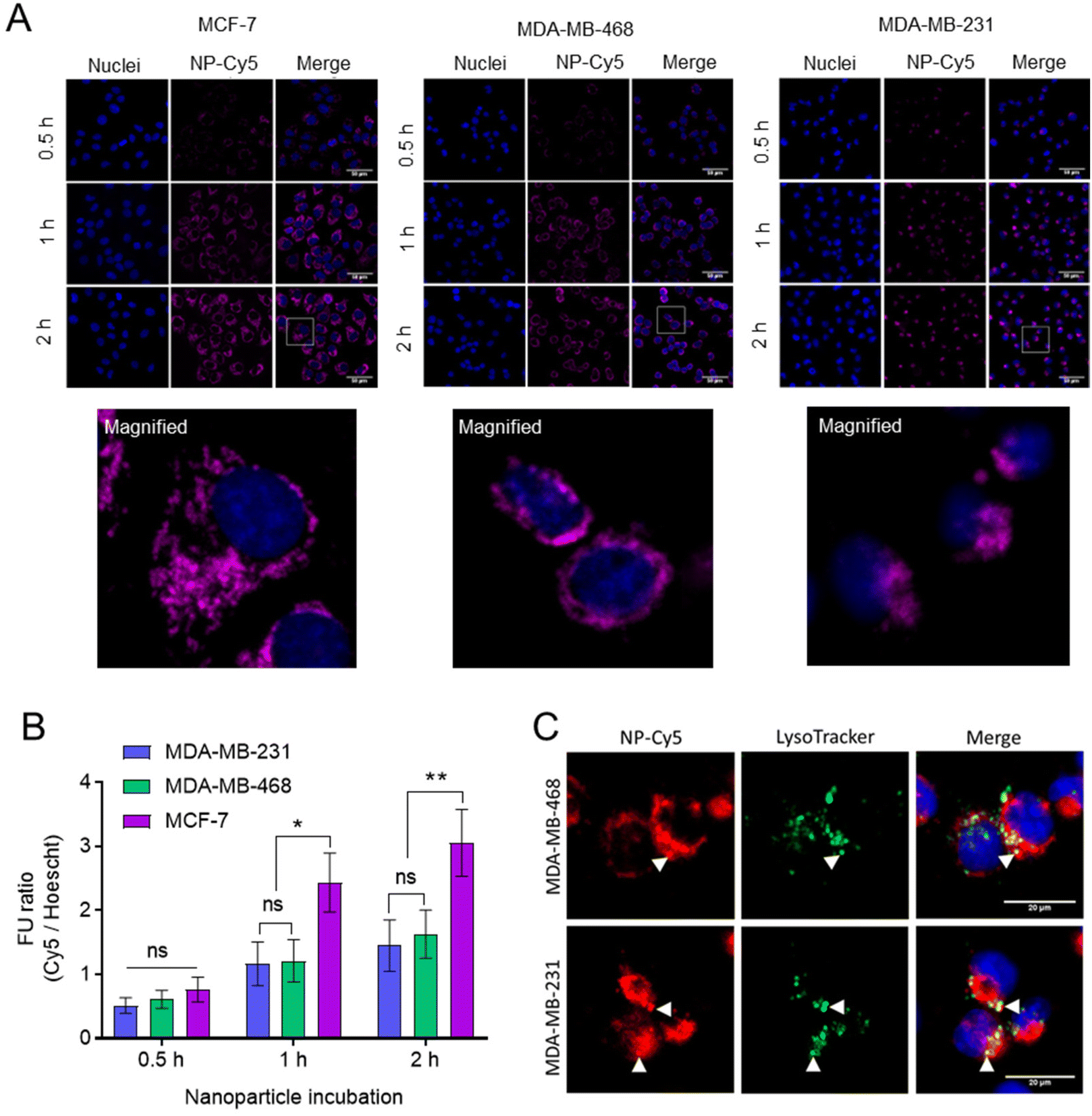 | ||
| Fig. 7 NP-Cy5 uptake characteristics. (A) NP uptake in BCCs following 0.5, 1 and 2 hours incubation. Scale bar is 50 μm. Magnified sections highlighted with white box are shown underneath. (B) Quantification of NP-Cy5 uptake images. Fluorescent unit (FU) ratio calculated via Cy5 signal normalisation to Hoechst nuclear signal. Data presented as mean ± S.D. (C) Lysosomal accumulation of NPs following 2 hours incubation. Staining of lysosomes performed with Lysotracker Green. Scale bar is 20 μm. White triangles indicate co-localisation of NP and lysosome signal. Statistical significance between cell lines tested using one-way ANOVA and Dunnett's multiple comparisons tests (*, p < 0.05; **, p < 0.005). | ||
Following confirmation that Cy5-NPs were readily internalised, the uptake of drug combination loaded NPs were studied. Fluorescent cell imaging was used to demonstrate that the drug-loaded NPs (mPEG-b-PLA-PCL-DTK 3 formulation; Table 1) were endocytosed and by monitoring the fluorescence of DOX it was possible to show the accumulation of DOX in breast cancer cell nuclei when administered either as free drug or in a polymer NP formulation (Fig. 8).
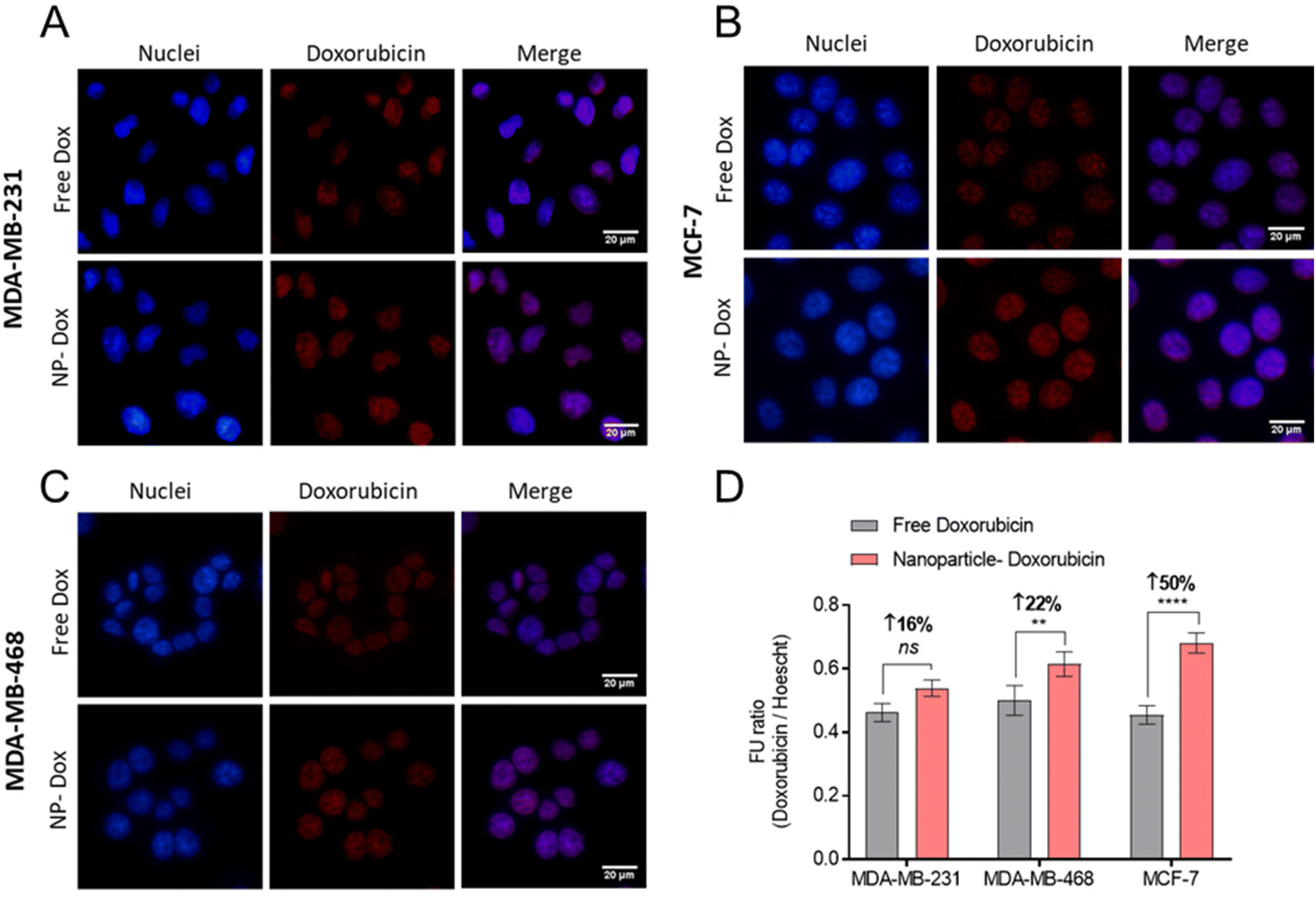 | ||
| Fig. 8 Doxorubicin delivery in BCCs using NPs. (A–C) Fluorescent micrographs of free doxorubicin and NP-doxorubicin. Doxorubicin treatments applied at 10 μM for 4 hours, followed by counter-staining with DAPI nuclear stain (blue), and PFA-fixation. Scale bars are 20 μM. (D) Quantification of intracellular DOX via fluorescence imaging ratioed across doxorubicin and DAPI signals. The NP-Dox formulation studied is mPEG-b-PLA-PCL-DTK 3 (Table 1). | ||
Image analysis and DOX signal quantification indicated increased DOX delivery via NPs to MCF-7 and MDA-MB-468 cell lines relative to free DOX (Fig. 8D). Moreover, the NPs’ enhancement of DOX delivery was higher in MCF-7 cells compared to MDA-MB-231 and MDA-MB-468, an observation likely linked to the overall increased Cy5-NPs uptake noted in MCF-7 cells (Fig. 7) and the increased basal ROS levels in this cell line relative to TNBC lines which may facilitate increased DOX release from NPs (Fig. S6†). These differences in DOX accumulation may also have been due to the different entry mechanisms for the free drug, via organic cation transporters,47,48 and the polymer carriers which were likely internalised through a range of clathrin-dependent and clathrin-independent pathways.49
The effects of DOX:OLA combination NPs were then evaluated in triple negative breast cancer spheroids (Fig. 9 and Table 3). The data demonstrated that increased potency was observed with NPs delivery of the DOX:OLA combinations relative to the unformulated combinations at the corresponding molar ratios. It can be noted that the enhanced potency with NPs was most prominent with combination molar ratios of 10:1 (DOX:OLA), with a 2.3 and 2.1-fold increase observed in MDA-MB-468 and MDA-MB-231 cells, respectively (Table 3). NPs containing a DOX:OLA ratio of 1:1 induced 1.9 and 1.7-fold increases in potency relative to unformulated combinations in MDA-MB-468 and MDA-MB-231 cells, respectively. No substantial change in potency was noted between NP formulations and unformulated control combinations with the previously determined antagonistic ratio of 1:10 in both cell lines tested.
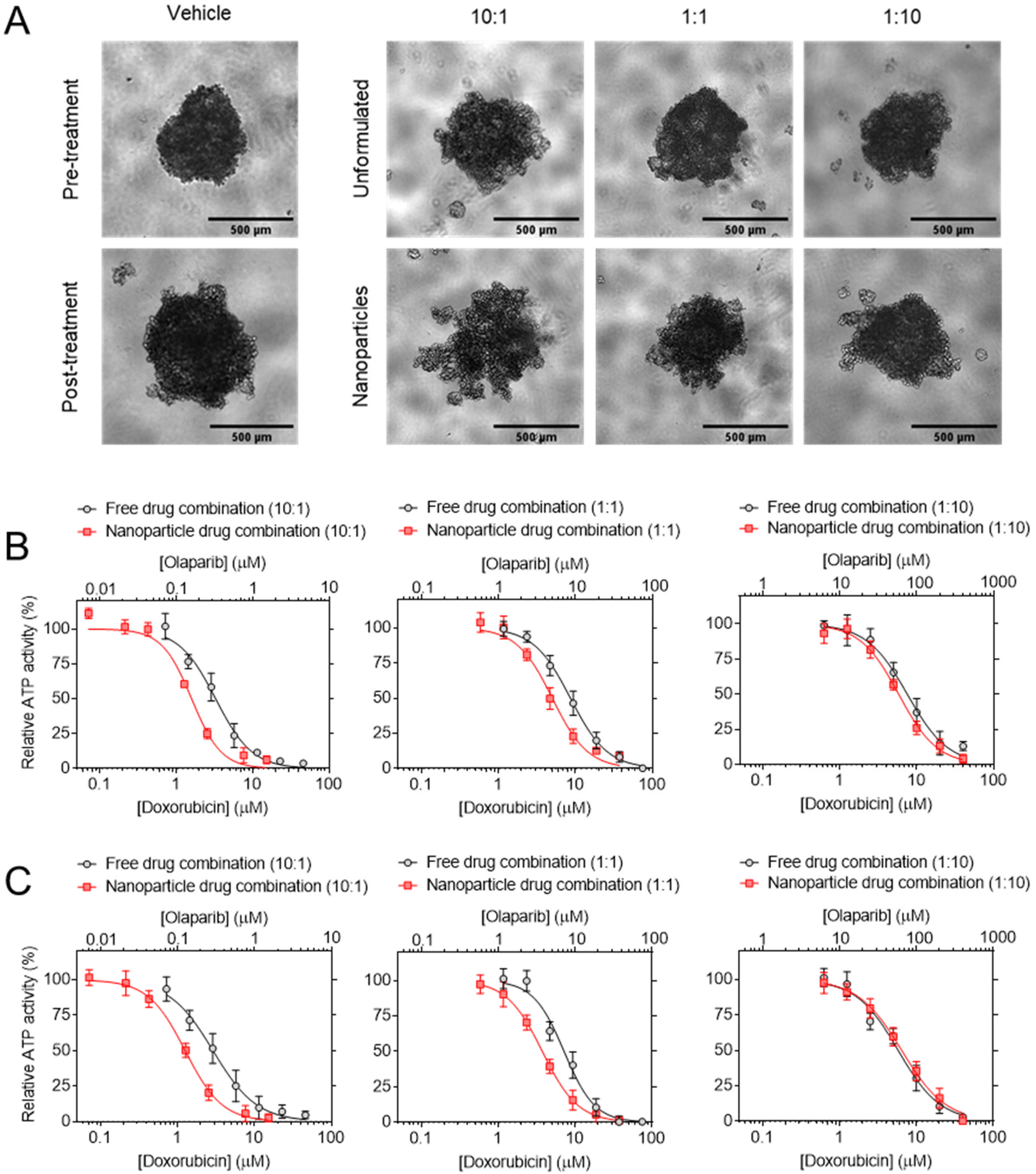 | ||
| Fig. 9 Cytotoxicity testing of combination NPs. (A) Potency of 10:1, 1:1 and 1:10 DOX:OLA combination NPs in (A) MDA-MB-231 and (B) MDA-MB-468 spheroids. (C) MDA-MB-231 spheroid imaging, scale bar is 500 μm. NPs are compared to their unformulated counterparts based on DOX:OLA molar ratio and calculated IC50 values (μM) are displayed in Table 3. Treatments applied for 72 h and assessed for cellular ATP levels using CellTiter Glo 3D assay. Values presented as mean ± S.D and come from three independent experiments. | ||
| Control | Nanoparticles | ||
|---|---|---|---|
| IC50 DOX (OLA) | IC50 DOX (OLA) | Fold-increase | |
| MDA-MB-231 | |||
| 10:1 |
3.28 ± 0.24 (0.25 ± 0.02) | 1.59 ± 0.16 (0.12 ± 0.02) | 2.1 |
| 1:1 |
8.63 ± 0.25 (10.3 ± 0.25) | 5.02 ± 0.42 (6.02 ± 0.42) | 1.7 |
| 1:10 |
7.55 ± 0.14 (113 ± 1.43) | 5.84 ± 0.27 (87.6 ± 2.71) | 1.3 |
| MDA-MB-468 | |||
| 10:1 |
2.91 ± 0.15 (0.22 ± 0.02) | 1.26 ± 0.12 (0.10 ± 0.01) | 2.3 |
| 1:1 |
7.20 ± 0.54 (8.64 ± 0.54) | 3.76 ± 0.03 (4.51 ± 0.03) | 1.9 |
| 1:10 |
5.80 ± 0.50 (87.0 ± 5.04) | 6.45 ± 0.29 (96.8 ± 2.91) | 0.9 |
Discussion
Molar ratio-dependent effects
The data gathered here demonstrates that combinations of DOX and OLA can generate synergistic or antagonistic effects in a manner dependent on the molar ratio applied. Thus, for excess DOX (100:1, 10:1) or equal (1:1) ratios, there were synergistic effects in terms of cytotoxic activity, whereas for excess OLA ratios (1:10, 1:100) there were antagonistic effects. Similar molar ratio dependent activity with these drugs has been observed previously in ovarian cell lines.50 These observations were consistent across both 2D and 3D in vitro models and with the range of assays conducted (Fig. 1 and 2, respectively). In 2D models, metabolic activity testing, used for IC50 and CI value calculation, was supported with colony survival studies, the latter testing effects on cell proliferation and colony formation over a series of cell generations. Similarly, in 3D spheroid models, IC50 and subsequent CI value determination assessed with ATP measurement (CellTiter Glo assay), correlated with spheroid morphology and size changes following drug treatment. Moreover, varying molar ratios of the drugs in combination were studied, as combination synergy and effects are known to be dependent on molar ratios in addition to distinct drug mechanism of actions.50,51
Luminal vs. triple negative breast cancer
Single treatment with OLA induced DNA damage and cell killing in both MDA-MB-231 and MCF-7 cells (Table 1 and Fig. 4); interestingly, however, the synergistic effects with DOX, including increased cell death and enhanced DNA damage, were manifest only in triple negative cell lines, and not in the luminal MCF-7 breast cancer cell line (Fig. 4). Further work is required to establish the reasons for this difference; however, it may be hypothesised that the DNA damage response associated with DOX treatments, even though this is not a primary mode of action for DOX, may differ between TNBC and luminal breast cancer, thus resulting in differential sensitisation of DOX by OLA treatment. Moreover, homologous recombination (HR) alterations or deficiencies in TNBC cells52–54 may further limit available repair mechanisms following DOX insult when combined with PARP inhibition (via OLA) and play a role in the synergy observed here. Indeed, several clinical trials are investigating PARP inhibition with OLA to induce synthetic lethality in HR deficient TNBC.55–58 A potential further explanation of differential DOX/OLA synergy across TNBC and luminal breast cancer cells may be centred around distinct regulation of the transcription repressor Snail, a protein associated with cancer cell survival and EMT. Treatment of MDA-MB-231 cells with DOX in combination with a PARP-inhibitor resulted in an increase in apoptosis compared to cells treated with DOX alone at the same dose. DOX induced significant increases in Snail expression in several breast cancer cell lines but inhibition of PARP-1 with the investigational compound ABT-888 reduced DOX-induced Snail expression, leading to subsequent increases in apoptotic cell death in these cases.59 In addition, several reports have demonstrated that cancer cells undergoing epithelial–mesenchymal transition (EMT) exhibit enhanced multidrug resistance.60–63 In ERα-positive cells, such as MCF-7 cells, resistance to DOX has been reported via DOX-induced upregulation of EMT-associated transcription factors, a mechanism shown to be absent in ERα-negative mesenchymal-like cells (MDA-MB-231 and MDA-MB-468).64 DOX treatment is also reported to induce expression of differing EMT-related proteins (E-cadherin, N-cadherin) in ER positive (MCF-7) and negative (MDA-MB-231, MDA-MB-468) cells, while OLA and other PARP inhibitors have been reported to reduce EMT.65 It is thus possible that the observed synergistic effects seen in our studies with DOX and OLA arose from a combination of enhanced apoptosis and reduced chemoresistance at specific dose ratios. However, antagonistic effects of the drug combinations, observed with cell viability assays (Fig. 1 and 2) were apparent also in MCF-7 cells. This suggests that the OLA-mediated mitochondrial effects noted in triple negative MDA-MB-231 cells (Fig. 5) may also be occurring in MCF-7 cells, although further assays would be required to validate this hypothesis.Mechanisms of synergistic anti-cancer activity
Induction of DNA double-strand breaks was assessed using immunostaining for the γH2AX foci.66 In the TNBC line, MDA-MB-231, significantly more γH2AX foci were observed with the synergistic combination ratios of 10:1 and 1:1 (DOX:OLA) compared to either drug as monotherapy (Fig. 4). At the antagonistic 1:10 (DOX:OLA) ratio, significantly decreased γH2AX foci were noted relative to the DOX monotherapy. Furthermore, at this antagonistic ratio foci numbers were not significantly different relative to those induced by OLA at the corresponding combination concentration (10 μM). Together, these observations suggest that the DNA damaging effects of DOX can be enhanced via OLA-mediated inhibition of PARP associated DNA repair and subsequently increase apoptosis and cell death. DOX is known to intercalate with DNA and inhibit topoisomerase II, in turn preventing DNA unwinding during transcription to halt DNA replication.67 In addition to topoisomerase II poisoning via trapping topoisomerase II at cleavage sites, DOX has been suggested to induce anticancer activity through inducing DNA strand breakage, either directly through DNA torsion resulting from intercalation68 or indirectly via the generation of ROS and subsequent oxidative stress.69 We have observed these mechanisms of DNA damage and generation of ROS in the current study (Fig. 4 and 5, respectively) and surmise that they may be responsible for the demonstrated synergy with the PARP1 inhibitor OLA. PARP1 is involved in various DNA damage responses70 and during the DNA repair process OLA inhibits PARP activity through trapping the enzyme at damaged DNA sites, subsequently preventing repair and additionally inducing DNA lesions through collision and collapse of replication forks.71
The use of high OLA concentrations appears however to abrogate DOX-induced DNA damage and this phenomenon may be driven in some part via mitochondrial-associated processes whereby DOX-induced ROS is reduced and mitochondrial membrane potential protected (Fig. 5). The current work investigated simultaneous application of drugs, reflecting how co-encapsulated drugs would be delivery via nanoparticles, however it is hypothesised synergy may be improved with a regime involving OLA pre-treatment prior to DOX in order to achieve pre-sensitisation.72
Nanoparticle uptake and potency
The increased potency of the NPs relative to unformulated combinations may be a result of increased drug delivery, as observed in Fig. 8. This observation of increased drug uptake was based on quantification of DOX, owing to its inherent fluorescence, and the enhanced delivery of this drug alone may have been responsible for the increased potency observed. DOX is a small molecule and can be rapidly transported into cells and enter its target site of action (nuclei) by organic cation transporters and also passive diffusion. Dox-loaded NPs rely however on endocytosis for cell internalisation and subsequence drug release prior to entry into the nuclei. Uptake of DOX into the cell nuclei is critical due to the mechanism of action for this molecule which involves intercalation with DNA to induce cell death. Previous reports indicate that this can be hindered by incomplete DOX release from particles, and this is typically associated with DOX-conjugates49,73 and nonresponsive NPs with high stability in the cellular environment.73–76 On the other hand, responsive polymeric materials for NP delivery of DOX have been shown to generate increased nuclear DOX delivery.75,76 Our studies using Cy5-NPs demonstrate rapid NP cellular uptake within 1 h and presence of the cytoplasmic Cy5-NP signal (Fig. 7). Moreover, this is coupled with DOX-NPs at 2 h showing increased DOX delivery relative to the free drug and a nuclear DOX localisation (Fig. 8). Thus, in the current work the use of oxidative-responsive materials to aid drug release alongside the rapidly internalised NPs are together likely responsible for the increased DOX delivery observed.A number of studies have demonstrated that the use of carriers for anti-cancer drugs can improve efficacy in vitro and/or therapeutic window in vivo compared to the free drugs, attributable to different internalisation mechanisms in vitro and also altered pharmacokinetic parameters in vivo.77–80 Although the full details of the endocytic mechanisms for the NPs in this study are not yet known, polymers of similar composition and size were internalised by both clathrin-and caveolin-mediated pathways in a range of breast cancer cells,49 and thus it is likely that these polymers entered by similar routes.
However, it can be hypothesised that OLA delivery is also enhanced with NP formulation, as OLA is subjected to similar drug resistance efflux pumps as DOX, which may be overcome via NP-mediated endocytosis.81 Interestingly, NP formulation of a 1:10 DOX:OLA combination resulted in a non-significant effect on potency. The reasons for this are unclear but may be related to the antagonistic effects (mitochondrial protection; Fig. 5) of high OLA concentrations. The theorised increased OLA delivery, and thus increased OLA intracellular accumulation with NP formulation, may be subsequently enhancing mitochondrial-mediated cell survival and protection from DOX cytotoxicity.
It is further possible that the more “DOX-rich” nanoparticles operated via a self-catalytic mechanism, in which any initial release of DOX caused the production of ROS species which in turn helped to cleave the oxidation-sensitive cross-links in the NPs, thus releasing more DOX. Confirming such a hypothesis would require in situ sensing of ROS generation, which is beyond the scope of this initial investigation, but the observation of enhanced combination potency of synergistic ratios (10:1 and 1:1) of these drugs in a responsive NP formulation suggests a potential advantage for these systems in future clinical practice. The differing pharmacokinetics of DOX and OLA when administered as free drugs mean that simultaneous delivery to target cells may be difficult to achieve, and thus tumours will be exposed to a range of drug:drug ratios, some of which will be antagonistic. In principle, maintenance of synergistic combination ratios is inherently simpler when the drugs are incorporated within the oxidation-responsive nanoparticles, as release will occur only on cleavage of the thioketal link. In practice, it may be that esterase activity in vivo also degrades the NPs to accelerate drug release, but such processes should be slower than those of cross-linker cleavage mediated NP breakdown, thus the systemic release of drug from these NPs is still less likely than the intended intracellular release.
Conclusions
In summary, we have shown that specific ratios of a topoisomerase II inhibitor and a DNA-damage repair inhibitor can exhibit potent and synergistic cytotoxicity in triple negative breast cancer cells in both 2D and 3D cultures. We suggest that the overall effects of the drugs are not simply additive and may arise from a range of different mechanisms, including a potential role of DOX in DNA damage in addition to its well-established topoisomerase II activity. In combination with OLA this resulted in an increase in DNA strand breakage, apoptosis, and cell death in TNBC cell lines. Furthermore, delivery via a polymeric carrier with an oxidation-sensitive cross-linker resulted in a further increase in cytotoxicity in 3D spheroids at specific drug:drug ratios, which we suggest is a function of the different entry mechanisms for small molecule drugs compared to nanoparticles. These experiments thus establish a further rationale for drug:drug combinations in breast cancer therapies, the use of nanoparticle formulations to enable controlled delivery of specific synergistic drug ratios and the potential advantages of responsive nanoparticle carriers for co-delivery of these agents.
Materials and methods
Materials
Standard reagents, solvents monomers and other materials for chemical synthesis were purchased from Sigma-Aldrich or ThermoFisher. High glucose DMEM (phenol red free, cat. # 31053-028) was acquired from Gibco (Life technologies). High glucose DMEM (cat. # D6546), Trypsin–EDTA (cat. # T3924), foetal bovine serum (FBS, cat. # F7524), penicillin–streptomycin antibiotic solution (10000 U penicillin and 10 mg mL−1 streptomycin, cat. # P0781), L-glutamine (200 mm, cat. # G7513) and DMSO (cat. # D2438) were purchased from Sigma-Aldrich. Hoechst 33342 (cat. # H1399) and Presto Blue 10× solution (cat. # A13262 100 mL) were acquired from Invitrogen. Cy5 alkyne was purchased from Sigma-Aldrich.
Monomer, cross-linker and polymer synthesis
1H NMR (300 MHz, CDCl3): δ 2.91 (m, 2H), 2.68 (m, 2H), 1.60 (s, 6H).
Polymer synthesis
Drug-loaded nanoparticles preparation
For a typical preparation, mPEG-b-poly(D,D-lactide-co-α-N3ε caprolactone) polymer (100 mg, ∼0.0015 mmol), 3,3′-(propane-2,2-diylbis(sulfanediyl))bis(N-(prop-2-yn-1-yl)propanamide) (3.8 mg, 0.012 mmol), with doxorubicin and olaparib in the desired molar ratios (10:1, 1:1, 1:10), were dissolved in DMSO (3 mL) and stirred or sonicated until completely dissolved. A solution of ascorbic acid (0.58 mg, 0.0029 mmol) in double-distilled water (10 mL), previously degassed with argon bubbling, was added dropwise via a syringe pump to the DMSO with vigorous stirring followed by copper(II) sulphate (0.47 mg, 0.0029 mmol) in 3 mL double-distilled water. The vial was flushed with argon while stirring at 37 °C, and the reaction was stirred overnight at 37 °C. The copper sulphate and non-entrapped drugs were removed via dialysis against double-distilled water containing EDTA (in excess relative to the amount of copper added) for 24 h at room temperature. Total drug loadings varied between 3–3.7 wt%, as determined by HPLC methods.
Quantification of drug loading in NPs
Standard curves for doxorubicin and olaparib were prepared using British Pharmacopoeia (BP) Reference Standards for both drugs, and dual standards were made to quantify two drugs together. Stock standards of DOX and OLA were prepared in DMSO at a concentration of 1 mg mL−1, and then working standards of combined DOX and OLA were prepared using a diluent similar to the mobile phase at time of injection (see below). The concentration of each drug in the standards was 50, 25, 10, 7.5, 2.5, 1 and 0.5 μg mL−1; separate working standards of DOX and OLA were prepared at 10 μg mL−1 in the diluent (80:20, water/ACN, pH 6.8) for comparison. Polymer samples were prepared by resuspending the dried sample (typically 3.5 mg) in 1 mL diluent with sonication and vortexing. The resultant solution was filtered through a 0.25 μm filter tip to remove any remaining polymer. OLA and DOX concentrations were determined on an Agilent Technologies 1200 Series HPLC system using an ACE Excel 5 C18 column (250 × 4.6 mm id) maintained at 25 °C. A gradient mobile phase of 10 mM sodium phosphate buffer (pH = 6.8) and acetonitrile was employed at a flow rate of 1 mL min−1. 80% aqueous buffer was applied for 5 minutes, linearly decreased to 40% over 15 minutes, before returning to initial conditions over 2 minutes and holding for a further 8 minutes. For OLA, the eluent was monitored using a variable wavelength detector at 254 nm with a peak eluting at a retention time of 15.0 minutes, while for DOX detection, a fluorescence detector at 485/590 nm (ex/em) and a peak eluted at a retention time of 14.1 minutes.
Cell culture
All cells were purchased from the American Type Culture Collection (ATCC; Manassas, Virginia). MDA-MB-231, MCF7 and MDA-MB-468 cells were used in a passage window of 15 and cultured in DMEM (Sigma-Aldrich) supplemented with 10% (v/v) foetal bovine serum (FBS; Sigma-Aldrich) and 2 mM L-glutamine. Culture conditions were maintained at 37 °C with 5% CO2 and 90% relative humidity.Cytotoxicity experiments
Drug and nanoparticle potency in 2D cultured cells was evaluated with the PrestoBlue cell viability assay (Thermo Fisher Scientific). All cell types were seeded at a density of 1 × 104 cells per well in a 96 well plate and cultured for 24 hours prior to assaying. Doxorubicin and Olaparib were exposed to cells for 48 hours applied in 100 μL phenol red free DMEM containing 10% (v/v) FBS. Triton X-100, applied at 1% (v/v) in phenol red free DMEM, was used as a cell death (positive) control and a vehicle control containing no drug used as a negative control. Following exposure, cells were washed twice with PBS and 100 μL 10% (v/v) PrestoBlue reagent diluted in phenol red free medium applied per well for 60 min at 37 °C. The resulting fluorescence was measured at 560/600 nm (λex/λem). Relative metabolic activity was calculated by setting values from the negative control as 100% and positive control values as 0% metabolic activity.3D spheroid assays
Corning 7007 ultra-low attachment (ULA) 96-well round bottom plates were used to culture the 3D spheroids. MDA-MB-231 and MDA-MB-468 cells were seeded at 2000 cells per well in the presence of Cultrex basement membrane extract (Cultrex-BME, 100 μg mL−1), and MCF-7 cells seeded at 5000 cells per well in the absence of extract. The plates were centrifuged at 300 RCF for 5 min and cultured for 3 days until spheroid formation was confirmed by visual inspection. For dosing, drugs and drug loaded nanoparticles were applied in phenol red free DMEM containing 10% (v/v) FBS and incubated with spheroids for 72 hours. Spheroid volume analysis, from images obtained with a Nikon Ti Eclipse inverted microscope, utilized an open source macro for the Fiji distribution of ImageJ written by Ivanov et al.83 Cellular ATP levels were measured as a proxy of spheroid viability, after 72 hours treatment incubation using the CellTiter-Glo 3D assay (Promega). In these experiments, 150 μL of cell culture media was removed per well and 50 μL CellTiter-Glo 3D reagent added to the remaining 50 μL well volume. The contents of the plate were mixed vigorously for 5 minutes, and the plate was incubated for 25 minutes at room temperature. Following this luminescence was measured on a Tecan Spark 10M plate reader using an integration time of 5000 ms.Calculation of combination index (CI) values
CI values were determined using the widely established Chou and Talalay method.38,39 To determine each CI value, cytotoxicity studies performed in either 2D or 3D spheroid BCC culture were conducted on (A) DOX applied as a single drug, (B) OLA as a single drug and (C) DOX:OLA combinations at a series of molar ratios. IC50 values were subsequently calculated and CI values determined for each combination based on the Chou and Talalay formula, where DSD is the IC50 concentration of DOX, DSO is the IC50 concentration of OLA, DCD is the IC50 concentration of DOX in combination with OLA and DCO is the IC50 concentration of OLA in combination with DOX.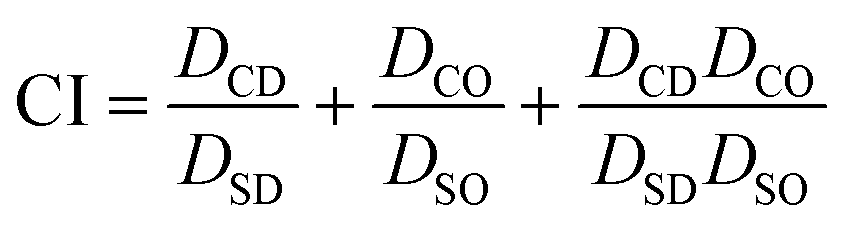
Using this method, CI values indicate synergy (<0.9), additive effect (0.9–0.1) or antagonism (>1.1).38,39 Graphpad prism (v7.05) was used to generate a heatmap using a three-colour system based on synergy represented by green, additive effects by yellow and antagonism by red.
Clonogenic cell survival assays
MCF-7 and MDA-MB-231 cells were seeded into 12 well plates at a density of 6 × 104 cells per well and cultured for 24 hours prior to treatment. Cells were exposed for 24 hours to doxorubicin (0.6–60 nM), olaparib (0.3–10 μM) or doxorubicin/olaparib combinations. Treatments were applied in DMEM containing 10% FBS, and this solution alone served as the vehicle control in all experiments. Cells were then harvested using trypsin, counted, and re-seeded at low density for colony formation in drug-free medium in 6 well plates. After 14 days colonies were fixed with 4% formalin solution in PBS, stained with 0.5% crystal violet and counted as a survivor if containing 50 or more cells. All experiments were performed at least thrice. Surviving fraction (SF) was calculated as: number of colonies/(number of cells plated × plating efficiency), where plating efficiency (PE) was defined as: number of control colonies obtained/number of control cells plated. The damage modification ratio (DMR) was calculated by dividing DOX concentration causing 90%, 50% or 10% cell survival in the absence of OLA treatment by the DOX concentration leading to 90%, 50% or 10% cell survival in the presence of OLA at set molar ratios (1:10, 1:1 or 10:1).
Immunofluorescence microscopy of γH2AX
MCF-7 and MDA-MB-231 cells were seeded on LabTek chamber slides (Thermo Fisher Scientific) with 3 × 104 per chamber well and cultured for 24 hours. Cells were then treated with doxorubicin, olaparib or combinations for 24 hours. Cells were fixed with 4% PFA for 15 minutes at room temperature, permeabilised with 0.1% (v/v) Triton X-100 for 15 minutes at room temperature and then blocked with 1.5% (v/v) bovine serum albumin (BSA) in PBS for 60 minutes at room temperature. Cells were then incubated for 2 hours at room temperature with (1:1000) mouse monoclonal anti-gH2AX/ser139, washed thrice with PBS and then incubated with (1:500) goat anti-mouse IgG AlexaFluor 488 for 1 hour at room temperature. The camber slides were also stained with 0.5 μg mL−1 DAPI to visualise nuclei. Slides were washed twice with PBS and then mounted with coverslips and ProLong™ Gold Antifade Mountant. Microscopy was performed using an EVOS M5000 fluorescent microscope, with subsequent image analysis and counting of foci using ImageJ software (v1.5).
Mitochondrial membrane potential
Alterations in mitochondrial membrane potential were assessed by a JC-1 (Biotium) aggregation assay. MDA-MB-231 cells were seeded in 96 well plates at a density of 1 × 104 cells per well, cultured for 24 hours and then incubated with doxorubicin (1 μM), olaparib (0.1, 1.0 or 10.0 μM), or combinations for 5–60 minutes. Following treatment cells were incubated with 5 μg mL−1 JC-1 dye for 15 minutes at 37 °C. Fluorescence was read at 550/600 nm (λex/λem) for detection of JC-1 aggregates, and 485/535 nm (λex/λem) for detection of JC-1 monomers. J-aggregate:monomer ratios were then normalized to values induced by the untreated control (set to a value of 1). For JC-1 imaging, live cell fluorescent microscopy was performed on a Nikon Ti Eclipse inverted microscope.
Intracellular ROS detection
MDA-MB-231 cells were seeded in 96 well plates with 1 × 104 cells per well, cultured for 24 hours, followed by incubation with doxorubicin (1 μM), olaparib (0.1, 1.0 or 10.0 μM), or combinations for 5–60 minutes. Intracellular ROS levels were then assessed using 10 μM CM-H2DCFDA probe (Thermo Fisher Scientific), incubated with cells for 30 minutes at 37 °C and applied in HBSS. Resulting fluorescence was measured at 492/520 nm (λex/λem). Measured values were then normalized to the untreated control (set as a value of 1).Detection of activated caspase-3/7
The CellEvent ® caspase-3/7 green detection reagent (Thermo Fisher Scientific) was employed to evaluate levels of activated caspase-3 or 7. MDA-MB-231 cells were cultured and treated as described above in 96 well plates. After treatment, 100 μl 2% (v/v) CellEvent probe in PBS was applied per well for 30 minutes at 37 °C. Fluorescent intensity was measured at 502/530 nm (λex/λem) and normalized to the untreated control (set as a value of 1).Live cell fluorescence microscopy
To assess cellular uptake of Cy5-NP, live cell fluorescence microscopy was used to image MDA-MB-231, MDA-MB-468 and MCF-7 cells. All cells were seeded in CellView™ 35 mm diameter glass-bottom cell culture dishes at a density of 2.5 × 105 cells per dish and cultured for 24 hours. Cy5-NPs (50 μg mL−1) were incubated with cells for 30, 60 and 120 minutes at 37 °C with 5% CO2. Following exposure, NP solutions were removed, and cells washed three times with ice-cold PBS. Cells were then stained with 10 μg mL−1 Hoechst 33342 (Thermo-Fisher) or 10 μg mL−1 Hoechst 33342 and 50 nM Lysotracker green DND-26 (Thermo-Fisher) applied in Hank's Balanced Salt Solution (HBSS) for 30 min. Staining solution was removed and cells washed twice with PBS. FluoroBrite DMEM was added to wells and cells were imaged on an inverted Nikon Eclipse TE 300 fluorescent microscope on DAPI, FITC and Cy5 filters. Images were processed using ImageJ software (1.52f).Conflicts of interest
The authors declare no conflicts of interest. Marianne Ashford and Patricia Monteiro are employees and shareholders of AstraZeneca.Acknowledgements
This work was supported by the Engineering and Physical Sciences Research Council (EPSRC)-UKRI [grant numbers EP/N006615/1; EP/N03371X/1]. This work was also funded by the Royal Society [Wolfson Research Merit Award WM150086] to CA and The Little Princess Trust in partnership with CCLG. The authors thank the Nanoscale and Microscale Research Centre (nmRC) for providing access to instrumentation, Paul Cooling and Douglas Crackett for expert technical help, and Sian Rankin-Turner and Carol Turrill for excellent administrative support. We also thank the School of Life Sciences imaging facility (SLIM) and their staff, particularly Dr Tim Self, for use of their facilities.References
- N. Harbeck, F. Penault-Llorca, J. Cortes, M. Gnant, N. Houssami, P. Poortmans, K. Ruddy, J. Tsang and F. Cardoso, Nat. Rev. Dis. Primers, 2019, 5, 66 CrossRef PubMed.
- G. Bianchini, C. De Angelis, L. Licata and L. Gianni, Nat. Rev. Clin. Oncol., 2022, 19, 91–113 CrossRef CAS PubMed.
- W. D. Foulkes, I. E. Smith and J. S. Reis-Filho, N. Engl. J. Med., 2010, 363, 1938–1948 CrossRef CAS PubMed.
- P. Boyle, Ann. Oncol., 2012, 23(Suppl 6), vi7–v12 CrossRef PubMed.
- G. Stockmans, K. Deraedt, H. Wildiers, P. Moerman and R. Paridaens, Curr. Opin. Oncol., 2008, 20, 614–620 CrossRef PubMed.
- M. De Laurentiis, D. Cianniello, R. Caputo, B. Stanzione, G. Arpino, S. Cinieri, V. Lorusso and S. De Placido, Cancer Treat. Rev., 2010, 36(Suppl 3), S80–S86 CrossRef CAS PubMed.
- G. Bianchini, J. M. Balko, I. A. Mayer, M. E. Sanders and L. Gianni, Nat. Rev. Clin. Oncol., 2016, 13, 674–690 CrossRef CAS PubMed.
- K. R. Bauer, M. Brown, R. D. Cress, C. A. Parise and V. Caggiano, Cancer, 2007, 109, 1721–1728 CrossRef PubMed.
- B. A. Kohler, R. L. Sherman, N. Howlader, A. Jemal, A. B. Ryerson, K. A. Henry, F. P. Boscoe, K. A. Cronin, A. Lake, A. M. Noone, S. J. Henley, C. R. Eheman, R. N. Anderson and L. Penberthy, J. Natl. Cancer Inst., 2015, 107, djv048 CrossRef PubMed.
- Y. Barenholz, J. Controlled Release, 2012, 160, 117–134 CrossRef CAS PubMed.
- J. K. Sicklick, S. Kato, R. Okamura, M. Schwaederle, M. E. Hahn, C. B. Williams, P. De, A. Krie, D. E. Piccioni, V. A. Miller, J. S. Ross, A. Benson, J. Webster, P. J. Stephens, J. J. Lee, P. T. Fanta, S. M. Lippman, B. Leyland-Jones and R. Kurzrock, Nat. Med., 2019, 25, 744–750 CrossRef CAS PubMed.
- J. A. Perez-Fidalgo, A. Cortés, E. Guerra, Y. García, M. Iglesias, U. Bohn Sarmiento, E. Calvo García, L. Manso Sánchez, A. Santaballa, A. Oaknin, A. Redondo, M. J. Rubio and A. González-Martín, ESMO Open, 2021, 6, 100212 CrossRef CAS PubMed.
- N. D. Donahue, H. Acar and S. Wilhelm, Adv. Drug Delivery Rev., 2019, 143, 68–96 CrossRef CAS PubMed.
- E. V. Batrakova, S. Li, A. M. Brynskikh, A. K. Sharma, Y. Li, M. Boska, N. Gong, R. L. Mosley, V. Y. Alakhov, H. E. Gendelman and A. V. Kabanov, J. Controlled Release, 2010, 143, 290–301 CrossRef CAS PubMed.
- F. Greco and M. J. Vicent, Adv. Drug Delivery Rev., 2009, 61, 1203–1213 CrossRef CAS PubMed.
- M. J. Vicent, F. Greco, R. I. Nicholson, A. Paul, P. C. Griffiths and R. Duncan, Angew. Chem., Int. Ed., 2005, 44, 4061–4066 CrossRef CAS PubMed.
- S. Ghosh, R. Lalani, V. Patel, D. Bardoliwala, K. Maiti, S. Banerjee, S. Bhowmick and A. Misra, J. Controlled Release, 2019, 296, 114–139 CrossRef CAS PubMed.
- S. Rawal and M. M. Patel, J. Controlled Release, 2019, 301, 76–109 CrossRef CAS PubMed.
- Q. Zhang, G. Xiang, Y. Zhang, K. Yang, W. Fan, J. Lin, F. Zeng and J. Wu, J. Pharm. Sci., 2006, 95, 2266–2275 CrossRef CAS PubMed.
- K. W. Wang, M. L. Jiang, J. L. Zhou, Y. S. Dong, Y. Liu, Q. Y. Zong, P. Sandbhor, N. D. P. Singh and Y. Y. Yuan, J. Controlled Release, 2022, 348, 1004–1015 CrossRef CAS PubMed.
- D. Zhou, Y. Cong, Y. Qi, S. He, H. Xiong, Y. Wu, Z. Xie, X. Chen, X. Jing and Y. Huang, Biomater. Sci., 2015, 3, 182–191 RSC.
- A. C. Begg, F. A. Stewart and C. Vens, Nat. Rev. Cancer, 2011, 11, 239–253 CrossRef CAS PubMed.
- H. Lu, D. Samanta, L. Xiang, H. Zhang, H. Hu, I. Chen, J. W. Bullen and G. L. Semenza, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E4600–E4609 CrossRef CAS PubMed.
- T. Miran, A. T. J. Vogg, N. Drude, F. M. Mottaghy and A. Morgenroth, FASEB J., 2018, 32, 2803–2813 CrossRef PubMed.
- A. Beatty, L. S. Fink, T. Singh, A. Strigun, E. Peter, C. M. Ferrer, E. Nicolas, K. Q. Cai, T. P. Moran, M. J. Reginato, U. Rennefahrt and J. R. Peterson, Mol. Cancer Ther., 2018, 17, 264–275 CrossRef CAS PubMed.
- F. L. Sarmiento-Salinas, A. Delgado-Magallon, J. B. Montes-Alvarado, D. Ramirez-Ramirez, J. C. Flores-Alonso, P. Cortes-Hernandez, J. Reyes-Leyva, I. Herrera-Camacho, M. Anaya-Ruiz, R. Pelayo, L. Millan-Perez-Pena and P. Maycotte, Front. Oncol., 2019, 9, 480 CrossRef PubMed.
- G. Barrera, ISRN Oncol., 2012, 2012, 137289 Search PubMed.
- I. I. C. Chio and D. A. Tuveson, Trends Mol. Med., 2017, 23, 411–429 CrossRef CAS PubMed.
- K. D. Tew and D. M. Townsend, Curr. Opin. Chem. Biol., 2011, 15, 156–161 CrossRef CAS PubMed.
- Y. Zhang and S. G. Martin, Clin. Oncol., 2014, 26, 289–300 CrossRef CAS PubMed.
- S. Cajot, D. Schol, F. Danhier, V. Preat, M. C. Gillet De Pauw and C. Jerome, Macromol. Biosci., 2013, 13, 1661–1670 CrossRef CAS PubMed.
- C. Lin, Z. Zhong, M. C. Lok, X. Jiang, W. E. Hennink, J. Feijen and J. F. J. Engbersen, J. Controlled Release, 2006, 116, 130–137 CrossRef CAS PubMed.
- G. Saito, J. A. Swanson and K.-D. Lee, Adv. Drug Delivery Rev., 2002, 55, 199–215 CrossRef PubMed.
- C. Lin, Z. Zhong, M. C. Lok, X. Jiang, W. E. Hennink, J. Feijen and J. F. J. Engbersen, Bioconjugate Chem., 2007, 18, 138–145 CrossRef CAS PubMed.
- C. Sun, Z. Wang, Z. Wang, L. Yue, Q. Cheng, Z. Ye, Q.-W. Zhang and R. Wang, Biomater. Sci., 2021, 9, 1355–1362 RSC.
- L. Yu, M. Zhang, F.-S. Du and Z.-C. Li, Polym. Chem., 2018, 9, 3762–3773 RSC.
- C. J. Lovitt, T. B. Shelper and V. M. Avery, BMC Cancer, 2018, 18, 41 CrossRef PubMed.
- T. C. Chou, Cancer Res., 2010, 70, 440–446 CrossRef CAS PubMed.
- T. C. Chou, Pharmacol. Rev., 2006, 58, 621–681 CrossRef CAS PubMed.
- E. Christidi and L. R. Brunham, Cell Death Dis., 2021, 12, 339 CrossRef CAS PubMed.
- A. M. Cseh, Z. Fabian, R. Quintana-Cabrera, A. Szabo, K. Eros, M. E. Soriano, F. Gallyas, L. Scorrano and B. Sumegi, Front. Physiol., 2019, 10, 538 CrossRef PubMed.
- D. B. Zorov, M. Juhaszova and S. J. Sollott, Biochim. Biophys. Acta, 2006, 1757, 509–517 CrossRef CAS PubMed.
- R. Riva, S. Schmeits, F. Stoffelbach, C. Jerome, R. Jerome and P. Lecomte, Chem. Commun., 2005, 5334–5336, 10.1039/B510282K.
- P. F. Monteiro, M. Gulfam, C. J. Monteiro, A. Travanut, T. F. Abelha, A. K. Pearce, C. Jerome, A. M. Grabowska, P. A. Clarke, H. M. Collins, D. M. Heery, P. Gershkovich and C. Alexander, J. Controlled Release, 2020, 323, 549–564 CrossRef CAS PubMed.
- N. Zheng, D. Xie, Z. Y. Zhang, J. Kuang, Y. B. Zheng, Q. Wang and Y. Li, J. Biomater. Appl., 2019, 34, 326–338 CrossRef CAS PubMed.
- F. Sodano, R. J. Cavanagh, A. K. Pearce, L. Lazzarato, B. Rolando, A. Fraix, T. F. Abelha, C. E. Vasey, C. Alexander, V. Taresco and S. Sortino, Biomater. Sci., 2020, 8, 1329–1344 RSC.
- H. H. Lee, B. F. Leake, R. B. Kim and R. H. Ho, Mol. Pharmacol., 2017, 91, 14–24 CrossRef CAS PubMed.
- M. Okabe, M. Unno, H. Harigae, M. Kaku, Y. Okitsu, T. Sasaki, T. Mizoi, K. Shiiba, H. Takanaga, T. Terasaki, S. Matsuno, I. Sasaki, S. Ito and T. Abe, Biochem. Biophys. Res. Commun., 2005, 333, 754–762 CrossRef CAS PubMed.
- V. Taresco, T. F. Abelha, R. J. Cavanagh, C. E. Vasey, A. B. Anane-Adjei, A. K. Pearce, P. F. Monteiro, K. A. Spriggs, P. Clarke, A. Ritchie, S. Martin, R. Rahman, A. M. Grabowska, M. B. Ashford and C. Alexander, Adv. Ther., 2020, 4, 2000103 CrossRef.
- S. Eetezadi, J. C. Evans, Y. T. Shen, R. De Souza, M. Piquette-Miller and C. Allen, Mol. Pharm., 2018, 15, 472–485 CrossRef CAS PubMed.
- A. Dicko, L. D. Mayer and P. G. Tardi, Expert Opin. Drug Delivery, 2010, 7, 1329–1341 CrossRef CAS PubMed.
- J. Staaf, D. Glodzik, A. Bosch, J. Vallon-Christersson, C. Reuterswärd, J. Häkkinen, A. Degasperi, T. D. Amarante, L. H. Saal, C. Hegardt, H. Stobart, A. Ehinger, C. Larsson, L. Rydén, N. Loman, M. Malmberg, A. Kvist, H. Ehrencrona, H. R. Davies, Å. Borg and S. Nik-Zainal, Nat. Med., 2019, 25, 1526 CrossRef CAS PubMed.
- N. C. Turner, N. Engl. J. Med., 2017, 377, 2490–2492 CrossRef PubMed.
- P. Polak, J. Kim, L. Z. Braunstein, R. Karlic, N. J. Haradhavala, G. Tiao, D. Rosebrock, D. Livitz, K. Kübler, K. W. Mouw, A. Kamburov, Y. E. Maruvka, I. Leshchiner, E. S. Lander, T. R. Golub, A. Zick, A. Orthwein, M. S. Lawrence, R. N. Batra, C. Caldas, D. A. Haber, P. W. Laird, H. Shen, L. W. Ellisen, A. D. D’Andrea, S. J. Chanock, W. D. Foulkes and G. Getz, Nat. Genet., 2017, 49, 1476–1486 CrossRef CAS PubMed.
- B. Kaufman, R. Shapira-Frommer, R. K. Schmutzler, M. W. Audeh, M. Friedlander, J. Balmaña, G. Mitchell, G. Fried, S. M. Stemmer, A. Hubert, O. Rosengarten, M. Steiner, N. Loman, K. Bowen, A. Fielding and S. M. Domchek, J. Clin. Oncol., 2015, 33, 244–250 CrossRef CAS PubMed.
- M. Robson, S. A. Im, E. Senkus, B. H. Xu, S. M. Domchek, N. Masuda, S. Delaloge, W. Li, N. Tung, A. Armstrong, W. T. Wu, C. Goessl, S. Runswick and P. Conte, N. Engl. J. Med., 2017, 377, 523–533 CrossRef CAS PubMed.
- M. E. Robson, S. A. Im, E. Senkus, B. H. Xu, S. M. Domchek, N. Masuda, S. Delaloge, N. D. E. Tung, A. Armstrong, M. Dymond, A. Fielding, A. Allen and P. Conte, Eur. J. Cancer, 2023, 184, 39–47 CrossRef CAS PubMed.
- J. J. J. Geenen, S. C. Linn, J. H. Beijnen and J. H. M. Schellens, Clin. Pharmacokinet., 2018, 57, 427–437 CrossRef CAS PubMed.
- G. Mariano, M. R. Ricciardi, D. Trisciuoglio, M. Zampieri, F. Ciccarone, T. Guastafierro, R. Calabrese, E. Valentini, A. Tafuri, D. Del Bufalo, P. Caiafa and A. Reale, Oncotarget, 2015, 6, 15008–15021 CrossRef PubMed.
- K. R. Fischer, A. Durrans, S. Lee, J. Sheng, F. Li, S. T. Wong, H. Choi, T. El Rayes, S. Ryu, J. Troeger, R. F. Schwabe, L. T. Vahdat, N. K. Altorki, V. Mittal and D. Gao, Nature, 2015, 527, 472–476 CrossRef CAS PubMed.
- X. Zheng, J. L. Carstens, J. Kim, M. Scheible, J. Kaye, H. Sugimoto, C. C. Wu, V. S. LeBleu and R. Kalluri, Nature, 2015, 527, 525–530 CrossRef CAS PubMed.
- T. Xu, J. Zhang, W. Chen, S. Pan, X. Zhi, L. Wen, Y. Zhou, B. W. Chen, J. Qiu, Y. Zhang, Q. Yang, X. Feng, X. Bai and T. Liang, Cancer Lett., 2016, 377, 140–148 CrossRef CAS PubMed.
- M. El Amrani, F. Corfiotti, M. Corvaisier, R. Vasseur, M. Fulbert, C. Skrzypczyk, A. C. Deshorgues, V. Gnemmi, D. Tulasne, F. Lahdaoui, A. Vincent, F. R. Pruvot, I. Van Seuningen, G. Huet and S. Truant, Mol. Carcinog., 2019, 58, 1985–1997 CrossRef CAS PubMed.
- X. Wan, J. Hou, S. Liu, Y. Zhang, W. Li, Y. Zhang and Y. Ding, Front. Cell Dev. Biol., 2021, 9, 583572 CrossRef PubMed.
- M. Schacke, J. Kumar, N. Colwell, K. Hermanson, G. A. Folle, S. Nechaev, A. Dhasarathy and L. Lafon-Hughes, Int. J. Mol. Sci., 2019, 20, 518 CrossRef CAS PubMed.
- L. J. Kuo and L. X. Yang, In Vivo, 2008, 22, 305–309 CAS.
- D. A. Gewirtz, Biochem. Pharmacol., 1999, 57, 727–741 CrossRef CAS PubMed.
- F. Yang, S. S. Teves, C. J. Kemp and S. Henikoff, Biochim. Biophys. Acta, 2014, 1845, 84–89 CAS.
- C. F. Thorn, C. Oshiro, S. Marsh, T. Hernandez-Boussard, H. McLeod, T. E. Klein and R. B. Altman, Pharmacogenet. Genomics, 2011, 21, 440–446 CrossRef CAS PubMed.
- A. Ray Chaudhuri and A. Nussenzweig, Nat. Rev. Mol. Cell Biol., 2017, 18, 610–621 CrossRef CAS PubMed.
- J. Murai, S. Y. Huang, B. B. Das, A. Renaud, Y. Zhang, J. H. Doroshow, J. Ji, S. Takeda and Y. Pommier, Cancer Res., 2012, 72, 5588–5599 CrossRef CAS PubMed.
- U. A. Matulonis and B. J. Monk, Ann. Oncol., 2017, 28, 443–447 CrossRef CAS PubMed.
- B. S. Chhikara, D. Mandal and K. Parang, J. Med. Chem., 2012, 55, 1500–1510 CrossRef CAS PubMed.
- W. L. Ye, J. B. Du, B. L. Zhang, R. Na, Y. F. Song, Q. B. Mei, M. G. Zhao and S. Y. Zhou, PLoS One, 2014, 9, e97358 CrossRef PubMed.
- C. Cui, Y. N. Xue, M. Wu, Y. Zhang, P. Yu, L. Liu, R. X. Zhuo and S. W. Huang, Biomaterials, 2013, 34, 3858–3869 CrossRef CAS PubMed.
- W. Chen, P. Zhong, F. Meng, R. Cheng, C. Deng, J. Feijen and Z. Zhong, J. Controlled Release, 2013, 169, 171–179 CrossRef CAS PubMed.
- M. Gulfam, T. Matini, P. F. Monteiro, R. Riva, H. Collins, K. Spriggs, S. M. Howdle, C. Jerome and C. Alexander, Biomater. Sci., 2017, 5, 532–550 RSC.
- C. J. Bowerman, J. D. Byrne, K. S. Chu, A. N. Schorzman, A. W. Keeler, C. A. Sherwood, J. L. Perry, J. C. Luft, D. B. Darr, A. M. Deal, M. E. Napier, W. C. Zamboni, N. E. Sharpless, C. M. Perou and J. M. DeSimone, Nano Lett., 2017, 17, 242–248 CrossRef CAS PubMed.
- G. Sahay, E. V. Batrakova and A. V. Kabanov, Bioconjugate Chem., 2008, 19, 2023–2029 CrossRef CAS PubMed.
- T. Minko, E. V. Batrakova, S. Li, Y. L. Li, R. I. Pakunlu, V. Y. Alakhov and A. V. Kabanov, J. Controlled Release, 2005, 105, 269–278 CrossRef CAS PubMed.
- H. L. Wong, R. Bendayan, A. M. Rauth, H. Y. Xue, K. Babakhanian and X. Y. Wu, J. Pharmacol. Exp. Ther., 2006, 317, 1372–1381 CrossRef CAS PubMed.
- S. Lenoir, R. Riva, X. Lou, C. Detrembleur, R. Jérôme and P. Lecomte, Macromolecules, 2004, 37, 4055–4061 CrossRef CAS.
- D. P. Ivanov, A. M. Grabowska and M. C. Garnett, Methods Mol. Biol., 2017, 1601, 43–59 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3bm01931d |
| This journal is © The Royal Society of Chemistry 2024 |